

Abstract
Ligands of the tumor necrosis factor superfamily (TNFSF) are appealing targets for immunotherapy research due to their integral involvement in stimulation or restriction of immune responses. TNFSF-targeted therapies are currently being developed to combat immunologically based diseases and cancer. A crucial determinant of effective TNFSF receptor binding and signaling is the trimeric quaternary structure of the ligand. Additionally, ligand multivalency is essential to propagate strong signaling in effector cells. Thus, designing a synthetic platform to display trimeric TNFSF ligands in a multivalent manner is necessary to further the understanding of ligand–receptor interactions. Viral nanocages have architectures that are amenable to genetic and chemical modifications of both their interior and exterior surfaces. Notably, the exterior surface of virus-like particles can be utilized as a platform for the modular multivalent presentation of target proteins. In this study, we build on previous efforts exploring the bacteriophage P22 virus-like particle for the exterior multivalent modular display of a potent immune-stimulating TNFSF protein, CD40 ligand (CD40L). Using a cell-based reporter system, we quantify the effects of tunable avidity on CD40 signaling by CD40L displayed on the surface of P22 nanocages. Multivalent presentation of CD40L resulted in a 53.6-fold decrease of the half maximal effective concentration (EC50) compared to free CD40L, indicating higher potency. Our results emphasize the power of using P22-based biomimetics to study ligand–receptor interactions within their proper structural context, which may contribute to the development of effective immune modulators.
Keywords: CD40L, CD40, multivalency, P22, VLP, signaling, biomimetics, TNFSF
Graphical Abstract

INTRODUCTION
Cell surface receptors are responsible for interacting with the extracellular environment and relaying information to the cytoplasmic signaling machinery. Structural elements of the interacting moieties including multivalency, proximity, shape, and availability determine the avidity of ligand–receptor interactions and ultimately the intensity of signal transduction. Binding affinity refers to the strength of the individual interaction of a ligand to its receptor, whereas binding avidity characterizes the overall strength of the collective affinities of multiple ligand–receptor binding partners. Cell signaling often requires multiple simultaneous interactions to effectively transmit a response, emphasizing the importance of ligand–receptor avidity.1 The ability to control the degree of ligand valency would enable manipulation of avidity as well as the duration and intensity of cellular signals. Because signaling proteins can become markedly less effective when taken out of their native multivalent structural context, controlling avidity in designed receptor–agonist systems is vital to achieve desirable levels of activity.2
Controlling target cellular signaling pathways for therapeutic uses is an extremely appealing strategy for addressing diseases linked to an inappropriately balanced immune system such as autoimmune diseases and cancer. Many targeted pathways involve proteins from the tumor necrosis factor superfamily (TNFSF), which consists of 19 ligands with various roles in cell proliferation, maturation, and death, particularly within immune processes.3–6 TNFSF members are type II membrane proteins that naturally exist as noncovalent homotrimers with an extracellular domain that assumes a conserved triangular pyramid-like structure.7 When in their natural membrane-bound trimeric form, these proteins can be potent modulators of immunity and controlled cell death. Furthermore, structure–function studies have demonstrated that a highly ordered geometric arrangement of TNFSF ligands and their receptors is required to activate signaling.8 The TNFSF member CD40 ligand (CD40L; also known as GP39, CD154, TRAP, TBAM) is a potent immunological costimulatory protein. For example, the interaction of CD40L on a CD4+ T cell with its receptor CD40 on a B cell is necessary for the maturation and antibody isotype switching of the B cell, leading to proliferation, differentiation, and survival of both plasma cells and memory B cells (Figure 1).9,10 Research continues to emerge that also implicates CD40L as a key regulator of T cells and antigen-presenting cells (APC) such as dendritic cells (DC) and macrophages.11–14 Because of its crucial positions at the gateway between the innate and adaptive immune systems and at the interface between tolerance and immunity, CD40L has become a promising candidate for applications as an adjuvant in vaccinations, as a potent immune enhancer against intracellular pathogens, and in cancer immunotherapy.3,5,15–19 However, the efficacies of some current CD40L-based therapeutics are limited by the lack of multivalency necessary for effective cellular signaling.17
Figure 1.

CD40L is at the gateway between the innate and adaptive immune systems. Antigen is processed by an antigen-presenting cell (APC), such as a B cell or a dendritic cell (DC). Peptides derived from the antigen are presented via major histocompatibility complex (MHC) II cell surface proteins, which engage a specific CD4+ T cell through its T cell receptor (TCR). The CD40L–CD40 costimulatory interaction triggers the APC to mature and become activated, leading to an effective adaptive immune response.
The receptors for TNFSF ligands are type 1 membrane proteins that naturally exist in a monomeric unbound state and rarely function as individual entities, instead requiring multimerization for effector functionality.20 The engagement of the trimeric ligand drives trimerization of the receptors and allows for signaling. The intensity of the signal is dictated by the number of ligand–receptor contacts between the participant cells, with efficient receptor signaling requiring the triggering of receptor multimers to cluster within the membranes of responding cells. These multireceptor signaling complexes foster compartmentalization of a subset of intracellular signal transduction proteins and likely prolong ligand binding, thus achieving spatial and temporal regulation. This is the case for the CD40–CD40L interaction with CD40L driving the trimerization and clustering of its cognate receptor (CD40) allowing for effective signaling with intensity being dependent on multivalency of the cell surface interactions.17,21–23 The need for multiple contacts within the close proximity on a cell membrane is fulfilled by the appropriate structural context of CD40L, specifically its trimeric quaternary structure, multivalency, C3 symmetry, and radial distribution of the CD40 binding motif.21,24,25 The binding of CD40 to structurally appropriate CD40L results in activation such as target gene expression.17,26–29 The importance of quaternary structure and valency is a common theme for TNFSF ligands as is demonstrated by B cell activating factor (BAFF), which naturally forms multivalent structures when its soluble signaling domain assembles into icosahedral cage architectures upon proteolytic cleavage.30
Efforts are ongoing to investigate CD40 agonists, such as CD40-stimulating antibodies and soluble CD40L, as alternatives to natural membrane-bound CD40L for immunological activation.31 Agonistic CD40 antibodies have been shown to induce an immune response by partial CD40 stimulation; however, the nature of bivalent antibody binding limits signaling since antibodies alone do not promote receptor clustering unless tethered and multivalent (e.g., bound to the surface of a cell).17,32 Soluble trimeric CD40L has lower avidity to CD40 than membrane-bound ligand trimers due to lack of multivalency leading to little or no CD40 clustering on effector cells.5,23,33 Previous efforts to increase the signaling efficiency of recombinant CD40L have utilized strategies to stabilize the trimeric structure of CD40L and increase valency with the highest valency reached being a display of four trimers of CD40L.23,33–38 Although these recombinant CD40L constructs lead to enhanced signaling, cytotoxic effects at higher doses are a concern.17,37,39,40 Naturally occurring monovalent CD40L may serve as a decoy ligand that competes with membrane-bound CD40L and could have a role in human carcinogenesis, which may ultimately limit the therapeutic usefulness of low-valency CD40L.41
A potential solution to overcome these limitations is to develop a highly multivalent, rigid platform for CD40L presentation. Virus-like particles (VLPs) provide regular multivalent scaffolds for the display of target proteins.42–45 Here, we utilize the viral capsid derived from bacteriophage P22 for the modular display of human CD40L (hCD40L). Figure 2 illustrates the display of Dec-fused hCD40L (DechCD40L) on P22 and the interaction of P22-bound DechCD40L with cell membrane-bound receptors (CD40).
Figure 2.
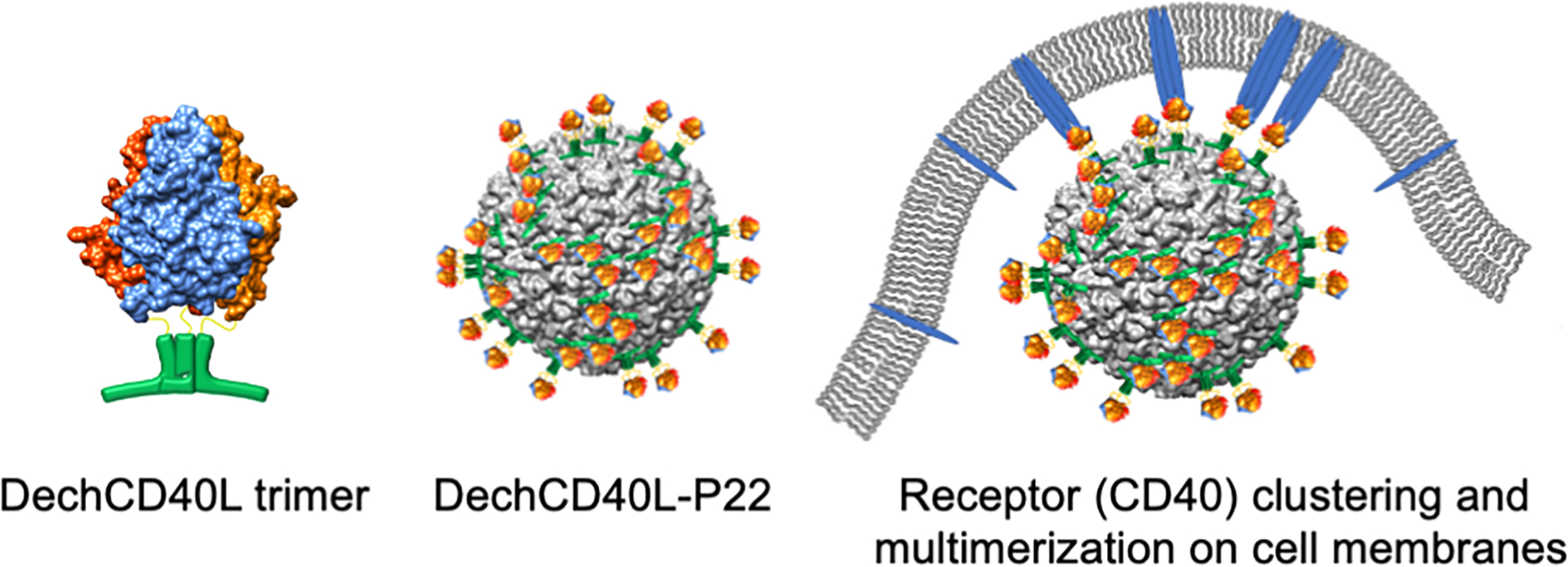
Strategy for multivalent DechCD40L presentation mediated by Dec binding to P22. Dec is genetically fused to hCD40L resulting in a soluble fusion protein, DechCD40L, which self-trimerizes and can be bound to P22 by simply mixing. The multivalent nature of the bound cargo allows for cross-linking of CD40 receptors on the surface of effector cells.
We previously demonstrated that the soluble region of murine CD40L can be bound to the P22 bacteriophage VLP via fusion with a decoration protein (Dec) derived from bacteriophage L.46–48 Dec only binds to the surface of P22 at specific sites formed during capsid expansion. The morphological change from a malleable and dynamic procapsid P22 to a rigid and angular expanded form can be achieved by heating. Dec binds to expanded P22 as a trimer at 60 symmetrically equivalent high-affinity sites located at the quasi threefold positions and 20 low-affinity sites located at the true threefold positions of the icosahedral capsid (Figure S1).49–51 Throughout the rest of this study, all reference to P22 refer to the expanded form unless specifically indicated. Previous structural studies examining the binding of Dec to the capsid have revealed that the trimeric protein is oriented such that the C-termini of the three monomers protrude away from the capsid as a cluster and thus provide an ideal site for the genetic fusion of C3 symmetric trimers for multivalent presentation.50
Genetic fusion of murine CD40L to Dec did not alter the affinity and stoichiometry of Dec binding to the capsid. Importantly, fused CD40L retained the ability to bind CD40 on the surface of primary murine B cells.46 While these data demonstrated that CD40L fused to Dec can be displayed multivalently, retain its quaternary structure, and bind its receptor, further examination is needed to quantify the avidity advantage of the system and characterize the structural determinants of these advantages. Here, we demonstrate the successful fusion of the soluble region of human CD40L to Dec, hereto referred to as DechCD40L. We examine the avidity behavior of DechCD40L displayed on P22 and show that occupancy on P22 can be controlled in a tunable fashion. Through cryo-transmission electron microscopy image reconstruction, we confirm that conjugated hCD40L is directed away from the capsid in a readily accessible position for receptor binding. Utilizing a model human cell line expressing CD40, we show that hCD40L presented on P22 at a stoichiometric ratio of 60 trimers/capsid results in more potent signaling capability, reflected by a 53.6 lower half maximal effective concentration (EC50) compared to free soluble CD40L. We also demonstrate that varying the degree of multivalency translates into changes in the level of cellular activation. Together, our data indicate that the DechCD40L-P22 system can be used to effectively amplify and tune the activity of CD40 through the multivalent presentation of CD40L with the correct quaternary structure. We report a simple system by which to study CD40L-CD40 signaling and to assess its dependency on CD40L valency. In a broader view, these results encourage further research regarding the potential for presentation and probing the effects of varying valency of other TNFSF members in the same manner.
RESULTS AND DISCUSSION
Expression of hCD40L Fused to Dec.
Based on previously demonstrated design principles for the fusion of murine CD40L to Dec, a codon-optimized gene for the extracellular region of human CD40L (hCD40L) was fused to the C-terminus of the Dec gene separated by a five amino acid (aa) linker consisting of flexible glycines and serines to form a continuous gene encoding DechCD40L (Figure 3). Expression of DechCD40L and purification via an N-terminal 6x histidine tag resulted in isolation of a protein that runs on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) near the expected molecular weight of 32.7 kDa for DechCD40L (Figure 4).
Figure 3.

Design of DechCD40L. N-terminus of the soluble portion of the human CD40L was fused to a flexible linker (GSSGS) preceded by Dec and a 6x histidine tag. Expressed DechCD40L self-assembles into homotrimers.
Figure 4.

DechCD40L can be purified as a soluble product and readily binds to P22. DechCD40L or Dec was mixed with P22 for 30 min at a ratio of 240 trimers per capsid followed by ultracentrifugation. Proteins successfully bound to P22 were detected by denaturing SDS polyacrylamide gel electrophoresis and Western blot analysis. (A) SDS-PAGE of nickel-affinity purified DechCD40L displaying a major band for DechCD40L near the expected molecular weight of 32.7 kDa. (B) SDS-PAGE of DechCD40L or Dec bound to P22. Arrows point to bands at the expected molecular weights for DechCD40L (32.7 kDa), P22 coat protein (CP, 47 kDa), and Dec (14.1 kDa). (C) Western blot analysis of DechCD40L bound to P22 compared to Dec bound to P22. The left blot is developed with an anti-P22 polyclonal mixture. The right blot is developed with an anti-Dec polyclonal mixture. (D) Purified DechCD40L developed with monoclonal anti-hCD40L. Soluble hCD40L is included for reference.
The molecular weight and quaternary structure of DechCD40L oligomers were assessed by size-exclusion chromatography (SEC). We observed two elution peaks that could not be independently resolved (Figure S2). The retention volume of DechCD40L, plotted against known molecular weight standards, indicates that the main DechCD40L peak corresponds to a DechCD40L trimer (98.1 kDa). The chromatogram also reveals a shoulder peak that corresponds to a dimer of trimers (196.2 kDa). An estimated ~8.2% of the total peak is accounted for by the shoulder multimer peak. This percentage is similar to that reported for a higher-molecular-weight component of a different CD40L fusion trimer, which had accounted for 10.4% of the total protein.17
Decoration of P22 with DechCD40L.
Fusion to Dec allows the trimer of hCD40L to be displayed on the P22 capsid surface, at as many as 80 Dec-binding sites, resulting in particles with multivalent ligand display for use in the CD40 cell experiments, as described later. Typically, we see near quantitative binding of Dec or DechCD40L to the P22 capsid, consistent with the very tight binding of Dec to P22. The high-affinity Dec-binding sites at quasi threefold axes of expanded P22 are spaced ~71 Å apart, which should provide adequate spacing for full occupancy binding of the DechCD40L fusion trimer (Figure S1).
DechCD40L was mixed with P22 in excess (>80 trimers per capsid) and purified by ultracentrifugation. SDS-PAGE of the recovered particles displayed bands near the expected molecular weights for DechCD40L (32.7 kDa) and P22 coat protein (47 kDa) (Figure 4B). Western blot analyses of DechCD40L bound to P22 compared to Dec bound to P22 show that DechCD40L stains positive with anti-Dec at the expected 32.7 kDa compared to the Dec control at 14.1 kDa (Figure 4C). Blotting with anti-CD40L confirms a DechCD40L band at 32.7 kDa and reveals evidence of some proteolytic cleavage of the DechCD40L construct by an additional band at ~16 kDa (Figure 4D), which is the approximate size of free hCD40L and suggests that the cleavage was localized to the flexible linker between the Dec and hCD40L domains.
CD40L Projects Away from the P22 Capsid Surface.
Previously, we have shown that P22 capsids decorated with Dec fused to murine CD40L bind to primary murine B cells displaying CD40 in high abundance.46 This suggests that murine CD40L, when presented on the surface of P22, is accessible for binding its cognate receptor CD40. However, the specific structural behavior of the Dec-CD40L fusion in the P22-bound state remains to be determined. Understanding how Dec-fused CD40L is structurally oriented on the capsid is essential in confirming the expected behavior of the construct and understanding the effects of multivalent display on CD40L–CD40 signaling.
Cryo-electron microscopy (cryo-EM) reconstructions were generated for P22 VLPs with and without bound DechCD40L. For the bound sample, an excess of DechCD40L was present to ensure full occupancy of the Dec-binding sites (Figure 5). Additional density was present on DechCD40L-decorated P22 at both the high-affinity and the low-affinity Dec-binding sites. Less density was present at the true threefold sites, which have been previously identified as lower-affinity Dec-binding sites, while more density protruded from the capsid surface at the high-affinity quasi threefold sites. At the center of each high-affinity binding site, a pillar of density extends away from the capsid terminating in a mushroom structure suggestive of hCD40L projecting away from the surface. The volume of additional density at each of the quasi threefold sites is 80–90 nm3. This experimentally determined volume is less than the expected volume for a 98.1 kDa DechCD40L trimer (118.9 nm3 assuming an average density of 1.37 g/cm3).52 However, 80–90 nm3 occupancy volume is still much larger than the expected 51.3 nm3 for a 42.3 kDa Dec trimer. This intermediate volume as well as the shape of the electron density suggests that hCD40L retains a degree of flexibility. This flexibility is likely provided, at least in part, by the glycine–serine linker that separates the Dec and CD40L domains.
Figure 5.

DechCD40L binds expanded P22 particles at the expected quasi threefold and true threefold sites. Additional density (red) can be seen at the anticipated Dec-binding sites when cryo-EM reconstructions of (A) expanded P22 particles were compared to (B) DechCD40L-decorated P22 particles. This added density has a threefold symmetric base with a central pillar extending away from the capsid surface and terminating in a poorly resolved globular head.
Partial Saturation of P22 with Dec Leads to Partially Decorated Capsids for Controlled Multivalent Presentation.
The icosahedral P22 cage provides a platform with 60 equivalent high-affinity binding sites for the trimeric Dec. Binding to the high-affinity site is characterized by a half-life of binding of at least 60 h indicative of stable association of Dec and P22.46 This stable binding interaction allows for potential stoichiometric control over the degree of multivalent presentation through partial occupancy of these sites. To verify the stability of decorated P22, we incubated P22 particles (no Dec) with fully decorated P22 in a 1:1 mixture and used size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) to characterize the sample (Figure S3A). The elution profile contained two peaks with distinctly different elution times, molecular weights, and radii with the first and second peaks corresponding to decorated and undecorated P22, respectively. This is clear evidence that Dec, once bound, did not equilibrate between undecorated and decorated P22 particles and suggests that the binding was mostly irreversible on the timescales considered here.
By varying the amount of Dec mixed with a fixed amount of P22 and analyzing the sample by SEC-MALS, we demonstrated that partial decoration of P22 with Dec is achievable and the resulting Dec-P22 molecular weight can be predicted by the input Dec:P22 stoichiometry (Figure 6A). Individual chromatograms are shown in Figure S3B, illustrating earlier elution times corresponding to higher amounts of Dec bound to the capsid. Line fit analysis reveals a linear relationship between the input number of Dec trimers and the calculated output number of Dec trimers per capsid (Figure 6B). Increasing amounts of Dec did not result in more binding past 60 Dec per capsid indicating saturation. Because the cryo-EM reconstructions demonstrated that DechCD40L binds to P22 at the known Dec-binding sites, we can assume that decoration of P22 with DechCD40L can also be controlled in the same stoichiometric manner although direct measurement of this was not possible due to strong interactions between the CD40L construct and the SEC-MALS column.
Figure 6.

Partial decoration of P22 with Dec. SEC-MALS was utilized to analyze Dec-P22 with multiple stoichiometries of the Dec trimer to P22. (A) Measured molecular weight of the particle versus number of Dec trimers. (B) Number of input Dec trimers plotted against the number of output Dec trimers derived from the molecular weight shows linear correlation (r = 0.64) between 0 and 80 trimers and no additional increase in molecular weight at greater than 80 Dec trimers.
Dec-P22 System Exhibits Tunable Signal Amplification through Controlled Multivalency.
To quantify the effects of multivalent display of human CD40L via the Dec-P22 system, the CD40L–CD40 cellular signaling HEK-Blue CD40L (InVivoGen) reporter assay was utilized. Briefly, HEK-Blue CD40L cells contain a vector for the increased display of human CD40 on the cell surface. This vector also encodes a secreted embryonic alkaline phosphatase (SEAP) gene driven by an NF-κB-sensitive promoter. Engagement of CD40 on the cell surface leads to NF-κB stimulation and quantifiable increases in the concentration of SEAP in the media, which can be measured with a simple colorimetric substrate.
To assess the signaling potential of DechCD40L as a soluble trimer (not bound to P22), a concentration series of DechCD40L ranging from 0.15 pM to 500 nM was incubated with cells for 24 h after which the extracellular activities of SEAP were measured. This assay revealed a concentration-dependent cellular response to DechCD40L with near-linear sensitivity from 0.01 to 10 nM. When this response was compared to soluble hCD40L at the same concentrations, DechCD40L displayed a much greater signal to concentration response as well as a higher maximum signal (Figure 7A). It is highly likely that fusion to trimeric Dec enforces the CD40L trimeric structure and could cause the formation of multi-trimers, which together would result in a higher valency of DechCD40L compared to hCD40L. A phenomenon termed domain swapping has been previously described in a similar recombinant CD40L fusion protein with both trimer-forming domains separated by a flexible linker.17 Based on our SEC data, it appears that DechCD40L likely undergoes domain swapping during expression and quaternary structure assembly, resulting in dimers of the trimer and possibly larger oligomers. Because the dimer of trimers cannot be separated from the trimers, it is not known how trimer multimers may bind P22 and whether bound multimers are a contributing factor to our experimental results.
Figure 7.

Controlled multivalent display of DechCD40L exhibits tunable signaling. Normalized cellular response is reported as percent activation compared to the maximum response at 1.5 nM DechCD40L-P22. Error bars indicate the standard error of the mean. (A) Concentration-dependent response was observed and shown to be strongest for DechCD40L displayed on P22 compared to unbound DechCD40L, hCD40L, and an admixture of P22 and hCD40L. Lines indicate Hill function fits. (B) Concentration of DechCD40L was held constant, while the amount of P22 varied leading to a range from low multivalency state (VLP oversaturated) to DechCD40L oversaturated (VLP starved), with the optimal stoichiometry (60 trimers/capsid) resulting in fully decorated particles and no unbound ligand. (C) Varying the amounts of P22 in constant 9 nM DechCD40L or hCD40L showed changes in signaling intensity only for DechCD40L. Lines are interpolations between points. (D) Percent activation of DechCD40L-P22 over the control hCD40L/P22 admixture at each ratio. Varying the background CD40L concentration reveals that the effect of changing CD40L valency is most notable at 3 and 9 nM. Lines are interpolations between points.
To elucidate the cellular effects of multivalent CD40L display, we tested the dose response of DechCD40L bound to P22 at a 180:1 molar stoichiometry (binding of 60 trimers at all quasi threefold sites). Multivalent display correlated with considerably increased signaling compared to free DechCD40L and hCD40L, with the most dramatic difference being between P22-bound DechCD40L and unbound hCD40L controls, where P22 admixed with hCD40L generated a response comparable to hCD40L alone, indicating that the VLP itself did not contribute to signaling. P22 alone, Dec alone, and Dec displayed on P22 were unable to stimulate notable signaling (Figure S5). We also tested Dec-fused mouse CD40L (DecmCD40L, from our previous study) in the same manner, which revealed that DecmCD40L elicited a signaling dose response comparable to that of DechCD40L, thus demonstrating that the homology between human and mouse CD40L allows for reactivity in this system. However, DecmCD40L multivalency on P22 did not amplify signaling, suggesting that there might be additional species-specific features necessary for a strong multivalent response (Figure S6).
The observed decreased signaling response at the highest doses (45–500 nM) of DechCD40L (bound and unbound to P22) is likely attributed to cellular toxicity. Incubation of cells with a very high concentration of DechCD40L resulted in overall cell death, as evidenced by cell rounding and detachment (Figure S4). High-dose CD40L cytotoxicity has been reported previously, highlighting the importance of developing a CD40L therapeutic that is effective at low doses.
EC50 values and binding coefficients were calculated using a Hill function fit without restraints for the P22-bound and unbound DechCD40L and compared to that of hCD40L alone or admixed with P22 (Table S1). The EC50 values for P22-bound and unbound DechCD40L were 0.03 and 0.23 nM, respectively, indicating that presentation of DechCD40L on P22 increased the signaling potency 9.4-fold. Comparing the EC50 values of P22-bound DechCD40L to free hCD40L indicates that hCD40L multivalency in combination with Dec fusion results in a 53.6-fold more potent CD40 signaling response. The Hill coefficient values across all samples assayed were at or near a value of 1 suggesting no cooperativity of binding in the fit to the experimental data. It should be noted that data from DechCD40L-P22 was not as well fit by a Hill function as data from other hCD40L samples. Efforts to restrict or modify starting values for fit parameters in the DechCD40L-P22 dataset demonstrated that the EC50 value is robust. However, the goodness of fit could not be improved by fit parameter restriction as measured by the χ2 value. To date, few tunable avidity systems have been available to test expected models of multivalent binding especially with the spatial control demonstrated here. Further work with DechCD40L and other constructs displayed on the surface of P22 will be needed in combination with binding model development to understand how the structural restrictions of both the multivalent ligand and the assay binding target lead to deviation from a Langmuir-style binding system.
Given our ability to control the degree of occupancy of Dec on the surface of P22, we tested the possibility that partially decorated capsids could generate intermediate levels of DechCD40L signaling. A series of DechCD40L to P22 stoichiometries were tested with the absolute concentration of CD40L held constant, varying only the concentration of P22 (Figure 7B). Varying the stoichiometries resulted in the number of available DechCD40L trimers per capsid ranging from 0.1 in a VLP-oversaturated state to 3000 in a VLP-starved state. Four different concentrations of CD40L were tested, representing the higher range of cellular activation, as previously determined by the concentration series. Figure 7C demonstrates the effect of CD40L multivalency on CD40 signaling when CD40L was held constant at 9 nM. As expected, the ratio of DechCD40L to P22 determined the level of CD40 signaling. Increasing CD40L multivalency resulted in higher levels of activity, with maximum signaling response centered near the stoichiometry corresponding to fully decorated capsids (60–80 trimers). At very high P22:DechCD40L ratios, where the degree of multivalent presentation is low, the weakest cell signaling was observed. With decreasing P22:DechCD40L ratios and increasing degree of multivalent presentation, the signal rises steadily to its maximum at the stoichiometry where DechCD40L fully saturates the binding sites of P22. Further decrease in the P22 concentration resulted in decreased signaling consistent with a “VLP-starved” state, such that a small number of P22 is fully decorated but all excess DechCD40L remains in an unbound form. The unbound DechCD40L contributes to a much lower cell signaling response and likely competes with fully decorated VLPs for receptor binding. These multivalent presentation experiments were repeated with three additional constant DechCD40L concentrations: 0.1, 3, and 90 nM. Figure 7D reveals the effect of multivalency as percent cellular activation adjusted by subtracting the unbound hCD40L (in an admixture with P22) from DechCD40L-P22 at each stoichiometry. Interestingly, the effect on cellular activation of varying valency at 3 nM hCD40L was similar to 9 nM hCD40L but appears to be muted at 0.3 and 90 nM hCD40L. Because the DechCD40L at 90 nM occurs at system saturation (Figure 7A), it is unsurprising that valency showed less of an effect at this high concentration of DechCD40L. At the lowest concentration (0.3 nM), the overall signal of 0.3 nM DechCD40L is higher than 0.3 nM hCD40L, but the effect of valency is lost, which may indicate the lower limit of sensitivity for this cellular based assay. Figure S7 shows the unadjusted data for all experiments.
These results illustrate the power of the P22-Dec display system to drastically amplify CD40L–CD40 signaling compared to monovalent CD40L and the ability to tune this amplification by simply changing the molar ratio of DechCD40L to P22 and therefore the occupancy and degree of multivalent presentation on the P22 surface. Although ligands fused to Dec-P22 are restricted to a regular distribution over the surface of the capsid, flexibility in the linker between Dec and the fusion protein will allow for some mobility, which may be crucial for CD40L–CD40 interactions. While it is possible that absence of fluidity (such as that of natural membrane-bound CD40L) could limit interactions with cell surface receptors, the ability to control valency will enable much more in-depth research of ligand–receptor signaling than that of soluble monovalent or low-valency ligands.
CONCLUSIONS
We have demonstrated cellular CD40 signal amplification through enforcement of the necessary quaternary structural elements to soluble CD40L. By utilizing the P22-Dec system, multivalent CD40L presentation is achieved via fusion to Dec and subsequent controlled stoichiometric binding to P22 VLPs. Structural analysis reveals that P22-bound CD40L projects away from the capsid and is in an ideal position to engage CD40. Cellular experiments demonstrate that this presentation is highly tunable and leads to a considerable increase in signaling over free soluble CD40L.
This sizeable net enhancement of signaling from the multivalent display is promising and demonstrates the value of the P22-Dec system. Further, a system that can be easily tuned surpasses the usefulness of a system that is either monovalent or has a fixed valency. The P22-Dec system offers the ability to tune the degree of multivalency by controlling the stoichiometry of Dec and P22. This tunability is useful in assessing the effects of varying signaling intensities on downstream cellular pathways and characteristics of the target receptor expressing cells, such as gene expression, differentiation, and proliferation.
There is often a disconnect between in vitro biochemical assessment of proteins and the examination of those proteins in a cellular context. Without preservation of necessary structural characteristics, binding avidity and activity studies may be misleading or inaccurate toward describing the behavior of a protein in its natural system. The relative ease of P22 VLP production and tunable nature of Dec binding makes Dec-P22 an appealing platform to design biomimetic materials, which can probe molecular and structural determinants of effector functions in signal transduction. This system also has the potential to impose avidity and clustering on receptors that do not naturally utilize those elements.
In summary, the P22-Dec system is a model for the tunable multivalent display of TNFSF proteins on a nanocage and demonstrates the usefulness of such a model to further explore ligand–receptor interactions and complex multireceptor networks. The trimeric structure of Dec is ideal for simultaneously enforcing the quaternary structure of TNFSF ligands and providing a method for binding trimeric ligands to the P22 protein nanocage. Several TNFSF ligands are currently being investigated to determine their potential usefulness in human medicine. Presumably, any trimeric ligand can be fused to Dec and displayed on P22 as we have done for CD40L. Building upon this design may possibly lead to the development of promising therapeutic candidates targeting cell surface signaling machinery.
MATERIALS AND METHODS
Materials.
DNA modifying enzymes were purchased from New England Biolabs and Promega. Oligonucleotides were purchased from Eurofins MWG Operon. Electrocompetent ClearColi cells were purchased from Lucigen. HEK-Blue CD40L cells, Quanti-Blue, and recombinant soluble human CD40L were purchased from InVivoGen. Antibodies used are as follows: anti-hCD40L from InVivoGen, anti-mouse-horseradish peroxidase (HRP) from Santa Cruz Biotechnology, anti-rabbit HRP from BioRad; and anti-P22 and anti-Dec were produced in-house. InstantBlue stain was purchased from Abcam. All other chemical reagents were purchased from Fisher Scientific.
Molecular Biology.
An N-terminal 6x histidine tag wild-type Dec gene, provided by Dr. Peter Prevelige, was ligated into a pETDuet vector via BamHI and SacI sites. The Dec-pETDuet plasmid was linearized by polymerase chain reaction (PCR) using the following primers, which removed the stop codon of Dec: 5′-GAGCTCGGCGCGCCTGCAGGTCGACAAGCTT-3′, 5′-GGATCCACTTCCTGATGTTGTTTCGATAGTC-3′.
A gene fragment coding for the soluble region of hCD40L (aa 114–261) with 25 base pair flanking regions matching the target plasmid insertion site was ordered from Integrated DNA Technologies, Inc. The DechCD40L construct was assembled using NEBuilder HiFi DNA Assembly Mix as per the manufacturer’s suggestions. DNA sequence confirmations were performed by Eurofins MWG Operon, Inc. The resulting DechCD40L fusion protein sequence is shown in Figure S8.
Expression, Lysis, and Purification.
P22 and DechCD40L constructs were transformed into ClearColi bacteria. Wild-type Dec was transformed into BL21(DE3) bacteria. Clones harboring expression vectors for DechCD40L, Dec, or for the P22 coat and scaffold proteins were grown in the Luria–Bertani (LB) medium at 37 °C in the presence of ampicillin to maintain selection for the plasmid. The expression of recombinant genes was induced by the addition of isopropyl β-d-thiogalactopyranoside to a final concentration of 0.5 mM once the cells reached mid-log phase (OD600 ~ 0.8). Cultures of DechCD40L were briefly cooled on ice at the point of induction and allowed to continue growing at room temperature for 4 h. Cultures of P22 and wild-type Dec were grown overnight at 37 °C after induction. Cells were harvested via centrifugation and cell pellets were stored overnight at −20 °C.
Thawed cell pellets were resuspended in sodium phosphate buffer (50 mM sodium phosphate, 100 mM sodium chloride, pH 7.0) with lysozyme, DNAse, and RNAse added and incubated at room temperature for 30 min. To minimize proteolytic cleavage, Pierce Protease Inhibitor was added to the buffer prior to resuspending the bacterial pellets. The cell suspension was lysed by sonication at 4 °C. Insoluble cellular components were removed by centrifugation at 12 000g for 45 min at 4 °C. After each column purification, fractions containing desired protein were determined by SDS-PAGE analysis. P22: P22 VLPs were pelleted from the post-lysis supernatant by ultracentrifugation through a 5 mL 35% (w/v) sucrose cushion. The resulting VLP pellet was resuspended in sodium phosphate buffer and centrifuged at 16 000g for 20 min to remove aggregates. The supernatant was filtered through 0.45 μm and run over an S-500 Sephadex (GE Healthcare Life Sciences) size-exclusion column using a Biorad Biologic Duoflow FPLC. Fractions containing P22 were concentrated by ultracentrifugation and the resulting VLP pellet was resuspended in an adequate volume of sodium phosphate buffer. Dec: Wild-type Dec and DechCD40L were purified using a 5 mL Roche cOmplete His-tag purification column in sodium phosphate buffer at pH 7.8 with imidazole concentrations as indicated. After filtering through 0.45 μm, the lysate was loaded onto the column at 2 mL/min and washed with 40 mL at 20 mM imidazole. Fractions were eluted in a two-step gradient from 5 to 250 mM followed by 250–500 mM imidazole. Pooled fractions containing DechCD40L were dialyzed overnight into sodium phosphate buffer without imidazole. The concentration of DechCD40L was achieved by rebinding to the column, washing with 40 mL at 20 mM imidazole, and stepwise elution with 250 mM imidazole followed by removal of imidazole by dialysis into sodium phosphate buffer (pH 7.0). DechCD40L fractions were pooled and further purified on a Superose 6 column at 0.3 mL/min. Saved fractions of DechCD40L did not demonstrate aggregation upon binding to expanded P22, as determined by UV–vis scanning. Concentrations of each construct were determined by UV absorption measured at 280 nm under denatured conditions (5 M guanidine hydrochloride) with extinction coefficients calculated using Protein Calculator v3.3 (Chris Putnam, Scripps).
P22 Expansion.
Procapsid P22 was treated with 1 mM dithiothreitol (DTT) for 2 h to limit capsid–capsid interactions, followed by ultracentrifugation to remove DTT. Expansion of P22 (1 mg/mL) was achieved by heating at 66 °C for 25 min. The successful expansion was confirmed via nondenaturing 1% agarose gel electrophoresis at 100 V for 1 h in 40 mM Tris, 20 mM acetate, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 8.0, followed by staining with InstantBlue.
SDS-PAGE and Western Blots.
Protein samples mixed with 4× loading buffer containing DTT were heated to 100 °C for 10 m and separated on a 15% acrylamide gel at a current of 35 mA for 45 m. Gels were stained with InstantBlue and rinsed with water. Gel images were captured using a UVP MultiDoc-IT Digital Imaging System. Repeats of the gels were transferred to nitrocellulose membranes followed by incubation with primary antibodies overnight at 4 °C and secondary antibody for 1 h at ambient temperature. Blots were developed with Opti-4CN reagent.
Size-Exclusion Chromatography.
Purified DechCD40L was evaluated using an analytical Superose-12 10/300 GL column (Sigma-Aldrich). The Igor Pro MultiPeak Fit analysis tool was used to fit a Gaussian curve for each peak and calculate the area under each curve. Known standards (BioRad Laboratories) were established with the same column to enable the calculation of the molecular weight corresponding to each DechCD40L peak.
Multiangle and Quasi-elastic Light Scattering.
Samples were separated over a WTC-200S5 (Wyatt Technologies) size-exclusion column using an Agilent 1200 high-performance liquid chromatography (HPLC) at 0.7 mL/min. The total run time was 30 m with an injection of 10 μL per run. Resultant peaks were detected using a UV–vis detector (Agilent), an HELEOS multi angle laser light scattering detector, and an Optilab rEX differential refractometer (Wyatt Technology Corporation). The average particle molecular weight (Mn) and the radius of hydration (rH) were calculated across each peak half max with Astra 5.3.14 software using a previously calculated dn/dc value of 0.185 mL/g.
Cryo-EM Analysis.
To prepare the specimen for cryo-EM imaging, P22 VLPs at 2 mg/mL in the expanded form were loaded with a 3× stoichiometric excess of 240 DechCD40L trimers per capsid. Approximately 4 μL of sample solution was then applied to a glow-discharged 200-mesh Quantifoil R2/2 holey carbon grid. The grid was plunged into a liquid ethane container cooled by liquid nitrogen using FEI Vitrobot Mark III. The frozen-hydrated cryo-EM grid was then transferred to a Gatan 626 cryo-holder and loaded into a JEOL JEM3200FS TEM. Images of P22 and DechCD40L-P22 were acquired at a nominal magnification of 60 000× using a Gatan UltraScan 4000 CCD camera with an energy filter slit opened at 30 eV. Individual particles were extracted from the cryo-EM images using EMAN2 (v2.12) software package and were processed using AUTO3DEM (v4.05). Undecorated P22 three-dimensional (3D) reconstruction was obtained using 225 micrographs containing 10 684 total particles. A total of 8549 particles were utilized in the final reconstruction, which reached 12.3 Å resolution based on a Fourier shell correlation with a cutoff of 0.5. DechCD40L-P22 3D reconstruction was calculated from 194 micrographs containing 11 170 particles, of which 8937 particles were used in the final model. The DechCD40L-P22 reconstruction reached a resolution of 14.3 Å. UCSF Chimera was used to render the 3D structures and calculate the volume.
HEK-Blue CD40L Cellular Assays.
Frozen HEK-Blue CD40L cells (1 mL) were thawed at 37 °C and immediately transferred to 14 mL of prewarmed growth media (Dulbecco’s modified Eagle’s medium (DMEM), 4.5 g/L glucose, 10% (v/v) fetal bovine serum, 50 U/mL penicillin, 50 mg/mL streptomycin, 2 mM L-glutamine, 100 μg/mL Normocin). Cells were centrifuged, resuspended in 1 mL of growth media, and transferred to 9 mL of growth media in a T-75 flask. Cells were passaged five times with selective antibiotics (30 μg/mL of Blasticidin and 100 μg/mL of Zeocin) being added after the second passage. For the experiments, cells were harvested and diluted in growth media (with 10% heat-inactivated fetal bovine serum, without Blasticidin and Zeocin) to a final concentration of ~300 000 cells/mL. For each assay, ~50 000 cells were added to each well of a 96-well cell culture plate containing samples as described below and incubated in the assay plate for 24 h at 37 °C and 5% CO2.
For the dosage study, samples were premixed and serially diluted such that the final concentrations in the wells after adding cells were as follows: DechCD40L, 500 nM to 0.15 pM and hCD40L, and 45 nM to 0.15 pM. Samples included were DechCD40L mixed with P22 at a 180:1 stoichiometry, DechCD40L without P22, and the following controls: P22, P22 with Dec, hCD40L alone, and as an admixture of hCD40L and P22. In addition, mouse CD40L fused to Dec was assayed in the same way (bound and unbound to P22) to assess species cross-reactivity.
For the variable multivalency study, P22 varying from 10 nM to 0.3 pM, such that the stoichiometry of DechCD40L trimers to capsids ranged from 0.1 to 3000, was first added to the wells. DechCD40L or hCD40L (9 nM final concentration in wells) was then added. The dilutions of P22 admixed with hCD40L served as controls. The experiments were repeated with three additional concentrations (0.3, 3, 90 mM DechCD40L or hCD40L).
To assess CD40 signaling, 20 μL of media was removed from each sample into a flat-bottom 96-well assay plate. QUANTI-Blue reagent suspension was added (180 μL) to each well and plates were incubated at 37 °C for 30 min. The absorbances at 635 nm were used to determine receptor activation, expressed as a percentage of the peak SEAP activity observed in the dosage experiment (1.5 nM DechCD40L-P22).
The Hill equation function within the Igor Pro software (base + (max − base))/(1 + (x1/2/x)rate) was used to fit the dosage data and to calculate the EC50 values, binding rates, and χ2 values.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ranjit Koliyatt for his initial experiments on the Dec loading of P22. This work was funded by a grant from the Human Frontier Science Program 4124801. E.S. was partially supported by the Graduate Training Program in Quantitative and Chemical Biology under Award T32 GM109825 and Indiana University. T.D. was supported in part by the National Science Foundation through grant 1720625. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsabm.1c00718.
Dec-binding locations on P22; SEC of DechCD40L; SEC and MALS of controlled Dec loading on P22; in vitro cellular toxicity of DechCD40L; in vitro assay controls; in vitro DecmCD40L assay; dosage curve fit analyses; in vitro multivalency assay with 0.3, 3, and 90 nM CD40L; DechCD40L annotated sequence (PDF)
Contributor Information
Cheri Peyton Goodall, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
Benjamin Schwarz, Laboratory of Bacteriology, National Institute of Allergy and Infectious Diseases, Hamilton, Montana 59840, United States.
Ekaterina Selivanovitch, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
John Avera, Walden Biosciences, Cambridge, Massachusetts 02139, United States.
Joseph Wang, Department of Microbiology and Immunology, The Pennsylvania State University College of Medicine, Hershey, Pennsylvania 17033, United States.
Heini Miettinen, Department of Microbiology and Immunology, Montana State University, Bozeman, Montana 59717, United States.
Trevor Douglas, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
REFERENCES
- (1).Mammen M; Choi SK; Whitesides GM Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem., Int. Ed. 1998, 37, 2754–2794. [DOI] [PubMed] [Google Scholar]
- (2).Kiessling LL; Gestwicki JE; Strong LE Synthetic multivalent ligands as probes of signal transduction. Angew. Chem., Int. Ed. 2006, 45, 2348–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kwa S; Lai L; Gangadhara S; Siddiqui M; Pillai VB; Labranche C; Yu T; Moss B; Montefiori DC; Robinson HL; Kozlowski PA; Amara RR CD40L-adjuvanted DNA/modified vaccinia virus Ankara simian immunodeficiency virus SIV239 vaccine enhances SIV-specific humoral and cellular immunity and improves protection against a heterologous SIVE660 mucosal challenge. J. Virol. 2014, 88, 9579–9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tansey MG; Szymkowski DE The TNF superfamily in 2009: new pathways, new indications, and new drugs. Drug Discovery Today 2009, 14, 1082–1088. [DOI] [PubMed] [Google Scholar]
- (5).Stone GW; Barzee S; Snarsky V; Santucci C; Tran B; Langer R; Zugates GT; Anderson DG; Kornbluth RS Nanoparticle-delivered multimeric soluble CD40L DNA combined with Toll-Like Receptor agonists as a treatment for melanoma. PLoS One 2009, 4, No. e7334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Grewal IS Overview of TNF superfamily: a chest full of potential therapeutic targets. Adv. Exp. Med. Biol. 2009, 647, 1–7. [DOI] [PubMed] [Google Scholar]
- (7).Bodmer JL; Schneider P; Tschopp J The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [DOI] [PubMed] [Google Scholar]
- (8).Vanamee ÉS; Faustman DL On the TRAIL of Better Therapies: Understanding TNFRSF Structure-Function. Cells 2020, 9, No. 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ara A; Ahmed KA; Xiang J Multiple effects of CD40-CD40L axis in immunity against infection and cancer. ImmunoTargets Ther. 2018, 7, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Clark EA; Ledbetter JA How B and T cells talk to each other. Nature 1994, 367, 425–428. [DOI] [PubMed] [Google Scholar]
- (11).Ma DY; Clark EA The role of CD40 and CD154/CD40L in dendritic cells. Semin. Immunol 2009, 21, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Smith CM; Wilson NS; Waithman J; Villadangos JA; Carbone FR; Heath WR; Belz GT Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat. Immunol. 2004, 5, 1143–1148. [DOI] [PubMed] [Google Scholar]
- (13).Suttles J; Stout RD Macrophage CD40 signaling: a pivotal regulator of disease protection and pathogenesis. Semin. Immunol 2009, 21, 257–264. [DOI] [PubMed] [Google Scholar]
- (14).van Kooten C; Banchereau J CD40-CD40 ligand. J. Leukocyte Biol. 2000, 67, 2–17. [DOI] [PubMed] [Google Scholar]
- (15).Banchereau J; Bazan F; Blanchard D; Briere F; Galizzi JP; van Kooten C; Liu YJ; Rousset F; Saeland S The CD40 antigen and its ligand. Annu. Rev. Immunol. 1994, 12, 881–922. [DOI] [PubMed] [Google Scholar]
- (16).Diehl L; den Boer AT; Schoenberger SP; van der Voort EI; Schumacher TN; Melief CJ; Offringa R; Toes RE CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat. Med. 1999, 5, 774–779. [DOI] [PubMed] [Google Scholar]
- (17).Kornbluth RS; Stempniak M; Stone GW Design of CD40 agonists and their use in growing B cells for cancer immunotherapy. Int. Rev. Immunol. 2012, 31, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Schönbeck U; Libby P The CD40/CD154 receptor/ligand dyad. Cell. Mol. Life Sci. 2001, 58, 4–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).van den Oord JJ; Maes A; Stas M; Nuyts J; Battocchio S; Kasran A; Garmyn M; De Wever I; De Wolf-Peeters C CD40 is a prognostic marker in primary cutaneous malignant melanoma. Am. J. Pathol. 1996, 149, 1953–1961. [PMC free article] [PubMed] [Google Scholar]
- (20).Lang I; Fullsack S; Wyzgol A; Fick A; Trebing J; Arana JA; Schafer V; Weisenberger D; Wajant H Binding Studies of TNF Receptor Superfamily (TNFRSF) Receptors on Intact Cells. J. Biol. Chem. 2016, 291, 5022–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Grassmé H; Jendrossek V; Bock J; Riehle A; Gulbins E Ceramide-rich membrane rafts mediate CD40 clustering. J. Immunol. 2002, 168, 298–307. [DOI] [PubMed] [Google Scholar]
- (22).Nadiri A; Polyak MJ; Jundi M; Alturaihi H; Reyes-Moreno C; Hassan GS; Mourad W CD40 translocation to lipid rafts: signaling requirements and downstream biological events. Eur. J. Immunol. 2011, 41, 2358–2367. [DOI] [PubMed] [Google Scholar]
- (23).Haswell LE; Glennie MJ; Al-Shamkhani A Analysis of the oligomeric requirement for signaling by CD40 using soluble multimeric forms of its ligand, CD154. Eur. J. Immunol. 2001, 31, 3094–3100. [DOI] [PubMed] [Google Scholar]
- (24).Trouche N; Wieckowski S; Sun W; Chaloin O; Hoebeke J; Fournel S; Guichard G Small multivalent architectures mimicking homotrimers of the TNF superfamily member CD40L: delineating the relationship between structure and effector function. J. Am. Chem. Soc. 2007, 129, 13480–13492. [DOI] [PubMed] [Google Scholar]
- (25).Fournel S; Wieckowski S; Sun W; Trouche N; Dumortier H; Bianco A; Chaloin O; Habib M; Peter JC; Schneider P; Vray B; Toes RE; Offringa R; Melief CJ; Hoebeke J; Guichard G C3-symmetric peptide scaffolds are functional mimetics of trimeric CD40L. Nat. Chem. Biol. 2005, 1, 377–382. [DOI] [PubMed] [Google Scholar]
- (26).Fanslow WC; Srinivasan S; Paxton R; Gibson MG; Spriggs MK; Armitage RJ Structural characteristics of CD40 ligand that determine biological function. Semin. Immunol. 1994, 6, 267–278. [DOI] [PubMed] [Google Scholar]
- (27).Kuhné MR; Robbins M; Hambor JE; Mackey MF; Kosaka Y; Nishimura T; Gigley JP; Noelle RJ; Calderhead DM Assembly and regulation of the CD40 receptor complex in human B cells. J. Exp. Med. 1997, 186, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).McWhirter SM; Pullen SS; Holton JM; Crute JJ; Kehry MR; Alber T Crystallographic analysis of CD40 recognition and signaling by human TRAF2. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 8408–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pullen SS; Labadia ME; Ingraham RH; McWhirter SM; Everdeen DS; Alber T; Crute JJ; Kehry MR High-affinity interactions of tumor necrosis factor receptor-associated factors (TRAFs) and CD40 require TRAF trimerization and CD40 multimerization. Biochemistry 1999, 38, 10168–10177. [DOI] [PubMed] [Google Scholar]
- (30).Mackay F; Schneider P Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [DOI] [PubMed] [Google Scholar]
- (31).Vonderheide RH The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gieffers C; Kluge M; Merz C; Sykora J; Thiemann M; Schaal R; Fischer C; Branschadel M; Abhari BA; Hohenberger P; Fulda S; Fricke H; Hill O APG350 induces superior clustering of TRAIL receptors and shows therapeutic antitumor efficacy independent of cross-linking via Fcgamma receptors. Mol. Cancer Ther. 2013, 12, 2735–2747. [DOI] [PubMed] [Google Scholar]
- (33).Wyzgol A; Muller N; Fick A; Munkel S; Grigoleit GU; Pfizenmaier K; Wajant H Trimer Stabilization, Oligomerization, and Antibody-Mediated Cell Surface Immobilization Improve the Activity of Soluble Trimers of CD27L, CD40L, 41BBL, and Glucocorticoid-Induced TNF Receptor Ligand. J. Immunol. 2009, 183, 1851–1861. [DOI] [PubMed] [Google Scholar]
- (34).Miconnet I; Pantaleo G A soluble hexameric form of CD40 ligand activates human dendritic cells and augments memory T cell response. Vaccine 2008, 26, 4006–4014. [DOI] [PubMed] [Google Scholar]
- (35).Matsuura JE; Morris AE; Ketchem RR; Braswell EH; Klinke R; Gombotz WR; Remmele RL Jr. Biophysical characterization of a soluble CD40 ligand (CD154) coiled-coil trimer: evidence of a reversible acid-denatured molten globule. Arch. Biochem. Biophys. 2001, 392, 208–218. [DOI] [PubMed] [Google Scholar]
- (36).Morris AE; Remmele RL; Klinke R; Macduff BM; Fanslow WC; Armitage RJ Incorporation of an isoleucine zipper motif enhances the biological activity of soluble CD40L (CD154). J. Biol. Chem. 1999, 274, 418–423. [DOI] [PubMed] [Google Scholar]
- (37).Jain A; Kovacs JA; Nelson DL; Migueles SA; Pittaluga S; Fanslow W; Fan XY; Wong DW; Massey J; Hornung R; Brown MR; Spinner JJ; Liu SY; Davey V; Hill HA; Ochs H; Fleisher TA Partial immune reconstitution of X-linked hyper IgM syndrome with recombinant CD40 ligand. Blood 2011, 118, 3811–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mazzei GJ; Edgerton MD; Losberger C; Lecoanet-Henchoz S; Graber P; Durandy A; Gauchat JF; Bernard A; Allet B; Bonnefoy JY Recombinant soluble trimeric CD40 ligand is biologically active. J. Biol. Chem. 1995, 270, 7025–7028. [DOI] [PubMed] [Google Scholar]
- (39).Brunekreeft KL; Strohm C; Gooden MJ; Rybczynska AA; Nijman HW; Grigoleit GU; Helfrich W; Bremer E; Siegmund D; Wajant H; de Bruyn M Targeted delivery of CD40L promotes restricted activation of antigen-presenting cells and induction of cancer cell death. Mol. Cancer 2014, 13, No. 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Vonderheide RH; Dutcher JP; Anderson JE; Eckhardt SG; Stephans KF; Razvillas B; Garl S; Butine MD; Perry VP; Armitage RJ; Ghalie R; Caron DA; Gribben JG Phase I study of recombinant human CD40 ligand in cancer patients. J. Clin. Oncol. 2001, 19, 3280–3287. [DOI] [PubMed] [Google Scholar]
- (41).Angelou A; Antoniou E; Garmpis N; Damaskos C; Theocharis S; Margonis GA The Role of Soluble CD40L Ligand in Human Carcinogenesis. Anticancer Res. 2018, 38, 3199–3201. [DOI] [PubMed] [Google Scholar]
- (42).Douglas T; Young M Viruses: making friends with old foes. Science 2006, 312, 873–875. [DOI] [PubMed] [Google Scholar]
- (43).Chackerian B Virus-like particles: flexible platforms for vaccine development. Expert Rev. Vaccines 2007, 6, 381–390. [DOI] [PubMed] [Google Scholar]
- (44).Franco D; Liu W; Gardiner DF; Hahn BH; Ho DD CD40L-Containing Virus-Like Particle as a Candidate HIV-1 Vaccine Targeting Dendritic Cells. J. Acquired Immune Defic. Syndr. 2011, 56, 393. [DOI] [PubMed] [Google Scholar]
- (45).Zhao L; Kopylov M; Potter CS; Carragher B; Finn MG Engineering the PP7 Virus Capsid as a Peptide Display Platform. ACS Nano 2019, 13, 4443–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Schwarz B; Madden P; Avera J; Gordon B; Larson K; Miettinen HM; Uchida M; LaFrance B; Basu G; Rynda-Apple A; Douglas T Symmetry Controlled, Genetic Presentation of Bioactive Proteins on the P22 Virus-like Particle Using an External Decoration Protein. ACS Nano 2015, 9, 9134–9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Newcomer RL; Schrad JR; Gilcrease EB; Casjens SR; Feig M; Teschke CM; Alexandrescu AT; Parent KN The phage L capsid decoration protein has a novel OB-fold and an unusual capsid binding strategy. Elife 2019, 8, No. e45345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Uchida M; LaFrance B; Broomell CC; Prevelige PE Jr.; Douglas T Higher order assembly of virus-like particles (VLPs) mediated by multi-valent protein linkers. Small 2015, 11, 1562–1570. [DOI] [PubMed] [Google Scholar]
- (49).Gilcrease EB; Winn-Stapley DA; Hewitt FC; Joss L; Casjens SR Nucleotide sequence of the head assembly gene cluster of bacteriophage L and decoration protein characterization. J. Bacteriol. 2005, 187, 2050–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Parent KN; Deedas CT; Egelman EH; Casjens SR; Baker TS; Teschke CM Stepwise molecular display utilizing icosahedral and helical complexes of phage coat and decoration proteins in the development of robust nanoscale display vehicles. Biomaterials 2012, 33, 5628–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tang L; Gilcrease EB; Casjens SR; Johnson JE Highly discriminatory binding of capsid-cementing proteins in bacteriophage L. Structure 2006, 14, 837–845. [DOI] [PubMed] [Google Scholar]
- (52).Fischer H; Polikarpov I; Craievich AF Average protein density is a molecular-weight-dependent function. Protein Sci. 2004, 13, 2825–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.