

Abstract

RNA is central to the proper function of cellular processes important for life on earth and implicated in various medical dysfunctions. Yet, RNA structural biology lags significantly behind that of proteins, limiting mechanistic understanding of RNA chemical biology. Fortunately, solution NMR spectroscopy can probe the structural dynamics of RNA in solution at atomic resolution, opening the door to their functional understanding. However, NMR analysis of RNA, with only four unique ribonucleotide building blocks, suffers from spectral crowding and broad linewidths, especially as RNAs grow in size. One effective strategy to overcome these challenges is to introduce NMR-active stable isotopes into RNA. However, traditional uniform labeling methods introduce scalar and dipolar couplings that complicate the implementation and analysis of NMR measurements. This challenge can be circumvented with selective isotope labeling. In this review, we outline the development of labeling technologies and their application to study biologically relevant RNAs and their complexes ranging in size from 5 to 300 kDa by NMR spectroscopy.
1. Introduction
RNA is central to medicine, chemical and structural biology, and basic research. For more than a half-century, it has been known that the code of life is imprinted in DNA sequences, following the so-called “sequence hypothesis”, usually wrongly labeled as the “central dogma” in the popular parlance.1 In the last several decades, it has become increasingly clear that the functions of cells are also transacted by DNA’s lesser-known relative, RNA.2 Indeed, the varied roles that RNAs play in both normal and dysfunctional cells have motivated RNA-based therapeutic development, as highlighted by the recent SARS COV-2 mRNA vaccines.3−9 Additionally, RNAs are central to the workings of molecular nanomachines such as the ribosome10−12 and the spliceosome13−15 to name a few. Moreover, thanks to the advent of genomic sequencing efforts, we now understand that the amount of RNA sequence transcribed in humans exceeds the number of protein sequences translated by at least 50-fold (Figure 1A).16 Paradoxically, the number of RNA-only structures deposited in the Protein Data Bank (PDB) remains below 1%, whereas the number of protein-only structures is a staggering 87% (Figure 1B). This paucity undercuts current understanding of RNA structure–function relationships.
Figure 1.

(A) Percentage of protein coding and nonprotein coding genomic material in selected genomes.16 Organismal complexity increases with RNA coding but decreases with protein coding capacity as a percentage of the DNA genomic output. (B) Percentage of RNA-only and protein-only structures deposited in the PDB. Given that this analysis excluded DNA-only structures and structures of protein–DNA/RNA complexes, the percentages do not sum to 100%. (C) Percentage of RNA-only and protein-only structures deposited in the Nucleic Acid Database (NDB) and PDB, sorted by structure determination technique. Given that this analysis is self-contained within categories, the percentages sum to 100%. NMR accounts for a larger fraction of RNA structures as compared to proteins. PDB and NDB statistics were accessed from https://www.rcsb.org/ and http://ndbserver.rutgers.edu/ in January 2022.
Nuclear magnetic resonance (NMR) spectroscopy accounts for ∼35% of the RNA structures deposited in the PDB and ∼7% of the protein structures, making it competitive with other biophysical tools such as X-ray crystallography and more recently cryo-electron microscopy (cryo-EM) (Figure 1C).17 Moreover, NMR spectroscopy provides high-resolution structural dynamic information in solution, rendering it an ideal tool to study RNA and its interactions with macromolecules or small drug-like compounds or both.18−25 However, unlike proteins, which are made up of 20 unique amino acid building blocks, RNAs are composed of only four aromatic nucleotides [i.e., adenosine (Ade or A), guanosine (Gua or G), cytidine (Cyt or C), and uridine (Uri or U)] that resonate over a very narrow chemical shift region. This poor chemical shift dispersion is further exacerbated with increasing RNA size. To overcome these limitations, novel isotope labeling strategies that incorporate atom-specific labels (e.g., uridine 13C6) or expand the number of NMR probes beyond the traditional 1H–15N and 1H–13C spin pairs (e.g., 13C–19F) have been developed.
In this review, we will outline the development of isotope labeling technologies for RNA NMR and some of the exciting new applications enabled by these labels to study small-to-large RNAs. Specifically, we will begin by detailing the benefits afforded by each common NMR-active isotope (Section 2). Next, we outline the various technologies that incorporate such labels into RNA building blocks and eventually into RNA (Section 3). This discussion will center around chemo-enzymatic labeling, a method that our group has extensively developed for the past near-decade. Next, we will examine how these labels benefit dynamics measurements (Section 4) and can be leveraged to study interactions involving large RNA systems (Section 5). Finally, to conclude, we will comment on how isotope labeling can advance the field of RNA chemical and structural biology (Section 6).
2. Stable Isotopes in NMR Spectroscopy
Frederick Soddy is credited with coining the word “isotope” from the Greek isos (i̋σος) and topos (τóπος) meaning “same place”,26 with the idea that stable isotopes are chemical elements that occupy the same position in the periodic table but differ in mass due to a different number of neutrons within the atomic nucleus. Stable isotopes have been used in a wide range of applications in industry, academia, and medicine.26 In particular, stable isotopes have significantly impacted methods such as NMR and mass spectrometry (MS). For this work, we will focus on how these probes impact RNA NMR spectroscopy, with special emphasis on proton (hydrogen-1 or 1H), deuterium (hydrogen-2 or 2H), carbon-13 (13C), nitrogen-15 (15N), fluorine-19 (19F), and phosphorus-31 (31P) (Table 1).
Table 1. Stable Isotopes Relevant to RNA NMR Spectroscopy27,28.
| isotope | natural abundance (%) | γ (rad Hz T–1) | spin |
|---|---|---|---|
| 1H | 99.99 | 26.752 × 107 | 1/2 |
| 2H | 0.01 | 4.107 × 107 | 1 |
| 12C | 98.90 | NMR inactive | NMR inactive |
| 13C | 1.10 | 6.728 × 107 | 1/2 |
| 14N | 99.63 | 1.934 × 107 | 1 |
| 15N | 0.37 | –2.713 × 107 | 1/2 |
| 19F | 100.00 | 25.181 × 107 | 1/2 |
| 31P | 100.00 | 10.839 × 107 | 1/2 |
2.1. Proton Isotope
The proton isotope has high natural abundance (∼100%) and the highest sensitivity of NMR receptive and stable nuclei (Table 1). Therefore, homonuclear two-dimensional (2D) 1H–1H NMR methods were attractive in the early days of NMR analysis. However, the very limited resolution of ribose and aromatic nucleobase resonances in the RNA 1H spectra restricted such studies to small RNAs (<5 kDa). Within the ribose, all protons with the exception of H1′ (i.e., H2′, H3′, H4′, H5′, and H5′′) are clustered within a narrow ∼0.6–0.8 ppm range (Figure 2A).29 Within the nucleobase, the chemical shift distribution of all protons is limited to 1 ppm or less, except for imino protons with a dispersion of ∼4 ppm (Figure 2A).30,31 Taken together, the distribution of proton resonances leads to severe chemical shift overlap that worsens as RNAs grow in size due to increased line broadening (Figure 2B). This, in part, explains the paucity of NMR structures of large RNAs (e.g., > 60 nt) (Figure 2C).
Figure 2.

(A) 1H NMR spectrum of a 61 nt RNA emphasize the narrow chemical shift dispersion of RNA protons. Here, bp and nc refer to canonical Watson–Crick base pair and noncanonical base pairs, respectively. A schematic of RNA ribose and nucleobase structures and numbering are shown above the spectrum. (B) Nucleobase region of 1H NMR spectra for RNAs of increasing size. Both signal overlap and broad linewidths worsen as RNAs grow in size. In fact, for the best visual representation, the signals corresponding to the 61 and 232 nt RNAs were increased to display them on a similar scale to that of the 14 nt RNA. (C) Histogram of RNA NMR structures in the NDB, sorted by RNA size (in nt, bin = 10 nt). Given the challenges faced by RNA NMR, there are only 23 NMR structures corresponding to RNAs > 60 nt. NDB statistics were accessed from http://ndbserver.rutgers.edu/ in January 2022.
2.2. Heteronuclear 15N and 13C Isotopes
Unlike protons, with a chemical shift span of 2–15 ppm, 15N and 13C nuclei in nucleic acids have larger chemical shift distributions among the various atomic sites. For example, 13C nuclei in RNA have chemical shifts from 61 (C5′-ribose) to 170 (nonprotonated pyrimidine nucleobase C4) ppm, and 15N nuclei from 70 (amino nitrogen) to 240 (nonprotonated purine nucleobase N7) ppm.29−31 Introduction of the 15N isotope (0.37%) into RNA nucleobases circumvents the extensive line broadening arising from the electric quadrupole moment of the naturally abundant 14N isotope (99.63%) (Table 1). Incorporation of the 15N isotope has several additional advantages. As a spin 1/2 nucleus with low gyromagnetic ratio (γ) (Table 1), the 15N isotope provides very narrow spectral lines. Nitrogen atoms, like protons and carbon, are distributed in nucleic acid major and minor grooves, and both grooves serve as important sites for metal, drug, or macromolecule interactions. However, given the wider chemical shift dispersion of 15N over the 1H nucleus and its narrower linewidths over 13C and 1H nuclei, 15N is more suited to monitor those grooves, especially in larger RNAs. However, nitrogen’s low-γ is also an “Achilles heel”. In the absence of appropriate NMR cryogenic probes and the availability of high magnetic fields, detecting low-γ nuclei such as 15N has been very unattractive. Increasing the availability of such probes is expected to reverse this trend. Nevertheless, these considerations suggest that the shortcomings of proton NMR can be overcome by heteronuclear NMR methods.32
Beginning in the 1980s, several groups introduced 15N, 2H, and 13C labels to facilitate NMR studies of RNAs and proteins.33−42 Depending on the scientific question, these labels were introduced uniformly or selectively using bacteria in vivo or enzyme catalyzed synthesis in vitro. Selective enrichment was achieved by growing auxotrophs on obligate chemically synthesized compounds. 13C-labeling of bacterial tRNAs33−35 and 15N-labeling of tRNA and 5S rRNA enabled various atomic sites in these RNAs to be monitored by NMR. Uniform 15N-labeling was also applied to 5S rRNA in vivo.36−39 To extend this labeling to additional RNAs, several research groups developed in vitro methods to convert ribonucleoside 5′-monophosphates isolated from bacteria grown on 15N-, 2H-, and 13C-sources into the corresponding triphosphates for in vitro transcription.43−47 These uniform 15N- and 13C-labeling technologies did extend the use of NMR to medium-sized RNAs (MW < 20 kDa). However, two perennial challenges of low signal-to-noise and decreased spectral resolution remained. The latter problem arises from the reintroduction of spectral overlap along the heteronuclear dimension as the RNA grows in size, and the former arises from increased relaxation that results from the slower overall tumbling of large biomolecules. The next section will describe recent labeling methods to overcome both problems.
2.3. Deuteration in Context of Heteronuclear 15N and 13C Isotopes
Deuteration (i.e., replacement of protons with deuterons) simplifies the multiplicity of spin–spin interactions, eliminates nonessential resonance lines, reduces spectral crowding, helps to identify coupling patterns, and improves calculation of coupling constants with precision.48 Given the smaller γ of the deuterium spin relative to proton (γD ≈ γH/6.5) (Table 1), the relaxation rates for deuterated nuclei are scaled proportionally by 2% [(γD/γH)2 ≈ 0.02]. By eliminating competing relaxation pathways of dipolar coupled protons, deuteration suppresses spin diffusion within a relaxation network, leading to smaller linewidths and higher signal-to-noise for the remaining protons and directly attached 13C and 15N nuclei.47−49 Given these advantages, 2H-labeling has played an important role in probing the structure, dynamics, and interactions of large RNAs by NMR.17,50−55
2.4. Fluorination in Context of 15N, 2H, and 13C Isotopes
In addition to 2H, magnetically active nuclei such as 19F have valuable spectroscopic properties that confer clear advantages in the study of macromolecular structure and conformational changes.56 These benefits include the 100% natural abundance of 19F (Table 1), a comparably large γ (94% of 1H) (Table 1), and a superior chemical shift dispersion that is ∼6-fold that of 1H.18,57 Furthermore, 19F is sensitive to changes in its local chemical environment, making it a useful probe of conformational changes.18,56,57 Finally, fluorine has an atomic radius (1.35 Å) slightly larger than that of a hydrogen (1.20 Å) but slightly smaller than that of a methyl group (2.00 Å). The 19F nuclei is therefore expected to substitute for either group without serious structural perturbations,58 making it a valuable tool for the in vitro study of medically important RNAs.59 Finally, 19F is virtually absent in biological systems and therefore offers 19F NMR a biorthogonal advantage of background-free drug screening.60 Taken together, 19F is an attractive probe for studying RNAs in solution. Details of new technologies developed to incorporate 19F into nucleobases will be presented in Section 3.1.1, and its utility to expand NMR studies to larger RNAs will be discussed in Section 5.
3. Preparation of 15N, 2H, 13C, and 19F Isotope-Labeled RNA
A number of companies (Cassia LLC, Cambridge Isotope Laboratories (CIL), INNotope, Sigma-Aldrich, and Silantes) offer isotope-labeled RNA building blocks with uniform and selective labeling. However, most comprehensive labels are made by academic laboratories using biochemical, biomass, chemical, and chemo-enzymatic approaches, as reviewed in the past.61−67 In this section, we outline promising developments in the chemical synthesis of isotope-labeled purine [i.e., adenine (Ade or A) and guanine (Gua or G)] and pyrimidine [i.e., cytosine (Cyt or C) and uracil (Ura or U) (in RNA) or thymine (Thy or T) (in DNA)] nucleobases and their incorporation into RNA. The main approaches to obtain isotope-labeled RNA are enzymatic or solid-phase chemical synthesis. The enzymatic approach involves DNA template-directed T7 RNA polymerase-based in vitro transcription using ribonucleoside 5′-triphosphates (rNTPs).44,45,68−76 The alternative method is chemical solid-phase synthesis using RNA phosphoramidites (amidites).77−80 Both methods can use unlabeled and isotope-labeled building blocks (rNTPs and amidites) to generate versatile RNA labeling patterns, as recently reviewed.61,62,66,67
3.1. Chemical Synthesis of Nucleobases
In this section, we give a general overview of the chemical synthetic methods to label RNA nucleobases at specific positions with 15N, 2H, 13C, and 19F isotopes. These nucleobases can then serve as the building blocks for the synthesis of the rNTPs or amidites that enable the eventual enzymatic or chemical production of labeled RNAs of defined sequence and length.
3.1.1. Specific 13C Labeling
3.1.1.1. Pyrimidine Synthesis with 15N, 2H, 13C, and 19F Labels
The uracil nucleobase is easily assembled using a method initially devised by Roberts and Poulter,81 later streamlined by SantaLucia and Tinoco and co-workers,71 and further improved by Kreutz and co-workers.82 In the original synthetic eight-step pathway described by Roberts and Poulter, the 13C label can be placed in any position of the six-membered ring simply by changing the 13C-source.81 SantaLucia and Tinoco and co-workers streamlined this to a three-step reaction scheme to make 13C-labeled cyanoacetyl urea from inexpensive commercially available 13C-labeled precursors.71 A slightly modified approach from Kreutz and co-workers uses bromoacetic rather than chloroacetic acid. Bromoacetic acid is the preferred starting material due to the lower costs and better handling of the cyanide reagent.74,82 Other methods with fewer steps exist such as condensation of malic or propiolic acid and urea.83,84 Even though these are straightforward two-step reactions, execution is not as convenient or cost-effective.
Using the Poulter-SantaLucia-Kreutz approach,71,74,81,82 [1-13C]- and [2-13C]-bromoacetic acid selectively incorporate 13C at uracil C4 and C5, respectively. Use of 13C-urea, on the other hand, delivers 13C at the C2 site, and that of 13C-potassium cyanide (13C-KCN) labels the C6 site. Finally, 15N-urea installs 15N at N1 and N3. All possible uracil heteroatom positions can therefore be labeled in good yields, and these reactions can be easily scaled to gram quantities.74,82 An example of a synthetic scheme using the Poulter-SantaLucia-Kreutz approach71,74,81,82 is shown for uracil C6 labeling (Scheme 1).82,85 In brief, bromoacetic acid 1 reacts with 13C-KCN and sodium carbonate (Na2CO3) in a Kolbe nitrile reaction to form 2-[cyano-13C]acetic acid 2. Treatment of 2 with urea in the presence of acetic anhydride (Ac2O) then yields a urea intermediate 3 that can be readily converted to [6-13C]-uracil 4 using a palladium catalyst (e.g., Pd/BaSO4) under hydrogen atmosphere (H2). Given that pyrimidine H5/H6 protons have three-bond scalar coupling (3JH5/H6 ≈ 8 Hz29) and strong dipolar coupling (H5–H6 distance of 2 Å) that complicate NMR experiments, selective and quantitative deuteration can be achieved by reacting 4 with triethylamine (TEA) to form the desired [6-13C, 5-2H]-uracil 5.85 Taken together, 5 was synthesized with four-steps in 63% overall yield (Scheme 1).82,85
Scheme 1. Synthetic Route to [6-13C, 5-2H]-Uracil82,85.

Given the valuable spectroscopic properties of 19F (Section 2.4), uracil can be fluorinated with the commercially available Selectfluor, as recently reported.18,57,86 This synthetic scheme is similar to that described for uracil C6 labeling (Scheme 1),82,85 except using [2-13C]-bromoacetic acid 6 as starting material. Kolbe nitrile reaction of 6 forms an intermediate 7 that reacts with 15N-urea and Ac2O to yield 8. Addition of Pd/BaSO4 in H2 to 8 then forms [5-13C, 1,3-15N2]-uracil 9, which can then be fluorinated with Selectfluor to yield [5-13C, 5-19F, 1,3-15N2]-uracil (5FU) 10. Again, selective and quantitative deuteration of H6 can remove coupling (3JH6F5 ≈ 7.1 Hz88) that complicates NMR spectra by heating 10 5FU in sodium deuteroxide (NaOD) to form [6-2H]-5FU 11.18,86,87 In summary, 11 was synthesized in five-steps with a total yield of 38% (Scheme 2).18,57,82,85,86
Scheme 2. Synthetic Route to [6-2H]-5FU18,57,82,85,86.

Finally, thymine C6 can be selectively labeled with a three-step synthesis in a manner similar to uracil labeling (Schemes 1 and 2).18,57,82,85,86 In brief, bromopropionic acid 12 is used in a Kolbe nitrile reaction followed by addition of urea and Ac2O to form intermediates 13 and 14.89,90 Then reaction of 14 with Pd/BaSO4 in H2 forms the desired [6-13C]-thymine 15 in 45% overall yield (Scheme 3).89
Scheme 3. Synthetic Route to [6-13C]-Thymine89.

3.1.1.2. Purine Synthesis with C8 Specific Labeling
As with pyrimidines, purine nucleobases can be selectively labeled with 13C and 15N isotopes using commercially available precursor compounds. In the early 1990s, SantaLucia and Tinoco and co-workers described an effective purine synthesis using 13C-formic acid to label purine C8.71 More recently, Kreutz and co-workers streamlined and improved the efficiency of such labeling in one-step reactions.75,85,91 Here, the condensation of 13C-formic acid 16 with morpholine forms morpholinium formate intermediate that immediately reacts with either 4,5,6-triaminopyrimidine 17 to yield [8-13C]-adenine 18 (Scheme 4) or 2,5,6-triaminopyrimidin-4-ol sulfate 19 to form [8-13C]-guanine 20 (Scheme 5) with 64% and 94% yield, respectively.75,85
Scheme 4. Synthetic Route to [8-13C]-Adenine75,85.

Scheme 5. Synthetic Route to [8-13C]-Guanine75,85.

3.1.1.3. Purine Synthesis with C2 Specific Labeling
As with purine C8 labeling, adenine C2 can be readily labeled. Labeling C2 is attractive because its chemical shift can monitor protonation at adenine N1,92 which cannot be achieved with 15N NMR experiments due to severe line broadening.92−94 Unlike the environments of single-stranded RNA, those in structured RNAs can shift the pKa values of protonated adenosine or cytidines significantly toward neutrality, serving both catalytic and structural functions in RNA enzymes.94−97 The 13C isotope can be incorporated at the purine C2 site starting with 5-aminoimidazole-4-carboxamide (AICA) and ethylsodium 13C-xanthate to form [2-13C]-hypoxanthine, [2-13C]-adenine, or [2-13C]-guanine.98
A preferred alternative for purine C2 labeling uses the method of Battaglia and Ouwerkerk and co-workers, wherein sodium ethoxide (C2H5ONa) mediates cyclization of ethyl cyanoacetate 21 with 13C-thiourea 22 to give [2-13C]-6-amino-2-thiouracil 23.99,100 Unlabeled sodium nitrite (NaNO2) is then used for nitrosylation (the 15N-labeled form can also be used to introduce a second isotope label) to form 24. Then sodium dithionite (Na2S2O4) mediates the reduction of the nitroso group to yield 25 followed by desulfurization over Raney-Nickel to form the diaminopyrimidine 26.101 Treatment of the product with sulfuric (H2SO4) and formic (HCOOH) acids yields [2-13C]-hypoxanthine 27.102 Subsequent reaction with phosphorus oxychloride (POCl3) and N,N-dimethylaniline (N,N-DMA) yields [2-13C]-6-chloropurine 28.103 In the final step, reaction with methanolic NH3 in a microwave reactor yields the desired [2-13C]-adenine 29 (Scheme 6).100 Alternative purine synthesis pathways have been devised to enable specific labeling of adenine C2 or any purine nitrogen position.98,100,102,104−108 We recently synthesized [7-15N]-labeled 29 through intermediates 21–23 and 15N-labeled intermediates 24–28 using the Battaglia-Ouwerkerk approach99,100 and demonstrated its utility in NMR analysis of RNA structure and dynamics (Scheme 6).104
Scheme 6. Synthetic Route to [2-13C]-Adenine.

Adapted with permission from Dayie and co-workers. Copyright 2020 Springer Nature.104 Adenine can be labeled at N7 by using 15N-labeled sodium nitrite in the second chemical step.
3.1.2. Specific 15N Labeling
Several approaches have been reported for the synthesis of atom-specific 15N-labeled nucleobases and nucleosides as well as their incorporation into the corresponding rNTPs and amidites for RNA synthesis.98,100−102,104−114 Here, we highlight those methods that allow streamlined 15N-labeled nucleobase synthesis in high yield. These labeling patterns permit direct monitoring of Watson–Crick base pairs or analysis of interconverting duplex, triplex, and quadruplex structures by multidimensional NMR.110−113
3.1.2.1. Pyrimidine N1, N3, and N4 Labeling
As described above, using the Poulter-SantaLucia-Kreutz approach,71,74,81,8215N-urea delivers 15N at uracil N1 and N3 sites. Cytosine labeling, on the other hand, occurs through uracil, given that the corresponding CTP can be built directly from enzymatic conversion (with ammonium chloride, NH4Cl) from UTP74,115 or by chemical synthesis from a transiently protected uridine amidite.85 In this way, all uracil isotope labeling patterns will be retained in CTP and cytidine amidites. Moreover, additional 15N-labeling of the cytidine N4 amino group can be achieved using 15NH4Cl in the enzymatic74 or chemical85 reaction, as will be described in Sections 3.2 and 3.3.
3.1.2.2. Purine N1, N3, N7, and N9 Labeling
Synthesis of adenine N1 labeling occurs in two-steps.101 Here, commercially available 5-aminoimidazole-4-carbonitrile 30 reacts with diethoxymethyl acetate (DEMA) to yield intermediate 31. Subsequent reaction of 31 with aqueous ammonia (NH3) readily forms the desired product [1-15N]-adenine 32 with a total yield of 60% (Scheme 7)101
Scheme 7. Synthetic Route to [1-15N]-Adenine101.

Adenine labeled at N3, on the other hand, can be synthesized in six steps.108 In brief, commercially available 4-imidazolecarboxylic acid 33 is nitrated with ammonium nitrate (NH415NO3) to afford 5-[nitro-15N]1H-imidazole-4-carboxylic acid 34. Activation of 34 with 1,1′-carbonyldiimidazole (CDI) in dimethylformamide (DMF) and excess NH3 forms carboxamide 35. Importantly, addition of 15NH4Cl in this step can also introduce a 15N label at the N1 site, permitting the eventual production of [1,3-15N2]-adenine.108 Catalytic reduction of 35 affords [5-15N]-AICA 36. Ring closure of 36 with triethyl orthoformate (HC(OC2H5)3) gives a hypoxanthine intermediate 37, which readily forms [3-15N]-6-chloropurine 38 upon chlorination with POCl3 and N,N-DMA. Finally, ammonolysis with ammonium hydroxide (NH4OH) yields the desired [3-15N]-adenine 39 with ∼47% total yield (Scheme 8).108
Scheme 8. Synthetic Route to [3-15N]-Adenine108.

Adenine N3 and its amino group can also be labeled at by 15NH4Cl and 15NH4OH in the second and final chemical steps, respectively.
In addition, purine N7 labeling is readily achieved and has been widely adapted.99,100,102,104,106,111 For example, synthesis of [7-15N]-guanine is achieved in three-steps. Nitrosylation of commercially available 2,6-diaminopyrimidin-4-ol 40 by Na15NO2 yields 2,6-diamino-5-[nitroso-15N]pyrimidin-4-ol 41. Reduction of 41 with sodium dithionite followed by acidification by H2SO4 forms 2,6-diamino-5-[amino-15N]pyrimidin-4-ol 42. In the final step, reflux with formamide (HCONH2) followed by HCOOH provides the desired [7-15N]-guanine 43 with a total yield of 65%500 (Scheme 9).
Scheme 9. Synthetic Route to [7-15N]-Guanine.

Dayie and co-workers.500
Several direct routes to 15N-labeled adenine initiate from commercially available aminopyrimidines.102,106 However, Micura and Kreutz and co-workers111 employed a sodium ethoxide mediated cyclization of 21 with 44 to form 6-amino-2-thiouracil 45.116 Subsequent nitrosylation of 45 installs the 15N label using Na15NO2 to yield the nitroso-containing 46.102 A sodium dithionite mediated reduction of the nitroso group forms 47 and desulfurization over Raney-Nickel affords 48.102 Subsequent treatment with H2SO4 and HCOOH yields hypoxanthine 49,102 which was then reacted with POCl3 and N,N-DMA to give [7-15N]-6-chloropurine 50.103 In the final step, reaction with methanolic NH3 in a microwave reactor gives the desired [7-15N]-adenine 51 with a total yield of 18% (Scheme 10).100,104,106,111 As mentioned above, we recently showcased the same synthetic scheme while also incorporating selective 13C2 labeling.104
Scheme 10. Synthetic Route to [7-15N]-Adenine.

Adapted with permission from Dayie and co-workers. Copyright 2020 Springer Nature.104 Adenine C2 can also be labeled if 13C-labeled thiourea is used as the starting material.
Finally, in the synthesis of N9-labeled adenine, 5-amino-4,6-dichloropyrimidine 52 is converted to a [9-15N]-6-chloropurine 53 using aqueous 15NH3 and DEMA.117 Then a reaction with aqueous NH3 yields the desired [9-15N]-adenine 54. This simple three-step reaction proceeds with an overall yield of 79% (Scheme 11).117
Scheme 11. Synthetic Route to [9-15N]-Adenine117.

3.1.3. Nucleobase Labels: Summary and Outlook
As described in Sections 3.1.1 and 3.1.2, and shown in Schemes 1–11, a wide range of isotope-labeled nucleobases (Table 2) are now available to the scientific community. Of all synthetic procedures, purine C8 sites are most readily labeled in one chemical step in a single day and with high yield (64–94%) (Table 2). Conversely, adenine N3 is the least readily labeled, taking 11 days (Figure 2). Adenine C2 and N7 have the lowest overall yields of 18% (Table 2). In future work, it would be advantageous to focus on improving yields and reducing the number of chemical steps. Nevertheless, these RNA labeling patterns are commonly chosen based on the experimental information required and less often dictated by the relative time and yield of the building blocks.
Table 2. Summary of All Nucleobase Labels As Outlined in Schemes 1–11.
| nucleobase label | time (days)a | chemical stepsb | yield (%) | ref |
|---|---|---|---|---|
| [8-13C]-adenine | 1 | 1 | 64 | (75), (85) |
| [8-13C]-guanine | 1 | 1 | 94 | (75), (85) |
| [2-13C]-adeninec | 2.5 | 7 (1) | 18 | (104) |
| [1-15N]-adenine | 2.5 | 2 (1) | 60 | (101) |
| [3-15N]-adenine | 11 | 6 (2) | 47 | (108) |
| [7-15N]-adeninec | 2.5 | 7 (1) | 18 | (104) |
| [7-15N]-guanined | 1.5 | 3 | 65 | |
| [9-15N]-adenine | 5.5 | 3 (3) | 79 | (117) |
| [6-13C, 5-2H]-uracil | 7 | 4 | 63 | (82), (85) |
| [5-13C, 5-19F, 6-2H]-uracil | 8 | 5 | 38 | (18), (57), (82), (85), (86) |
| [6-13C]-thymine | 2.5 | 3 | 45 | (89) |
Total reaction time was based on the time required for all chemical steps. In addition, 16 h were added for any explicit mention of overnight procedures, and 24 h were added for any chromatographic purifications.
Number in parentheses represents the number of chromatographic purification steps.
All data for [2-13C]-adenine and [7-15N]-adenine labeling came from the same doubly labeled [2-13C, 7-15N]-adenine labeling scheme.104
This synthetic procedure is from Dayie and co-workers.500
3.2. Chemo-enzymatic Labeling
With chemically synthesized isotope-labeled nucleobases in-hand, this section outlines the various enzymatic methods that can be used to build them into isotope-labeled rNTPs (and dNTPs). Alternatively, this can be accomplished using Escherichia coli(45,118−120) or Methylophilus methylotrophus(44) grown on 13C- or 15N-enriched media, as reviewed elsewhere.62,66
3.2.1. Enzymatic Coupling of Nucleobase and Ribose Sources
The first enzymatic approach to prepare isotope-labeled rNTPs was the Gilles-Schramm-Williamson pentose phosphate pathway method,65,121−124 which uses isotope-labeled d-glucoses as the precursor and requires 14 enzymes (Table 3) and several coenzymes. This method is appealing for uniform ribose labeling using commercially available uniformly 13C- or 2H-labeled d-glucoses.
Table 3. Enzymes of Glycolysis, Pentose Phosphate, and Nucleotide Biosynthesis and Salvage Pathway for rNTP Synthesis.
| enzymea | abbreviation | EC number | source |
|---|---|---|---|
| Gilles-Schramm-Williamson and Co-workers65,121−124 | |||
| Hexokinase | HXK | 2.7.1.1 | Baker’s yeast |
| Glucose-6-phosphate isomerase | PGI1 | 5.3.1.9 | Baker’s yeast |
| Glucose-6-phosphate dehydrogenase | ZWF | 1.1.1.49 | L. mesenteroides |
| Phosphogluconate dehydrogenase | GND | 1.1.1.44 | Torula yeast |
| Ribose-5-phosphate isomerase | RPI1 | 5.3.1.6 | Spinach |
| Phosphoribosylpyrophosphate synthetase | PRPPS | 2.7.6.1 | E. coli |
| Adenine phosphoribosyltransferase | APRT | 2.4.2.7 | JM109/pTTA6 |
| Uracil phosphoribosyltransferase | UPRT | 2.4.2.9 | JM109/pTTU2 |
| Xanthine-guanine phosphoribosyltransferase | XGPRT | 2.4.2.22 | JM109/pTTG2 |
| Nucleoside-monophosphate kinase | NMPK | 2.7.4.4 | Bovine liver |
| Myokinase (Adenylate kinase) | MK | 2.7.4.3 | Rabbit muscle |
| Guanylate kinase | GK | 2.7.4.8 | Porcine brain |
| 3-Phosphoglycerate mutase | YIBO | 5.4.2.1 | Rabbit muscle |
| Enolase | ENO | 4.2.1.11 | Baker’s yeast |
| Pyruvate kinase | PYKF | 2.7.1.40 | Rabbit muscle |
| Glutamate dehydrogenase (NAD(P)+) | GLUD | 1.4.1.3 | Bovine liver |
| CTP synthase | CTPS | 6.3.4.2 | JM109/pMW5 |
| l-Lactate dehydrogenase | LDH | 1.1.1.27 | Rabbit muscle |
| Dayie and Co-workers74,75,128 | |||
| Ribokinase | RK | 2.7.1.15 | E. coli |
| Creatine kinase | CK | 2.7.3.2 | Chicken muscle |
| UMP kinase | UMPK | 2.7.4.22 | E. coli |
| Serianni and Co-workers129 | |||
| Purine nucleoside phosphorylase | PNPase | 2.4.2.1 | E. coli |
| Xanthine oxidase | XO | 1.1.3.22 | Buttermilk |
| Catalase | CT | 1.11.1.6 | Bovine liver |
| Uridine phosphorylase | UPase | 2.4.2.3 | E. coli |
Given that there is overlap in the enzymes used in the methods of Schramm-Williamson and co-workers65,121−124 and Dayie and co-workers,74,75,128 only the unique enzymes are listed for the latter. All enzymes are commercially available except APRT, UPRT, XGPRT, CTPS, and RK.128 These are currently only available in a few academic laboratories. At some point, these plasmids would be available at Addgene.
In brief, hexokinase (HXK) (EC 2.7.1.1) phosphorylates 13C-labeled d-glucose 55 at its O6 position to yield glucose-6-phosphate 56. Then glucose-6-phosphate dehydrogenase (ZWF) (EC 1.1.1.49) oxidizes 56 to 6-phosphogluconate 57, and phosphogluconate dehydrogenase (GND) (EC 1.1.1.44) further oxides 57 to 58. Finally, ribose-5-phosphate isomerase (RPI1) (EC 5.3.1.6) isomerizes 58 to ribose-5-phosphate 59. Following isomerization, phosphoribosylpyrophosphate synthetase (PRPPS) (EC 2.7.6.1) pyrophosphorylates 59 at its O1′ site to yield 60. Then, adenine (APRT) (EC 2.4.2.7), guanine (XGPRT) (EC 2.4.2.22), or uridine (UPRT) (2.4.2.9) phosphoribosyl transferases facilitate the nucleophilic attack of the adenine or guanine N9 or uracil N1 to the C1′ of 60 to yield 5′-monophosphates 61–63, respectively. Adenylate (MK) (EC 2.7.4.3), guanylate (GK) (EC 2.7.4.8), or nucleoside monophosphate (NMPK) (EC 2.7.4.4) kinases phosphorylate 61–63 to form the 5′-diphosphates 64–66, respectively. Pyruvate kinase (PYKF) (EC 2.7.1.40) then catalyzes the final phosphorylation to form the 5′-triphosphates 67–69 (Scheme 12).65,121−124 Finally, UTP 69 can be converted to CTP 70 by CTP synthase (CTPS) (EC 6.3.4.2) (Scheme 12).65,121−124 Importantly, 15N-labeling of the cytidine amino group can be achieved by using 15NH3 in the final step (Scheme 12).65,121−124
Scheme 12. Enzymatic Synthesis of Isotope-Labeled rNTPs from d-Glucose Sources65,121−124.

Moreover, Williamson and Hennig and co-workers demonstrated that the Gilles-Schramm-Williamson method65,121−124 is compatible with 19F-labeled nucleobases58,125,126 by synthesizing [2-19F]-ATP,126 [5-19F]-UTP,125 and [5-19F]-CTP.125 However, d-ribose is a more cost-effective labeled precursor than d-glucose for the selective 13C- or 2H-ribose labeling of rNTPs.127
On the basis of earlier work by Whitesides and co-workers,130−132 our group truncated the relatively complex Gilles-Schramm-Williamson method65,121−124 to use 10 enzymes instead of 18, and two cofactor regeneration systems (dATP and creatine phosphate) (Table 3). This chemo-enzymatic labeling74,75,128 is a versatile technology to couple nucleobase to ribose followed by subsequent phosphorylation to the rNTP in a one-pot enzymatic reaction.74,75,128 The nucleobase and ribose building blocks can be unlabeled, isotope-labeled, chemically synthesized, or commercially available. This method therefore permits a diverse set of labeling patterns. Moreover, this approach has many advantages over previously reported de novo(72,73) or chemical133−137 synthesis methods including fewer enzymes, fewer synthetic steps, and greater yields. This method affords the facile coupling of chemically synthesized uniformly 15N- and 13C/15N-labeled uracil (Scheme 1)82,85 to commercially available unlabeled d-ribose and 13C-labeled d-ribose. The resulting uniformly 15N-labeled and uniformly 13C/15N-labeled UTP provided 338- and 14-fold savings over the commercially available material from CIL, respectively. However, the main advantage of chemo-enzymatic synthesis is the ability to generate noncommercially available atom-specific labeling patterns.
We showcased the power of this method with the synthesis of [1′,5′,6-13C3, 1,3-15N2]-pyrimidine rNTPs using six enzymes (Table 3).74 We also used this method to synthesize [1′,8-13C2]-, or [2′,8-13C2]-, or [1′,5′,8-13C3]-ATPs and -GTPs with five enzymes (Table 3).75 First, 13C-labeled D-ribose 71 was phosphorylated at its O5 position by ribokinase (RK) (EC 2.7.1.15) to yield ribose-5-phosphate 72 followed by pyrophosphorylation at the O1 site by PRPPS to afford 73. Then APRT, XGPRT, or UPRT catalyzed the nucleophilic attack of the adenine or guanine N9 or uracil N1 to the C1′ of 73 to yield 5′-monophosphates 74–76, respectively. Phosphorylation of 74–76 is achieved by MK, GK, or UMP kinase (UMPK) (EC 2.7.4.22) to form the 5′-diphosphates 77–79, respectively. Creatine kinase (CK) (EC 2.7.3.2) then facilitates the final phosphorylation to afford the 5′-triphosphates 80–82 (Scheme 13).74,75,128 Similar to the Gilles-Schramm-Williamson method,65,121−124 a final 15N label can be introduced at the CTP 83 amino group if 15NH4Cl is used alongside CTPS in the final enzymatic step (Scheme 13).74,75,128 These atom-specifically labeled rNTPs can then be used with in vitro transcription to make RNAs without any size limit. Importantly, these labeling patterns reduced spectral crowding, increased signal-to-noise ratios, facilitated direct carbon detection experiments, and eliminated 13C–13C scalar and dipolar couplings.63,74,75,86,104
Scheme 13. Enzymatic Synthesis of Isotope-Labeled rNTPs from d-Ribose Sources74,75,128.

As with the Gilles-Schramm-Williamson method,65,121−124 the approach developed by Dayie and co-workers74,75,128 is also compatible with 19F-labeled nucleobases (e.g., [2-19F]-adenine and [5-19F]-uracil18,86). It is worth noting that Serianni and co-workers have also developed a complementary approach to enzymatically couple nucleobase and ribose sources using four enzymes (Table 3).129 Their method uses hypoxanthine 84 and 1-O-acetyl-2,3,5-tri-O-benzoyl-α-d-ribofuranoside (ATBR) 85 in a Vorbrüggen reaction (detailed in Scheme 15) to yield inosine 86. Then purine nucleoside phosphorylase (PNPase) (EC 2.4.2.1) replaces the hypoxanthine moiety on the C1 position of 86 with a phosphate group to give α-d-ribofuranosyl-1-phosphate sodium salt (αR1P) 87 (Table 3).129 Then 87 is glycosylated by PNPase with adenine or guanine or by UPase (EC 2.4.3.2) with uracil to form nucleosides 88–90, respectively (Scheme 14).129 Products 88–90 can then be converted to the desired rNTP or amidite with further enzymatic or chemical synthesis.
Scheme 15. Synthetic Route to [6-13C, 5-2H]-Uridine 2′-O-TOM Amidite85.

Scheme 14. Enzymatic Synthesis of Isotope-Labeled Nucleosides from Inosine129.

3.2.2. Enzymatic Methods for Position-Specific Labeling
While these chemo-enzymatic methods enable straightforward atom-specific labeling, they rely solely on DNA template-directed T7 RNA polymerase-based in vitro transcription and are therefore unable to incorporate these labels position-specifically (e.g., nucleotide 5). Fortunately, there are two alternative enzymatic methods capable of such position-specific labeling, both of which are compatible with the isotope-labeled rNTP building blocks described above. Wang and co-workers developed a hybrid solid–liquid phase transcription technique that employs an automated robotic platform known as position-selective labeling of RNA (PLOR).138 In PLOR, the DNA template is attached to beads and RNA synthesis is initiated by the addition of T7 RNA polymerase and a mixture of three of the four rNTP building blocks (e.g., ATP, GTP, and CTP). The beads are then washed and a new rNTP mixture is added, this time containing the previously omitted building block. Thus, PLOR can incorporate any isotope-labeled rNTP (e.g., [6-13C, 5-2H]-UTP) position-specifically, assuming the desired labeling site (e.g., uridine 10) does not coincide with a stretch of identical nucleotides (e.g., UUU). While isotope labeling by PLOR has aided NMR studies of RNA,138−140 its widespread use is still limited due to the requisite equipment needed and its laborious nature.
Schwalbe and co-workers developed an alternative chemo-enzymatic approach for position-specific labeling.141 Importantly, this method uses standard laboratory equipment and commercially available enzymes T4 RNA ligase 1 (EC 6.5.1.3), recombinant shrimp alkaline phosphatase (rSAP) (EC 3.1.3.1), and T4 RNA ligase 2 (EC 6.5.1.3), making it more accessible than PLOR. In their method, a modified nucleoside 3′,5′-biphosphate is incorporated at the 3′-end of an RNA fragment by T4 RNA ligase 1 followed by dephosphorylation by rSAP and DNA-splinted ligation by T4 RNA ligase 2. This technique has been used to introduce modified nucleosides (i.e., photocaged, photoswitchable, and isotope-labeled) into RNAs up to 392 nts. While this method holds great promise for NMR applications, low yields of bis-phosphorylation (6–22%) and ligation (9–49%) reactions are a major drawback.141 More recent efforts by Schwalbe and co-workers to improve this technology include the addition of magnetic streptavidin beads as a solid-support and 5′-biotinylated RNA.142
3.2.3. rNTP Labels: Summary and Outlook
As described in Section 3.2.1 and shown in Scheme 13, the chemo-enzymatic labeling method developed by Dayie and co-workers74,75,128 permits the synthesis of a versatile assortment of rNTPs with atom-specific isotope labels (Table 4). While there are other enzymatic methods to generate both atom-specific (e.g., the Gilles-Schramm-Williamson65,121−124 or Serriani129 methods shown in Schemes 12 and 14, respectively) and position-specific (e.g., PLOR138 and the Schwalbe method141,142) labels, no other technique offers the versatility and simplicity that is afforded by the Dayie method. Our one-pot chemo-enzymatic approach can produce isotope-labeled purine and pyrimidine rNTPs in a few days and with high yield (75–95%) (Table 4). The main disadvantage of this method is the need to express and purify five noncommercial enzymes in-house (Table 3). However, providing these plasmids to Addgene will make our method widely accessible to the field.
Table 4. Summary of rNTP Labels Made from Chemo-enzymatic Synthesis74,75,128.
| rNTP labela | time (days)b | enzymatic stepsc | yield (%) | ref |
|---|---|---|---|---|
| [8-13C]-ATP | 1.5 | 1 (1) | 90 | (75) |
| [8-13C]-GTP | 1.5 | 1 (1) | 75 | (75) |
| [1′,5′,6-13C3, 1,3-15N2]-CTP | 3 | 3 (2) | 95 | (74) |
| [1′,5′,6-13C3, 1,3-15N2]-UTP | 2.5 | 2 (2) | 90 | (74) |
[8-13C]-adenine and -guanine were coupled to [1-13C]-, or [2-13C]-, or [1,5-13C2]-d-ribose to generate a variety of ATPs and GTPs.75 The [6-13C, 1,3-15N2]-uracil and -cytosine nucleobases, on the other hand, were coupled to [1′,5′-13C2]-d-ribose only.74 Nevertheless, the reported times, enzymatic steps, and yields are representative of all ATP, GTP, CTP, and UTP reactions made with this method.
Total reaction time was based on the time required for all chemical steps. In addition, 24 h were added for any chromatographic purification.
3.3. Synthesis of Labeled RNA Phosphoramidites
While the enzymatic production of RNA with isotope-labeled rNTPs44,45,69−75 is the most widely used approach to obtain labeled RNA, an attractive alternative is to use isotope-labeled amidites and solid-phase synthesis. Like PLOR introduced by Wang and co-workers138 and the chemo-enzymatic approach developed by Schwalbe and co-workers,141,142 the amidite method offers the advantage of position-specific RNA labeling. However, even though amidite labeling is currently the most effective and widely used method for position-specific labeling, its utility for NMR studies is limited to RNAs ≈ 60 nt.
3.3.1. 15N and 13C Labeling
The Kreutz and Micura groups have used isotope-labeled nucleobases to prepare 2′-O-tert-butyldimethylsilyl (tBDMS) and 2′-O-[(triisopropylsilyl)oxy]methyl (TOM) phosphoramidites for NMR studies,57,82,85,89,110,111,143,144 as recently reviewed.61 A representative example of [6-13C, 5-2H]-pyrimidine 2′-O-TOM amidite syntheses is shown in Schemes 15 and 16.85 In brief, [6-13C, 5-2H]-uracil 5 is coupled to ATBR under Vorbrüggen conditions137 to give the 2′,3′,5′-O-benzoyl (Bz)-protected 91, which is then fully deprotected to nucleoside 92 after treatment with methylamine (CH3NH2) in ethanol (C2H5OH). Addition of 4,4′-dimethoxytrityl chloride (DMT-Cl) and TOM-Cl protects the 5′- and 2′-hydroxyl (OH) to form 93 and 94, respectively. Finally, phosphitylation of the 3′-OH of 94 with 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (CEP-Cl) and N,N-diisopropylethylamine (DiPEA) yields the desired [6-13C, 5-2H]-uridine 2′-O-TOM amidite 95 with five-steps in 22% total yield (Scheme 15).85
Scheme 16. Synthetic Route to [6-13C, 5-2H]-N4–Ac-Cytidine 2′-O-TOM Amidite85.

The corresponding cytidine derivative is obtained from 94 in four additional steps (Scheme 16).85 First, the 3′-OH of 94 is transiently acetylated with Ac2O to afford 96. Then treatment with 2,4,6-triisopropylbenzenesulfonyl chloride (TiBSC) and TEA yields the 5′-O-DMT-2′-O-TOM cytidine 97, which is immediately N4-acetylated (Ac) with Ac2O to form 98. Finally, 3′-OH phosphitylation yields the desired [6-13C, 5-2H]-N4–Ac-cytidine 2′-O-TOM amidite 99. Starting from uracil 5, this cytidine synthesis has an overall yield of 14% (Scheme 16).85
In contrast to pyrimidines, the starting purine is protected before beginning the nucleosidation reaction. Representative examples of [8-13C]-purine 2′-O-TOM amidite syntheses are shown in Schemes 17 and 18.85 Starting with [8-13C]-adenine 18, N6-Bz-protected adenine 100 is formed with a yield of 86%. A subsequent Vorbrüggen reaction137 gives the 2′,3′,5′-O-Bz-protected 101, which is readily 2′,3′,5′-O-deprotected to nucleoside 102 after treatment with sodium hydroxide (NaOH) in pyridine and C2H5OH. Then, 5′-OH tritylation, 2′-OH TOM protection, and 3′-OH phosphitylation yields 103, 104, and 105, respectively. Taken together, [8-13C]-N6-Bz-adenosine 2′-O-TOM amidite 105 was synthesized with 17% total yield (Scheme 17).85
Scheme 17. Synthetic Route to [8-13C]-N6-Bz-Adenosine 2′-O-TOM Amidite85.

Scheme 18. Synthetic Route to [8-13C]-N2-iBu-Guanosine 2′-O-TOM Amidite85.

Guanosine synthesis, on the other hand, proceeds from a N2-isobutyryl (iBu) protected guanine 106 made from [8-13C]-guanine 20 with a yield of 77%. From there, however, synthesis proceed as with adenine. That is, 106 is reacted under Vorbrüggen conditions137 to form 107, which is then 2′,3′,5′-O-deprotected to nucleoside 108. Again, 5′-OH tritylation, 2′-OH TOM protection, and 3′-OH phosphitylation yields 109, 110, and 111, respectively. In summary, [8-13C]-N2-iBu-guanosine 2′-O-TOM amidite 111 was synthesized with an overall yield of 18% (Scheme 18).85 Importantly, Schemes 15−18 can be adapted to prepare 2′-O-tBDMS amidites simply by altering the 2′-OH protection reaction steps.
However, these 2′-O-tBDMS or 2′-O-TOM amidites are not suitable for producing RNAs > 60 nts. Instead, amidites with 2-cyanoethoxymethyl (CEM) as the 2′-OH protecting group145,146 are used, due to its increased coupling efficiency, which rivals that in DNA synthesis.80 Using a protocol developed by Yano and co-workers,145,146 Kreutz and co-workers prepared [6-13C, 5-2H]-pyrimidine, [8-13C]-purine, and the modified [1,3-15N2]-dihydrouridine and [2,8-13C2]-inosine 2′-O-CEM amidites.91 While the benefits of the CEM amidite method are attractive for obvious reasons, it has not gained widespread use due to the commercial unavailability of both unlabeled and isotope-labeled CEM amidites.
3.3.2. 19F Labeling and Post-transcriptional Modifications
Another benefit of labeling with amidites is the position-specific incorporation of modified building blocks. Indeed, many epigenetic and post-transcriptional modifications modulate the structure, dynamics, and folding of RNAs, and NMR is providing new insights into their functions.147 These studies have been greatly aided by the synthesis of 13C- or 15N-labeled amidites bearing modifications such as uridine 5-oxyacetic acid (cmo5U)148 and N6-methyladenine (m6A).149,150 In collaboration with the Al-Hashimi group, Kreutz and co-workers synthesized a 15N-labeled cmo5U amidite.148 Their synthetic route begins from bromoacetic acid 1 and through intermediates 112 and 113 to assemble [1,3-15N2]-uracil 114, as in Schemes 1(82,85) and 2.18,57,82,86 Then 114 was coupled to ATBR under Vorbrüggen conditions, 2′,3′,5′-O-deprotected, and hydroxylated at the C5 position to yield 115, 116, and 117, respectively. Addition of para-toluene sulfonic acid (pTSA) and dimethoxypropane ((CH3)2C(OCH3)2) then formed the 2′,3′,5′-O-protected nucleoside 118. Reacting 118 with ethyl-2-iodo acetate in C2H5OH and NaOH transformed the 5-OH into an ethylcarboxymethoxy group while also deprotecting the 5′-OH to afford 119. After transient 2′,3′-O-deprotection of 119 to form 120, the 3′- and 5′-OH were immediately protected along with 2′-O-tBDMS protection to yield 121 by adding di-tert-butylsilyl bis(trifluoromethanesulfonate) (DtBS) and tBDMS-Cl. Addition of pyridine and CH3OH to 121 forms 122, and subsequent treatment with nitrophenyl ethanol (NPE), N-dimethyl aminopyridine (DMAP), and N-ethyl-N′-(3-dimethyl aminopropyl) carbodiimide (EDC) construct the NPE-protected cmo5 group to yield 123. Reaction of 123 with hydrogen fluoride (HF) affords the 3′,5′-O-deprotected 124, which can then be 5′-O-tritylatyed to yield 125. Finally, phosphitylation of the 3′-OH of 125 with 2-cyanoethyl N,N,N′,N′-tetraisopropylphosphorodiamidite (TiPCEP) yields 126 (Scheme 19).148 Taken together, [1,3-15N2]-cmo5U 2′-O-tBDMS amidite 126 was synthesized with 15 steps in 1% total yield (Scheme 19).148
Scheme 19. Synthetic Route to [1,3-15N2]-cmo5-Uridine 2′-O-tBDMS Amidite148.

Another example from the Al-Hashimi and Kreutz groups showcases the synthesis of a 13C-labeled m6A amidite.149 Their synthetic route begins with ethyl cyanoacetate 21 and 13C-thiourea 22 and through intermediates 23–25 to assemble [2-13C]-5,6-diamino-4-pyrimidinone 26, as in Scheme 6.104 In contrast to Scheme 6, however, H13COOH was used with H2SO4 to introduce a second 13C label and form [2,8-13C2]-hypoxanthine 127. Then the familiar Vorbrüggen reaction of 127 with ATBR yields the 2′,3′,5′-O-Bz-protected 128 followed by addition of sulfuryl chloride (SO2Cl2) to yield 6-chloropurine nucleoside 129. Sequential addition of CH3NH2 in C2H5OH and then H2O affords the m6A nucleoside 130. Again, the synthetic route ends with 2′-O-tBDMS protection, 5′-O-tritylation, and 3′-O-phosphitylation to yield 131, 132, and 133, respectively (Scheme 20).149 In summary, [2,8-13C2]-N6-methyladenosine 2′-O-tBDMS amidite 133 was synthesized in 11 steps with an overall yield of 4% (Scheme 20).149
Scheme 20. Synthetic Route to [2,8-13C2]-N6-Methyladenosine 2′-O-tBDMS Amidite149.

Commercially, INNotope has 13C-labeled N1-methyladenine, m6A, and N3-methylcytidine 2′-O-tBDMS amidites available. Finally, [1,3-15N2]-pseudouridine (Ψ) amidites can be made from 15N-labeled uracil with 11 steps in 6% total yield.151
Additionally, building on the work shown in Scheme 2,18,57,82,85,86 Kreutz and co-workers showcased new methods to incorporate 19F–13C into the pyrimidine nucleobase of amidites.18,57,86 Starting from [6-13C]-uracil 4, fluorination is achieved with Selectfluor to yield 5FU 134, as in Scheme 2.18,57,82,85,86 The remaining chemical steps are similar for other 2′-O-tBDMS amidite syntheses (Schemes 19148 and 20(149)). That is, 134 is coupled to ATBR under Vorbrüggen conditions, 2′,3′,5′-O-deprotected, and then 3′,5′-O-protected and 2′-O-tBDMS protected to yield 135, 136, and 137, respectively. Finally, 137 is 5′-O-tritylated, and 3′-O-phosphitylated to yield 138 and 139, respectively (Scheme 21).57 Taken together, [5-13C, 5-19F]-uridine 2′-O-tBDMS amidite 139 was synthesized with six-steps in 8% total yield (Scheme 21).57 The corresponding cytidine derivative is obtained from 137 through intermediates 140–142 to afford the desired 143 (Scheme 22),57 as in Scheme 16.85 In summary, [5-13C, 5-19F]-N4-Ac-cytidine 2′-O-tBDMS amidite 143 was synthesized in eight-steps with an overall yield of 4% (Scheme 22).57 These labeling topologies not only capitalize on the beneficial spectroscopic properties of the 19F nuclei (Section 2.4) but also open the door to NMR studies of large RNAs, as will be discussed in greater detail in Section 5.
Scheme 21. Synthetic Route to [5-13C, 5-19F]-Uridine 2′-O-tBDMS Amidite57.

Scheme 22. Synthetic Route to [5-13C, 5-19F]-Cytidine 2′-O-tBDMS Amidite57.

3.3.3. Synergy between Phosphoramidites and Chemo-enzymatic Labeling
In principle, any nucleobase labeling scheme described in Section 3.1 can be coupled to any commercially available 13C- or 2H-labeled d-ribose (from Omicron Biochemicals or CIL) with the chemo-enzymatic method (Section 3.2) and built into an amidite with a variety of 2′-OH protecting groups (Section 3.3). Indeed, our group recently made [1′,8-13C2]-N6-Bz-adenosine 2′-O-tBDMS144 and [1′,6-13C2, 5-2H]-uridine 2′-O-CEM152 amidites via chemo-enzymatic synthesis, dephosphorylation with rSAP, and chemical synthesis. These amidites can then be used to make RNA via solid-phase synthesis. Given that the Kreutz and Micura groups have implemented a wide variety of atom-specific labeling schemes into the nucleobase of RNAs,57,61,82,85,89,110,111,143,144 this hybrid approach is only needed if ribose labeling is desired in a position-specific manner. However, INNotope and Silantes have [1′,2,8-13C3]-N6-Ac-adenosine, [1′,8-13C2]-adenosine, [1′,8-13C2]-N2-Ac-guanosine, [1′,6-13C2, 5-2H]-uridine, and [1′,6-13C2, 5-2H]-N4-Ac-cytidine 2′-O-tBDMS amidites available.
3.3.4. Phosphoramidite Labels: Summary and Outlook
As described in Sections 3.3.1 and 3.3.2, and shown in Schemes 15–22, again, a wide range of isotope-labeled amidites (Table 5) are becoming available to the scientific community. For all synthetic protocols, pyrimidine C6/C5 and purine C8 sites are most readily labeled. The production of these 2′-O-TOM amidites is streamlined85 and proceeds quickly (∼1 week) and with adequate yields (14–18%) (Table 5). The introduction of 19F labels and post-transcriptional modifications, on the other hand, dramatically increases the time of synthesis (i.e., up to 10 days) and reduces the overall reaction yields (i.e., as low as 1%) (Table 5). Nevertheless, the benefits afforded by the position-specific incorporation of these labels into RNA more than offsets these shortcomings. As with nucleobase labeling, researchers are typically motivated by the scientific question they are pursuing rather than the relative yields of each labeling reaction. Still, improvements in reaction yields and reduction in chemical steps would be advantageous for future work.
Table 5. Summary of All RNA Phosphoramidite Labels As Outlined in Schemes 15–22.
| RNA phosphoramidite labela | time (days)b | chemical stepsc | yield (%) | ref |
|---|---|---|---|---|
| [8-13C]-N6-Bz-adenosine (TOM) | 4.5 | 5 (4) | 17 | (85) |
| [2,8-13C2]-N6-methyladenosine (tBDMS) | 8 | 11 (5) | 4 | (149) |
| [8-13C]-N2-Ac-guanosine (TOM) | 5 | 5 (4) | 18 | (85) |
| [6-13C, 5-2H]-N4-Ac-cytidine (TOM) | 8 | 8 (6) | 34 | (85) |
| [5-13C, 5-19F]-N4-Ac-cytidine (tBDMS) | 10 | 8 (6) | 4 | (57) |
| [6-13C, 5-2H]-uridine (TOM) | 4 | 5 (3) | 22 | (85) |
| [5-13C, 5-19F]-uridine (tBDMS) | 7.5 | 6 (4) | 8 | (57) |
| [1,3-15N2]-cmo5-uridine (tBDMS) | 8 | 15 (3) | 1 | (148) |
The 2′-OH protecting groups are listed in the parentheses.
Total reaction time was based on the time required for all chemical steps. In addition, 16 h were added for any explicit mention of overnight procedures and 24 h were added for any chromatographic purifications.
Reactions for amidites harboring post-transcriptional modifications begin with isotope-labeled precursors whereas reactions for unmodified amidites begin with isotope-labeled protected nucleobase. Also, the number in parentheses represents the number of chromatographic purification steps.
3.4. Current State of RNA Labeling: Where We Are and Where We Are Headed
Despite the synergy between the synthesis of nucleobases (Section 3.1), rNTPs (Section 3.2), and amidites (Section 3.3), and their contribution to RNA labeling for applications with solution NMR spectroscopy, a number of insurmountable limitations remain for RNAs prepared enzymatically (using, e.g., T7 RNA polymerase) and chemically (i.e., solid-phase synthesis). The former is incapable of position-specific labeling and the latter is size limited, even though both methods can install isolated 1H–13C spin pairs into RNA that remove the 13C–13C scalar and dipolar couplings that are normally present in uniformly labeled RNA, as will be detailed in Section 4.
Again, unlike DNA template-directed in vitro transcription, a tremendous advantage to the field is that amidite labeling and solid-phase synthesis can provide direct read-outs of the biophysical consequences of post-transcriptional modifications. This will be discussed in greater detail in Section 4.2.2.3. However, despite this strength, the “size problem” of solid-phase synthesis limits the production of RNAs to ∼60 nt, beyond which it is exceedingly difficult to prepare RNA in high yield and sufficient purity for NMR studies. Even though the 2′-O-CEM91,145,146 protecting group initially held promise for synthesizing larger RNAs, it has not gained widespread use. Conversely, while much larger RNAs can be transcribed enzymatically, larger RNAs always carry with them more extensive signal overlap and broader linewidths. These complications make NMR analysis of RNAs > 60 nt extremely difficult, even when atom-specific labeling is used. However, introducing 13C–19F spin pairs into RNA,18,57,86 leveraging the spectral properties of the 15N nuclei,53,153 or combining selective deuteration with 1H NMR17,53−55 all hold promise to lessen the burden imposed by overlap and broad lines. This will be discussed in detail in Section 5.
It is clear that elucidating the structure, interactions, and dynamics of large RNAs and their complexes (e.g., those implicated in viral transcription, splicing, nuclear export, translation, packaging, and particle assembly) requires developing breakthrough technologies and new experimental strategies to solve the structures of such large RNAs rapidly and accurately. While the advances in the synthesis of atom-specific isotope-labeled rNTPs and amidites are essential first steps in this direction, the ability to incorporate these labels position-specifically will be a game changer for RNA structural and chemical biology. Overnight, it would transform our ability to perform position-specific readouts in vitro and in vivo. Moreover, it would enable scientists to peer directly into the active site of RNA enzymes, visualize the binding pockets of RNA–drug complexes, and exquisitely map out the interfaces of RNA–protein, RNA–RNA, or RNA–DNA–RNA hybrids. At least that is the dream. While we await these technological advances, the availability of these isotope-labeled RNA building blocks with diverse labeling topologies (Figure 3) still bodes well to address structural dynamic features of RNAs with NMR spectroscopy as well as MS or small angle neutron/X-ray scattering. The remaining sections highlight how the labels described in Section 3 can be exploited to study RNA structure, interactions, and dynamics by NMR spectroscopy.
Figure 3.
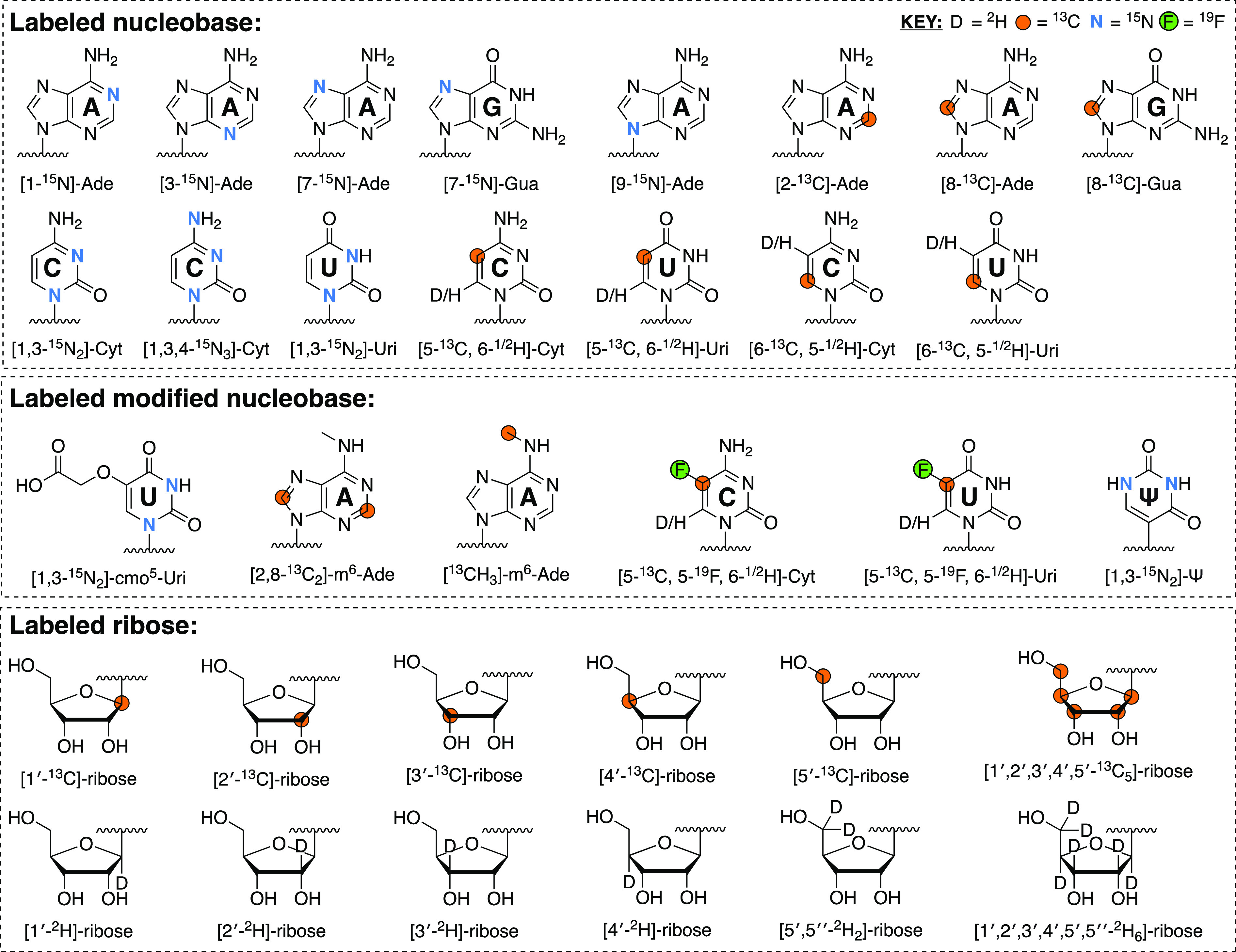
List of possible atom-specifically isotope-labeled nucleobase and ribose labeling patterns. These can be coupled to form rNTPs via chemo-enzymatic synthesis but also converted into amidites with further chemical synthesis. Nucleobase labeling patterns (unmodified and modified) are based on the synthetic schemes described in Sections 3.1 and 3.3. These need not be mutually exclusive, and some labeled sites can be incorporated simultaneously. Labeled ribose, on the other hand, is available from commercial sources (Omicron Biochemicals and CIL).
4. NMR Probes of Macromolecular Dynamics
Originating more than 45 years ago, early investigations of RNA dynamics were limited to the study of bacterial tRNAs using one-dimensional (1D) NMR methods.154 More than a decade later, development of 1D and 2D heteronuclear polarization transfer schemes to measure heteronuclear relaxation rates155−157 uniquely positioned solution NMR spectroscopy to probe protein158−161 and RNA162−166 dynamics. With multidimensional NMR spectroscopy, we can measure the dynamics of ribose, nucleobase, and phosphorus nuclei distributed along the entire RNA structure.167−171 We can especially characterize motions that range from picosecond to seconds and visualize conformers that are transient and sparsely populated (Figure 4). For these low populated states, we can extract chemical shifts (structure), rates (kinetics), and populations (thermodynamics) under various physiological conditions of temperature, salt, pH, and cellular environment. Finally, we can examine how the cellular milieu modulates the structure, dynamics, and interactions of RNA in real time.
Figure 4.

Dynamic processes in RNA and corresponding NMR methods and RNA nuclei that can be used to characterize such motions. The highlighted 15N and 13C sites have been used extensively in NMR spin relaxation and relaxation dispersion experiments,147 whereas 31P169,170 and 2H171 sites are probed less frequently. Alternative time charts can be found elsewhere.147,168,172,173
4.1. Probing Fast Motions with Uniform and Selective Labels
On the picosecond (ps)-to-nanosecond (ns) (ps-ns) time scales, spin relaxation provides information about the amplitude and time scale of motions powered by the bond vectors (e.g., 15N–1H, 13C–1H, 13C–19F, 1H–1H) reorienting relative to the external applied magnetic field (Figure 4).168,174−176 Longitudinal relaxation describes the return to the equilibrium distribution of spins along the z-axis, with a characteristic exponential time constant T1 (or rate constant R1 = 1/T1). Transverse relaxation, on the other hand, describes the decay of magnetization in the transverse xy-plane, with a characteristic decay time constant T2 (or rate constant R2 = 1/T2). Larger R2 values produce broader peaks and lower peak heights in an NMR experiment. The linewidth, defined as full-width at half-height (given in Hz), is Δν1/2 = R2/π. The heteronuclear Overhauser effect (hNOE) measures the enhancement of the heteroatom magnetization that arises from saturating the proton magnetization, and is mediated by their dipolar interaction.
For an isolated pair of spin-1/2 nuclei S and I (here, S is 15N, 13C, 31P, 19F; and I is 1H), R1, R2, and the hNOE of nucleus S are related to the rotational diffusion tensor of the molecule according to well-known relations:177,178
| 1 |
| 2 |
| 3 |
| 4,5 |
where  , σx = σ33 – σ11, σy =
σ33 – σ22, σ11, σ22, and σ33 are the principal
components of
the chemical shielding anisotropy (CSA) tensor,179,180J(ω) is a spectral density function, which
is assumed to be a Lorentzian (e.g., simplest form is
, σx = σ33 – σ11, σy =
σ33 – σ22, σ11, σ22, and σ33 are the principal
components of
the chemical shielding anisotropy (CSA) tensor,179,180J(ω) is a spectral density function, which
is assumed to be a Lorentzian (e.g., simplest form is 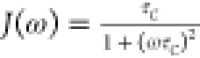 ), γi is the gyromagnetic ratio of spin i, rSI is the distance between spins I
and S, h is Planck’s constant, and Rex is the exchange contribution to R2 due to slow (i.e., microsecond-to-millisecond, μs-ms)
motions. The raw data represented by the three relaxation parameters
(R1, R2, and hNOE) reveal the nucleotide level variation of the dynamic
motions encoded in the RNA primary sequence. Additional motional variables
such as the overall correlation time (τC) and generalized
order parameter (S) can be fit within a Model Free formalism181,182 to describe fast (i.e., ps-ns) motions. Though, for reasons enumerated
below, this becomes problematic for large uniformly labeled RNAs.183
), γi is the gyromagnetic ratio of spin i, rSI is the distance between spins I
and S, h is Planck’s constant, and Rex is the exchange contribution to R2 due to slow (i.e., microsecond-to-millisecond, μs-ms)
motions. The raw data represented by the three relaxation parameters
(R1, R2, and hNOE) reveal the nucleotide level variation of the dynamic
motions encoded in the RNA primary sequence. Additional motional variables
such as the overall correlation time (τC) and generalized
order parameter (S) can be fit within a Model Free formalism181,182 to describe fast (i.e., ps-ns) motions. Though, for reasons enumerated
below, this becomes problematic for large uniformly labeled RNAs.183
The RNA motions reported by R1, R2, and hNOE are easily probed by 13C162−165,183−188 and 15N162,166,189 nuclei. 15N sites are present in the four nucleobases at the following sites: adenosine (Ade)-H2-N1, Ade-H2-N3, Ade-H8-N7, and Ade-H8-N9, guanosine (Gua)-H1-N1, Gua-H8-N7, and Gua-H8-N9, uridine (Uri)-H3-N3, and Uri-H6-N1, and cytidine (Cyt)-H6-N1 (Figures 2 and 4). These are suitable reporters of hydrogen-bonding and non-hydrogen-bond dynamics that occur in base-paired and nonbase-paired regions. However, solvent exposed imino regions are usually broadened beyond detection. Nonprotonated nitrogen sites such as Ade-N1 and Ade-N3, purine (Pur)-N7 and Pur-N9, and pyrimidine (Pyr)-N1 remain underutilized. The limited availability of directly protonated imino nitrogen probes has made protonated carbons an attractive alternative for probing RNA relaxation. These sites are found in both the ribose (C1′–C5′) and nucleobase (Ade-C2, Pur-C8, Pyr-C5, and Pyr-C6) moieties (Figures 2 and 4).
Despite the greater number of detectable 13C nuclei in RNA, complications arise for measurements and analysis of 13C relaxation. First, the carbon sites are linked by intricate multibond couplings (i.e., to 15N, 13C, and 1H nuclei) that are proximally positioned within 3 Å or less. Therefore, 13C spins do not approximate an isolated two-spin system. In uniformly labeled samples, these extensive dipolar couplings complicate 13C R1 rate measurements and analysis119,120,184,185,187,188,190−194 in biopolymers of large size (τC > 7 ns). Given this fact, our group has developed pulse schemes (based on the isolated 1H–15N backbone amide spin pair in proteins195) to leverage the isolated 1H–13C spin pairs afforded by our atom-specifically labeled RNA samples (Figure 5).
Figure 5.

Pulse scheme for transverse relaxation optimized spectroscopy (TROSY)-detected experiments for measuring (A) rotating-frame (R1ρ) (from which R2 can be calculated185,195) and (B) 13C R1 rates in selectively labeled RNA, adapted from previous reports.195 Quadrature detection and sensitivity-enhanced/gradient-selection is implemented using the Rance-Kay196,197 echo/antiecho scheme with the polarity of G1 inverted and phase Φ4 and Φ5 incremented 180° for each second FID of the quadrature pair.
Theoretical simulations of R1 rates for Pyr-C5 and Pyr-C6, ribose C1′, Ade-C2, and Pur-C8 in uniformly and selectively labeled RNAs suggest that the various 1H–13C, 13C–13C, and 13C–15N dipolar couplings (Figure 6A) present in uniformly labeled samples lead to overestimated R1 rates (Figure 6B). Moreover, this discrepancy, measured by the R1 difference (where R1 difference = [100 × (R1,uni – R1,sel)/R1,uni)]), increases with higher molecular weights and magnetic field strengths (Figure 6B). Experimental measurements with our customized pulse sequence (for selectively labeled RNA) (Figure 5) and those of others185 (for uniformly labeled RNA), corroborated our simulations, suggesting that these discrepancies in R1 cannot be wholly ignored, even for fairly isolated Ade-C2 and Pur-C8 sites.187,188 Taken together, the contribution of 13C–13C dipolar interactions needs to be explicitly taken into consideration in data analysis of uniformly labeled RNA. Spin relaxation measurements on uniformly labeled RNA from Al-Hashimi and co-workers185 demonstrate that this is not an insurmountable hurdle. Nevertheless, the focus of our discussion on RNA dynamics will center on slower conformational exchange motions, which will be discussed in Section 4.2.
Figure 6.

Dipolar couplings complicate dynamics measurements in uniformly labeled RNA. (A) Nucleobase and ribose structures shown to highlight dipolar coupling networks to nuclei of interest (i.e., Ade-C2 and Ade-C8, Uri-C6, and ribose C1′). Distances are shown in units of Å. (B) Simulated R1 rates and R1 difference (defined as above) for the nuclei highlighted in panel A. R1 simulations were carried out for 800 MHz field and R1 difference simulations were run at multiple magnetic fields. All simulations were carried out at various τC values, and additional details can be found in the original works.187,188
4.2. Probing Slow Motions with Uniform and Selective Labels: Relaxation Dispersion and Saturation Transfer Methods
Spin-1/2 nuclei with a positive gyromagnetic ratio either align parallel (α, high-populated, favorable energetic state) to the static NMR magnetic field (B0) or antiparallel (β, low-populated, unfavorable state). The net bulk magnetization, oriented parallel to B0, can be realigned with radiofrequency (RF) pulses along a direction perpendicular to B0. The magnetization then precesses about B0 at a resonant Larmor frequency (ω) characteristic of the nucleus. When Fourier transformed, this detectable oscillating time-domain signal yields a frequency-domain NMR spectrum with signals at characteristic frequencies for each nucleus. When referenced against a standard frequency (e.g., sodium-3-(trimethylsilyl)-1-propanesulfonate (DSS) for 1H), we obtain a field-independent chemical shift that is directly proportional to the energy difference between the α and β states.
For RNA exchanging between two states A and B, the chemical shift difference (Δω) between the two states and the exchange rate constant [kex, sum of the forward (kAB) and reverse (kBA) rate constants] or the exchange lifetime (τex = 1/kex) determine if two distinct NMR peaks are observed and what signal intensity and linewidth are obtained for a given nucleus.198,199 In the slow exchange regime, two distinct peaks are detected at the chemical shifts of the individual states, and the peak intensities are proportional to the populations of each state. In the fast exchange regime, kex is much larger than Δω, and therefore, a single peak is observed at the population-weighted average chemical shift. In the intermediate exchange regime, which, as its name implies, lies between the fast and slow time scales, kex ≈ Δω.
Regardless of the exchange regime, if chemical exchange is present, R2 increases by Rex, which depends on kex and Δω and can therefore be modulated by magnetic field strength.198−202 Dynamics on the intermediate and slow time scales (i.e., μs-ms) can be characterized with relaxation dispersion (RD) using R1ρ,202,203 Carr–Purcell–Meiboom–Gill (CPMG),204−206 or chemical exchange saturation transfer (CEST)207 experiments (Figure 4). Moreover, even processes slower than seconds can be studied with real-time NMR (Figure 4).208
For two-site exchange, a general expression for the R2 rate constant (RCPMG(τcp)) for state A (where pA > pB), that encompasses all conformational exchange time scales, is given by the Carver-Richards equation:198,209
| 6 |
| 7 |
| 8 |
| 9 |
| 10 |
where R2A/B and pA/B are the R2 rate and relative populations of the A/B state, respectively. A main disadvantage of the CPMG experiment is that only the magnitude (and not the sign) of Δω is obtained. Still, this disadvantage of the CPMG experiment is offset by the relative ease of its implementation and data analysis. That is, conformational exchange is easily detected by a nonflat CPMG curve when plotting R2,eff versus vCPMG (Figure 7A). Nonexchanging nuclei, on the other hand, have no dependence of R2,eff on vCPMG and therefore appear as flat curves (Figure 7A).
Figure 7.

Simulated NMR RD experiments. (A) CPMG curves for two nuclei: one in exchange (in red, Rex > 0) using the parameters kex = 794 s–1, pB = 8.7%, and Δω = 228 Hz (150 MHz 13C-Larmor frequency) and one without (in black, Rex = 0, or Δω = 0, or both), based on published data.210 (B) CEST profile for a given nuclei showing evidence of two states A and B. Calculations assumed kex = 121 s–1, pB = 10.8%, γ(1H)B0/2π = 600 MHz, Δω = −4 ppm, R1A = R1, T = 0.3 s, and the B1 fields specified on the figure. (C) R1ρ profile for a given nuclei showing evidence of two states A and B. Calculations used the same parameters as in (B) but with different B1 fields, which are again specified on the figure. As seen in the CEST and R1ρ profiles, at higher B1 fields, linewidths broaden to the point where state B becomes increasingly difficult to detect. CEST and R1ρ profiles are based on published data.211
R1ρ and CEST experiments provide more robust information regarding the chemical shifts of state B. For a two-site model, Δω, kex, and pB can be extracted from CEST profiles using the Bloch-McConnell 7 × 7 matrix (including the equilibrium magnetization terms).212−214 By combining all data sets, global kex and pB values can be fit numerically for all the CEST profiles, plotted as I/I0 versus spin-lock offset (in Hz) (Figure 7B). The 7 × 7 two-site Bloch-McConnell equation is derived from the relaxation matrix and the kinetic rate matrix for an exchanging two-site system:207,211,213,214
 |
11 |
where R1A/B, ωA/B, and ω1 are the R1 rate of the A/B state, the offset of the B1 spin-lock field from the peaks in the A/B state (in rad s–1), and the B1 field strength (in rad s–1), respectively. The evolution of magnetization for the peak in state A during the CEST spinlock period is given by
| 12 |
Similarly, under the R1ρ model for two-site exchange, the R1ρ value for state A magnetization is given by215
| 13 |
and
 |
14 |
 |
15,16 |
where Ω = ωrf – Ωobs is the difference between the resonance frequency of the observed nucleus (Ωobs) and the spinlock transmitter frequency (ωrf). For R1ρ experiments, conformational exchange can be detected by plotting R2,eff versus Ω/2π (Figure 7C). The expression for CEST and R1ρ (eqs 11–16) provide insight into the parameters that are important for acquiring useful data. For example, higher B1 fields decrease chemical shift resolution between states and also broadens linewidths (Figure 7B,C).
While almost all RD studies involve two-site systems, expressions for CPMG, R1ρ, and CEST models for characterizing N-site exchange have been described by Arthur Palmer III and co-workers.198 Indeed, work from Al-Hashimi and co-workers on Watson–Crick mismatches and base pair reshuffling in RNA feature R1ρ and CEST data that described three-site exchange.216
4.2.1. Slow Motions: Are Selective Labels Needed?
As with spin relaxation, the scalar and dipolar couplings present in uniformly labeled samples can lead to complications in RD and CEST experiments. As we have discussed elsewhere,75 numerous spectroscopic solutions have been proposed to circumvent the problems that arise from 13C–13C couplings that exist in uniformly labeled RNA. These advances include constant time evolution,217−220 adiabatic band selective decoupling,221−223 and selective cross-polarization with weak RF fields.224−226 These solutions have benefited RD and CEST experiments to varying degrees in RNA. Specifically, 13C–13C scalar couplings (e.g., C1′–C2′ or C5–C6) complicate CPMG experiments227,228 to a much larger degree than both CEST and R1ρ. However, these couplings still pose a problem to CEST229,230 and R1ρ(211) and oscillations are sometimes observed in the decay profiles of C1′ and C6 nuclei. Moreover, as with spin relaxation, these couplings must be explicitly taken into consideration in data analysis. The number of coupled homogeneous differential equations (n) is equal to (2 × 4m) – 1, where m is the number of weakly coupled nuclear spins in an m-spin system. Therefore, for 1-, 2-, and 3-spin systems, n = 7, 31, and 127, respectively.213,214,230 This transforms the CEST matrix (eq 11) from 7 × 7 to 31 × 31 for 13C–13C scalar coupled spin pairs found in the nucleobase and ribose moieties. Atom-specific labeling (Section 3), on the other hand, circumvents this problem entirely, and dramatically simplifies NMR spectra, especially when incorporated position-specifically via solid-phase synthesis (Section 3.3). However, a drawback for selective labels is the obvious reduction of probe sites.
Nevertheless, using both selective and uniformly labeled RNA, CEST and R1ρ experiments have now been applied to the protonated nucleobase (Pyr-C5 and Pyr-C6, Pur-C8, and Ade-C2) and ribose (C1′-C5′) carbons, the nucleobase imino (Gua-N1 and Thy/Uri-N3) and amino (Gua-N2) nitrogen, nucleobase (Uri-H3, Gua-H1, Ade-H2, Pur-H8, Pyr-H5, and Pyr-H6) and ribose H1′ protons, as well as nonprotonated (Gua-N7, Ade-N1, and Pur-N7) and amino (Cyt-N4) nitrogen sites (Figure 4).75,147,167,211,231−233 In practice, CPMG experiments are solely implemented on selectively labeled RNA, and mainly from our group75,144,227 and the Kreutz group,82,85,89,210,234 though not exclusively.193 CEST and R1ρ, on the other hand, have been used to great success with uniformly labeled RNA by the Al-Hashimi,31,147,167,235−245 Petzold,246,247 and Zhang211,231,248−250 groups. Moreover, Petzold and co-workers have developed a SELective Optimized Proton Experiment (SELOPE) approach251 that can be implemented with R1ρ and CEST252 experiments using unlabeled samples. The rest of this section will highlight recent examples of RD experiments on labeled (selectively and uniformly) and unlabeled RNA.
4.2.2. Examples of Relaxation Dispersion Experiments in Selectively Labeled RNA
As highlighted above, implementation of RD experiments on selectively labeled RNA circumvents all complications from strong 13C–13C scalar couplings and permits straightforward data analysis. The following sections will be devoted to showcasing examples of CPMG, CEST, and R1ρ experiments performed on selectively labeled RNAs. Specifically, we will highlight recent work from our group227,233 using isotope-labeled rNTPs and from Kreutz and Al-Hashimi and co-workers150 using isotope-labeled amidites with post-transcriptional modifications.
4.2.2.1. CPMG in Atom-Specifically Labeled RNA
Until recently, CPMG experiments to measure the chemical shifts of nucleobase methine 1H and ribose methylene C5′(H2) in a low populated, transient state (i.e., state B) were not available. This gap existed, in part, due to complications from 13C–13C scalar couplings. To fill this knowledge gap, our group adapted single-quantum (SQ) 1H CPMG experiments previously designed for methyl groups in protein side-chains253,254 to obtain CPMG data for the selectively labeled ([2′,8-13C2]-ATP, [1′,6-13C2]-CTP, [1′,8-13C2]-GTP, and [2′,6-13C2]-UTP) bacterial A-site RNA (Figures 8A,B).227
Figure 8.

(A) Pulse scheme for SQ 1H CPMG experiment for selectively labeled RNA,227 adapted from previous reports.253,254 (B) Secondary structure of the 27 nt bacterial A-site RNA with all nucleotides harboring isotope labels shown bolded in orange. Nucleotides that were found to be in exchange are circled. Exchange parameters were extracted from a global fit of the CPMG data (i.e., G19-C8 and A21-C8). (C) Pulse scheme for methylene CH21H–13C TROSY-detected CPMG experiment for selectively labeled RNA,227 adapted from previous reports.256 (D) Secondary structure of the 29 nt iron-responsive element (IRE) RNA with isotope labels and nucleotides in exchange presented as in panel B. Exchange parameters were extracted from a global fit of the CPMG data (i.e., C18–C5′, C18–C1′, and C18–C6) and likely refer to a structural rearrangement in the IRE triloop. Orange circles and D refer to 13C and 2H nuclei, respectively. Additional details can be found in the original work.227
The SQ 1H CPMG experiment was amenable to Pur-H8 sites, detecting exchange in G19 and A21. The extracted exchange rate (kex = 4000 ± 100 s–1) from a global fit was consistent with that determined from a standard 1H–13C TROSY CPMG experiment (kex = 3000 ± 800 s–1), demonstrating that these new experiments are feasible for RNA (Figure 8B).227 Moreover, these data agree with R1ρ measurements on uniformly labeled RNA from Al-Hashimi and co-workers, which suggests that each measurement, using various methods and labeling techniques, is picking up fundamental motions within this RNA.255 In addition, these SQ experiments could provide important data on 1H chemical shifts, which are currently lacking, such as ribose H1′ and Pyr-H6. In the latter case, however, the presence of Pyr-H5 can cause dispersive CPMG patterns for the H6 site.227 Fortunately, Pyr-H5 deuteration is easily achieved (Scheme 1),85 and therefore, this experiment can be readily implemented to obtain data for Pyr-H6 sites.
Our group also designed a CH21H–13C TROSY-detected CPMG pulse sequence (Figure 8C)227,256 to leverage the isolated 13C spin at the ribose C5′ position (Figure 4) afforded by our chemo-enzymatic labeling (Sections 3.1 and 3.2).74,75,128 This new CPMG experiment was implemented using the selectively labeled ([1′,5′,6-13C3, 5-2H]-CTP) iron-responsive element (IRE) RNA and detected exchange in C18–C5′ (Figure 8D).227 These data were then globally fit with additional CPMG data from other nuclei to obtain chemical shift (Δω = 2.5 ± 0.2 ppm), population (pB = 1.7 ± 0.2%), and exchange rate (kex = 3600 ± 300 s–1) information that suggests a significant structural rearrangement in the IRE triloop (Figure 8D).227
4.2.2.2. CEST in Atom- and Position-Specifically Labeled RNA
In addition to using selective labels to benefit CPMG experiments, they can also be used to simplify CEST experiments. Specifically, our group combined enzymatic ligation, chemo-enzymatic labeling, and newly developed CEST experiments (Figure 9A) to study the conformational equilibria of the SAM-II riboswitch in the apo (ligand-free) state.233 To understand the formation of the SAM metabolite-binding pocket, a SAM-II RNA was constructed via DNA splinted ligation with T4 DNA ligase (EC 6.5.1.1) of two RNA fragments: an unlabeled 31 nt acceptor fragment and a [1′,6-13C2, 5-2H]-CTP labeled 21 nt donor fragment. This strategy enabled position-specific labeling, given that there was only one cytidine (C43) in the donor sequence and therefore permitted direct monitoring of the G22–C43 base pair interaction in the SAM binding pocket. Moreover, the isolated spin pair labeling topology enabled the design of a 1H CEST experiment, and simplified setup and analysis of 1H and 13C CEST experiments without complications from 13C–13C couplings to Cyt-C1′ and Cyt-C6 sites.229
Figure 9.

(A) Pulse scheme for 13C and 1H CEST experiments with temperature compensation (TC) and 1H decoupling (1H Dec) for selectively labeled RNA,233 adapted from previous reports.211,213 (B) Secondary structure of the 52 nt SAM-II riboswitch RNA. C43 position-specific labeling is shown bolded in orange and circled to indicate that it was the subject of CEST experiments. Exchange parameters were extracted from a global fit of the CEST data (i.e., C43–C1′ and C43–C6) and reveal a transition from an open to a closed conformation that resembles the SAM-bound form. Orange circles and D refer to 13C and 2H nuclei, respectively. Additional details can be found in the original work.233
To leverage the labeling scheme, our group designed a new 13C CEST experiment based on previous pulse schemes211,213 and used it on the apo SAM-II riboswitch (Figure 9A). The CEST profiles of C43–C1′ and C43–C6 indicated two states of the free SAM-II riboswitch: one that matched the resonance of the ligand-free, highly populated conformation (i.e., state A) and another that matched the ligand-bound, transient conformation (i.e., state B) (Figure 9B).233 We then used our new 1H CEST experiment (Figure 9A) to indirectly obtain the C43–H1′ chemical shift of state A and B.233 In agreement with the 13C data, the 1H chemical shift of state B matched the ligand-bound SAM-II (Figure 9B).233 Taken together, these results suggest that the apo SAM-II exists in a dynamic equilibrium (kex = 36 ± 3 s–1) between an open (highly populated, pA = 90.5 ± 0.5%) and a partially closed (transient, pB = 9.5 ± 0.5%) state (Figure 9B).233 Moreover, these results underscore the emerging consensus that transient, low populated states likely enhance rapid ligand recognition and therefore play a potentially ubiquitous role in RNA recognition and signaling.
4.2.2.3. R1ρ and CEST in Atom- and Position-Specifically Labeled RNA Harboring Post-transcriptional Modifications
Perhaps the greatest benefit of selective labeling is the ability to monitor the structural dynamic consequences of epigenetic and post-transcriptional modifications. Using labels created by Kreutz and co-workers, the Al-Hashimi group has been at the forefront of exploring how these modifications alter the dynamic ensembles of nucleic acids.148−150,232,257−260 One such example is m6A, an abundant RNA post-transcriptional modification that modulates gene expression,261−263 viral lifecycles,264−270 and other biological phenomena.271−274 Recent work from the Al-Hashimi group demonstrated that m6A preferentially slows RNA duplex annealing with minimal effect on the rate of duplex melting.149 The effect of m6A on hybridization kinetics stands in contrast to the effect of mismatches. Mismatches also slow the rate of duplex annealing but dramatically increase the rate of duplex melting.275−277 Of critical importance, the methylamino group of the m6A nucleobase can form two rotational isomers that interconvert on the millisecond time scale278,279 (Figure 10A). The preferred syn isomer (i.e., high-populated, state A) cannot form a canonical Watson–Crick base pair with uridine due to a steric clash between the uridine keto group and the methylamino278−280 and is therefore mismatch-like (Figure 10A). Instead, when base-paired with uridine, the methylamino rotates into the anti isomer (i.e., transient, state B) to form a canonical Watson–Crick m6A:U base pair (Figure 10A).
Figure 10.

(A) Equilibrium between syn:anti conformations of the m6A nucleobase and the types of base pairing that each conformation can adopt.278−280 (B) Secondary structure of the 9 and 18 nt ssRNA and dsRNA that were position-specifically labeled with isotope-labeled m6A, as shown bolded in orange and circled to indicate that it was the subject of RD and CEST experiments. RNA samples harboring m6A were either made with [2,8-13C2]-m6A (top) or [13CH3]-m6A (bottom) labels to obtain 13C RD and CEST data for CH3 (methyl), C2, or C8 sites. (C) Schematic of the four-state CS-and-IF kinetic model with rate constants shown from RD and CEST data collected at 65 °C.150 Orange circles refer to 13C. Additional details can be found in the original work.150
Kinetic mechanisms that involve binding and conformational change can occur via pathways wherein the conformational change occurs prior to (conformational selection, CS) or post (induced fit, IF) binding. Al-Hashimi and co-workers employed their recently developed RD-based and CEST experiments31,147,167,236−245 to measure hybridization kinetics of single- and double-stranded RNA (ssRNA and dsRNA, respectively) harboring atom- and position-specifically labeled m6A probes (i.e., [2,8-13C2]-m6A or [13CH3]-m6A) (Figure 10B) to determine how m6A modulates hybridization.150 In this way, they had direct readouts of the effects of the m6A isomers on Watson-Crick or mismatch-like hybridizations. They showed that m6A with the methylamino group in the anti conformation forms a Watson–Crick base pair with uridine that transiently isomerizes on the millisecond time scale to a singly hydrogen-bonded (pB ≈ 1%) mismatch-like conformation, with the methylamino group in the syn conformation.150 This rapid interconversion between Watson–Crick and mismatch forms, combined with different syn:anti preferences in ssRNA and dsRNA states, hints at how m6A slows duplex annealing without affecting melting via two pathways in which isomerization occurs before (CS) or after (IF) duplex annealing (Figure 10C).150
4.2.3. Examples of Relaxation Dispersion Experiments without Selectively Labeled RNA
While RD experiments work well with selective labels, it is not a prerequisite, as long as care is taken to either minimize strong 13C–13C scalar couplings (i.e., probe nuclei where these are minimized) or take them into consideration in data analysis. The following sections will be devoted to showcasing examples of CEST and R1ρ experiments performed without selectively labeled RNA. We will highlight recent work from the Zhang249 and Petzold246 groups using uniformly 13C/15N-labeled rNTPs and also new experiments from the Petzold251 and Al-Hashimi252 groups that require no labels at all.
4.2.3.1. CEST and R1ρ Experiments in Uniformly Labeled RNA
RNA dynamics can regulate biological processes from transcription to translation. One such example is the Bacillus cereus fluoride riboswitch RNA (Figure 11A), which has been characterized extensively by Zhang and co-workers.249 Here, they showed that the riboswitch aptamer adopts a near-identical solution structure249 with (holo) and without (apo) the fluoride ligand, in agreement with X-ray crystal structures (Figure 11B).281 Moreover, these states also undergo very similar dynamic motions across a wide range of time scales, as determined from 13C spin relaxation rates and residual dipolar couplings (RDCs).249 However, functional assays indicate that transcription activation is fluoride-dependent and kinetically driven.281,282 What is more, mutational studies suggest that a prefolded “holo-like” apo state lowers the kinetic barrier for ligand binding, enabling efficient fluoride sensing to activate transcription below or near the toxicity threshold. Until recently, the mechanism by which this holo-like apo state achieves the “transcription–off” state remained unknown.249
Figure 11.

(A) Secondary structure of the 48 nt fluoride riboswitch aptamer RNA with domains labeled by color. (B) Solution NMR structure249 of the apo aptamer (B. cereus) (PDB ID, 5KH8) (left) compared to crystal structures281 of the apo (PDB ID, 4ENC) and holo (PDB ID, 3VRS) aptamers (T. petrophila). In solution, the aptamer adopts near-identical structures in the apo and holo forms, in agreement with crystallography.249,281 (C) Schematic of the equilibrium between the highly populated apo state (i.e., State A) and the transient “holo-like” conformation of the apo state (i.e., State B). Exchange parameters were extracted from a global fit of the CEST data. The transient “holo-like” conformation of the apo state (i.e., State B) occludes the formation of a reverse Hoogsteen base pair in the highly populated conformation of the apo state (i.e., State A) to signal transcription termination. Additional details can be found in the original work.249
To shed light on this mechanism, 13C CEST experiments were implemented on uniformly 13C/15N-GTP- and uniformly 13C/15N -ATP/UTP labeled aptamer RNA. For the holo state, CEST profiles consistently showed a single, highly populated conformation (i.e., state A).249 A subset of CEST profiles of the apo state, on the other hand, revealed the presence of conformational exchange to a transient state (i.e., state B).249 The nucleotides that undergo chemical exchange were localized to the junction of P3, J13, J23, and the 3′-tail, suggesting a concerted transition (Figure 11A,B). A global fit of the CEST data determined the population (pB = 1.4 ± 0.1%) and lifetime (τB = 3.2 ± 0.3 ms) of the holo-like conformation of the apo state. This fleeting process differentiates the apo and holo states. Rapid transition to the holo-like conformation of the apo state, which unlocks the highly conserved reverse Hoogsteen base pair located at the interface between the aptamer domain and the expression platform, promotes strand invasion and provides a path to transcription termination (Figure 11C).249 Conversely, fluoride binding allosterically suppresses access to the holo-like conformation of the apo state, ensuring continued gene transcription.249
RNA can also regulate the initial steps of translational silencing. This process begins when a mature miRNA binds to the human Argonaute (Ago2) protein to form the RNA-induced silencing complex (RISC).283 Here, translational silencing is predominantly controlled by base pair complementarity between the “seed” region of the miRNA and the target mRNA.283−288 Interestingly, data from bioinformatics,289 structural,290 and mutational291 studies all suggest that RNA dynamics within the central bulge of miRNA–mRNA duplex likely controls mRNA fate. To test this hypothesis, Petzold and co-workers used R1ρ experiments coupled with molecular dynamics simulations to investigate the structural dynamics of the interaction between miR-34a and its miRNA recognition element in the 3′-UTR of silent information regulator 1 mRNA (mSirt1) (Figure 12A).246 Using these experiments, the authors detected chemical exchange in nucleotides surrounding the central bulge of the miR-34a–mSirt1 duplex (Figure 12A).246 In this structural rearrangement, the gG8:tC17 base pair (‘g’ refers to the guide miRNA and ‘t’ refers to the target mRNA) interconverts from a highly populated (i.e., state A) to a transient (i.e., state B) conformation. A global fit of the R1ρ data determined the exchange rate (kex = 1008 ± 12 s–1) and population (pB = 0.9 ± 0.2%) of the unfavorable state (Figure 12B),246 and the chemical shift data246 from 1H (Δω −2.20 ± 0.02 ppm) and 15N (Δω −3.8 ± 0.1 ppm) R1ρ experiments suggest formation of a gG8:tU21 wobble pair (Figure 12B),246 a motif seen in other miRNAs.292,293 Taken together, the miR-34a–mSirt1 binding site is in equilibrium between a highly populated 7-mer-A1 and a transient 8-mer-GU (Figure 12B).
Figure 12.

(A) Secondary structure of the mir-34a–mSirt1 duplex.246 Nucleotides that were found to be in exchange are circled. (B) Schematic of the equilibrium between the highly populated 7-mer-A1 and transient 8-mer-GU mir-34a–mSirt1 duplex. Exchange parameters were extracted from a global fit of the R1ρ data (i.e., gG8-H1, gG8-N1, gG8-C8, tC17-C1′, tA19-C8, tU20-C1′, tU21-C6, and tA22-C8). The boxed nucleotides represent the critical switch from the gG8:tC17 to gG8:tU21 base pair. (C) Replotted functional data246 showing the percentage of target repression for each miR-34a duplex. The transient 8-mer-GU reduces target mRNA levels ∼2-fold compared to the highly populated 7-mer-A1. The 8-mer-GU duplex therefore represents a “catalytically competent RISC”. Additional details can be found in the original work.246
Next, Petzold and co-workers sought to investigate the functional relevance of the 8-mer-GU unfavorable state using a functional assay and simulated complexes of human Ago with 7-mer-A1 and 8-mer-GU 34a–mSirt1 duplexes. Interestingly, the switch to the 8-mer-GU state causes coaxial stacking of the seed and supplementary helix fitting into Ago2, reminiscent of an active state in prokaryotic Ago.294,295 Moreover, this state enhances repression of the target mRNA, revealing the importance of this dynamic miRNA–mRNA structure (Figure 12C).
4.2.3.2. CEST and R1ρ Experiments in Unlabeled RNA
After highlighting RD experiments in selectively and uniformly labeled RNA, we will conclude this section with a brief description of two pulse schemes that permit R1ρ(251) and CEST252 experiments in unlabeled RNA. In the first, Petzold and co-workers developed a SELOPE homonuclear NMR method by combining the selective excitation of specific groups of protons and reduction of spectral crowding using coherence transfer among scalar coupled protons. These coherence transfers take advantage of uniform homonuclear three bond scalar coupling between H5 and H6 for pyrimidine bases (3JH5H6 ∼ 8–10 Hz) or between H1' and H2' for ribose in C2'-endo conformation (3JH1'H2' ∼ 8Hz). Taken together, SELOPE permits well-resolved 1D and 2D spectra of unlabeled RNA. To demonstrate the utility of this method to probe RNA transient states, Petzold and co-workers adapted the SELOPE pulse scheme to include a spinlock (Figure 13A).251 As proof-of-concept, this new 1H R1ρ SELOPE experiment was used to detect chemical exchange in the central bulge region of the GUG RNA (Figure 13B).251 Importantly, this method enables the use of lower spinlock strengths to measure slower exchange time scales.251
Figure 13.

(A) Pulse scheme for 1H R1ρ experiment on unlabeled RNA using a SELOPE readout.251 (B) Secondary structure of the 25 nt GUG RNA. Nucleotides that were found to be in exchange are circled, and representative exchange parameters for U7–H6 (shaded green) are shown. (C) Pulse scheme for 1H CEST experiment on unlabeled RNA again with a SELOPE readout.252 (D) Equilibrium between Watson–Crick and Hoogsteen A:T and G:C base pairs is depicted. Exchange rates and populations are shown based on previous reports,236 and reporter imino protons are shaded red. Additional details can be found in the original works.251,252
Building on this work, Al-Hashimi and co-workers introduced a high-power 1H CEST SELOPE experiment to target imino protons (Figure 13C).252 To showcase the utility of this method, Watson–Crick to Hoogsteen exchange of G:C and A:T base pairs in DNA were monitored (Figure 13D).252 Importantly, Al-Hashimi and co-workers showed that short relaxation delays could be used to characterize fast exchange events that effectively minimize NOE effects that complicate 1H RD experiments.213,252,296−301 Moreover, their approach also takes advantage of high-power RF fields recently shown to extend the time scale sensitivity of CEST to include faster exchange processes that were traditionally only detectable by R1ρ.252,302 While both of these exciting new advancements hold promise, they are inherently limited to small RNAs. However, RNA biology is increasingly moving toward larger and larger RNAs. This important topic will be the focus of the next section.
5. Exploring Large Molecular Weight Nucleic Acids
Until now, most studies of RNA dynamics have focused on relatively small systems. However, RNA structural biology is increasingly moving toward larger RNAs, especially as cryo-EM advances in resolution and popularity.303−305 Solution NMR spectroscopy, unlike X-ray crystallography and cryo-EM, is the only biophysical technique capable of probing nucleic acid conformational dynamics on a wide range of time scales in a physiologically relevant environment. Moreover, four technological advances have expanded the types of problems that NMR can tackle in studies of molecular nanomachines on the order of 1 MDa: (1) commercial availability of high-field magnets, up to 1.2 GHz 1H Larmor frequency (28.2 T),306 (2) specialized probes (e.g., cryo-probes) that minimize noise associated with the NMR signals,307 (3) new isotope labeling technologies (described in Section 3), and (4) the design of new NMR experiments that are tailored to the isotope labeling used (described in Section 4). Our final section will describe how new labeling efforts can be leveraged to study large RNAs by NMR.
Taking inspiration from protein labeling,308 our group installed 19F directly next to a 13C spin in UTP (Scheme 2)18,86 and showed that, compared to the 13C–1H spin pair, 13C–19F had better sensitivity, ∼6-times wider chemical shift dispersion and ∼2-times more favorable relaxation properties in 2D TROSY experiments (Figure 14A,B).18 Importantly, the high sensitivity of the 19F nucleus enabled clear delineation of helical and nonhelical regions as well as G:U wobble and Watson–Crick base pairs (Figure 14C).18,57 In parallel, the Kreutz group incorporated 13C–19F into both cytidine and uridine 2′-O-tBDMS amidites (Schemes 21 and 22) to show the same effect in RNAs made by solid-phase synthesis.57 These findings suggest that structural insights are possible even in the absence of complete resonance assignment, which is a substantial bottleneck for large RNAs. Moreover, these labeling schemes can be readily adapted to exploit 19F CEST and R1ρ experiments, which have been described for proteins up to 360 kDa.309−314
Figure 14.

(A) Simulated 13C R2 rates (linewidths) at various magnetic fields in RNAs of various molecular weights (as measured by τC) to compare the relative TROSY effects of 13C–1H or 13C–19F spin pairs, which are shown on the left. (B) Same simulated rates as in A for each spin pair but only at the magnetic fields corresponding to the narrowest linewidths (smallest R2) (600 and 950 MHz for 13C–19F or 13C–1H, respectively, as shown by the gray lines in panel A).18 (B) Representative 19F–13C TROSY spectrum to highlight the dispersion of resonances based on secondary structure (i.e., G:U wobble base pairs, nonhelical nucleotides, and helical A:U base pairs). Spectral regions are colored to match the respective uridines on the corresponding RNA. Additional details can be found in the original works.18,57
An alternative approach to heteronuclear correlation experiments that include nuclei with large CSAs such as 13C and 19F, which broaden the lines of nearby protons, was recently described by Bax and Summers and co-workers.53 This approach capitalizes on the favorable relaxation properties of 15N nuclei within RNA nucleobases. Here, they employed 1H–15N heteronuclear multiple quantum coherence (HMQC) experiments to measure 15N R1ρ rates and RDCs in a large 232 nt (∼78 kDa) RNA by selectively transferring magnetization from Ade-H2 to Ade-N1/N3 via the two-bond scalar coupling (2JHN ≈ 15 Hz29) (Figure 15). Extending this method in the same 232 nt RNA, Marchant and Tjandra and co-workers measured pseudocontact shifts using the two-bond scalar coupling of Ade-H8-N7 (2JH8N7 ≈ 11 Hz29) and Ade-H8-N9 (2JH8N9 ≈ 8 Hz) for coherence transfer.315 Importantly, both experiments would benefit by atom-specific labeling. That is, selective 15N labeling of Ade-N1 or Ade-N3 (described in Section 3.1.2.2) (Schemes 7 and 8) would reduce crowding considerably and direct magnetization transfer uniquely from Ade-H2 rather than splitting it between both sites, as in uniformly 15N-labeled RNA (Figure 15). In the same way, selective 15N labeling of Pur-N7 or Pur-N9 (described in Section 3.1.2.2) (Schemes 9–11) would again reduce crowding and direct coherence transfer uniquely from Pur-H8 (Figure 15). However, selective pulses can be deployed to affect the same decrowding and directed transfer. These labeling topologies could then be leveraged to probe two-bond 15N CEST in large RNAs, as recently described by Zhang and co-workers.231
Figure 15.

Examples of possible routes for coherence transfer between two-bond scalar couplings between Ade–H2-N1 and Ade–H2-N3 (2JH2N1 ≈ 15 Hz29), Pur–H8-N7 (2JH8N7 ≈ 11 Hz29) and Pur–H8-N9 (2JH8N9 ≈ 8 Hz29) in uniformly and selectively labeled RNA.
Our final example of harnessing the versatility of the 15N nuclei is one that exploits the narrow linewidths in 1H–15N TROSY experiments compared to its 1H–13C counterpart (Figure 16A). Here, Fürtig and Schwalbe and co-workers investigated several reconstituted complexes between an adenine-sensing riboswitch and the 30S ribosome by NMR spectroscopy.153 In particular, they implemented the 1H–15N BEST-TROSY pulse scheme316,317 to obtain incredible spectra for a massive-sized complex (>800 kDa) (Figure 16B). Taken together, Fürtig and Schwalbe and co-workers succeed in illuminating the dynamic network that links the riboswitch RNA regulator, adenine ligand inducer, and ribosome protein S1 modulator during translation initiation.153
Figure 16.

(A) Simulated TROSY-detected R2 rates (linewidths) for 13C and 15N nuclei at 800 MHz. The 15N nuclei has significantly narrower linewidths (smaller R2) than that of 13C. (B) Structural model of the >800 kDa complex of adenine-sensing riboswitch bound to the 30S ribosomal complex (structural model built from PDB IDs 1Y26 and 5MLN).153 Additional details can be found in the original work.153
6. Conclusion
In humans, RNA transcripts exceed the number of proteins decoded by more than 50-fold, and yet the number of RNA structures remains below 1%, preventing a detailed understanding of RNA function (Figure 1). It is therefore essential to characterize RNA structural dynamics and interactions at atomic resolution to fill this critical knowledge gap. Over the past two decades, NMR spectroscopy has assumed a central role in RNA structure determination and probing dynamics on functionally relevant time scales in solution. In this review, we have summarized some of the many contributions of solution NMR studies to our knowledge of RNA structure, dynamics, and interactions, as facilitated by isotope labeling. We have presented a detailed overview of the prominent role stable isotopes continue to play in NMR analysis of nucleic acids (Section 2), how to synthesize these labels and introduce them into RNA (Section 3), and how these labels benefit NMR analysis. Of great interest, selective isotope labeling alleviates spectral crowding and removes dipolar and scalar couplings to simplify NMR dynamics measurements and data interpretation (Section 4). Moreover, recent advances in labeling open the door to study large RNA systems in a manner previously thought impossible (Section 5). As new orthogonal technologies are developed to better characterize the functional relevance of RNA, their structural dynamics will become increasingly important to better understand the cellular basis of RNA-based dysfunction that leads to various diseases. We anticipate that several imminent breakthrough technologies, some described herein, will enable NMR spectroscopy to continue to play a pivotal role in shining light on the structure, dynamics, and function of the important “dark matter of the genome”, RNA in vitro, in cellulo, and in vivo.
Acknowledgments
T.K.D. acknowledges support from NIH (U54AI50470) and NSF (1808705 and DBI1040158). The authors would like to thank Hashim Al-Hashimi (Duke University), Jeffery Davis (University of Maryland, College Park), Christoph Kreutz (University of Innsbruk, Austria), and Lyle Isaacs (University of Maryland, College Park) for helpful discussions. The authors extend a special thanks to Christoph Kreutz for an ongoing collaboration.
Glossary
Abbreviations
- ABR
1-O-acetyl-2,3,5-tri-O-benzoyl-α-d-ribofuranoside
- Ac
acetyl
- Ac2O
acetic anhydride
- Ade
adenine
- Ago2
Argonaute 2
- AICA
5-aminoimidazole-4-carboxamide
- amidites
phosphoramidites
- APRT
adenine phosphoribosyltransferase
- ATBR
1′-O-acetyl-(2′,3′,5′-O-tribenzoyl)-β-d-ribofuranose
- ATP
adenosine 5′-triphosphate
- B0
reference magnetic field strength
- B1
radio frequency field applied during RD or CEST
- Bz
benzoyl
- (CH3)2C(OCH3)2
dimethoxypropane
- C2H5OH
ethanol
- C2H5ONa
sodium ethoxide
- CH3NH2
methylamine
- CDI
1,1′-carbonyldiimidazole
- CEM
2-cyanoethoxymethyl
- CEP-Cl
2-cyanoethyl N,N-diisopropylchlorophosphoramidite
- CEST
chemical exchange saturation transfer
- CIL
Cambridge Isotope Laboratories
- CK
creatine kinase
- cmo5U
5-oxyacetic acid
- CPMG
Carr–Purcell–Meiboom–Gill
- Cryo-EM
cryo-electron microscopy
- CS
conformational selection
- CTP
cytidine 5′-triphosphate
- CTPS
CTP synthase
- Cyt
cytosine
- DEMA
diethoxymethyl acetate
- DiPEA
N,N-diisopropylethylamine
- DMAP
N-dimethyl aminopyridine
- DMF
dimethylformamide
- DMT
4,4′-dimethoxytrityl
- DMT-Cl
4,4′-dimethoxytrityl chloride
- dsRNA
double-stranded RNA
- DSS
sodium-3-(trimethylsilyl)1-propanesulfonate
- DtBS
di-tert-butylsilyl bis(trifluoromethanesulfonate)
- EDC
N-ethyl-N′-(3-dimethyl aminopropyl) carbodiimide
- GK
guanylate kinase
- GND
phosphogluconate dehydrogenase
- GTP
guanosine 5′-triphosphate
- Gua
guanine
- H2SO4
sulfuric acid
- HC(OC2H5)3
triethyl orthoformate
- HCOOH
formic acid
- HCOONH2
formamide
- HF
hydrogen fluoride
- HMQC
heteronuclear multiple quantum coherence
- hNOE
heteronuclear Overhauser effect
- HXK
hexokinase
- iBu
isobutyryl
- IF
induced fit
- KCN
potassium cyanide
- kAB
forward reaction rate
- kBA
reverse reaction rate
- kex
exchange rate
- MK
myokinase
- mRNA
messenger RNA
- MS
mass spectrometry
- ms
millisecond
- mSirt1
silent information regulator 1 mRNA
- m6A
N6-methyladenine
- Na2CO3
sodium carbonate
- NaNO2
sodium nitrite
- Na2S2O4
sodium dithionite
- NaOD
sodium deuteroxide
- NaOH
sodium hydroxide
- NDB
nucleic acid database
- NH3
ammonia
- NH4Cl
ammonium chloride
- NH415NO2
15N-labeled ammonium nitrate
- NH4OH
ammonium hydroxide
- NMPK
nucleoside-monophosphate kinase
- NMR
nuclear magnetic resonance
- N,N-DMA
N,N-dimethylaniline
- NPE
nitrophenyl ethanol
- ns
nanosecond
- nt
nucleotide
- OH
hydroxyl
- pA
population of state A
- pB
population of state B
- PDB
protein data bank
- Pd/BaSO4
palladium catalyst
- PNPase
purine nucleoside phosphorylase
- POCl3
phosphorus oxychloride
- PRPPS
phosphoribosylpyrophosphate synthetase
- ps
picosecond
- pTSA
para toluene sulfonic acid
- Pur
purine
- PYKF
pyruvate kinase
- Pyr
pyrimidine
- RD
relaxation dispersion
- RDC
residual dipolar coupling
- Rex
chemical exchange contribution to R2
- RF
radiofrequency
- RISC
RNA-induced silencing complex
- RK
ribokinase
- rNTPs
ribonucleoside 5′-triphosphates
- RPI1
ribose-5-phosphate isomerase 1
- rSAP
recombinant shrimp alkaline phosphatase
- R1
longitudinal relaxation rate
- R1ρ
rotating-frame transverse relaxation rate
- R2
transverse relaxation rate
- S
order parameter
- SELOPE
selective optimized proton experiment
- SO2Cl2
sulfuryl chloride
- SQ
single quantum
- ssRNA
single-stranded RNA
- tBDMS
2′-O-tert-butyldimethylsilyl
- TC
temperature compensation
- TEA
triethylamine
- TiBSC
2,4,6-triisopropylbenzenesulfonyl chloride
- TiPCEP
2-cyanoethyl N,N,N′,N′-tetraisopropylphosphorodiamidite
- TOM
2′-O-[(triisopropylsilyl)oxy]methyl
- TROSY
transverse relaxation optimized spectroscopy
- T1
longitudinal relaxation time
- T2
transverse relaxation time
- UMPK
uridine monophosphate kinase
- UPRT
uracil phosphoribosyltransferase
- Ura
uracil
- UTP
uridine 5′-triphosphate
- XGPRT
xanthine-guanine phosphoribosyltransferase
- ZWF
glucose-6-phosphate dehydrogenase
- αR1P
α-d-ribofuranose 1-phosphate
- Δω
chemical shift difference
- ω
Larmor frequency
- τC
overall correlation time
- τex
exchange lifetime
- μs
microsecond
- 1D
one-dimensional
- 2D
two-dimensional
- 5FU
[5-13C, 5-19F, 6-2H]-uracil
Biographies
Theodore K. Dayie obtained his International Baccalaureate from the United World Colleges of the Atlantic (Llantwit Major, Wales), his Bachelor of Arts in Physics (Hamilton College, NY), and studied biophysics at Harvard University and obtained his PhD in 1996 with Gerhard Wagner. He was a Jane Coffin Childs postdoctoral fellow with Jamie Williamson at the Massachusetts Institute of Technology from 1996–1998, and then at The Scripps Research Institute (TSRI) from 1998–2000. After work at TSRI, he became an assistant researcher at the Lerner Research Institute of the Cleveland Clinic from 2000–2008. In 2008, he accepted an offer to become an associate professor at the University of Maryland, College Park. He is currently a full professor in the Department of Chemistry and Biochemistry with research focused on NMR spectroscopy of RNA molecules and their complexes. His group continues to make contributions to NMR studies of RNA structure, dynamics, and interactions.
Lukasz T. Olenginski graduated Phi Beta Kappa from Franklin and Marshall (F&M) College with degrees in Biochemistry and Molecular Biology and Applied Mathematics in 2015. At F&M, he worked under the dual mentorship of Christine Phillips-Piro and Scott Brewer to investigate the biophysical properties of unnatural amino acid-harboring proteins. After F&M, Lukasz spent the next two years working under Peggy Hsieh at the National Institutes of Health to expand his interests into the molecular and cellular fields by characterizing the mitotic roles of nuclear membrane proteins. Returning to the biophysical side, he joined the group of Theodore K. Dayie in 2018. Since then, he has dived head-first into the RNA-world, balancing his time between the development of new RNA labeling strategies and probing the conformational dynamics of viral RNAs.
Kehinde M. Taiwo earned a Biochemistry degree from Bowen University in Nigeria in 2011. She then went on to earn a Master’s degree in Clinical Neuropsychiatry from the University of Birmingham in the United Kingdom in 2013 and another Master’s degree in Biochemistry from California State University, Long Beach (CSULB) in 2017. While at CSULB, she received the Maria Erlinda Co Sarna Scholarship for prospects in research. In 2018, she joined the group of Theodore K. Dayie and has been involved in biophysical research using NMR and other techniques to probe the structures and conformational dynamics of RNAs to understand their functions as well as to develop RNA-based therapeutics. She is a member of the National Organization for the Professional Advancement of Black Chemists and Chemical Engineers (NOBCChE).
The authors declare no competing financial interest.
References
- Crick F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- Cech T. R.; Steitz J. A. The Noncoding RNA Revolution-Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Zhang N. N.; Li X. F.; Deng Y. Q.; Zhao H.; Huang Y. J.; Yang G.; Huang W. J.; Gao P.; Zhou C.; Zhang R. R.; et al. Thermostable mRNA Vaccine Against COVID-19. Cell 2020, 182, 1271–1283. 10.1016/j.cell.2020.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho W.; Gao M.; Li F.; Li Z.; Zhang X. Q.; Xu X. Next-Generation Vaccines: Nanoparticle-Mediated DNA and mRNA Delivery. Adv. Healthc. Mater. 2021, 10, e2001812 10.1002/adhm.202001812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandbrink J. B.; Shattock R. J. RNA Vaccines: A Suitable Platform for Tackling Emerging Pandemics?. Front. Immunol. 2020, 11, 608460. 10.3389/fimmu.2020.608460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J. S.; O’Halloran J. A.; Kalaidina E.; Kim W.; Schmitz A. J.; Zhou J. Q.; Lei T.; Thapa M.; Chen R. E.; Case J. B.; et al. SARS-CoV-2 mRNA Vaccines Induce Persistent Human Germinal Centre Responses. Nature 2021, 596, 109–113. 10.1038/s41586-021-03738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett K. S.; Edwards D. K.; Leist S. R.; Abiona O. M.; Boyoglu-Barnum S.; Gillespie R. A.; Himansu S.; Schäfer A.; Ziwawo C. T.; DiPiazza A. T.; et al. SARS-CoV-2 mRNA Vaccine Design Enabled by Prototype Pathogen Preparedness. Nature 2020, 586, 567–571. 10.1038/s41586-020-2622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L. A.; Anderson E. J.; Rouphael N. G.; Roberts P. C.; Makhene M.; Coler R. N.; McCullough M. P.; Chappell J. D.; Denison M. R.; Stevens L. J.; et al. An mRNA Vaccine against SARS-CoV-2 — Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan M. J.; Lyke K. E.; Kitchin N.; Absalon J.; Gurtman A.; Lockhart S.; Neuzil K.; Raabe V.; Bailey R.; Swanson K. A.; et al. Phase I/II Study of COVID-19 RNA Vaccine BNT162b1 in Adults. Nature 2020, 586, 589–593. 10.1038/s41586-020-2639-4. [DOI] [PubMed] [Google Scholar]
- Ban N.; Nissen P.; Hansen J.; Moore P. B.; Steitz T. A. The Complete Atomic Structure of the Large Ribosomal Subunit at 2.4 Å Resolution. Science 2000, 289, 905–920. 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- Jobe A.; Liu Z.; Gutierrez-Vargas C.; Frank J. New Insights into Ribosome Structure and Function. Cold Spring Harb. Perspect. Biol. 2019, 11, a032615 10.1101/cshperspect.a032615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson M. R.; Lessen L. N.; Wang J.; Prabhakar A.; Corsepius N. C.; Green R.; Puglisi J. D. Mechanisms That Ensure Speed and Fidelity in Eukaryotic Translation Termination. Science 2021, 373, 876–882. 10.1126/science.abi7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang J.; Wan R.; Yan C.; Shi Y. Structural Basis of Pre-mRNA Splicing. Science 2015, 349, 1191–1198. 10.1126/science.aac8159. [DOI] [PubMed] [Google Scholar]
- Wilkinson M. E.; Charenton C.; Nagai K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. 10.1146/annurev-biochem-091719-064225. [DOI] [PubMed] [Google Scholar]
- Wan R.; Bai R.; Zhan X.; Shi Y. How Is Precursor Messenger RNA Spliced by the Spliceosome?. Annu. Rev. Biochem. 2020, 89, 333–358. 10.1146/annurev-biochem-013118-111024. [DOI] [PubMed] [Google Scholar]
- Alexander R. P.; Fang G.; Rozowsky J.; Snyder M.; Gerstein M. B. Annotating Non-Coding Regions of the Genome. Nat. Rev. Genet. 2010, 11, 559–571. 10.1038/nrg2814. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Keane S. C.; Su Z.; Irobalieva R. N.; Chen M.; Van V.; Sciandra C. A.; Marchant J.; Heng X.; Schmid M. F.; et al. Structure of the 30 KDa HIV-1 RNA Dimerization Signal by a Hybrid Cryo-EM, NMR and Molecular Dynamics Approach. Structure 2018, 26, 490–498. 10.1016/j.str.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becette O. B.; Zong G.; Chen B.; Taiwo K. M.; Case D. A.; Dayie T. K. Solution NMR Readily Reveals Distinct Structural Folds and Interactions in Doubly 13C- and 19F-Labeled RNAs. Sci. Adv. 2020, 6, eabc6572 10.1126/sciadv.abc6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abulwerdi F. A.; Xu W.; Ageeli A. A.; Yonkunas M. J.; Arun G.; Nam H.; Schneekloth J. S.; Dayie T. K.; Spector D.; Baird N.; Le Grice S. F. J. Selective Small-Molecule Targeting of a Triple Helix Encoded by the Long Noncoding RNA, MALAT1. ACS Chem. Biol. 2019, 14, 223–235. 10.1021/acschembio.8b00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind K. E.; Du Z.; Fujinaga K.; Peterlin B. M.; James T. L. Structure-Based Computational Database Screening, In Vitro Assay, and NMR Assessment of Compounds That Target TAR RNA. Chem. Biol. 2002, 9, 185–193. 10.1016/S1074-5521(02)00106-0. [DOI] [PubMed] [Google Scholar]
- Johnson E. C.; Feher V. A.; Peng J. W.; Moore J. M.; Williamson J. R. Application of NMR SHAPES Screening to an RNA Target. J. Am. Chem. Soc. 2003, 125, 15724–15725. 10.1021/ja037499s. [DOI] [PubMed] [Google Scholar]
- Thompson R. D.; Baisden J. T.; Zhang Q. NMR Characterization of RNA Small Molecule Interactions. Methods 2019, 167, 66–77. 10.1016/j.ymeth.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binas O.; Jesus V. de; Landgraf T.; Völklein A. E.; Martins J.; Hymon D.; Bains J. K.; Berg H.; Biedenbänder T.; Fürtig B.; et al. 19F NMR-Based Fragment Screening for 14 Different Biologically Active RNAs and 10 DNA and Protein Counter-Screens. ChemBioChem. 2021, 22, 423–433. 10.1002/cbic.202000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc R. M.; Kasprzak W. K.; Longhini A. P.; Olenginski L. T.; Abulwerdi F.; Ginocchio S.; Shields B.; Nyman J.; Svirydava M.; Del Vecchio C.; et al. Structural Insights of the Conserved “Priming Loop” of Hepatitis B Virus Pre-Genomic RNA. J. Biomol. Struct. Dyn. 2021, 1–13. 10.1080/07391102.2021.1934544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker A.; Weigand J. E.; Akabayov S. R.; Altincekic N.; Bains J. K.; Banijamali E.; Binas O.; Castillo-Martinez J.; Cetiner E.; Ceylan B.; et al. Secondary Structure Determination of Conserved SARS-CoV-2 RNA Elements by NMR Spectroscopy. Nucleic Acids Res. 2020, 48, 12415–12435. 10.1093/nar/gkaa1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson D. J. Historical and Contemporary Stable Isotope Tracer Approaches to Studying Mammalian Protein Metabolism. Mass Spectrom. Rev. 2018, 37, 57–80. 10.1002/mas.21507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein M. A.; King K. F.; Zhou X. J.. Handbook of MRI Pulse Sequences; Elsevier Inc., 2004. [Google Scholar]
- Harris R. K.; Becker E. D.; Cabral de Menezes S. M.; Goodfellow R.; Granger P. NMR Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts. IUPAC Recommendations 2001. International Union of Pure and Applied Chemistry. Physical Chemistry Division. Commission on Molecular Structure and Spectroscopy. Magn. Reson. Chem. 2002, 40, 489–505. 10.1002/mrc.1042. [DOI] [PubMed] [Google Scholar]
- Wijmenga S. S.; van Buuren B. N. M. The Use of NMR Methods for Conformational Studies of Nucleic Acids. Prog. Nucl. Magn. Reson. Spectrosc. 1998, 32, 287–387. 10.1016/S0079-6565(97)00023-X. [DOI] [Google Scholar]
- Wang Y.; Han G.; Jiang X.; Yuwen T.; Xue Y. Chemical Shift Prediction of RNA Imino Groups: Application toward Characterizing RNA Excited States. Nat. Commun. 2021, 12, 1595. 10.1038/s41467-021-21840-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y.; Kellogg D.; Kimsey I. J.; Sathyamoorthy B.; Stein Z. W.; McBrairty M.; Al-Hashimi H. M. Characterizing RNA Excited States Using NMR Relaxation Dispersion. Methods Enzymol. 2015, 558, 39–73. 10.1016/bs.mie.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnieders R.; Keyhani S.; Schwalbe H.; Fürtig B. More than Proton Detection—New Avenues for NMR Spectroscopy of RNA. Chem.-Eur. J. 2020, 26, 102–113. 10.1002/chem.201903355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt P. G.; Sierzputowska-Gracz H.; Agris P. F. Internal Motions in Yeast Phenylalanine Transfer RNA from 13C NMR Relaxation Rates of Modified Base Methyl Groups: A Model-Free Approach. Biochemistry 1987, 26, 8529–8534. 10.1021/bi00400a006. [DOI] [PubMed] [Google Scholar]
- Oslen J. I.; Schweizer M. P.; Walkiw I. J.; Hamill W. D.; Horton W. J.; Grant D. M. Carbon-13 NMR Relaxation Studies of Pre-Melt Structural Dynamics in [4-13C-Uracil] Labeled E. Coli Transfer RNA1Val*. Nucleic Acids Res. 1982, 10, 4449–4464. 10.1093/nar/10.14.4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeyuki Y.; Usuki K. M.; Ziro Y.; Susumu N.; Tatsuo M. Tertiary Structures of Escherichia Coli tRNA as Studied by NMR Spectroscopy with 13C-Labeling Method. FEBS Lett. 1980, 119, 77–80. 10.1016/0014-5793(80)81001-5. [DOI] [PubMed] [Google Scholar]
- Davis D. R.; Yamaizumi Z.; Nishimura S.; Poulter C. D. 15N-labeled 5S RNA. Identification of uridine base pairs in Escherichia coli 5S RNA by 1H-15N multiple quantum NMR. Biochemistry 1989, 28, 4105–4108. 10.1021/bi00435a072. [DOI] [PubMed] [Google Scholar]
- Griffey R. H.; Davis D.; Yamaizumi Z.; et al. 15N-Labeled Escherichia Coli tRNAfMet, tRNAGlu, tRNATyr, and tRNAPhe. Double Resonance and Two-Dimensional NMR of N1-Labeled Pseudouridine. J. Biol. Chem. 1985, 260, 9734–9741. 10.1016/S0021-9258(17)39300-6. [DOI] [PubMed] [Google Scholar]
- Roy S.; Papastavros M. Z.; Redfield A. G.; Sanchez V. Nitrogen-15-Labeled Yeast tRNAPhe: Double and Two-Dimensional Heteronuclear NMR of Guanosine and Uracil Ring NH Groups. Biochemistry 1984, 23, 4395–4400. 10.1021/bi00314a024. [DOI] [PubMed] [Google Scholar]
- Gewirth D. T.; Abo S. R.; Leontis N. B.; Moore P. B. Secondary Structure of 5S RNA: NMR Experiments on RNA Molecules Partially Labeled with Nitrogen-15. Biochemistry 1987, 26, 5213–5220. 10.1021/bi00390a047. [DOI] [PubMed] [Google Scholar]
- Torchia D. A.; Sparks S. W.; Bax A. Staphylococcal Nuclease: Sequential Assignments and Solution Structure. Biochemistry 1989, 28, 5509–5524. 10.1021/bi00439a028. [DOI] [PubMed] [Google Scholar]
- LeMaster D. M.; Richards F. M. NMR Sequential Assignment of Escherichia Coli Thioredoxin Utilizing Random Fractional Deuteriation. Biochemistry 1988, 27, 142–150. 10.1021/bi00401a022. [DOI] [PubMed] [Google Scholar]
- Kime M. J.; Moore P. B. Escherichia Coli Ribosomal 5S RNA-Protein L25 Nucleoprotein Complex: Effects of RNA Binding on the Protein Structure and the Nature of the Interaction. Biochemistry 1984, 23, 1688–1695. 10.1021/bi00303a017. [DOI] [PubMed] [Google Scholar]
- Santoro J.; King G. C. A Constant-Time 2D Overbodenhausen Experiment for Inverse Correlation of Isotopically Enriched Species. J. Magn. Reson. 1992, 97, 202–207. 10.1016/0022-2364(92)90250-B. [DOI] [Google Scholar]
- Batey R. T.; Inada M.; Kujawinski E.; Puglisi J. D.; Williamson J. R. Preparation of Isotopically Labeled Ribonucleotides for Multidimensional NMR Spectroscopy of RNA. Nucleic Acids Res. 1992, 20, 4515–4523. 10.1093/nar/20.17.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonowicz E. P.; Sirr A.; Legault P.; Jucker F. M.; Baer L. M.; Pardi A. Preparation of 13C and 15N Labelled RNAs for Heteronuclear Multi-Dimensional NMR Studies. Nucleic Acids Res. 1992, 20, 4507–4513. 10.1093/nar/20.17.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonowicz E. P.; Pardi A. Three-Dimensional Heteronuclear NMR Studies of RNA. Nature 1992, 355, 184–186. 10.1038/355184a0. [DOI] [PubMed] [Google Scholar]
- Nikonowicz E. P.; Michnicka M.; Kalurachchi K.; Dejong E. Preparation and Characterization of a Uniformly 2H/15N-Labeled RNA Oligonucleotide for NMR Studies. Nucleic Acids Res. 1997, 25, 1390–1396. 10.1093/nar/25.7.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földesi A.; Trifonova A.; Kundu M. K.; Chattopadhyaya J. The Synthesis of Deuterionucleosides. Nucleosides, Nucleotides and Nucleic Acids 2000, 19, 1615–1656. 10.1080/15257770008045450. [DOI] [PubMed] [Google Scholar]
- Sattler M.; Fesik S. W. Use of Deuterium Labeling in NMR: Overcoming a Sizeable Problem. Structure 1996, 4, 1245–1249. 10.1016/S0969-2126(96)00133-5. [DOI] [PubMed] [Google Scholar]
- Kang M.; Eichhorn C. D.; Feigon J. Structural Determinants for Ligand Capture by a Class II PreQ1 Riboswitch. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E663–E671. 10.1073/pnas.1400126111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd P. S.; Brown J. B.; Brown J. D.; Catazaro J.; Chaudry I.; Ding P.; Dong X.; Marchant J.; O’Hern C. T.; Singh K.; Swanson C.; Summers M. F.; Yasin S. NMR Studies of Retroviral Genome Packaging. Viruses 2020, 12, 1115. 10.3390/v12101115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotar A.; Foley H. N.; Baughman K. M.; Keane S. C. Advanced Approaches for Elucidating Structures of Large RNAs Using NMR Spectroscopy and Complementary Methods. Methods 2020, 183, 93–107. 10.1016/j.ymeth.2020.01.009. [DOI] [PubMed] [Google Scholar]
- Marchant J.; Bax A.; Summers M. F. Accurate Measurement of Residual Dipolar Couplings in Large RNAs by Variable Flip Angle NMR. J. Am. Chem. Soc. 2018, 140, 6978–6983. 10.1021/jacs.8b03298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane S. C.; Heng X.; Lu K.; Kharytonchyk S.; Ramakrishnan V.; Carter G.; Barton S.; Hosic A.; Florwick A.; Santos J.; et al. Structure of the HIV-1 RNA Packaging Signal. Science 2015, 348, 917–921. 10.1126/science.aaa9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza V.; Dey A.; Habib D.; Summers M. F. NMR Structure of the 101-Nucleotide Core Encapsidation Signal of the Moloney Murine Leukemia Virus. J. Mol. Biol. 2004, 337, 427–442. 10.1016/j.jmb.2004.01.037. [DOI] [PubMed] [Google Scholar]
- Rastinejad F.; Evilia C.; Lu P. Studies of Nucleic Acids and Their Protein Interactions by 19F NMR. Methods Enzymol. 1995, 261, 560–575. 10.1016/S0076-6879(95)61025-1. [DOI] [PubMed] [Google Scholar]
- Nußbaumer F.; Plangger R.; Roeck M.; Kreutz C. Aromatic 19F-13C TROSY—[19F, 13C]-Pyrimidine Labeling for NMR Spectroscopy of RNA. Angew. Chem., Int. Ed. 2020, 59, 17062–17069. 10.1002/anie.202006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L. G.; Hennig M. 19F-Site-Specific-Labeled Nucleotides for Nucleic Acid Structural Analysis by NMR. Methods Enzymol. 2016, 566, 59–87. 10.1016/bs.mie.2015.05.015. [DOI] [PubMed] [Google Scholar]
- Huang W.; Varani G.; Drobny G. P. 13C/15N-19F Intermolecular REDOR NMR Study of the Interaction of TAR RNA with Tat Peptides. J. Am. Chem. Soc. 2010, 132, 17643–17645. 10.1021/ja1051439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutz C.; Kählig H.; Konrat R.; Micura R. A General Approach for the Identification of Site-Specific RNA Binders by 19F NMR Spectroscopy: Proof of Concept. Angew. Chem., Int. Ed. 2006, 45, 3450–3453. 10.1002/anie.200504174. [DOI] [PubMed] [Google Scholar]
- Becette O.; Olenginski L. T.; Dayie T. K. Solid-Phase Chemical Synthesis of Stable Isotope-Labeled RNA to Aid Structure and Dynamics Studies by NMR Spectroscopy. Molecules 2019, 24, 3476. 10.3390/molecules24193476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asadi-Atoi P.; Barraud P.; Tisne C.; Kellner S. Benefits of Stable Isotope Labeling in RNA Analysis. Biol. Chem. 2019, 400, 847–865. 10.1515/hsz-2018-0447. [DOI] [PubMed] [Google Scholar]
- Dayie K. T. Key Labeling Technologies to Tackle Sizeable Problems in RNA Structural Biology. Int. J. Mol. Sci. 2008, 9, 1214–1240. 10.3390/ijms9071214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K.; Miyazaki Y.; Summers M. F. Isotope Labeling Strategies for NMR Studies of RNA. J. Biomol. NMR 2010, 46, 113–125. 10.1007/s10858-009-9375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L. G.; Tolbert T. J.; Williamson J. R. Preparation of Specifically 2H- and 13C-Labeled Ribonucleotides. Methods Enzymol. 2000, 317, 18–38. 10.1016/S0076-6879(00)17004-1. [DOI] [PubMed] [Google Scholar]
- Olenginski L. T.; Taiwo K. M.; LeBlanc R. M.; Dayie T. K. Isotope-Labeled RNA Building Blocks for NMR Structure and Dynamics Studies. Molecules 2021, 26, 5581. 10.3390/molecules26185581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchanka A.; Kreutz C.; Carlomagno T. Isotope Labeling for Studying RNA by Solid-State NMR Spectroscopy. J. Biomol. NMR 2018, 71, 151–164. 10.1007/s10858-018-0180-7. [DOI] [PubMed] [Google Scholar]
- Helmling C.; Keyhani S.; Sochor F.; Fürtig B.; Hengesbach M.; Schwalbe H. Rapid NMR Screening of RNA Secondary Structure and Binding. J. Biomol. NMR 2015, 63, 67–76. 10.1007/s10858-015-9967-y. [DOI] [PubMed] [Google Scholar]
- Milligan J. F.; Uhlenbeck O. C. Synthesis of Small RNAs Using T7 RNA Polymerase. Methods Enzymol. 1989, 180, 51–62. 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- Milligan J. F.; Groebe D. R.; Witherell G. W.; Uhlenbeck O. C. Oligoribonucleotide Synthesis Using T7 RNA Polymerase and Synthetic DNA Templates. Nucleic Acids Res. 1987, 15, 8783–8798. 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaLucia J. Jr; Shen L. X.; Cai Z.; Lewis H.; Tinoco I. Jr Synthesis and NMR of RNA with Selective Isotopic Enrichment in the Bases. Nucleic Acids Res. 1995, 23, 4913–4921. 10.1093/nar/23.23.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheisz H. L.; Szymczyna B. R.; Scott L. G.; Williamson J. R. Enzymatic De Novo Pyrimidine Nucleotide Synthesis. J. Am. Chem. Soc. 2011, 133, 297–304. 10.1021/ja1059685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheisz H. L.; Szymczyna B. R.; Scott L. G.; Williamson J. R. Pathway Engineered Enzymatic De Novo Purine Nucleotide Synthesis. ACS Chem. Biol. 2008, 3, 499–511. 10.1021/cb800066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarado L. J.; LeBlanc R. M.; Longhini A. P.; Keane S. C.; Jain N.; Yildiz Z. F.; Tolbert B. S.; D’Souza V. M.; Summers M. F.; Kreutz C.; et al. Regio-Selective Chemical-Enzymatic Synthesis of Pyrimidine Nucleotides Facilitates RNA Structure and Dynamics Studies. Chembiochem 2014, 15, 1573–1577. 10.1002/cbic.201402130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhini A. P.; LeBlanc R. M.; Becette O.; Salguero C.; Wunderlich C. H.; Johnson B. A.; D’Souza V. M.; Kreutz C.; Dayie T. K. Chemo-Enzymatic Synthesis of Site-Specific Isotopically Labeled Nucleotides for Use in NMR Resonance Assignment, Dynamics and Structural Characterizations. Nucleic Acids Res. 2016, 44, e52 10.1093/nar/gkv1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C.; Zheng M.; Rüdisser S. A Simple and Efficient Method to Reduce Nontemplated Nucleotide Addition at the 3′ Terminus of RNAs Transcribed by T7 RNA Polymerase. RNA 1999, 5, 1268–1272. 10.1017/S1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese C. B. The Chemical Synthesis of Oligo- and Poly-Ribonucleotides. Nucleic Acids and Molecular Biology 1989, 3, 164–181. 10.1007/978-3-642-83709-8_11. [DOI] [Google Scholar]
- Ogilvie K. K.; Sadana K. L.; Thompson E. A.; Quilliam M. A.; Westmore J. B. The Use of Silyl Groups in Protecting the Hydroxyl Functions of Ribonucleosides. Tetrahedron Lett. 1974, 15, 2861–2863. 10.1016/S0040-4039(01)91763-0. [DOI] [Google Scholar]
- Ogilvie K. K.; Theriault N.; Sadana K. L. Synthesis of Oligoribonucleotides. J. Am. Chem. Soc. 1977, 99, 7741–7743. 10.1021/ja00465a073. [DOI] [PubMed] [Google Scholar]
- Beaucage S. L.; Caruthers M. H. Deoxynucleoside Phosphoramidites—A New Class of Key Intermediates for Deoxypolynucleotide Synthesis. Tetrahedron Lett. 1981, 22, 1859–1862. 10.1016/S0040-4039(01)90461-7. [DOI] [Google Scholar]
- Roberts J. L.; Poulter C. D. 2′,3′,5′-Tri-O-Benzoyl[4-13C]Uridine. An Efficient, Regiospecific Synthesis of the Pyrimidine Ring. J. Org. Chem. 1978, 43, 1547–1550. 10.1021/jo00402a014. [DOI] [Google Scholar]
- Wunderlich C. H.; Spitzer R.; Santner T.; Fauster K.; Tollinger M.; Kreutz C. Synthesis of (6-13C)Pyrimidine Nucleotides as Spin-Labels for RNA Dynamics. J. Am. Chem. Soc. 2012, 134, 7558–7569. 10.1021/ja302148g. [DOI] [PubMed] [Google Scholar]
- Niu C. H. Synthesis of [4-15NH2]- and [1,3-15N2]Cytidine Derivatives for Use in NMR-Monitored Binding Tests. Anal. Biochem. 1984, 139, 404–407. 10.1016/0003-2697(84)90025-3. [DOI] [PubMed] [Google Scholar]
- Davidson D.; Baudisch O. The Preparation of Uracil From Urea. J. Am. Chem. Soc. 1926, 48, 2379–2383. 10.1021/ja01420a020. [DOI] [Google Scholar]
- Juen M. A.; Wunderlich C. H.; Nußbaumer F.; Tollinger M.; Kontaxis G.; Konrat R.; Hansen D. F.; Kreutz C. Excited States of Nucleic Acids Probed by Proton Relaxation Dispersion NMR Spectroscopy. Angew. Chem., Int. Ed. 2016, 55, 12008–12012. 10.1002/anie.201605870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiwo K. M.; Becette O. B.; Zong G.; Chen B.; Zavalij P. Y.; Dayie T. K. Chemo-Enzymatic Synthesis of 13C- and 19F-Labeled Uridine-5′-Triphosphate for RNA NMR Probing. Monatsh. Chem. 2021, 152, 441–447. 10.1007/s00706-021-02757-z. [DOI] [Google Scholar]
- Cushley R. J.; Lipsky S. R.; Fox J. J. Reactions of 5-Fluorouracil Derivatives with Sodium Deuteroxide. Tetrahedron Lett. 1968, 9, 5393–5396. 10.1016/S0040-4039(00)89787-7. [DOI] [Google Scholar]
- Hennig M.; Munzarová M. L.; Bermel W.; Scott L. G.; Sklenár V.; Williamson J. R. Measurement of Long Range 1H-19F Scalar Coupling Constants and Their Glycosidic Torsion Dependence in 5-Fluoropyrimidine-Substituted RNA. J. Am. Chem. Soc. 2006, 128, 5851–5858. 10.1021/ja060165t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nußbaumer F.; Juen M. A.; Gasser C.; Kremser J.; Müller T.; Tollinger M.; Kreutz C. Synthesis and Incorporation of 13C-Labeled DNA Building Blocks to Probe Structural Dynamics of DNA by NMR. Nucleic Acids Res. 2017, 45, 9178–9192. 10.1093/nar/gkx592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. R.; Boxer S. G. Synthesis of a Thymidine Phospboramidite Labelled with 13C at C6: Relaxation Studies of the Loop Region in a 13C Labelled DNA Hairpin. Nucleic Acids Res. 1988, 16, 1529–1540. 10.1093/nar/16.4.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremser J.; Strebitzer E.; Plangger R.; Juen M. A.; Nußbaumer F.; Glasner H.; Breuker K.; Kreutz C. Chemical Synthesis and NMR Spectroscopy of Long Stable Isotope Labelled RNA. Chem. Commun. 2017, 53, 12938–12941. 10.1039/C7CC06747J. [DOI] [PubMed] [Google Scholar]
- Legault P.; Pardi A. In Situ Probing of Adenine Protonation in RNA by 13C NMR. J. Am. Chem. Soc. 1994, 116, 8390–8391. 10.1021/ja00097a066. [DOI] [Google Scholar]
- Wolter A. C.; Weickhmann A. K.; Nasiri A. H.; Hantke K.; Ohlenschläger O.; Wunderlich C. H.; Kreutz C.; Duchardt-Ferner E.; Wöhnert J. A Stably Protonated Adenine Nucleotide with a Highly Shifted pKa Value Stabilizes the Tertiary Structure of a GTP-Binding RNA Aptamer. Angew. Chem., Int. Ed. 2017, 56, 401–404. 10.1002/anie.201609184. [DOI] [PubMed] [Google Scholar]
- Legault P.; Pardi A. Unusual Dynamics and pKa Shift at the Active Site of a Lead-Dependent Ribozyme. J. Am. Chem. Soc. 1997, 119, 6621–6628. 10.1021/ja9640051. [DOI] [Google Scholar]
- Thaplyal P.; Bevilacqua P. C. Experimental Approaches for Measuring pKa’s in RNA and DNA. Methods Enzymol. 2014, 549, 189–219. 10.1016/B978-0-12-801122-5.00009-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C. L.; Alexov E.; Pyle A. M.; Honig B. Calculation of pKas in RNA: On the Structural Origins and Functional Roles of Protonated Nucleotides. J. Mol. Biol. 2007, 366, 1475–1496. 10.1016/j.jmb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Veenis A. J.; Li P.; Soudackov A. V.; Hammes-Schiffer S.; Bevilacqua P. C. Investigation of the pKa of the Nucleophilic O2′ of the Hairpin Ribozyme. J. Phys. Chem. B 2021, 125, 11869–11883. 10.1021/acs.jpcb.1c06546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad J. L.; Gaffney B. L.; Jones R. A. 15N-Multilabeled Adenine and Guanine Nucleosides. Syntheses of [1,3,NH2-15N3]- and [2-13C-1,3,NH2-15N3]-Labeled Adenosine, Guanosine, 2′-Deoxyadenosine, and 2′-Deoxyguanosine. J. Org. Chem. 1999, 64, 6575–6582. 10.1021/jo982372k. [DOI] [PubMed] [Google Scholar]
- Ouwerkerk N.; Van Boom J.; Lugtenburg J.; Raap J. Synthesis of [1′,2′,5′,2-13C 4]-2′-Deoxy-D-Adenosine by a Chemoenzymatic Strategy to Enable Labelling of Any of the 215 Carbon-13 and Nitrogen-15 Isotopomers. Eur. J. Org. Chem. 2002, 2002, 2356–2362. . [DOI] [Google Scholar]
- Battaglia U.; Long J. E.; Searle M. S.; Moody C. J. 7-Deazapurine Biosynthesis: NMR Study of Toyocamycin Biosynthesis in Streptomyces Rimosus Using 2-13C-7-15N-Adenine. Org. Biomol. Chem. 2011, 9, 2227–2232. 10.1039/c0ob01054e. [DOI] [PubMed] [Google Scholar]
- Sethi S.; Gupta S. P.; Jenkins E. E.; Whitehead C. W.; Townsend L. B.; McCloskey J. A. Mass Spectrometry of Nucleic Acid Constituents. Electron Ionization Spectra of Selectively Labeled Adenines. J. Am. Chem. Soc. 1982, 104, 3349–3353. 10.1021/ja00376a017. [DOI] [Google Scholar]
- Pagano A. R.; Lajewski W. M.; Jones R. A. Syntheses of [6,7-15N]-Adenosine, [6,7-15N]-2′-Deoxyadenosine, and [7-15N]-Hypoxanthine. J. Am. Chem. Soc. 1995, 117, 11669–11672. 10.1021/ja00152a006. [DOI] [Google Scholar]
- Bendich A.; Russell P. J. Jr; Fox J. J. The Synthesis and Properties of 6-Chloropurine and Purine. J. Am. Chem. Soc. 1954, 76, 6073–6077. 10.1021/ja01652a062. [DOI] [Google Scholar]
- Olenginski L. T.; Dayie T. K. Chemo-Enzymatic Synthesis of [2-13C, 7-15N]-ATP for Facile NMR Analysis of RNA. Monatsh. Chem. 2020, 151, 1467–1473. 10.1007/s00706-020-02667-6. [DOI] [Google Scholar]
- Barrio M. D. C. G.; Scopes D. I. C.; Holtwick J. B.; Leonard N. J. Syntheses of All Singly Labeled [15N]Adenines: Mass Spectral Fragmentation of Adenine. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 3986–3988. 10.1073/pnas.78.7.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolli M. L.; Medana C.; Romagnano S.; Castoldi F.; Pozzoli S.; Vago F.; Fanelli R.; Airoldi L. Synthesis of Labelled [15N3]-6-Bromopurine, a Useful Precursor of 15N-Labelled O6-Alkylguanines, to be Used as Internal Standards for Quantitative GC-MS Analyses. J. Label. Compd. and Radiopharm. 1998, 41, 243–252. [Google Scholar]
- Zhao H.; Pagano A. R.; Wang W.; Shallop A.; Gaffney B. L.; Jones R. A. Use of a 13C Atom To Differentiate Two 15N-Labeled Nucleosides. Syntheses of [15NH2]-Adenosine, [1,NH2-15N2]- and [2-13C-1,NH2-15N2]-Guanosine, and [1,7,NH2-15N3]- and [2-13C-1,7,NH2-15N3]-2′-Deoxyguanosine. J. Org. Chem. 1997, 62, 7832–7835. 10.1021/jo971206u. [DOI] [Google Scholar]
- Jain M. L.; Tsao Y. P.; Ho N. L.; Cheng J. W. A Facile Synthesis of [N1,NH2-15N2]-, [N3,NH2-15N2]-, and [N1,N3,NH2-15N3]-Labeled Adenine. J. Org. Chem. 2001, 66, 6472–6475. 10.1021/jo010253q. [DOI] [PubMed] [Google Scholar]
- Wachowius F.; Höbartner C. Chemical RNA Modifications for Studies of RNA Structure and Dynamics. ChemBioChem. 2010, 11, 469–480. 10.1002/cbic.200900697. [DOI] [PubMed] [Google Scholar]
- Neuner S.; Santner T.; Kreutz C.; Micura R. The “Speedy” Synthesis of Atom-Specific 15N Imino/Amido-Labeled RNA. Chem.-Eur. J. 2015, 21, 11634–11643. 10.1002/chem.201501275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner S.; Kreutz C.; Micura R. The Synthesis of 15N(7)-Hoogsteen Face-Labeled Adenosine Phosphoramidite for Solid-Phase RNA Synthesis. Monatsh. Chem. 2017, 148, 149–155. 10.1007/s00706-016-1882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santner T.; Rieder U.; Kreutz C.; Micura R. Pseudoknot Preorganization of the PreQ1 Class I Riboswitch. J. Am. Chem. Soc. 2012, 134, 11928–11931. 10.1021/ja3049964. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Gaffney B. L.; Jones R. A. 15N NMR of a Specifically Labeled RNA Fragment Containing Intrahelical GU Wobble Pairs. J. Am. Chem. Soc. 1997, 119, 6432–6433. 10.1021/ja970370w. [DOI] [Google Scholar]
- Weissman B. P.; Li N. S.; York D.; Harris M.; Piccirilli J. A. Heavy Atom Labeled Nucleotides for Measurement of Kinetic Isotope Effects. Biochim. Biophys. Acta - Proteins Proteomics 2015, 1854, 1737–1745. 10.1016/j.bbapap.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn F. A.; MacDonnell J. E.; Bearne S. L. Structural Requirements for the Activation of Escherichia Coli CTP Synthase by the Allosteric Effector GTP Are Stringent, but Requirements for Inhibition Are Lax. J. Biol. Chem. 2008, 283, 2010–2020. 10.1074/jbc.M707803200. [DOI] [PubMed] [Google Scholar]
- Taiwo K. M.; Olenginski L. T.; Nußbaumer F.; Nam H.; Hilber S.; Kreutz C.; Dayie T. K. Synthesis of [7-15N]-GTPs for RNA structure and dynamics by NMR spectroscopy. Monatsh. Chem. 2022, 10.1007/s00706-022-02892-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masjost B.; Ballmer P.; Borroni E.; Zürcher G.; Winkler F. K.; Jakon-Roetne R.; Diederich F. Structure-Based Design, Synthesis, and in vitro Evaluation of Bisubstrate Inhibitors for Catechol O-Methyltransferase (COMT). Chem.-Eur. J. 2000, 6, 971–982. . [DOI] [PubMed] [Google Scholar]
- Orji C. C.; Kelly J.; Ashburn D. A.; Silks L. A. First Synthesis of β-2′-Deoxy[9-15N]Adenosine. J. Chem. Soc. Perkin Trans. 1 1996, 7, 595–597. 10.1039/P19960000595. [DOI] [Google Scholar]
- Hoffman D. W.; Holland J. A. Preparation of Carbon-13 Labeled Ribonucleotides Using Acetate as an Isotope Source. Nucleic Acids Res. 1995, 23, 3361–3362. 10.1093/nar/23.16.3361-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur C. S.; Dayie T. K. Asymmetry of 13C Labeled 3-Pyruvate Affords Improved Site Specific Labeling of RNA for NMR Spectroscopy. J. Biomol. NMR 2012, 52, 65–77. 10.1007/s10858-011-9582-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur C. S.; Luo Y.; Chen B.; Eldho N. V.; Dayie T. K. Biomass Production of Site Selective 13C/15N Nucleotides Using Wild Type and a Transketolase E. Coli Mutant for Labeling RNA for High Resolution NMR. J. Biomol. NMR 2012, 52, 103–114. 10.1007/s10858-011-9586-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin D. W.; Leung H. B.; Schramm V. L. Synthesis of Nucleotides with Specific Radiolabels in Ribose. Primary 14C and Secondary 3H Kinetic Isotope Effects on Acid-Catalyzed Glycosidic Bond Hydrolysis of AMP, DAMP, and Inosine. J. Biol. Chem. 1984, 259, 9411–9417. 10.1016/S0021-9258(17)42716-5. [DOI] [PubMed] [Google Scholar]
- Gilles A. M.; Cristea I.; Palibroda N.; Hilden I.; Jensen K. F.; Sarfati R. S.; Namane A.; Ughetto-Monfrin J.; Bârzu O. Chemienzymatic Synthesis of Uridine Nucleotides Labeled with [15N] and [13C]. Anal. Biochem. 1995, 232, 197–203. 10.1006/abio.1995.0007. [DOI] [PubMed] [Google Scholar]
- Rising K. A.; Schramm V. L. Enzymic Synthesis of NAD+ with the Specific Incorporation of Atomic Labels. J. Am. Chem. Soc. 1994, 116, 6531–6536. 10.1021/ja00094a006. [DOI] [Google Scholar]
- Parkin D. W.; Schramm V. L. Catalytic and Allosteric Mechanism of AMP Nucleosidase from Primary, β-Secondary, and Multiple Heavy Atom Kinetic Isotope Effects. Biochemistry 1987, 26, 913–920. 10.1021/bi00377a036. [DOI] [PubMed] [Google Scholar]
- Hennig M.; Scott L. G.; Sperling E.; Bermel W.; Williamson J. R. Synthesis of 5-Fluoropyrimidine Nucleotides as Sensitive NMR Probes of RNA Structure. J. Am. Chem. Soc. 2007, 129, 14911–14921. 10.1021/ja073825i. [DOI] [PubMed] [Google Scholar]
- Scott L. G.; Geierstanger B. H.; Williamson J. R.; Hennig M. Enzymatic Synthesis and 19F NMR Studies of 2-Fluoroadenine-Substituted RNA. J. Am. Chem. Soc. 2004, 126, 11776–11777. 10.1021/ja047556x. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Zhao S.; Serianni A. S. Labeling Monosaccharides With Stable Isotopes. Methods Enzymol. 2015, 565, 423–458. 10.1016/bs.mie.2015.06.016. [DOI] [PubMed] [Google Scholar]
- Arthur P. K.; Alvarado L. J.; Dayie T. K. Expression, Purification and Analysis of the Activity of Enzymes from the Pentose Phosphate Pathway. Protein Expr. Purif. 2011, 76, 229–237. 10.1016/j.pep.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Turney T.; Surjancev I.; Serianni A. S. Enzymatic Synthesis of Ribo- and 2′-Deoxyribonucleosides from Glycofuranosyl Phosphates: An Approach to Facilitate Isotopic Labeling. Carbohydr. Res. 2017, 449, 125–133. 10.1016/j.carres.2017.07.006. [DOI] [PubMed] [Google Scholar]
- Hirschbein B. L.; Mazenod F. P.; Whitesides G. M. Synthesis of Phosphoenolypyruvate and Its Use in ATP Cofactor Regeneration. J. Org. Chem. 1982, 47, 3765–3766. 10.1021/jo00140a036. [DOI] [Google Scholar]
- Simon E. S.; Grabowski S.; Whitesides G. M. Convenient Syntheses of Cytidine 5′-Triphosphate, Guanosine 5′-Triphosphate, and Uridine 5′-Triphosphate and Their Use in the Preparation of UDP-Glucose, UDP-Glucuronic Acid, and GDP-Mannose. J. Org. Chem. 1990, 55, 1834–1841. 10.1021/jo00293a030. [DOI] [Google Scholar]
- Gross A.; Abril O.; Lewis J. M.; Geresh S.; Whitesides G. M. Practical Synthesis of 5-Phospho-D-Ribosyl α-1-Pyrophosphate (PRPP): Enzymatic Routes from Ribose 5-Phosphate or Ribose. J. Am. Chem. Soc. 1983, 105, 7428–7435. 10.1021/ja00363a037. [DOI] [Google Scholar]
- Duker J. M.; Serianni A. S. (13C)-Substituted Sucrose: 13C-1H and 13C-13C Spin Coupling Constants to Assess Furanose Ring and Glycosidic Bond Conformations in Aqueous Solution. Carbohydr. Res. 1993, 249, 281–303. 10.1016/0008-6215(93)84096-O. [DOI] [PubMed] [Google Scholar]
- Cook G. P.; Greenberg M. M. A General Synthesis of C2′-Deuterated Ribonucleosides. J. Org. Chem. 1994, 59, 4704–4706. 10.1021/jo00095a060. [DOI] [Google Scholar]
- Toyama A.; Takino Y.; Takeuchi H.; Harada I. Ultraviolet Resonance Raman Spectra of Ribosyl C(1′)-Deuterated Purine Nucleosides: Evidence of Vibrational Coupling between Purine and Ribose Rings. J. Am. Chem. Soc. 1993, 115, 11092–11098. 10.1021/ja00077a005. [DOI] [Google Scholar]
- Földesi A.; Nilson F. P. R.; Glemarec C.; Gioeli C.; Chattopadhyaya J. Synthesis of 1′,2′,3′,4′,5′,5″-2H6-β-D-Ribonucleosides and 1′,2′,2″,3′,4′,5′,5″-2H7-β-D-2′-Deoxyribonucleosides for Selective Suppression of Proton Resonances in Partially-Deuterated Oligo-DNA, Oligo-RNA and in 2,5A Core (1H-NMR Window). Tetrahedron 1992, 48, 9033–9072. 10.1016/S0040-4020(01)82001-9. [DOI] [Google Scholar]
- Vorbrüggen H.; Höfle G. Nucleoside Syntheses, XXIII1) On the Mechanism of Nucleoside Synthesis. Chem. Ber. 1981, 114, 1256–1268. 10.1002/cber.19811140405. [DOI] [Google Scholar]
- Liu Y.; Holmstrom E.; Zhang J.; Yu P.; Wang J.; Dyba M. A.; Chen D.; Ying J.; Lockett S.; Nesbitt D. J.; et al. Synthesis and Applications of RNAs with Position-Selective Labelling and Mosaic Composition. Nature 2015, 522, 368–372. 10.1038/nature14352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Holmstrom E.; Yu P.; Tan K.; Zuo X.; Nesbitt D. J.; Sousa R.; Stagno J. R.; Wang Y. X. Incorporation of Isotopic, Fluorescent, and Heavy-Atom-Modified Nucleotides into RNAs by Position-Selective Labeling of RNA. Nat. Protoc. 2018, 13, 987–1005. 10.1038/nprot.2018.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Yu P.; Dyba M.; Sousa R.; Stagno J. R.; Wang Y. X. Applications of PLOR in Labeling Large RNAs at Specific Sites. Methods 2016, 103, 4–10. 10.1016/j.ymeth.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyhani S.; Goldau T.; Blümler A.; Heckel A.; Schwalbe H. Chemo-Enzymatic Synthesis of Position-Specifically Modified RNA for Biophysical Studies Including Light Control and NMR Spectroscopy. Angew. Chem., Int. Ed. 2018, 130, 12193–12197. 10.1002/ange.201807125. [DOI] [PubMed] [Google Scholar]
- Blümler A.; Schwalbe H.; Heckel A. Solid-Phase-Supported Chemoenzymatic Synthesis of a Light-Activatable tRNA Derivative. Angew. Chem., Int. Ed. 2022, 61, e202111613 10.1002/anie.202111613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich C. H.; Juen M. A.; LeBlanc R. M.; Longhini A. P.; Dayie T. K.; Kreutz C. Stable Isotope-Labeled RNA Phosphoramidites to Facilitate Dynamics by NMR. Methods Enzymol. 2015, 565, 461–494. 10.1016/bs.mie.2015.06.013. [DOI] [PubMed] [Google Scholar]
- Chen B.; Longhini A. P.; Nußbaumer F.; Kreutz C.; Dinman J. D.; Dayie T. K. CCR5 RNA Pseudoknots: Residue and Site-Specific Labeling Correlate Internal Motions with MicroRNA Binding. Chem.-Eur. J. 2018, 24, 5462–5468. 10.1002/chem.201705948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba Y.; Masuda H.; Watanabe N.; Ego T.; Takagaki K.; Ishiyama K.; Ohgi T.; Yano J. Chemical Synthesis of a Very Long Oligoribonucleotide with 2-Cyanoethoxymethyl (CEM) as the 2′-O-Protecting Group: Structural Identification and Biological Activity of a Synthetic 110mer Precursor-MicroRNA Candidate. Nucleic Acids Res. 2007, 35, 3287–3296. 10.1093/nar/gkm202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgi T.; Kitagawa H.; Yano J. Chemical Synthesis of Oligoribonucleotides with 2′-O-(2-Cyanoethoxymethyl)-Protected Phosphoramidites. Curr. Protoc. Nucleic Acid Chem. 2008, 34, 2.15.1–2.15.19. 10.1002/0471142700.nc0215s34. [DOI] [PubMed] [Google Scholar]
- Liu B.; Shi H.; Al-Hashimi H. M. Developments in Solution-State NMR Yield Broader and Deeper Views of the Dynamic Ensembles of Nucleic Acids. Curr. Opin. Struct. Biol. 2021, 70, 16–25. 10.1016/j.sbi.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strebitzer E.; Rangadurai A.; Plangger R.; Kremser J.; Juen M. A.; Tollinger M.; Al-Hashimi H. M.; Kreutz C. 5-Oxyacetic Acid Modification Destabilizes Double Helical Stem Structures and Favors Anionic Watson-Crick like Cmo5U-G Base Pairs. Chem.-Eur. J. 2018, 24, 18903–18906. 10.1002/chem.201805077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; Liu B.; Nussbaumer F.; Rangadurai A.; Kreutz C.; Al-Hashimi H. M. NMR Chemical Exchange Measurements Reveal That N6-Methyladenosine Slows RNA Annealing. J. Am. Chem. Soc. 2019, 141, 19988–19993. 10.1021/jacs.9b10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Shi H.; Rangadurai A.; Nussbaumer F.; Chu C. C.; Erharter K. A.; Case D. A.; Kreutz C.; Al-Hashimi H. M. A Quantitative Model Predicts How m6A Reshapes the Kinetic Landscape of Nucleic Acid Hybridization and Conformational Transitions. Nat. Commun. 2021, 12, 5201. 10.1038/s41467-021-25253-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desaulniers J. P.; Chang Y. C.; Aduri R.; Abeysirigunawardena S. C.; SantaLucia J.; Chow C. S. Pseudouridines in RRNA Helix 69 Play a Role in Loop Stacking Interactions. Org. Biomol. Chem. 2008, 6, 3892–3895. 10.1039/b812731j. [DOI] [PubMed] [Google Scholar]
- Olenginski L. T.; Becette O. B.; Beaucage S. L.; Dayie T. K. Synthesis of Atom-Specific Nucleobase and Ribose Labeled Uridine Phosphoramidite for NMR Analysis of Large RNAs. Monatsh. Chem. 2021, 152, 1361–1367. 10.1007/s00706-021-02851-2. [DOI] [Google Scholar]
- de Jesus V.; Qureshi N. S.; Warhaut S.; Bains J. K.; Dietz M. S.; Heilemann M.; Schwalbe H.; Fürtig B. Switching at the Ribosome: Riboswitches Need rProteins as Modulators to Regulate Translation. Nat. Commun. 2021, 12, 4723. 10.1038/s41467-021-25024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komoroski R. A.; Allerhand A. Observation of Resonances from Some Minor Bases in the Natural-Abundance Carbon-13 Nuclear Magnetic Resonance Spectrum of Unfractionated Yeast Transfer Ribonucleic Acid. Evidence for Fast Internal Motion of the Dihydrouracil Rings. Biochemistry 1974, 13, 369–372. 10.1021/bi00699a023. [DOI] [PubMed] [Google Scholar]
- Nirmala N. R.; Wagner G. Measurement of 13C Relaxation Times in Proteins by Two-Dimensional Heteronuclear 1H-13C Correlation Spectroscopy. J. Am. Chem. Soc. 1988, 110, 7557–7558. 10.1021/ja00230a057. [DOI] [Google Scholar]
- Sklenář V. Í.; Torchia D.; Bax A. Measurement of Carbon-13 Longitudinal Relaxation Using 1H Detection. J. Magn. Reson. 1987, 73, 375–379. 10.1016/0022-2364(87)90214-9. [DOI] [Google Scholar]
- Kay L. E.; Jue T. L.; Bangerter B.; Demou P. C. Sensitivity Enhancement of 13C T1Measurements via Polarization Transfer. J. Magn. Reson. 1987, 73, 558–564. 10.1016/0022-2364(87)90024-2. [DOI] [Google Scholar]
- Farrow N. A.; Muhandiram R.; Singer A. U.; Pascal S. M.; Kay C. M.; Gish G.; Shoelson S. E.; Pawson T.; Forman-Kay J. D.; Kay L. E. Backbone Dynamics of a Free and a Phosphopeptide-Complexed Src Homology 2 Domain Studied by 15N NMR Relaxation. Biochemistry 1994, 33, 5984–6003. 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- Dayie K. T.; Wagner G. Relaxation-Rate Measurements for 15N-1H Groups with Pulsed-Field Gradients and Preservation of Coherence Pathways. J. Magn. Reson. 1994, 111, 121–126. 10.1006/jmra.1994.1236. [DOI] [Google Scholar]
- Peng J. W.; Wagner G. Mapping of Spectral Density Functions Using Heteronuclear NMR Relaxation Measurements. J. Magn. Reson. 1992, 98, 308–332. 10.1016/0022-2364(92)90135-T. [DOI] [Google Scholar]
- Kay L. E.; Torchia D. A.; Bax A. Backbone Dynamics of Proteins as Studied by 15N Inverse Detected Heteronuclear NMR Spectroscopy: Application to Staphylococcal Nuclease. Biochemistry 1989, 28, 8972–8979. 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- Dayie K. T.; Brodsky A. S.; Williamson J. R. Base Flexibility in HIV-2 TAR RNA Mapped by Solution 15N, 13C NMR Relaxation. J. Mol. Biol. 2002, 317, 263–278. 10.1006/jmbi.2001.5424. [DOI] [PubMed] [Google Scholar]
- Hoogstraten C. G.; Wank J. R.; Pardi A. Active Site Dynamics in the Lead-Dependent Ribozyme. Biochemistry 2000, 39, 9951–9958. 10.1021/bi0007627. [DOI] [PubMed] [Google Scholar]
- Boisbouvier J.; Brutscher B.; Simorre J.-P.; Marion D. 13C Spin Relaxation Measurements in RNA: Sensitivity and Resolution Improvement Using Spin-State Selective Correlation Experiments. J. Biomol. NMR 1999, 14, 241–252. 10.1023/A:1008365712799. [DOI] [Google Scholar]
- Hall K. B.; Tang C. 13C Relaxation and Dynamics of the Purine Bases in the Iron Element RNA Hairpin. Biochemistry 1998, 37, 9323–9332. 10.1021/bi9805285. [DOI] [PubMed] [Google Scholar]
- Akke M.; Fiala R.; Jiang F.; Patel D.; Palmer III A. G. Base Dynamics in a UUCG Tetraloop RNA Hairpin Characterized by 15N Spin Relaxation: Correlations with Structure and Stability. RNA 1997, 3, 702–709. [PMC free article] [PubMed] [Google Scholar]
- Rangadurai A.; Szymaski E. S.; Kimsey I. J.; Shi H.; Al-Hashimi H. M. Characterizing Micro-to-Millisecond Chemical Exchange in Nucleic Acids Using Off-Resonance R1ρ Relaxation Dispersion. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 112–113, 55–102. 10.1016/j.pnmrs.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marušič M.; Schlagnitweit J.; Petzold K. RNA Dynamics by NMR Spectroscopy. ChemBioChem. 2019, 20, 2685–2710. 10.1002/cbic.201900072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozinovic S.; Richter C.; Rinnenthal J.; Fürtig B.; Duchardt-Ferner E.; Weigand J. E.; Schwalbe H. Quantitative 2D and 3D Γ-HCP Experiments for the Determination of the Angles α and ζ in the Phosphodiester Backbone of Oligonucleotides. J. Am. Chem. Soc. 2010, 132, 10318–10329. 10.1021/ja910015n. [DOI] [PubMed] [Google Scholar]
- Rinnenthal J.; Richter C.; Nozinovic S.; Fürtig B.; Lopez J. J.; Glaubitz C.; Schwalbe H. RNA Phosphodiester Backbone Dynamics of a Perdeuterated CUUCGg Tetraloop RNA from Phosphorus-31 NMR Relaxation Analysis. J. Biomol. NMR 2009, 45, 143–155. 10.1007/s10858-009-9343-x. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P.; Kay L. E. A Suite of 2H NMR Spin Relaxation Experiments for the Measurement of RNA Dynamics. J. Am. Chem. Soc. 2005, 127, 6893–6901. 10.1021/ja0427799. [DOI] [PubMed] [Google Scholar]
- Bothe J. R.; Nikolova E. N.; Eichhorn C. D.; Chugh J.; Hansen A. L.; Al-Hashimi H. M. Characterizing RNA Dynamics at Atomic Resolution Using Solution-State NMR Spectroscopy. Nat. Methods 2011, 8, 919–931. 10.1038/nmeth.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hashimi H. M.; Walter N. G. RNA Dynamics: It Is about Time. Curr. Opin. Struct. Biol. 2008, 18, 321–329. 10.1016/j.sbi.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J. W. Cross-Correlated 19F Relaxation Measurements for the Study of Fluorinated Ligand-Receptor Interactions. J. Magn. Reson. 2001, 153, 32–47. 10.1006/jmre.2001.2422. [DOI] [PubMed] [Google Scholar]
- Palmer III A. G. NMR Characterization of the Dynamics of Biomacromolecules. Chem. Rev. 2004, 104, 3623–3640. 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- Ishima R.; Torchia D. A. Protein Dynamics from NMR. Nat. Struct. Biol. 2000, 7, 740–743. 10.1038/78963. [DOI] [PubMed] [Google Scholar]
- Fushman D.; Cowburn D. Nuclear Magnetic Resonance Relaxation in Determination of Residue-Specific 15N Chemical Shift Tensors in Proteins in Solution: Protein Dynamics, Structure, and Applications of Transverse Relaxation Optimized Spectroscopy. Methods Enzymol. 2001, 339, 109–122. 10.1016/S0076-6879(01)39312-6. [DOI] [PubMed] [Google Scholar]
- Dayie K. T.; Wagner G.; Lefevre J. F. Theory and Practice of Nuclear Spin Relaxation In Proteins. Annu. Rev. Phys. Chem. 1996, 47, 243–282. 10.1146/annurev.physchem.47.1.243. [DOI] [PubMed] [Google Scholar]
- Fushman D.; Tjandra N.; Cowburn D. Measurement of 15N Chemical Shift Anisotropy in Solution. J. Am. Chem. Soc. 1998, 120, 10947–10952. 10.1021/ja981686m. [DOI] [Google Scholar]
- Ying J.; Grishaev A.; Bryce D. L.; Bax A. Chemical Shift Tensors of Protonated Base Carbons in Helical RNA and DNA from NMR Relaxation and Liquid Crystal Measurements. J. Am. Chem. Soc. 2006, 128, 11443–11454. 10.1021/ja061984g. [DOI] [PubMed] [Google Scholar]
- Lipari G.; Szabo A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 1. Theory and Range of Validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. 10.1021/ja00381a009. [DOI] [Google Scholar]
- Lipari G.; Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982, 104, 4559–4570. 10.1021/ja00381a010. [DOI] [Google Scholar]
- Shajani Z.; Varani G. 13C NMR Relaxation Studies of RNA Base and Ribose Nuclei Reveal a Complex Pattern of Motions in the RNA Binding Site for Human U1A Protein. J. Mol. Biol. 2005, 349, 699–715. 10.1016/j.jmb.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Shajani Z.; Drobny G.; Varani G. Binding of U1A Protein Changes RNA Dynamics as Observed by 13C NMR Relaxation Studies. Biochemistry 2007, 46, 5875–5883. 10.1021/bi602658x. [DOI] [PubMed] [Google Scholar]
- Hansen A. L.; Al-Hashimi H. M. Dynamics of Large Elongated RNA by NMR Carbon Relaxation. J. Am. Chem. Soc. 2007, 129, 16072–16082. 10.1021/ja0757982. [DOI] [PubMed] [Google Scholar]
- Blad H.; Reiter N. J.; Abildgaard F.; Markley J. L.; Butcher S. E. Dynamics and Metal Ion Binding in the U6 RNA Intramolecular Stem-Loop as Analyzed by NMR. J. Mol. Bio. 2005, 353, 540–555. 10.1016/j.jmb.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Nam H.; Becette O.; LeBlanc R. M.; Oh D.; Case D. A.; Dayie T. K. Deleterious Effects of Carbon-Carbon Dipolar Coupling on RNA NMR Dynamics. J. Biomol. NMR 2020, 74, 321–331. 10.1007/s10858-020-00315-z. [DOI] [PubMed] [Google Scholar]
- Olenginski L. T.; Dayie T. K. Quantifying the Effects of Long-Range 13C-13C Dipolar Coupling on Measured Relaxation Rates in RNA. J. Biomol. NMR 2021, 75, 203–211. 10.1007/s10858-021-00368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Sun X.; Watt E. D.; Al-Hashimi H. M. Resolving the Motional Modes That Code for RNA Adaptation. Science 2006, 311, 653–656. 10.1126/science.1119488. [DOI] [PubMed] [Google Scholar]
- Boisbouvier J.; Wu Z.; Ono A.; Kainosho M.; Bax A. Rotational Diffusion Tensor of Nucleic Acids from 13C NMR Relaxation. J. Biomol. NMR 2003, 27, 133–142. 10.1023/A:1024931619957. [DOI] [PubMed] [Google Scholar]
- Johnson J. E.; Julien K. R.; Hoogstraten C. G. Alternate-Site Isotopic Labeling of Ribonucleotides for NMR Studies of Ribose Conformational Dynamics in RNA. J. Biomol. NMR 2006, 35, 261–274. 10.1007/s10858-006-9041-x. [DOI] [PubMed] [Google Scholar]
- Yamazaki T.; Muhandiram R.; Kay L. E. NMR Experiments for the Measurement of Carbon Relaxation Properties in Highly Enriched, Uniformly 13C,15N-Labeled Proteins: Application to 13Cα Carbons. J. Am. Chem. Soc. 1994, 116, 8266–8278. 10.1021/ja00097a037. [DOI] [Google Scholar]
- Johnson J. E. Jr; Hoogstraten C. G. Extensive Backbone Dynamics in the GCAA RNA Tetraloop Analyzed Using 13C NMR Spin Relaxation and Specific Isotope Labeling. J. Am. Chem. Soc. 2008, 130, 16757–16769. 10.1021/ja805759z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayie K. T.; Wagner G. Carbonyl Carbon Probe of Local Mobility in 13C,15N-Enriched Proteins Using High-Resolution Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1997, 119, 7797–7806. 10.1021/ja9633880. [DOI] [Google Scholar]
- Lakomek N. A.; Ying J.; Bax A. Measurement of 15N relaxation rates in perdeuterated proteins by TROSY-based methods. J. Biomol. NMR 2012, 53, 209–221. 10.1007/s10858-012-9626-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer III A. G.; Cavanagh J.; Wright P. E.; Rance M. Sensitivity Improvement in Proton-Detected Two-Dimensional Heteronuclear Correlation NMR Spectroscopy. J. Magn. Reson. 1991, 93, 151–170. 10.1016/0022-2364(91)90036-S. [DOI] [Google Scholar]
- Kay L.; Keifer P.; Saarinen T. Pure Absorption Gradient Enhanced Heteronuclear Single Quantum Correlation Spectroscopy with Improved Sensitivity. J. Am. Chem. Soc. 1992, 114, 10663–10665. 10.1021/ja00052a088. [DOI] [Google Scholar]
- Palmer III A. G.; Koss H. Chemical Exchange. Methods Enzymol. 2019, 615, 177–236. 10.1016/bs.mie.2018.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A. G.; Kroenke C. D.; Loria J. P. Nuclear Magnetic Resonance Methods for Quantifying Microsecond-to-Millisecond Motions in Biological Macromolecules. Methods Enzymol. 2001, 339, 204–238. 10.1016/S0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- Anthis N. J.; Clore G. M. Visualizing Transient Dark States by NMR Spectroscopy. Q. Rev. Biophys. 2015, 48, 35–116. 10.1017/S0033583514000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer III A. G.; Grey M. J.; Wang C. Solution NMR Spin Relaxation Methods for Characterizing Chemical Exchange in High-Molecular-Weight Systems. Methods Enzymol. 2005, 394, 430–465. 10.1016/S0076-6879(05)94018-4. [DOI] [PubMed] [Google Scholar]
- Palmer A. G. III; Massi F. Characterization of the Dynamics of Biomacromolecules Using Rotating-Frame Spin Relaxation NMR Spectroscopy. Chem. Rev. 2006, 106, 1700–1719. 10.1021/cr0404287. [DOI] [PubMed] [Google Scholar]
- Peng J. W.; Thanabal V.; Wagner G. H. 2D Heteronuclear NMR Measurements of Spin-Lattice Relaxation Times in the Rotating Frame of X Nuclei in Heteronuclear HX Spin Systems. J. Magn. Reson. 1991, 94, 82–100. 10.1016/0022-2364(91)90296-6. [DOI] [Google Scholar]
- Luz Z.; Meiboom S. Nuclear Magnetic Resonance Study of the Protolysis of Trimethylammonium Ion in Aqueous Solution—Order of the Reaction with Respect to Solvent. J. Chem. Phys. 1963, 39, 366–370. 10.1063/1.1734254. [DOI] [Google Scholar]
- Meiboom S.; Gill D. Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 29, 688–691. 10.1063/1.1716296. [DOI] [Google Scholar]
- Carr H. Y.; Purcell E. M. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys. Rev. 1954, 94, 630–638. 10.1103/PhysRev.94.630. [DOI] [Google Scholar]
- Vallurupalli P.; Bouvignies G.; Kay L. E. Studying “Invisible” Excited Protein States in Slow Exchange with a Major State Conformation. J. Am. Chem. Soc. 2012, 134, 8148–8161. 10.1021/ja3001419. [DOI] [PubMed] [Google Scholar]
- Schanda P.; Brutscher B. Very Fast Two-Dimensional NMR Spectroscopy for Real-Time Investigation of Dynamic Events in Proteins on the Time Scale of Seconds. J. Am. Chem. Soc. 2005, 127, 8014–8015. 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- Carver J. P.; Richards R. E. A General Two-Site Solution for the Chemical Exchange Produced Dependence of T2 upon the Carr-Purcell Pulse Separation. J. Magn. Reson. 1972, 6, 89–105. 10.1016/0022-2364(72)90090-X. [DOI] [Google Scholar]
- Strebitzer E.; Nußbaumer F.; Kremser J.; Tollinger M.; Kreutz C. Studying Sparsely Populated Conformational States in RNA Combining Chemical Synthesis and Solution NMR Spectroscopy. Methods 2018, 148, 39–47. 10.1016/j.ymeth.2018.05.007. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Hansen A. L.; Zhang Q. Characterizing Slow Chemical Exchange in Nucleic Acids by Carbon CEST and Low Spin-Lock Field R1ρ NMR Spectroscopy. J. Am. Chem. Soc. 2014, 136, 20–23. 10.1021/ja409835y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell H. M. Reaction Rates by Nuclear Magnetic Resonance. J. Chem. Phys. 1958, 28, 430–431. 10.1063/1.1744152. [DOI] [Google Scholar]
- Bouvignies G.; Kay L. E. Measurement of Proton Chemical Shifts in Invisible States of Slowly Exchanging Protein Systems by Chemical Exchange Saturation Transfer. J. Phys. Chem. B 2012, 116, 14311–14317. 10.1021/jp311109u. [DOI] [PubMed] [Google Scholar]
- Helgstrand M.; Härd T.; Allard P. Simulations of NMR Pulse Sequences during Equilibrium and Non-Equilibrium Chemical Exchange. J. Biomol. NMR 2000, 18, 49–63. 10.1023/A:1008309220156. [DOI] [PubMed] [Google Scholar]
- Korzhnev D. M.; Orekhov V. Y.; Kay L. E. Off-Resonance R1ρ NMR Studies of Exchange Dynamics in Proteins with Low Spin-Lock Fields: An Application to a Fyn SH3 Domain. J. Am. Chem. Soc. 2005, 127, 713–721. 10.1021/ja0446855. [DOI] [PubMed] [Google Scholar]
- Kimsey I. J.; Szymanski E. S.; Zahurancik W. J.; Shakya A.; Xue Y.; Chu C. C.; Sathyamoorthy B.; Suo Z.; Al-Hashimi H. M. Dynamic Basis for dG-dT Misincorporation via Tautomerization and Ionization. Nature 2018, 554, 195–201. 10.1038/nature25487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Ven F. J. M.; Philippens V. E. P. Optimization of Constant-Time Evolution in Multidimensional NMR Experiments. J. Magn. Reson. 1992, 97, 637–644. 10.1016/0022-2364(92)90045-9. [DOI] [Google Scholar]
- Grzesiek S.; Bax A. Improved 3D Triple-Resonance NMR Techniques Applied to a 31 KDa Protein. J. Magn. Reson. 1992, 96, 432–440. 10.1016/0022-2364(92)90099-S. [DOI] [Google Scholar]
- Bax A.; Freeman R. Investigation of Complex Networks of Spin-Spin Coupling by Two-Dimensional NMR. J. Magn. Reson. 1981, 44, 542–561. 10.1016/0022-2364(81)90287-0. [DOI] [Google Scholar]
- Bax A.; Mehlkopf A. F.; Smidt J. Homonuclear Broadband-Decoupled Absorption Spectra, with Linewidths Which Are Independent of the Transverse Relaxation Rate. J. Magn. Reson. 1979, 35, 167–169. 10.1016/0022-2364(79)90088-X. [DOI] [Google Scholar]
- Dayie K. T. Resolution Enhanced Homonuclear Carbon Decoupled Triple Resonance Experiments for Unambiguous RNA Structural Characterization. J. Biomol. NMR 2005, 32, 129–139. 10.1007/s10858-005-5093-6. [DOI] [PubMed] [Google Scholar]
- Brutscher B.; Boisbouvier J.; Kupče E.; Tisné C.; Dardel F.; Marion D.; Simorre J.-P. Base-Type-Selective High-Resolution 13C Edited NOESY for Sequential Assignment of Large RNAs. J. Biomol. NMR 2001, 19, 141–151. 10.1023/A:1008340210079. [DOI] [PubMed] [Google Scholar]
- Kupče E.; Wagner G. Multisite Band-Selective Decoupling in Proteins. J. Magn. Reson. 1996, 110, 309–312. 10.1006/jmrb.1996.0048. [DOI] [PubMed] [Google Scholar]
- Ferrage F.; Eykyn T. R.; Bodenhausen G. Frequency-Switched Single-Transition Cross-Polarization: A Tool for Selective Experiments in Biomolecular NMR. ChemPhysChem 2004, 5, 76–84. 10.1002/cphc.200300905. [DOI] [PubMed] [Google Scholar]
- Pelupessy P.; Chiarparin E.; Bodenhausen G. Excitation of Selected Proton Signals in NMR of Isotopically Labeled Macromolecules. J. Magn. Reson. 1999, 138, 178–181. 10.1006/jmre.1999.1715. [DOI] [PubMed] [Google Scholar]
- Chiarparin E.; Pelupessy P.; Bodenhausen G. Selective Cross-Polarization in Solution State NMR. Mol. Phys. 1998, 95, 759–767. 10.1080/002689798166396. [DOI] [Google Scholar]
- LeBlanc R. M.; Longhini A. P.; Tugarinov V.; Dayie T. K. NMR Probing of Invisible Excited States Using Selectively Labeled RNAs. J. Biomol. NMR 2018, 71, 165–172. 10.1007/s10858-018-0184-3. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P.; Scott L.; Williamson J. R.; Kay L. E. Strong Coupling Effects during X-Pulse CPMG Experiments Recorded on Heteronuclear ABX Spin Systems: Artifacts and a Simple Solution. J. Biomol. NMR 2007, 38, 41–46. 10.1007/s10858-006-9139-1. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Yang D. 13Cα CEST Experiment on Uniformly 13C-Labeled Proteins. J. Biomol. NMR 2015, 61, 89–94. 10.1007/s10858-014-9888-1. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Yang D. Effects of J Couplings and Unobservable Minor States on Kinetics Parameters Extracted from CEST Data. J. Magn. Reson. 2014, 249, 118–125. 10.1016/j.jmr.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Baisden J. T.; Zhang Q. Probing Excited Conformational States of Nucleic Acids by Nitrogen CEST NMR Spectroscopy. J. Magn. Reson. 2020, 310, 106642. 10.1016/j.jmr.2019.106642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangadurai A.; Kremser J.; Shi H.; Kreutz C.; Al-Hashimi H. M. Direct Evidence for (G)O6···H 2-N4(C) + Hydrogen Bonding in Transient G(Syn)-C + and G(Syn)-m 5 C + Hoogsteen Base Pairs in Duplex DNA from Cytosine Amino Nitrogen off-Resonance R1ρ Relaxation Dispersion Measurements. J. Magn. Reson. 2019, 308, 106589. 10.1016/j.jmr.2019.106589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; LeBlanc R.; Dayie T. K. SAM-II Riboswitch Samples at Least Two Conformations in Solution in the Absence of Ligand: Implications for Recognition. Angew. Chem., Int. Ed. 2016, 55, 2724–2727. 10.1002/anie.201509997. [DOI] [PubMed] [Google Scholar]
- Kloiber K.; Spitzer R.; Tollinger M.; Konrat R.; Kreutz C. Probing RNA Dynamics via Longitudinal Exchange and CPMG Relaxation Dispersion NMR Spectroscopy Using a Sensitive 13C-Methyl Label. Nucleic Acids Res. 2011, 39, 4340–4351. 10.1093/nar/gkq1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A. L.; Nikolova E. N.; Casiano-Negroni A.; Al-Hashimi H. M. Extending the Range of Microsecond-to-Millisecond Chemical Exchange Detected in Labeled and Unlabeled Nucleic Acids by Selective Carbon R1ρ NMR Spectroscopy. J. Am. Chem. Soc. 2009, 131, 3818–3819. 10.1021/ja8091399. [DOI] [PubMed] [Google Scholar]
- Alvey H. S.; Gottardo F. L.; Nikolova E. N.; Al-Hashimi H. M. Widespread Transient Hoogsteen Base Pairs in Canonical Duplex DNA with Variable Energetics. Nat. Commun. 2014, 5, 4786. 10.1038/ncomms5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimsey I. J.; Petzold K.; Sathyamoorthy B.; Stein Z. W.; Al-Hashimi H. M. Visualizing Transient Watson-Crick-like Mispairs in DNA and RNA Duplexes. Nature 2015, 519, 315–320. 10.1038/nature14227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y.; Gracia B.; Herschlag D.; Russell R.; Al-Hashimi H. M. Visualizing the Formation of an RNA Folding Intermediate Through a Fast Highly Modular Secondary Structure Switch. Nat. Commun. 2016, 7, ncomms11768 10.1038/ncomms11768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangadurai A.; Szymanski E. S.; Kimsey I.; Shi H.; Al-Hashimi H. M. Probing Conformational Transitions Towards Mutagenic Watson-Crick-like G·T Mismatches Using Off-Resonance Sugar Carbon R1ρ Relaxation Dispersion. J. Biomol. NMR 2020, 74, 457–471. 10.1007/s10858-020-00337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriman D. K.; Xue Y.; Yang S.; Kimsey I. J.; Shakya A.; Clay M.; Al-Hashimi H. M. Shortening the HIV-1 TAR RNA Bulge by a Single Nucleotide Preserves Motional Modes Over a Broad Range of Time Scales. Biochemistry 2016, 55, 4445–4456. 10.1021/acs.biochem.6b00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski E. S.; Kimsey I. J.; Al-Hashimi H. M. Direct NMR Evidence That Transient Tautomeric and Anionic States in dG·dT Form Watson-Crick-like Base Pairs. J. Am. Chem. Soc. 2017, 139, 4326–4329. 10.1021/jacs.7b01156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay M. C.; Ganser L. R.; Merriman D. K.; Al-Hashimi H. M. Resolving Sugar Puckers in RNA Excited States Exposes Slow Modes of Repuckering Dynamics. Nucleic Acids Res. 2017, 45, e134 10.1093/nar/gkx525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; Clay M. C.; Rangadurai A.; Sathyamoorthy B.; Case D. A.; Al-Hashimi H. M. Atomic Structures of Excited State A-T Hoogsteen Base Pairs in Duplex DNA by Combining NMR Relaxation Dispersion, Mutagenesis, and Chemical Shift Calculations. J. Biomol. NMR 2018, 70, 229–244. 10.1007/s10858-018-0177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Dethoff E. A.; Al-Hashimi H. M. Invisible RNA State Dynamically Couples Distant Motifs. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 9485–9490. 10.1073/pnas.1407969111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe J. R.; Stein Z. W.; Al-Hashimi H. M. Evaluating the Uncertainty in Exchange Parameters Determined from Off-Resonance R1ρ Relaxation Dispersion for Systems in Fast Exchange. J. Magn. Reson. 2014, 244, 18–29. 10.1016/j.jmr.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baronti L.; Guzzetti I.; Ebrahimi P.; Friebe Sandoz S.; Steiner E.; Schlagnitweit J.; Fromm B.; Silva L.; Fontana C.; Chen A. A.; et al. Base-Pair Conformational Switch Modulates miR-34a Targeting of Sirt1 mRNA. Nature 2020, 583, 139–144. 10.1038/s41586-020-2336-3. [DOI] [PubMed] [Google Scholar]
- Steiner E.; Schlagnitweit J.; Lundström P.; Petzold K. Capturing Excited States in the Fast-Intermediate Exchange Limit in Biological Systems Using 1H NMR Spectroscopy. Angew. Chem., Int. Ed. 2016, 128, 16101–16104. 10.1002/ange.201609102. [DOI] [PubMed] [Google Scholar]
- Baisden J. T.; Boyer J. A.; Zhao B.; Hammond S. M.; Zhang Q. Visualizing a Protonated RNA State That Modulates MicroRNA-21 Maturation. Nat. Chem. Biol. 2021, 17, 80–88. 10.1038/s41589-020-00667-5. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Guffy S. L.; Williams B.; Zhang Q. An Excited State Underlies Gene Regulation of a Transcriptional Riboswitch. Nat. Chem. Biol. 2017, 13, 968–974. 10.1038/nchembio.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B.; Zhang Q. Measuring Residual Dipolar Couplings in Excited Conformational States of Nucleic Acids by CEST NMR Spectroscopy. J. Am. Chem. Soc. 2015, 137, 13480–13483. 10.1021/jacs.5b09014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlagnitweit J.; Steiner E.; Karlsson H.; Petzold K. Efficient Detection of Structure and Dynamics in Unlabeled RNAs: The SELOPE Approach. Chem.-Eur. J. 2018, 24, 6067–6070. 10.1002/chem.201800992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Rangadurai A.; Shi H.; Al-Hashimi H. M. Rapid Assessment of Watson-Crick to Hoogsteen Exchange in Unlabeled DNA Duplexes Using High-Power SELOPE Imino 1H CEST. Magn. Reson. 2021, 2, 715–731. 10.5194/mr-2-715-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten R.; Villali J.; Kern D.; Mulder F. A. A. Probing Microsecond Time Scale Dynamics in Proteins by Methyl 1H Carr-Purcell-Meiboom-Gill Relaxation Dispersion NMR Measurements. Application to Activation of the Signaling Protein NtrCr. J. Am. Chem. Soc. 2010, 132, 17004–17014. 10.1021/ja107410x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A. L.; Lundström P.; Velyvis A.; Kay L. E. Quantifying Millisecond Exchange Dynamics in Proteins by CPMG Relaxation Dispersion NMR Using Side-Chain 1H Probes. J. Am. Chem. Soc. 2012, 134, 3178–3189. 10.1021/ja210711v. [DOI] [PubMed] [Google Scholar]
- Dethoff E. A.; Petzold K.; Chugh J.; Casiano-Negroni A.; Al-Hashimi H. M. Visualizing Transient Low-Populated Structures of RNA. Nature 2012, 491, 724–728. 10.1038/nature11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miclet E.; Williams D. C.; Clore G. M.; Bryce D. L.; Boisbouvier J.; Bax A. Relaxation-Optimized NMR Spectroscopy of Methylene Groups in Proteins and Nucleic Acids. J. Am. Chem. Soc. 2004, 126, 10560–10570. 10.1021/ja047904v. [DOI] [PubMed] [Google Scholar]
- Abou Assi H.; Rangadurai A. K.; Shi H.; Liu B.; Clay M. C.; Erharter K.; Kreutz C.; Holley C. L.; Al-Hashimi H. M. 2′-O-Methylation Can Increase the Abundance and Lifetime of Alternative RNA Conformational States. Nucleic Acids Res. 2020, 48, 12365–12379. 10.1093/nar/gkaa928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan P. P.; AlSadhan I.; Merriman D. K.; Al-Hashimi H.; Herschlag D. Pseudouridine and N6-Methyladenosine Modifications Weaken PUF Protein/RNA Interactions. RNA 2017, 23, 611–618. 10.1261/rna.060053.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Merriman D. K.; Choi S. H.; Schumacher M. A.; Plangger R.; Kreutz C.; Horner S. M.; Meyer K. D.; Al-Hashimi H. M. A Potentially Abundant Junctional RNA Motif Stabilized by m6A and Mg2+. Nat. Commun. 2018, 9, 2761. 10.1038/s41467-018-05243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.-C.; Liu B.; Plangger R.; Kreutz C.; Al-Hashimi H. M. m6A Minimally Impacts the Structure, Dynamics, and Rev ARM Binding Properties of HIV-1 RRE Stem IIB. PLoS One 2019, 14, e0224850 10.1371/journal.pone.0224850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S.; Ries R. J.; Jaffrey S. R. Reading, Writing and Erasing mRNA Methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- Roundtree I. A.; Evans M. E.; Pan T.; He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Dominissini D.; Rechavi G.; He C. Gene Expression Regulation Mediated through Reversible m6A RNA Methylation. Nat. Rev. Genet. 2014, 15, 293–306. 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- Kim G. W.; Siddiqui A. N6-Methyladenosine Modification of HCV RNA Genome Regulates Cap-Independent IRES-Mediated Translation via YTHDC2 Recognition. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2022024118 10.1073/pnas.2022024118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G. W.; Siddiqui A. Hepatitis B Virus X Protein Recruits Methyltransferases to Affect Cotranscriptional N6-Methyladenosine Modification of Viral/Host RNAs. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2019455118 10.1073/pnas.2019455118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam H.; Khan M.; Gokhale N. S.; McIntyre A. B.; Kim G. W.; Jang J. Y.; Kim S. J.; Mason C. E.; Horner S. M.; Siddiqui A. N6-Methyladenosine Modification of Hepatitis B Virus RNA Differentially Regulates the Viral Life Cycle. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 8829–8834. 10.1073/pnas.1808319115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesser C. R.; Karijolich J.; Dominissini D.; He C.; Glaunsinger B. A. N6-Methyladenosine Modification and the YTHDF2 Reader Protein Play Cell Type Specific Roles in Lytic Viral Gene Expression During Kaposi’s Sarcoma-Associated Herpesvirus Infection. PLoS Pathog. 2018, 14, e1006995 10.1371/journal.ppat.1006995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumuru N.; Zhao B. S.; Lu W.; Lu Z.; He C.; Wu L. N6-Methyladenosine of HIV-1 RNA Regulates Viral Infection and HIV-1 Gag Protein Expression. Elife 2016, 5, e15528 10.7554/eLife.15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichinchi G.; Zhao B. S.; Wu Y.; Lu Z.; Qin Y.; He C.; Rana T. M. Dynamics of Human and Viral RNA Methylation During Zika Virus Infection. Cell Host Microbe 2016, 20, 666–673. 10.1016/j.chom.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale N. S.; McIntyre A. B. R.; McFadden M. J.; Roder A. E.; Kennedy E. M.; Gandara J. A.; Hopcraft S. E.; Quicke K. M.; Vazquez C.; Willer J.; et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. 10.1016/j.chom.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Yang Y.; Sun B.-F.; Shi Y.; Yang X.; Xiao W.; Hao Y.-J.; Ping X.-L.; Chen Y.-S.; Wang W.-J.; et al. FTO-Dependent Demethylation of N6-Methyladenosine Regulates mRNA Splicing and is Required for Adipogenesis. Cell Res. 2014, 24, 1403–1419. 10.1038/cr.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Lu Z.; Gomez A.; Hon G. C.; Yue Y.; Han D.; Fu Y.; Parisien M.; Dai Q.; Jia G.; et al. N6-Methyladenosine-Dependent Regulation of Messenger RNA Stability. Nature 2014, 505, 117–120. 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W.; Adhikari S.; Dahal U.; Chen Y. S.; Hao Y. J.; Sun B. F.; Sun H. Y.; Li A.; Ping X. L.; Lai W. Y.; et al. Nuclear m6A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Weng Y.-L.; Wang X.; An R.; Cassin J.; Vissers C.; Liu Y.; Liu Y.; Xu T.; Wang X.; Wong S. Z. H.; et al. Epitranscriptomic m6A Regulation of Axon Regeneration in the Adult Mammalian Nervous System. Neuron 2018, 97, 313–325. 10.1016/j.neuron.2017.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawa K.; Knoll W. Mismatching Base-pair Dependence of the Kinetics of DNA-DNA Hybridization Studied by Surface Plasmon Fluorescence Spectroscopy. Nucleic Acids Res. 2004, 32, 2372–2377. 10.1093/nar/gkh572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Zhan J.; Man B.; Jiang S.; Yue W.; Gao S.; Guo C.; Liu H.; Li Z.; Wang J.; Zhou Y. Real-Time Reliable Determination of Binding Kinetics of DNA Hybridization Using a Multi-Channel Graphene Biosensor. Nat. Commun. 2017, 8, 14902. 10.1038/ncomms14902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse I. I.; Kim H.; Ha T. A Rule of Seven in Watson-Crick Base Pairing of Mismatched Sequences. Nat. Struct. Mol. Biol. 2012, 19, 623–627. 10.1038/nsmb.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J. D.; Von Hippel P. H. Effects of Methylation on the Stability of Nucleic Acid Conformations. Studies at the Polymer Level. J. Biol. Chem. 1978, 253, 927–961. 10.1016/S0021-9258(17)38193-0. [DOI] [PubMed] [Google Scholar]
- Engel J. D.; von Hippel P. H. Effects of Methylation on the Stability of Nucleic Acid Conformations: Studies at the Monomer Level. Biochemistry 1974, 13, 4143–4158. 10.1021/bi00717a013. [DOI] [PubMed] [Google Scholar]
- Roost C.; Lynch S. R.; Batista P. J.; Qu K.; Chang H. Y.; Kool E. T. Structure and Thermodynamics of N6-Methyladenosine in RNA: A Spring-Loaded Base Modification. J. Am. Chem. Soc. 2015, 137, 2107–2115. 10.1021/ja513080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren A.; Rajashankar K. R.; Patel D. J. Fluoride Ion Encapsulation by Mg2+ Ions and Phosphates in a Fluoride Riboswitch. Nature 2012, 486, 85–89. 10.1038/nature11152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. L.; Sudarsan N.; Weinberg Z.; Roth A.; Stockbridge R. B.; Breaker R. R. Widespread Genetic Switches and Toxicity Resistance Proteins for Fluoride. Science 2012, 335, 233–235. 10.1126/science.1215063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D. P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle N. T.; MacRae I. J. The Crystal Structure of Human Argonaute2. Science 2012, 336, 1037–1040. 10.1126/science.1221551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K.; Weinberg D. E.; Bartel D. P.; Patel D. J. Structure of Yeast Argonaute with Guide RNA. Nature 2012, 486, 368–374. 10.1038/nature11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A.; Farh K. K. H.; Johnston W. K.; Garrett-Engele P.; Lim L. P.; Bartel D. P. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Mol. Cell 2007, 27, 91–105. 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen C. B.; Shomron N.; Sandberg R.; Hornstein E.; Kitzman J.; Burge C. B. Determinants of Targeting by Endogenous and Exogenous MicroRNAs and siRNAs. RNA 2007, 13, 1894–1910. 10.1261/rna.768207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkayam E.; Kuhn C.-D.; Tocilj A.; Haase A. D.; Greene E. M.; Hannon G. J.; Joshua-Tor L. The Structure of Human Argonaute-2 in Complex with miR-20a. Cell 2012, 150, 100–110. 10.1016/j.cell.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W.; Bhattacharyya S. N.; Sonenberg N. Mechanisms of Post-Transcriptional Regulation by MicroRNAs: Are the Answers in Sight?. Nat. Rev. Genet. 2008, 9, 102–114. 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Sheu-Gruttadauria J.; Xiao Y.; Gebert L. F.; MacRae I. J. Beyond the Seed: Structural Basis for Supplementary MicroRNA Targeting by Human Argonaute2. EMBO J. 2019, 38, e101153 10.15252/embj.2018101153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee L. M.; Flores-Jasso C. F.; Salomon W. E.; Zamore P. D. Argonaute Divides Its RNA Guide into Domains with Distinct Functions and RNA-Binding Properties. Cell 2012, 151, 1055–1067. 10.1016/j.cell.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancati G.; Großhans H. An Interplay of miRNA Abundance and Target Site Architecture Determines miRNA Activity and Specificity. Nucleic Acids Res. 2018, 46, 3259–3269. 10.1093/nar/gky201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton J. P.; Lovci M. T.; Huang J. L.; Yeo G. W.; Pasquinelli A. E. Pairing Beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. 10.1016/j.molcel.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng G.; Zhao H.; Wang J.; Rao Y.; Tian W.; Swarts D. C.; van der Oost J.; Patel D. J.; Wang Y. Structure-Based Cleavage Mechanism of Thermus Thermophilus Argonaute DNA Guide Strand-Mediated DNA Target Cleavage. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 652–657. 10.1073/pnas.1321032111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Juranek S.; Li H.; Sheng G.; Wardle G. S.; Tuschl T.; Patel D. J. Nucleation, Propagation and Cleavage of Target RNAs in Ago Silencing Complexes. Nature 2009, 461, 754–761. 10.1038/nature08434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuwen T.; Huang R.; Kay L. E. Probing Slow Timescale Dynamics in Proteins Using Methyl 1H CEST. J. Biomol. NMR 2017, 68, 215–224. 10.1007/s10858-017-0121-x. [DOI] [PubMed] [Google Scholar]
- Yuwen T.; Sekhar A.; Kay L. E. Separating Dipolar and Chemical Exchange Magnetization Transfer Processes in 1H-CEST. Angew. Chem., Int. Ed. 2017, 56, 6122–6125. 10.1002/anie.201610759. [DOI] [PubMed] [Google Scholar]
- Sekhar A.; Rosenzweig R.; Bouvignies G.; Kay L. E. Hsp70 Biases the Folding Pathways of Client Proteins. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E2794–E2801. 10.1073/pnas.1601846113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundström P.; Akke M. Off-Resonance Rotating-Frame Amide Proton Spin Relaxation Experiments Measuring Microsecond Chemical Exchange in Proteins. J. Biomol. NMR 2005, 32, 163–173. 10.1007/s10858-005-5027-3. [DOI] [PubMed] [Google Scholar]
- Eichmüller C.; Skrynnikov N. R. A New Amide Proton R1ρ Experiment Permits Accurate Characterization of Microsecond Time-Scale Conformational Exchange. J. Biomol. NMR 2005, 32, 281–293. 10.1007/s10858-005-0658-y. [DOI] [PubMed] [Google Scholar]
- Ishima R.; Wingfield P. T.; Stahl S. J.; Kaufman J. D.; Torchia D. A. Using Amide 1H and 15N Transverse Relaxation To Detect Millisecond Time-Scale Motions in Perdeuterated Proteins: Application to HIV-1 Protease. J. Am. Chem. Soc. 1998, 120, 10534–10542. 10.1021/ja981546c. [DOI] [Google Scholar]
- Rangadurai A.; Shi H.; Al-Hashimi H. M. Extending the Sensitivity of CEST NMR Spectroscopy to Micro-to-Millisecond Dynamics in Nucleic Acids Using High-Power Radio-Frequency Fields. Angew. Chem., Int. Ed. 2020, 59, 11262–11266. 10.1002/anie.202000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z.; Zhang K.; Kappel K.; Li S.; Palo M. Z.; Pintilie G. D.; Rangan R.; Luo B.; Wei Y.; Das R.; et al. Cryo-EM Structures of Full-Length Tetrahymena Ribozyme at 3.1 Å Resolution. Nature 2021, 596, 603–607. 10.1038/s41586-021-03803-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel K.; Zhang K.; Su Z.; Watkins A. M.; Kladwang W.; Li S.; Pintilie G.; Topkar V. V.; Rangan R.; Zheludev I. N.; et al. Accelerated Cryo-EM-Guided Determination of Three-Dimensional RNA-Only Structures. Nat. Methods 2020, 17, 699–707. 10.1038/s41592-020-0878-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla S. L.; Sherlock M. E.; MacFadden A.; Kieft J. S. A Viral RNA Hijacks Host Machinery Using Dynamic Conformational Changes of a tRNA-like Structure. Science 2021, 374, 955–960. 10.1126/science.abe8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchinat E.; Barbieri L.; Cremonini M.; Banci L. Protein In-Cell NMR Spectroscopy at 1.2 GHz. J. Biomol. NMR 2021, 75, 97–107. 10.1007/s10858-021-00358-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs H.; Moskau D.; Spraul M. Cryogenically Cooled Probes—A Leap in NMR Technology. Prog. Nucl. Magn. Reson. Spectrosc. 2005, 46, 131–155. 10.1016/j.pnmrs.2005.03.001. [DOI] [Google Scholar]
- Boeszoermenyi A.; Chhabra S.; Dubey A.; Radeva D. L.; Burdzhiev N. T.; Chanev C. D.; Petrov O. I.; Gelev V. M.; Zhang M.; Anklin C.; et al. Aromatic 19F-13C TROSY: A Background-Free Approach to Probe Biomolecular Structure, Function, and Dynamics. Nat. Methods 2019, 16, 333–340. 10.1038/s41592-019-0334-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitevski-LeBlanc J. L.; Hoang J.; Thach W.; Larda S. T.; Prosser R. S. 19F NMR Studies of a Desolvated Near-Native Protein Folding Intermediate. Biochemistry 2013, 52, 5780–5789. 10.1021/bi4010057. [DOI] [PubMed] [Google Scholar]
- Kim T. H.; Mehrabi P.; Ren Z.; Sljoka A.; Ing C.; Bezginov A.; Ye L.; Pomès R.; Prosser R. S.; Pai E. F. The Role of Dimer Asymmetry and Protomer Dynamics in Enzyme Catalysis. Science 2017, 355, eeag2355 10.1126/science.aag2355. [DOI] [PubMed] [Google Scholar]
- Hoang J.; Prosser R. S. Conformational Selection and Functional Dynamics of Calmodulin: A 19F Nuclear Magnetic Resonance Study. Biochemistry 2014, 53, 5727–5736. 10.1021/bi500679c. [DOI] [PubMed] [Google Scholar]
- Aramini J. M.; Hamilton K.; Ma L.-C.; Swapna G. V. T.; Leonard P. G.; Ladbury J. E.; Krug R. M.; Montelione G. T. 19F NMR Reveals Multiple Conformations at the Dimer Interface of the Nonstructural Protein 1 Effector Domain from Influenza A Virus. Structure 2014, 22, 515–525. 10.1016/j.str.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeck J. H.; Kremer W.; Sprangers R. A Suite of 19F Based Relaxation Dispersion Experiments to Assess Biomolecular Motions. J. Biomol. NMR 2020, 74, 753–766. 10.1007/s10858-020-00348-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebau J.; Tersa M.; Trastoy B.; Patrick J.; Rodrigo-Unzueta A.; Corzana F.; Sparrman T.; Guerin M. E.; Mäler L. Unveiling the Activation Dynamics of a Fold-Switch Bacterial Glycosyltransferase by 19F NMR. J. Biol. Chem. 2020, 295, 9868–9878. 10.1074/jbc.RA120.014162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland M.; Catazaro J.; Rajasekaran R.; Strub M.-P.; O’Hern C.; Bermejo G. A.; Summers M. F.; Marchant J.; Tjandra N. Long-Range RNA Structural Information via a Paramagnetically Tagged Reporter Protein. J. Am. Chem. Soc. 2019, 141, 1430–1434. 10.1021/jacs.8b11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier A.; Brutscher B. Recovering Lost Magnetization: Polarization Enhancement in Biomolecular NMR. J. Biomol. NMR 2011, 49, 9–15. 10.1007/s10858-010-9461-5. [DOI] [PubMed] [Google Scholar]
- Solyom Z.; Schwarten M.; Geist L.; Konrat R.; Willbold D.; Brutscher B. BEST-TROSY Experiments for Time-Efficient Sequential Resonance Assignment of Large Disordered Proteins. J. Biomol. NMR 2013, 55, 311–321. 10.1007/s10858-013-9715-0. [DOI] [PubMed] [Google Scholar]