

Abstract
Paramagnetic chemical probes have been used in electron paramagnetic resonance (EPR) and nuclear magnetic resonance (NMR) spectroscopy for more than four decades. Recent years witnessed a great increase in the variety of probes for the study of biological macromolecules (proteins, nucleic acids, and oligosaccharides). This Review aims to provide a comprehensive overview of the existing paramagnetic chemical probes, including chemical synthetic approaches, functional properties, and selected applications. Recent developments have seen, in particular, a rapid expansion of the range of lanthanoid probes with anisotropic magnetic susceptibilities for the generation of structural restraints based on residual dipolar couplings and pseudocontact shifts in solution and solid state NMR spectroscopy, mostly for protein studies. Also many new isotropic paramagnetic probes, suitable for NMR measurements of paramagnetic relaxation enhancements, as well as EPR spectroscopic studies (in particular double resonance techniques) have been developed and employed to investigate biological macromolecules. Notwithstanding the large number of reported probes, only few have found broad application and further development of probes for dedicated applications is foreseen.
1. Paramagnetic NMR for Biomolecular Studies
1.1. General Introduction
Nuclear magnetic resonance (NMR) spectroscopy is the most widely used spectroscopic technique to obtain structural information with atomic resolution. The NMR observable most frequently used for structure determination is the nuclear Overhauser effect (NOE), which measures dipolar interactions between protons separated by up to about 5 Å and, thus, provides internuclear distance restraints.1,2 Owing to the much larger magnetic moment of unpaired electrons compared with nuclei (about 658 times greater for a lone electron than for a proton), dipolar interactions involving unpaired electrons, such as in metalloproteins containing paramagnetic metal ions, can be detected over a much greater distance range. As the paramagnetic effects of unpaired electrons on the NMR spectrum tend to be very large and are readily detected, potentially over distances up to 100 Å from the paramagnetic center, they present valuable long-range structural information. In fact, 50 years ago and prior to the invention of two-dimensional NMR spectroscopy, paramagnetic effects were thought to be the key to 3D structure determinations of proteins and other biomolecules in solution.3,4 The paramagnetic effects are in many instances anisotropic, contributing not only distance but also orientational restraints.
Apart from uses in NMR spectroscopy, paramagnetic labels also feature most prominently in electron paramagnetic resonance (EPR) spectroscopy, where they have gained in importance by their capacity to accurately measure distances between paramagnetic centers on the nanometer scale. This Review will focus on applications in NMR spectroscopy, while also acknowledging the performance of tags for distance measurements by EPR spectroscopy, specifically in double-electron–electron resonance (DEER) experiments. Furthermore, europium(III) and terbium(III) are paramagnetic metal ions which, in conjunction with an aromatic antenna group, can yield intense luminescence suitable for distance measurements by Förster resonance energy transfer (FRET).5,6 The same probes can thus be suitable for NMR and FRET applications, but luminescence applications are not discussed in this Review which focuses on paramagnetism and NMR spectroscopy.
The field of paramagnetic NMR has been reviewed before. In particular, the theory of paramagnetic NMR and the resulting spectral effects have been described in many reviews and books, covering magnetic susceptibility,7−11 relaxation,12 and effects in solids13 in great detail. This Review presents an integrated formalism of the various effects and includes the most recent effects reported.
Initially, paramagnetic NMR spectroscopy of biological macromolecules took advantage of paramagnetic metal ions present in proteins,14,15 and this approach is still finding application for the study of metalloproteins and also in research into metal trafficking.16 Also metal substitution has been used extensively.17 With the arrival of paramagnetic tags, the application of the tool box of paramagnetic NMR spectroscopy became available for biomolecules that lack a natural paramagnetic center, expanding its potential greatly. Several reviews give a general overview of the possibilities of employing pseudocontact shifts (PCSs), paramagnetic relaxation enhancements (PREs), and residual dipolar couplings (RDCs) (terms to be explained below) or focus on the range of different tags available and their applications.18−26
Specific applications have been the topic of dedicated reviews. One example is the use of paramagnetism for partial alignment to generate RDCs for the study of structure and dynamics.27 PCSs provide useful data for the structure determination of proteins and protein–protein or protein–ligand complexes, as sole restraints, or in combination with other data, such as X-ray diffraction and small-angle X-ray scattering.10,26,28−30 PREs are useful in particular because of their ability to report on minor, “invisible” or “dark” states present in a sample.31−34 PREs generated by soluble probes, that is, paramagnetic molecules not covalently linked to the molecule of interest, are referred to as solvent PREs and can provide information on molecular surfaces and changes in surface exposure upon complex formation.35,36 A recent review describes the use of paramagnetic NMR in drug discovery.37 Tags have been mostly designed for and applied to proteins, but applications to other biomolecules have started to appear, such as oligosaccharides,38,39 and a significant body of work exists describing oligonucleotides with paramagnetic centers for EPR measurements.40−47
The main aim of this Review is to describe the state-of-the-art of paramagnetic tags for biological macromolecules. We wish to illustrate the diversity of tags and discuss their pros and cons for various applications. Contrary to most other reviews, particular attention is paid to the chemical synthesis of the tags because the chemistry of the more advanced cyclen-based lanthanoid tags is not trivial and in some cases a limiting factor, due to demanding synthetic routes or limited chemical stability under physiological conditions and in the presence of proteins. Paramagnetic compounds have many more applications, e.g., as contrast agents in magnetic resonance imaging (MRI) and polarizing agents for dynamic nuclear polarization (DNP). These topics are considered outside the scope of this Review, and the reader is referred to excellent, recently published reviews.48,49 Paramagnetic tags attached to proteins exclusively for the purpose of studies by EPR spectroscopy have been comprehensively discussed previously.50 Similarly, our discussion omits EPR spin labels attached via a long and flexible tether, such as resulting from tagging noncanonical amino acids.51 In contrast, our account attempts to give a comprehensive overview over paramagnetic tags designed specifically for structure studies of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) oligomers by NMR and EPR spectroscopy. Finally, various paramagnetic compounds have been devised as sample additives to enhance longitudinal relaxation rates in solid-state NMR spectroscopy.52,53 As far as these compounds are not chemically attached to biological macromolecules, the reader is again referred to a recent review for a more complete compilation.54
As paramagnetic probes elicit a multitude of effects depending on their chemical, physical, and dynamic properties, an increasing number of probes have been synthesized with the aim of facilitating their installation in biological macromolecules and maximizing the information content that can be gathered from the observation of the paramagnetic effects. About half of the published paramagnetic probes were described for the first time in the past five years. To assess their utility for different purposes, it is necessary to understand the origin and manifestation of the paramagnetic effects. Many of the most stringent tag requirements stem from applications in paramagnetic NMR spectroscopy.
The terms spin label, paramagnetic tag and paramagnetic probe are frequently found in the literature and also used throughout this Review. They all refer to paramagnetic compounds that are introduced and used to generate paramagnetic effects to study a system of interest. In general, these terms can be considered synonyms. The term “spin label” is probably the oldest and has been widely used in the field of EPR for many years. It refers to labeling a system with an unpaired electron spin. “Paramagnetic tag” emphasizes the quality of a small label to give information about the bearer, that is, provide site-specific information about the tagged molecule. The term “paramagnetic probe” relates more generally to the function, that is, probing the molecule under investigation and includes also soluble compounds that are not bound to specific sites.
1.2. Qualitative Description of Paramagnetic Effects in NMR Spectroscopy
Paramagnetic effects detected in NMR spectra depend on the dipolar fields generated by unpaired electrons. If the electron spins relax slowly compared with the rotational correlation time of the electron–nucleus vector, the dipolar field of the electron at the site of the nuclear spin averages to zero, provided that the molecule reorientates isotropically in solution. In this case, the chemical shift of the nuclear spin does not change, but the time fluctuation of the dipolar field can greatly enhance the nuclear relaxation. Electron spins that relax rapidly compared with the rotational correlation time of the electron–nucleus vector lead to a Curie spin, which is the time-averaged net magnetic moment of the electron spin aligned with the external magnetic field. The Curie spin constitutes a molecular magnetic susceptibility. In this situation, it is useful to describe the magnetic susceptibility associated with the paramagnetic center, χ, by a tensor that defines its magnitude as a function of the molecular orientation in the external magnetic field. In general, the χ tensor of a paramagnetic center with rapidly relaxing electrons is anisotropic and its anisotropic component is commonly referred to as Δχ tensor.
Chemical shift changes observed in NMR spectra due to the presence of a paramagnetic center are referred to as hyperfine shifts. For paramagnetic centers with a Curie spin, hyperfine shifts comprise two parts, the contact shift and PCS. Contact shifts are a consequence of the delocalization of unpaired electron spin density across chemical bonds and thus are only observed for nuclear spins fairly close to the paramagnetic center. In contrast to the contact shift, the PCS is a through-space interaction, which arises from the time-averaged dipolar interaction between the unpaired electron spins and the nuclear spin. PCSs are observable over much greater distances and, as they are independent of bond angles, can be described by fewer parameters, which pertain to the Δχ tensor and the location of the nuclear spin relative to the Δχ tensor. In this way, PCSs deliver long-range distance and orientation information, which can readily be interpreted.
Anisotropic magnetic susceptibilities also cause weak alignment of the paramagnetic molecule in the external magnetic field, leading to the observation of RDCs between nuclear spins. In as far as the molecule tumbles in solution as a single rigid entity, the alignment affects the entire molecule irrespective of the location of the paramagnetic center and, therefore, RDCs do not depend on the distance of the nuclear spins from the paramagnetic center. Conveniently, the size and orientation of the alignment tensor are proportional to the size and orientation of the Δχ tensor.
A paramagnetic center with Curie spin generates a magnetic dipolar field at the site of a nuclear spin. The mathematical description of the effect of this dipolar field on the magnetic shielding of the nucleus is closely similar to that of chemical shift anisotropy (CSA). To highlight this similarity, the term dipolar shielding anisotropy (DSA) has been coined for this paramagnetic effect. The similarity extends to cross-correlated relaxation (CCR) effects between DSA and dipole–dipole (DD) relaxation, which is manifested in differential relaxation rates of multiplet components, just like the CSA/DD cross-correlation effects that form the basis of transverse relaxation optimized spectroscopy (TROSY).55,56 DSA/DD cross-correlated relaxation can be used to evaluate the angles between an internuclear bond and the principal axes of the DSA tensor. As the magnitude of the DSA tensor depends on the distance of the nuclear spin from the paramagnetic center, conversion of CCR effects into structural restraints is more involved than for RDCs. Nonetheless, these CCR effects are readily observable and can provide useful distinctions between different pairs of coupled nuclear spins that otherwise display similar paramagnetic effects.57
A different type of CCR effect also occurs between CSA and DSA relaxation. This CCR effect can significantly affect the nuclear net relaxation rates, either enhancing or reducing the relaxation of nuclear magnetization in the paramagnetic state compared with the corresponding diamagnetic state.58
In general, paramagnetic centers enhance the relaxation rates of nuclear spins, which is referred to as PRE. A PRE is the direct consequence of dipole–dipole interactions between the paramagnetic center and a nuclear spin. It decreases with the sixth power of the distance between them. The large magnitude of the PRE at short distances and its steep distance dependence render the PRE a sensitive tool to detect minor states that position the nucleus close to the unpaired electrons, as may occur in dynamic proteins carrying a paramagnetic tag. Paramagnetic centers with slow electronic relaxation (∼109 s–1), such as presented by nitroxide radicals, Mn(II), and Gd(III) ions, generate PREs by dipole–dipole relaxation, referred to as Solomon–Bloembergen–Morgan (SBM) or Solomon relaxation.59 In contrast, SBM relaxation is much less efficient for unpaired electrons with fast electronic relaxation rates (>1011 s–1) and Curie spin relaxation becomes more prominent,60,61 especially for long rotational correlation times of the molecule and at increased magnetic field strength. In general, the SBM mechanism is the sole contribution to PREs in isotropic paramagnetic species and the Curie spin mechanism adds an additional component in systems characterized by paramagnetic anisotropy, which can nonetheless become the dominant contribution to PREs.
Many transition metal ions contain unpaired electrons, leading to paramagnetism. The paramagnetism of 3d block and 4f block ions has been studied most thoroughly.29,62 Mn(II) ions have a long electronic relaxation time, causing no PCSs but strong PREs. High-spin Co(II) features a Curie spin and can generate large PCSs with weak PREs, compared with other 3d block ions. In aqueous solution, iron is stable in the oxidation states +2 and +3. Depending on its ligands, Fe(II) can be paramagnetic or diamagnetic. Similarly, the paramagnetism of Fe(III) can be either large (high spin) or small (low spin), depending on the coordination environment. The 4f block elements are referred to as lanthanoids,63 with the general symbol Ln. Unlike 3d block ions, lanthanoids are seldom involved in biological processes, with the first examples of lanthanoenzymes discovered only in 2011 in methanotrophic bacteria.64 Lutetium(III) and lanthanum(III) have no unpaired electrons, while all other Ln(III) ions of the series are paramagnetic. Gd(III) ions are unique for featuring a strong isotropic magnetic susceptibility, which is generated by seven unpaired electrons. Gd(III) ions yield exceptionally strong PREs without generating PCSs. Other Ln(III) ions have Curie spins of different magnitude, causing both PCS and PRE effects. Stable organic radicals such as nitroxides or the trityl radical can also be used to create a paramagnetic center. They do not cause PCSs, but yield sizable PREs.
Quantification of the paramagnetic effects is commonly achieved by comparison with a diamagnetic species. Therefore, the appropriate choice of a diamagnetic reference as similar to the paramagnetic sample as possible is very important. Lanthanoids are chemically similar to each other, so that a diamagnetic reference can be obtained easily by substitution of the paramagnetic ion with Lu(III) or La(III). Furthermore, the ionic radius of Y(III) is practically the same as that of Ho(III), making it an excellent diamagnetic reference for the heavy lanthanoid ions ranging from Tb(III) to Yb(III). For this reason, the present article makes no formal distinction between Ln(III) and Y(III) ions. In the case of nitroxide radicals, the diamagnetic reference is usually obtained by chemical reduction to the corresponding hydroxylamine, which is readily achieved with ascorbic acid. Alternatively, diamagnetic control probes (such as hydroxylamine derivatives) can be used that are chemically similar to the nitroxide probes.65 As the properties of 3d block ions vary more than those of Ln(III) ions, it is more difficult to identify suitable diamagnetic references. Zn(II) is commonly used as a diamagnetic reference for Co(II) and Mn(II) because charge and size are similar, and Ga(III) has been successfully used as a diamagnetic reference for high-spin Fe(III).66,67
1.3. Quantitative Description of Paramagnetic Effects in NMR Spectroscopy
Mathematical descriptions of the different paramagnetic effects are well established and enable their quantitative interpretation. Hyperfine shifts, such as PCSs, are reported as the change in chemical shift (measured in ppm) caused by the presence of a paramagnetic center. RDCs are measured in hertz, reporting on the change in multiplet splitting observed due to weak paramagnetic alignment of the molecules with the magnetic field. PREs and cross-correlated relaxation effects are measured in s–1, presenting the paramagnetic enhancement in longitudinal (R1) or transverse (R2) relaxation rates of nuclear spins over a corresponding diamagnetic reference, with R1 and R2 being the inverse of the T1 and T2 relaxation times. The basic equations governing the various paramagnetic effects have been treated in numerous review articles and books.7−9,11,13,19,68 In the following, we present a summary to highlight the salient features together with correction terms that may become relevant in special circumstances.
1.3.1. Pseudocontact Shift (PCS)
The PCS of a nuclear spin can be described by eq 1(7)
| 1 |
| 2 |
where r is the distance between the paramagnetic center and the nuclear spin, and the angles θ and ϕ are the polar angles describing the location of the nucleus with respect to the principal axes of the χ tensor. The axial component of the magnetic susceptibility anisotropy, Δχax, and the rhombic component Δχrh are defined by eq 2, where χx, χy, and χz denote the values of magnetic susceptibility along the respective principal axes of the χ tensor (Figure 1A). Being defined as the anisotropic component of the χ tensor, the Δχ tensor is spanned by principal axes that are aligned with those of the χ tensor. Use of eq 1 for calculating the PCS of a nuclear spin in a given molecular structure requires the prior knowledge of the location of the paramagnetic center and the orientation of the Δχ tensor relative to the molecule. The orientation of the Δχ tensor is commonly described by three Euler angles (α, β, and γ) and, in the case of proteins, reported relative to coordinates deposited in the protein data bank (PDB). Together with the Δχax and Δχrh values, eight parameters are thus required to define the Δχ tensor relative to a set of atomic coordinates. They can be obtained by using the experimentally measured PCS data of at least eight different nuclear spins to fit the Δχ tensor to the molecular structure. A number of software packages69−73 are available to perform this fit. In practice, good fits require at least three times more PCS data than the minimum of eight, in which case it is also possible to obtain a measure of the uncertainties of the Δχ tensor parameters by Monte Carlo random variation of the data input.
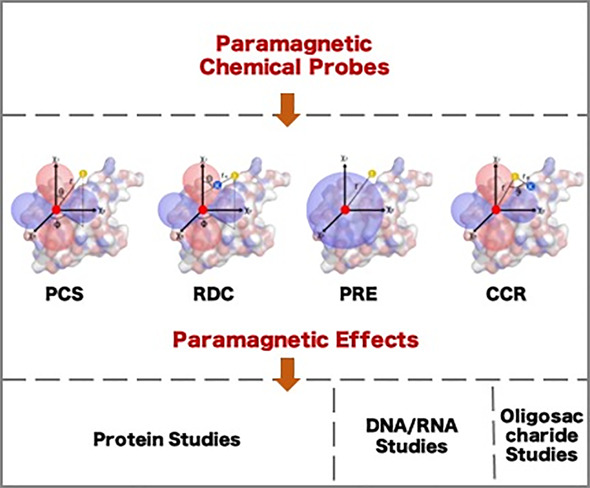
Figure 1.

Schematic representation of the paramagnetic effects of PCS, RDC, PRE, and CCR, respectively, where CCR refers to cross-correlation between DSA and dipole–dipole relaxation. The top panel depicts the geometric dependencies relative to the frame of a χ tensor. The bottom panel illustrates the effects in the NMR spectra of paramagnetic (red) versus diamagnetic (black) samples.
To fit Δχ tensors to molecular structures and calculate correction terms arising from cross-correlation effects and molecular alignment, it is most convenient to use a matrix representation of the χ tensor. The isotropic component of the χ tensor is given by
| 3 |
where μ0 is the induction constant and ge and S are the electronic g factor and spin, respectively; their values are replaced by gJ and J for lanthanoid ions.74 μB is the Bohr magneton; kB denotes the Boltzmann constant, and T is the absolute temperature. Including the anisotropic components of the χ tensor, a concise mathematical description of the dipolar shift tensor σ at a given position r and distance r from the paramagnetic center can be written as
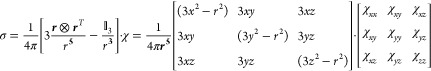 |
4 |
where  3 denotes the 3 × 3 identity
matrix, ⊗ denotes the Kronecker product, and x, y, and z the coordinates of the
nucleus relative to the origin of the χ tensor.7,73 (Note that the dipolar shift tensor is the negative of the dipolar
shielding tensor.) The full χ tensor including its isotropic
component is needed to calculate PREs, but the Δχ tensor
suffices to calculate PCSs. The Δχ tensor in matrix representation
is the traceless part of the χ tensor (i.e., the same as the
χ tensor, except that each diagonal element is reduced by their
average). In the representation of eq 4, the PCS is given by the trace of the shielding tensor
3 denotes the 3 × 3 identity
matrix, ⊗ denotes the Kronecker product, and x, y, and z the coordinates of the
nucleus relative to the origin of the χ tensor.7,73 (Note that the dipolar shift tensor is the negative of the dipolar
shielding tensor.) The full χ tensor including its isotropic
component is needed to calculate PREs, but the Δχ tensor
suffices to calculate PCSs. The Δχ tensor in matrix representation
is the traceless part of the χ tensor (i.e., the same as the
χ tensor, except that each diagonal element is reduced by their
average). In the representation of eq 4, the PCS is given by the trace of the shielding tensor
| 5 |
which can also be expressed in terms of Δχ tensor elements
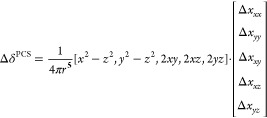 |
6 |
Eq 6 illustrates how separating the position coordinates of the PCS equation (eq 5) from the Δχ tensor leads to a linear form of the equation with 5 unique elements in a column vector defining the tensor. Equation 6 thus allows Δχ tensor fits for a fixed position by singular value decomposition. Combined with the three coordinates of the metal center relative to the coordinates of the molecule, a Δχ tensor fit thus requires determining eight parameters.
1.3.2. Residual Dipolar Coupling (RDC)
In isotropic solutions, internuclear dipolar couplings average to zero due to fast molecular tumbling, but RDCs re-emerge upon partial alignment of the molecules in a magnetic field. RDCs are most easily measured for one-bond scalar couplings, where they manifest in altered distance (measured in Hz) between well-separated multiplet components.27 If the molecular alignment is caused by a paramagnetic center with anisotropic magnetic susceptibility, the alignment tensor A is directly proportional to the Δχ tensor, with the same axes directions7
| 7 |
where B0 is the magnetic field strength. The residual dipolar coupling between two nuclear spins I and K is described by7,75
| 8 |
where γI and γK denote the gyromagnetic ratios of the nuclear spins I and K, respectively, rIK is the internuclear distance, h Planck’s constant, and the angles Θ and Φ determine the orientation of the I–K vector relative to the Δχ tensor (Figure 1B). The RDC, thus, yields information about the orientation of the internuclear vector in the frame of the Δχ tensor independent of the distance of the nuclei from the paramagnetic center. RDCs allow fitting the alignment tensor to a protein structure and, once the tensor frame has been determined, the RDCs can be used to establish bond vector orientations within a protein structure. As reorientational motions of a bond vector reduce the RDC by averaging over different orientations, RDC measurements can be used to study protein dynamics.27,76
1.3.3. Residual anisotropic chemical shift (RACS)
The weak molecular alignment in the magnetic field caused by anisotropic magnetic susceptibilities not only produces RDCs, but also changes the chemical shifts of nuclear spins that feature significant CSA tensor anisotropies by rendering the averaging over different molecular orientations incomplete. The change in chemical shift caused by such RACS effects poses a limit on the accuracy with which PCSs can be measured. If the structure of the molecule is known, the CSA tensors associated with individual nuclei can be taken into account by estimating a RACS correction for each PCS measured. The corrections can be significant at high magnetic field strength,77 as the degree of molecular alignment depends on the square of the magnetic field (eq 7).
1.3.4. Residual Anisotropic Dipolar Shift (RADS)
Residual anisotropic dipolar shifts present another effect by which experimentally measured PCS data may have to be corrected to obtain the pure PCS described by eq 1. Like the RACS effect, the RADS effect arises from weak molecular alignment in the magnetic field, which leads to incomplete averaging of anisotropic chemical shifts. Paramagnetic centers with a Curie spin create a dipolar shielding tensor at the site of the nuclear spin, which is generally anisotropic and similar to a CSA tensor. As molecular alignment results in nonuniform averaging of the dipolar shielding, the PCS produced by the effective DSA tensor is no longer fully represented by eq 1. Fortunately, RADS corrections can be calculated and, importantly, are barely measurable in practice.7
1.3.5. Saturation Effect
Finally, saturation of magnetic moments at high magnetic field strength renders the Δχ tensor and, hence PCSs, field dependent. This effect is small even at 18.8 T (800 MHz for 1H NMR) but may become more important at much higher field strengths.78
1.3.6. Paramagnetic Relaxation Enhancement (PRE)
Spin–spin interactions are an important source of nuclear relaxation. Due to the large magnetic moment of unpaired electrons, electron–nuclear spin interactions easily provide a dominant source of nuclear relaxation. The SBM relaxation mechanism59,79,80 applies to all paramagnetic centers. For paramagnetic centers with unpaired electrons that relax rapidly within the rotational correlation time of the molecule, the Curie spin mechanism60,61 provides an additional and often dominant source of PREs. SBM relaxation describes the effect from dipole–dipole coupling between electron and nuclear spins, as it drives nuclear relaxation by the variation in dipolar fields due to molecular reorientation and changes in the electronic spin state due to longitudinal electron relaxation. The paramagnetic enhancements in longitudinal (R1SBM) and transverse (R2) relaxation rates can be described by
| 9 |
| 10 |
| 11 |
where rIS is the distance between nuclear and electron spin and ωI and ωS are the nuclear and electron Larmor frequencies, respectively. Electronic spin flips are accounted for by the effective correlation time τc, which is composed of the rotational correlation time τr and the electronic lifetime τs as shown by eq 11. Terms in eqs 9 and 10 that depend on the electron frequencies can usually be neglected unless the lifetime of the electronic spin states is sufficiently short to approach the inverse of the electron precession frequency. The steep distance dependence of the PRE makes it very sensitive to changes in rIS, which has been exploited to detect little populated conformations and protein–ligand complexes in solution.81−83
Curie spin relaxation likewise results from dipole–dipole coupling between electron and nuclear spins, but it arises from the dipolar field created by the average electronic polarization due to higher populations of lower-energy as opposed to higher-energy electronic spin states.84 Curie spin relaxation applies to systems in which electron relaxation occurs in a time short compared with the rotational correlation time of the molecule. Nuclear relaxation is insensitive to magnetic fields fluctuating at rates that are orders of magnitude faster than the Larmor frequency, such as the very fast electronic spin flips associated with Curie spins. Curie spin relaxation is driven by rotational tumbling, with the rotational correlation time τr characterizing the time-dependent modulation. Because of a quadratic dependence on the nuclear Larmor frequency, Curie spin relaxation can outweigh SBM relaxation for high-molecular weight systems in a strong magnetic field85,86 (note that the calculations of Table 2 in ref (85) were performed for τr = 10 ns instead of τr = 1 ns as stated in the original) while decreasing noticeably with increasing temperature.9 A quantitative description of PREs by Curie spin relaxation is given by eqs 12 and 13 for longitudinal (R1Curie) and transverse (R2) relaxation rates.
| 12 |
| 13 |
Eqs 9–13 describe the situation of isotropic magnetic moments associated with the paramagnetic center. In general, however, magnetic moments often are anisotropic. At the atomic level, such anisotropies usually arise when the magnetic moment of a paramagnetic metal ion depends not only on the electronic spin but also on spin–orbit couplings and therefore on the ligand field. This situation is particularly prominent for paramagnetic lanthanoid ions (except Gd(III)) and therefore their time-averaged magnetic moment is best described by an anisotropic molecular magnetic susceptibility tensor. For complexes with Sm(III) and Eu(III) ions, the situation is further complicated by a significant population of low-lying excited electronic states with different paramagnetic characteristics, which is manifested by a temperature dependence that is less steep than predicted by eqs 12 and 13.74
An extension to the SBM theory has recently been described, which accounts for anisotropic dipolar spectral density in terms of a spectral power density tensor.87 As this tensor usually cannot be derived theoretically, however, this extended theory depends on a greater number of parameters to be fitted to the experimental data. The anisotropic SBM theory is relevant for SBM relaxation generated by lanthanoid ions with anisotropic χ tensors. For these ions, however, Curie spin relaxation often exceeds SBM relaxation, making it difficult to detect the anisotropy effects in the SBM relaxation contribution.
The impact of anisotropic χ tensors on Curie spin relaxation can be calculated.61 Using the matrix formalism of eq 4, the R1Curie and R2 rates can be written
| 14 |
| 15 |
| 16 |
| 17 |
where ω refers to the Larmor frequency of the nuclear spin. The anisotropy effects tend to be small and not usually apparent in PRE measurements. Limited evidence has been found in cross-correlation effects.88
1.3.7. Cross-correlated Relaxation (CCR)
1.3.7.1. DSA/CSA Cross-correlation
For systems with Curie spins, the dipolar field emanating from the paramagnetic center contributes a variable magnetic field at the site of the nuclear spin, which drives nuclear relaxation in a way akin to chemical shift anisotropy. This analogy is highlighted by describing Curie spin relaxation as DSA relaxation. The matrix formalism of eq 4 automatically includes the effect from cross-correlation between DSA and CSA relaxation, as the effective shielding tensor at the site of the nuclear spin, σeff, is the sum of the DSA and CSA tensors. Experimentally, the PRE including cross-correlated relaxation is obtained as usual by subtracting the relaxation rate measured for the diamagnetic reference, R(σCSA), from the relaxation rate in the paramagnetic state, R(σeff), where both terms can be calculated using eqs 14–17.
| 18 |
For nuclei for which CSA relaxation is the main relaxation mechanism in the diamagnetic state, the contribution by the DSA/CSA cross-correlation effect can be dominant. In this case, it is possible that the σeff tensor becomes more isotropic than the CSA tensor because of fortuitous compensation by the DSA tensor, which is manifested by lesser relaxation rates in the paramagnetic than in the diamagnetic state. This was predicted in 200485 and experimentally observed for the first time in 2016.58 The effect was demonstrated with negative PREs of 15N nuclei.
1.3.7.2. DSA/DD Cross-correlation
Just as CSA/DD cross-correlation effects give rise to differential broadening effects of individual multiplet components (which has been exploited, for example, in TROSY spectra), DSA/DD cross-correlation effects also create differential broadening effects of multiplet components.89 Using the matrix representation of eq 4, the cross-correlation effect can be calculated for the example of a 1H spin that is bonded to a 15N nucleus by
| 19 |
| 20 |
| 21 |
| 22 |
where σN denotes the shift tensor at the site of the 1H spin, I, that originates from the dipolar field of the 15N spin and adds either positively or negatively to the full shielding tensor of the 1H spin, depending on the spin state of the 15N nucleus (as described by σ↑ and σ↓). With the complete shift tensors at hand, the differential line broadening observed between the doublet components of the 1H spin, RCurie·DD, is readily calculated by using eqs 4, 14–17, and 19–22.
CSA/DD cross-correlation effects only appear in NMR spectra recorded without broadband decoupling. The value of the associated structural information has been demonstrated in a 3D structure of calculation of cytochrome c′ from Rhodobacter capsulatus using paramagnetic restraints only.90
1.3.7.3. Accuracy of Δχ Tensor Determination
The matrix representation of the Δχ tensor of eq 6 facilitates the fitting of Δχ tensors to the atomic coordinates of the macromolecular structure by a linear least-squares fit, using experimentally observed PCSs. The quality of the fit is commonly described by a quality factor Q, where a low value indicates a good fit:
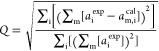 |
23 |
where aexp and acal are the experimental and calculated PCSs values, respectively, the index m indicates ensemble averaging of spins that are common between different models of the molecular structure, and the index i is for summation over all spins of the molecule. Alternative Q factors have been proposed.91,92 The Q factor proposed by Bashir et al.91 uses sums of experimental and calculated values in the denominator of eq 23 and, therefore, tends to be 2 times smaller. This definition has the advantage that it does not bias for calculated values that are smaller over those that are larger than the experimental values, which matters in the case of poor fits with large differences between observed and calculated data, as can be the case with PREs. For example, for (aexp, acal) of (10, 20) Q = 1, but for (20, 10), Q = 0.5. The adjusted Q factor yields 0.33 in both cases. Importantly, Q factors are meaningful only, if the number of fitted data greatly exceeds the number of variables, as very small values in the denominator can render its calculation unstable. Therefore, fitting algorithms usually attempt to minimize the root-mean-square deviation (RMSD) between calculated and experimental values rather than Q factor.
In the case of paramagnetic metal probes attached to the target molecule via a long and flexible linker, the range of positions assumed by the metal ion relative to the target calls for an equal range of Δχ tensors to be fitted. Fitting multiple tensors, however, usually is not possible as each Δχ tensor determination requires at least eight PCSs to fit the tensor parameters and accurate fits require significantly more PCSs. The attempt to fit more than a single Δχ tensor to the limited data available would easily lead to problems with overfitting. The convention to fit single effective Δχ tensors has two consequences. First, the location of the paramagnetic center obtained by the fit does not correspond to its real position. In fact, it is quite possible that, due to temporary close proximity of the paramagnetic metal ion, the PCSs observed for a range of nuclear spins near the attachment site of the paramagnetic tag are larger than expected for a single immobile metal position. In this case, fitting of the data by a single effective Δχ tensor tends to increase the Δχ tensor and indicate a metal position that is further away from the surface of the target molecule than expected. This does not invalidate the use of effective Δχ tensors to back-calculate PCSs, but the predictive value of PCSs decreases with increasing distance from the nuclear spins, whose PCS data were used to fit the Δχ tensor.93 Notably, exceptionally good correlations between experimental and back-calculated PCSs can be obtained even with tags attached via flexible linkers.94
The difficulty to determine accurate Δχ tensors for flexible probes makes it difficult to assign the magnitudes of reported Δχ tensors to intrinsic probe characteristics. When a large number of PCSs has been used for the Δχ tensor fit, fits performed for rigid probes tend to produce low Q factors. An alternative way of assessing the flexibility of a paramagnetic probe is to compare the alignment tensor obtained by fitting RDCs with the Δχ tensor obtained by fitting PCSs, as both should be proportional to each other (eq 7). In practice, however, the alignment tensor is almost always smaller than that derived from PCSs because (i) RDCs are very sensitive to the accuracy of the molecular coordinates and (ii) molecules are not rigid and the orientations of the internuclear vectors determining the RDCs are averaged due to molecular dynamics as reflected by order parameters below one.
1.4. Paramagnetic NMR of Biomolecules in the Solid State
Solid-state NMR spectroscopy of biological macromolecules such as microcrystalline, fibrillar and membrane proteins is a growing area of interest largely driven by recent advancements in magic angle spinning (MAS) technology.95,96 Rotation speeds beyond 100 kHz have been shown to allow the acquisition of 1H-detected multidimensional NMR spectra with good resolution and sensitivity without the need of protein perdeuteration.97,98 The first paramagnetic macromolecules to be investigated by solid-state NMR were metalloproteins, such as Co(II) substituted matrix metalloproteinase-12, for which PCSs were measured to characterize protein structure.99−101 Co(II)-substituted superoxide dismutase was also investigated by PCSs and PREs with 1H detected ultrafast MAS to determine molecular structure.102,103 Paramagnetic tags attached to biomolecules offer similarly useful long-range structural information in the solid state as in solution, but there are differences in the paramagnetic effects.53,52
1.4.1. Paramagnetic NMR Effects in Static and Rotating Solids
An unordered paramagnetic compound in the solid state features molecular orientations distributed uniformly. As the dipolar shielding tensor arising from the paramagnetic center (see eq 4) renders the chemical shift for the nuclear spin dependent on the orientation of the molecule, the resulting powder pattern in the NMR spectrum reflects the principal axes of the σ tensor.
For the case of rotating solids, MAS of the paramagnetic species achieves coherent averaging of the σ tensor. At low spinning rates, this tends to split the powder pattern into spinning sidebands.104 When the spinning rate is greater than the dipolar shielding term, however, only the isotropic component of the σ tensor, which includes the PCSs, remains observable in the NMR spectrum and can be described by eq 1.105
While incoherent averaging of the σ tensor in solution NMR underpins the Curie spin relaxation mechanism as described in eqs 12–13, the coherent averaging by MAS effectively removes this pathway as a source of PREs in solid-state NMR.106 Therefore, PREs of nuclear spins are unaffected by the Curie mechanism (except for nuclei in close proximity to the paramagnetic center, where the σ tensor term becomes larger than the MAS rate) and SBM relaxation (eqs 9–10) presents the dominant source of PREs in solids. SBM relaxation is governed by the incoherent electronic correlation time T1e and independent of the MAS rate.59
2. Sources of Paramagnetism
This section presents a brief overview of the most frequently used paramagnetic metal ions and nitroxide radicals.
2.1. 3d Block Transition Metal Ions
Transition metals in the 3d block of the periodic table can be in various oxidation states with different numbers of unpaired electrons occupying the d-orbitals.107 Therefore, these cations are great candidates for generating various paramagnetic effects. They are frequently found in proteins. It is estimated that more than 25% of the known proteins contain one or more transition metal ions.108 Paramagnetic NMR has long been applied to investigate the metal binding sites of metalloproteins.109−111 The first 3D structure determination of a metalloprotein was performed on a heme protein containing a low-spin Fe(III) ion.112 Proteins containing many other paramagnetic transition metal ions have been studied, with iron, cobalt, manganese, copper, and nickel ions figuring most frequently. These ions are discussed hereafter.
Transition metal ion complexes of 3d block elements are stable with 4–6 electron donor sites. Depending on the number of coordination sites, transition metal ions feature mainly three types of coordination geometries, which are tetrahedral, square pyramidal, and octahedral (Figure 2). The magnetic properties of 3d block complexes can be strongly affected by the ligands, the environments, and the coordination geometries. Some general properties are shown in Table 1. The unpaired electrons usually are partially delocalized to the ligand orbitals, which affects the magnetic properties. Iron is a good example. Fe(III) and Fe(II) are the most stable and commonly found oxidation states of iron. Both of them can be in paramagnetic high-spin states and Fe(III) is also paramagnetic in the low-spin state. With weak axial ligands, Fe(III) is in a high-spin state, whereas Fe(III) assumes a low-spin state with strong ligands. High-spin Fe(III) contains five unpaired electrons, and its electronic relaxation time can vary from 10–9 to 10–11 s.113,114 Therefore, it can generate sizable PCSs and PREs, depending on the ligands. The magnetic properties of low-spin state Fe(III) are quite different due to its short electronic relaxation time, which is below 10–11 s.113,114 As a result, the paramagnetism of low-spin Fe(III) is most prominently manifested in PCSs. Fe(II) has six electrons occupying d-orbitals, so when these electrons are paired, Fe(II) is diamagnetic. Its high-spin state has up to four unpaired electrons with a short electronic relaxation time (10–12 s), which causes PCSs.107 Proteins containing iron ions have been well studied by paramagnetic NMR, for example cytochrome c, rubredoxin, and hemoglobin.62
Figure 2.
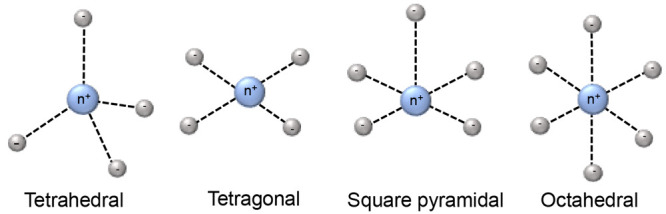
Schematic diagrams of 3d block ions coordination geometries.
Table 1. Magnetic Properties of 3d Block and 4f Block Ions.
| ion | conf.a | Jb | τsc | ranged | PCSse | PREse | RDCse |
|---|---|---|---|---|---|---|---|
| Fe(III) (HS) | [Ar]3d5 | 5/2 | 10–9–10–13 | 20 | + | ++ | |
| Fe(III) (LS) | [Ar]3d5 | 1/2 | 10–11–10–13 | 15 | ++ | + | |
| Mn(II) | [Ar]3d5 | 5/2 | 10–8 | 25 | ++ | ||
| Fe(II) (HS) | [Ar]3d6 | 2 | 10–12 | 15 | + | + | |
| Co(II) (HS) | [Ar]3d7 | 3/2 | 10–12 | 25 | ++ | + | |
| Ni(II) | [Ar]3d8 | 1 | 10–10–10–12 | 15 | + | + | |
| Cu(II) | [Ar]3d9 | 1/2 | 10–8–10–9 | 20 | + | ++ | |
| Ce(III) | [Xe]4f1 | 5/2 | 10–13 | 10 | + | + | |
| Pr(III) | [Xe]4f2 | 4 | 10–13 | 20 | + | + | |
| Nd(III) | [Xe]4f3 | 9/2 | 10–13 | 10 | + | + | |
| Sm(III) | [Xe]4f5 | 5/2 | 10–13 | 7 | + | + | |
| Eu(III) | [Xe]4f6 | 0 | 10–13 | 15 | + | + | |
| Gd(III) | [Xe]4f7 | 7/2 | 10–8 | 25 | +++ | ||
| Tb(III) | [Xe]4f8 | 6 | 10–13 | 45 | ++++ | + | +++ |
| Dy(III) | [Xe]4f9 | 15/2 | 10–13 | 45 | ++++ | + | +++ |
| Ho(III) | [Xe]4f10 | 8 | 10–13 | 35 | +++ | + | ++ |
| Er(III) | [Xe]4f11 | 15/2 | 10–13 | 30 | ++ | + | + |
| Tm(III) | [Xe]4f12 | 6 | 10–12–10–13 | 50 | +++ | ++ | ++ |
| Yb(III) | [Xe]4f13 | 7/2 | 10–13 | 25 | ++ | + | + |
| Nitr.f | 1/2 | 10–7 | 15 | ++ |
The electronic configuration of Co(II) is 3d7, which can be in a high-spin state containing three unpaired electrons or in a low-spin state containing a single unpaired electron. The electronic relaxation times of low-spin Co(II) are longer (10–9–10–10 s) than for the high-spin state (∼10–12 s), thus low-spin Co(II) causes strong PREs and small PCSs.107 In contrast, high-spin Co(II) can generate the largest PCSs among 3d block ions combined with weak PREs. Mn(II) harbors one of its five unpaired electrons in each of the d-orbitals and has a long electronic relaxation time, causing the strongest PREs of all 3d block ions. Cu(II) is the most stable ionic state of copper. Three types of Cu(II) ions are found in proteins, which are distinguished by the Cu(II) ion coordination.15,62 In type-I copper(II) proteins, the ion is coordinated with trigonal (or distorted tetrahedral) geometry, involving at least one sulfur atom of cysteine and two nitrogens of two histidine residues.115 In type-II copper(II) proteins, the copper ion is coordinated by nitrogen and oxygen atoms with tetrahedral geometry. In the third type, two Cu(II) ions are antiferromagnetically coupled, decreasing the net magnetic susceptibility and electron relaxation time.116 Consequently, various paramagnetic effects were observed for Cu(II). Cu(II) contains one unpaired electron and generates small PCSs, but the PREs are strong, because of its relatively long electronic relaxation time (10–8–10–9 s). Therefore, Cu(II) is outstanding for PREs and it is widely used in EPR spectroscopy.107,117,118 The divalent ion of nickel has two unpaired electrons, generating small PCSs with sizable PREs.107
2.2. Lanthanoid Ions
Lanthanoid ions were introduced into NMR studies of proteins decades ago, as agents for chemical shift changes and line broadening.119,120 The chemical properties of all Ln(III) ions are similar because the 4f-orbitals are shielded by 5s and 5p subshells, and their unpaired electrons do not participate substantially in different coordinating interactions with ligands. Ligands that bind Ln(III) ions thus have similar affinities for all lanthanoid ions. An overview of the paramagnetic properties of paramagnetic lanthanoid ions (excluding promethium, which is an unstable radioactive element) is presented in Table 2.
Table 2. Paramagnetic Properties of Paramagnetic and Nonradioactive Lanthanoid Ionsa.

The radii of the yellow spheres correspond to the distance, where paramagnetic relaxation enhancement is predicted to broaden a 1H NMR signal by 80 Hz on a 800 MHz NMR spectrometer, assuming a protein with a rotational correlation time of 15 ns and a temperature of 25 °C, calculated using eqs 10 and 13. The isotropic component of the χ tensors (χiso, in 10–32 m3) were calculated using eq 3 with values for S and ge taken from ref (74). The Δχ tensors reported for calbindin D9k125 are represented by isosurfaces of the PCSs drawn at ±5 ppm. The values of the Δχ tensors are given in the unique tensor representation,69 with their relative orientations corresponding to those reported for calbindin D9k. The electronic longitudinal relaxation times were calculated for a magnetic field strength of 0.5 T following ref (737). They are not strongly field dependent.738 The T1e time of gadolinium increases with the rotational correlation time and the square of the magnetic field strength.739 The scale bar at the left indicates a distance of 20 Å.
Gd(III) is unique because its seven unpaired electrons are distributed equally among the f-orbitals, producing a magnetic dipole moment that is independent of molecular orientation in an external magnetic field. As its electronic relaxation time is long (>10–8 s) at field strengths >3 T,29,86 Gd(III) generates large PREs and enjoys increasing popularity in EPR as spin label.121−123 Although the remaining paramagnetic lanthanoid ions generate anisotropic χ tensors and produce all the paramagnetic effects discussed above, it has recently been shown that PRE measurements with Er(III), which generates large PREs associated with relatively small PCSs, can yield more accurate distance measurements than commonly used paramagnetic agents with long electronic relaxation times.124 In general, Tb(III) and Dy(III) generate the largest Δχ tensors and, hence, cause the largest PCSs, while Tm(III) and Ho(III) produce medium-sized PCSs. Sizeable PCSs can also be observed with Yb(III) and Er(III). This general ordering, which follows theoretical expectations based on gJ factor and J quantum number74 and has been experimentally verified for calbindin D9k with one of the native calcium ions replaced by a Ln(III) ion,125 is not always maintained as, for reasons not understood at present, some complexes of Tm(III) have been found to produce larger Δχ tensors than the same complexes with Dy(III) or Tb(III).126−129 Other lanthanoid ions are much less frequently used for paramagnetic NMR of proteins because their Δχ tensors are smaller.86
2.3. Nitroxide Radicals
Nitroxide probes are organic molecules with five- or six-membered heterocyclic rings and a radical that is protected against homodimerization by bulky chemical groups (Figure 3). Because of their relatively small size combined with a long electronic relaxation time of a single unpaired electron and its reasonably well-defined localization, nitroxides are the most frequently used paramagnetic compounds in NMR to generate PRE distance restraints up to 20–25 Å.130 The stability of the radical depends on the size of the ring and the substituents in the α position. In general, nitroxides in a five-membered ring are chemically more stable than in six-membered rings and bulkier groups in the α position promote stability by improved steric shielding. MTSL ((1-oxyl-2,2,5,5-tetramethyl-d-pyrroline-3-methyl)-methanethiosulfonate) is commercially available and the most popular nitroxide for protein labeling (Figure 3).
Figure 3.

General structure of nitroxides. R represents an alkyl group.
3. General Overview of Natural and Chemically Generated Paramagnetic Centers
Paramagnetism of biomolecules can exist naturally or be introduced artificially by different strategies. Metalloproteins containing paramagnetic ions can readily be studied by NMR techniques tailored to paramagnetic samples. In some instances of diamagnetic metalloproteins, a diamagnetic metal ion can be replaced by a paramagnetic ion. Not all of the diamagnetic metalloproteins are suitable for metal ion substitution, however, and this approach does not work for nonmetalloproteins. Consequently, to gain access to the long-range structural restraints associated with paramagnetism, various methods have been devised to introduce paramagnetic centers. Two types of approaches can be distinguished. Paramagnetic centers can be added to the solution, resulting in solvent PREs. In this case, the PREs of protein nuclei are caused by nonspecific interactions with the paramagnetic relaxation agent in the solution. Such agents are designed to tumble freely and independently of the protein molecules, so that any anisotropy of the paramagnetic center will average to zero, thus yielding only PREs and suppressing PCSs or RDCs. The probes for generating solvent-PREs are usually chemically synthesized.35,36
Alternatively, a paramagnetic center can be attached covalently at a specific site on the protein. The two main methods of the covalent approach are the introduction of a genetically encoded metal binding site in the target protein and the chemical attachment of a paramagnetic center to the protein. Several principles need to be followed in the design of a suitable protein paramagnetic center. (i) The structure and properties of the target protein should be maintained. A highly charged or hydrophobic paramagnetic center as well as improper attachment sites can cause unfolding of the target protein and result in precipitation. Thus, small probes with low charge and high water solubility are preferred. (ii) To exploit the effects associated with paramagnetic metal ions featuring anisotropic χ tensors, the metal ion needs to be attached rigidly to the target protein, as the anisotropic effects decrease dramatically with the mobility of a paramagnetic center relative to the protein. Furthermore, movements of the paramagnetic center lead to averaging of the paramagnetic effects, which hampers the translation into structural information. (iii) The paramagnetic center should assume a single conformation. Many metal complexes show flexibility of parts of the coordinating cage, including exchange of coordinating groups. Different coordination states usually result in different orientations of the Δχ tensor and are thus prone to producing more than a single set of PCSs or RDCs (in the slow exchange limit) or line broadening (in the intermediate exchange regime). (iv) For probes that are linked via two identical tethers (e.g., to a pair of cysteine residues in the target protein), C2 symmetry is important to avoid that attachment results in different isomers, each with its own Δχ tensor. (v) The metal affinity needs to be high and the probe needs to be stable and easily available, either commercially or by straightforward chemical synthesis.
3.1. Metalloproteins
3.1.1. Paramagnetic Metalloproteins
Among the metalloproteins with paramagnetic properties, iron and copper proteins are the best studied by paramagnetic NMR. Iron often occurs in either iron–sulfur clusters, such as in ferredoxin,113,131 or coordinated to heme rings, like in cytochromes.132 In FeS clusters, iron is bound to sulfurs from cysteine and inorganic sulfur.62 In heme, the macrocycle acts as a tetradentate ligand for the iron and provides space for additional axial ligands. Bertini and co-workers extensively studied iron proteins by paramagnetic NMR.112,133−139 The structure of oxidized Saccharomyces cerevisiae iso-1-cytochrome c, containing a low-spin Fe(III) ion, was the first paramagnetic metalloprotein for which the structure was refined with paramagnetic restraints.139 The strategy used started with a known structure based on other restraints, such as NOEs, to fit the Δχ tensor of the paramagnetic center. This allowed using the PCSs as additional restraints in new rounds of structure calculations and refinement of the Δχ tensor based on the new structure. This iterative method has proven successful in yielding a more accurate 3D structure of the target protein.112,139
3.1.2. Metalloproteins with Paramagnetic Substitution
In many cases, metals are either not paramagnetic or have inconvenient paramagnetic properties. In this situation, it may be possible to substitute the natural metal ion with another one characterized by different paramagnetic properties. For example, Cu(II) is usually ligated by histidine, cysteine, aspartic acid, or tyrosine residues, or a sulfide.115,140 As Cu(II) mainly generates PREs, the NMR signals of the coordinating residues are very broad. By substitution of Cu(II) with Co(II), Donaire et al. studied coordination in the blue copper protein azurin141 and Bertini et al. investigated the metal binding site of stellacyanin.142 Also Ln(III) ions have been used for substitution of metal ions of similar ionic radius and coordination chemistry.143 Calcium binding sites have been particularly successful in this respect and Ln(III) ions have frequently been successfully substituted for Ca(II) in the EF-hand motif.119,144 Most calcium proteins feature a pair of EF-hand motives, but simultaneous substitution of both Ca(II) ions with two Ln(III) ions is unfavorable due to electrostatic repulsion. Therefore, calbindin D9k, which possesses two Ca(II) binding sites in EF-hand motives, is suitable for selective Ln(III) substitution into a single one of the Ca(II) binding sites.145 Using calbindin D9k as a model protein, the whole lanthanoid group was incorporated in this way, yielding a useful comparative study of their paramagnetic effects (Table 2).125 Similarly, calmodulin, which harbors four calcium binding sites in four EF-hand motives, has successfully been studied by paramagnetic NMR146 and the Asn60Asp mutant was shown to promote the Ln(III) affinity and specificity of a specific Ca(II) binding site.145,147,148
3.2. Genetically Encoded Metal Binding Sites
3.2.1. Natural Amino Acids or Peptides
Metal-generated paramagnetic structure restraints were initially explored in detail with paramagnetic metalloproteins and, having realized their exceptional value for 3D structure determinations of proteins, subsequently extended to diamagnetic metalloproteins by substitution with paramagnetic metal ions. In nonmetalloproteins, paramagnetic metal binding sites can be created by chemical modification of the target protein. Inspired by the strategy of substituting Ln(III) ions into EF-hands, lanthanoid-binding peptides (LBPs) have been proposed for adding a paramagnetic center to a protein (Table 3). Initially, a LBP with an EF-hand like motif was fused to the target protein at its N-terminus.149 Subsequently, LBPs with improved lanthanoid ion binding affinity were identified by the Imperiali group and the number of residues reduced to 17.150−152 Double-lanthanoid-binding tags were shown to enable the binding of two Ln(III) ions simultaneously in a peptide with less than 40 residues.153,154 Because of the high mobility of terminal fusion tags, however, the paramagnetic effects in the target protein were greatly reduced by averaging. This situation was improved substantially by inserting an LBP into protein loops, as demonstrated for three different loops of interleukin-1β (IL1β) and confirmation of the tag structure by X-ray crystallography.155 Coordinated with Gd(III) ions, these constructs can also be used for Gd–Gd distance measurement by EPR spectroscopy.154 Disadvantages of this approach are that it limits paramagnetic centers to termini or loop regions of the protein and any isotope labeling of the protein also labels the LBPs, which complicates the NMR spectra especially for the diamagnetic reference. In addition, this approach requires that the structure of the target protein is known, while the exact location of the lanthanoid ion is still difficult to predict in advance.
Table 3. Frequently Used Lanthanoid Binding Peptides.
To overcome these drawbacks, LBPs were developed that can be attached by linking them to the protein chemically. Generally, these tags contain free thiol groups for attachment to cysteine residues.156 They can be introduced anywhere by introducing a cysteine residue on the protein surface. Su et al. designed a series of LBPs with a cysteine residue for attachment to cysteine in the protein via a disulfide bond.157 The Δχ tensor could be varied by including either a d- or l-cysteine in the LBP or varying the position of the cysteine residue in the LBP. Using the N-terminal domain of the Escherichia coli arginine repressor (ArgN) as model protein, large paramagnetic effects were obtained for all Ln(III) loaded LBPs with different tensor orientations for each of the tags,157 but the mobility of the LBPs relative to the protein decreased the anisotropic paramagnetic effects similar to the LBP fusion method.154 To reduce the mobility of the LBP tag, it has been proposed to anchor the tag at two sites.158,159 The first double-anchored LBP was designed by Saio et al. and combined a N-terminal fusion with a chemical linkage to the target protein via a disulfide bond.158 Following fusion of the LBP to the N-terminus of the B1 immunoglobulin binding domain of protein G (GB1), a cysteine residue in the LBP was linked to a cysteine residue in GB1 by thiol activation using 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) to form the second linkage. The double linkage was referred to as L2GB. As expected for a more rigid tag attachment, larger Δχ tensors were observed compared with the same LBP attached by fusion only (referred to as L1GB).158
An alternative straightforward approach is to use the metal binding propensity of canonical amino acids like histidine, aspartic acid, and tyrosine to bind metal ions. For example, His6 tags are routinely installed for protein purification and have been explored also for paramagnetic NMR. Unfortunately, like N-terminal LBP fusions, this tag proved too mobile to yield good properties for paramagnetic NMR beyond generating PREs.160 In contrast, dihistidine motives can generate better defined metal ion binding sites and have been used for EPR distance measurements between two Cu(II) ions.161−163 This approach was recently extended successfully to paramagnetic NMR studies, where a Co(II) ion bound to a dihistidine motif generated sizable PCSs in various proteins.164 It was shown that dihistidine motives with good metal binding affinity can be installed either in α-helices or β-sheets.164
3.2.2. Noncanonical Amino Acid and Their General Synthetic Approaches
Well over 100 noncanonical amino acids (ncAAs) with a wide variety of side chains have been incorporated into proteins by genetic encoding.165−167 Among these, some are capable of binding metal ions. For example, bipyridylalanine (BpyAla, Figure 4)168 has successfully been used to endow proteins with new hydrolytic function or greater stability enabled by its capacity to bind divalent metal ions.169,170 Paramagnetic transition metal ions, such as Co(II), which prefer nitrogen ligands and require fewer coordination sites than Ln(III) ions, can be coordinated by BpyAla. A West Nile virus NS2B-NS3 protease (WNVpro) mutant containing a BpyAla residue in a loop was shown to generate PCSs following coordination with a Co(II) ion, but additional coordination was required from proximal side chains, as was demonstrated by mutation of nearby residues.171 2-Amino-3-(8-hydroxyquinolin-3-yl)propanoic acid (HQA, Figure 4)172 has equally been explored as the basis for metal ion binding motives.173−178 Unfortunately, Ln(III) ions coordinated by HQA generally lead to quantitative protein precipitation,178 but Mn(II) captured by HQA has successfully been used to generate PREs in membrane proteins.173
Figure 4.
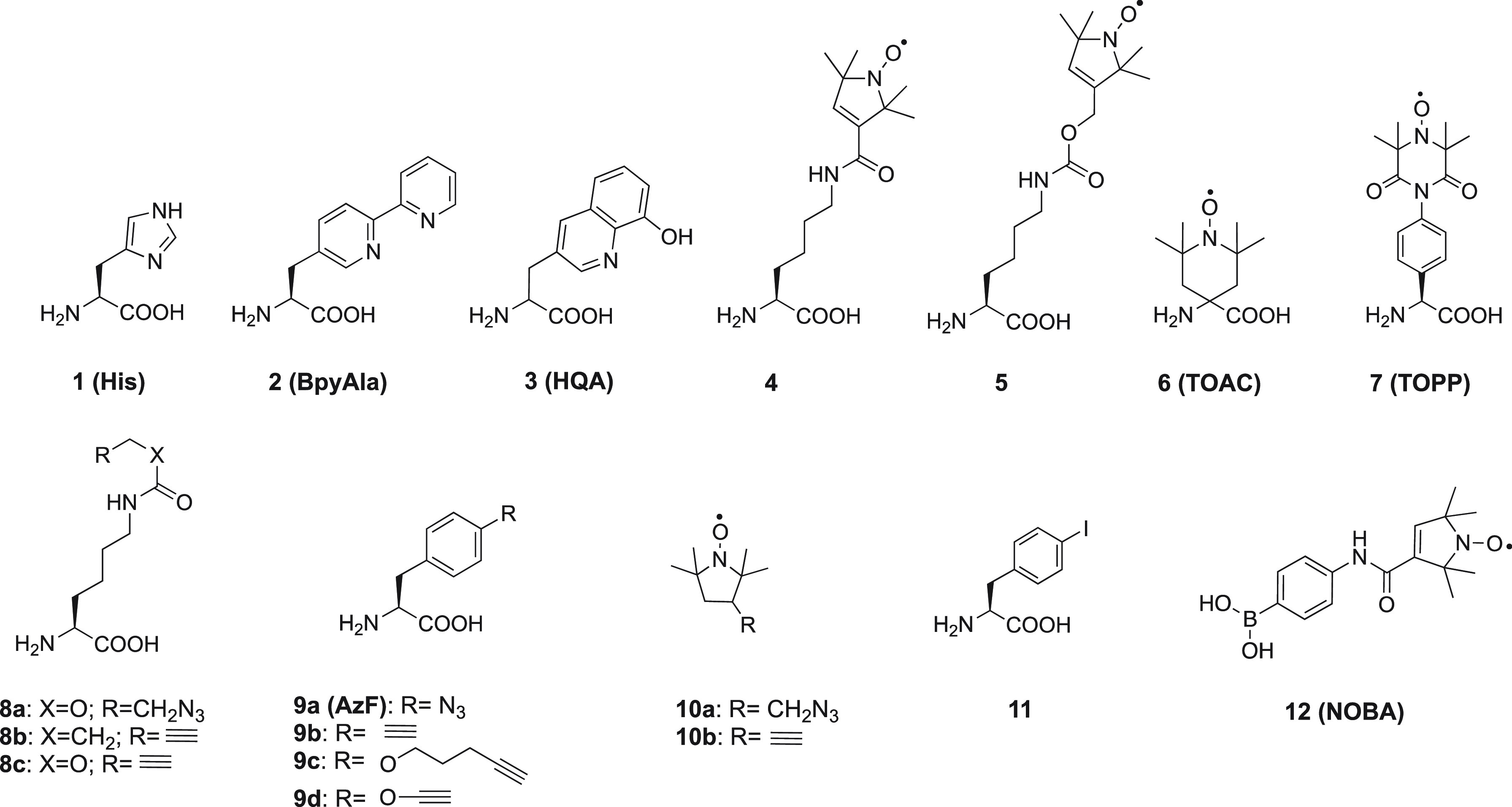
Chemical structures of some natural (1), noncanonical (2–9, 11) amino acids, and nitroxide radicals (10, 12).
A related approach uses site-specific incorporation of one or two phosphoserine residues, which is a naturally occurring amino acid produced by post-translational modification, but which can also be incorporated as a ncAA in response to an amber stop codon, owing to a recently developed genetic encoding system.179−181 A phosphoserine residue in conjunction with an aspartate or glutamate residue, or two phosphoserine residues together with a glutamate residue can generate Ln(III) binding sites that position the metal ion very precisely on the target protein, generating substantial Δχ tensors.182 Unfortunately, the close proximity of charged amino acid side chains produces significant electrostatic repulsion, which can lead to unfolding of the protein and poor protein yields.
As discussed in section 2, not only paramagnetic metal ions but also nitroxide radicals constitute useful paramagnetic centers for generating PREs. Noncanonical amino acids containing a nitroxide radical have been synthesized (Figure 4, compounds 4–7)183−185 and some of them have been incorporated into proteins by genetic encoding.51,183 Alternatively, a nitroxide radical can be attached to a ncAA after incorporation in the target protein. In one method, an azido or alkynyl-bearing ncAA (Figure 4, 8a/9a or 8b/c/9b)186−188 is installed in the target protein and reacted with an alkynyl or azido-functionalized nitroxide radical (Figure 4, 10a or 10b),188 using the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction189 to form a triazole linker. A similar strategy employs the Suzuki–Miyaura coupling reaction, where an iodide ncAA (Figure 4, 11) is reacted with a boronic acid spin label (NOBA, Figure 4, 12).190 For ncAAs containing a nitroxide group from the beginning, reduction of the radical by the reducing conditions inside E. coli cells limits the yield of successful incorporation. Among the nitroxide radical ncAAs mentioned above, only TOAC was used as a paramagnetic center for NMR studies, to generate PREs in the complex between a peptide derived from focal adhesion kinase (FAK) and Src homology 3 (SH3) domain of Src kinase.191 Others were only used for EPR studies.51,183,188,192−195
Two main strategies are followed in the synthesis of ncAA, which involve either the side chain modification of a natural AA or alkylation of a glycine equivalent (Scheme 1).196−198 Recently, additional strategies for ncAA synthesis have been published.199−201 Method A of Scheme 1 is most frequently applied to synthesize ncAA probes. p-Azido-l-phenylalanine, which is one of the most popular ncAAs, is synthesized by the first strategy with l-phenylalanine as the starting compound. Two approaches give the product in two steps (Scheme 2A).202−204 Similarly, compound 4 is synthesized by coupling reaction between l-asparagine and a nitroxide radical derivative (Scheme 2B).183BpyAla and HQA are ncAAs that can directly coordinate metal ions. Their synthetic routes differ from those mentioned above. Instead of starting from a natural AA, their synthesis starts from the compounds possessing the metal binding ability, which are modified with amido and carboxyl groups to form the final ncAA, as shown in Scheme 2C and D.168,172,205
Scheme 1. Synthetic Strategies for Modification of Amino Acids.

Scheme 2. Synthetic Routes of p-Azido-l-phenylalanine (A),202−204 Compound 4 (B),183BpyAla (C),205 and HQA (D)174,
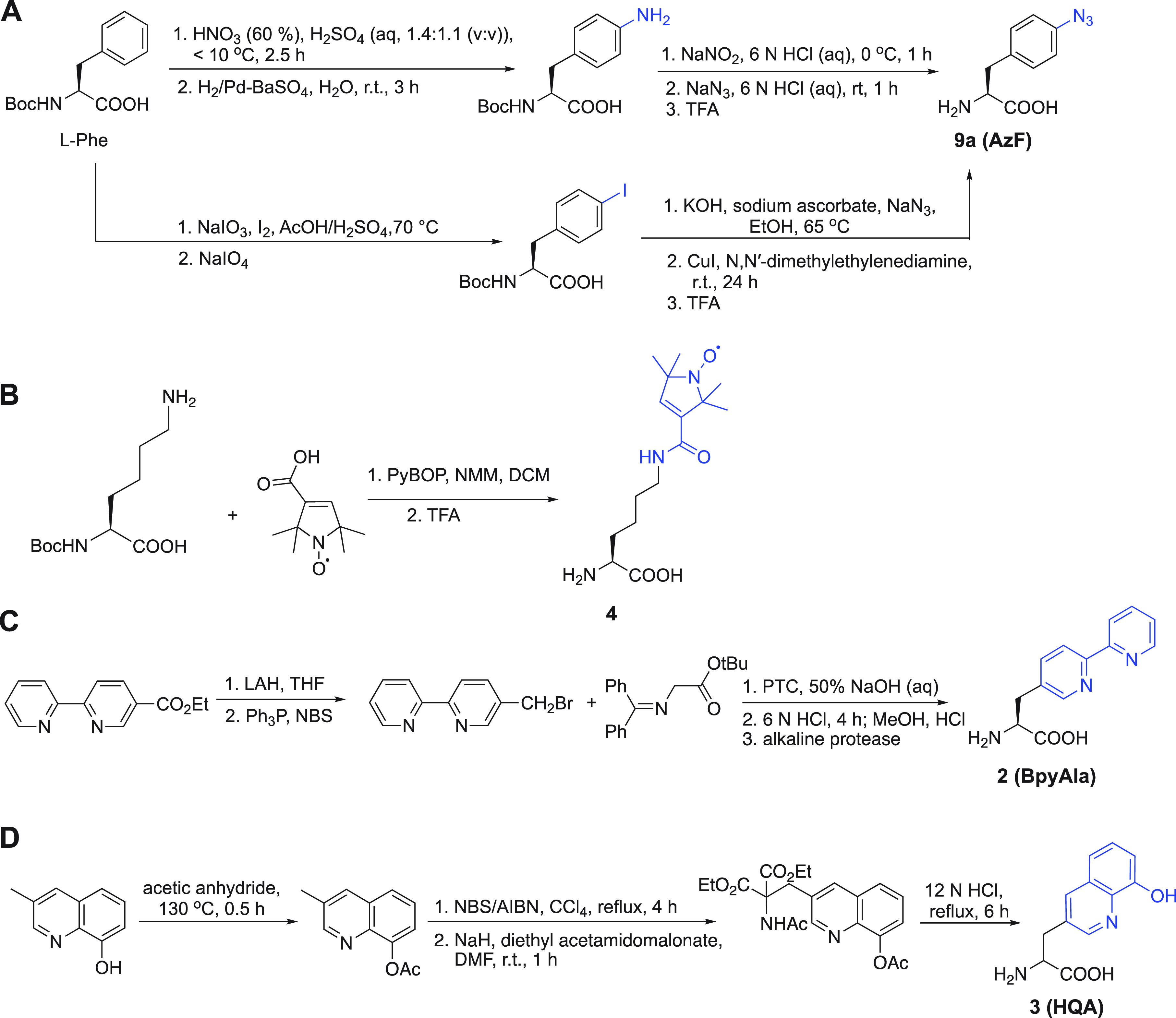
The functional groups of the ncAAs are colored in blue.
3.3. Synthetic Tags for Proteins
To obtain useful paramagnetic results, other than solvent PREs or RDCs, it is critical that the paramagnetic center is at a fixed distance and orientation relative to the nuclear spin and its molecular frame. Rapid distance and orientation variation lead to nonlinear averaging of the paramagnetic effects, making interpretation more complicated or impossible. Thus, the linkage between the paramagnetic center and the molecule needs to be as short and rigid as possible, contrary to, for example, the longer linkers that are often used in fluorescence spectroscopy. Second, it is critical that a paramagnetic center is attached to only a single, well-defined site on the molecule. The presence of more than one center greatly complicates the interpretation of PCS and PRE data. Similarly, if the tagging site is undetermined, the information content of PCS and PRE data becomes uncertain. As a consequence, tagging of proteins is best achieved on a residue with unique chemical reactivity on the accessible surface. For this reason, cysteine is by far the most popular residue type targeted for tagging, as many proteins lack surface exposed cysteine residues, so that unique sites can be engineered by site-directed mutagenesis. If exposed cysteines are present in the native protein, they can be replaced first by alanine or serine, usually without consequence for the structure or function of the protein. The general procedure requires reduction of any disulfide bridges using a reducing agent such as dithiothreitol, and removal of the reductant, before the tagging reaction can take place.
3.3.1. Nitroxide Probes
3.3.1.1. Nitroxide Probes for Studies of Biomolecular Structure
Nitroxides are extensively used as spin-labels for structural studies of proteins, protein–DNA complexes, and protein–RNA complexes by EPR and NMR spectroscopy. Two main factors need to be considered for the application of nitroxides in biological systems. The radical character must be stable with respect to the chemistry used to attach the tag to the target molecule. Nitroxides are stable under most nonreducing conditions but can nonetheless be prone to oxidation to an oxammonium cation or reduction to a hydroxylamine (see Scheme 3). Several ways have been reported to stabilize nitroxides.206 Besides increased chemical stability associated with five- versus six-membered heterocycles and bulky substituents adjacent to the nitroxide group, a nitroxide equipped with cyloalkanyl groups (Figure 5, 13) also tends to possess a longer electronic relaxation time than MTSL (Figure 3).207
Scheme 3. Two Synthetic Approaches toward Nitroxide Radicals.

(a) Oxidation with m-CPBA or H2O2/cat. Na2WO2. (b) Oxidation with MnO2.
Figure 5.
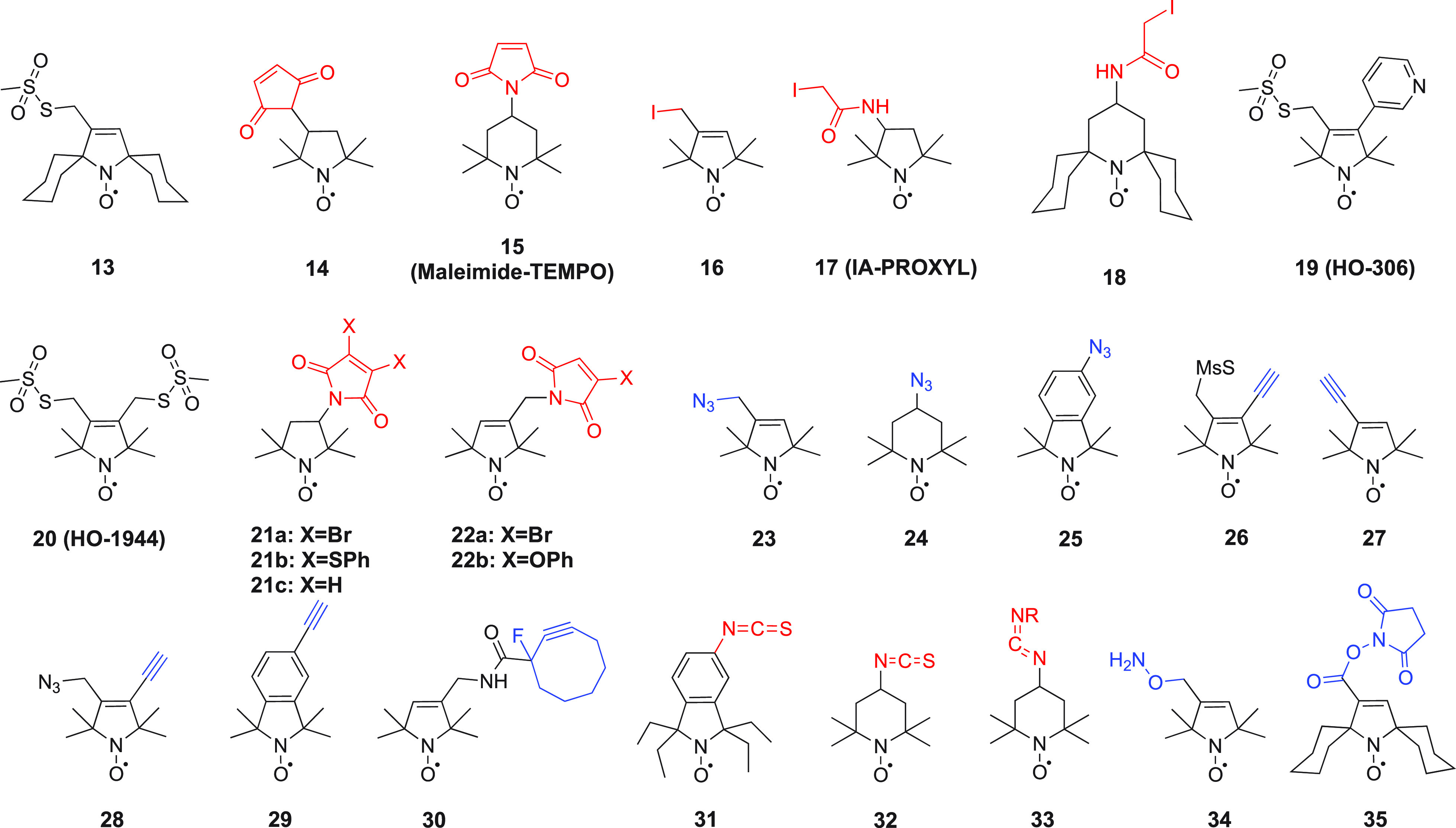
Structures of nitroxide probes. The functional groups for covalent attachment to cysteine via a thioether are highlighted in red and the functional groups for covalent attachment to a ncAA are highlighted in blue. Probe 13(207) is derived from MTSL; 14(211) and 15(212) are maleimide functionalized nitroxides; probes 16,21317,213 and 18(391) are iodide functionalized nitroxides; probes 20,22121,221,223 and 22(224) are double-anchored nitroxides; probes 23,22524,225 and 25(225) are azide functionalized nitroxides; probes 26,22727,22728,22729,228 and 30(229) are alkyne functionalized nitroxides; probes 31(230) and 32(231) are iso(thio)cyanide functionalized nitroxides; probe 33(231) is a carbodiimide functionalized nitroxide; probe 34(232) is a hydroxylamine ether functionalized nitroxide; probe 35(207) is a nitroxide functionalized with a hydroxysuccinimide ester.
The most efficient way of connecting a nitroxide label to a biomolecule is to use an activated thiol, such as methanethiosulfonate (MST)208,209 or a pyridylthio group,210 which form disulfide bonds with cysteine residues. Disulfide bonds, however, are easily cleaved under reducing conditions. Therefore, maleimide211,212 and iodide212−214 nitroxide probes have been developed (Figure 5, 14–18) to form thioether bonds with cysteine residues, which are more stable under reducing conditions than disulfide bonds.
An important consideration is that any flexibility of the linker (Figure 6A) leads to averaging of the spin–spin interactions over a range of distances, reducing the accuracy of any distance measurements.215−218 Probe mobility can be restricted by adding a bulky chemical group on the heterocycle (Figure 5, 19),219 introducing a second attachment group (Figure 5, 20–22),220−224 or incorporating a free radical containing unnatural amino acid into a sterically crowded site of the target protein (Figure 4, 4–7).183,195 In addition, nitroxides with alternative reactive functionalities have been reported, such as the azide (Figure 5, 23–25)225,226 and alkyne (Figure 5, 26–30)227−229 containing probes amenable to “click” chemistry for attachment to, for example, p-azido-l-phenylalanine (AzF, Figure 4, 9a). Others contain an isothiocyanate (Figure 5, 31–32)230,231 or carbodiimide (Figure 5, 33)231 group for reaction with a glutamate side chain, the hydroxylamine ether probe (Figure 5, 34)232 to generate an oxime ether upon reaction with proteins containing a p-acetylphenylalanine residue, or a hydroxysuccinimide ester (Figure 5, 35)207 for reaction with a lysine side chain. These probes contain bulky groups in the vicinity of the nitroxide group and are well suited to minimize the movement of the radical relative to the protein frame.
Figure 6.

(A) Reaction of MTSL with a cysteine residue of a protein generates a flexible linkage with five rotatable bonds (red arrows). (B) Schematic representation of two different enantiomeric EDTA complexes produced by different coordination of the metal ion. M denotes the metal ion. O denotes the carboxyl groups. Curved lines represent the ethylene groups and dashed lines trace the octahedral coordination.
3.3.1.2. General Synthetic Approaches toward Nitroxide Probes
Several methods have been established for the preparation of nitroxides.206 In general, the nitroxide is formed by oxidation of a secondary amine with meta-chloroperbenzoic acid (m-CPBA) or H2O2 as oxidant (Scheme 3). The latter method requires a catalyst. In both methods, the amine group is first oxidized to form a hydroxylamine intermediate that is subsequently further oxidized to an oxammonium salt intermediate to finally yield the nitroxide. Alternatively, a mild oxidant, such as MnO2, can be applied to oxidize a hydroxylamine to give the desired nitroxide (Scheme 3). The m-CPBA oxidation is fast because less polar solvents can be used and compounds with electron-deficient double bonds are tolerated under these conditions.233 In contrast, H2O2 in the presence of a catalyst (tungstate salt) requires the use of a polar solvent system, in which lipophilic substrates are poorly soluble.
As discussed in section 3.3.1, there are two factors that need to be considered for improving the stability of nitroxides, one being the ring size and the other the substituents in the α positions.206,234 In the following, we discuss the synthesis of different types of nitroxides with five- or six-membered rings and various substituents in the α-positions.
Tetramethylpiperidone (Scheme 4, 36), which is commercially available, is the most commonly used starting material for the synthesis of nitroxide probes, due to its reactive ketone group. It can readily be modified to provide water-soluble TEMPO derivatives (see Scheme 4) or embellished with a reactive functionality (Scheme 4, 37a–d).225,231,235 Sakai et al. successfully developed a mild method to exchange the tetramethyl groups of compound 36 by more bulky six–membered heterocyclic or homocyclic rings (Scheme 4, 38a–b).236 In this reaction ammonium chloride acts as catalyst and, by using 15NH4Cl, the authors were able to study the mechanism of this interesting exchange reaction that appears to involve two cross-aldol reactions, two fragmentations and two Michael additions, one of which acts as the final cyclization step.236 Similarly, the ketone group of compound 38a was transformed to produce compounds 39a–b (Scheme 4).225 The sulfur–carbon bonds of compound 38c can be removed by Raney nickel to give a tetraethyl substituted derivative (Scheme 4, 40).237 After oxidation of the NH of compound 40 to a nitroxide, the hydroxyl group can be converted to different functional groups (compounds 41a–c).225,238,239
Scheme 4. Preparation of Various Nitroxides from Tetramethylpiperidone.
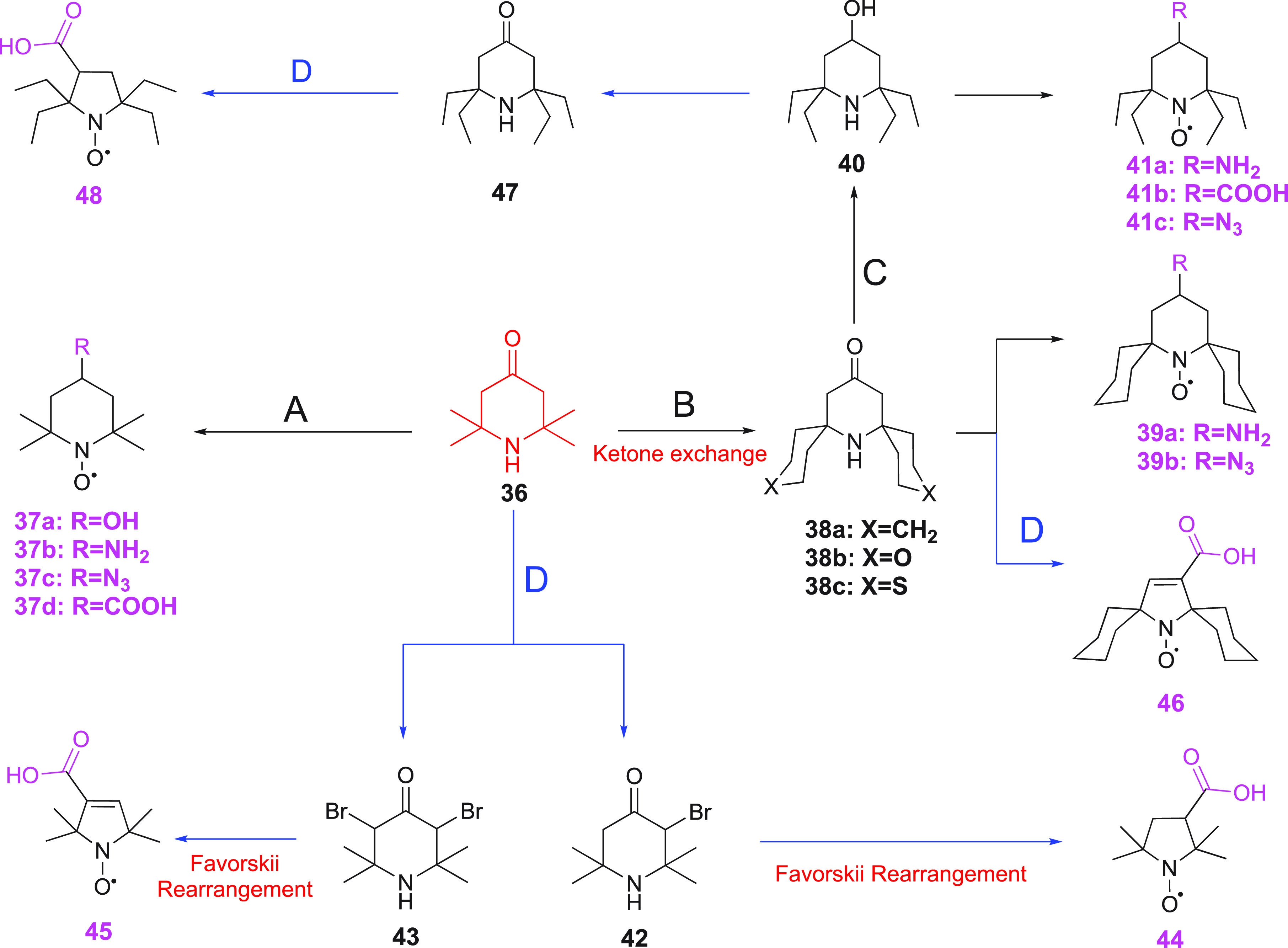
(A) i. NaBH4, H2O2/cat. Na2WO2 (to give 37a);225 ii. reductive amination with NaBH3CN (to give 37b);231 iii. active amine with methanesulfonyl to substitute with NaN3 (to give 37c);225 iv. carboxylation with tosylmethylisocyanide (to give 37d).235 (B) Cyclohexanone, NH4Cl, H2O2/cat. Na2WO2 (to give 38a–c).236 (C) Raney–nickel (to give 40).237 (D) i. Bromination (to give 42, 43); ii. Favorskii rearrangement (to give 44, 45).225,240 Blue colored arrows indicate the synthetic route to five-membered ring nitroxides.
Tetramethylpiperidone is also a precursor for the synthesis of five-membered ring nitroxides via a Favorskii ring contraction.225,240 Tetramethylpiperidone 36 is first brominated to yield compound 42 or 43. Under basic conditions, compounds 42 and 43 give the pyrrolidine derivative 44 and the pyrroline 45, respectively (Scheme 4). A similar sequence of steps leads to compounds 46 and 48 (Scheme 4).225,233,237
Another method based on the Horner–Wadsworth–Emmons reaction for making α-tetrasubstituted piperidones from a bisphosphonate has been reported (Scheme 5).241 In this procedure, a monoenone (Scheme 5, compound 49) and a dienone intermediate (Scheme 5, compound 50a) were generated. The monoenone was further reacted to give nonsymmetric dienone (Scheme 5, compound 50b). Both dienones (Scheme 5, compounds 50a and 50b) further reacted with hydroxylamine through a double aza-Michael addition reaction to yield the corresponding tetra-substituted piperidones 51a and 51b, which were oxidized to give the nitroxides (Scheme 5, compounds 52–54).
Scheme 5. Synthetic Route to α-Tetrasubstituted Piperidone from a Bisphosphonate Precursor.

(a) i. LDA (lithium diisopropylamide) at 0 °C. ii. BuLi (butyllithium) at −35 °C. iii. excess 3-pentanone. (b) i. LDA, THF, ii. acetone. (c) i. NH4OH at 105 °C. ii. m-CPBA. (d) Na2WO4, H2O2. (e) i. TMSCHN2 ((diazomethyl)trimethylsilane), BF3·OEt2; ii. Na2WO4, H2O2..
Isoindolinyl nitroxides are of special interest due to their benzannulated scaffold with diminished flexibility as compared to aliphatic nitroxide derivatives.225N-benzylphthalimide (Scheme 6, compound 55) is the most popular starting material for the synthesis of isoindolinyl nitroxides reported in the early 1980s, via the use of excess Grignard reagent to give 57.242 A more efficient procedure than a quadruple Grignard addition was found later, which proceeded via isolation of the monoaddition product (compound 56) and subsequent conversion into compound 57 (Scheme 6).243 The further transformation of the benzylamine to a nitroxide was achieved by debenzylation and oxidation (Scheme 6, 58).244−246 The latter compound can readily be transformed into a probe, such as compound 59a/b (Scheme 6), which can be attached to a thiol group of a biological macromolecule.244 Several other routes toward functionalized isoindolinyl nitroxides have been reported. One route involves bromination of the aromatic ring and removal of the benzyl protection group of compound 55, followed by substitution of the bromide by a cyano or carboxyl group (Scheme 6, 59c/d).247 Another route involves the construction of the functionalized aromatic ring via a Diels–Alder reaction on a diene nitroxide221 or following a higher yielding stepwise procedure using a disubstituted pyrroline nitroxide as starting material, leading to compound 59d.247
Scheme 6. Synthetic Routes Towards Isoindolinyl Nitroxide.

(a) RMgBr. (b) Excess RMgBr. (c) i. Pd–C/H2; ii. m-CPBA.
3.3.2. Aminopoly(Carboxylic Acid)-Based Probes
Aminopoly(carboxylic acid)s have the ability to bind metals strongly and they can easily be modified with functional groups to enable linkage to biomolecules. In this section, the most commonly applied aminopoly(carboxylic acid) based probes and their synthetic strategies are discussed.
3.3.2.1. EDTA-Based Probes
EDTA (ethylenediaminetetraacetic acid) is a widely used chelating agent for a broad range of metal ions, due to its high metal ion binding affinity with four carboxylic acids and two amine groups. Its complexes with paramagnetic ions can be linked to proteins via a disulfide bond by functionalizing them with MTS or pyridylthio groups. The first EDTA based paramagnetic probe for proteins, SPy-EDTA (Figure 7, 60), was reported by Ebright et al. in 1992.248 This probe soon became commercially available and its various metal ion complexes were used for protein paramagnetic NMR studies.249−251 These studies revealed two sets of PCSs generated by probe 60 loaded with Co(II) or Dy(III), as the metal complex produces two enantiomeric forms, which leads to diastereomers after linkage with the protein (Figure 6B).249,251 Subsequent introduction of chiral centers and a rigid attachment group to Ln(III)-EDTA resulted in single sets of PCSs, but the Δχ tensor was relatively small (Figure 7, 61).251,252 Diastereomer formation is not a critical problem for DEER distance measurements and Su et al. (2015) designed EDTA based EPR probes with short linkers as Mn(II) tags (Figure 7, 62–65), which yielded a narrow distribution of distances.253
Figure 7.
Structures of aminopoly(carboxylic acid) based probes. Functional groups for covalent attachment to a cysteine residue in the target protein via thioether formation are highlighted in red. Alkyne groups for attachment to an azide group in a ncAA via “click” reaction are highlighted in blue. Probes 60–65249−253 are EDTA based. Probe 61 has two chiral centers. Probe 66(255) is double-anchored. Probes 73–77263−266 are PyMTA based. The asterisk identifies one of the chiral carbon atoms.
3.3.2.2. DTPA-, DTTA-, and TAHA-Based Probes
Diethylenetriaminepentaacetic acid (DTPA) has a structure similar to EDTA, but features a higher metal binding affinity.254 DTPA functionalized with two MST groups, CLaNP-1 (caged lanthanoid NMR probe 1, Figure 7, 66), was designed as an NMR probe for proteins, using Yb(III) and Y(III) as paramagnetic and diamagnetic metal ions, respectively.255CLaNP-1 loaded with Yb(III) generated large PCSs, but multiple sets of PCSs were observed for each nuclear spin observed in the protein, because the coordination cage assumes multiple conformations.255 DTPA coordinates lanthanoids in an octadentate fashion and forms a pair of enantiomers.256 In 2020, Su and co-workers showed that a combination of DPTA and l-cysteine (Py-l-Cys-DTPA (Figure 7, 67)257 yields an open chain lanthanoid chelator that shows no exchange between conformations on the low millisecond time. Upon attachment to various sites on ubiquitin, large PCS and RDC were obtained, which were dependent on the site of attachment, likely due to a different degree of mobility for this single-arm tag. The probe Cys-Ph-TAHA (Figure 7, 68)258 was based on C3-symmetric TAHA (triaminohexaacetate). The paramagnetic properties of lanthanoid loaded Cys-Ph-TAHA were determined by using two ubiquitin mutants (T12C and S57C). PCSs and RDCs were observed. Two minor species were observed for Cys-Ph-TAHA labeled ubiquitin T12C, but not for ubiquitin S57C.258
Two DTTA (diethylene-triamine-tetraacetate) based protein probes (DTTA-C3-yne and DTTA-C4-yne, Figure 7, 69 and 70)259 were reported, which possess an alkynyl group for “click” ligation to a genetically encoded ncAA containing an azide group, with formation of a triazole linker (Figure 4, 9a). Tb(III) or Tm(III) loaded probes conjugated to ubiquitin mutants generated single sets of PCSs with a good correlation between experimental and back-calculated values. The triazole ring in the linker limits the number of rotatable bonds as it can form a conjugated double-bond system with the phenyl ring of the noncanonical residue side chain. DTTA-C3-yne, which has a shorter linker, showed a larger Δχ tensor than did DTTA-C4-yne. DTTA-C3-yne was used in the structure determination of a transient protein complex.259 Chen et al. reported phenylsulfonated pyridine modified DTTA probes (Figure 7, 71–72), which can react with cysteine with formation of a thioether bond shown to be stable in a cell lysate.2604PS-6M-PyDTTA (Figure 7, 72) has an extra methyl group in position six of the pyridine ring. This small difference proved to alter the speed of probe ligation, lanthanoid ion binding affinity, and Δχ tensor. 4PS-6M-PyDTTA showed a higher reactivity toward ubiquitin S57C and the Staphylococcus aureus sortase A (SrtA) mutant D82C.260 Following attachment, 6M-PyDTTA exhibited a lower metal binding affinity but larger Δχ tensor than PyDTTA. Interestingly, the two probes loaded with Tm(III) gave PCSs of opposite sign, indicating different orientations of the Δχ tensor.260
3.3.2.3. PyMTA-Based Probes
In contrast to the linear structure of EDTA, 2,2′,2″,2‴-((pyridine-2,6-diylbis(methylene))bis(azanetriyl))tetraacetic acid (PyMTA) has a pyridine core with diazanediyldiacetic acid pendants in positions 2 and 6. Its Ln(III) complexes have been used for luminescence experiments and as contrast agents in MRI.261,2624MMPyMTA (Figure 7, 73) was designed for chemical stability and successfully used as a paramagnetic lanthanoid probe to study the structures of minor protein species.2634V-PyMTA (Figure 7, 74)264 was reported as a paramagnetic Ln(III) probe with a vinyl group in position 4, which forms a thioether bond with cysteine. After successful attachment of 4V-PyMTA to ubiquitin, small PCSs were observed.264 Following substitution of position 3 of the pyridine ring with bromide to obtain probe 75, the reactivity was improved, but not the paramagnetic effects.25 Further work introduced phenylsulfonated in pyridine position 4 as the protein conjugation group (Figure 7, 76).265 Compared with probes 73 and 74, probe 76 yielded larger PCSs due to the short and rigid thioether attachment, but with a low reaction rate, even under fairly harsh conditions.265 Subsequently, probe 77 (Figure 7)266 was constructed, which features both good reactivity and selectivity for solvent exposed cysteine residues.
3.3.2.4. General Synthetic Strategies of Aminopoly(Carboxylic Acid)-Based Probes
There are two main strategies for the synthesis of aminopoly(carboxylic acid) based probes. The first involves either the direct modification of the carboxylic acid with attachment groups by a coupling reaction, such as applied for probes 60 (derived from EDTA) and 66 (derived from DTPA), or functionalization of the central amine with an attachment group as in, for example, probes 69 and 70 (derived from protected DTTA). The second strategy starts from the core structure of the probe, introduces the attachment group, and subsequently modifies the intermediate with a polyamine to introduce the carboxylic acid pendants. For example, the synthesis of probes 71 and 72 starts from a mono- or dimethylpyridine derivative, to which the attachment group (benzenesulfonyl) is added after several modification steps. Thus, after N-oxidation, nitration, rearrangement to their trifluoroacetate esters, and in situ mild hydrolysis of α-picoline or 2,6-lutidine, a nucleophilic aromatic substitution provided the sulfones. By alkylation with a halogenated pyridine derivative (Scheme 7, structure colored in red), the polyamine intermediate (Scheme 7, structure colored in magenta) is generated after deprotection and finally the poly(carboxylic acid)s are introduced to give the probe (Scheme 7, structure colored in blue).260
Scheme 7. Synthetic Route to Probes 71 (4PS-PyDTTA) and 72 (4PS-6M-PyDTTA).260.
3.3.3. Cyclen-Based Tags
Cyclen(1,4,7,10-tetraazacyclododecane) is a cyclic ring molecule of tetraethyleneamine, which can be modified to obtain various metal binding complexes, which are particularly suited for lanthanoid ions. DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) is one of the most frequently used cyclen derivatives. It was first synthesized in 1976.267 DOTA complexes are thermally stable and bind Ln(III) ions tightly.268 It is important to note that Ln(III)-DOTA complexes undergo conformational exchange involving cyclen ring flips combined with reorientation of the tetracarboxylic acid pendants (Figure 8). The pendant arms can coordinate clockwise (Λ) or anticlockwise (Δ) and the cyclen ring has two conformations (λλλλ and δδδδ). Consequently, Ln(III)-DOTA forms two pairs of enantiomeric species (square antiprism, SAP, and twisted square antiprism, TSAP) in solution, which tend to produce different magnetic effects in the case of anisotropic paramagnetism and averaging between them due to conformational exchange.269−271 Therefore, the designs of the cyclen probes described below aim to lock the cyclen ring in a single conformation to obtain uniform paramagnetic effects, by incorporating substituents on the cyclen ring and/or the pendant arms with the appropriate chirality.
Figure 8.
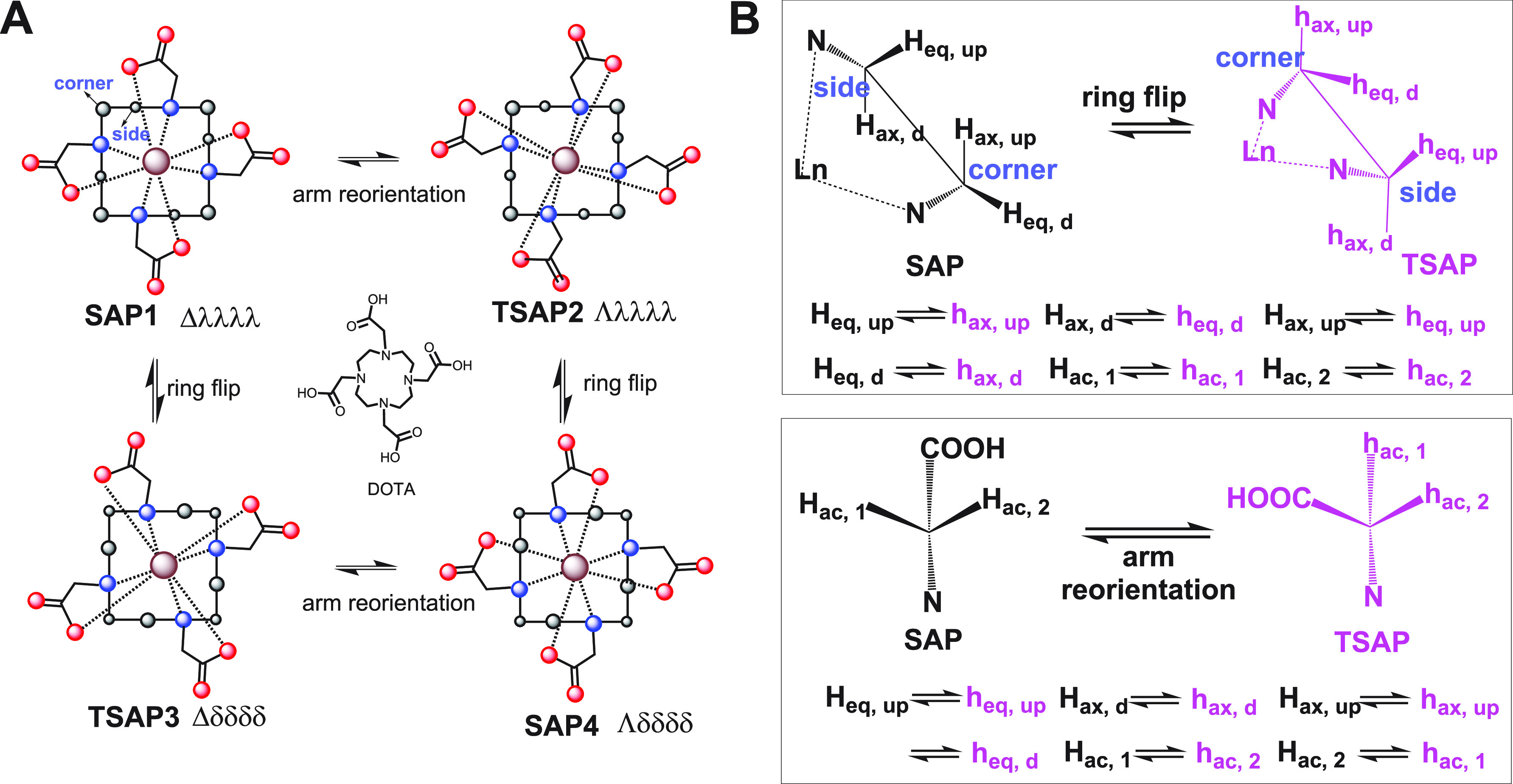
(A) Schematic representation of four different stereoisomers populated by DOTA lanthanoid complexes and their conformational exchange. The cyclen ring is drawn as a square with solid lines; nitrogen, oxygen, and carbon atoms are shown as blue, red, and black spheres, respectively; the metal ion is represented by a brown sphere. (B) Newman projections showing the effect of the ring flip on the positions of the hydrogen atoms of two neighboring carbon atoms. Adapted from Q. Miao (2019) Design, synthesis and application of paramagnetic NMR probes for protein structure studies. PhD Thesis, Leiden University.
3.3.3.1. Double-Anchored Attachment
The first reported cyclen based protein paramagnetic probe was CLaNP-3 (Figure 9, 78)272 in complex with paramagnetic Ln(III) ions. By functionalizing two opposing tetracarboxylic acid arms of DOTA with MTS groups, CLaNP-3 can be anchored to a protein via two disulfide-bridges for maximum immobilization of the metal ion relative to the protein. For pseudoazurin (Paz) with mutations E51C and E54C tagged with Yb(III)-CLaNP-3, PCSs were detected for nuclear spins up to 35 Å from the paramagnetic center. Some residues, however, displayed peak doubling, suggesting that the complex populated more than a single conformation with different Δχ tensors.272
Figure 9.
Chemical structures of cyclen-based paramagnetic probes. The functional groups for attachment to cysteine via thioether formation are highlighted in red. The functional groups for attachment to an azide group in ncAA via “click” reaction are highlighted in blue. The cyclen rings with substituents are shown in magenta. 78,27279,127,27380,27681,27782,27883,279 and 84(280) are double-armed probes. Asterisks identify chiral carbon atoms.
Subsequently, CLaNP-5 (Figure 9, 79)127,273 was produced and became one of the most used lanthanoid probes. In CLaNP-5, the two acetate pendant arms of CLaNP-3 were replaced by pyridyl-N-oxides, which disfavors the population of the TSAP conformation of its lanthanoid complexes. For Yb(III)-CLaNP-5 linked to Paz E51C/E54C, a single set of large PCSs was observed. In contrast, CLaNP-5 attached via a single arm yielded only small PCSs due to averaging caused by the flexibility of the attachment.273 Further studies demonstrated that magnetic properties of CLaNP-5 are insensitive to the chemical environment of the attachment site, allowing the prediction of the Δχ tensor parameters and Ln position for a known protein structure with reasonable accuracy.127CLaNP-5, thus, is highly suitable for structural studies of proteins and protein complexes. It turned out, however, that the detection of little populated protein states by relaxation dispersion (RD) experiments is compromised by the presence of minor conformations of the complex that can lead to probe-induced RD effects.274,275 Other disadvantages of CLaNP-5 are its charge (+3), making it less favorable for some proteins and protein complexes, and the disulfide bonds formed with the protein are liable to reduction, which can lead to loss of the probe over a period of several weeks.
To decrease the charge, CLaNP-7 (Figure 9, 80)276 contains two phenolic groups, lowering the net charge to +1 by deprotonation of the phenol hydroxyls. CLaNP-7 is yellow in color, which is convenient during the tag labeling reaction and protein purification. Similar to CLaNP-5, it induced a single set of PCSs in Paz E51C/E54C, with a Δχ tensor that was only slightly smaller than that of CLaNP-5.276 Unexpectedly, two sets of PCSs were observed specifically for Yb(III)-CLaNP-7 linked cytochrome c and this peak doubling was pH-dependent. By mutation of His39, which is adjacent to the probe attachment site, to alanine, it was demonstrated that the peak doubling was caused by an interaction with this residue, suggesting that the hydration water molecule, which coordinates the Ln(III) ion in its ninth coordination site, forms a hydrogen bond with the imidazole ring of His39 and in this way breaks the C2 symmetry of the probe. The effect was pH dependent due to protonation and deprotonation of the imidazole ring. Since disulfide bonds can easily be reduced, CLaNP-9 (Figure 9, 81)277 was designed as a variant of CLaNP-5, featuring two bromoacetanilide groups to react with cysteine residues by formation of chemically stable thioether bonds. Using T4 lysozyme (T4Lys) N55C/V57C and Paz E51C/E54C as model proteins, Yb(III) loaded CLaNP-9 yielded single sets of PCSs under reducing conditions, indicating the high stability of this probe. Because of the rapid hydrolysis of the bromoacetanilide groups in aqueous solutions, however, quantitative tagging proved difficult to achieve.
The tags T1 and T2 (Figure 9, 82)278 similarly are double-anchored cyclen based probes using pyridylthio groups as attachment groups. T1 and T2 are enantiomers, as their two free coordination arms are chiral isopropanol moieties. Large Δχ tensors were found for these lanthanoid complexes linked to Staphylococcus aureus 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK) K76C/C80, and exceptionally narrow distance distributions were obtained by DEER measurements using the tags loaded with Gd(III).278 The paramagnetic effects induced by other paramagnetic Ln(III) ions, however, are sensitive to the pH, with large PCSs and RDCs detected at high pH. Another pair of enantiomeric probes, T7 and T8 (Figure 9, 83),279 were designed with two shorter chiral attachment arms. Although large Δχ tensors were determined for a wide range of pH values for both tags, the χ tensor orientation changed between high pH (8) and low pH (6.5). It was noted that T7 produced incomplete ligation yields for one of the test proteins (HPPK K76C/C80).279
The tags described above are all lanthanoid based cyclen probes. Recently, several double-arm 3d block ion probes (transition metal ion NMR probes, TraNPs) were reported.280 Among them, TraNP-1 (Figure 9, 84)280 can tightly bind Co(II) and Mn(II) ions. Its Co(II) complex generates a single set of PCSs and yields a medium sized tensor, which is suitable for application in small proteins or localized studies in larger proteins or protein complexes. The authors compared the tensor orientation between Co(II)-TraNP-1 and Yb(III)-CLaNP-5 ligated to T4Lys K147C/T151C by Δχ tensor fits using experimentally measured PCSs. The tags delivered significantly different tensor orientations as evidenced by the PCS iso-surfaces shown in Figure 10.280
Figure 10.

Comparison of Co(II)-TraNP-1-SS and Yb(III)-CLaNP-5 attached to T4Lys K147C/T151C. (A) Metal positions and tensor orientations. (B) PCS iso-surfaces (±0.4 ppm for Yb(III)-CLaNP-5, ±0.2 ppm for Co(II)-TraNP-1-SS). (C) Models of the protein–probe structures. The protein is shown in ribbon representation; the probes and cysteine residues are shown as sticks and the metal ions as spheres. Reproduced without changes from ref (280). Copyright 2019 the authors, under CCA license (https://creativecommons.org/licenses/by/4.0/).
Recently, the double-anchored probe CLaNP-13 (Figure 9, 85) was designed for DEER studies in living cells.281 By virtue of two maleimide groups, CLaNP-13 forms thioether bonds with cysteine residues, which in a cellular environment are more stable than disulfide bonds. As Michael additions of thiols to maleimides generate enantiomers, NMR measurements showed peak doubling for the Yb(III) complex attached to T4Lys double-cysteine mutants. DEER measurements, however, yielded a narrow distance distribution for CLaNP-13 attached to the quadruple mutant T4Lys N55C/V57C/K147C/T151C, and the probe was also shown to be suitable for in-cell DEER measurements.
3.3.3.2. Single Site Attachment
DOTA-M8 (Figure 9, 86)282 is a single-arm lanthanoid probe, which produces Δχ tensors of remarkable size. Its structure is based on M4DOTMA.283 Because of the eight chiral methyl substitutions on the cyclen ring and acetate pendant arms, M4DOTMA loaded with Yb(III) exhibits a single conformation, which is rigidified by steric crowding.283 A set of large PCSs was reported for Dy(III)-DOTA-M8 labeled ubiquitin S57C. Dy(III) is tightly bound to DOTA-M8, withstanding harsh chemical and physical conditions.282 About 15–20% of a minor species appeared in the NMR spectra, however, and this ratio increased to 50% by heating the tagged protein to 323 K. Subsequent studies revealed slow conformational exchange between two coordination geometries of Ln(III)-DOTA-M8, which produce different Δχ tensors, with the exchange rate depending on the radius of the lanthanoid ion.271,284
As DOTA-M8 is ligated to the protein via a disulfide bond, the ligation is not stable under reducing conditions. This prompted efforts to link DOTA-M8 to proteins by different attachment chemistries. By replacing the pyridylthio linker arm in DOTA-M8 by a propyliodoacetamide (−C3H6NHCOCH2I) group, a chemically more stable thioether bond is formed in the reaction with cysteine. This new tag was referred to as M8-CAM-I (Figure 9, 87) and its usefulness for in-cell NMR measurements was demonstrated, but the longer linker resulted in a smaller Δχ tensor than for DOTA-M8.285 By using 4-(phenylsulfonyl)pyridine as the attachment group, another DOTA-M8 derivative, referred to as M7Py-DOTA (Figure 9, 88), forms a more rigid and shorter linkage to the protein, which is also suitable for in-cell NMR.286 As the reactivity of the 4-(phenylsulfonyl)pyridine group is relatively low, the CF3-substituted phenyl analogues (Figure 9, 89 and 90) were designed for enhanced reactivity, but this approach did not increase the rate of protein tagging because the rate-limiting step appears to be the formation of a Meisenheimer-type complex rather than the dissociation of the pyridine-sulfone moiety.287 Consequently, the probes M7FPy-DOTA (Figure 9, 91)287 and M7PyThiazole-DOTA (Figure 9, 92)287 were produced, both of which proved to react with cysteine with high selectivity and efficiency. The authors found that M7FPy-DOTA can react also with tris(2-carboxyethyl)phosphine (TCEP) to release benzenesulfinic acid, which can catalyze protein dimerization by intermolecular disulfide bridge formation between cysteine residues. M7PyThiazole-DOTA performs better, as it is more reactive toward cysteine than TCEP and generates an even larger Δχ tensor than DOTA-M8.287 Recent work introduced a 3-nitro-substituted 2-(methylsulfonyl)pyridine moiety as attachment group to design a new DOTA-M8 based probe named M7-Nitro (Figure 9, 93).288 This probe possesses high protein ligation rates (more than 95% ligation in 30 minutes at room temperature) and a very large Δχ tensor (Δχax = 94.3 × 10–32 m3) was measured with Dy(III)-M7-Nitro labeled ubiquitin S57C. The authors attributed the improvements in ligation to the methylsulfone leaving group in the ortho position and the presence of the nitro substituent in the meta position.
To further improve the rigidity of the cyclen-based probes, DOTA derivatives with more crowded substituents, P4M4-DOTA (Figure 9, 94)126 and P4T-DOTA (Figure 9, 95)289 were synthesized. Only a single conformation, Λ(δδδδ), was detected in ROESY spectra of the Lu(III)-P4M4-DOTA complex.126 Linking lanthanoid ion loaded P4M4-DOTA to ubiquitin S75C and human carbonic anhydrase II (hCA II) S166C delivered single sets of PCSs, but some of the Q factors of the Δχ tensor fits were relatively large owing to the length and flexibility of the attachment arm. As expected, P4T-DOTA performs better than P4M4-DOTA because of a shorter and more rigid linker. P4T-DOTA also forms a stable thioether bond with cysteine.
C1 (Figure 9, 96) is another early example of a DOTA based probe for protein studies by paramagnetic NMR, which is relatively easy to synthesize.290 In this tag, the metal ion is coordinated by amides instead of carboxyl groups. Loading of C1 with Ln(III) ions requires harsh conditions and the ions are tightly bound thereafter. Three chiral bulky pendant arms ensure that the C1 tag populates a single conformation as indicated by 1H NMR spectra of Yb(III) loaded C1 and single sets of PCSs were observed with all proteins tested. C2 (Figure 9, 96) is the enantiomer of C1 and generates a slightly different tensor orientation.291 On the basis of the structure of probe C1, the protein EPR probe C9 (Figure 9, 97)292 was designed with a chiral tagging arm. Variants of C1 with alkyne attachment groups, C3 and C4 (Figure 9, 98, 99),293 were developed for ligation to the ncAA AzF (Figure 4, 9a) by “click” chemistry.293 The PCSs obtained with probe C4 are smaller than those of probe C3 due to the longer linker of the former. The Cu(I) catalyzed ligation reaction was shown to be selective also in the presence of cysteine residues in the target protein and thus suitable for proteins containing cysteine residues of structural and functional importance. Subsequent experience showed, however, that many proteins are prone to precipitation in the presence of Cu(I) catalysts.182 For this reason, the recently published C12 probe (Figure 9, 100)294 reacts with cysteine by formation of a thioether bond to its aromatic ring linkage group. Complete tagging yields of cysteine were obtained at room temperature overnight. In contrast, the tag reacts with selenocysteine in minutes, providing a route to selective tagging of proteins in the presence of native cysteine residues.294
Ln(III) complexes of C1 and its variants are relatively large, and although experimental evidence is scarce,295 their hydrophobic aromatic rings may result in unexpected specific interactions with the protein surface. Therefore, a number of alternative cyclen probes were produced with hydrophilic pendants, where the single conformation of the cyclen ring is ensured by the chirality of the pendant arms. The probe C5 (Figure 9, 101)129 contains three chiral (S)-2-hydroxypropyl groups as pendant arms. Variants of C5 with different attachment linkers were also explored, including C6 (Figure 9, 102),129 which contains a rigid picolinic acid linker, C7 (Figure 9, 103),129 which contains a chiral alcohol in the attachment arm instead of an amide, and C8 (Figure 9, 103),129 which is the enantiomer of C7. Like C1 and C2, these tags can generate large Δχ tensors. A comparison of three proteins and different attachment sites showed, however, that the Δχ tensor produced by C7 is highly sensitive to the protein environment, which is unusual for lanthanoid based cyclen probes.129 Probe C10 (Figure 9, 104)296 was designed for DNA studies (discussed in section 3.4). Ligated to ubiquitin A28C, it produced Δχ tensors of medium size, as expected for the relatively long and flexible attachment arm. Recently, DO3MA-Py (Figure 9, 105),297DO3MA-6MePy (Figure 9, 106),297DO3MA-3BrPy (Figure 9, 107),298 and BrPSPy-6M-DO3M(S)A (Figure 9, 108)299 were reported, which contain methyl substituted carboxylic acid pendants and phenylsulfonated pyridine with different substituents on the pyridine ring as functional group for attachment. These lanthanoid ion complexes carry zero net charge and form thioether bonds with cysteine residues. Large PCSs were observed with these probes. While the ligation rates of DO3MA-Py and DO3MA-6MePy are slow even at high pH and temperature, the bromide substituent in DO3MA-3BrPy decreased the reaction time to 6 h at ambient temperature. Sizeable Δχ tensors were obtained with the Yb(III) complex of DO3MA-3BrPy attached to ubiquitin D39C and ubiquitin E64C, and DO3MA-3BrPy loaded with Gd(III) was also successfully used for in-cell distance measurements by EPR, although the distance distribution obtained was relatively broad.298 Its companion BrPSPy-6M-DO3M(S)A has an extra methyl group on the pyridine and the authors reported that this design improved the rigidity of the tag.300 By replacing bromine in DO3MA-3BrPy for a fluorine atom, Su and co-workers synthesized the probes 4PS-5F-Py-DO3MA (Figure 9, 109) and 4PS-5F-Py-DO3A (Figure 9, 110) to analyze the conformational exchanges of DOTA-Ln-like probes using 1D and 2D EXSY NMR spectroscopy.301 They demonstrated that even the presence of chiral centers in three of the pendants of a DO3A-type cyclen tag does not prevent the population of a minor conformational species, which persists following ligation of the lanthanoid complexes to ubiquitin G47C.301 Notably, a second conformational species has also been reported for DOTA-M8, although this probe contains four additional stereocenters in the cyclen ring.287 The relative conformational stabilities of SAP and TSAP conformations of DOTA-type cyclen-Lu(III) complexes with different chiralities of methylated cyclen and carboxylate pendants has recently been rationalized computationally.302
Another recent approach used a photocatalyzed reaction to conjugate a vinyl probe to proteins.303 Using 2,2-dimethoxy-2-phenylacetophenone (DPAP) as a photoactivated radical initiator, the ligation rate of the vinylpicolinic acid modified probe 111 (Figure 9) with GB1 T53C was greatly accelerated, but the Δχ tensors obtained were small.303
3.3.3.3. General Synthetic Approaches of Cyclen Based Probes
DOTA and its analogues have been used as metal ion ligands for decades and the thermodynamic properties, synthesis, and applications of their metal complexes have been investigated in detail.268,304−308 Commensurate with the wide range of applications for cyclen and DOTA compounds in many different fields, numerous synthetic routes have been reported. It should be noted that DOTA based probes with chiral substituents to ensure conformational rigidity, require the execution of a multistep synthetic scheme. In addition, the protective group strategy has to be chosen carefully to allow the introduction of one or two attachment arms. Among the different synthetic methods reported for large-scale cyclen preparation,307,309−314 three main strategies stand out for modification of the cyclen ring. The first is based on one of the synthetic procedures of cyclen (Scheme 8A). This approach was explored by Jacques et al., who used substituted ethylenediamine derivatives and chloroacetyl chloride derivatives as starting materials to form a linear diamide intermediate (1c in Scheme 8A), which was further reacted with an ethylenediamine derivative to yield the cyclic product 1d. Finally, the amide groups of the ring were reduced to give the core macrocyclic structure 113 (Scheme 8A).315,316 On the basis of this method, symmetric and asymmetric functionalized cyclen derivatives were produced, such as cyclohexyl, cyclohexene and tetraline substituted cyclens 113a–c,304 and the diol substituted compounds 113d–e.315,316 Later on, the same group developed a new method to synthesize cyclen derivatives with C4 symmetry, where, instead of using a stepwise cyclization, a more straightforward synthetic route was followed by oligomerization of N-substituted aziridine (2a in Scheme 8B) in the presence of p-toluenesulfonic acid (PTSA, Scheme 8B).283,317 This method directly gives the N-protected cyclen derivatives (2b in Scheme 8B), albeit in low yield, and, after deprotection, the desired macrocycle 114. The cyclen based probes DOTA-M8, M7Py-DOTA, M7FPy-DOTA, P4T-DOTA as well as M7PyThiazole-DOTA (Figure 9) were synthesized using this approach and several other groups subsequently used this approach for cyclen based ligand synthesis.313,314 Anelli et al. employed both approaches for the synthesis of more hydrophilic macrocyclic compounds.318 Recently, Häussinger and co-workers reported a new method for synthesizing modified cyclens (Scheme 8C),126,319 which starts from chiral aminoaldehydes (3c in Scheme 8C) derived from their respective amino acid precursors. Via reductive amination, two suitably protected linear alkylated amino acid derivatives (3e and 3f) are produced, which are readily coupled to each other and cyclized by standard solution peptide coupling under high dilution. Compared to the other two strategies, this newly reported route provides a high yield in the cyclization step and the overall yield of macrocycles 115 has been reported around 22%.319
Scheme 8. Three Strategies for Cyclen Ring Modulation.
The magenta colored structures show the final cyclized ring with different substituents. The compounds in magenta are applied for paramagnetic probe design.
3.3.4. Small Molecule Tags
Small molecules like dipicolinic acid (DPA), iminodiacetic acid (IDA), nitrilotriacetic acid (NTA), and [2,2’:6′,2″]terpyridine-6,6″-dicarboxylic acid (TDA) have also been explored for the design of paramagnetic NMR probes. Their attraction derives from their small size, which minimizes the potential impact on the protein structure, and their relative ease of synthesis. In complexes with Ln(III) ions, these small probes leave several coordination sites of the Ln(III) ion free for coordination by solvent molecules or additional protein side chains. With these tags, the metal complexes are formed by titration with metal ions after probe attachment, which opens the possibility of inaccurate metal to protein ratios and binding of metals to other, undesired sites.
3.3.4.1. DPA, TDA, and HQA Tags
DPA coordinates metal ions in nonchiral geometry, thus reducing the potential for different enantiomeric forms of metal coordination and peak doubling in NMR spectra. Despite presenting only a tridentate ligand, the affinity of DPA for lanthanoid ions is in the low nanomolar range.320,3214MMDPA (Figure 11, 116)322 was the first DPA tag developed. It was attached to a cysteine residue following activation with DTNB, which results in a disulfide linkage, and shown to generate PCSs following attachment to Cys68 of the ArgN protein, in which the coordination of the paramagnetic lanthanoid ion appears to be supported by coordination to a glutamate side chain.322 Despite the limited coordination of the lanthanoid ion, the dissociation rate of the metal ion was slow at 25 °C (0.1 s–1), but the Δχ tensor was smaller than that found with cyclen based tags. Loaded with Gd(III) ions, the tag was also demonstrated to be suitable for Gd–Gd distance measurements by EPR spectroscopy in different proteins.122 To shorten the linker length, the probes 3MDPA (Figure 11, 117)323 and 4MDPA (Figure 11, 118)324 were produced, which feature the sulfur atom right on the pyridine ring. 3MDPA, which has a thiol group in position 3, was ligated to ArgN like 4MMDPA via a disulfide bond following activation of its cysteine residue with DTNB. In complex with paramagnetic Ln(III) ions, Δχ tensor orientations were obtained different from those with 4MMDPA and metal coordination still appeared to be assisted by Glu21, but the magnitudes of the Δχ tensors were small. Compared to the 3MDPA tag, the disulfide dimer of 4MDPA is a more convenient tag for ligation, as it reacts spontaneously and quantitatively with cysteines by disulfide exchange. While it displayed relatively small Δχ tensors with ArgN, larger Δχ tensors were obtained with the intracellular domain of the p75 neurotrophin receptor (p75 ICD). The tensor orientations differed substantially from those obtained with the corresponding 4MMDPA complexes for ArgN but not for p75 ICD, suggesting important contributions by additional coordinating amino acid side chains. The DPA tags also display good affinity for Co(II), as demonstrated first for ArgN with 3MDPA tag.323
Figure 11.

Chemical structures of small molecule probes. Functional groups designed for attachment via thioether formation are highlighted in red. 116,322117,323118,324121,326122,326 and 124(265) are DPA-based probes. 119 and 120 are TDA based probes. 123(176) is based on 8-hydroxyquinoline. 125(327) is an NTA based probe. 126(328) and 127(329) are IDA based probes.
Using DPA as starting materials, TDA based probes, 4MTDA (probe 119, Figure 11)325 and 4MMTDA (probe 120, Figure 11)325 were synthesized. Large PCS were detected after their Ln ion loaded complexes were ligated to ubiquitin T22C or A28C. They also exhibit fluorescent properties. For both mutants, ubiquitin side chains are involved in metal coordination. The authors also showed that 4MTDA exchanges lanthanide ions more slowly than DPA, as 2D 1H–1H EXSY spectra showed no observable chemical exchange. The ligation yields were reported to be relatively low.
Probe 121 (Figure 11)326 contains a vinyl group to link the probe to the protein via Michael addition to form a thioether linker. The vinyl group avoids the problem of maleimide functionalized tags (Figure 11, 122), which create a new chiral center upon reaction with a thiol group.326 A vinyl group was also used to ligate the 8-hydroxyquinoline based probe 2V-8HQ (Figure 11, 123)176 to a cysteine. While the tag is not suitable for binding lanthanoid ions, it binds divalent transition metal ions such as Mn(II), Co(II), Ni(II), Cu(II), and Zn(II) with micromolar affinities, and a large Δχ tensor was observed for the Co(II) complex of 2V-8HQ bound to ubiquitin A28C/E24H, where the binding of the metal ion was assisted by the histidine residue.176 A DPA derivative with a phenylsulfonated pyridine functional group (Figure 11, 124)265 was shown to generate a stable, rigid and short tether with a thioether bond to a cysteine, but Δχ tensor parameters were not reported.
3.3.4.2. NTA and IDA Tags
Similar to DPA and 8HQ tags, NTA-based probes immobilize metal ions on target proteins depending on the assistance from additional coordinating groups provided by other amino acid residues. The first NTA-based probe suitable for binding lanthanoid ions, NTA-SH (Figure 11, 125),327 was obtained by modifying cysteine with two acetate groups. Single sets of PCSs were reported upon complexation with paramagnetic lanthanoid ions following attachment of the probe to either one or two cysteine residues of the target protein. For a single NTA-SH tag, much larger Δχ tensors were observed with ArgN than ubiquitin A28C, suggesting that the protein environment plays a critical role in immobilizing the Ln(III) ion. This difficult-to-predict variability can be reduced by simultaneously attaching NTA-SH tags at positions of i and i + 4 of an α-helix to create a metal binding site with coordination by both NTA moieties. This approach can also deliver very large Δχ tensors.327 As the metal ion affinity is not as high as for DOTA ligands, the metal ion can be exchanged by washing with EDTA and subsequently introducing another metal ion.
An even smaller tag is based on IDA, which only contains two carboxylates. The IDA-SH tag (Figure 11, 126)328 and its activated form (Figure 11, 127)329 were reported to yield a single set of PCSs after chelating lanthanoid ions. This tag can yield large Δχ tensors, if the Ln(III) ion can be additionally coordinated by a protein side chain as observed for ubiquitin A28C, where Asp32 is located in the same α-helix as residue 28.328 By ligating two IDA-SH tags to cysteine residues in positions i and i + 4 of an α-helix, a Co(II) ion can be coordinated in an octahedral complex and produces sizable PCSs (Figure 11, 127).329 In ubiquitin E24C/A28C, however, this tag arrangement performed best with substoichiometric quantities of Co(II) and Zn(II) ions, whereas lanthanoid ions produced multiple species. Subsequent study of this tag and metal complexation mode in the same protein and in T4Lys K147C/T151C revealed additional species also with Co(II) ions and difficulties to achieve complete saturation of the metal binding site.280 A systematic exploration of the best binding partner for an IDA-SH or NTA-SH tag attached to a cysteine residue in position i of an α helix indicated that the IDA-SH tag forms the best Ln(III) binding sites together with an aspartate residue in position i + 4, while the sterically more demanding NTA-SH tag produces the best sites in combination with a glutamate residue in position i – 4.330 As secondary structure predictions identify amphipathic α helices with good reliability, this approach is of interest for proteins of unknown 3D structure.
3.3.4.3. Hybrid Double-Arm Attachment
Nitsche et al. recently introduced a new tagging strategy, where a small metal binding tag is attached to two cysteine residues, but additional coordination of the lanthanoid ion by a negatively charged side chain of the protein is still required to immobilize the metal ion.331 The concept was demonstrated with the Zika virus NS2B-NS3 protease mutated to contain cysteine residues in positions 101 and 131. The side-chains of the cysteine residues are in close proximity and avidly bind the pnictogens arsenic, antimony, and bismuth in oxidation state III. The third coordination site of the pnictogen is available for binding a mercaptomethylaryl tag, such as 4MMDPA to capture a Ln(III) ion, which was shown to generates sizable Δχ tensors in conjunction with additional coordination by a glutamate residue. The fundamental advantage of the concept is its selectivity for two cysteines in close proximity. As single thiol groups bind pnictogens with negligible affinity, it allowed tagging of the protein in the presence of the native, solvent exposed single cysteine residues.
3.3.5. Cosolute Paramagnetic Probes
Cosolute paramagnetic NMR probes do not chemically modify the targeted biological macromolecule. A structurally diverse class of paramagnetic molecules has been designed and investigated for their noncovalent interaction with the solvent exposed surface residues of oligopeptides and proteins. Cosolute probes can be as simple as salts of paramagnetic transition metals. The approach is based on the idea that large compounds and charged ions do not readily access the interior of folded proteins, so that the NMR signals of surface residues experience stronger PREs than buried residues.332 Mn(II) ions are well suited for this type of NMR experiments.333−335
Owing to its diradical character, molecular oxygen is paramagnetic and one of the simplest molecular probes used in NMR. As oxygen dissolves more easily in the hydrophobic interior of membranes than in aqueous solution, this solubility gradient has been used to probe the structure and positioning of a membrane protein,336 and a number of related NMR experiments have been reported that exploit the unique properties of oxygen.337−341 In early work, Williams and co-workers obtained cosolute paramagnetic effects by directly adding paramagnetic metal ions, such as Gd(III), Eu(III), and Mn(II), to protein solutions.110,120,342 Using [Cr(CN)6]3–, [Fe(CN)6]3–, and [Co(CN)6]3– complexes in 1D 1H NMR spectroscopy experiments, Williams and co-workers studied the competition with negatively charged substrate molecules for binding to phosphoglycerate kinase.111
Organic nitroxide radicals present a more varied class of paramagnetic cosolute probes. Early results showed that di-tert-butylnitroxide generates contact shifts with several small organic molecules in carbon tetrachloride.343 On the basis of these results, Kopple and Schamper344 prepared 3-oxyl-2,2,4,4-tetramethyloxazolidine (Figure 12) that is soluble in chloroform and methanol, and these solvents and this probe were used to detect line broadening in the 1H NMR spectrum of amides of the solvent exposed amino acid residues in the C2 symmetric cyclic decapeptide gramicidin S, while the amides of the amino acid residues involved in intramolecular H-bonding were not affected. The same research group reported that a water-soluble variant (HyTEMPO or TEMPOL, Figure 12) can serve as a tool to identify hydrophobic surface residues of proteins, using the attenuation of antiphase COSY cross-peaks as a sensitive method to detect small line-broadening effects caused by the presence of the paramagnetic agent.345 Building on these results, Fesik and co-workers showed that quantitative measurement of R1(1H) relaxation rate enhancements generated by TEMPOL allowed accurate identification of the solvent exposed regions of cyclosporin A bound to cyclophilin.346TEMPOL also proved useful to probe the accessibility of amino acid residues in protein unfolding experiments347 and studies of conformational change.348 The attempt to probe electrostatic potentials using the charged derivatives 4-carboxy-TEMPO and 4-amino-TEMPO (Figure 12) showed the expected trends but also demonstrated limitations attributed to local variations in translational diffusion constants.347−349 Stronger solvent PREs can be generated by Gd(III) ions and the uncharged and highly water-soluble Gd(III)DTPA-BMA probe (Figure 13) can perform better than TEMPOL, because it is less hydrophobic, stable with regard to redox-active compounds, and the paramagnetic center less accessible.350 Solvent PREs generated by Gd(III)DTPA-BMA were shown to deliver powerful restraints for 3D structure determinations of proteins and peptides,35,351,352 which prompted the design of Gd(III)TTHA-TMA(353,354) as a more spherical Gd(III) probe without any hydration water bound to the Gd(III) ion. The use of alternative uncharged Gd(III) probes such as Gd2(L7)(H2O)2,355Gd(DO3A), and Gd(HP-DO3A),356 as well as complexes with increasing charge, such as [Gd(EDTA)]1–,357[Gd(DTPA)]2–,358 and [Gd(DOTP)]5−,359 has also been explored (Figure 13). The strong PRE effects generated by Gd(III) complexes have also proven useful to shorten the T1 relaxation time of H2O360 and protein protons361 without greatly increasing the line widths of protein NMR signals, enabling sensitivity enhancements by faster scan repetition rates. It is important to note, however, that some proteins may harbor specific binding sites for some of these complexes, which can be made apparent by PCSs generated by complexes with paramagnetic lanthanoid ions other than Gd(III). For example, the cosolute probe [Tb(DOTP)]5– was found to generate specific PCSs in the DNA binding protein fd gene 5359 and [Ln(DPA)3]3– (with Ln = Tm, Yb, or Tb, Figure 13) was found to generate significant PCSs in ArgN and several other proteins.362,363 The Δχ tensor generated in this way can be tuned by using DPA derivatives carrying different substituents (−Br, −COOH, or −CH2OH) on the aromatic ring, which form lanthanoid complexes like the parent [Ln(DPA)3]3– and bind to the same site on the protein.363 As rapid reorientation of the complexes averages PCSs to zero,350 the observation of PCSs can be taken as positive proof of specific binding. Natural binding sites for [Ln(DPA)3]3– are infrequent in proteins and it has been shown that binding sites specific for this complex can be engineered by positioning the side chains of two positively charged residues (lysine or arginine) in close proximity, which, at the same time, need to be sufficiently far from negatively charged carboxylates to prevent their engagement in salt bridges.364 In contrast to covalently bound paramagnetic probes, specific binding sites for complexes such as [Gd(DPA)3]3– offer the opportunity to measure PREs for nuclear spins very close to the binding site, as the PRE magnitude can be tuned by the amount of relaxation agent added.365 PREs generated by the cyclen complexes Gd(DO3A), Fe(DO3A), and Ni(DO2A) (Figure 13) have also been explored. The Gd(III) ion in Gd(DO3A) has two accessible coordination sites available and specifically targets carboxylates on protein surfaces.366Fe(DO3A) has been used to speed up data acquisition by shortening the T1 relaxation time of an intrinsically disordered protein.367Ni(DO2A) has been reported to be particularly suitable for shortening the T1 relaxation times of H2O and protein signals without broadening them significantly.368 As the ligand fills all coordination sites of the Ni(II) ion, it is unlikely to bind to specific sites on proteins. At the same time, it is a water-soluble probe without carrying a net charge.
Figure 12.

Chemical structures of nitroxide based cosolute probes.
Figure 13.
Chemical structures of metal ion based cosolute probes.
3.3.6. Development of Cysteine Based Probe Attachment Chemistry
Historically, the development of chemical approaches for the selective modification of proteins has its origin in studies of amino acid chemistry.369 Further important insights on site-specific chemical modification came from studies of post-translationally modified proteins.370 New chemistry for site-specific ligation reactions were opened by the development of methods to incorporate noncanonical amino acids either by genetic encoding or the use of chemically misacylated tRNAs.371−373
The thiol group of cysteine has unique nucleophilic characteristics, as compared to the side chains of any of the other canonical amino acid residues. Because of the higher acidity of RCH2SH as compared to RCH2OH, cysteine residues in proteins can be deprotonated at slightly basic pH, which increases their reactivity374−383 toward soft electrophiles. With little chemical activation of the paramagnetic probes, the reaction conditions used for the attachment of the probes can be mild. Figure 14 provides an outline of the most relevant cysteine modifications with respect to the attachment of probes amenable for NMR and EPR.384−386
Figure 14.
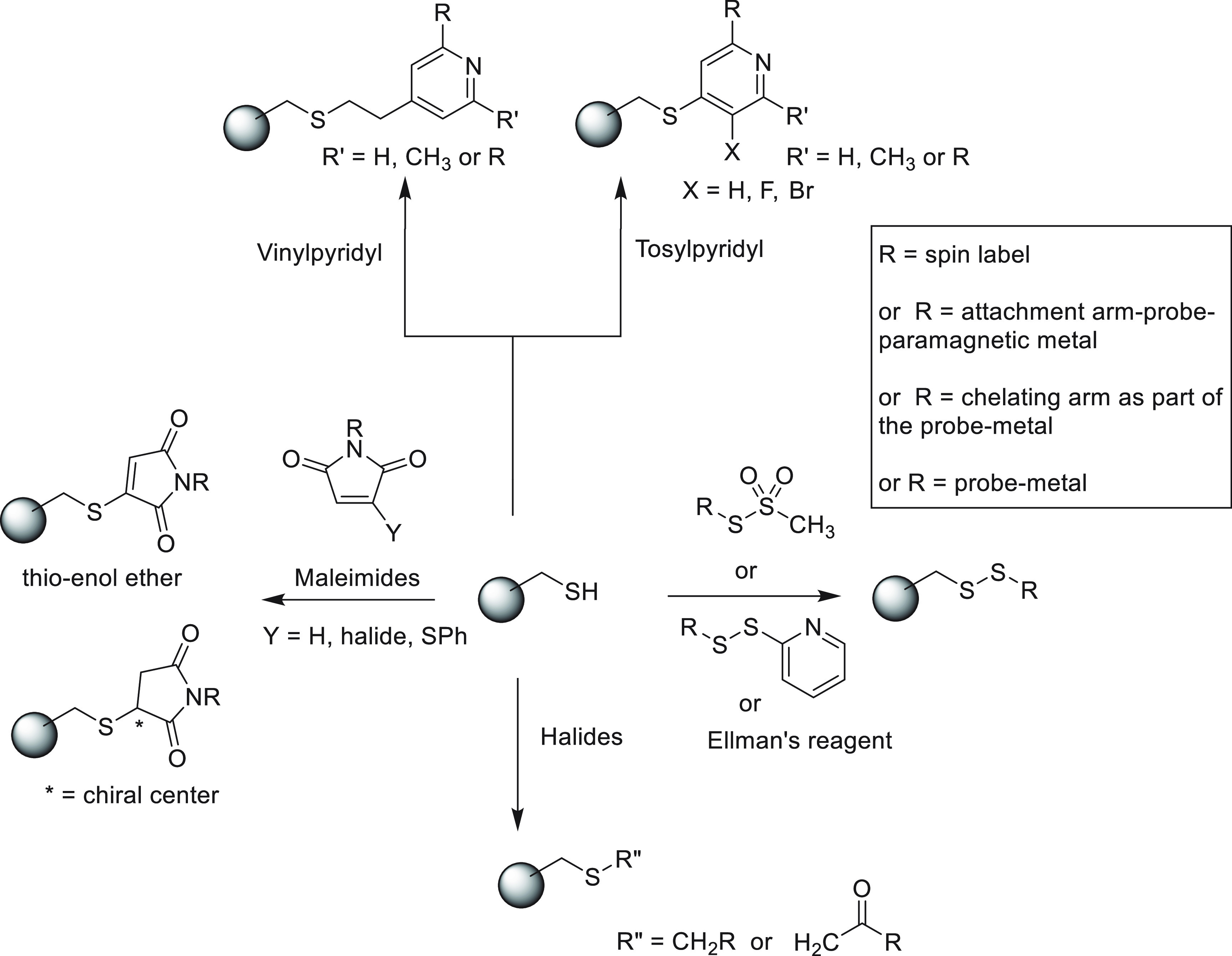
Most relevant chemical methods to connect NMR or EPR probes to one or more cysteine residues in proteins. The aim of the figure is to provide an overview of the currently used chemistry for the modification of cysteine residues, suitable for the attachment of a chemically sensitive paramagnetic center. The use of maleimide as reactive group results in the generation of a chiral center, and therefore, this type of linkage is not recommended in NMR studies.
A standard, selective and mild, method for attaching a probe to a cysteine residue is by disulfide formation. One of the most frequently used reactive functionalities to accomplish this is the MTS or S-mesyl group. This group is stable under acidic conditions, which is advantageous, if acid labile protective groups need to be removed at a late stage of chemical probe synthesis.280 At physiological pH the thiol of cysteine forms a disulfide linkage with the RS-moiety of the S-mesyl compound with the release of methane and SO2 in a practically irreversible reaction. Several other reactive functionalities are known to generate S–S bonds,387 in particular the 2-pyridyldisulfanyl group (as in the probes 60,24882,27883,27986,28294,12696,29097,292101,129103,129112,388 and 127(329)). Disulfide bonds are also made by activation of a cysteine residue of a protein using a mild oxidizing agent such as Ellman’s reagent (DTNB) and addition of the probe as a thiol.322,323,327,328,389 Similarly, a chelating moiety was introduced by reaction of a cysteine residue with ArS-SAr (Ar = PyMTA266 or Ar = DPA324). Several procedures are available to produce a thioether, which is chemically more stable than a disulfide linkage. α-Iodo or α-bromo carbonyl reagents (XCH2C(=O)R) have traditionally been used to generate thioethers with cysteine residues, as the reaction is fast and selective at the physiological pH.369,390 This reactive functionality has successfully been installed in a number of probes, such as in probes 16,21317,39181,27787,285 and 104.296 Furnishing DOTA type probes with two α-bromo-carbonyl functionalities, however, has proven challenging because of the high reactivity of this functionality.277
Another standard reactive functionality is the maleimide group, which has a high reactivity and selectivity for cysteine residues around neutral pH.392,393 Maleimide probes react with cysteines via Michael addition, forming a new stereocenter211,212,281 and thus giving rise to different stereoisomers which complicate PCS assignments. The use of a maleimide functionality to attach a probe for NMR experiments is therefore not recommended. Monobromomaleimide probes223,394 do not have this problem because the Michael addition is followed by an elimination reaction resulting in a thio-enol ether linkage (Figure 14), and Michael reactions with vinylyridine probes (probes 64,25365,25374,26475,25121,326 and 123(176)) do not generate new chiral centers either. Tosylpyridine probes (62,25363,25371,26072,26076,26577,26688,28689,28790,28791,28793,288105,297106,297107,298108,300109,301110,301 and 124(265)) form thioethers. The electrophilicity of these probes is enhanced by the presence of a halide substituent on the pyridyl ring. Similar to the tosyl group, a nitro group on the pyridine ring can act as a leaving group.294
3.4. Paramagnetic Probes for DNA and RNA
Studying oligonucleotides with paramagnetic probes can provide valuable insights into the structure and dynamics of DNA and RNA as well as their interactions with proteins.40−47 Labeling methods of oligonucleotides can be classified into four main categories. First, noncovalent spin labels can be selectively introduced at an abasic site without covalent attachment of the probe to the nucleotide. Second, spin labels can be selectively attached to the nucleotide backbone via reaction with modified ribose or phosphate groups in the oligonucleotide. Third, spin labels can be selectively attached to a single base via reaction with a modified purine or pyrimidine base. Fourth, a fully synthetic and already labeled nucleotide can be introduced into oligonucleotides by standard solid-phase oligonucleotide synthesis. Occasionally, a synthetic method may allow the attachment of probes to the nucleotide prior to, during, or post oligonucleotide synthesis. All four strategies are reviewed in the following sections with a particular emphasis on the attachment chemistries.395
3.4.1. Noncovalent Spin Labels
Paramagnetic cosolutes, which have been used intensively for the study of proteins (section 3.3.5), have also been used to study oligonucleotides by paramagnetic NMR spectroscopy. For example, the nitroxide radical TEMPOL (Figure 12) and the Gd(III) complex of TTHA-TMA (Figure 13) have been used to study the surface accessibility of RNA and refine its 3D structure.396,397
To probe biomolecules more precisely, it is necessary to introduce the spin label at a defined position as rigidly as possible. In the case of proteins, this is usually achieved by site-selective mutation and subsequent covalent bond formation between probe and amino acid side chains (section 3.3). Selective and rigid noncovalent labeling of proteins is not straightforward and achievable only in special cases, requiring the availability of a suitably labeled protein ligand.398 In the case of oligonucleotides, noncovalent spin labeling is a much more generally applicable technique that appeals by its simplicity. Synthetic spin labels can mimic a purine or pyrimidine base and specifically bind to an abasic site in double-stranded DNA or RNA oligomers, thus circumventing the need to synthesize the entire spin-labeled nucleotides. Currently available noncovalent spin labels are all nitroxide radicals, which allow applications in NMR (PRE) and EPR. Noncovalent oligonucleotide tags containing a paramagnetic metal ion may be suitable for PCS measurements but are yet to be explored.
The first site-directed noncovalent spin label, which binds to an abasic site in DNA, was reported by Lhomme and co-workers (Figure 15, 128).399,400 Compound 128 combines the base adenine with the DNA intercalator acridine, which are connected via a flexible linker. Nakatani and co-workers developed spin label NCD-TEMPO (Figure 15, 129) and a nitronyl nitroxide analogue NCD-NN (Figure 15, 130), which can specifically bind to G–G mismatches in DNA duplexes, allowing for the construction of tandem arrays and three-dimensional assemblies of electron spins.401−404 Spin label NA-TEMPO (Figure 15, 131) was designed in this work to clarify the effect of orthogonal sequence selectivity on the electron-spin arrangement.402
Figure 15.
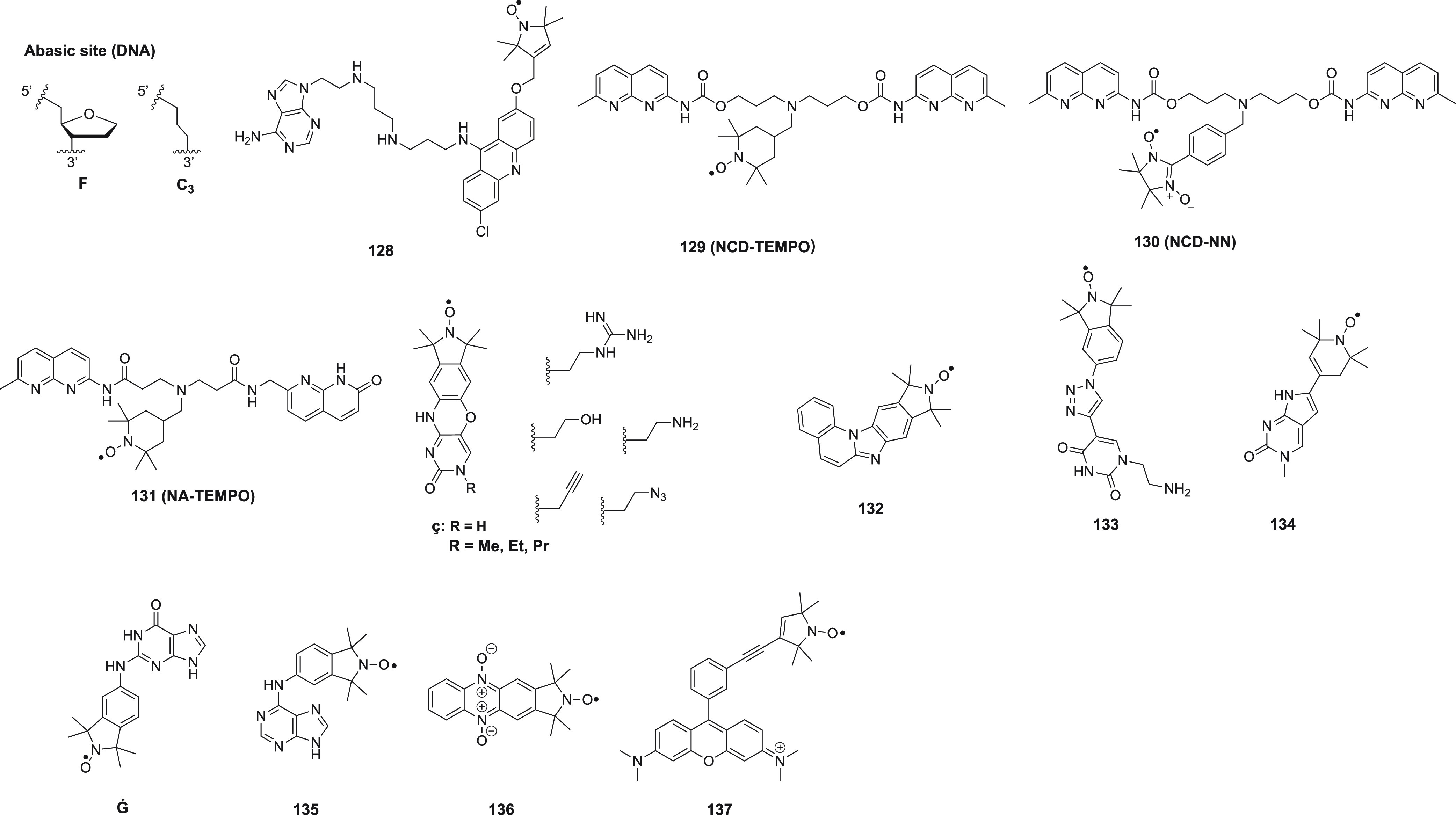
Site-directed noncovalent spin labels for DNA and RNA.
Sigurdsson and co-workers introduced the noncovalent DNA spin label ç, which allows labeling without intercalation or specific sequence requirements.405 Its synthesis from 5-bromouracil is shown in Scheme 9. Mimicking a cytosine, ç binds best to a DNA duplex if paired with a guanosine nucleotide (Kd ∼ 5 μM at −10 °C).405 Binding of ç (Figure 15) does not only depend on the pairing base, but also on the flanking sequence, with a 5′-d(DçT) sequence affording the highest affinity.406 The abasic site should also be located at least four base pairs from the end of the DNA duplex.406 A DNA duplex labeled with two ç labels enabled DEER distance measurements up to 70 Å. Binding of a double-stranded lac operator DNA sequence to the lac repressor protein has also been probed by DEER. Bending of the double helix was manifested in a distance decrease between both ç spin labels by about 5 Å, which, however, is close to the resolution limit of this method.407 The introduction of substituents at the N3 nitrogen of ç affected the affinity, with large substituents compromising the affinity most.406,408 Derivatives containing an amino or guanidino group displayed slightly increased affinity and improved solubility.408 The anucleoside C3 (Figure 15) was explored as an alternative abasic site over the tetrahydrofuran analogue F (Figure 15) with limited impact on the affinity of ç to the DNA.406
Scheme 9. Synthesis of the Noncovalent DNA Spin Label ç from 5-Bromouracil.
5-Bromouracil is regioselective benzylated, subsequently converted into its O4-sulfonylated derivative (TPS = 2,4,6-triisopropylbenzenesulfonyl) and coupled to an isoindol aminophenol derivative, followed by ring-closure. The benzyl group is removed and N-oxidation gives the nitroxide.
The nitroxide radical 132 (Figure 15) was developed as an alternative noncovalent spin label that binds to abasic sites in RNA and DNA duplexes.409 Best pairing was observed with cytosine which does not interact well with ç. Although spin labeling of RNA and DNA was confirmed by EPR spectroscopy, 132 has not been used in any further applications. Another campaign screened ten pyrimidine based spin labels for their suitability to occupy abasic sites in DNA and RNA duplexes.410 Triazole linked (e.g., 133 (Figure 15)) and pyrrolocytosine-based (Figure 15, 134) labels showed very promising results. Spin label 133 displayed increased solubility and affinity (DNA) compared to its N1-methylated analogue (not shown). It was speculated that the increased affinity may result from a salt bridge formed between the ethylamino group and the phosphate backbone. The DNA label ç has insufficient affinity to RNA (∼30%).410 In contrast, 133 shows much higher affinity (100% under identical conditions) to an abasic RNA site; it also binds to an unmodified RNA duplex, which, however, compromises specificity.410
The isoindoline-nitroxide derivative Ǵ overcomes previous limitations and binds with high affinity and specificity to abasic sites in RNA duplexes. Ǵ pairs with a cytosine and is suitable for DEER distance measurements when combined with an RNA duplex containing two abasic sites.411Ǵ can be prepared from commercially available materials in a single straightforward synthetic step (Scheme 10), facilitating its broad use.411Ǵ can also bind to abasic sites in DNA-RNA hybrids, either in the RNA or DNA strand, and, similar to ç, must be positioned at least four residues from the end of the duplex to achieve optimal binding.412 The adenine derived spin label 135 complements the guanidine derived Ǵ (Figure 15); 135 was identified from a series of purine derived nitroxides and can be introduced at abasic sites in DNA and RNA duplexes where it pairs with thymine or uracil, respectively.412Ǵ has been successfully applied to measure distances in both DNA and RNA duplexes by DEER.413 Combined with molecular dynamics simulations, these experiments indicate strong Watson–Crick base pairing, no disturbance of the overall helical structure (minor local disturbances occur) and rigid binding of Ǵ. Recently, Ǵ was also used to label DNA and RNA triplexes containing a single abasic site.414 The N-oxide phenazine 136 is the latest noncovalent spin label that can bind to an abasic site in DNA but is unsuitable for RNA.415
Scheme 10. Synthesis of the Noncovalent RNA Spin label Ǵ from 2-Bromohypoxanthine via a Nucleophilic Aromatic Substitution.

If the spin label can directly bind to RNA or DNA, no chemical modifications of oligonucleotides (not even the introduction of an abasic site) are required. Despite the principal appeal of this strategy, however, achieving the necessary specificity of the interactions between label and oligonucleotide can be challenging, as observed for intercalating spin labels based on chlorpromazine,416 ethidium bromide, or other polyaromatic carcinogens,417 Ru(II)-phenanthroline complexes,418 or acridine.419 Bifunctional cross-linking agents are also of limited specificity.420,421 One solution is presented by the use of an aptamer. For example, the malachite green RNA aptamer (a 38-nucleotide sequence) is known to bind malachite green or closely related dyes such as tetramethylrosamine (TMR). If TMR is synthetically combined with a spin label as in 137 (Figure 15), it can be used to selectively label the RNA aptamer.422 Combination with a spin label that is covalently attached to the RNA backbone (145, see section below) allowed DEER measurements. Similarly, a ternary complex consisting of a DNA oligomer, the molecule chromomycin-A3 and paramagnetic Co(II), where Co(II) was bound to two oxygens of the chromophore, has been used to measure PCSs and improve the structure of the complex.423,424
3.4.2. Probe Attachment to the Sugar–Phosphate Backbone
Some paramagnetic spin labels can be attached to the phosphate backbone of DNA and RNA. It has been noted that spin labels attached to phosphodiesters interfere less with duplex formation than other labels, as they are located at the edges of the polynucleotide helices.43 The greatest disadvantage of this approach is that labeling of phosphodiesters inevitably results in the formation of diastereomers, which need to be separated by chromatography and identified, if unambiguous structural information is to be obtained from paramagnetic NMR experiments or EPR studies.296,425,426 Recently, a synthetic strategy for the chiral synthesis of phosphorothioates has been described, which may help to overcome these challenges.427 In addition, it is important to note that, when labeling RNA, the 2′ hydroxyl group adjacent to the labeled phosphodiester needs to be protected as a 2′-OMe group or replaced by a hydrogen atom to prevent strand cleavage through 2′,3′-transesterification.428
The introduction of a hydrogen phosphonate group during oligonucleotide synthesis enables the selective attachment of 4-amino-TEMPO via H-phosphonate chemistry yielding a phosphoramidate (Figure 16A).429,430 Using protected oligonucleotides, 4-hydroxy-TEMPO can be attached to the 3′ or 5′ end of DNA via a phosphoester bond (not shown),431 which has been used to study protein DNA interactions by NMR.432 Similarly, the increased reactivity of terminal phosphate groups can be exploited by selective activation of the 3′ and 5′ ends of unprotected DNA and subsequent reaction with 4-amino-TEMPO to form phosphoramidates.433 A fully protected TEMPO-phosphoramidite building block (Figure 16B, 138) compatible with oligomer synthesis has been used to form an ester between 4-hydroxy-TEMPO and the phosphate at the 5′ end of RNA.434
Figure 16.
Strategies for the attachment of probes to the phosphodiester backbone or terminal phosphate groups of oligonucleotides.
A frequently used functional group for site-directed labeling of the phosphate backbone is presented by phosphorothioate. Selective incorporation of spin labels at the 5′ end of RNA is possible directly by transcription from DNA in a two-step process where, first, a DNA template is transcribed in the presence of NTPs (nucleotidetriphosphates) and a phosphorothioate nucleotide and, second, a spin label is attached to the 5′ phosphorothioate group. The approach has been demonstrated with guanosine monophosphorothioate (139), T7 RNA polymerase, and spin label 140, which was attached via disulfide formation (Figure 16C).435
Alternatively, a phosphorothioate moiety can be enzymatically installed in the 5′ position of RNA or DNA using T4 polynucleotide kinase, prior to alkylation with the spin label 16 (Figure 16).436 The iodoacetamide based spin label 17 (Figure 16) can be ligated to internal phosphorothioate nucleotides in DNA (Figure 16E).437 The rigid spin label 16 has been used more frequently.438 It has been attached to RNA and DNA to study, for example, the GAAA tetraloop/receptor interaction,439 the human telomeric G-quadruplex,440 riboswitch dynamics,441 CRISPR-Cas9,442 DNA structure and dynamics,443 distances by DEER,444 and protein DNA interactions.445 The 4-bromo-substituted analogue R5a has been used to study ribozymes, CRISPR-Cas9, and the structure and dynamics of DNA.426,446−449 The double-alkylating analogue R5c links two adjacent phosphorothioates to give a conformationally highly constrained cyclic spin label for RNA and DNA (Figure 16D).220 Graham and co-workers developed the first lanthanide tag (Figure 9, C10) that can be attached to phosphorothioate, enabling the observation of PCSs in NMR spectra of DNA.296
Alternatively, paramagnetic spin labels can be attached to the sugar (ribose) part of the backbone. The 2′ position in oligonucleotides is the only position readily available for modification. Nucleotides bearing a 2′ amino group can be site-specifically installed in oligonucleotides using standard oligonucleotide synthesis (2′ amino-modified phosphoramidites of pyrimidine-based nucleotides are commercially available).450,451 The most commonly used spin label is 4-isocyanato-TEMPO (Figure 17, 142), which is readily accessible from 4-amino-TEMPO and diphosgene.450,452 The isocyanate group forms a stable and rigid urea moiety between the 2′ position of the nucleotide and the TEMPO spin label, which has been used in numerous applications involving EPR and NMR to study RNA and its interactions.453−461 An alternative succinimide-based spin label (Figure 17, 143) was found to affect the thermostability of RNA helices.462 It can also be introduced at a 5′ amine or piperazine group (Figure 17).463 The isoindoline-based spin labels 144 (Figure 17) and 145 (Figure 17) bear an isothiocyanate group to form a thiourea linkage with the 2′ amino group.230 The tetraethyl derivative 145 (Figure 17) displayed significant stability toward reducing conditions.
Figure 17.
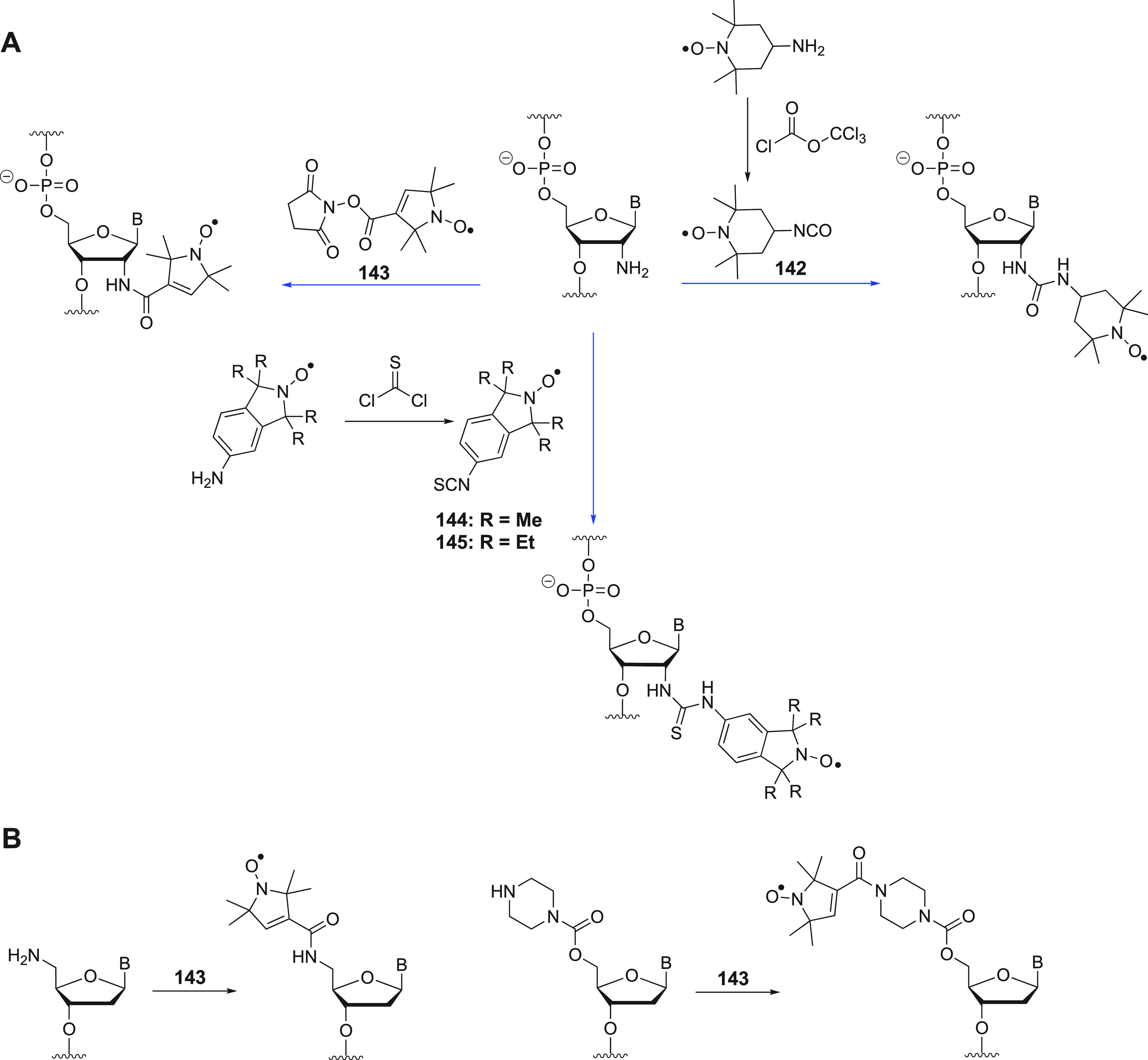
Probe attachment to (A) a 2′ amino group of internal nucleotides (blue arrows indicate the labeling reactions) and (B) alternative 5′ modifications.
Attaching a probe to the 2′ position via “click” chemistry has also been studied extensively (Figure 18). Attachment of probe 24 (Figure 18) to a 2′ propargyl ether was the first example described; however, the construct produced broad distance distributions in DEER experiments due to a rather long and flexible linker.43,464 A much shorter linker can be established, if the alkynyl group is directly connected to the 2’ carbon as in 2′-alkynylnucleotides (Figure 18).225,465 Azide based spin labels 23–25 (Figure 18) and 146–148 (Figure 18) can be attached selectively via “click” chemistry and have been used to measure distances in DNA by DEER.225 A crystal structure of double-stranded DNA with label 24 has been solved (Figure 19).465 Combined with EPR experiments and MD simulations, the results indicate only minimal perturbance of the DNA structure because of the presence of the spin label. Royzen and co-workers developed alkyne substituted Cu(II) (Figure 18, 149) and Co(II) (Figure 18, 150) binding ligands that can be attached to a linker-azide functional group in the 2′ position (Figure 18).466,467 Both tags were successfully used to study binding of the HIV-1 nucleocapsid protein 7 to an RNA pentanucleotide by PRE measurements using NMR spectroscopy.466,467 In the case of the cobalt tag 150, observation of PCSs was not reported, which potentially relates to the flexibility of the linker between probe and RNA.467 The “click” reaction between a 2′ azido GTP and an alkyne-TEMPO derivative produces a spin labeled nucleotide that can be used for site-specific post-transcriptional labeling of RNA (not shown).468 The GTP analogue is a substrate for a Tb3+-deoxyribozyme that installs it specifically on an internal adenine nucleotide via a 2′,5′-phosphodiester linkage.468
Figure 18.

Probe attachment via “click” chemistry to the 2′ position.
Figure 19.
X-ray crystal structure (PDB code 6QJS) of a 12 base pair DNA duplex containing the spin label 24 (highlighted in pink) attached in the 2′ position via “click” chemistry. The phosphate backbone is shown as a ribbon. The distance between both nitroxide radicals determined by DEER is 30 Å.
Finally, spin labels can be selectively attached to the 5′ or 3′ hydroxy ends of nucleosides. Selective 5′ labeling can be achieved via carbamate or amide formation with TEMPO,430143 (Figure 17B),463 or the large trityl radical 151 (TAM) (Figure 20).469 Attachment to both 5′ nucleotides of a DNA duplex allows distance measurements by DEER. Although trityl radicals offer advantages over nitroxide radicals, such as narrow spectral width and stability under reducing conditions,43 their size and hydrophobicity limit applications with biomolecules. Recently, alternative linkers were developed that allow the attachment of TAM anywhere in the DNA sequence using an achiral non-nucleoside phosphoramidite (Figure 20).470 Selective modification of the 3′ end of RNAs with TEMPO has also been demonstrated.471−473
Figure 20.
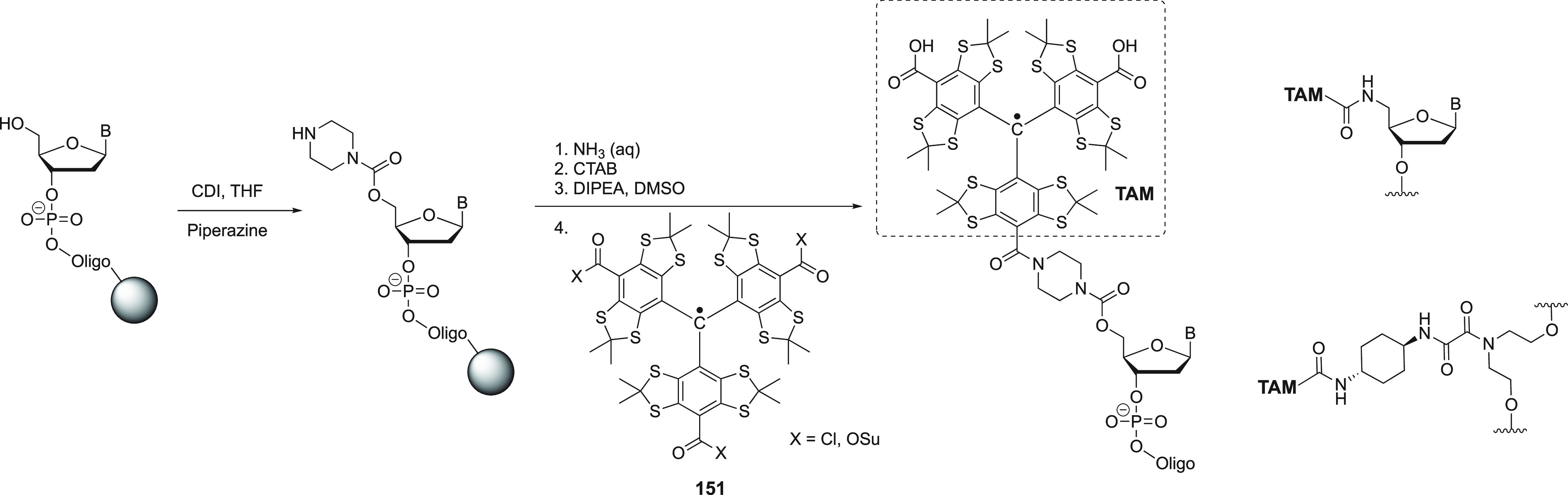
Attachment of the triarylmethyl (TAM)-based spin label 151 to the 5′ end of DNA. The immobilized oligonucleotide is treated with CDI and piperazine and cleaved from the support using standard conditions. The resulting oligonucleotide is treated with cetyltrimethylammonium bromide (CTAB), taken up in DMSO and treated with 151 as the chloride in the presence of base. The two remaining acid chloride functions were hydrolyzed. Two alternative TAM attachments strategies are shown.
3.4.3. Probe Attachment to Modified Bases
The majority of paramagnetic probes used to study RNA and DNA are attached to purine or pyrimidine bases. Labels can be fully introduced by synthetic chemistry to furnish labeled nucleotides that can subsequently be used directly in solid-phase oligonucleotide synthesis (covered in the next section). Alternatively, labels can be introduced postsynthetically (by chemical synthesis or transcription) by attachment to a modified base either prior or post removal of protection groups (in the case of chemical synthesis). The postsynthetic approach can help minimize possible decomposition of the nitroxide radical during synthesis, but it may result in incomplete labeling or side reactions with other functional groups.43
A functional group frequently employed for postsynthetic modification is presented by sulfur substituents in purine and pyrimidine bases (Figure 21). 4-Thiouracil has been used most frequently. 4-Thiouridine can be found in natural tRNAs of E. coli and has been selectively labeled with spin label 17/141 (Figure 16) without the need for any synthetic modifications.474
Figure 21.

Postsynthetic probe attachment to thiopyrimidines and thiopurines.
Subsequently, 4-thiouridine was installed in synthetic RNA and labeled with 17/141 to study a double-stranded RNA-binding domain in the Staufen protein by NMR spectroscopy.475 A similar protocol476 was also used to determine the solution structure of large RNAs and protein RNA complexes,477 for example, to study E. coli protein sequestration by the noncoding RNA RsmZ by NMR and EPR.478 A very similar system using deoxy-4-thiouridine and 17/141 in synthetic DNAs (installed at a 5′ overhang) allowed the study of DNA and the modulator recognition factor 2 (Mrf2) by NMR measurements of PREs.479 As an alternative to the formation of thioethers, thiouridine can form disulfide bonds with spin labels 152–154 (Figure 21), which has been used to study RNA structures.480 2-Thiocytidine has been incorporated into tRNA using tRNA nucleotidyl transferase and reacted with spin label 155.481 The rare nucleotide 2-thio-5-(N-methylaminomethyl)-uridine from the anticodon region of E. coli tRNAGlu was acylated with spin label 156.482,483 The thiopurine nucleotide 6-thioguanosine can be enzymatically incorporated into RNA via a DNA-splint mediated ligation strategy and subsequently reacted with spin label 17 to study long RNAs by NMR spectroscopy (Figure 21).484,485
Convertible nucleotides present yet another strategy to introduce a functional group during oligonucleotide synthesis that can selectively react with spin labels postsynthetically. This strategy uses aromatic ethers or fluorine as leaving groups and amino groups in spin labels, such as 4-amino-TEMPO, as nucleophiles (Figure 22).486−489 Both pyrimidine and purine analogues have been described, and the approach has been employed in several studies involving RNA and DNA.490−495
Figure 22.

Convertible nucleotides for postsynthetic transformation into spin labels. The 2-fluoro-hypoxanthine derivative has also been used as O-NPE protected version.
Alkynyl bases, such as the commonly used 5-ethynyl-2′-deoxyuridine (EdU), have been developed to allow for the selective reaction with azido based spin labels (e.g., 24, 25, 148) via “click” chemistry during postsynthetic modification of DNA and RNA (Figure 23A).496−503 The tags have been demonstrated to be particularly useful for distance measurements by DEER.496,497,499,500,502,503 The strategy also enables the labeling of very long RNAs (>400 nucleotides) and distance measurements within them.500 NHS esters 143 and 159 (Figure 23B) have been used to acetylate nucleotides that contain aliphatic amines (Figure 23B).504−507 Selective labeling of guanine involves a multistep process including the use of a complementary DNA reagent. Despite the length of the resulting linkers, the strategy successfully enabled DEER distance measurements in RNA duplexes. An analogue of cisplatin (160, Figure 23C) has also been used to deliver spin labels to DNA by ligand exchange with guanine (Figure 23C),508 enabling paramagnetic NMR measurements (PRE) that confirmed bending of the DNA duplex induced by the presence of the platinum complex.509 Using an on-column strategy, spin label 27 (Figure 23D) was coupled to iodo-nucleotides in RNA by the Sonogashira reaction.510 The approach for 2-iodo-adenine is shown in Figure 23. Nucleotide modification via Diels–Alder reactions was also successfully investigated but not applied for EPR or NMR spectroscopy of DNA or RNA.511
Figure 23.

Various strategies for postsynthetic probe attachment to nucleotide bases.
The selective posttranscriptional labeling of RNA can be achieved by using an expanded genetic alphabet based on the unnatural base pair dTPT3-dNaM.512 Subsequently, the cyclopropene residue in the unnatural RNA nucleotide can be selectively modified with tetrazine reagents via a [4 + 2] cycloaddition with nitrogen release. If the tetrazine is linked to a spin label, as in 161 (Figure 23E), this approach can be used to introduce a paramagnetic tag into RNA after transcription from DNA.513 Despite the very long linker, a distance in a self-complementary RNA duplex could be determined by DEER. Alternatively, the dTPT3-dNAM system can be used to directly incorporate the spin-labeled nucleotide TPT3NO into RNA by in vitro transcription, resulting in a much shorter linker and without the need for ligation chemistry (Figure 23E).514
Different from proteins, lanthanoid tags for oligonucleotides are scarce. One reason is the catalytic effect of lanthanoid ions on hydrolytic cleavage of the phosphodiester bond if the metal ion is not fully protected by the complexation agent. Consequently, cyclen based tags, such as C10, are the preferred option.296 Recently, two TAHA based lanthanide tags were reported, which allowed the measurements of PCSs and RDCs in NMR spectra of double-stranded DNA.515 Both tags 68 (Figure 7) and 162 (Figure 23F) can be attached via disulfide bond formation to a thiophenyl substituent in a selectively incorporated nucleotide derived from EdU (Figure 23F).
3.4.5. Fully Synthetic Paramagnetic Nucleotide Probes
Fully synthetic nucleotides are usually synthesized as their phosphoramidite analogues for subsequent use in standard solid-phase oligonucleotide synthesis. A summary of paramagnetic derivatives is given in Figure 24. TEMPO-labeled derivatives of 2′-deoxycytidine (Figure 24, 163), 5-methyl-2′-deoxycytidine (Figure 24, 164), 2′- deoxyadenosine (Figure 24, 165), and 2-amino-2′-deoxyadenosine (Figure 24, 166) were used in automated oligonucleotide synthesis, generating EPR-active DNAs.516−518 Because of the covalent link of the TEMPO group with the base, its mobility serves as a sensitive probe of the microenvironment, which was employed to detect single-base mismatches in duplex DNA by EPR using nucleotide 163.519 TEMPO has also been attached via a semiflexible urea linker in analogues 167 (Figure 24) and 168 (Figure 24).520
Figure 24.

Paramagnetic nucleotides for incorporation during oligonucleotide synthesis.
The C5 position of pyrimidines is an alternative site for spin labeling that has been studied and applied intensively (Figure 24). Using phosphotriester chemistry, TEMPO nucleotides (Figure 24, 169 and 170) and probe 171 (Figure 24) were incorporated into short DNA oligomers.521,522 Longer linkers between pyrimidine bases and nitroxide spin labels were also explored.47,523,524 The first spin-labeled phosphoramidite for incorporation into DNA was the uridine analogue 172, which was prepared by Sonogashira cross-coupling,525 which can be performed during solid-phase synthesis.526 The alkynyl linkage limits the flexibility of this probe, making it valuable for DEER applications to measure long-range distances with narrow distance distributions.527,528 The cytidine analogue 173 was also described.529 Sonogashira coupling yielding 172 and 173 can also be performed directly during the synthesis of oligonucleotides,527,530,531 similar to the attachment of 27 to 2-iodo-adenine (Figure 23).510 Nucleotide 174(532,533) is based on a very similar design and proven to be useful in DEER experiments.
As an alternative, the isoindoline based spin labels ImU, OxU, and ExlmU have been described (Figure 24).534,535 Their nitroxide group is aligned with the axis of the rotatable linker to the nucleoside, which has obvious advantages for immobilization and distance measurements by DEER.536 A 2′-methoxy derivative, ImUm, has also been described and used for distance measurements in RNA duplexes.537 Recently, the tetraethyl nitroxide version EImUm was developed, which is much more resistant to reduction and was, thus, successfully used to study RNA duplexes in oocytes.538
Hopkins and co-workers developed the nucleotide Q (Figure 24).539,540 Pairing of Q with the noncanonical base 2-aminopyridine (2AP) results in a rigid spin label that has been used to study the sequence-dependent dynamics of duplex DNAs.541,542 Sigurdsson and co-workers developed this concept further by designing the rigid spin label Ç, which does not require a noncanonical base for pairing, broadening its range of applications (Figure 24).543,544Ç represents the fusion of a cytosine with a nitroxide featured isoindole via an oxazine linkage. It is identical with the noncovalent spin label ç (Figure 15) except for the linkage to the ribose. Ç prefers to pair with guanine and does not perturb the structure of a DNA duplex, as observed in a crystal structure (Figure 25).545 A ribo-analogue with a methoxy modification in the 2′ position (Çm) has been developed for use in RNA.546 The synthetic incorporation of Ç and Çm into DNA and RNA was recently improved to circumvent partial reduction during oligonucleotide synthesis.547 Both labels deliver accurate distances in DNA and RNA and have been used in numerous applications.548−558 Recently, the tetraethyl nitroxide versions EÇ and EÇm have been developed and incorporated into DNA and RNA.559 They are more inert toward reduction by ascorbate and thus potentially allow in-cell EPR measurements, as recently demonstrated for EImUm.538
Figure 25.
X-ray crystal structure (PDB code 3OT0)545 of a 10 base pair DNA duplex containing the Ç spin label (highlighted in pink). Two uridine residues are 2′-methylated. The phosphate backbone is shown as a ribbon. The distance between both nitroxide radicals (18 Å) is indicated. Hydrogen bonds between guanine and Ç are indicated by dashed lines.
Commercially available EDTA-derivatized deoxythymidine (EDTA-C2-dT) can be incorporated into DNA during oligonucleotide synthesis.560 Loaded with a Fe(II) or Mn(II) ion, it has been used to study protein DNA interactions by NMR measurements of PREs.560,561 The long and flexible linker and the occurrence of enantiomeric forms of metal–EDTA complexes challenge applications that require the measurement of PCSs. The design of a wider range of oligonucleotide tags that can be used to measure PCSs is still a little explored area of research.
Saxena and co-workers incorporated the Cu(II)-binding and commercially available 2,2′-dipicolylamine (dPA) phosphoramidite in lieu of a nucleotide into DNA (Figure 24).562−564 Its use in DNA duplexes requires an abasic site in the complementary strand and allows distance measurements by EPR spectroscopy. A related spin label (L) was used to label DNA quadruplexes with Cu(II) forming a Cu(pyridine)4 complex (Figure 24).565,566 While initial studies used the racemic form of the tag, later studies employed the (S) isomer566 for distance measurements. A DNA quadruplex can also be labeled with a large dinuclear platinum(II) complex linked to TEMPO.567
3.5. Paramagnetic Probes for Oligosaccharides
Carbohydrates present an additional class of biological macromolecules that have been studied with various paramagnetic tags. To date, the variety of tags and attachment chemistries has been more limited than for proteins and oligonucleotides.38,39 Most strategies capitalize on the possibility of selective amination at the reducing terminus using ammonium formate and subsequent formation of an amide bond between tag and oligosaccharide amine (Figure 26). Following this approach, TEMPO has been installed at the reducing end of N-acetyllactosamine (Figure 26A, 175) to study its interaction with proteins by NMR spectroscopy.568,569 Subsequently, a more complex high-mannose-type oligosaccharide was studied with the same approach.570
Figure 26.
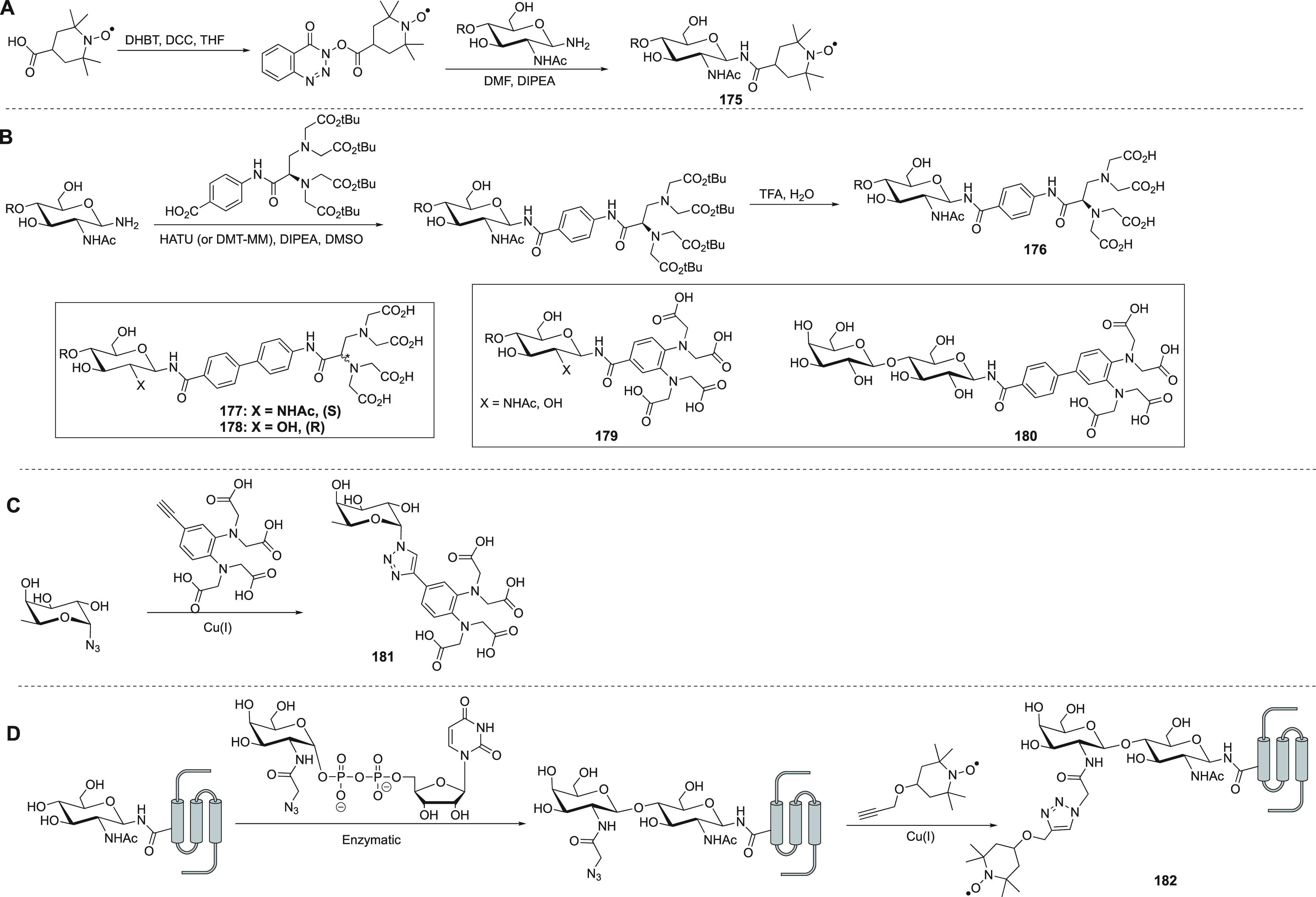
Carbohydrate probes and their synthesis.
An EDTA-based lanthanide tag was first attached to the aminated reducing terminus of N,N′-diacetylchitobiose (Figure 26B, 176) to enable paramagnetic NMR measurements with an excellent correlation between experimental and predicted PCSs.571 Subsequently, this tagging strategy was also applied to high-mannose-type oligosaccharides.572,573 Biphenyl variants of this tag in (S) (Figure 26B, 177)574 and (R) (Figure 26B, 178)575 configurations were also described.
Kato and co-workers developed a phenylenediamine based lanthanide chelating tag that can be coupled to the aminated reducing terminus of oligosaccharides using standard amide bond formation (i.e., HATU (2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate), DIPEA (N,N-diisopropylethylamine), and DMF (N,N-dimethylformamide)) yielding 179 (Figure 26B).576 This tag has since been used in various applications to study carbohydrates and their interactions with proteins by paramagnetic NMR spectroscopy.577−580 A biphenyl variant attached to lactose (Figure 26B, 180) has also been used to study lactose conformations and its interactions with a receptor protein.581
Another report described the use of an l-fucose azide and an alkyne based tag for direct “click” reaction to furnish compound 181 (Figure 26C).582 It provided an elegant way to obtain a model for the binding site of the fucose on a 70 kDa receptor protein by the use of intermolecular PREs and PCSs to position the lanthanoid ion on the carbohydrate with respect to the protein. In a partially enzymatic and partially synthetic approach, an azide bearing saccharide was selectively introduced to a glycosylation site in a protein and subsequently reacted with an alkyne based label to yield the spin-labeled glycoprotein 182 (Figure 26D).583
4. Complications in PRE-to-Distance Conversion
The PRE is very strong for a nucleus I close to a paramagnetic center S and drops off rapidly with increasing distance rIS because of its rIS–6 dependence (eqs 9–13). Therefore, PREs are powerful tools for the detection of minor states or the characterization of ensembles of conformations, such as found in intrinsically disordered proteins or encounter complexes.33,34,584 At the same time, the strong distance dependence of the PRE adds uncertainties to distance measurements of major conformations, when minor conformations or alternative interactions featuring short distances rIS contribute disproportionally to the observed PRE. Minor species populated as low as 0.1% can be manifested in the observed PRE in this way.
Most obviously, problems arise when proteins are prone to aggregation, but the potential for intermolecular associations may also be introduced by the chemical tag. In principle, intermolecular effects can be detected by measuring the PREs at different concentrations of the tagged molecule,365 but accurate measurements of PREs at low concentration may be very time-consuming for biological macromolecules. Intermolecular PREs can be suppressed by dilution of the paramagnetically labeled molecule with the diamagnetic reference, but as the chemical shifts are conserved in the presence of paramagnetic centers with slow electronic relaxation times, the superimposition of NMR signals from diamagnetic and paramagnetic molecules makes it difficult to disentangle the intra- and intermolecular PREs.
A second complication arises from incomplete paramagnetic tagging. Tags with limited metal binding affinity are easily under- or overtitrated with metal ions and nitroxide tags may be subject to partial reduction or oxidation to diamagnetic species, leading to mixtures of paramagnetic and diamagnetic species even following quantitative chemical ligation. These complications may explain, why the correlation of PREs with distance has been disappointing in many published examples, despite the steep distance dependence of the PREs, compromising the accurate conversion of PREs into distances.130,365,434,459,461,585−587
Finally, the PRE-to-distance conversion is compromised by any flexibility of the paramagnetic tag as well as uncertainties in electronic relaxation rates. In the case of tags containing Gd(III), Mn(II), or Cu(II) ions as the paramagnetic center, the PRE is entirely or predominantly driven by the SBM mechanism, which depends on the effective correlation time of the vector connecting the nuclear spin with the paramagnetic center. In addition, the electronic relaxation rate depends on the ligand-field of the metal complex. In practice, conversion of the PREs into distances, thus, requires calibration against known distances to atoms in the target molecule.249,588 In the case of nitroxide tags, the electronic relaxation time is very long (of the order of 100 ns),589 but the generally hydrophobic nature of these tags makes the assumption of absence of intra- and intermolecular associations more tenuous.
The problem of incomplete paramagnetic tagging and uncontrolled intermolecular PREs can be solved by using metal tags with anisotropic magnetic susceptibility tensor. In this case, the NMR signals of paramagnetic and diamagnetic species are separated by PCSs. Furthermore, intermolecular PREs can readily be accounted for by mixing the paramagnetically tagged molecule with the diamagnetic reference. In the mixture, the relaxation rates of diamagnetic and paramagnetic species are equally affected by intermolecular PREs, so that the intermolecular effects drop out when calculating the difference in relaxation rates between paramagnetic and diamagnetic species. The concept was explored and demonstrated with calbindin D9k containing Er(III) or other paramagnetic lanthanoid ions as the paramagnetic center and Lu(III) as the diamagnetic reference.124 The results showed that PREs extracted from longitudinal relaxation rates were less sensitive to intermolecular effects than transverse PREs but that more accurate distance restraints were obtained by measuring transverse relaxation rates as measurements of R1(1H) relaxation rates are affected by multiexponential relaxation due to cross-relaxation. Specifically, transverse PREs measured with the Er(III) sample delivered reliable distances in the range between 12 and 25 Å with an RMSD below 1 Å, if RDCs in the paramagnetic state were controlled by using short relaxation delays and a magnetic field strength not above 14.1 T. In the case of flexible tags, however, distance variations and difficulties to determine the effective correlation time remain compromising factors in quantitative PRE-to-distance conversions.
5. Examples of Applications of Paramagnetic NMR in Protein Studies
Paramagnetic effects provide structural information, which can be used further to assess the dynamics of biological macromolecules, such as domain reorientation or the exchange with minor conformational species. The following section serves to give examples that illustrate the range of applications of paramagnetic probes, including protein resonance assignment, 3D structure determination, studies of protein–protein and protein–ligand complexes, and the identification of lowly populated states. Section 6 adds further examples for an overview of the breadth of applications for the different types of probes.
5.1. Protein Structure Studies
5.1.1. NMR Resonance Assignments in the Paramagnetic and Diamagnetic States
Sequence-specific resonance assignments are the starting point for any protein structural study by NMR spectroscopy. If the protein is not too large and amenable to uniform labeling with 15N, 13C, and 2H, the resonance assignment generally is easiest by heteronuclear multidimensional (3D/4D) NMR experiments. For large proteins, complete resonance assignments become challenging because of increasing line width and spectral overlap. PCSs offer a way to increase the spectral dispersion and thus resolve spectral overlap. This is particularly useful for correlation spectra of methyl groups, which otherwise show limited spectral resolution.358
If the 3D structure of the target protein is known in advance, PCSs generated by a site-specifically attached probe can be used to assist the resonance assignments.590 Fundamentally, measuring PCSs requires attributing the chemical shift observed for a nucleus in the paramagnetic state to its chemical shift in the diamagnetic state. Different methods have been devised to achieve this.591 Most commonly, 2D correlation spectra, such as heteronuclear single-quantum correlation (HSQC) spectra, are used, where the cross-peaks of paramagnetic and diamagnetic samples are displaced along approximately parallel lines. This observation holds true in particular for paramagnetic metal ions featuring large Δχ tensors as, in this case, PREs broaden the signals of nuclear spins near the metal ion beyond detection and PCSs can be observed only for nuclei beyond a minimal distance from the paramagnetic center. As the distance between two adjacent nuclear spins is much shorter than the distance from the paramagnetic center, very similar PCSs are observed for both spins involved in a cross-peak in 2D correlation spectra or spectra of higher dimensionality, leading to the cross-peak displacements along approximately parallel lines. This greatly facilitates assigning the paramagnetic peaks to their parents in the spectrum recorded of the diamagnetic reference and the attribution can further be underpinned by using two paramagnetic samples produced with the same probe but different paramagnetic metal ions, which generate PCSs of different magnitude. In this case, all three peaks (two paramagnetic, one diamagnetic) produced between a pair of nuclear spins can be expected to lie on the same line. It is further helpful that, for many probes, the Δχ tensors of Tb(III) and Tm(III) ions tend to shift the cross-peaks in opposite directions relative to the diamagnetic reference (Table 2).290,158,258,265 Because the PCSs differ for different protons in a molecule, spectral overlap makes it very difficult to measure proton PCSs by 1D NMR spectroscopy.
In an unbiased way, the assignment of paramagnetic peaks to their diamagnetic parent can be achieved by a spectrum that generates cross-peaks between both. This requires a sample simultaneously containing both the paramagnetic and diamagnetic species, where the probes installed bind the metal ions in a way that allows chemical exchange between paramagnetic and diamagnetic metal ions in the slow-exchange regime (exchange rate between about 1 and 100 s–1). These stringent criteria have been fulfilled in some examples, such as probes 125(327) and 126,328 but are difficult to design in advance.330,592,593 Notably, the situation of fast chemical exchange also provides a straightforward way of measuring PCSs as, in this case, they increase continuously with increasing concentration of the paramagnetic metal probe.365,591 Lesser binding affinity, however, comes with the risk of lesser binding specificity, which can make it difficult to interpret the PCSs.
Having measured a sufficient number of PCSs to fit a Δχ tensor to the 3D structure of the biological macromolecule, the Δχ tensor can be used to predict further PCSs to assist with the resonance assignment. When using a double-armed probe like CLaNP-5 (Figure 9, 79) that is relatively rigid relative to the protein, it is also possible to predict an initial Δχ tensor with good accuracy.127 As nuclei located at different positions relative to the paramagnetic center can have the same PCS, the distinction needs to be made by additional data, such as PREs, RDCs, and CCR effects,57 or by PCSs generated from more paramagnetic centers installed at different sites of the protein. The software packages PLATYPUS,57 Echidna,594 Possum,595 and PARAssign596 were developed specifically for resonance assignment using paramagnetic restraints.
An instructive example is the assignment of the diamagnetic resonances of methyl groups of the N-terminal domain of Hsp90 with CLaNP-5 tags (Figure 9, 79) installed at three different sites. The protein contains 76 methyl groups in leucine, isoleucine, and valine residues, and over 60% of them could be assigned based on the protein structure and 1H PCSs measured in 13C-HSQC spectra with Yb(III) as the paramagnetic metal ion, using PARAssign without the use of any prior assignments from the diamagnetic sample.597 For the remaining methyl groups the choice of possible assignments was greatly reduced. As the Δχ tensor orientations of the S50C/D54C and T149C/I187C mutants happened to be almost parallel, the information content was greatly reduced and the assignment was predominantly based on two Δχ tensors only. If the Δχ tensor has been determined independently, for example, from 15N-HSQC spectra of backbone amides, PCSs from a single site with a single paramagnetic metal ion can be sufficient to assign 75% of the methyl resonances in the diamagnetic state using the program Possum, as shown for the 30 kDa complex between the N-terminal exonuclease domain ε186 and the subunit θ of the E. coli DNA polymerase III (ε186/θ).595 By starting from a X-ray crystal structure, the work also highlighted the fundamental problem of this approach, which arises when subtle differences exist between crystal and solution structures.
5.1.2. Protein Structure Determination
PCSs arguably deliver the most useful paramagnetic structure restraints for 3D structure determinations of proteins, because they are readily measured with high accuracy. In addition, RDCs obtained by paramagnetic alignment convey useful information about relative domain orientations. Paramagnetically generated molecular alignment and measurement of RDCs also is of interest for studies of protein dynamics,598,599 which rely on multiple different alignment tensors, as it can be challenging to obtain a larger number of significantly different alignments with the help of conventional alignment media, which act mostly by steric or electrostatic effects.600 Software packages like CYANA,601 Xplor-NIH,70,602 and Rosetta603−607 have been extended with modules for protein structure computation with paramagnetic restraints.
In the 1990s, Bertini and co-workers refined 3D structures of proteins with the help of paramagnetic data, demonstrating that the structural restraints gained by paramagnetism can outweigh the loss of NMR signals from 1H spins in the vicinity of the paramagnetic center.112,139 More recently, the benefit of paramagnetic data for 3D structure determinations was highlighted by Nietlispach and co-workers, who applied PCS restraints to refine the structure of the phototactic receptor sensory rhodopsin II (pSRII), a membrane protein with over 240 residues from the family of seven transmembrane helix proteins.608 In previous work, the authors had determined the structure of pSRII in a micelle using NOEs.609 By labeling pSRII at four different positions with the paramagnetic probe C2 (Figure 9, 96) loaded with different paramagnetic metal ions, the global fold of the protein could be derived by the exclusive use of PCS restraints. By combining the PCS and NOE data, the structure was refined to a backbone RMSD of 1.5 Å to the mean.608
Besides delivering unique long-range structural restraints, the chemical shift measurements needed to measure PCSs can be performed very quickly. This has recently been exploited to elucidate the structure of the transient enzyme intermediate formed by SrtA with its substrate peptide.266 The enzyme–substrate complex is an unstable intermediate that contains a labile thioester bond with the active-site cysteine residue (Cys184) and presents only a minor species in solution. Nonetheless, comparison with the chemical shifts of a stable disulfide-bond-linked analogue enabled the resonance assignment of the thioester intermediate and attachment of the probes 76 and 77 (Figure 7) loaded with one of four lanthanoids, Dy(III), Tb(III), Tm(III), and Lu(III) to SrtA delivered PCSs. Using the PCSs as input for Xplor-NIH, the structure of the thioester intermediate was calculated as an ensemble of conformers with a RMSD of 1.1 Å.266
The sensitivity with which PCSs can be measured is also of great advantage for measurements at low concentrations, such as encountered in in-cell studies of protein structure. Using GB1 as a model protein, Müntener et al. attached the M7Py-DOTA tag (Figure 9, 88) loaded with one of three lanthanoids, Tb(III), Tm(III), and Lu(III), and recorded PCSs and RDCs at about 50 μM protein concentration after transferring the labeled protein into Xenopus laevis oocytes.286 The paramagnetic restraints measured by in-cell NMR assisted the 3D structure determination using GPS-Rosetta.606 The same protein at the same low concentration was targeted at the same time by Pan et al., using PCSs measured in X. laevis oocytes.610 To measure PCSs, this group attached 4PS-PyMTA (Figure 7, 76) loaded with Tb(III), Tm(III), Yb(III), or Y(III) ions and calculated the structure without RDCs using GPS-Rosetta. The authors were careful to point out, however, that Rosetta611 is one of the most powerful software packages for 3D structure predictions of small proteins from the amino acid sequence alone.610 Specifically, they showed that the fold of GB1 is already correctly predicted by CS-Rosetta,603 when backbone chemical shifts are provided as the only experimental data.610
The conversion of PCS data into structure restraints requires knowledge of the Δχ tensor, which is best obtained by fitting to 3D structure coordinates. For proteins of unknown structure, the powerful model building algorithm provided by Rosetta thus is an excellent starting point where models that do not allow good Δχ tensor fits can be discarded early in the structure calculation.603 In this way, PCS data not only improve the quality of the final structures but also accelerate the convergence of the calculations. The value of PCS restraints for protein fold determination, even if backbone chemical shifts and PCSs from backbone amide protons are the only experimental data provided, is very clear for larger proteins.604,605,607,612
Because of their long-range nature, PCSs are particularly suited to determine full structures in situations, where the structure of a rigid protein domain is known while the conformation or orientation of another part is not. In this situation, even sparse PCS data have been shown to be sufficient to fit Δχ tensors to the rigid part and use the PCSs of the unknown part to determine its structure or relative orientation.147,291,612−619
It is instructive to compare the PCS structure restraints with the way, in which the global positioning system (GPS) identifies the location of objects on Earth. Just as the measurement of the distances to several satellites identifies a single location on the surface of Earth, the PCSs of a nuclear spin can be measured for different protein samples prepared with tags positioned at different sites. Each PCS defines an isosurface and the intersection of four PCS isosurfaces defines a single point in space.606,620 The concept was demonstrated with the 3D structure determination of the C-terminal domain of ERp29 tagged at four different sites either with IDA-SH (Figure 11, 126) or C1 tags (Figure 9, 96), using Tm(III) and Tb(III) as the paramagnetic ions.606 The same concept was applied to determine the structure of this protein with the help of PCSs measured with Co(II) ions bound to double-histidine motives in four different helices, one at a time.592 Notably, PCSs of the backbone amide protons alone were sufficient to obtain the correct protein fold. While PCSs from an increasing number of tagging sites increases the level of structural information available, structural details on the conformations of amino acid side chains can already be obtained with two different tagging sites, as the side chain locations are restricted by their link to the backbone.94,621
5.1.3. Protein–Protein Interactions
With a paramagnetic probe on one of the interacting proteins in a protein–protein complex, the paramagnetic effects in the partner protein can be recorded to determine the relative position and orientation of the two proteins in a rigid body docking approach, which assumes that the structures of the individual binding partners are conserved in the complex (Figure 27). The first example of this approach, from 1998, used the paramagnetic heme in cytochrome f to induce PCS in the electron transfer partner plastocyanin and model the structure of the complex.622−624 In an early example using lanthanoid ions, the structure of the ε186/θ complex was modeled with the help of PCSs measured with Dy(III), Ho(III), or Er(III) data in the natural metal binding site of ε186, using a rigid body docking approach, although the PCSs were also used to refine the structure of θ in its bound state.69,625
Figure 27.
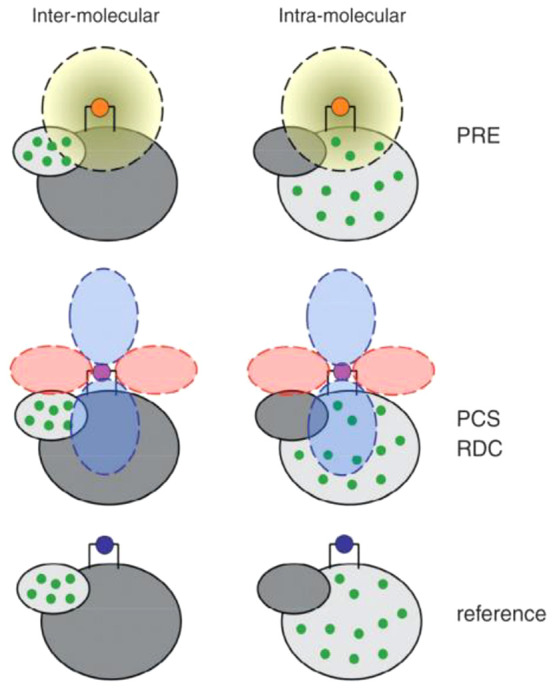
Strategy for using PREs, PCSs, and RDCs for structure determination of protein–protein complexes using differently labeled samples. PREs are measured using a tag with slow electronic relaxation (e.g., Gd(III)) or a paramagnetic center with little χ tensor anisotropy (e.g., Er(III)). PCSs and RDCs are obtained using the tag with an anisotropic paramagnetic center (e.g., Tm(III)). The data are measured with respect to a diamagnetic reference produced with the tag containing, for example, a Lu(III) ion. To simplify the NMR spectra and distinguish between intermolecular and intramolecular effects, the complex can be prepared with isotope labeling (symbolized by green dots) of one of the proteins only. The intramolecular restraints are used to fit the position of the paramagnetic center and orient the Δχ tensor. 2D NMR spectra (typically [15N,1H]-HSQC spectra) can be sufficient to obtain a complete set of inter- and intramolecular PREs, RDCs, and PCSs, which allow determining the relative position and orientation of the two proteins in a rigid body docking approach. Reproduced with permission from ref (30). Copyright 2014 Elsevier.
PCSs were also successfully used to model the structure of a homodimer. In this case, two mutants of p62 PB1, named DR and KE, were produced to prevent oligomerization beyond dimer formation.159 A two-point anchored LBP was incorporated at the N-terminus of DR to coordinate with paramagnetic lanthanoid ions. The lanthanoid position relative to the DR mutant was determined by fitting the Δχ tensors and PCSs were measured in the KE mutant. Using the measured PCS and chemical shift changes observed at the interface of the KE mutant, rigid body minimization protocol in Xplor-NIH70,602 delivered the structure of the dimer. It was noted, however, that multiple solutions were possible even with the availability of PCS data sets from different lanthanoid ions as the Δχ tensor axes tended to be similar. The degeneracy could be resolved by modifying the LBP.626
Protein–protein interactions (PPI) are based on noncovalent forces, such as charge–charge attractions, hydrophobic interactions, and hydrogen bonds, and paramagnetic effects are excellent tools to investigate very weak protein–protein interactions such as observed in encounter complexes. As the protein partners approach each other in solution, the formation of an encounter complex precedes the formation of the specific protein–protein complex. These weak and dynamic complexes are essential for rapid complex formation, yet difficult to characterize by traditional techniques.627,628 PREs are particularly suited for investigating such weak PPIs, as they are insensitive to the relative orientation of the proteins and sensitive to minor states in which a nucleus is closer to a paramagnetic center than in the major state, which usually is the final specific complex. Complementary information can be obtained by PCSs and RDCs, provided that the orientation of the Δχ tensor changes only over a limited angular range, when the proteins populate an ensemble of complex structures. Averaging over a greater range is evidenced by averaging of the PCSs and RDCs to zero.
The electron-transfer complex of yeast iso-1-cytochrome c (Cc) and yeast cytochrome c peroxidase (CcP) is an example of a weak PPI comprehensively characterized by paramagnetic NMR. Both proteins contain a paramagnetic heme group, but the intermolecular PCSs are too small to be suitable for structural studies. Following the attachment of a paramagnetic tag (MTSL, Figure 3) at different locations on the CcP surface, PREs were recorded for Cc and utilized to dock Cc on CcP. The resulting model of the complex was similar to the crystal structure. In this case, the encounter complex represented no less than 30% of the complex and its interface was preferentially located near that of the final specific complex.65,91 CcP variants with added negative patches were used to study the role of charge distribution and PREs showed that cytochrome c visits such new patches in the encounter complex.83
In another example, the complex between cytochrome P450cam (cytP450cam) and putidaredoxin (Pdx) was investigated using CLaNP-7 (Figure 9, 80) loaded with Tm(III) or Gd(III) to obtain intermolecular PCSs, RDCs, and PREs.629 The position of putidaredoxin on cytP450cam found with these restraints was in good agreement with crystal structures obtained independently.629,630 The study highlighted the value in combining different types of paramagnetic information to obtain a good structure. In addition, the NMR data provided evidence for the presence of a lowly populated encounter complex. Using additional paramagnetic restraints and the maximum occurrence of regions methodology,631 it could be shown that in this encounter complex Pdx visits a large area of the surface of cytP450cam.
With increasing molecular weight of the system, it becomes increasingly difficult to obtain the specific NMR resonance assignments required to fit Δχ tensors. In this situation, site-specific incorporation of a probe that can easily be observed in the NMR spectrum can be combined with a paramagnetic probe to monitor the PRE on the NMR probe and, thus, derive distance restraints. The concept has recently been demonstrated by Abdelkader et al., who used genetic encoding for site-specific incorporation of an unnatural amino acid carrying a trimethylsilyl (TMS) group at the end of the amino acid side chain to obtain a readily observable 1H NMR probe (an intense singlet near 0 ppm).632 Using a 71 kDa homodimeric p-cyano-l-phenylalanyl-tRNA synthetase as an example, a mixture of TMS labeled protein and paramagnetically labeled protein (using either MTSL or the C1 tag loaded with Gd(III) attached to a cysteine residue) revealed a PRE across the dimer interface with Gd(III) but not with MTSL.632 Taking into account the flexibilities of the tags and TMS amino acid side chain, the data could be interpreted as a distance restraint between about 10 and 20 Å.
5.1.4. Protein–Ligand Interactions
NMR spectroscopy is uniquely suited to study protein–ligand interactions in drug discovery campaigns because NMR works in solution and can provide atomic level information about the binding site and ligand orientation. It is useful to distinguish between the scenarios where the exchange rate of the ligand is fast or slow on the time scale of the NMR experiment. Rapid and slow ligand binding and dissociation generally correlates with relatively weak and strong ligand affinities, respectively. Paramagnetic NMR opens new approaches for studies in either limit.
Fragment-based ligand screening is a method that searches for small molecules that bind to proteins weakly. The use of paramagnetic NMR for fragment-based ligand screening has been explored by Saio et al. with a double-anchored LBP.633 Binding of the ligand was reported by PREs generated by a Gd(III) ion in the LBP, where the PREs were detected as increased transverse relaxation rates of the 1H signals in 1D NMR spectra of the ligand, using T1ρ relaxation experiments. In the limit where the ligand exchanges between free and bound state faster than the transverse relaxation rate, the relaxation enhancement experienced in the bound state due to the proximity of the paramagnetic center in the target protein is transferred to the spectrum of the free ligand. In analogy to the transferred NOEs and saturation transfer, the effect is referred to as transferred PRE (tPRE). The size of a tPRE reflects the distance between the nuclear spin of the ligand in the bound state and the paramagnetic center attached to the target protein. In the same way, also transferred PCSs (tPCS) can be observed and used to assess the structure of protein–ligand complexes.633
In another example, Guan et al. demonstrated that the location and orientation of a ligand can be determined solely using tPCSs, provided that the ligand is in fast exchange between bound and free states relative to the size of the PCS (expressed in rad s–1).620 In the fast exchange limit, the experiments allow using the free ligand in excess, that is, only a small fraction of the ligand is bound and the PCSs measured are scaled by the bound fraction. Although the PCSs measured are thus relatively small, the smaller size is more than compensated by the narrow line width observed for the ligand, which is dominated by the fraction of free ligand. Placing the paramagnetic center, Ln(III)-CLaNP-5 (Figure 9, 79), at three positions, one at a time, on the target protein FKBP12 (Figure 28) enabled a PCS-based triangulation approach to capture the ligand’s site of binding and orientation relative to the protein. The structure of the complex obtained from tPCSs agreed with the restraints obtained from NOEs but was easier to obtain in the absence of extensive 1H NMR resonance assignments of the protein.
Figure 28.

Transferred PCSs identify the binding site and binding orientation of ligand molecules that are in fast exchange with excess free ligand. (A) tPCSs (the difference between the resonance positions for the paramagnetic (para) and diamagnetic (dia) samples) observed in the 1H 1D NMR spectra of excess ligand in complex with the protein, which is tagged with either paramagnetic or diamagnetic ions (one site at a time). The dashed and solid lines identify the ligand resonances in the diamagnetic and paramagnetic samples, respectively. (B) Superposition of the averaged NOE structure (in green) of the FKBP12–1 complex and the best five structures (in orange) calculated using tPCSs. The protein backbone is represented as a gray ribbon except for the residues Asp37 and Tyr82 (yellow). The average RMSD of the ligand from PCS calculations relative to the NOE calculation is 2.8 ± 0.4 Å. Reproduced with permission from ref (620). Copyright 2013. American Chemical Society.
In an early example, John et al. showed that tPCSs generated by a single lanthanide binding site can be sufficient to determine the binding site and orientation of a weakly bound ligand, if steric restrictions imposed by the protein invalidate any of the alternative structural solutions of the tPCS data set.634 In this example, the lanthanide ion was bound deep inside the metal binding pocket of the ε186/θ complex. The ligand (thymidine) bound with millimolar binding affinity and tPCSs were observable in 1D 1H and 13C NMR spectra of the ligand present in over 500-fold excess over the protein complex. The sensitivity of the approach is illustrated by the speed (<0.5 h) with which the 13C NMR spectra were recorded at natural isotopic abundance. The binding mode coincided closely with that observed in the crystal structure of the ε186–thymidine monophosphate complex.
A caveat associated with tPRE and tPCS effects is the possibility that the weakly binding ligand temporarily associates with the paramagnetic tag rather than the target molecule. False positives arising in this way cannot adequately be addressed by a control experiment probing the direct interaction between ligand and free paramagnetic tag, as unexpected interactions are likely promoted by the formation of binding pockets between tag and target molecule. The strong distance dependence of PCSs and PREs notably amplifies the effects of minor populations of direct ligand–tag interactions.
As ligand binding tends to affect the global structure of the target molecule little or not at all, changes in PCSs observed locally in the target molecule can be used to determine local structural changes arising from the interaction with a ligand, such as changes in amino acid side chains. A recent study labeled the protein FKBP12 with Ln(III)-CLaNP-5 (Figure 9, 79) at three sites, and valine and leucine residues were selectively isotope labeled with 13CH3 groups.621 Significant changes in PCSs were observed between the free and ligand-bound protein for methyl groups located near the ligand binding site, indicating displacements of the methyl groups in the 1–5 Å range.
In the slow exchange limit, it is not meaningful to use ligand in excess over the target protein, which makes it much harder to discriminate the NMR signals of the ligand against the background of protein signals, especially as the chemical shifts of the bound ligand usually differ from those in the free state. While isotope labeling of the ligand enables its selective detection by NMR spectroscopy, the chemical synthesis of isotope labeled ligands is cumbersome in practice. Chen et al. demonstrated that chemical labeling of the ligand with a tert-butyl group presents a synthetically more practical alternative.635 As the 1H NMR signal of the tert-butyl group is an intense signal, it can readily be detected by 1D 1H NMR. The specific example used was the 27 kDa complex between the dengue virus NS2B-NS3 protease, tagged at three different sites with C2 tags loaded with Tm(III), Tb(III), or Y(III), and a covalently binding peptide inhibitor labeled with a tert-butyl group.635 NOESY spectra revealed PCSs not only of the tert-butyl group but also of ligand resonances of protons near the tert-butyl group, which enabled positioning and orienting the ligand in the bound state. Although the structural information obtained for the ligand in this way appears limited, it proved decisive in identifying the correct pose obtained by independent modeling of the protein–ligand complex.
5.1.5. Structures of Minor States
Protein folding, enzyme catalysis, and other functions require conformational exchange, involving excited states with low populations and short lifetimes (microseconds to milliseconds). The structures of these lowly populated states can usually not be solved by X-ray crystallography. Relaxation dispersion measurements using the Carr–Purcell–Meiboom–Gill (CPMG) sequence and T1ρ experiments, as well as chemical exchange saturation transfer (CEST) experiments are NMR methods, which have proven uniquely suitable for the study of such high-energy states of proteins.636−638
Relaxation dispersion experiments can yield the absolute difference in chemical shifts (Δδ) of nuclear spins between the minor and major states. In combination with RDC restraints obtained in an external alignment medium, the structure of a minor state can be determined in favorable cases.639,640 It is still difficult, however, to convert the limited experimental information into a 3D structure. In this situation, PCSs of the minor state would provide powerful structural restraints. If the PCSs of the minor state differ from those of the major state, they can be extracted from a relaxation dispersion experiment, where the diamagnetic control sample delivers the chemical shift change in the diamagnetic state and the paramagnetic sample delivers the Δδ values including the PCSs.275 For this application, rigidity of the paramagnetic center is critically important to obtain accurate structural restraints. Earlier relaxation dispersion studies of cardiac troponin C substituted with lanthanoids (Ce(III) or Pr(III)) indicated that the PCSs observed most likely reflect motions of the paramagnetic center.641 To avoid this problem, both the attachment site and the probe must be rigid on the millisecond time scale. When CLaNP-5 loaded with Tm(III), Yb(III), or Lu(III) was linked to Paz and Cc, which are known to be rigid proteins, 1H CPMG relaxation dispersion experiments revealed dispersion effects likely reflecting movements of the tag rather than protein.275 When CLaNP-5 was attached to adenylate kinase, relaxation dispersion experiments similarly indicated the presence of a minor conformational state of the probe, in which the Δχ tensor is rotated by 20° relative to the major state. This work illustrated the difficulty of finding a probe without any exchange phenomena on the millisecond time scale. In particular, ring flips of the cyclen ring are hard to suppress.274
Alternatively, PCS restraints for determining the structure of a minor state can be derived from CEST experiments. For a proof of concept of PCS-CEST experiments, Ma et al. used Abp1p SH3–Ark1p, which is a protein–peptide complex with a slow off-rate of the bound peptide, as a model system.2634MMPyMTA (Figure 7, 73) loaded with Tb(III) was attached to the SH3 domain and the minor state was produced by adding only 0.03 equiv of Ark1p peptide. The 15N PCSs of the minor species determined by the CEST measurements proved to be in agreement with the 1:1 Abp1p SH3–Ark1p complex. The same paramagnetic tag was attached to the FF domain of HYPA/FBP11, which is in equilibrium with <2% of unfolded conformation. The PCS-CEST experiment reproduced these results. In particular, the experiments showed that the PCSs of the minor species were small, as expected for a less-ordered conformation.
5.2. Biomolecules with Paramagnetic Tags in the Solid State
The protein GB1 has been investigated extensively as a model protein to test the effect of cysteine ligated paramagnetic tags by MAS solid-state NMR.642 Longitudinal R1 PREs of 15N spins in GB1 were measured in the presence of SPy-EDTA (Figure 7, 60)248 loaded with Cu2+,643 and also with Mn2+.644 The PRE data proved to be good indicators of distance and were measured for 6 tagging sites and used as structural restraints for computationally solving the protein structure,645 while also accounting for tag flexibility.646 A less flexible short-armed cyclen based tag TETAC (1-(2-(pyridin-2-yldisulfanyl)ethyl)-1,4,7,10-tetraazacyclododecane) (Figure 9, 112) was also investigated with Cu2+ in GB1.388 While the relatively short T1e time of Cu2+ ions results in appreciable R1 PREs by the SBM mechanism, R2 PREs are often small, which presents the basis for increasing the sensitivity of NMR experiments by accelerating the recovery of longitudinal magnetization between scans with little increase in line width.647R1 PREs of 1H spins are challenging to measure in solids due to spin diffusion effects, but R2 PREs of 1H can offer long-range distance restraints.648R2 PREs by the SBM mechanism are largest for paramagnetic centers with slowly relaxing electrons such as the nitroxide spin label, which has also been demonstrated in GB1 by ligating cysteine residues with MTSL.649 Measurement of PCSs generated by paramagnetic metal tags is more difficult in solids than in solution, because different orientations of the metal complex relative to the protein would lead to a range of different PCSs. Nonetheless, PCSs have successfully been measured in GB1 ligated with 4MMDPA tag (Figure 11, 116) loaded with Co2+ and lanthanoids and were used to help solve the protein structure with Rosetta.389
Because of the close proximity of molecules in the solid state, intermolecular paramagnetic effects can be substantial. To minimize these, in most investigations of paramagnetic GB1 in the solid state the sample was diluted with diamagnetic protein. Alternatively, intermolecular paramagnetic effects in a purely paramagnetic sample can be accounted for by methods of NMR crystallography that consider paramagnetic effects in a periodic lattice.650
Aside from solid proteins in the microcrystalline state, paramagnetic effects have also been investigated by solid-state NMR of membrane proteins. For example, the seven-helix transmembrane protein sensory rhodopsin from Anabaena tagged at two sites with MTSL nitroxide spin labels was investigated in lipid bilayers.651,652 Intermolecular PREs arising from the nitroxide spin label revealed the oligomerization interface of the monomer. Another membrane system, lactose transport protein from Streptococcus thermophilus tagged with 3-maleimido-proxyl (Figures 5 and 14),211 was investigated in native membranes.653 Here, PREs were measured for a 13C-labeled substrate to probe the proximity of an interhelix loop to the protein active site. Finally, PREs have also been utilized to solve the structure of a precipitated protein–RNA complex, named L7Ae, by using IA-PROXYL spin label (Figures 5 and 17)213 at various tagging sites of the protein and measuring intermolecular PREs in isotope labeled RNA.654
6. Applications of Different Types of Paramagnetic Probes
Table 4 presents an overview of synthetic probes that have been successfully applied to protein studies by NMR spectroscopy. It provides an impression of the range of applications, where the various probes have been found to be useful. The table also includes some of the probes developed with the aim to obtain narrow distance distributions in DEER experiments. This section briefly summarizes the applications of the different probes listed. Examples that were already mentioned in section 5 are not discussed in detail again here.
Table 4. Applications of Synthetic Probes in Protein NMR Studies.
| probe | applications in protein NMR studies | comments |
|---|---|---|
| MTSL (Figure 3) | protein global folding and structure determination130,586,649,652,656,668,669,671 | small tag generating sizable PRE effects; easy to handle and commercially available; in NMR experiments, this tag produces PREs as the only paramagnetic effect; the nitroxide group is sensitive to reduction or oxidation; reduction of the disulfide bond formed with the target leads to loss of the tag |
| protein–protein interaction65,83,91,658 | ||
| protein–ligand interaction661−663 | ||
| protein dynamic studies664−667,670,730 | ||
| protein distance measurements by 19F-PRE587 | ||
| maleimide-TEMPO (Figure 5, 15) | protein conformation studies667 | commercially available and attached to protein via stable thioether linker |
| SPy-EDTA and analogues (Figure 7, 60–61) | protein structure determination249,250,643,644,674,675 | commercially available, but exists in multiple conformations |
| protein binding target determination676 | ||
| protein alignment251 | ||
| 4MMPyMTA and analogues (Figure 7, 73–77) | protein structure determination266 and in-cell protein structure determination610 | lanthanoid based probe for attachment to single cysteine residues; small, hydrophilic, and carrying a low net charge; forming a stable thioether linkage with cysteine; complete ligation yields can be difficult to obtain |
| protein–peptide complex263 | ||
| protein–ligand interactions678,679 | ||
| protein complex NMR signal assignment677 | ||
| CLaNP-5 (Figure 9, 79) | methyl group assignment597,621 | frequently used lanthanoid-based cyclen probe, suitable for PCS measurements; the double-armed linkage to the target results in high rigidity; it carries a net charge of 3+ and some of its pendants are fairly hydrophobic; the disulfide linkage between probe and target is prone to reduction |
| protein–ligand interaction studies76,620,617,641 | ||
| protein structure determination616,682,731 | ||
| protein dynamic studies274,275 | ||
| protein–protein complex studies680 | ||
| protein–peptide complex studies681 | ||
| CLaNP-7 (Figure 9, 80) | protein complex studies629,631,685−687 | similar to CLaNP-5, but less charged and with yellow color |
| protein structure determination629 | ||
| protein conformation studies685 | ||
| DOTA-M8 (Figure 9, 86) | intrinsically disordered proteins; increasing NMR spectral dispersion694 | binds Ln(III) tightly, produces largeΔχ tensors, and has zero net charge; disulfide bond after attachment to the protein may be prone to reduction; two conformations were reported. |
| protein domain studies695,696 | ||
| protein alignment697 | ||
| protein–ligand interaction698 | ||
| RNA structure analysis699 | ||
| protein structure refinement701 | ||
| M8-CAM-I (Figure 9, 87) | in-cell protein structure determination285 | shares the core structure with DOTA-M8 but a propyliodoacetamide attachment replaces the pyridylthio linker to form a more stable C–S linkage; a long tether to the protein backbone reduces its Δχ tensor magnitudes |
| M7Py-DOTA (Figure 9, 88) | in-cell protein structures from 2D NMR experiments286 | an analogue of DOTA-M8, with a 4-(phenylsulfonyl)pyridine moiety yielding attachment with a short and rigid linker but the attachment reactivity is low |
| M7PyThiazol-DOTA (Figure 9, 92) | homotrimeric protein structure investigation700 | a newer version of M7Py-DOTA; reacts with cysteine with higher selectivity and efficiency; generates a Δχ tensor even larger than DOTA-M8 |
| protein–ligand interaction698 | ||
| protein structure refinement701 | ||
| C1/C2 (Figure 9, 96) | methyl group assignment689 | lanthanoid based cyclen probe for attachment to single cysteine residues; suitable for PCS measurements but with high net charge and bulky hydrophobic pendants; forming a disulfide linkage between probe and target; C1 and C2 are enantiomers |
| protein structure determination94,604,606,612 | ||
| protein–protein interactions and protein–ligand interactions291,618,619,690 | ||
| DNA–protein domain binding studies693 | ||
| C3 (Figure 9, 98) | protein structure studies702 | similar to the C1 and C2 tags but for attachment to p-azido-l-phenylalanine by Cu(I)-catalyzed click chemistry, which is orthogonal to cysteine chemistry |
| TEMPOL (Figure 12) | protein accessible surface characterizations345,346,355,711−714,732,733 | commercially available and frequently used but somewhat hydrophobic; unstable under reducing conditions |
| BPTI aggregation715 | ||
| protein folding347 | ||
| protein dynamics studies664−667,670,730 | ||
| protein distance measurements by19F-PRE587 | ||
| Gd(III)DTPA-BMA (Figure 13) | protein surface accessible regions characterizations350,716,734 | high solubility, stability, and low protein binding affinity; zero net charge |
| BPTI aggregation715 | ||
| protein structure determination352,721 | ||
| protein complexes studies719,723 | ||
| protein domain studies722 | ||
| spectral editing726 | ||
| Gd(III)TTHA-TMA (Figure 13) | protein dynamic determinations353 | zero net charge |
| [Ln(III)(DOTP)]5– (Figure 13) | protein–protein interaction718 | high negative charge; can generate both PCSs (loaded with Tb(III)) and PREs (loaded with Gd(III)) in some cases |
| [Ln(III)(DPA)3]3– (Figure 13) | multisite protein–ligand binding interactions724 | can generate both PCSs (loaded with Tb(III)) and PREs (loaded with Gd(III)) in some cases |
| cationic peptides structure determination725 | ||
| [Ln(III)(DTPA)]2– (Figure 13) | spectral editing358 | negatively charged |
| Gd(III)(DO3A) (Figure 13) | identification of residues close to carboxylates366 | commercially available and zero net charged |
| [Ln(III)(EDTA)]1– (Figure 13) | protein–protein interface mapping357 | commercially available and negatively charged |
| spectral editing358 |
6.1. Applications of Nitroxide Probes
Commercially available MTSL (Figure 3) has been used successfully in a wide range of protein NMR studies, making it impossible to compile all usage in a comprehensive way. Methods have been devised to speed up accurate 1H PRE measurements655 and been applied to demonstrate that MTSL derivatized with a bulky group sufficiently limits the internal motions of the tag to enable the PRE data analysis with the assumption of a single nitroxide position relative to the protein.219 The potential of PREs generated by MTSL for global fold determination of proteins was assessed by Battiste and Wagner, who showed their value for determining the fold of eukaryotic translation initiation factor 4E (eIF4E) in combination with HN–HN NOEs and chemical shift data.130 Choosing Bacillus amyloliquefaciens barnase as the model protein, Gaponenko et al. also provided an early demonstration of the value of MTSL-generated PREs in improving the accuracy and ease of global folding determination.656 The long-range nature of PREs (generated by MTSL) was shown to be particularly valuable for determining the structure of a loop-rich protein, SAPK-interacting protein 1 (Sin1) in combination with limited NOE data.657
Examples for the use of MTSL in NMR studies of protein–protein complexes have been mentioned in section 5.65,91,658 In a recent example, Olivieri et al. used a mixture of 15N- and MTSL-labeled ubiquitin with 15N/13C-labeled ubiquitin to distinguish intra- and intermolecular PREs in the transient diubiquitin complex in a single sample.659MTSL has also been used more generally in NMR studies of complexes between proteins and smaller ligands. Gochin and co-workers employed paramagnetic NMR in a second-site screening campaign660 to identify ligands binding in the hydrophobic pocket of HIV-1 gp41.661 By labeling a short peptide, which was known to bind at the edge of the pocket, with MTSL or cysteaminyl-EDTA loaded with either Fe(II) or Co(II), the authors measured the paramagnetic effects on the low-molecular weight ligand as it was bound to the protein, thus detecting its binding site.661 The MTSL labeled peptide alone provided sufficient information as the same group, in a subsequent study, used only MTSL labeled peptide to determine the binding site of a different indole ligand binding to HIV-1 gp41.662 PREs measured with Rho guanine-nucleotide dissociation inhibitor 2 (RhoGDI2) labeled with MTSL provided Ruan and co-workers with key structural information to characterize the binding mode of three hits identified from a fragment library of 890 compounds.663
The strong distance dependence of PREs generated by MTSL has been successfully exploited in studies of protein dynamics, including studies of the interdomain movements of cardiac troponin C664 and calmodulin.665 Using a mixture of MTSL labeled and isotope labeled protein, Yang et al. monitored the slow exchange of monomeric units in the homodimer Dsy0195 from Desulfitobacterium hafniense Y51 by paramagnetic NMR and DEER experiments.666 Investigation of the open-to-closed transition of maltose-binding protein with PREs generated by the probe maleimide-TEMPO (Figure 5, 15) revealed that the apo state of the protein populates a major open form and a minor closed species.667
MTSL has also been utilized in NMR studies of proteins in the solid state,649,652,668,669 where structural restraints from PREs can amount to a sizable fraction of the data available for structure calculations. Especially in the case of membrane proteins, MTSL has proven useful to assess their dynamics,670 structure,586,671 and function.672
Using the MTSL tagged and cyanobacterial lectin CV–N-containing fluorinated amino acids as an example, Matei and Gronenborn demonstrated that the high resolution of 19F NMR spectra allows facile measurement of the distances between 19F spins and the paramagnetic center by 19F PREs, which proved to be in a good agreement with the results from 1H PRE measurements.587 Following labeling of a Myc peptide with a CF3 compound, 19F PREs were also shown to provide a convenient way of assessing proximity to MTSL labeled Max peptide, both in the disordered state and the structurally defined complex with DNA.673
6.2. Applications of Aminopoly(Carboxylic Acid)-Based Probes
Dvoretsky et al.674 and Vadim et al.250 showed that SPy-EDTA (Figure 7, 60) complexed with different paramagnetic metal ions enables structural studies of proteins by paramagnetic NMR. Because of the different enantiomeric forms of EDTA–metal complexes, the tag is suited for PRE measurements but is less so for PCS measurements, as demonstrated in a structure refinement of the protein domain ArgN using the SPy-EDTA-Cu(II) complex.249 Nonetheless, SPy-EDTA loaded with different lanthanoid ions proved successful in aligning a helical transmembrane protein for RDC measurements.675 As already discussed in section 5.2, SPy-EDTA loaded with Mn(II)644 and Cu(II)643 was also successfully applied to probe the structure of GB1 by solid-state NMR. Using solid-state NMR, Wang et al. identified glucuronoarabinoxylan, which is the major matrix polysaccharide in maize cell walls, as the binding target of β-expansins, by tagging the protein with Mn(II)-SPy-EDTA and measuring 1H and 13C PREs in 13C enriched maize cell walls.676
4MMPyMTA (Figure 7, 73), 4PS-PyMTA (Figure 7, 76), and 4PS-BrPyMTA (Figure 7, 77) are three other frequently used probes employed in protein NMR research. 4PS-PyMTA and 4PS-BrPyMTA were used for elucidating the 3D structure of a protein in cells610 and in pure protein solution (section 5.1.2).266 Su and co-workers labeled SrtA with 4PS-BrPyMTA to assign NMR signals of an unstable intermediate by using PCSs and residue-selective isotope labeling.6774MMPyMTA was used to detect the excited state of a protein–peptide complex by the new PCS-CEST method (section 5.1.5),263 and this approach was recently extended to 19F NMR, referred to as 19F PCS-CEST, to gain PCS restraints for protein–ligand interactions in the intermediate exchange regime.678 In a similar vein, 1H PCSs of a protein–ligand complex presenting a minor species in solution were extracted from relaxation dispersion experiments (1H PCS-RD experiments) to gain insight into the binding mode of a ligand to a bromodomain labeled with a 4MMPyMTA–Tm(III) tag.679 The approach elegantly circumvents excessive line broadening in the intermediate exchange regime by detecting the PCSs of the minor species in the NMR signals of the ligand added in large excess.679
6.3. Applications of Cyclen-Based Probes
Section 5 gave some examples of applications of cyclen based probes in protein studies. Further examples are summarized below.
6.3.1. Double-Anchored Probes
The encounter complex between Cc and bovine adrenodoxin (Adx) was characterized by measuring PCSs and RDCs generated by the double-arm probe Yb(III)-CLaNP-5 (Figure 9, 79) on Cc.76 Loaded with different lanthanoid ions, CLaNP-5 also delivered highly informative PCSs and PREs to model the structure of the little soluble 65 kDa complex between membrane associated Adx and adrenodoxin reductase (AdR).680
Saio et al. used Yb(III)-CLaNP-5 to explore ligand-driven conformation changes of the multidomain protein MurD, where the PCSs identified a novel semiclosed conformation of the protein.617 Venkata et al. used PREs generated by Gd(III)-CLaNP-5 and MTSL to provide compelling evidence for two fundamentally different orientations of polyproline helices when bound to the Src SH3 domain, which are populated to different degrees.681
The CLaNP-5 tag has also proven useful to investigate conformational changes of proteins. In a maximum occurrence (MO) analysis of the different relative domain orientations of CaM, PCS, and PRE data generated by Ln(III)-CLaNP-5 tags attached to either domain of CaM significantly improved the definition of the conformational space populated by the protein.682 This tag was also used to study domain orientation in matrix metalloproteinase-1.616 More recently, Yb(III)-CLaNP-5 was used to investigate the conformational states of the single-domain GH11 glycosidase from Bacillus circulans (BCX)), which proved to be in full agreement with crystal structures683 and only locally affected by site-directed mutagenesis.684
The double-anchored probe CLaNP-7 (Figure 9, 80) proved similarly useful for the determination of the structure and encounter state of the complex between cytP450cam and putidaredoxin (Pdx), as discussed in section 5.1.3.629,631 Paramagnetic data recorded with the Yb(III)-CLaNP-7 tag further demonstrated that the structure of the cytP450cam–Pdx complex predominantly populates a closed conformation in solution in contradiction to the open state reported by other crystal structures.685CLaNP-7 also proved useful to generate paramagnetic structure restraints of the heat-shock protein 90 (Hsp90) in the presence of proteins bound to the chaperone.686,687
Except for a recent study on domain motions in diubiquitin,614 the use of other double-arm probes, such as T1/T2 (Figure 9, 82)688 and CLaNP-13 (Figure 9, 85)281 has only been explored for DEER distance measurements between two Gd(III) ions, with the aim to obtain narrow distance distributions by restricting the flexibility of the tags. In either case, the widths of the distance distributions obtained was not narrower than those obtained with some of the singly linked probes.
6.3.2. Applications of Single-Arm Cyclen Probes
Among the single-arm cyclen based probes, DOTA-M8 (Figure 9, 86) and C1/C2 (Figure 9, 96) have been applied most frequently for protein structure investigations. The probe C2 loaded with Dy(III) and Yb(III) attached at different sites was used to assign the methyl resonances of a four-helix bundle SNARE complex by measuring PCSs in 13C-HMQC spectra.689 PCSs induced in the C2B domain of synaptotagmin-1 bound to the SNARE complex provided clear evidence for a highly dynamic binding mode between the two components.690
The dengue virus serotype 2 NS2B-NS3 protease (NS2B-NS3pro) is a recognized drug target but yields an overlapped and difficult to assign NMR spectrum. The interaction of NS2B with NS3pro was investigated by PCSs generated with probes C1 and C2 loaded with Tm(III) and Tb(III). The PCSs revealed that the enzyme predominantly assumes a closed conformation that is stabilized by the presence of positively charged active-site inhibitors.291,618 The same probes were used to investigate a NS2B-NS3pro construct without covalent linkage, which confirmed the closed conformation in the absence of inhibitors.619 A similar situation was documented for a linked construct of the related Zika virus NS2B-NS3 protease, where PCSs were induced in NS2B by Tb(III)-C2 and Tm(III)-C2 attached to NS3pro, revealing the closed conformation and stabilization by the presence of inhibitor.691
PCSs generated by the C1 and C2 tags also provided the basis for the development of algorithms to determine the 3D structure of proteins, using the PCSs of backbone amides and backbone chemical shifts as the sole experimental restraints.94,604,606,612 Specifically, Pearce et al. demonstrated that two tagging sites can suffice to structurally define large segments of a protein, provided PCSs are available for multiple homo- and heteronuclear spins of the polypeptide backbone.94 Using the combined information from PCSs and RDCs, the same study concluded that the molecular dynamics ensemble representation of ubiquitin (PDB ID 2KOX)692 fitted the paramagnetic NMR data better than any single 3D structure available of the protein. Finally, the study showed that 1H PCSs from tags at two sites in many cases suffices to determine the side-chain conformations of amino acids with methyl groups. In general, however, a complete 3D structure determination of a protein requires at least three, if not four, different probe tagging sites.
The C1 tag loaded with Yb(III) and Tb(III) ions was also used to assess the binding mode of 15N/13C-labeled dGMP to the nucleotide binding R3H domain from human Sμbp-2, as well as the structural changes in the R3H domain in response to nucleotide binding.693 As an alternative to isotope labeling, a chemical tag, such as a tert-butyl group, also enables the selective detection of 1H NMR signals of stably bound ligands and has been used to characterize the binding mode of a ligand to dengue virus NS2B-NS3pro by PCSs generated with C2 loaded with Tm(III) and Tb(III) and detected in NOESY spectra.635
The DOTA-M8 tag (Figure 9, 86) has been used in a particularly diverse range of applications, including the improvement of NMR spectral resolution of intrinsically disordered proteins (IDP),694 generation of RDCs to elucidate the structure of a transmembrane helix695 and an α-helix from myosin-VI that is remarkably stable in solution as a single helix,696 generation of RDCs in the ribosomal stalk protein bL12 to probe the orientational independence of N- and C-terminal domains,697 determining the fluorine coordinates of ligands bound to hCA II by PCSs measured in 19F NMR spectra recorded with four different tag attachment sites (including PCSs generated with M7PyThiazole-DOTA),698 and generation of PCSs in RNA.699 Specifically, Göbl et al. demonstrated that the relatively weak paramagnetism of Yb(III) in DOTA-M8 was sufficient to improve spectral resolution in two different IDPs and enable additional resonance assignments,694 whereas Chiliveri et al. used Tm(III)-DOTA-M8 to generate sizable molecular alignment of the HIV-1 gp41 transmembrane helix.695 Similarly, Wang et al. used Tm(III)-DOTA-M8 to assess the domain alignment of ribosome-bound bL12 monomers and dimers by RDC measurements,697 Zimmermann et al. used the Tm(III)-DOTA-M8 tag to generate PCSs in 19F NMR spectra of fluorine containing ligands bound to hCA II,698 and Strickland et al. showed that the RNA-binding protein U1A tagged with Tm(III)-DOTA-M8 is capable of generating sizable PCSs in an RNA hairpin equipped with a U1A binding loop, which would otherwise be difficult to label with a paramagnetic tag.699
M7PyThiazole-DOTA (Figure 9, 92) is a newer probe than DOTA-M8, which offers a more rigid tether between lanthanide ion and protein backbone. Loaded with Tm(III) and attached to each monomer of the homotrimeric protein Skp, a single set of PCSs was generated by M7PyThiazole-DOTA, which could be fitted by Δχ tensors positioned at three sites related by C3 symmetry.700 Using DOTA-M8 and M7PyThiazole-DOTA as paramagnetic tags, Cucuzza et al. developed a new iterative procedure for structure refinement of repeat proteins based on backbone amide PCSs. The authors successfully determined the fold of the protein YM4A in solution.701 The related probes M7Py-DOTA (section 5.1.2)286 and M8-CAM-I,285 which likewise generate stable thioether linkages with cysteine, have been deployed for in-cell measurements of PCSs to obtain structural restraints with Tm(III) and Dy(III) ions, respectively, in GB1 and ubiquitin.
Lanthanoid tags can be deployed in DEER experiments when loaded with Gd(III), while generating PCSs in NMR experiments when loaded with other paramagnetic lanthanoid ions. Both approaches were combined by Abdelkader et al. in a structural analysis of the E. coli aspartate/glutamate binding protein with the C3 tag ligated to AzF.702 Multiple sites tagged pairwise with Gd(III)-C3 delivered accurate positions of the metal ion relative to the crystal structure of the protein, which subsequently allowed fitting Δχ tensors using PCSs measured with Tm(III)-C3 and Tb(III)-C3 in 5-parameter fits that relied on the metal coordinates derived from the DEER measurements. The approach is of interest for proteins of limited solubility, where resonance assignment by 3D NMR experiments is difficult but 2D NMR spectra are accessible of samples prepared with selective isotope labeling, which can be assigned by high-throughput site-directed mutagenesis.
Gd(III) complexes of the above-mentioned probes and other single arm probes have been explored for distance measurements by DEER experiments in multiple EPR studies.298,703−709 Specifically the probes DO3MA-3BrPy (Figure 9, 107) and C3 (Figure 9, 98) were deployed for distance measurements in proteins embedded in cells.708,709
6.4. Applications of Small Chemical Probes
Compared with the aforementioned probes, small chemical based probes have been less frequently used in paramagnetic NMR of proteins, which can be attributed to the inconvenience of having to titrate with paramagnetic metal ions after the tagging reaction and the risk of populating alternative binding sites with stray metal ions, which do not bind at the intended site either due to weak binding affinities to the probe or an excess of metal ion. Among the small chemical Gd(III) probes, 4MMDPA (Figure 11, 116) and the IDA-SH (Figure 11, 126) tag have been used for DEER distance measurements in proteins.688,710
6.5. Applications of Cosolute Paramagnetic Probes
Cosolute paramagnetic probes are popular tools for detecting solvent accessible protein surfaces. Petros et al. explored the use of TEMPOL and [Gd(III)(DTPA)]2– for the identification of solvent exposure of ubiquitin and hen egg white lysozyme (HEWL) by double-quantum-filtered proton correlation spectra (DQF-COSY), concluding that the two probes preferentially interacted with hydrophobic amino acid side chains and carboxylate groups, respectively.345 Fesik et al. subsequently showed that longitudinal 1H PREs generated by TEMPOL identified the solvent-exposed residues of 13C-labeled cyclosporin bound to cyclophilin.346 The same group confirmed this approach using a 13C-labeled FK-506 analog bound to FKBP.711 Molinari et al. concluded that TEMPOL makes no specific interactions with bovine pancreatic trypsin inhibitor (BPTI), allowing interpretation of signal attenuations in homo- and heteronuclear NMR spectra in terms of solvent exposure.712 This was followed by a string of paramagnetic perturbation studies of a number of proteins, including Tendamistat,713 MNEI,346 HEWL,347,355 and Sso7d (from Sulfolobus solfataricus),714 using TEMPOL, as well as soluble Gd(III) complexes as the paramagnetic agents. Bernini et al. investigated protein aggregation by monitoring the attenuation of 1H NMR signals of BPTI in the presence of TEMPOL and Gd(III)DTPA-BMA as a function of protein concentration.715TEMPOL-induced paramagnetic perturbation was shown to be broadly compatible with results obtained on the solvent exposure of aromatic amino acid side chains by photochemically induced dynamic nuclear polarization (photo-CIDNP) experiments.347 The introduction of Gd(III)DTPA-BMA provided an alternative uncharged cosolute probe for paramagnetic perturbation studies, which is less hydrophobic than TEMPOL and was shown to deliver better correlations between calculated and experimental solvent PREs350 but in the case of α-bungarotoxin was reported to still show uneven attenuations of [13C,1H]-HSQC cross-peaks of CαH groups across the protein,716 which were reproduced by the much larger probe Gd2L7.717 To identify the interaction surface of a protein–protein complex by comparing cross-peak attenuations of the complex versus those of the individual proteins, a charged probe like [Gd(III)(EDTA)]1– was shown to be sufficient.357 Almeida et al. further demonstrated that also the highly charged probes [Gd(III)(DOTP)]5– and [Gd(III)-DOTAM]3+ allow probing for protein–protein interactions.718 It has been noted, however, that Gd(III) complexes with free coordination sites, such as in Gd(III)(DO3A), can favor transient associations with carboxylate groups on the protein surface.366
Different algorithms have been developed to exploit PREs from paramagnetic cosolutes for determining the 3D structures of proteins352 and protein complexes,719 as well as gaining information on protein dynamics.353 Madl et al. selected Gd(III)DTPA-BMA to aid protein structure determination, using a “second-sphere interaction model” to translate PREs into distance restraints between the NMR-active nuclei and the protein surface.352 This became the basis of a strategy to integrate solvent PRE data generated by Gd(III)DTPA-BMA into the program HADDOCK720 to elucidate the structure of a 150 kDa ternary protein complex. Gd(III)DTPA-BMA also provided useful PRE restraints for the structure determination of the homodimeric protein Sam68,721 and the probe clearly demonstrated how farnesylation alters the solvent accessibility of the C-terminal polypeptide segment of peroxisomal biogenesis factor 19 (PEX19).722 By measuring transverse PREs of amide protons with Gd(III)DTPA-BMA as a function of protein concentration and taking into account local molecular crowding as an impediment to translational diffusion, Jens Led and co-workers identified a method to distinguish specific from nonspecific contacts in weakly self-associating human growth hormone.723 To eliminate exchange effects arising from rapid relaxation of Gd(III)-coordinated hydration water, the group of Chun Tang developed Gd(III)TTHA-TMA as a cosolute probe without free coordination sites on the Gd(III) ion, which allowed determining the populations of open and closed conformations from solvent PREs by comparison with theoretical expectations based on crystal structures and molecular dynamics simulations.353
As discussed in section 3.3.5, the [Ln(III)(DPA)3]3– probe can generate measurable PCSs and its Gd(III) complex can produce PREs. Ma et al. investigated the binding of [Ln(III)(DPA)3]3– probes (Ln = Eu, Gd, Tb, Tm, or Yb) to multiple sites in HEWL, which had been identified in a crystal structure of the protein with [Eu(III)(DPA)3]3–.724 While the paramagnetic effects observed could not be fully accounted for by the crystal structure, an MD simulation reproduced the highest affinity binding site and correlated small PCS magnitudes with increased local protein dynamics. More recently, Swarbrick et al. used PCSs generated by [Tm(III)(DPA)3]3– to improve the spectral resolution of NOESY spectra of lipopeptide polymyxin B (PMXB) and the peptide MSI-594 bound to micelles.725 The PCSs originated from transient association of the probe with the peptides, but were not used as structural restraints. Instead, the concentration of the probe was tuned to improve the spectral resolution while limiting PREs, and the structures were determined from trNOEs.
Finally, it has been pointed out that, at a fundamental NMR-technical level, paramagnetic solvent probes can be used to simplify NMR spectral interpretation. For example, Schwarzinger and co-workers showed that difference spectra calculated for HSQC or NOESY experiments recorded in the presence and absence of Gd(III)DTPA-BMA can be used to focus on signals with little accessibility to the probe.726 Similarly, Sattler and Fesik proposed the use of high concentrations of [Dy(III)(DTPA)]2– to obtain substantial chemical shift changes in NMR spectra.358
7. Conclusions and Prospects
The invention and further development of paramagnetic and, in particular, site-specific probes has opened a wide range of protein structure investigation methods both by NMR spectroscopy and EPR spectroscopy. Although nitroxide spin labels have long been used with great success for PRE measurements and in EPR studies, many more structure restraints beyond distances become accessible with paramagnetic metal ions associated with anisotropic molecular magnetic susceptibility tensors. Lanthanoid ions stand out for their intrinsically large Δχ tensors and synthetic chemists have worked hard to optimize lanthanoid binding probes. A number of broad conclusions can be drawn from comparing the performance of these probes. (i) Site-specific attachment is difficult to attain without one or more covalent bonds. (ii) Rigid attachment of a lanthanoid ion is important for obtaining useful Δχ tensors and more easily achieved by double-arm attachment. In the case of proteins, immobilization can be achieved by additional coordination by carboxylate groups of amino acid side chains. (iii) Probes synthesized with the lanthanoid ion in place are more user-friendly and less prone to artifacts than probes that need to be titrated with metal ions following ligation with the target molecule. Therefore, cyclen based probes continue to be attractive despite the multistep protocols of their synthesis.
Significant challenges remain. Most covalently binding probes target cysteine residues and thus are incompatible with proteins containing natural cysteine residues that are accessible and functionally important. To avoid the dependence on cysteine residues, schemes for site-specific incorporation of noncanonical amino acid capable of binding lanthanoid probes have been developed. A generally satisfying solution has not yet been found, however, because either the resulting linker to the target protein is quite long (as in the case of, for example, probes attached by click chemistry), the unnatural amino acid disrupts the protein structure (as in the case of, for example, phosphoserine), or the amino acid causes protein precipitation upon metal binding (as in the case of the bipyridyl and 8-hydroxy-quinoline amino acids). An emerging solution may be offered by the site-specific incorporation of photocaged selenocysteine, which, following decaging by UV irradiation, delivers a site that is much more reactive toward alkylating probes than cysteine thiol groups.727,728 The potential of harnessing this strategy for attaching double-armed probes is clear but has not yet been explored. Also the incorporation of an unnatural amino acid containing a stable nitroxide radical has not yet been achieved in a way that would dethrone the MTSL tag as the most popular spin label, partly because nitroxides are sensitive to reduction in live cells and partly because no aminoacyl-tRNA synthetase is available that is specific for a nitroxide amino acid featuring a short and rigid linker between nitroxide group and protein backbone.
Moving from lanthanoid ions to transition metal ions reduces the size of complexation cages and enables good binding affinities with nitrogen ligands but abandons the large and varied range of paramagnetic effects afforded by different lanthanoid ions. Attachment modes that leave vacant coordination sites on the metal ion risk leading to metal mediated protein dimerization. While this has not been observed routinely, it is a distinct possibility for many of the very small probes and also when a transition metal ion binding site is engineered by a pair of histidine residues.
With the advent of completely rigid probes, exciting prospects are opened for high-accuracy protein structure analysis, including determining the 3D structures of minor, little populated protein states by PCSs. With chemically stable ligation chemistry, such studies will foreseeably be possible also in living cells. Therefore, finding new rigid chelators remains an important goal for the future.
Finally, paramagnetic metal probes are becoming increasingly popular in EPR spectroscopy for measuring metal–metal distances in the nanometer range, where the metal ions can be Gd(III), Mn(II), or Cu(II) ions.121,162,163,729 Both NMR and EPR are based on spin interactions, and it is only logical that paramagnetic systems bring these two fields closer together. The combination of these spectroscopies provides a more complete picture of the world of biological macromolecules.
Acknowledgments
Q.M. acknowledges support from the Chinese Scholarship Council (CSC grant to Q.M., No. 201506870013). C.N. acknowledges funding from the Australian Research Council (DE190100015, DP200100348). Financial support by the Australian Research Council for a Laureate Fellowship to G.O. (FL170100019) and through a Centre of Excellence (CE200100012) is also gratefully acknowledged.
Glossary
Abbreviations
- 2AP
2-aminopyridine
- AdR
adrenodoxin reductase
- ArgN
Escherichia coli arginine repressor
- BCX
single-domain GH11 glycosidase from Bacillus circulans
- BPTI
bovine pancreatic trypsin inhibitor
- BpyAla
bipyridyl alanine
- BuLi
butyllithium
- CAP
catabolite gene activator protein
- Cc
yeast iso-1-cytochrome c
- CcP
yeast cytochrome c peroxidase
- CCR
cross-correlated relaxation
- CDI
N,N′-carbonyldiimidazole
- CEST
chemical exchange saturation transfer
- CLaNP
caged lanthanoid NMR probe
- COSY
correlation spectroscopy
- CPMG
Carr–Purcell–Meiboom–Gill
- CSA
chemical shift anisotropy
- CTAB
cetyltrimethylammonium bromide
- CuAAC
copper-catalyzed azide–alkyne cycloaddition
- Cyclen
1,4,7,10-tetraazacyclododecane
- Cys-Ph-TAHA
cysteinyl-phenyl-triaminohexaacetate
- cytP450cam
cytochrome P450cam
- DCM
dichloromethane
- DD
dipole–dipole
- DEER
double-electron–electron resonance
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- DNA
desoxyribonucleic acid
- DNP
dynamic nuclear polarization
- DOTA
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
- dPA
2,2′-dipicolylamine
- DPA
dipicolinic acid
- DPAP
2,2-dimethoxy-2-phenylaceto-phenone
- DQF-COSY
double-quantum-filtered COSY
- DSA
dipolar shielding anisotropy
- DTNB
5,5′-dithio-bis(2-nitrobenzoic acid)
- DTPA
diethylenetriaminepentaacetic acid
- DTTA
diethylene-triamine-tetraacetate
- EDTA
ethylenediaminetetraacetic acid
- EdU
5-ethynyl-2′-deoxyuridine
- eIF4E
eukaryotic translation initiation factor 4E
- EPR
electron paramagnetic resonance
- FAK
focal adhesion kinase
- FRET
Förster resonance energy transfer
- GB1
B1 immunoglobulin binding domain of protein G
- GPS
global positioning system
- HATU
2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- hCA II
human carbonic anhydrase II
- HEWL
hen egg white lysozyme
- HPPK
Staphylococcus aureus 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase
- HQA
2-amino-3-(8-hydroxyquinolin-3-yl)propanoic acid
- Hsp90
heat-shock protein 90
- HSQC
heteronuclear single-quantum correlation
- IDA
iminodiacetic acid
- IDP
intrinsically disordered protein
- IL1β
interleukin-1β
- LBP
lanthanoid-binding peptide
- LDA
lithium diisopropylamide
- Ln(III)
trivalent lanthanoid ion
- Ln
lanthanoid
- m-CPBA
meta-chloroperbenzoic acid
- MAS
magic angle spinning
- MO
maximum occurrence
- MRI
magnetic resonance imaging
- MST
methanethiosulfonate
- MTSL
(1-oxy-,2,2,5,5-tetramethyl-D-pyrroline-3-methyl)-methanethiosulfonate
- ncAA
noncanonical amino acid
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- NOESY
nuclear Overhauser effect spectroscopy
- NS2B-NS3pro
dengue virus serotype 2 NS2B-NS3 protease
- NTA
nitrilotriacetic acid
- NTP
nucleotide triphosphate
- p75 ICD
p75 neurotrophin receptor
- PCS
pseudocontact shift
- PDB
protein data bank
- Pdx
putidaredoxin
- PEX19
C-terminal polypeptide segment of peroxisomal biogenesis factor 19
- photo-CIDNP
photochemically induced dynamic nuclear polarization
- PPI
protein–protein interactions
- PRE
paramagnetic relaxation enhancement
- PyMTA
2,2′,2″,2‴-((pyridine-2,6-diylbis(methylene))bis(azanetriyl))tetraacetic acid
- RACS
residual anisotropic chemical shift
- RADS
residual anisotropic dipolar shift
- RD
relaxation dispersion
- RDC
residual dipolar coupling
- RhoGDI2
Rho guanine-nucleotide dissociation inhibitor 2
- RMSD
root-mean-square deviation
- RNA
ribonucleic acid
- SAP
square antiprism
- SBM
Solomon–Bloembergen–Morgan
- SH3
Src homology 3
- SrtA
Staphylococcus aureus sortase A
- PMXB
lipopeptide polymyxin B
- T4Lys
T4 lysozyme
- TAHA
triaminohexaacetate
- TCEP
tris(2-carboxyethyl)phosphine
- TETAC
1-(2-(pyridin-2-yldisulfanyl)ethyl)-1,4,7,10-tetraazacyclododecane
- THF
tetrahydrofuran
- TMR
tetramethylrosamine
- TMS
trimethylsilyl
- TMSCHN2
(diazomethyl)trimethylsilane
- tPCS
transferred PCS
- tPRE
transferred PRE
- TPS
2,4,6-triisopropylbenzenesulfonyl
- TraNP
transition metal ion NMR probe
- TROSY
transverse relaxation optimized spectroscopy
- TSAP
twisted square antiprism
Symbols
- χ
magnetic susceptibility tensor
- χx
magnetic susceptibility value along the tensor framework x axis
- χy
magnetic susceptibility value along the tensor framework y axis
- χz
magnetic susceptibility value along the tensor framework z axis
- Δχ
anisotropic magnetic susceptibility tensor
- Δχax
axial component of the Δχ tensor
- Δχrh
rhombic component of the Δχ tensor
- μB
Bohr magneton
- kB
Boltzmann constant
- T
absolute temperature
- μ0
induction constant
- ge
electronic g factor
- S
total electron spin quantum number
- gJ
electronic g factor of lanthanoid ions
- J
total angular momentum quantum number
- σ
dipolar shift tensor
- r
vector connecting paramagnetic center and nuclear spin
- r
|r|
-
 3
3
3 × 3 identity matrix
- ⊗
Kronecker product
- A
alignment tensor
- B0
magnetic field strength
- γI
gyromagnetic ratio of the nuclear spin I
- γK
gyromagnetic ratio of the nuclear spin K
- rIK
internuclear distance between I and K
- h
Planck’s constant
- R1SBM
paramagnetic enhancement of the longitudinal relaxation rate due to SBM mechanism
- R2SBM
paramagnetic enhancement of the transverse relaxation rate due to SBM mechanism
- ωI
nuclear Larmor frequency
- ωS
electron Larmor frequency
- τc
correlation time
- τr
rotational correlation time
- τs
electronic lifetime
- R1Curie
paramagnetic enhancement of the longitudinal relaxation rate due to Curie mechanism
- R2Curie
paramagnetic enhancement of the transverse relaxation rate due to Curie mechanism
- Ω
Larmor frequency of the nuclear spin
- σN
1H spin shift tensor originated by the dipolar field of the 15N spin
- σ↑
positive 1H spin shift tensor originated by the dipolar field of the 15N spin
- σ↓
negative 1H spin shift tensor originated by the dipolar field of the 15N spin
Biographies
Qing Miao completed her Ph.D. under the supervision of Prof. Marcellus Ubbink and Dr. Mark Overhand in 2019 at Leiden University. During her Ph.D. studies, she focused on paramagnetic NMR probe design, synthesis, and applications. Currently, she works as a junior research fellow at Shaanxi University of Science & Technology and is interested in natural abundance isotope analysis research.
Christoph Nitsche studied chemistry and business administration and received his Ph.D. in medicinal chemistry from Heidelberg University in 2014. He worked as a Feodor Lynen Fellow at the Australian National University and as a Rising Star Fellow at the Free University of Berlin. In 2020, he was appointed at the Australian National University, where he is currently Senior Lecturer and ARC DECRA Fellow in the Research School of Chemistry. His research program in medicinal chemistry and chemical biology focuses on drug discovery against infectious diseases, biocompatible chemistry, and peptide and protein modification.
Henry Orton completed his Ph.D. in 2020 at the Australian National University with a focus on experiment and software development for paramagnetic relaxation in biomolecular NMR spectroscopy. He is currently a postdoctoral fellow with Professor Gottfried Otting with interests in ultrafast magic angle spinning of biological solids.
Mark Overhand obtained a M. Sc. (Dec. 1989) from Leiden University (oligosaccharide synthesis, supervisors G.J. Boons, G. van der Marel, and J.H. van Boom) and a Ph.D. (Jan. 1995) from the University of Virginia, US (natural product synthesis, advisor S.M. Hecht). He did a postdoc. with a Swiss stipend at the ETH in Switzerland in the group of D.S. Seebach (design and synthesis of novel beta-hexapeptide helix) and returned to Leiden where he remains (1996–present): postdoc, assistant prof. (UD), associate prof. (UHD). Stanford: sabbatical (summer 2004). Awards: Golden spatula 1986 (B. sc. research), best teacher faculty of science 2006, Leiden University.
Gottfried Otting studied chemistry at the universities of Heidelberg and Freiburg (Germany) and received his Ph.D. in protein NMR spectroscopy from the ETH-Zürich (Switzerland) in 1987. He continued at the ETH-Zürich as a research assistant until 1992, when he was appointed professor of Molecular Biophysics at the Karolinska Institute in Stockholm (Sweden). In 2002, he was appointed professor at the Research School of Chemistry of the Australian National University in Canberra (Australia) on a Federation Fellowship by the Australian Research Council (ARC). He currently is an ARC Laureate Fellow at the same place. His research program focuses on method developments for protein NMR spectroscopy, including techniques for the production of protein samples with labels for structure analysis and drug discovery.
Marcellus Ubbink obtained a Ph.D. in Chemistry from Leiden University in 1994, working on paramagnetic properties of metalloproteins. During his postdoc research at the University of Cambridge (UK), he developed a method to determine the structure of two proteins using paramagnetic NMR, based in intermolecular paramagnetic effects caused by a heme group. He returned to Leiden University in 1997 as an assistant professor and was appointed to associate professor (2004) and full professor (2010). Since 2019, he is also Programme Director of the bachelor Life Science and Technology. His current research interests comprise protein–protein interactions, enzymology, protein evolution, and paramagnetic protein NMR spectroscopy.
The authors declare no competing financial interest.
References
- Overhauser A. W. Polarization of Nuclei in Metals. Phys. Rev. 1953, 92, 411–415. 10.1103/PhysRev.92.411. [DOI] [Google Scholar]
- Brüschweiler R.; Case D. A. Charaterization of Biomolecular Structure and Dynamics by NMR Cross Relaxation. Prog. Nucl. Magn. Reson. Spectrosc. 1994, 26, 27–58. 10.1016/0079-6565(94)80003-0. [DOI] [Google Scholar]
- Barry C. D.; North A. C. T.; Glasel J. A.; Williams R. J. P.; Xavier A. V. Quantitative Determination of Mononucleotide Conformations in Solution Using Lanthanide Ion Shift and Broadening NMR Probes. Nature 1971, 232, 236–245. 10.1038/232236a0. [DOI] [PubMed] [Google Scholar]
- Campbell I. D.; Dobson C. M.; Williams R. J. P.; Xavier A. V. The Determination of the Structure of Proteins in Solution: Lysozyme. Ann. N. Y. Acad. Sci. 1973, 222, 163–174. 10.1111/j.1749-6632.1973.tb15259.x. [DOI] [PubMed] [Google Scholar]
- Sculimbrene B. R.; Imperiali B. Lanthanide-Binding Tags as Luminescent Probes for Studying Protein Interactions. J. Am. Chem. Soc. 2006, 128, 7346–7352. 10.1021/ja061188a. [DOI] [PubMed] [Google Scholar]
- Delbianco M.; Sadovnikova V.; Bourrier E.; Mathis G.; Lamarque L.; Zwier J. M.; Parker D. Bright, Highly Water-Soluble Triazacyclononane Europium Complexes To Detect Ligand Binding with Time-Resolved FRET Microscopy. Angew. Chem., Int. Ed. 2014, 53, 10718–10722. 10.1002/anie.201406632. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G. Magnetic Susceptibility in Paramagnetic NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 40, 249–273. 10.1016/S0079-6565(02)00002-X. [DOI] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G.; Ravera E.. NMR of Paramagnetic Molecules: Applications to Metallobiomolecules and Modelsetallobiomolecules and Models; Elsevier Science & Technology: Oxford, 2017; pp 1–495. [Google Scholar]
- Parigi G.; Ravera E.; Luchinat C. Magnetic Susceptibility and Paramagnetism-Based NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 114–115, 211–236. 10.1016/j.pnmrs.2019.06.003. [DOI] [PubMed] [Google Scholar]
- Ravera E.; Parigi G.; Luchinat C. What Are the Methodological and Theoretical Prospects for Paramagnetic NMR in Structural Biology? A Glimpse into the Crystal Ball. J. Magn. Reson. 2019, 306, 173–179. 10.1016/j.jmr.2019.07.027. [DOI] [PubMed] [Google Scholar]
- Paramagnetism in Experimental Biomolecular NMR; Luchinat C., Parigi G., Ravera E., Eds.; The Royal Society of Chemistry: Cambridge, 2018; pp 1–316. [Google Scholar]
- Kowalewski J.; Kruk D.; Parigi G. NMR Relaxation in Solution of Paramagnetic Complexes: Recent Theoretical Progress for S ≥ 1. Adv. Inorg. Chem. 2005, 57, 41–104. 10.1016/S0898-8838(05)57002-8. [DOI] [Google Scholar]
- Pell A. J.; Pintacuda G.; Grey C. P. Paramagnetic NMR in Solution and the Solid State. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 111, 1–271. 10.1016/j.pnmrs.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C. New Applications of Paramagnetic NMR in Chemical Biology. Curr. Opin. Chem. Biol. 1999, 3, 145–151. 10.1016/S1367-5931(99)80026-X. [DOI] [PubMed] [Google Scholar]
- Arnesano F.; Banci L.; Piccioli M. NMR Structures of Paramagnetic Metalloproteins. Q. Rev. Biophys. 2005, 38, 167–219. 10.1017/S0033583506004161. [DOI] [PubMed] [Google Scholar]
- Piccioli M.; Turano P. Transient Iron Coordination Sites in Proteins: Exploiting the Dual Nature of Paramagnetic NMR. Coord. Chem. Rev. 2015, 284, 313–328. 10.1016/j.ccr.2014.05.007. [DOI] [Google Scholar]
- Ravera E.; Cerofolini L.; Fragai M.; Parigi G.; Luchinat C. Characterization of Lanthanoid-Binding Proteins Using NMR Spectroscopy. Methods Enzymol. 2021, 651, 103–137. 10.1016/bs.mie.2021.01.039. [DOI] [PubMed] [Google Scholar]
- Otting G. Prospects for Lanthanides in Structural Biology by NMR. J. Biomol. NMR 2008, 42, 1–9. 10.1007/s10858-008-9256-0. [DOI] [PubMed] [Google Scholar]
- Koehler J.; Meiler J. Expanding the Utility of NMR Restraints with Paramagnetic Compounds: Background and Practical Aspects. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 360–389. 10.1016/j.pnmrs.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saio T.; Ishimori K. Accelerating Structural Life Science by Paramagnetic Lanthanide Probe Methods. Biochim. Biophys. Acta, Gen. Subj. 2020, 1864, 129332. 10.1016/j.bbagen.2019.03.018. [DOI] [PubMed] [Google Scholar]
- Su X.-C.; Otting G. Paramagnetic Labelling of Proteins and Oligonucleotides for NMR. J. Biomol. NMR 2010, 46, 101–112. 10.1007/s10858-009-9331-1. [DOI] [PubMed] [Google Scholar]
- Keizers P. H. J.; Ubbink M. Paramagnetic Tagging for Protein Structure and Dynamics Analysis. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 88–96. 10.1016/j.pnmrs.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Liu W.-M.; Overhand M.; Ubbink M. The Application of Paramagnetic Lanthanoid Ions in NMR Spectroscopy on Proteins. Coord. Chem. Rev. 2014, 273–274, 2–12. 10.1016/j.ccr.2013.10.018. [DOI] [Google Scholar]
- Joss D.; Häussinger D. Design and Applications of Lanthanide Chelating Tags for Pseudocontact Shift NMR Spectroscopy with Biomacromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 114–115, 284–312. 10.1016/j.pnmrs.2019.08.002. [DOI] [PubMed] [Google Scholar]
- Su X.-C.; Chen J.-L. Site-Specific Tagging of Proteins with Paramagnetic Ions for Determination of Protein Structures in Solution and in Cells. Acc. Chem. Res. 2019, 52, 1675–1686. 10.1021/acs.accounts.9b00132. [DOI] [PubMed] [Google Scholar]
- Nitsche C.; Otting G. Pseudocontact Shifts in Biomolecular NMR Using Paramagnetic Metal Tags. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 98–99, 20–49. 10.1016/j.pnmrs.2016.11.001. [DOI] [PubMed] [Google Scholar]
- Tolman J. R.; Ruan K. NMR Residual Dipolar Couplings as Probes of Biomolecular Dynamics. Chem. Rev. 2006, 106, 1720–1736. 10.1021/cr040429z. [DOI] [PubMed] [Google Scholar]
- Pintacuda G.; John M.; Su X.-C.; Otting G. NMR Structure Determination of Protein–Ligand Complexes by Lanthanide Labeling. Acc. Chem. Res. 2007, 40, 206–212. 10.1021/ar050087z. [DOI] [PubMed] [Google Scholar]
- Otting G. Protein NMR Using Paramagnetic Ions. Annu. Rev. Biophys. 2010, 39, 387–405. 10.1146/annurev.biophys.093008.131321. [DOI] [PubMed] [Google Scholar]
- Hass M. A. S.; Ubbink M. Structure Determination of Protein-Protein Complexes with Long-Range Anisotropic Paramagnetic NMR Restraints. Curr. Opin. Struct. Biol. 2014, 24, 45–53. 10.1016/j.sbi.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Clore G. M.; Iwahara J. Theory, Practice, and Applications of Paramagnetic Relaxation Enhancement for the Characterization of Transient Low-Population States of Biological Macromolecules and Their Complexes. Chem. Rev. 2009, 109, 4108–4139. 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore G. M. Exploring Sparsely Populated States of Macromolecules by Diamagnetic and Paramagnetic NMR Relaxation. Protein Sci. 2011, 20, 229–246. 10.1002/pro.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilder J. T.; Hass M. A. S.; Keizers P. H. J.; Ubbink M.Chapter 6: Paramagnetic NMR Spectroscopy and Lowly Populated States. In Recent Developments in Biomolecular NMR; Clore G. M., Potts J., Eds.; The Royal Society of Chemistry: Cambridge, 2012; pp 130–150. [Google Scholar]
- Ubbink M.; Di Savino A.. Chapter 5: Protein–Protein Interactions. In Paramagnetism in Experimental Biomolecular NMR; Luchinat C., Parigi G., Ravera E., Eds.; The Royal Society of Chemistry: Cambridge, 2018; pp 134–162. [Google Scholar]
- Hocking H. G.; Zangger K.; Madl T. Studying the Structure and Dynamics of Biomolecules by Using Soluble Paramagnetic Probes. ChemPhysChem 2013, 14, 3082–3094. 10.1002/cphc.201300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z.; Schwieters C. D.; Tang C. Theory and Practice of Using Solvent Paramagnetic Relaxation Enhancement to Characterize Protein Conformational Dynamics. Methods 2018, 148, 48–56. 10.1016/j.ymeth.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softley C. A.; Bostock M. J.; Popowicz G. M.; Sattler M. Paramagnetic NMR in Drug Discovery. J. Biomol. NMR 2020, 74, 287–309. 10.1007/s10858-020-00322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K.; Yamaguchi T. Paramagnetic NMR Probes for Characterization of the Dynamic Conformations and Interactions of Oligosaccharides. Glycoconjugate J. 2015, 32, 505–513. 10.1007/s10719-015-9599-1. [DOI] [PubMed] [Google Scholar]
- Valverde P.; Quintana J. I.; Santos J. I.; Ardá A.; Jiménez-Barbero J. Novel NMR Avenues to Explore the Conformation and Interactions of Glycans. ACS Omega 2019, 4, 13618–13630. 10.1021/acsomega.9b01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa G. Z.; Qin P. Z. Site-Directed Spin Labeling Studies on Nucleic Acid Structure and Dynamics. Prog. Nucleic Acid Res. Mol. Biol. 2008, 82, 147–197. 10.1016/S0079-6603(08)00005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin P. Z.; Dieckmann T. Application of NMR and EPR Methods to the Study of RNA. Curr. Opin. Struct. Biol. 2004, 14, 350–359. 10.1016/j.sbi.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Sigurdsson S. T. Nitroxides and Nucleic Acids: Chemistry and Electron Paramagnetic Resonance (EPR) Spectroscopy. Pure Appl. Chem. 2011, 83, 677–686. 10.1351/PAC-CON-10-09-28. [DOI] [Google Scholar]
- Shelke S. A.; Sigurdsson S. T. Site-Directed Spin Labeling for EPR Studies of Nucleic Acids. Nucleic Acids Mol. Biol. 2016, 31, 159–187. 10.1007/978-3-319-27111-8_8. [DOI] [Google Scholar]
- Shelke S. A.; Sigurdsson S. T. Site-Directed Spin Labelling of Nucleic Acids. Eur. J. Org. Chem. 2012, 2012, 2291–2301. 10.1002/ejoc.201101434. [DOI] [Google Scholar]
- Duss O.; Yulikov M.; Allain F. H. T.; Jeschke G. Combining NMR and EPR to Determine Structures of Large RNAs and Protein–RNA Complexes in Solution. Methods Enzymol. 2015, 558, 279–331. 10.1016/bs.mie.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Cekan P.; Sigurdsson S. T.; Qin P. Z. Studying RNA Using Site-Directed Spin-Labeling and Continuous-Wave Electron Paramagnetic Resonance Spectroscopy. Methods Enzymol. 2009, 469, 303–328. 10.1016/S0076-6879(09)69015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyes R. S.; Bobst A. M.. Spin-Labeled Nucleic Acids. In Biological Magnetic Resonance; Berliner L. J., Ed.; Springer: Boston, 2002; pp 283–338. [Google Scholar]
- Wahsner J.; Gale E. M.; Rodríguez-Rodríguez A.; Caravan P. Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem. Rev. 2019, 119, 957–1057. 10.1021/acs.chemrev.8b00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly Thankamony A. S.; Wittmann J. J.; Kaushik M.; Corzilius B. Dynamic Nuclear Polarization for Sensitivity Enhancement in Modern Solid-State NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 102–103, 120–195. 10.1016/j.pnmrs.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Bagryanskaya E. G.; Krumkacheva O. A.; Fedin M. V.; Marque S. R. A. Development and Application of Spin Traps, Spin Probes, and Spin Labels. Methods Enzymol. 2015, 563, 365–396. 10.1016/bs.mie.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Braun T.; Drescher M.; Summerer D. Expanding the Genetic Code for Site-Directed Spin-Labeling. Int. J. Mol. Sci. 2019, 20, 373. 10.3390/ijms20020373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroniec C. P. Solid-State Nuclear Magnetic Resonance Structural Studies of Proteins Using Paramagnetic Probes. Solid State Nucl. Magn. Reson. 2012, 43–44, 1–13. 10.1016/j.ssnmr.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Jaroniec C. P. Structural Studies of Proteins by Paramagnetic Solid-State NMR Spectroscopy. J. Magn. Reson. 2015, 253, 50–59. 10.1016/j.jmr.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocman V.; Di Mauro G. M.; Veglia G.; Ramamoorthy A. Use of Paramagnetic Systems to Speed-up NMR Data Acquisition and for Structural and Dynamic Studies. Solid State Nucl. Magn. Reson. 2019, 102, 36–46. 10.1016/j.ssnmr.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhu P. K.; Grandori R.; Hohenthanner K.; Mandal P. K.; Müller N. Geometry Dependent Two-Dimensional Heteronuclear Multiplet Effects in Paramagnetic Proteins. J. Biomol. NMR 2001, 20, 31–37. 10.1023/A:1011292410478. [DOI] [PubMed] [Google Scholar]
- Pervushin K.; Riek R.; Wider G.; Wüthrich K. Attenuated T2 Relaxation by Mutual Cancellation of Dipole–Dipole Coupling and Chemical Shift Anisotropy Indicates an Avenue to NMR Structures of Very Large Biological Macromolecules in Solution. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 12366–12371. 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintacuda G.; Keniry M. A.; Huber T.; Park A. Y.; Dixon N. E.; Otting G. Fast Structure-Based Assignment of 15N HSQC Spectra of Selectively 15N-Labeled Paramagnetic Proteins. J. Am. Chem. Soc. 2004, 126, 2963–2970. 10.1021/ja039339m. [DOI] [PubMed] [Google Scholar]
- Orton H. W.; Kuprov I.; Loh C.-T.; Otting G. Using Paramagnetism to Slow Down Nuclear Relaxation in Protein NMR. J. Phys. Chem. Lett. 2016, 7, 4815–4818. 10.1021/acs.jpclett.6b02417. [DOI] [PubMed] [Google Scholar]
- Solomon I. Relaxation Processes in a System of Two Spins. Phys. Rev. 1955, 99, 559–565. 10.1103/PhysRev.99.559. [DOI] [Google Scholar]
- Gueron M. Nuclear Relaxation in Macromolecules by Paramagnetic Ions: A Novel Mechanism. J. Magn. Reson. 1975, 19, 58–66. 10.1016/0022-2364(75)90029-3. [DOI] [Google Scholar]
- Vega A. J.; Fiat D. Nuclear Relaxation Processes of Paramagnetic Complexes the Slow-Motion Case. Mol. Phys. 1976, 31, 347–355. 10.1080/00268977600100261. [DOI] [Google Scholar]
- Bertini I.; Turano P.; Vila A. J. Nuclear Magnetic Resonance of Paramagnetic Metalloproteins. Chem. Rev. 1993, 93, 2833–2932. 10.1021/cr00024a009. [DOI] [Google Scholar]
- Connelly N. G. C.; Damhus T.; Hartshorn R. M.; Hutton A. T.. Nomenclature of Inorganic Chemistry: IUPAC Recommendations 2005; The Royal Society of Chemistry: Cambridge, 2005. [Google Scholar]
- Cotruvo J. A. The Chemistry of Lanthanides in Biology: Recent Discoveries, Emerging Principles, and Technological Applications. ACS Cent. Sci. 2019, 5, 1496–1506. 10.1021/acscentsci.9b00642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov A. N.; Worrall J. A. R.; Holtzmann E.; Ubbink M. Solution Structure and Dynamics of the Complex between Cytochrome c and Cytochrome c Peroxidase Determined by Paramagnetic NMR. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 18945–18950. 10.1073/pnas.0603551103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers T. J.; Carr P. D.; Loh C.-T.; Jackson C. J.; Otting G. Iron(III) Located in the Dinuclear Metallo-β-Lactamase IMP-1 by Pseudocontact Shifts. Angew. Chem., Int. Ed. 2014, 53, 14269–14272. 10.1002/anie.201408693. [DOI] [PubMed] [Google Scholar]
- Xu X.; Scanu S.; Chung J.-S.; Hirasawa M.; Knaff D. B.; Ubbink M. Structural and Functional Characterization of the Ga-Substituted Ferredoxin from Synechocystis Sp. PCC6803, a Mimic of the Native Protein. Biochemistry 2010, 49, 7790–7797. 10.1021/bi100712g. [DOI] [PubMed] [Google Scholar]
- Parigi G.; Benda L.; Ravera E.; Romanelli M.; Luchinat C. Pseudocontact Shifts and Paramagnetic Susceptibility in Semiempirical and Quantum Chemistry Theories. J. Chem. Phys. 2019, 150, 144101. 10.1063/1.5037428. [DOI] [PubMed] [Google Scholar]
- Schmitz C.; Stanton-Cook M. J.; Su X.-C.; Otting G.; Huber T. Numbat: An Interactive Software Tool for Fitting Δχ-Tensors to Molecular Coordinates Using Pseudocontact Shifts. J. Biomol. NMR 2008, 41, 179–189. 10.1007/s10858-008-9249-z. [DOI] [PubMed] [Google Scholar]
- Schwieters C. D; Kuszewski J. J; Tjandra N.; Marius Clore G The Xplor-NIH NMR Molecular Structure Determination Package. J. Magn. Reson. 2003, 160, 65–73. 10.1016/S1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Meiler J.; Peti W.; Griesinger C. DipoCoup: A Versatile Program for 3D-Structure Homology Comparison Based on Residual Dipolar Couplings and Pseudocontact Shifts. J. Biomol. NMR 2000, 17, 283–294. 10.1023/A:1008362931964. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Cavallaro G.; Giachetti A.; Luchinat C.; Parigi G. Paramagnetism-Based Restraints for Xplor-NIH. J. Biomol. NMR 2004, 28, 249–261. 10.1023/B:JNMR.0000013703.30623.f7. [DOI] [PubMed] [Google Scholar]
- Orton H. W.; Huber T.; Otting G. Paramagpy: Software for Fitting Magnetic Susceptibility Tensors Using Paramagnetic Effects Measured in NMR Spectra. Magn. Reson. 2020, 1, 1–12. 10.5194/mr-1-1-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleaney B. Nuclear Magnetic Resonance Shifts in Solution Due to Lanthanide Ions. J. Magn. Reson. 1972, 8, 91–100. 10.1016/0022-2364(72)90027-3. [DOI] [Google Scholar]
- Tolman J. R.; Flanagan J. M.; Kennedy M. A.; Prestegard J. H. Nuclear Magnetic Dipole Interactions in Field-Oriented Proteins: Information for Structure Determination in Solution. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 9279–9283. 10.1073/pnas.92.20.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Keizers P. H. J.; Reinle W.; Hannemann F.; Bernhardt R.; Ubbink M. Intermolecular Dynamics Studied by Paramagnetic Tagging. J. Biomol. NMR 2009, 43, 247–254. 10.1007/s10858-009-9308-0. [DOI] [PubMed] [Google Scholar]
- John M.; Park A. Y.; Pintacuda G.; Dixon N. E.; Otting G. Weak Alignment of Paramagnetic Proteins Warrants Correction for Residual CSA Effects in Measurements of Pseudocontact Shifts. J. Am. Chem. Soc. 2005, 127, 17190–17191. 10.1021/ja0564259. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Felli I. C.; Luchinat C. High Magnetic Field Consequences on the NMR Hyperfine Shifts in Solution. J. Magn. Reson. 1998, 134, 360–364. 10.1006/jmre.1998.1507. [DOI] [PubMed] [Google Scholar]
- Solomon I. 200 and More NMR Experiment. Phys. Rev. 1955, 99, 559. 10.1103/PhysRev.99.559. [DOI] [Google Scholar]
- Bloembergen N.; Morgan L. O. Proton Relaxation Times in Paramagnetic Solutions. Effects of Electron Spin Relaxation. J. Chem. Phys. 1961, 34, 842–850. 10.1063/1.1731684. [DOI] [Google Scholar]
- Clore G. M. Visualizing Lowly-Populated Regions of the Free Energy Landscape of Macromolecular Complexes by Paramagnetic Relaxation Enhancement. Mol. BioSyst. 2008, 4, 1058–1069. 10.1039/b810232e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madl T.; Felli I. C.; Bertini I.; Sattler M. Structural Analysis of Protein Interfaces from 13C Direct-Detected Paramagnetic Relaxation Enhancements. J. Am. Chem. Soc. 2010, 132, 7285–7287. 10.1021/ja1014508. [DOI] [PubMed] [Google Scholar]
- Di Savino A.; Foerster J. M.; La Haye T.; Blok A.; Timmer M.; Ullmann G. M.; Ubbink M. Efficient Encounter Complex Formation and Electron Transfer to Cytochrome c Peroxidase with an Additional, Distant Electrostatic Binding Site. Angew. Chem., Int. Ed. 2020, 59, 23239–23243. 10.1002/anie.202010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abragam A.Thermal Relaxation in Liquids and Cases. In The Principles of Nuclear Magnetism; Clarendon Press: Oxford, 1961; pp 264–353. [Google Scholar]
- Pintacuda G.; Kaikkonen A.; Otting G. Modulation of the Distance Dependence of Paramagnetic Relaxation Enhancements by CSAxDSA Cross-Correlation. J. Magn. Reson. 2004, 171, 233–243. 10.1016/j.jmr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G.; Ravera E.. Chapter 8: Lanthanoids and Actinoids: Shift and Relaxation. In NMR of Paramagnetic Molecules: Applications to Metallobiomolecules and Modelsetallobiomolecules and Models; Elsevier Science & Technology: Oxford, 2017; pp 255–276. [Google Scholar]
- Suturina E. A.; Mason K.; Geraldes C. F. G. C.; Chilton N. F.; Parker D.; Kuprov I. Lanthanide-Induced Relaxation Anisotropy. Phys. Chem. Chem. Phys. 2018, 20, 17676–17686. 10.1039/C8CP01332B. [DOI] [PubMed] [Google Scholar]
- Pintacuda G.; Hohenthanner K.; Otting G.; Müller N. Angular Dependence of Dipole-Dipole-Curie-Spin Cross-Correlation Effects in High-Spin and Low-Spin Paramagnetic Myoglobin. J. Biomol. NMR 2003, 27, 115–132. 10.1023/A:1024926126239. [DOI] [PubMed] [Google Scholar]
- Ghose R.; Prestegard J. H. Electron Spin-Nuclear Spin Cross-Correlation Effects on Multiplet Splittings in Paramagnetic Proteins. J. Magn. Reson. 1997, 128, 138–143. 10.1006/jmre.1997.1227. [DOI] [PubMed] [Google Scholar]
- Hus J.-C.; Marion D.; Blackledge M. De Novo Determination of Protein Structure by NMR Using Orientational and Long-Range Order Restraints. J. Mol. Biol. 2000, 298, 927–936. 10.1006/jmbi.2000.3714. [DOI] [PubMed] [Google Scholar]
- Bashir Q.; Volkov A. N.; Ullmann G. M.; Ubbink M. Visualization of the Encounter Ensemble of the Transient Electron Transfer Complex of Cytochrome c and Cytochrome c Peroxidase. J. Am. Chem. Soc. 2010, 132, 241–247. 10.1021/ja9064574. [DOI] [PubMed] [Google Scholar]
- Clore G. M.; Garrett D. S. R-Factor, Free R, and Complete Cross-Validation for Dipolar Coupling Refinement of NMR Structures. J. Am. Chem. Soc. 1999, 121, 9008–9012. 10.1021/ja991789k. [DOI] [Google Scholar]
- Shishmarev D.; Otting G. How Reliable Are Pseudocontact Shifts Induced in Proteins and Ligands by Mobile Paramagnetic Metal Tags? A Modelling Study. J. Biomol. NMR 2013, 56, 203–216. 10.1007/s10858-013-9738-6. [DOI] [PubMed] [Google Scholar]
- Pearce B. J. G.; Jabar S.; Loh C.-T.; Szabo M.; Graham B.; Otting G. Structure Restraints from Heteronuclear Pseudocontact Shifts Generated by Lanthanide Tags at Two Different Sites. J. Biomol. NMR 2017, 68, 19–32. 10.1007/s10858-017-0111-z. [DOI] [PubMed] [Google Scholar]
- McDermott A. Structure and Dynamics of Membrane Proteins by Magic Angle Spinning Solid-State NMR. Annu. Rev. Biophys. 2009, 38, 385–403. 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- Tycko R. Solid State NMR Studies of Amyloid Fibril Structure. Annu. Rev. Phys. Chem. 2011, 62, 279–299. 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreas L. B.; Jaudzems K.; Stanek J.; Lalli D.; Bertarello A.; Le Marchand T.; Cala-De Paepe D.; Kotelovica S.; Akopjana I.; Knott B.; et al. Structure of Fully Protonated Proteins by Proton-Detected Magic- Angle Spinning NMR. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 9187–9192. 10.1073/pnas.1602248113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbet-Massin E.; Pell A. J.; Retel J. S.; Andreas L. B.; Jaudzems K.; Franks W. T.; Nieuwkoop A. J.; Hiller M.; Higman V.; Guerry P.; et al. Rapid Proton-Detected NMR Assignment for Proteins with Fast Magic Angle Spinning. J. Am. Chem. Soc. 2014, 136, 12489–12497. 10.1021/ja507382j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balayssac S.; Bertini I.; Bhaumik A.; Lelli M.; Luchinat C. Paramagnetic Shifts in Solid-State NMR of Proteins to Elicit Structural Information. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17284–17289. 10.1073/pnas.0708460105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Emsley L.; Lelli M.; Luchinat C.; Mao J.; Pintacuda G. Ultrafast MAS Solid-State NMR Permits Extensive 13C and 1H Detection in Paramagnetic Metalloproteins. J. Am. Chem. Soc. 2010, 132, 5558–5559. 10.1021/ja100398q. [DOI] [PubMed] [Google Scholar]
- Luchinat C.; Parigi G.; Ravera E.; Rinaldelli M. Solid-State NMR Crystallography through Paramagnetic Restraints. J. Am. Chem. Soc. 2012, 134, 5006–5009. 10.1021/ja210079n. [DOI] [PubMed] [Google Scholar]
- Knight M. J.; Felli I. C.; Pierattelli R.; Bertini I.; Emsley L.; Herrmann T.; Pintacuda G. Rapid Measurement of Pseudocontact Shifts in Metalloproteins by Proton-Detected Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2012, 134, 14730–14733. 10.1021/ja306813j. [DOI] [PubMed] [Google Scholar]
- Knight M. J.; Pell A. J.; Bertini I.; Felli I. C.; Gonnelli L.; Pierattelli R.; Herrmann T.; Emsley L.; Pintacuda G. Structure and Backbone Dynamics of a Microcrystalline Metalloprotein by Solid-State NMR. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11095–11100. 10.1073/pnas.1204515109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricq M. M.; Waugh J. S. NMR in Rotating Solids. J. Chem. Phys. 1979, 70, 3300–3316. 10.1063/1.437915. [DOI] [Google Scholar]
- Knight M. J.; Felli I. C.; Pierattelli R.; Emsley L.; Pintacuda G. Magic Angle Spinning NMR of Paramagnetic Proteins. Acc. Chem. Res. 2013, 46, 2108–2116. 10.1021/ar300349y. [DOI] [PubMed] [Google Scholar]
- Kervern G.; Steuernagel S.; Engelke F.; Pintacuda G.; Emsley L. Absence of Curie Relaxation in Paramagnetic Solids Yields Long 1H Coherence Lifetimes. J. Am. Chem. Soc. 2007, 129, 14118–14119. 10.1021/ja075319z. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G.; Ravera E.. Chapter 7: Transition Metal Ions: Shift and Relaxation. In NMR of Paramagnetic Molecules: Applications to Metallobiomolecules and Modelsetallobiomolecules and Models; Elsevier Science & Technology: Oxford, 2017; pp 175–253. [Google Scholar]
- Andreini C.; Bertini I.; Rosato A. A Hint to Search for Metalloproteins in Gene Banks. Bioinformatics 2004, 20, 1373–1380. 10.1093/bioinformatics/bth095. [DOI] [PubMed] [Google Scholar]
- Aguiar A.; de Haas G. H.; Jansen E. H. J. M.; Slotboom A. J.; Williams R. J. P. Proton-Nuclear-Magnetic-Resonance/pH-Titration Studies of the Histidines of Pancreatic Phospholipase A2. Eur. J. Biochem. 1979, 100, 511–518. 10.1111/j.1432-1033.1979.tb04196.x. [DOI] [PubMed] [Google Scholar]
- Cookson D. J.; Levine B. A.; Williams R. J. P.; Jontell M.; Linde A.; de Bernard B. Cation Binding by the Rat-Incisor-Dentine Phosphoprotein: A Spectroscopic Investigation. Eur. J. Biochem. 1980, 110, 273–278. 10.1111/j.1432-1033.1980.tb04865.x. [DOI] [PubMed] [Google Scholar]
- Fairbrother W. J.; Graham H. C.; Williams R. J. P. An NMR Study of Anion Binding to Yeast Phosphoglycerate Kinase. Eur. J. Biochem. 1990, 190, 161–169. 10.1111/j.1432-1033.1990.tb15560.x. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Bren K. L.; Cremonini M. A.; Gray H. B.; Luchinat C.; Turano P. The Use of Pseudocontact Shifts to Refine Solution Structures of Paramagnetic Metalloproteins: Met80Ala Cyano-Cytochrome c as an Example. JBIC, J. Biol. Inorg. Chem. 1996, 1, 117–126. 10.1007/s007750050030. [DOI] [Google Scholar]
- Sands R. H.; Dunham W. R. Spectroscopic Studies on Two-Iron Ferredoxins. Q. Rev. Biophys. 1974, 7, 443–504. 10.1017/S0033583500001517. [DOI] [PubMed] [Google Scholar]
- Bryar T. R.; Daughney C. J.; Knight R. J. Paramagnetic Effects of Iron(III) Species on Nuclear Magnetic Relaxation of Fluid Protons in Porous Media. J. Magn. Reson. 2000, 142, 74–85. 10.1006/jmre.1999.1917. [DOI] [PubMed] [Google Scholar]
- Nersissian A. M.; Shipp E. L. Blue Copper-Binding Domains. Adv. Protein Chem. 2002, 60, 271–340. 10.1016/S0065-3233(02)60056-7. [DOI] [PubMed] [Google Scholar]
- Murthy N. N.; Karlin K. D.; Bertini I.; Luchinat C. NMR and Electronic Relaxation in Paramagnetic Dicopper(II) Compounds. J. Am. Chem. Soc. 1997, 119, 2156–2162. 10.1021/ja961555q. [DOI] [Google Scholar]
- Andersson K. K.; Schmidt P. P.; Katterle B.; Strand K. R.; Palmer A. E.; Lee S. K.; Solomon E. I.; Gräslund A.; Barra A.-L. Examples of High-Frequency EPR Studies in Bioinorganic Chemistry. JBIC, J. Biol. Inorg. Chem. 2003, 8, 235–247. 10.1007/s00775-002-0429-0. [DOI] [PubMed] [Google Scholar]
- Fielding A. J.; Concilio M. G.; Heaven G.; Hollas M. A. New Developments in Spin Labels for Pulsed Dipolar EPR. Molecules 2014, 19, 16998–17025. 10.3390/molecules191016998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L.; Sykes B. D. Use of Lanthanide-Induced Nuclear Magnetic Resonance Shifts for Determination of Protein Structure in Solution: EF Calcium Binding Site of Carp Parvalbumin. Biochemistry 1983, 22, 4366–4373. 10.1021/bi00288a004. [DOI] [PubMed] [Google Scholar]
- Dwek R. A.; Richards R. E.; Morallee K. G.; Nieboer E.; Williams R. J. P.; Xavier A. V. The Lanthanide Cations as Probes in Biological Systems: Proton Relaxation Enhancement Studies for Model Systems and Lysozyme. Eur. J. Biochem. 1971, 21, 204–209. 10.1111/j.1432-1033.1971.tb01457.x. [DOI] [PubMed] [Google Scholar]
- Goldfarb D. Gd3+ Spin Labeling for Distance Measurements by Pulse EPR Spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 9685–9699. 10.1039/c3cp53822b. [DOI] [PubMed] [Google Scholar]
- Potapov A.; Yagi H.; Huber T.; Jergic S.; Dixon N. E.; Otting G.; Goldfarb D. Nanometer-Scale Distance Measurements in Proteins Using Gd3+ Spin Labeling. J. Am. Chem. Soc. 2010, 132, 9040–9048. 10.1021/ja1015662. [DOI] [PubMed] [Google Scholar]
- Qi M.; Groß A.; Jeschke G.; Godt A.; Drescher M. Gd(III)-PyMTA Label Is Suitable for In-Cell EPR. J. Am. Chem. Soc. 2014, 136, 15366–15378. 10.1021/ja508274d. [DOI] [PubMed] [Google Scholar]
- Orton H. W.; Otting G. Accurate Electron-Nucleus Distances from Paramagnetic Relaxation Enhancements. J. Am. Chem. Soc. 2018, 140, 7688–7697. 10.1021/jacs.8b03858. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Janik M. B. L.; Lee Y. M.; Luchinat C.; Rosato A. Magnetic Susceptibility Tensor Anisotropies for a Lanthanide Ion Series in a Fixed Protein Matrix. J. Am. Chem. Soc. 2001, 123, 4181–4188. 10.1021/ja0028626. [DOI] [PubMed] [Google Scholar]
- Joss D.; Bertrams M.-S.; Häussinger D. A Sterically Overcrowded, Isopropyl-Substituted, Lanthanide-Chelating Tag for Protein Pseudocontact Shift NMR Spectroscopy: Synthesis of Its Macrocyclic Scaffold and Benchmarking on Ubiquitin S57C and HCA II S166C. Chem. - Eur. J. 2019, 25, 11910–11917. 10.1002/chem.201901692. [DOI] [PubMed] [Google Scholar]
- Keizers P. H. J.; Saragliadis A.; Hiruma Y.; Overhand M.; Ubbink M. Design, Synthesis, and Evaluation of a Lanthanide Chelating Protein Probe: CLaNP-5 Yields Predictable Paramagnetic Effects Independent of Environment. J. Am. Chem. Soc. 2008, 130, 14802–14812. 10.1021/ja8054832. [DOI] [PubMed] [Google Scholar]
- Joss D.; Walliser R. M.; Zimmermann K.; Häussinger D. Conformationally Locked Lanthanide Chelating Tags for Convenient Pseudocontact Shift Protein Nuclear Magnetic Resonance Spectroscopy. J. Biomol. NMR 2018, 72, 29–38. 10.1007/s10858-018-0203-4. [DOI] [PubMed] [Google Scholar]
- Lee M. D.; Loh C.-T.; Shin J.; Chhabra S.; Dennis M. L.; Otting G.; Swarbrick J. D.; Graham B. Compact, Hydrophilic, Lanthanide-Binding Tags for Paramagnetic NMR Spectroscopy. Chem. Sci. 2015, 6, 2614–2624. 10.1039/C4SC03892D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiste J. L.; Wagner G. Utilization of Site-Directed Spin Labeling and High-Resolution Heteronuclear Nuclear Magnetic Resonance for Global Fold Determination of Large Proteins with Limited Nuclear Overhauser Effect Data. Biochemistry 2000, 39, 5355–5365. 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- Fukuyama K. Structure and Function of Plant-Type Ferredoxins. Photosynth. Res. 2004, 81, 289–301. 10.1023/B:PRES.0000036882.19322.0a. [DOI] [PubMed] [Google Scholar]
- Reedy C. J.; Gibney B. R. Heme Protein Assemblies. Chem. Rev. 2004, 104, 617–650. 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G. Hyperfine Shifts in Low-Spin Iron(III) Hemes: A Ligand Field Analysis. Eur. J. Inorg. Chem. 2000, 2000, 2473–2480. . [DOI] [Google Scholar]
- Antonkine M. L.; Bentrop D.; Bertini I.; Luchinat C.; Shen G.-Z.; Bryant D. A.; Stehlik D.; Golbeck J. H. Paramagnetic 1H NMR Spectroscopy of the Reduced, Unbound Photosystem I Subunit PsaC: Sequence-Specific Assignment of Contact-Shifted Resonances and Identification of Mixed- and Equal-Valence Fe-Fe Pairs in [4Fe-4S] Centers FA- and FB-. JBIC, J. Biol. Inorg. Chem. 2000, 5, 381–392. 10.1007/PL00010667. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Turano P.; Battaini G.; Casella L. The Magnetic Properties of Myoglobin as Studied by NMR Spectroscopy. Chem. - Eur. J. 2003, 9, 2316–2322. 10.1002/chem.200204562. [DOI] [PubMed] [Google Scholar]
- Dilg A. W. E.; Capozzi F.; Mentler M.; Iakovleva O.; Luchinat C.; Bertini I.; Parak F. G. Comparison and Characterization of the [Fe4S4]2+/3+ Centre in the Wild-Type and C77S Mutated HiPIPs from Chromatium Vinosum Monitored by Mossbauer, 57Fe ENDOR and EPR Spectroscopies. JBIC, J. Biol. Inorg. Chem. 2001, 6, 232–246. 10.1007/s007750000191. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Felli I. C.; Sarrou J. Backbone-Only Restraints for Fast Determination of the Protein Fold: The Role of Paramagnetism-Based Restraints. Cytochrome B562 as an Example. J. Magn. Reson. 2005, 172, 191–200. 10.1016/j.jmr.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Macinai R.; Martinuzzi S.; Pierattelli R.; Silvia Viezzoli M. Isolation and Characterization of Cytochrome c2 from Rhodopseudomonas Palustris. Inorg. Chim. Acta 1998, 269, 125–134. 10.1016/S0020-1693(97)05778-2. [DOI] [Google Scholar]
- Banci L.; Bertini I.; Bren K. L.; Gray H. B.; Sompornpisut P.; Turano P. Solution Structure of Oxidized Saccharomyces Cerevisiae Iso-1-Cytochrome c. Biochemistry 1997, 36, 8992–9001. 10.1021/bi963025c. [DOI] [PubMed] [Google Scholar]
- Adman E. T. Copper Protein Structures. Adv. Protein Chem. 1991, 42, 145–197. 10.1016/S0065-3233(08)60536-7. [DOI] [PubMed] [Google Scholar]
- Donaire A.; Salgado J.; Moratal J.-M. Determination of the Magnetic Axes of Cobalt(II) and Nickel(II) Azurins from 1H NMR Data: Influence of the Metal and Axial Ligands on the Origin of Magnetic Anisotropy in Blue Copper Proteins. Biochemistry 1998, 37, 8659–8673. 10.1021/bi971974f. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Fernández C. O.; Karlsson B. G.; Leckner J.; Luchinat C.; Malmström B. G.; Nersissian A. M.; Pierattelli R.; Shipp E.; Valentine J. S.; et al. Structural Information through NMR Hyperfine Shifts in Blue Copper Proteins. J. Am. Chem. Soc. 2000, 122, 3701–3707. 10.1021/ja992674j. [DOI] [Google Scholar]
- Pidcock E.; Moore G. R. Structural Characteristics of Protein Binding Sites for Calcium and Lanthanide Ions. JBIC, J. Biol. Inorg. Chem. 2001, 6, 479–489. 10.1007/s007750100214. [DOI] [PubMed] [Google Scholar]
- Snyder E. E.; Buoscio B. W.; Falke J. J. Calcium(II) Site Specificity: Effect of Size and Charge on Metal Ion Binding to an EF-Hand-like Site. Biochemistry 1990, 29, 3937–3943. 10.1021/bi00468a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Donaire A.; Jiménez B.; Luchinat C.; Parigi G.; Piccioli M.; Poggi L. Paramagnetism-Based versus Classical Constraints: An Analysis of the Solution Structure of Ca Ln Calbindin D9k. J. Biomol. NMR 2001, 21, 85–98. 10.1023/A:1012422402545. [DOI] [PubMed] [Google Scholar]
- Biekofsky R. R.; Muskett F. W.; Schmidt J. M.; Martin S. R.; Browne J. P.; Bayley P. M.; Feeney J. NMR Approaches for Monitoring Domain Orientations in Calcium-Binding Proteins in Solution Using Partial Replacement of Ca2+ by Tb3+. FEBS Lett. 1999, 460, 519–526. 10.1016/S0014-5793(99)01410-6. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Gupta Y. K.; Luchinat C.; Parigi G.; Peana M.; Sgheri L.; Yuan J. Paramagnetism-Based NMR Restraints Provide Maximum Allowed Probabilities for the Different Conformations of Partially Independent Protein Domains. J. Am. Chem. Soc. 2007, 129, 12786–12794. 10.1021/ja0726613. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Gelis I.; Katsaros N.; Luchinat C.; Provenzani A. Tuning the Affinity for Lanthanides of Calcium Binding Proteins. Biochemistry 2003, 42, 8011–8021. 10.1021/bi034494z. [DOI] [PubMed] [Google Scholar]
- Ma C.; Opella S. J. Lanthanide Ions Bind Specifically to an Added “EF-Hand” and Orient a Membrane Protein in Micelles for Solution NMR Spectroscopy. J. Magn. Reson. 2000, 146, 381–384. 10.1006/jmre.2000.2172. [DOI] [PubMed] [Google Scholar]
- Franz K. J.; Nitz M.; Imperiali B. Lanthanide-Binding Tags as Versatile Protein Coexpression Probes. ChemBioChem 2003, 4, 265–271. 10.1002/cbic.200390046. [DOI] [PubMed] [Google Scholar]
- Nitz M.; Franz K. J.; Maglathlin R. L.; Imperiali B. A Powerful Combinatorial Screen to Identify High-Affinity Terbium(III)-Binding Peptides. ChemBioChem 2003, 4, 272–276. 10.1002/cbic.200390047. [DOI] [PubMed] [Google Scholar]
- Nitz M.; Sherawat M.; Franz K. J.; Peisach E.; Allen K. N.; Imperiali B. Structural Origin of the High Affinity of a Chemically Evolved Lanthanide-Binding Peptide. Angew. Chem., Int. Ed. 2004, 43, 3682–3685. 10.1002/anie.200460028. [DOI] [PubMed] [Google Scholar]
- Silvaggi N. R.; Martin L. J.; Schwalbe H.; Imperiali B.; Allen K. N. Double-Lanthanide-Binding Tags for Macromolecular Crystallographic Structure Determination. J. Am. Chem. Soc. 2007, 129, 7114–7120. 10.1021/ja070481n. [DOI] [PubMed] [Google Scholar]
- Barthelmes D.; Gränz M.; Barthelmes K.; Allen K. N.; Imperiali B.; Prisner T.; Schwalbe H. Encoded Loop-Lanthanide-Binding Tags for Long-Range Distance Measurements in Proteins by NMR and EPR Spectroscopy. J. Biomol. NMR 2015, 63, 275–282. 10.1007/s10858-015-9984-x. [DOI] [PubMed] [Google Scholar]
- Barthelmes K.; Reynolds A. M.; Peisach E.; Jonker H. R. A.; DeNunzio N. J.; Allen K. N.; Imperiali B.; Schwalbe H. Engineering Encodable Lanthanide-Binding Tags into Loop Regions of Proteins. J. Am. Chem. Soc. 2011, 133, 808–819. 10.1021/ja104983t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X.-C.; Huber T.; Dixon N. E.; Otting G. Site-Specific Labelling of Proteins with a Rigid Lanthanide-Binding Tag. ChemBioChem 2006, 7, 1599–1604. 10.1002/cbic.200600142. [DOI] [PubMed] [Google Scholar]
- Su X.-C.; McAndrew K.; Huber T.; Otting G. Lanthanide-Binding Peptides for NMR Measurements of Residual Dipolar Couplings and Paramagnetic Effects from Multiple Angles. J. Am. Chem. Soc. 2008, 130, 1681–1687. 10.1021/ja076564l. [DOI] [PubMed] [Google Scholar]
- Saio T.; Ogura K.; Yokochi M.; Kobashigawa Y.; Inagaki F. Two-Point Anchoring of a Lanthanide-Binding Peptide to a Target Protein Enhances the Paramagnetic Anisotropic Effect. J. Biomol. NMR 2009, 44, 157–166. 10.1007/s10858-009-9325-z. [DOI] [PubMed] [Google Scholar]
- Saio T.; Yokochi M.; Kumeta H.; Inagaki F. PCS-Based Structure Determination of Protein-Protein Complexes. J. Biomol. NMR 2010, 46, 271–280. 10.1007/s10858-010-9401-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. R.; Lauritzen C.; Dahl S. W.; Pedersen J.; Led J. J. Binding Ability of a HHP-Tagged Protein towards Ni2+ Studied by Paramagnetic NMR Relaxation: The Possibility of Obtaining Long-Range Structure Information. J. Biomol. NMR 2004, 29, 175–185. 10.1023/B:JNMR.0000019251.09648.c4. [DOI] [PubMed] [Google Scholar]
- Voss J.; Salwiński L.; Kaback H. R.; Hubbell W. L. A Method for Distance Determination in Proteins Using a Designed Metal Ion Binding Site and Site-Directed Spin Labeling: Evaluation with T4 Lysozyme. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 12295–12299. 10.1073/pnas.92.26.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham T. F.; Putterman M. R.; Desai A.; Horne W. S.; Saxena S. The Double-Histidine Cu2+-Binding Motif: A Highly Rigid, Site-Specific Spin Probe for Electron Spin Resonance Distance Measurements. Angew. Chem., Int. Ed. 2015, 54, 6330–6334. 10.1002/anie.201501968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawless M. J.; Ghosh S.; Cunningham T. F.; Shimshi A.; Saxena S. On the Use of the Cu2+-Iminodiacetic Acid Complex for Double Histidine Based Distance Measurements by Pulsed ESR. Phys. Chem. Chem. Phys. 2017, 19, 20959–20967. 10.1039/C7CP02564E. [DOI] [PubMed] [Google Scholar]
- Bahramzadeh A.; Jiang H.; Huber T.; Otting G. Two Histidines in an α-Helix: A Rigid Co2+ -Binding Motif for PCS Measurements by NMR Spectroscopy. Angew. Chem., Int. Ed. 2018, 57, 6226–6229. 10.1002/anie.201802501. [DOI] [PubMed] [Google Scholar]
- Wang L.; Schultz P. G. Expanding the Genetic Code. Angew. Chem., Int. Ed. 2005, 44, 34–66. 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- Wan W.; Tharp J. M.; Liu W. R. Pyrrolysyl-TRNA Synthetase: An Ordinary Enzyme but an Outstanding Genetic Code Expansion Tool. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 1059–1070. 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z.; Johnston W. A.; Alexandrov K. Cell-Free Approach for Non-Canonical Amino Acids Incorporation Into Polypeptides. Front. Bioeng. Biotechnol. 2020, 10.3389/fbioe.2020.01031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. S.; Schultz P. G. Biosynthesis of a Site-Specific DNA Cleaving Protein. J. Am. Chem. Soc. 2008, 130, 13194–13195. 10.1021/ja804653f. [DOI] [PubMed] [Google Scholar]
- Xie J.; Liu W.; Schultz P. G. A Genetically Encoded Bidentate, Metal-Binding Amino Acid. Angew. Chem., Int. Ed. 2007, 46, 9239–9242. 10.1002/anie.200703397. [DOI] [PubMed] [Google Scholar]
- Luo X.; Wang T. S. A.; Zhang Y.; Wang F.; Schultz P. G. Stabilizing Protein Motifs with a Genetically Encoded Metal-Ion Chelator. Cell Chem. Biol. 2016, 23, 1098–1102. 10.1016/j.chembiol.2016.08.007. [DOI] [PubMed] [Google Scholar]
- Nguyen T. H. D.; Ozawa K.; Stanton-Cook M.; Barrow R.; Huber T.; Otting G. Generation of Pseudocontact Shifts in Protein NMR Spectra with a Genetically Encoded Cobalt(II)-Binding Amino Acid. Angew. Chem. 2011, 123, 718–720. 10.1002/ange.201005672. [DOI] [PubMed] [Google Scholar]
- Lee H. S.; Spraggon G.; Schultz P. G.; Wang F. Genetic Incorporation of a Metal-Ion Chelating Amino Acid into Proteins as a Biophysical Probe. J. Am. Chem. Soc. 2009, 131, 2345–2481. 10.1021/ja808340b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. H.; Wang V. S.; Radoicic J.; De Angelis A. A.; Berkamp S.; Opella S. J. Paramagnetic Relaxation Enhancement of Membrane Proteins by Incorporation of the Metal-Chelating Unnatural Amino Acid 2-Amino-3-(8-Hydroxyquinolin-3-Yl)Propanoic Acid (HQA). J. Biomol. NMR 2015, 61, 185–196. 10.1007/s10858-014-9884-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. S.; Spraggon G.; Schultz P. G.; Wang F. Genetic Incorporation of a Metal-Ion Chelating Amino Acid into Proteins as a Biophysical Probe. J. Am. Chem. Soc. 2009, 131, 2481–2483. 10.1021/ja808340b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W.; Guo J. Expanding the Chemistry of Fluorescent Protein Biosensors through Genetic Incorporation of Unnatural Amino Acids. Mol. BioSyst. 2013, 9, 2961–2970. 10.1039/c3mb70204a. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Huang F.; Huber T.; Su X.-C. Site-Specific Tagging Proteins with a Rigid, Small and Stable Transition Metal Chelator, 8-Hydroxyquinoline, for Paramagnetic NMR Analysis. J. Biomol. NMR 2016, 64, 103–113. 10.1007/s10858-016-0011-7. [DOI] [PubMed] [Google Scholar]
- Park N.; Ryu J.; Jang S.; Lee H. S. Metal Ion Affinity Purification of Proteins by Genetically Incorporating Metal-Chelating Amino Acids. Tetrahedron 2012, 68, 4649–4654. 10.1016/j.tet.2012.04.019. [DOI] [Google Scholar]
- Jones D. H.; Cellitti S. E.; Hao X.; Zhang Q.; Jahnz M.; Summerer D.; Schultz P. G.; Uno T.; Geierstanger B. H. Site-Specific Labeling of Proteins with NMR-Active Unnatural Amino Acids. J. Biomol. NMR 2010, 46, 89–100. 10.1007/s10858-009-9365-4. [DOI] [PubMed] [Google Scholar]
- Yang A.; Ha S.; Ahn J.; Kim R.; Kim S.; Lee Y.; Kim J.; Söll D.; Lee H. Y.; Park H. S. A Chemical Biology Route to Site-Specific Authentic Protein Modifications. Science 2016, 354, 623–626. 10.1126/science.aah4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Oh S.; Yang A.; Kim J.; Söll D.; Lee D.; Park H.-S. A Facile Strategy for Selective Incorporation of Phosphoserine into Histones. Angew. Chem., Int. Ed. 2013, 52, 5771–5775. 10.1002/anie.201300531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirman N. L.; Barber K. W.; Aerni H. R.; Ma N. J.; Haimovich A. D.; Rogulina S.; Isaacs F. J.; Rinehart J. A Flexible Codon in Genomically Recoded Escherichia Coli Permits Programmable Protein Phosphorylation. Nat. Commun. 2015, 6, 8130. 10.1038/ncomms9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekkattu Tharayil S.; Mahawaththa M. C.; Loh C.-T.; Adekoya I.; Otting G. Phosphoserine for the Generation of Lanthanide-Binding Sites on Proteins for Paramagnetic Nuclear Magnetic Resonance Spectroscopy. Magn. Reson. 2021, 2, 1–13. 10.5194/mr-2-1-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. J.; Borbas J.; Drescher M.; Summerer D. A Genetically Encoded Spin Label for Electron Paramagnetic Resonance Distance Measurements. J. Am. Chem. Soc. 2014, 136, 1238–1241. 10.1021/ja411535q. [DOI] [PubMed] [Google Scholar]
- Stoller S.; Sicoli G.; Baranova T. Y.; Bennati M.; Diederichsen U. TOPP: A Novel Nitroxide-Labeled Amino Acid for EPR Distance Measurements. Angew. Chem., Int. Ed. 2011, 50, 9743–9746. 10.1002/anie.201103315. [DOI] [PubMed] [Google Scholar]
- Marchetto R.; Schreier S.; Nakaie C. R. A Novel Spin-Labeled Amino Acid Derivative for Use in Peptide Synthesis: (9-Fluorenylmethyloxycarbonyl)-2,2,6,6- Tetramethylpiperidine-N-Oxyl-4-Amino-4-Carboxylic Acid. J. Am. Chem. Soc. 1993, 115, 11042–11043. 10.1021/ja00076a093. [DOI] [Google Scholar]
- Nguyen D. P.; Lusic H.; Neumann H.; Kapadnis P. B.; Deiters A.; Chin J. W. Genetic Encoding and Labeling of Aliphatic Azides and Alkynes in Recombinant Proteins via a Pyrrolysyl-TRNA Synthetase/TRNACUA Pair and Click Chemistry. J. Am. Chem. Soc. 2009, 131, 8720–8721. 10.1021/ja900553w. [DOI] [PubMed] [Google Scholar]
- Chin J. W.; Santoro S. W.; Martin A. B.; King D. S.; Wang L.; Schultz P. G. Addition of P-Azido-L-Phenylalanine to the Genetic Code of Escherichia Coli. J. Am. Chem. Soc. 2002, 124, 9026–9027. 10.1021/ja027007w. [DOI] [PubMed] [Google Scholar]
- Widder P.; Berner F.; Summerer D.; Drescher M. Double Nitroxide Labeling by Copper-Catalyzed Azide-Alkyne Cycloadditions with Noncanonical Amino Acids for Electron Paramagnetic Resonance Spectroscopy. ACS Chem. Biol. 2019, 14, 839–844. 10.1021/acschembio.8b01111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Kugele A.; Braun T. S.; Widder P.; Williams L.; Schmidt M. J.; Summerer D.; Drescher M. Site-Directed Spin Labelling of Proteins by Suzuki-Miyaura Coupling via a Genetically Encoded Aryliodide Amino Acid. Chem. Commun. 2019, 55, 1923–1926. 10.1039/C8CC09325C. [DOI] [PubMed] [Google Scholar]
- Lindfors H. E.; de Koning P. E.; Drijfhout J. W.; Venezia B.; Ubbink M. Mobility of TOAC Spin-Labelled Peptides Binding to the Src SH3 Domain Studied by Paramagnetic NMR. J. Biomol. NMR 2008, 41, 157–167. 10.1007/s10858-008-9248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. J.; Fedoseev A.; Bücker D.; Borbas J.; Peter C.; Drescher M.; Summerer D. EPR Distance Measurements in Native Proteins with Genetically Encoded Spin Labels. ACS Chem. Biol. 2015, 10, 2764–2771. 10.1021/acschembio.5b00512. [DOI] [PubMed] [Google Scholar]
- Halbmair K.; Wegner J.; Diederichsen U.; Bennati M. Pulse EPR Measurements of Intramolecular Distances in a TOPP-Labeled Transmembrane Peptide in Lipids. Biophys. J. 2016, 111, 2345–2348. 10.1016/j.bpj.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roser P.; Schmidt M. J.; Drescher M.; Summerer D. Site-Directed Spin Labeling of Proteins for Distance Measurements in vitro and in Cells. Org. Biomol. Chem. 2016, 14, 5468–5476. 10.1039/C6OB00473C. [DOI] [PubMed] [Google Scholar]
- Wegner J.; Valora G.; Halbmair K.; Kehl A.; Worbs B.; Bennati M.; Diederichsen U. Semi-Rigid Nitroxide Spin Label for Long-Range EPR Distance Measurements of Lipid Bilayer Embedded β-Peptides. Chem. - Eur. J. 2019, 25, 2203–2207. 10.1002/chem.201805880. [DOI] [PubMed] [Google Scholar]
- Narancic T.; Almahboub S. A.; O’Connor K. E. Unnatural Amino Acids: Production and Biotechnological Potential. World J. Microbiol. Biotechnol. 2019, 35, 67. 10.1007/s11274-019-2642-9. [DOI] [PubMed] [Google Scholar]
- Talukder P.; Chen S.; Roy B.; Yakovchuk P.; Spiering M. M.; Alam M. P.; Madathil M. M.; Bhattacharya C.; Benkovic S. J.; Hecht S. M. Cyanotryptophans as Novel Fluorescent Probes for Studying Protein Conformational Changes and DNA-Protein Interaction. Biochemistry 2015, 54, 7457–7469. 10.1021/acs.biochem.5b01085. [DOI] [PubMed] [Google Scholar]
- Hopkins C. D.; Malinakova H. C. Synthesis of Unnatural α-Amino Acid Derivatives via a Three-Component Coupling Method Utilizing an Allylpalladium Umpolung. Synthesis (Stuttg). 2007, 3558–3566. 10.1055/s-2007-990845. [DOI] [Google Scholar]
- Dwulet N. C.; Zolfaghari T. A.; Brown M. L.; Cannon J. S. Diastereoselective Synthesis of Unnatural Amino Acids by Alkylation of α-tert-Butanesulfinamide Auxiliary-Bound Enolates. J. Org. Chem. 2018, 83, 11510–11518. 10.1021/acs.joc.8b01379. [DOI] [PubMed] [Google Scholar]
- Inada H.; Furukawa K.; Shibuya M.; Yamamoto Y. One-Pot, Two-Step Synthesis of Unnatural α-Amino Acids Involving the Exhaustive Aerobic Oxidation of 1,2-Diols. Chem. Commun. 2019, 55, 15105–15108. 10.1039/C9CC07889D. [DOI] [PubMed] [Google Scholar]
- Aycock R. A.; Pratt C. J.; Jui N. T. Aminoalkyl Radicals as Powerful Intermediates for the Synthesis of Unnatural Amino Acids and Peptides. ACS Catal. 2018, 8, 9115–9119. 10.1021/acscatal.8b03031. [DOI] [Google Scholar]
- Katz B. M.; Lundquist L. J.; Walsh D. A.; Glass D. B. Synthesis, Characterization and Inhibitory Activities of (4-N3[3,5-3H]Phe10)PKI(6–22)Amide and Its Precursors: Photoaffinity Labeling Peptides for the Active Site of Cyclic AMP-Dependent Protein Kinase. Int. J. Pept. Protein Res. 1989, 33, 439–445. 10.1111/j.1399-3011.1989.tb00220.x. [DOI] [PubMed] [Google Scholar]
- Richardson M. B.; Brown D. B.; Vasquez C. A.; Ziller J. W.; Johnston K. M.; Weiss G. A. Synthesis and Explosion Hazards of 4-Azido- l -Phenylalanine. J. Org. Chem. 2018, 83, 4525–4536. 10.1021/acs.joc.8b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Ma D. Synthesis of Aryl Azides and Vinyl Azides via Proline-Promoted CuI-Catalyzed Coupling Reactions. Chem. Commun. 2004, 7, 888–889. 10.1039/b400878b. [DOI] [PubMed] [Google Scholar]
- Imperiali B.; Prins T. J.; Fisher S. L. Chemoenzymatic Synthesis of 2-Amino-3-(2,2′-Bipyridinyl)Propanoic Acids. J. Org. Chem. 1993, 58, 1613–1616. 10.1021/jo00058a058. [DOI] [Google Scholar]
- Haugland M. M.; Lovett J. E.; Anderson E. A. Advances in the Synthesis of Nitroxide Radicals for Use in Biomolecule Spin Labelling. Chem. Soc. Rev. 2018, 47, 668–680. 10.1039/C6CS00550K. [DOI] [PubMed] [Google Scholar]
- Kirilyuk I. A.; Polienko Y. F.; Krumkacheva O. A.; Strizhakov R. K.; Gatilov Y. V.; Grigor’ev I. A.; Bagryanskaya E. G. Synthesis of 2,5-Bis(Spirocyclohexane)-Substituted Nitroxides of Pyrroline and Pyrrolidine Series, Including Thiol-Specific Spin Label: An Analogue of MTSSL with Long Relaxation Time. J. Org. Chem. 2012, 77, 8016–8027. 10.1021/jo301235j. [DOI] [PubMed] [Google Scholar]
- Smith D. J.; Miggio E. T.; Kenyon G. L. Simple Alkanethiol Groups for Temporary Blocking of Sulfhydryl Groups of Enzymes. Biochemistry 1975, 14, 766–771. 10.1021/bi00675a019. [DOI] [PubMed] [Google Scholar]
- Gaffney B. J.The Chemistry of Spin Labe. In Spin Labeling: Theory and Applications; Berliner L. J., Ed.; Academic Press, 1976; pp 183–238. [Google Scholar]
- Zecherle G. N.; Oleinikov A.; Traut R. R. The Proximity of the C-Terminal Domain of Escherichia Coli Ribosomal Protein L7/L12 to L10 Determined by Cysteine Site-Directed Mutagenesis and Protein-Protein Cross-Linking. J. Biol. Chem. 1992, 267, 5889–5896. 10.1016/S0021-9258(18)42637-3. [DOI] [PubMed] [Google Scholar]
- Griffith O. H.; McConnell H. M. A Nitroxide-Maleimide Spin Label. Proc. Natl. Acad. Sci. U. S. A. 1966, 55, 8–11. 10.1073/pnas.55.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berliner L. J.; Grunwald J.; Hankovszky H. O.; Hideg K. A Novel Reversible Thiol-Specific Spin Label: Papain Active Site Labeling and Inhibition. Anal. Biochem. 1982, 119, 450–455. 10.1016/0003-2697(82)90612-1. [DOI] [PubMed] [Google Scholar]
- Hankovszky H. O.; Hideg K.; Lex L. Nitroxyls: VII1. Synthesis and Reactions of Highly Reactive 1-Oxyl-2,2,5,5-Tetramethyl-2,5-Dihydro-Pyrrole-3-Ylmethyl Sulfonates. Synthesis (Stuttg). 1980, 914–916. 10.1055/s-1980-29269. [DOI] [Google Scholar]
- Meyer V.; Swanson M. A.; Clouston L. J.; Boratyński P. J.; Stein R. A.; Mchaourab H. S.; Rajca A.; Eaton S. S.; Eaton G. R. Room-Temperature Distance Measurements of Immobilized Spin-Labeled Protein by DEER/PELDOR. Biophys. J. 2015, 108, 1213–1219. 10.1016/j.bpj.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Columbus L.; Hubbell W. L. A New Spin on Protein Dynamics. Trends Biochem. Sci. 2002, 27, 288–295. 10.1016/S0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- Lietzow M. A.; Hubbell W. L. Motion of Spin Label Side Chains in Cellular Retinol-Binding Protein: Correlation with Structure and Nearest-Neighbor Interactions in An Antiparallel β-Sheet. Biochemistry 2004, 43, 3137–3151. 10.1021/bi0360962. [DOI] [PubMed] [Google Scholar]
- Columbus L.; Kálai T.; Jekö J.; Hideg K.; Hubbell W. L. Molecular Motion of Spin Labeled Side Chains in α-Helices: Analysis by Variation of Side Chain Structure. Biochemistry 2001, 40, 3828–3846. 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- Mchaourab H. S.; Lietzow M. A.; Hideg K.; Hubbell W. L. Motion of Spin-Labeled Side Chains in T4 Lysozyme. Correlation with Protein Structure and Dynamics. Biochemistry 1996, 35, 7692–7704. 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- Fawzi N. L.; Fleissner M. R.; Anthis N. J.; Kálai T.; Hideg K.; Hubbell W. L.; Clore G. M. A Rigid Disulfide-Linked Nitroxide Side Chain Simplifies the Quantitative Analysis of PRE Data. J. Biomol. NMR 2011, 51, 105–114. 10.1007/s10858-011-9545-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. H.; Popova A. M.; Hideg K.; Qin P. Z. A Nucleotide-Independent Cyclic Nitroxide Label for Monitoring Segmental Motions in Nucleic Acids. BMC Biophys. 2015, 8, 6. 10.1186/s13628-015-0019-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kálai T.; Balog M.; Jekö J.; Hideg K. Synthesis and Reactions of a Symmetric Paramagnetic Pyrrolidine Diene. Synthesis (Stuttg). 1999, 973–980. 10.1055/s-1999-3502. [DOI] [Google Scholar]
- Fleissner M. R.; Bridges M. D.; Brooks E. K.; Cascio D.; Kálai T.; Hideg K.; Hubbell W. L. Structure and Dynamics of a Conformationally Constrained Nitroxide Side Chain and Applications in EPR Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16241–16246. 10.1073/pnas.1111420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher F. F.; Sanchania V. A.; Tolner B.; Wright Z. V. F.; Ryan C. P.; Smith M. E. B.; Ward J. M.; Caddick S.; Kay C. W. M.; Aeppli G.; et al. Homogeneous Antibody Fragment Conjugation by Disulfide Bridging Introduces “Spinostics.. Sci. Rep. 2013, 3, 1525. 10.1038/srep01525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjaj B.; Shah A.; Bell S.; Shirran S. L.; Botting C. H.; Slawin A. M. Z.; Hulme A. N.; Lovett J. E. Synthesis of Next-Generation Maleimide Radical Labels. Synlett 2016, 27, 2357–2361. 10.1055/s-0035-1562451. [DOI] [Google Scholar]
- Haugland M. M.; El-Sagheer A. H.; Porter R. J.; Peña J.; Brown T.; Anderson E. A.; Lovett J. E. 2′-Alkynylnucleotides: A Sequence- and Spin Label-Flexible Strategy for EPR Spectroscopy in DNA. J. Am. Chem. Soc. 2016, 138, 9069–9072. 10.1021/jacs.6b05421. [DOI] [PubMed] [Google Scholar]
- Kucher S.; Korneev S.; Tyagi S.; Apfelbaum R.; Grohmann D.; Lemke E. A.; Klare J. P.; Steinhoff H.-J.; Klose D. Orthogonal Spin Labeling Using Click Chemistry for in Vitro and in Vivo Applications. J. Magn. Reson. 2017, 275, 38–45. 10.1016/j.jmr.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Úr G.; Kálai T.; Balog M.; Bognár B.; Gulyás-Fekete G.; Hideg K. Synthesis of New Pyrroline Nitroxides with Ethynyl Functional Group. Synth. Commun. 2015, 45, 2122–2129. 10.1080/00397911.2015.1066391. [DOI] [Google Scholar]
- Keddie D. J.; Fairfull-Smith K. E.; Bottle S. E. The Palladium-Catalysed Copper-Free Sonogashira Coupling of Isoindoline Nitroxides: A Convenient Route to Robust Profluorescent Carbon-Carbon Frameworks. Org. Biomol. Chem. 2008, 6, 3135–3143. 10.1039/b806963h. [DOI] [PubMed] [Google Scholar]
- Kálai T.; Fleissner M. R.; Jekö J.; Hubbell W. L.; Hideg K. Synthesis of New Spin Labels for Cu-Free Click Conjugation. Tetrahedron Lett. 2011, 52, 2747–2749. 10.1016/j.tetlet.2011.03.077. [DOI] [Google Scholar]
- Saha S.; Jagtap A. P.; Sigurdsson S. T. Site-Directed Spin Labeling of 2′-Amino Groups in RNA with Isoindoline Nitroxides That Are Resistant to Reduction. Chem. Commun. 2015, 51, 13142–13145. 10.1039/C5CC05014F. [DOI] [PubMed] [Google Scholar]
- Gölz J. P.; Bockelmann S.; Mayer K.; Steinhoff H.-J.; Wieczorek H.; Huss M.; Klare J. P.; Menche D. EPR Studies of V-ATPase with Spin-Labeled Inhibitors DCC and Archazolid: Interaction Dynamics with Proton Translocating Subunit c. ChemMedChem 2016, 11, 420–428. 10.1002/cmdc.201500500. [DOI] [PubMed] [Google Scholar]
- Fleissner M. R.; Brustad E. M.; Kálai T.; Altenbach C.; Cascio D.; Peters F. B.; Hideg K.; Peuker S.; Schultz P. G.; Hubbell W. L. Site-Directed Spin Labeling of a Genetically Encoded Unnatural Amino Acid. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 21637–21642. 10.1073/pnas.0912009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Paletta J. T.; Berg K.; Reinhart E.; Rajca S.; Rajca A. Synthesis of Unnatural Amino Acids Functionalized with Sterically Shielded Pyrroline Nitroxides. Org. Lett. 2014, 16, 5298–5300. 10.1021/ol502449r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett J. E.; Haugland M. M.; Anderson E. A.Tuning the Properties of Nitroxide Spin Labels for Use in Electron Paramagnetic Resonance Spectroscopy through Chemical Modification of the Nitroxide Framework. In Electron Paramagnetic Resonance, Vol. 25; Chechik V., Murphy D. M., Eds.; The Royal Society of Chemistry: Cambridge, 2017; pp 1–34. [Google Scholar]
- Guo W.-W.; Zhang C.; Ye J.; Liu Z.-K.; Chen K.; Wu C.-D. Suspending Ion Electrocatalysts in Charged Metal–Organic Frameworks to Improve the Conductivity and Selectivity in Electroorganic Synthesis. Chem. - Asian J. 2019, 14, 3627–3634. 10.1002/asia.201900640. [DOI] [PubMed] [Google Scholar]
- Sakai K.; Yamada K.; Yamasaki T.; Kinoshita Y.; Mito F.; Utsumi H. Effective 2,6-Substitution of Piperidine Nitroxyl Radical by Carbonyl Compound. Tetrahedron 2010, 66, 2311–2315. 10.1016/j.tet.2010.02.004. [DOI] [Google Scholar]
- Paletta J. T.; Pink M.; Foley B.; Rajca S.; Rajca A. Synthesis and Reduction Kinetics of Sterically Shielded Pyrrolidine Nitroxides. Org. Lett. 2012, 14, 5322–5325. 10.1021/ol302506f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gölz J. P.; NejatyJahromy Y.; Bauer M.; Muhammad A.; Schnakenburg G.; Grimme S.; Schiemann O.; Menche D. Design, Synthesis, EPR-Studies and Conformational Bias of Novel Spin-Labeled DCC-Analogues for the Highly Regioselective Labeling of Aliphatic and Aromatic Carboxylic Acids. Chem. - Eur. J. 2016, 22, 9591–9598. 10.1002/chem.201600528. [DOI] [PubMed] [Google Scholar]
- Schulte B.; Tsotsalas M.; Becker M.; Studer A.; De Cola L. Dynamic Microcrystal Assembly by Nitroxide Exchange Reactions. Angew. Chem., Int. Ed. 2010, 49, 6881–6884. 10.1002/anie.201002851. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Noble B. B.; Mater A. C.; Monteiro M. J.; Coote M. L.; Jia Z. Effect of Heteroatom and Functionality Substitution on the Oxidation Potential of Cyclic Nitroxide Radicals: Role of Electrostatics in Electrochemistry. Phys. Chem. Chem. Phys. 2018, 20, 2606–2614. 10.1039/C7CP07444A. [DOI] [PubMed] [Google Scholar]
- Wetter C.; Gierlich J.; Knoop C. A.; Müller C.; Schulte T.; Studer A. Steric and Electronic Effects in Cyclic Alkoxyamines—Synthesis and Applications as Regulators for Controlled/Living Radical Polymerization. Chem. - Eur. J. 2004, 10, 1156–1166. 10.1002/chem.200305427. [DOI] [PubMed] [Google Scholar]
- Griffiths P. G.; Moad G.; Rizzardo E. Synthesis of the Radical Scavenger 1,1,3,3-Tetramethylisoindolin-2-Yloxyl. Aust. J. Chem. 1983, 36, 397–401. 10.1071/CH9830397. [DOI] [Google Scholar]
- Jayawardena V. C.; Fairfull-Smith K. E.; Bottle S. E. Improving the Yield of the Exhaustive Grignard Alkylation of N-Benzylphthalimide. Aust. J. Chem. 2013, 66, 619–625. 10.1071/CH12528. [DOI] [Google Scholar]
- Braun T. S.; Widder P.; Osswald U.; Groß L.; Williams L.; Schmidt M.; Helmle I.; Summerer D.; Drescher M. Isoindoline-Based Nitroxides as Bioresistant Spin Labels for Protein Labeling through Cysteines and Alkyne-Bearing Noncanonical Amino Acids. ChemBioChem 2020, 21, 958–962. 10.1002/cbic.201900537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.-S.; Mak K. W.; Tse M. K.; Yeung S. K.; Li B.-H.; Chan Y.-W. Reactions of Nitroxides with Metalloporphyrin Alkyls Bearing Beta Hydrogens: Aliphatic Carbon–Carbon Bond Activation by Metal Centered Radicals. J. Organomet. Chem. 2008, 693, 399–407. 10.1016/j.jorganchem.2007.11.009. [DOI] [Google Scholar]
- Chan K. S.; Li X. Z.; Lee S. Y. Ligand-Enhanced Aliphatic Carbon–Carbon Bond Activation of Nitroxides by Rhodium(II) Porphyrin. Organometallics 2010, 29, 2850–2856. 10.1021/om1000869. [DOI] [Google Scholar]
- Thomas K.; Chalmers B. A.; Fairfull-Smith K. E.; Bottle S. E. Approaches to the Synthesis of a Water-Soluble Carboxy Nitroxide. Eur. J. Org. Chem. 2013, 2013, 853–857. 10.1002/ejoc.201201551. [DOI] [Google Scholar]
- Ebright Y. W.; Chen Y.; Pendergrast P. S.; Ebright R. H. Incorporation of an EDTA-Metal Complex at a Rationally Selected Site within a Protein: Application to EDTA-Iron DNA Affinity Cleaving with Catabolite Gene Activator Protein (CAP) and Cro. Biochemistry 1992, 31, 10664–10670. 10.1021/bi00159a004. [DOI] [PubMed] [Google Scholar]
- Pintacuda G.; Moshref A.; Leonchiks A.; Sharipo A.; Otting G. Site-Specific Labelling with a Metal Chelator for Protein-Structure Refinement. J. Biomol. NMR 2004, 29, 351–361. 10.1023/B:JNMR.0000032610.17058.fe. [DOI] [PubMed] [Google Scholar]
- Gaponenko V.; Altieri A. S.; Li J.; Byrd R. A. Breaking Symmetry in the Structure Determination of (Large) Symmetric Protein Dimers. J. Biomol. NMR 2002, 24, 143–148. 10.1023/A:1020948529076. [DOI] [PubMed] [Google Scholar]
- Ikegami T.; Verdier L.; Sakhaii P.; Grimme S.; Pescatore B.; Saxena K.; Fiebig K. M.; Griesinger C. Novel Techniques for Weak Alignment of Proteins in Solution Using Chemical Tags Coordinating Lanthanide Ions. J. Biomol. NMR 2004, 29, 339–349. 10.1023/B:JNMR.0000032611.72827.de. [DOI] [PubMed] [Google Scholar]
- Leonov A.; Voigt B.; Rodriguez-Castañeda F.; Sakhaii P.; Griesinger C. Convenient Synthesis of Multifunctional EDTA-Based Chiral Metal Chelates Substituted with an S-Mesylcysteine. Chem. - Eur. J. 2005, 11, 3342–3348. 10.1002/chem.200400907. [DOI] [PubMed] [Google Scholar]
- Martorana A.; Yang Y.; Zhao Y.; Li Q.-F.; Su X.-C.; Goldfarb D. Mn(II) Tags for DEER Distance Measurements in Proteins via C-S Attachment. Dalt. Trans. 2015, 44, 20812–20816. 10.1039/C5DT04123F. [DOI] [PubMed] [Google Scholar]
- Hart J. R. Ethylenediaminetetraacetic Acid and Related Chelating Agents. Ullmann’s Encycl. Ind. Chem. 2011, 13, 573–578. 10.1002/14356007.a10_095.pub2. [DOI] [Google Scholar]
- Prudêncio M.; Rohovec J.; Peters J. A.; Tocheva E.; Boulanger M. J.; Murphy M. E. P.; Hupkes H. J.; Kosters W.; Impagliazzo A.; Ubbink M. A Caged Lanthanide Complex as a Paramagnetic Shift Agent for Protein NMR. Chem. - Eur. J. 2004, 10, 3252–3260. 10.1002/chem.200306019. [DOI] [PubMed] [Google Scholar]
- Parker D.; Dickins R. S.; Puschmann H.; Crossland C.; Howard J. A. K. Being Excited by Lanthanoid Coordination Complexes: Aqua Species, Chirality, Excited-State Chemistry, and Exchange Dynamics. Chem. Rev. 2002, 102, 1977–2010. 10.1021/cr010452+. [DOI] [PubMed] [Google Scholar]
- Chen J.-L.; Li B.; Li X.-Y.; Su X.-C. Dynamic Exchange of the Metal Chelating Moiety: A Key Factor in Determining the Rigidity of Protein–Tag Conjugates in Paramagnetic NMR. J. Phys. Chem. Lett. 2020, 11, 9493–9500. 10.1021/acs.jpclett.0c02196. [DOI] [PubMed] [Google Scholar]
- Peters F.; Maestre-Martinez M.; Leonov A.; Kovačič L.; Becker S.; Boelens R.; Griesinger C. Cys-Ph-TAHA: A Lanthanide Binding Tag for RDC and PCS Enhanced Protein NMR. J. Biomol. NMR 2011, 51, 329–337. 10.1007/s10858-011-9560-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W.-X.; Gu X.-H.; Dong X.; Tang C. Lanthanoid Tagging via an Unnatural Amino Acid for Protein Structure Characterization. J. Biomol. NMR 2017, 67, 273–282. 10.1007/s10858-017-0106-9. [DOI] [PubMed] [Google Scholar]
- Chen J.-L.; Zhao Y.; Gong Y.-J.; Pan B.-B.; Wang X.; Su X.-C. Stable and Rigid DTPA-like Paramagnetic Tags Suitable for in Vitro and in Situ Protein NMR Analysis. J. Biomol. NMR 2018, 70, 77–92. 10.1007/s10858-017-0160-3. [DOI] [PubMed] [Google Scholar]
- Bonnet C. S.; Buron F.; Caillé F.; Shade C. M.; Drahoš B.; Pellegatti L.; Zhang J.; Villette S.; Helm L.; Pichon C.; et al. Pyridine-Based Lanthanide Complexes Combining MRI and NIR Luminescence Activities. Chem. - Eur. J. 2012, 18, 1419–1431. 10.1002/chem.201102310. [DOI] [PubMed] [Google Scholar]
- Pellegatti L.; Zhang J.; Drahos B.; Villette S.; Suzenet F.; Guillaumet G.; Petoud S.; Tóth É. Pyridine-Based Lanthanide Complexes: Towards Bimodal Agents Operating as near Infrared Luminescent and MRI Reporters. Chem. Commun. 2008, 6591–6593. 10.1039/b817343e. [DOI] [PubMed] [Google Scholar]
- Ma R.-S.; Li Q.-F.; Wang A.-D.; Zhang J.-H.; Liu Z.-J.; Wu J.-H.; Su X.-C.; Ruan K. Determination of Pseudocontact Shifts of Low-Populated Excited States by NMR Chemical Exchange Saturation Transfer. Phys. Chem. Chem. Phys. 2016, 18, 13794–13798. 10.1039/C6CP01127F. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Li Q.-F.; Cao C.; Huang F.; Su X.-C. Site-Specific Labeling of Proteins with a Chemically Stable, High-Affinity Tag for Protein Study. Chem. - Eur. J. 2013, 19, 1097–1103. 10.1002/chem.201202495. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Wang J.-T.; Pei Y.-Y.; Su X.-C. Site-Specific Tagging Proteins via a Rigid, Stable and Short Thiolether Tether for Paramagnetic Spectroscopic Analysis. Chem. Commun. 2015, 51, 2824–2827. 10.1039/C4CC08493D. [DOI] [PubMed] [Google Scholar]
- Chen J.-L.; Wang X.; Yang F.; Cao C.; Otting G.; Su X.-C. 3D Structure Determination of an Unstable Transient Enzyme Intermediate by Paramagnetic NMR Spectroscopy. Angew. Chem., Int. Ed. 2016, 55, 13744–13748. 10.1002/anie.201606223. [DOI] [PubMed] [Google Scholar]
- Stetter H.; Frank W. Complex Formation with Tetraazac Ycloalkane-N, N’, N”, N”’-Tetraacetic Acids as a Function of Ring Size. Angew. Chem., Int. Ed. Engl. 1976, 15, 686. 10.1002/anie.197606861. [DOI] [Google Scholar]
- Izatt R. M.; Pawlak K.; Bradshaw J. S.; Bruening R. L. Thermodynamic and Kinetic Data for Macrocycle Interaction with Cations and Anions. Chem. Rev. 1991, 91, 1721–2085. 10.1021/cr00008a003. [DOI] [Google Scholar]
- Purgel M.; Baranyai Z.; De Blas A.; Rodríguez-Blas T.; Bányai I.; Platas-Iglesias C.; Tóth I. An NMR and DFT Investigation on the Conformational Properties of Lanthanide(III) 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetate Analogues Containing Methylenephosphonate Pendant Arms. Inorg. Chem. 2010, 49, 4370–4382. 10.1021/ic100177n. [DOI] [PubMed] [Google Scholar]
- Jacques V.; Desreux J. F. Quantitative Two-Dimensional EXSY Spectroscopy and Dynamic Behavior of a Paramagnetic Lanthanide Macrocyclic Chelate: YbDOTA (DOTA = 1,4,7,10-Tetraazacyclododecane-N,N’,N”,N”’-Tetraacetic Acide). Inorg. Chem. 1994, 33, 4048–4053. 10.1021/ic00096a033. [DOI] [Google Scholar]
- Opina A. C. L.; Strickland M.; Lee Y.-S.; Tjandra N.; Byrd R. A.; Swenson R. E.; Vasalatiy O. Analysis of the Isomer Ratios of Polymethylated-DOTA Complexes and the Implications on Protein Structural Studies. Dalt. Trans. 2016, 45, 4673–4687. 10.1039/C5DT03210E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasie M. D.; Comuzzi C.; van den Nieuwendijk A. M. C. H.; Prudêncio M.; Overhand M.; Ubbink M. Long-Range-Distance NMR Effects in a Protein Labeled with a Lanthanide-DOTA Chelate. Chem. - Eur. J. 2007, 13, 1715–1723. 10.1002/chem.200600916. [DOI] [PubMed] [Google Scholar]
- Keizers P. H. J.; Desreux J. F.; Overhand M.; Ubbink M. Increased Paramagnetic Effect of a Lanthanide Protein Probe by Two-Point Attachment. J. Am. Chem. Soc. 2007, 129, 9292–9293. 10.1021/ja0725201. [DOI] [PubMed] [Google Scholar]
- Hass M. A. S.; Liu W.-M.; Agafonov R. V.; Otten R.; Phung L. A.; Schilder J. T.; Kern D.; Ubbink M. A Minor Conformation of a Lanthanide Tag on Adenylate Kinase Characterized by Paramagnetic Relaxation Dispersion NMR Spectroscopy. J. Biomol. NMR 2015, 61, 123–136. 10.1007/s10858-014-9894-3. [DOI] [PubMed] [Google Scholar]
- Hass M. A. S.; Keizers P. H. J.; Blok A.; Hiruma Y.; Ubbink M. Validation of a Lanthanide Tag for the Analysis of Protein Dynamics by Paramagnetic NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 9952–9953. 10.1021/ja909508r. [DOI] [PubMed] [Google Scholar]
- Liu W.-M.; Keizers P. H. J.; Hass M. A. S.; Blok A.; Timmer M.; Sarris A. J. C.; Overhand M.; Ubbink M. A PH-Sensitive, Colorful, Lanthanide-Chelating Paramagnetic NMR Probe. J. Am. Chem. Soc. 2012, 134, 17306–17313. 10.1021/ja307824e. [DOI] [PubMed] [Google Scholar]
- Liu W.-M.; Skinner S. P.; Filippov D. V.; Blok A.; Ubbink M.; Timmer M.; Overhand M.; Hass M. A. S. A Two-Armed Lanthanoid-Chelating Paramagnetic NMR Probe Linked to Proteins via Thioether Linkages. Chem. - Eur. J. 2014, 20, 6256–6258. 10.1002/chem.201400257. [DOI] [PubMed] [Google Scholar]
- Lee M. D.; Dennis M. L.; Swarbrick J. D.; Graham B. Enantiomeric Two-Armed Lanthanide-Binding Tags for Complementary Effects in Paramagnetic NMR Spectroscopy. Chem. Commun. 2016, 52, 7954–7957. 10.1039/C6CC02325H. [DOI] [PubMed] [Google Scholar]
- Lee M. D.; Dennis M. L.; Graham B.; Swarbrick J. D. Short Two-Armed Lanthanide-Binding Tags for Paramagnetic NMR Spectroscopy Based on Chiral 1,4,7,10-Tetrakis(2-Hydroxypropyl)-1,4,7,10-Tetraazacyclododecane Scaffolds. Chem. Commun. 2017, 53, 13205–13208. 10.1039/C7CC07961C. [DOI] [PubMed] [Google Scholar]
- Miao Q.; Liu W.-M.; Kock T.; Blok A.; Timmer M.; Overhand M.; Ubbink M. A Double Armed, Hydrophilic, Transition Metal Complex as Paramagnetic NMR Probe. Angew. Chem., Int. Ed. 2019, 58, 13093–13100. 10.1002/anie.201906049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Q.; Zurlo E.; de Bruin D.; Wondergem J. A. J.; Timmer M.; Blok A.; Heinrich D.; Overhand M.; Huber M.; Ubbink M. A Two-Armed Probe for In-Cell DEER Measurements on Proteins. Chem.—Eur. J. 2020, 26, 17128–17133. 10.1002/chem.202002743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häussinger D.; Huang J.-R.; Grzesiek S. DOTA-M8: An Extremely Rigid, High-Affinity Lanthanide Chelating Tag for PCS NMR Spectroscopy. J. Am. Chem. Soc. 2009, 131, 14761–14767. 10.1021/ja903233w. [DOI] [PubMed] [Google Scholar]
- Ranganathan R. S.; Raju N.; Fan H.; Zhang X.; Tweedle M. F.; Desreux J. F.; Jacques V. Polymethylated DOTA Ligands. 2. Synthesis of Rigidified Lanthanide Chelates and Studies on the Effect of Alkyl Substitution on Conformational Mobility and Relaxivity. Inorg. Chem. 2002, 41, 6856–6866. 10.1021/ic025695e. [DOI] [PubMed] [Google Scholar]
- Strickland M.; Schwieters C. D.; Göbl C.; Opina A. C. L.; Strub M. P.; Swenson R. E.; Vasalatiy O.; Tjandra N. Characterizing the Magnetic Susceptibility Tensor of Lanthanide-Containing Polymethylated-DOTA Complexes. J. Biomol. NMR 2016, 66, 125–139. 10.1007/s10858-016-0061-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikone Y.; Hirai G.; Mishima M.; Inomata K.; Ikeya T.; Arai S.; Shirakawa M.; Sodeoka M.; Ito Y. A New Carbamidemethyl-Linked Lanthanoid Chelating Tag for PCS NMR Spectroscopy of Proteins in Living HeLa Cells. J. Biomol. NMR 2016, 66, 99–110. 10.1007/s10858-016-0059-4. [DOI] [PubMed] [Google Scholar]
- Müntener T.; Häussinger D.; Selenko P.; Theillet F.-X. In-Cell Protein Structures from 2D NMR Experiments. J. Phys. Chem. Lett. 2016, 7, 2821–2825. 10.1021/acs.jpclett.6b01074. [DOI] [PubMed] [Google Scholar]
- Müntener T.; Kottelat J.; Huber A.; Häussinger D. New Lanthanide Chelating Tags for PCS NMR Spectroscopy with Reduction Stable, Rigid Linkers for Fast and Irreversible Conjugation to Proteins. Bioconjugate Chem. 2018, 29, 3344–3351. 10.1021/acs.bioconjchem.8b00512. [DOI] [PubMed] [Google Scholar]
- Joss D.; Winter F.; Häussinger D. A Novel, Rationally Designed Lanthanoid Chelating Tag Delivers Large Paramagnetic Structural Restraints for Biomolecular NMR. Chem. Commun. 2020, 56, 12861–12864. 10.1039/D0CC04337K. [DOI] [PubMed] [Google Scholar]
- Joss D.; Häussinger D. P4T-DOTA – a Lanthanide Chelating Tag Combining a Sterically Highly Overcrowded Backbone with a Reductively Stable Linker. Chem. Commun. 2019, 55, 10543–10546. 10.1039/C9CC04676C. [DOI] [PubMed] [Google Scholar]
- Graham B.; Loh C.-T.; Swarbrick J. D.; Ung P.; Shin J.; Yagi H.; Jia X.; Chhabra S.; Barlow N.; Pintacuda G.; et al. DOTA-Amide Lanthanide Tag for Reliable Generation of Pseudocontact Shifts in Protein NMR Spectra. Bioconjugate Chem. 2011, 22, 2118–2125. 10.1021/bc200353c. [DOI] [PubMed] [Google Scholar]
- de la Cruz L.; Nguyen T. H. D.; Ozawa K.; Shin J.; Graham B.; Huber T.; Otting G. Binding of Low Molecular Weight Inhibitors Promotes Large Conformational Changes in the Dengue Virus NS2b-NS3 Protease: Fold Analysis by Pseudocontact Shifts. J. Am. Chem. Soc. 2011, 133, 19205–19215. 10.1021/ja208435s. [DOI] [PubMed] [Google Scholar]
- Abdelkader E. H.; Lee M. D.; Feintuch A.; Cohen M. R.; Swarbrick J. D.; Otting G.; Graham B.; Goldfarb D. A New Gd3+ Spin Label for Gd3+-Gd3+ Distance Measurements in Proteins Produces Narrow Distance Distributions. J. Phys. Chem. Lett. 2015, 6, 5016–5021. 10.1021/acs.jpclett.5b02451. [DOI] [PubMed] [Google Scholar]
- Loh C.-T.; Ozawa K.; Tuck K. L.; Barlow N.; Huber T.; Otting G.; Graham B. Lanthanide Tags for Site-Specific Ligation to an Unnatural Amino Acid and Generation of Pseudocontact Shifts in Proteins. Bioconjugate Chem. 2013, 24, 260–268. 10.1021/bc300631z. [DOI] [PubMed] [Google Scholar]
- Herath I. D.; Breen C.; Hewitt S.; Berki T.; Kassir A.; Dodson C.; Judd M.; Jabar S.; Cox N.; Otting G.; et al. A Chiral Lanthanide Tag for Stable and Rigid Attachment to Single Cysteine Residues in Proteins for NMR, EPR and Time-resolved Luminescence Studies. Chem. - Eur. J. 2021, 27, 13009. 10.1002/chem.202101143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelkader E. H.; Feintuch A.; Yao X.; Adams L. A.; Aurelio L.; Graham B.; Goldfarb D.; Otting G. Protein Conformation by EPR Spectroscopy Using Gadolinium Tags Clicked to Genetically Encoded P-Azido-L-Phenylalanine. Chem. Commun. 2015, 51, 15898–15901. 10.1039/C5CC07121F. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Lee M. D.; Carruthers T. J.; Szabo M.; Dennis M. L.; Swarbrick J. D.; Graham B.; Otting G. New Lanthanide Tag for the Generation of Pseudocontact Shifts in DNA by Site-Specific Ligation to a Phosphorothioate Group. Bioconjugate Chem. 2017, 28, 1741–1748. 10.1021/acs.bioconjchem.7b00202. [DOI] [PubMed] [Google Scholar]
- Yang F.; Wang X.; Pan B.-B.; Su X.-C. Single-Armed Phenylsulfonated Pyridine Derivative of DOTA Is Rigid and Stable Paramagnetic Tag in Protein Analysis. Chem. Commun. 2016, 52, 11535–11538. 10.1039/C6CC06114A. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Yang F.; Gong Y.-J.; Chen J.-L.; Goldfarb D.; Su X.-C. A Reactive, Rigid GdIII Labeling Tag for In-Cell EPR Distance Measurements in Proteins. Angew. Chem., Int. Ed. 2017, 56, 2914–2918. 10.1002/anie.201611051. [DOI] [PubMed] [Google Scholar]
- Ma B.; Chen J.-L.; Cui C.-Y.; Yang F.; Gong Y.-J.; Su X.-C. Rigid, Highly Reactive and Stable DOTA-like Tags Containing a Thiol-Specific Phenylsulfonyl Pyridine Moiety for Protein Modification and NMR Analysis. Chem. - Eur. J. 2021, 27, 16145. 10.1002/chem.202102495. [DOI] [PubMed] [Google Scholar]
- Ma B.; Chen J.-L.; Cui C.-Y.; Yang F.; Gong Y.-J.; Su X.-C. Rigid, Highly Reactive and Stable DOTA-like Tags Containing a Thiol-specific Phenylsulfonyl Pyridine Moiety for Protein Modification and NMR Analysis. Chem.—Eur. J. 2021, 27, 16145–16152. 10.1002/chem.202102495. [DOI] [PubMed] [Google Scholar]
- Chen J.-L.; Chen B. G.; Li B.; Yang F.; Su X.-C. Assessing Multiple Conformations of Lanthanide Binding Tags for Proteins Using a Sensitive19F-Reporter. Chem. Commun. 2021, 57, 4291–4294. 10.1039/D1CC00791B. [DOI] [PubMed] [Google Scholar]
- Lee Y.-S.; Mou Z.; Opina A. C. L.; Vasalatiy O. Origin of the Isomer Stability of Polymethylated DOTA Chelates Complexed with Ln3+ Ions. Eur. J. Inorg. Chem. 2021, 2021, 1428–1440. 10.1002/ejic.202100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis M.; Softley C.; Giuntini S.; Gentili M.; Ravera E.; Parigi G.; Fragai M.; Popowicz G.; Sattler M.; Luchinat C.; et al. The Photocatalyzed Thiol-Ene Reaction: A New Tag to Yield Fast, Selective and Reversible Paramagnetic Tagging of Proteins. ChemPhysChem 2020, 21, 863–869. 10.1002/cphc.202000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comblin V.; Gilsoul D.; Hermann M.; Humblet V.; Jacques V.; Mesbahi M.; Sauvage C.; Desreux J. F. Designing New MRI Contrast Agents: A Coordination Chemistry Challenge. Coord. Chem. Rev. 1999, 185–186, 451–470. 10.1016/S0010-8545(99)00028-4. [DOI] [Google Scholar]
- Richman J. E.; Atkins T. J. Nitrogen analogs of crown ethers. J. Am. Chem. Soc. 1974, 96 (7), 2268–2270. 10.1021/ja00814a056. [DOI] [Google Scholar]
- Rashid H. U.; Martines M. A. U.; Jorge J.; de Moraes P. M.; Umar M. N.; Khan K.; Rehman H. U. Cyclen-Based Gd3+complexes as MRI Contrast Agents: Relaxivity Enhancement and Ligand Design. Bioorg. Med. Chem. 2016, 24, 5663–5684. 10.1016/j.bmc.2016.09.069. [DOI] [PubMed] [Google Scholar]
- Athey P. S.; Kiefer G. E. A New, Facile Synthesis of 1, 4,7,10-Tetraazacyclododecane: Cyclen. J. Org. Chem. 2002, 67, 4081–4085. 10.1021/jo016111d. [DOI] [PubMed] [Google Scholar]
- Hansen G. R.; Burg T. E. Unique Synthesis of 1,4,7,10-tetraazacyclododecane. J. Heterocycl. Chem. 1968, 5, 305. 10.1002/jhet.5570050232. [DOI] [Google Scholar]
- Hervé G.; Bernard H.; Le Bris N.; Yaouanc J.-J.; Handel H.; Toupet L. A New Route to Cyclen, Cyclam and Homocyclen. Tetrahedron Lett. 1998, 39, 6861–6864. 10.1016/S0040-4039(98)01497-X. [DOI] [Google Scholar]
- Koike T.; Gotoh T.; Aoki S.; Kimura E.; Shiro M. A New Macrocyclic Tetraamine, 2,4-Dinitrophenylcyclen (= 1-(2,4-Dinitrophenyl)-1,4,7,10-Tetraazacyclododecane): Synthesis, Cation Reporter Properties, and Recognition of 1-Methylthymine by Its Zinc (II) Complex. Inorg. Chim. Acta 1998, 270, 424–432. 10.1016/S0020-1693(97)05998-7. [DOI] [Google Scholar]
- Weisman G. R.; Reed D. P. A New Synthesis of Cyclen (1,4,7,10-Tetraazacyclododecane). J. Org. Chem. 1996, 61, 5186–5187. 10.1021/jo9606665. [DOI] [PubMed] [Google Scholar]
- Kaden T. A.Ten-Membered Rings or Larger with One or More Nitrogen Atoms. In Comprehensive Heterocyclic Chemistry II: A Review of the Literature 1982–1995; 1996; Vol. 9, pp 789–807. [Google Scholar]
- Jebasingh B.; Alexander V. Microwave-Assisted Synthesis of 1,4,7,10-Tetraazacyclododecane. Synth. Commun. 2006, 36, 653–657. 10.1080/15459620500408850. [DOI] [Google Scholar]
- Kizaki K.; Uehara M.; Fuyuhiro A.; Fukuda T.; Ishikawa N. Synthesis of a Series of Monophthalocyaninato Cyclen Heavy Lanthanide(III) Complexes with Tetragonal Symmetry. Inorg. Chem. 2018, 57, 668–675. 10.1021/acs.inorgchem.7b02505. [DOI] [PubMed] [Google Scholar]
- Jacques V.; Gilsoul D.; Comblin V.; Desreux J. F. Rigidified Macrocyclic Lanthanide Chelates for Magnetic Resonance Imaging. J. Alloys Compd. 1997, 249, 173–177. 10.1016/S0925-8388(96)02529-7. [DOI] [Google Scholar]
- Vanasschen C.; Bouslimani N.; Thonon D.; Desreux J. F. Gadolinium DOTA Chelates Featuring Alkyne Groups Directly Grafted on the Tetraaza Macrocyclic Ring: Synthesis, Relaxation Properties, “Click” Reaction, and High-Relaxivity Micelles. Inorg. Chem. 2011, 50, 8946–8958. 10.1021/ic2010997. [DOI] [PubMed] [Google Scholar]
- Ranganathan R. S.; Pillai R. K.; Raju N.; Fan H.; Nguyen H.; Tweedle M. F.; Desreux J. F.; Jacques V. Polymethylated DOTA Ligands. 1. Synthesis of Rigidified Ligands and Studies on the Effects of Alkyl Substitution on Acid–Base Properties and Conformational Mobility. Inorg. Chem. 2002, 41, 6846–6855. 10.1021/ic025657v. [DOI] [PubMed] [Google Scholar]
- Anelli L. P.; Beltrami A.; Franzini M.; Paoli P.; Rossi P.; Uggeri F.; Virtuani M. Gd (III) Complexes of Poly (Hydroxymethyl) Substituted Derivatives of 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid. Inorg. Chim. Acta 2001, 317, 218–229. 10.1016/S0020-1693(01)00364-4. [DOI] [Google Scholar]
- Müntener T.; Thommen F.; Joss D.; Kottelat J.; Prescimone A.; Häussinger D. Synthesis of Chiral Nine and Twelve-Membered Cyclic Polyamines from Natural Building Blocks. Chem. Commun. 2019, 55, 4715–4718. 10.1039/C9CC00720B. [DOI] [PubMed] [Google Scholar]
- Grenthe I. Stability Relationships Among the Rare Earth Dipicolinates. J. Am. Chem. Soc. 1961, 83, 360–364. 10.1021/ja01463a024. [DOI] [Google Scholar]
- Pettinari C.; Marchetti F.; Drozdov A.. Higher Denticity Ligands. In Comprehensive Coordination Chemistry II; 2004; Vol. 1, pp 211–251. [Google Scholar]
- Su X.-C.; Man B.; Beeren S.; Liang H.; Simonsen S.; Schmitz C.; Huber T.; Messerle B. A.; Otting G. A Dipicolinic Acid Tag for Rigid Lanthanide Tagging of Proteins and Paramagnetic NMR Spectroscopy. J. Am. Chem. Soc. 2008, 130, 10486–10487. 10.1021/ja803741f. [DOI] [PubMed] [Google Scholar]
- Man B.; Su X.-C.; Liang H.; Simonsen S.; Huber T.; Messerle B. A.; Otting G. 3-Mercapto-2,6-Pyridinedicarboxylic Acid: A Small Lanthanide-Binding Tag for Protein Studies by NMR Spectroscopy. Chem. - Eur. J. 2010, 16, 3827–3832. 10.1002/chem.200902904. [DOI] [PubMed] [Google Scholar]
- Jia X.; Maleckis A.; Huber T.; Otting G. 4,4’-Dithiobisdipicolinic Acid: A Small and Convenient Lanthanide Binding Tag for Protein NMR Spectroscopy. Chem. - Eur. J. 2011, 17, 6830–6836. 10.1002/chem.201003573. [DOI] [PubMed] [Google Scholar]
- Huang F.; Pei Y.-Y.; Zuo H.-H.; Chen J.-L.; Yang Y.; Su X.-C. Bioconjugation of Proteins with a Paramagnetic NMR and Fluorescent Tag. Chem. - Eur. J. 2013, 19, 17141–17149. 10.1002/chem.201302273. [DOI] [PubMed] [Google Scholar]
- Li Q.-F.; Yang Y.; Maleckis A.; Otting G.; Su X.-C. Thiol-Ene Reaction: A Versatile Tool in Site-Specific Labelling of Proteins with Chemically Inert Tags for Paramagnetic NMR. Chem. Commun. 2012, 48, 2704–2706. 10.1039/c2cc17900h. [DOI] [PubMed] [Google Scholar]
- Swarbrick J. D.; Ung P.; Su X.-C.; Maleckis A.; Chhabra S.; Huber T.; Otting G.; Graham B. Engineering of a Bis-Chelator Motif into a Protein α-Helix for Rigid Lanthanide Binding and Paramagnetic NMR Spectroscopy. Chem. Commun. 2011, 47, 7368–7370. 10.1039/c1cc11893e. [DOI] [PubMed] [Google Scholar]
- Swarbrick J. D.; Ung P.; Chhabra S.; Graham B. An Iminodiacetic Acid Based Lanthanide Binding Tag for Paramagnetic Exchange NMR Spectroscopy. Angew. Chem., Int. Ed. 2011, 50, 4403–4406. 10.1002/anie.201007221. [DOI] [PubMed] [Google Scholar]
- Swarbrick J. D.; Ung P.; Dennis M. L.; Lee M. D.; Chhabra S.; Graham B. Installation of a Rigid EDTA-like Motif into a Protein α-Helix for Paramagnetic NMR Spectroscopy with Cobalt(II) Ions. Chem. - Eur. J. 2016, 22, 1228–1232. 10.1002/chem.201503139. [DOI] [PubMed] [Google Scholar]
- Yagi H.; Maleckis A.; Otting G. A Systematic Study of Labelling an α-Helix in a Protein with a Lanthanide Using IDA-SH or NTA-SH Tags. J. Biomol. NMR 2013, 55, 157–166. 10.1007/s10858-012-9697-3. [DOI] [PubMed] [Google Scholar]
- Nitsche C.; Mahawaththa M. C.; Becker W.; Huber T.; Otting G. Site-Selective Tagging of Proteins by Pnictogen-Mediated Self-Assembly. Chem. Commun. 2017, 53, 10894–10897. 10.1039/C7CC06155B. [DOI] [PubMed] [Google Scholar]
- Lane A. N.; Jardetzky O. Identification of Surface Residues in the Trp Repressor of Escherichia Coli. Eur. J. Biochem. 1985, 152, 411–418. 10.1111/j.1432-1033.1985.tb09212.x. [DOI] [PubMed] [Google Scholar]
- Porcelli F.; Buck B.; Lee D.-K.; Hallock K. J.; Ramamoorthy A.; Veglia G. Structure and Orientation of Pardaxin Determined by NMR Experiments in Model Membranes. J. Biol. Chem. 2004, 279, 45815–45823. 10.1074/jbc.M405454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schievano E.; Calisti T.; Menegazzo I.; Battistutta R.; Peggion E.; Mammi S.; Palù G.; Loregian A. pH-Dependent Conformational Changes and Topology of a Herpesvirus Translocating Peptide in a Membrane-Mimetic Environment. Biochemistry 2004, 43, 9343–9351. 10.1021/bi0496438. [DOI] [PubMed] [Google Scholar]
- Niccolai N.; Tiezzi E.; Valensin G. Manganese(II) as Magnetic Relaxation Probe in the Study of Biomechanisms and of Biomacromolecules. Chem. Rev. 1982, 82, 359–384. 10.1021/cr00050a002. [DOI] [Google Scholar]
- Luchette P. A.; Prosser R. S.; Sanders C. R. Oxygen as a Paramagnetic Probe of Membrane Protein Structure by Cysteine Mutagenesis and 19F NMR Spectroscopy. J. Am. Chem. Soc. 2002, 124, 1778–1781. 10.1021/ja016748e. [DOI] [PubMed] [Google Scholar]
- Hernández G.; Teng C.-L.; Bryant R. G.; LeMaster D. M. O2 Penetration and Proton Burial Depth in Proteins: Applicability to Fold Family Recognition. J. Am. Chem. Soc. 2002, 124, 4463–4472. 10.1021/ja017340k. [DOI] [PubMed] [Google Scholar]
- Sakakura M.; Noba S.; Luchette P. A.; Shimada I.; Prosser R. S. An NMR Method for the Determination of Protein-Binding Interfaces Using Dioxygen-Induced Spin–Lattice Relaxation Enhancement. J. Am. Chem. Soc. 2005, 127, 5826–5832. 10.1021/ja047825j. [DOI] [PubMed] [Google Scholar]
- Evanics F.; Hwang P. M.; Cheng Y.; Kay L. E.; Prosser R. S. Topology of an Outer-Membrane Enzyme: Measuring Oxygen and Water Contacts in Solution NMR Studies of PagP. J. Am. Chem. Soc. 2006, 128, 8256–8264. 10.1021/ja0610075. [DOI] [PubMed] [Google Scholar]
- Bezsonova I.; Evanics F.; Marsh J. A.; Forman-Kay J. D.; Prosser R. S. Oxygen as a Paramagnetic Probe of Clustering and Solvent Exposure in Folded and Unfolded States of an SH3 Domain. J. Am. Chem. Soc. 2007, 129, 1826–1835. 10.1021/ja065173o. [DOI] [PubMed] [Google Scholar]
- Prosser R. S.; Evanics F.; Kitevski J. L.; Patel S. The Measurement of Immersion Depth and Topology of Membrane Proteins by Solution State NMR. Biochim. Biophys. Acta, Biomembr. 2007, 1768, 3044–3051. 10.1016/j.bbamem.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Campbell I. D.; Dobson C. M.; Williams R. J. P.; Xavier A. V. Resolution Enhancement of Protein PMR Spectra Using the Difference between a Broadened and a Normal Spectrum. J. Magn. Reson. 1973, 11, 172–181. 10.1016/0022-2364(73)90004-8. [DOI] [Google Scholar]
- Morishima I.; Endo K.; Yonezawa T. Studies on Nuclear Magnetic Resonance Contact Shifts Induced by Hydrogen Bonding with Organic Radicals. I. 1H and 13C Contact Shifts of Protic Molecules in the Presence of the Nitroxide Radical. J. Am. Chem. Soc. 1971, 93, 2048–2050. 10.1021/ja00737a035. [DOI] [Google Scholar]
- Kopple K. D.; Schamper T. J. Proton Magnetic Resonance Line Broadening Produced by Association with a Nitroxide Radical in Studies of Amide and Peptide Conformation. J. Am. Chem. Soc. 1972, 94, 3644–3646. 10.1021/ja00765a074. [DOI] [PubMed] [Google Scholar]
- Petros A. M.; Mueller L.; Kopple K. D. NMR Identification of Protein Surfaces Using Paramagnetic Probes. Biochemistry 1990, 29, 10041–10048. 10.1021/bi00495a005. [DOI] [PubMed] [Google Scholar]
- Fesik S. W.; Gemmecker G.; Olejniczak E. T.; Petros A. M. Identification of Solvent-Exposed Regions of Enzyme-Bound Ligands by Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1991, 113, 7080–7081. 10.1021/ja00018a080. [DOI] [Google Scholar]
- Improta S.; Molinari H.; Pastore A.; Consonni R.; Zetta L. Probing Protein Structure by Solvent Perturbation of NMR Spectra. Eur. J. Biochem. 1995, 227, 87–96. 10.1111/j.1432-1033.1995.tb20362.x. [DOI] [PubMed] [Google Scholar]
- Yuan T.; Ouyang H.; Vogel H. J. Surface Exposure of the Methionine Side Chains of Calmodulin in Solution. A Nitroxide Spin Label and Two-Dimensional NMR Study. J. Biol. Chem. 1999, 274, 8411–8420. 10.1074/jbc.274.13.8411. [DOI] [PubMed] [Google Scholar]
- Teng C.-L.; Bryant R. G. Spin Relaxation Measurements of Electrostatic Bias in Intermolecular Exploration. J. Magn. Reson. 2006, 179, 199–205. 10.1016/j.jmr.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Pintacuda G.; Otting G. Identification of Protein Surfaces by NMR Measurements with a Paramagnetic Gd(III) Chelate. J. Am. Chem. Soc. 2002, 124, 372–373. 10.1021/ja016985h. [DOI] [PubMed] [Google Scholar]
- Respondek M.; Madl T.; Göbl C.; Golser R.; Zangger K. Mapping the Orientation of Helices in Micelle-Bound Peptides by Paramagnetic Relaxation Waves. J. Am. Chem. Soc. 2007, 129, 5228–5234. 10.1021/ja069004f. [DOI] [PubMed] [Google Scholar]
- Madl T.; Bermel W.; Zangger K. Use of Relaxation Enhancements in a Paramagnetic Environment for the Structure Determination of Proteins Using NMR Spectroscopy. Angew. Chem., Int. Ed. 2009, 48, 8259–8262. 10.1002/anie.200902561. [DOI] [PubMed] [Google Scholar]
- Gong Z.; Gu X.-H.; Guo D.-C.; Wang J.; Tang C. Protein Structural Ensembles Visualized by Solvent Paramagnetic Relaxation Enhancement. Angew. Chem., Int. Ed. 2017, 56, 1002–1006. 10.1002/anie.201609830. [DOI] [PubMed] [Google Scholar]
- Gu X.-H.; Gong Z.; Guo D.-C.; Zhang W.-P.; Tang C. A Decadentate Gd(III)-Coordinating Paramagnetic Cosolvent for Protein Relaxation Enhancement Measurement. J. Biomol. NMR 2014, 58, 149–154. 10.1007/s10858-014-9817-3. [DOI] [PubMed] [Google Scholar]
- Bernini A.; Spiga O.; Venditti V.; Prischi F.; Bracci L.; Tong A. P.-L.; Wong W.-T.; Niccolai N. NMR Studies of Lysozyme Surface Accessibility by Using Different Paramagnetic Relaxation Probes. J. Am. Chem. Soc. 2006, 128, 9290–9291. 10.1021/ja062109y. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Friedman J. I.; Stivers J. T. Cosolute Paramagnetic Relaxation Enhancements Detect Transient Conformations of Human Uracil DNA Glycosylase (HUNG). Biochemistry 2011, 50, 10724–10731. 10.1021/bi201572g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam S.; Hemme C. L.; Yoshida N.; Suzuki K.; Nagase H.; Berjanskii M.; Wu B.; Van Doren S. R. TIMP-1 Contact Sites and Perturbations of Stromelysin 1 Mapped by NMR and a Paramagnetic Surface Probe. Biochemistry 1998, 37, 9650–9657. 10.1021/bi980128h. [DOI] [PubMed] [Google Scholar]
- Sattler M.; Fesik S. W. Resolving Resonance Overlap in the NMR Spectra of Proteins from Differential Lanthanide-Induced Shifts. J. Am. Chem. Soc. 1997, 119, 7885–7886. 10.1021/ja971356m. [DOI] [Google Scholar]
- Dick L. R.; Geraldes C. F. G. C.; Sherry A. D.; Gray C. W.; Gray D. M. 13C NMR of Methylated Lysines of Fd Gene 5 Protein: Evidence for a Conformational Change Involving Lysine 24 upon Binding of a Negatively Charged Lanthanide Chelate. Biochemistry 1989, 28, 7896–7904. 10.1021/bi00445a052. [DOI] [PubMed] [Google Scholar]
- Otting G.; Liepinsh E. Selective Excitation of the Water Signal by a Q-Switched Selective Pulse. J. Magn. Reson., Ser. B 1995, 107, 192–196. 10.1006/jmrb.1995.1078. [DOI] [Google Scholar]
- Sibille N.; Bellot G.; Wang J.; Déméné H. Low Concentration of a Gd-Chelate Increases the Signal-to-Noise Ratio in Fast Pulsing BEST Experiments. J. Magn. Reson. 2012, 224, 32–37. 10.1016/j.jmr.2012.07.022. [DOI] [PubMed] [Google Scholar]
- Su X.-C.; Liang H.; Loscha K. V.; Otting G. [Ln(DPA)3]3- Is a Convenient Paramagnetic Shift Reagent for Protein NMR Studies. J. Am. Chem. Soc. 2009, 131, 10352–10353. 10.1021/ja9034957. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Yang Y.; Li Q.-F.; Huang F.; Zuo H.-H.; Su X.-C. Noncovalent Tagging Proteins with Paramagnetic Lanthanide Complexes for Protein Study. Chem. - Eur. J. 2013, 19, 5758–5764. 10.1002/chem.201204152. [DOI] [PubMed] [Google Scholar]
- Jia X.; Yagi H.; Su X.-C.; Stanton-Cook M.; Huber T.; Otting G. Engineering [Ln(DPA)3]3- Binding Sites in Proteins: A Widely Applicable Method for Tagging Proteins with Lanthanide Ions. J. Biomol. NMR 2011, 50, 411–420. 10.1007/s10858-011-9529-x. [DOI] [PubMed] [Google Scholar]
- Yagi H.; Loscha K. V.; Su X.-C.; Stanton-Cook M.; Huber T.; Otting G. Tunable Paramagnetic Relaxation Enhancements by [Gd(DPA)3]3- for Protein Structure Analysis. J. Biomol. NMR 2010, 47, 143–153. 10.1007/s10858-010-9416-x. [DOI] [PubMed] [Google Scholar]
- Aime S.; D’Amelio N.; Fragai M.; Lee Y. M.; Luchinat C.; Terreno E.; Valensin G. A Paramagnetic Probe to Localize Residues next to Carboxylates on Protein Surfaces. JBIC, J. Biol. Inorg. Chem. 2002, 7, 617–622. 10.1007/s00775-002-0340-8. [DOI] [PubMed] [Google Scholar]
- Oktaviani N. A.; Risør M. W.; Lee Y.-H.; Megens R. P.; de Jong D. H.; Otten R.; Scheek R. M.; Enghild J. J.; Nielsen N. C.; Ikegami T.; et al. Optimized Co-Solute Paramagnetic Relaxation Enhancement for the Rapid NMR Analysis of a Highly Fibrillogenic Peptide. J. Biomol. NMR 2015, 62, 129–142. 10.1007/s10858-015-9925-8. [DOI] [PubMed] [Google Scholar]
- Cai S.; Seu C.; Kovacs Z.; Sherry A. D.; Chen Y. Sensitivity Enhancement of Multidimensional NMR Experiments by Paramagnetic Relaxation Effects. J. Am. Chem. Soc. 2006, 128, 13474–13478. 10.1021/ja0634526. [DOI] [PubMed] [Google Scholar]
- Means G. E.; Feeney R. E. Chemical Modifications of Proteins: History and Applications. Bioconjugate Chem. 1990, 1, 2–12. 10.1021/bc00001a001. [DOI] [PubMed] [Google Scholar]
- Walsh C. T.; Garneau-Tsodikova S.; Gatto G. J. Jr. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angew. Chem., Int. Ed. 2005, 44, 7342–7372. 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem. Rev. 2014, 114, 4764–4806. 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- Kim C. H.; Axup J. Y.; Schultz P. G. Protein Conjugation with Genetically Encoded Unnatural Amino Acids. Curr. Opin. Chem. Biol. 2013, 17, 412–419. 10.1016/j.cbpa.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckler T. G.; Chang L.-H.; Zama Y.; Naka T.; Chorghade M. S.; Hecht S. M. T4 RNA Ligase Mediated Preparation of Novel “Chemically Misacylated” TRNAPhes. Biochemistry 1984, 23, 1468–1473. 10.1021/bi00302a020. [DOI] [PubMed] [Google Scholar]
- Xu L.; Kuan S. L.; Weil T. Contemporary Approaches for Site-Selective Dual Functionalization of Proteins. Angew. Chem., Int. Ed. 2021, 60, 13757–13777. 10.1002/anie.202012034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T.; Hamachi I. Chemistry for Covalent Modification of Endogenous/Native Proteins: From Test Tubes to Complex Biological Systems. J. Am. Chem. Soc. 2019, 141, 2782–2799. 10.1021/jacs.8b11747. [DOI] [PubMed] [Google Scholar]
- Rawale D. G.; Thakur K.; Adusumalli S. R.; Rai V. Chemical Methods for Selective Labeling of Proteins. Eur. J. Org. Chem. 2019, 2019, 6749–6763. 10.1002/ejoc.201900801. [DOI] [Google Scholar]
- Reddy N. C.; Kumar M.; Molla R.; Rai V. Chemical Methods for Modification of Proteins. Org. Biomol. Chem. 2020, 18, 4669–4691. 10.1039/D0OB00857E. [DOI] [PubMed] [Google Scholar]
- Nolan M. D.; Scanlan E. M. Applications of Thiol-Ene Chemistry for Peptide Science. Front. Chem. 2020, 8, 1060. 10.3389/fchem.2020.583272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt E. A.; Cal P. M. S. D.; Oliveira B. L.; Bernardes G. J. L. Contemporary Approaches to Site-Selective Protein Modification. Nat. Rev. Chem. 2019, 3, 147–171. 10.1038/s41570-019-0079-1. [DOI] [Google Scholar]
- Asiimwe N.; Al Mazid M. F.; Murale D. P.; Kim Y. K.; Lee J.-S. Recent Advances in Protein Modifications Techniques for the Targeting N-Terminal Cysteine. Pept. Sci. 2021, e24235 10.1002/pep2.24235. [DOI] [Google Scholar]
- Ochtrop P.; Hackenberger C. P. R. Recent Advances of Thiol-Selective Bioconjugation Reactions. Curr. Opin. Chem. Biol. 2020, 58, 28–36. 10.1016/j.cbpa.2020.04.017. [DOI] [PubMed] [Google Scholar]
- Sakamoto S.; Hamachi I. Recent Progress in Chemical Modification of Proteins. Anal. Sci. 2019, 35, 5–27. 10.2116/analsci.18R003. [DOI] [PubMed] [Google Scholar]
- Spicer C. D.; Pashuck E. T.; Stevens M. M. Achieving Controlled Biomolecule–Biomaterial Conjugation. Chem. Rev. 2018, 118, 7702–7743. 10.1021/acs.chemrev.8b00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. L. Site-Selective Protein-Modification Chemistry for Basic Biology and Drug Development. Nat. Chem. 2016, 8, 103–113. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Gunnoo S. B.; Madder A. Chemical Protein Modification through Cysteine. ChemBioChem 2016, 17, 529–553. 10.1002/cbic.201500667. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Bernardes G. J. L.; Lin Y. A.; Davis B. G. Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chem. - Asian J. 2009, 4, 630–640. 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- Witt D. Recent Developments in Disulfide Bond Formation. Synthesis 2008, 2008, 2491–2509. 10.1055/s-2008-1067188. [DOI] [Google Scholar]
- Sengupta I.; Gao M.; Arachchige R. J.; Nadaud P. S.; Cunningham T. F.; Saxena S.; Schwieters C. D.; Jaroniec C. P. Protein Structural Studies by Paramagnetic Solid-State NMR Spectroscopy Aided by a Compact Cyclen-Type Cu(II) Binding Tag. J. Biomol. NMR 2015, 61, 1–6. 10.1007/s10858-014-9880-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Pilla K. B.; Li Q.; Zhang Z.; Su X.; Huber T.; Yang J. Magic Angle Spinning NMR Structure Determination of Proteins from Pseudocontact Shifts. J. Am. Chem. Soc. 2013, 135, 8294–8303. 10.1021/ja4021149. [DOI] [PubMed] [Google Scholar]
- Goddard D. R.; Michaelis L. Derivatives of Keratin. J. Biol. Chem. 1935, 112, 361–371. 10.1016/S0021-9258(18)74993-4. [DOI] [Google Scholar]
- Meyer V.; Swanson M. A.; Clouston L. J.; Boratyński P. J.; Stein R. A.; McHaourab H. S.; Rajca A.; Eaton S. S.; Eaton G. R. Room-Temperature Distance Measurements of Immobilized Spin-Labeled Protein by DEER/PELDOR. Biophys. J. 2015, 108, 1213–1219. 10.1016/j.bpj.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J. E.; Ward W. H. Cross-Linking of Bovine Plasma Albumin and Wool Keratin. J. Am. Chem. Soc. 1956, 78, 2414–2418. 10.1021/ja01592a020. [DOI] [Google Scholar]
- Massa S.; Xavier C.; De Vos J.; Caveliers V.; Lahoutte T.; Muyldermans S.; Devoogdt N. Site-Specific Labeling of Cysteine-Tagged Camelid Single-Domain Antibody-Fragments for Use in Molecular Imaging. Bioconjugate Chem. 2014, 25, 979–988. 10.1021/bc500111t. [DOI] [PubMed] [Google Scholar]
- Smith M. E. B.; Schumacher F. F.; Ryan C. P.; Tedaldi L. M.; Papaioannou D.; Waksman G.; Caddick S.; Baker J. R. Protein Modification, Bioconjugation, and Disulfide Bridging Using Bromomaleimides. J. Am. Chem. Soc. 2010, 132, 1960–1965. 10.1021/ja908610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosik K.; Debiec K.; Czarnecka A.; Sochacka E.; Leszczynska G. Synthesis of Nucleobase-Modified RNA Oligonucleotides by Post-Synthetic Approach. Molecules 2020, 25, 3344. 10.3390/molecules25153344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z.; Yang S.; Yang Q.-F.; Zhu Y.-L.; Jiang J.; Tang C. Refining RNA Solution Structures with the Integrative Use of Label-Free Paramagnetic Relaxation Enhancement NMR. Biophys. Rep. 2019, 5, 244–253. 10.1007/s41048-019-00099-2. [DOI] [Google Scholar]
- Venditti V.; Niccolai N.; Butcher S. E. Measuring the Dynamic Surface Accessibility of RNA with the Small Paramagnetic Molecule TEMPOL. Nucleic Acids Res. 2008, 36, e20–e20. 10.1093/nar/gkm1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brath U.; Swamy S. I.; Veiga A. X.; Tung C.-C.; Van Petegem F.; Erdélyi M. Paramagnetic Ligand Tagging to Identify Protein Binding Sites. J. Am. Chem. Soc. 2015, 137, 11391–11398. 10.1021/jacs.5b06220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont P.; Chapelle C.; Demeunynck M.; Michon J.; Michon P.; Lhomme J. Introduction of a Nitroxide Group on Position 2 of 9-Phenoxyacridine: Easy Access to Spin Labelled DNA-Binding Conjugates. Bioorg. Med. Chem. Lett. 1998, 8, 669–674. 10.1016/S0960-894X(98)00089-4. [DOI] [PubMed] [Google Scholar]
- Thomas F.; Michon J.; Lhomme J. Interaction of a Spin-Labeled Adenine–Acridine Conjugate with a DNA Duplex Containing an Abasic Site Model. Biochemistry 1999, 38, 1930–1937. 10.1021/bi981770e. [DOI] [PubMed] [Google Scholar]
- Atsumi H.; Maekawa K.; Nakazawa S.; Shiomi D.; Sato K.; Kitagawa M.; Takui T.; Nakatani K. Noncovalent Assembly of TEMPO Radicals Pair-Wise Embedded on a DNA Duplex. Chem. Lett. 2010, 39, 556–557. 10.1246/cl.2010.556. [DOI] [Google Scholar]
- Atsumi H.; Maekawa K.; Nakazawa S.; Shiomi D.; Sato K.; Kitagawa M.; Takui T.; Nakatani K. Tandem Arrays of TEMPO and Nitronyl Nitroxide Radicals with Designed Arrangements on DNA. Chem. - Eur. J. 2012, 18, 178–183. 10.1002/chem.201102693. [DOI] [PubMed] [Google Scholar]
- Atsumi H.; Nakazawa S.; Dohno C.; Sato K.; Takui T.; Nakatani K. Ligand-Induced Electron Spin-Assembly on a DNA Tile. Chem. Commun. 2013, 49, 6370–6372. 10.1039/c3cc41801d. [DOI] [PubMed] [Google Scholar]
- Maekawa K.; Nakazawa S.; Atsumi H.; Shiomi D.; Sato K.; Kitagawa M.; Takui T.; Nakatani K. Programmed Assembly of Organic Radicals on DNA. Chem. Commun. 2010, 46, 1247–1249. 10.1039/b913061f. [DOI] [PubMed] [Google Scholar]
- Shelke S. A.; Sigurdsson S. T. Noncovalent and Site-Directed Spin Labeling of Nucleic Acids. Angew. Chem., Int. Ed. 2010, 49, 7984–7986. 10.1002/anie.201002637. [DOI] [PubMed] [Google Scholar]
- Shelke S. A.; Sigurdsson S. T. Structural Changes of an Abasic Site in Duplex DNA Affect Noncovalent Binding of the Spin Label ç. Nucleic Acids Res. 2012, 40, 3732–3740. 10.1093/nar/gkr1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginsson G. W.; Shelke S. A.; Rouillon C.; White M. F.; Sigurdsson S. T.; Schiemann O. Protein-Induced Changes in DNA Structure and Dynamics Observed with Noncovalent Site-Directed Spin Labeling and PELDOR. Nucleic Acids Res. 2013, 41, e11–e11. 10.1093/nar/gks817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelke S. A.; Sigurdsson S. T. Effect of N3Modifications on the Affinity of Spin Label ç for Abasic Sites in Duplex DNA. ChemBioChem 2012, 13, 684–690. 10.1002/cbic.201100728. [DOI] [PubMed] [Google Scholar]
- Chalmers B. A.; Saha S.; Nguyen T.; McMurtrie J.; Sigurdsson S. T.; Bottle S. E.; Masters K.-S. TMIO-PyrImid Hybrids Are Profluorescent, Site-Directed Spin Labels for Nucleic Acids. Org. Lett. 2014, 16, 5528–5531. 10.1021/ol502003a. [DOI] [PubMed] [Google Scholar]
- Shelke S. A.; Sandholt G. B.; Sigurdsson S. T. Nitroxide-Labeled Pyrimidines for Non-Covalent Spin-Labeling of Abasic Sites in DNA and RNA Duplexes. Org. Biomol. Chem. 2014, 12, 7366–7374. 10.1039/C4OB01095G. [DOI] [PubMed] [Google Scholar]
- Kamble N. R.; Gränz M.; Prisner T. F.; Sigurdsson S. T. Noncovalent and Site-Directed Spin Labeling of Duplex RNA. Chem. Commun. 2016, 52, 14442–14445. 10.1039/C6CC08387K. [DOI] [PubMed] [Google Scholar]
- Kamble N. R.; Sigurdsson S. T. Purine-Derived Nitroxides for Noncovalent Spin-Labeling of Abasic Sites in Duplex Nucleic Acids. Chem. - Eur. J. 2018, 24, 4157–4164. 10.1002/chem.201705410. [DOI] [PubMed] [Google Scholar]
- Heinz M.; Erlenbach N.; Stelzl L. S.; Thierolf G.; Kamble N. R.; Sigurdsson S. T.; Prisner T. F.; Hummer G. High-Resolution EPR Distance Measurements on RNA and DNA with the Non-Covalent Ǵ Spin Label. Nucleic Acids Res. 2020, 48, 924–933. 10.1093/nar/gkz1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamble N.; Wolfrum M.; Halbritter T.; Sigurdsson S. T.; Richert C. Noncovalent Spin-Labeling of DNA and RNA Triplexes. Chem. Biodivers. 2020, 17, e1900676 10.1002/cbdv.201900676. [DOI] [PubMed] [Google Scholar]
- Juliusson H. Y.; Sigurdsson S. T. Nitroxide-Derived N-Oxide Phenazines for Noncovalent Spin-Labeling of DNA. ChemBioChem 2020, 21, 2635–2642. 10.1002/cbic.202000128. [DOI] [PubMed] [Google Scholar]
- Ohnishi S.; McConnell H. M. Interaction of the Radical Ion of Chlorpromazine with Deoxyribonucleic Acid. J. Am. Chem. Soc. 1965, 87, 2293. 10.1021/ja01088a040. [DOI] [PubMed] [Google Scholar]
- Hong S.; Piette L. H. Electron Spin Resonance Spin-Label Studies of Intercalation of Ethidium Bromide and Aromatic Amine Carcinogens in DNA. Cancer Res. 1976, 36, 1159–1171. [PubMed] [Google Scholar]
- Ottaviani M. F.; Ghatlia N. D.; Bossmann S. H.; Barton J. K.; Dürr H.; Turro N. J. Nitroxide-Labeled Ruthenium(II)-Polypyridyl Complexes as EPR Probes to Study Organized Systems. 2. Combined Photophysical and EPR Investigations of B-DNA. J. Am. Chem. Soc. 1992, 114, 8946–8952. 10.1021/ja00049a027. [DOI] [Google Scholar]
- Sinha B. K.; Chignell C. F. Acridine Spin Labels as Probes for Nucleic Acids. Life Sci. 1975, 17, 1829–1836. 10.1016/0024-3205(75)90466-X. [DOI] [PubMed] [Google Scholar]
- Spielmann H. P.; Chi D.-Y.; Hunt N. G.; Klein M. P.; Hearst J. E. Spin-Labeled Psoralen Probes for the Study of DNA Dynamics. Biochemistry 1995, 34, 14801–14814. 10.1021/bi00045a022. [DOI] [PubMed] [Google Scholar]
- Raikova E.; Ivanov I.; Kaffalieva D.; Demirov G.; Raikov Z. Spin-Labelling of DNA with Hydrazine Mustard Spin Label (HMSL). Int. J. Biochem. 1982, 14, 41–46. 10.1016/0020-711X(82)90174-4. [DOI] [PubMed] [Google Scholar]
- Saha S.; Hetzke T.; Prisner T. F.; Sigurdsson S. T. Noncovalent Spin-Labeling of RNA: The Aptamer Approach. Chem. Commun. 2018, 54, 11749–11752. 10.1039/C8CC05597A. [DOI] [PubMed] [Google Scholar]
- Gochin M. Nuclear Magnetic Resonance Studies of a Paramagnetic Metallo DNA Complex. J. Am. Chem. Soc. 1997, 119, 3377–3378. 10.1021/ja9633169. [DOI] [Google Scholar]
- Tu K.; Gochin M. Structure Determination by Restrained Molecular Dynamics Using NMR Pseudocontact Shifts as Experimentally Determined Constraints. J. Am. Chem. Soc. 1999, 121, 9276–9285. 10.1021/ja9904540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant G. P. G.; Popova A.; Qin P. Z. Diastereomer Characterizations of Nitroxide-Labeled Nucleic Acids. Biochem. Biophys. Res. Commun. 2008, 371, 451–455. 10.1016/j.bbrc.2008.04.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova A. M.; Qin P. Z. A Nucleotide-Independent Nitroxide Probe Reports on Site-Specific Stereomeric Environment in DNA. Biophys. J. 2010, 99, 2180–2189. 10.1016/j.bpj.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse K. W.; DeGruyter J. N.; Schmidt M. A.; Zheng B.; Vantourout J. C.; Kingston C.; Mercer S. E.; Mcdonald I. M.; Olson R. E.; Zhu Y.; et al. Unlocking P(V): Reagents for Chiral Phosphorothioate Synthesis. Science 2018, 361, 1234–1238. 10.1126/science.aau3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gish G.; Eckstein F. DNA and RNA Sequence Determination Based on Phosphorothioate Chemistry. Science 1988, 240, 1520–1522. 10.1126/science.2453926. [DOI] [PubMed] [Google Scholar]
- Makino K.; Murakami A.; Nagahara S.; Nakatsuji Y.; Takeuchi T. A Study on Spin-Labelled Oligonucleotide Synthesis and Its Electron Spin Resonance Behavior in Solution. Free Radical Res. Commun. 1989, 6, 311–316. 10.3109/10715768909055156. [DOI] [PubMed] [Google Scholar]
- Murakami A.; Mukae M.; Nagahara S.; Konishi Y.; Ide H.; Makino K. Oligonucleotides Site-Specifically Spin-Labeled at 5′-Terminal or Internucleotide Linkage and Their Use in Gene Analyses. Free Radical Res. Commun. 1993, 19, s117–s128. 10.3109/10715769309056s117. [DOI] [PubMed] [Google Scholar]
- Claesen C. A. A.; Daemen C. J. M.; Tesser G. I. Synthesis of Spin-Labelled Di- and Tri-2-Deoxyadenylates. Recl. Trav. Chim. Pays-Bas 1986, 105, 116–123. 10.1002/recl.19861050404. [DOI] [Google Scholar]
- Folkers P. J. M.; van Duynhoven J. P. M.; van Lieshout H. T. M.; Harmsen B. J. M.; van Boom J. H.; Tesser G. I.; Konings R. N. H.; Hilbers C. W. Exploring the DNA Binding Domain of Gene V Protein Encoded by Bacteriophage M13 with the Aid of Spin-Labeled Oligonucleotides in Combination with 1H-NMR. Biochemistry 1993, 32, 9407–9416. 10.1021/bi00087a020. [DOI] [PubMed] [Google Scholar]
- Kuznetsov N. A.; Milov A. D.; Koval V. V.; Samoilova R. I.; Grishin Y. A.; Knorre D. G.; Tsvetkov Y. D.; Fedorova O. S.; Dzuba S. A. PELDOR Study of Conformations of Double-Spin-Labeled Single- and Double-Stranded DNA with Non-Nucleotide Inserts. Phys. Chem. Chem. Phys. 2009, 11, 6826–6832. 10.1039/b904873a. [DOI] [PubMed] [Google Scholar]
- Wunderlich C. H.; Huber R. G.; Spitzer R.; Liedl K. R.; Kloiber K.; Kreutz C. A Novel Paramagnetic Relaxation Enhancement Tag for Nucleic Acids: A Tool to Study Structure and Dynamics of RNA. ACS Chem. Biol. 2013, 8, 2697–2706. 10.1021/cb400589q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko J. C.; Pio M. S.; Tinoco I.; Shin Y. K. A Novel 5′ Displacement Spin-Labeling Technique for Electron Paramagnetic Resonance Spectroscopy of RNA. RNA 1999, 5, 1158–1166. 10.1017/S1355838299990830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant G. P. G.; Qin P. Z. A Facile Method for Attaching Nitroxide Spin Labels at the 5′ Terminus of Nucleic Acids. Nucleic Acids Res. 2007, 35, e77 10.1093/nar/gkm240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidanza J. A.; McLaughlin L. W. Introduction of Reporter Groups at Specific Sites in DNA Containing Phosphorothioate Diesters. J. Am. Chem. Soc. 1989, 111, 9117–9119. 10.1021/ja00207a028. [DOI] [PubMed] [Google Scholar]
- Qin P. Z.; Haworth I. S.; Cai Q.; Kusnetzow A. K.; Grant G. P. G.; Price E. A.; Sowa G. Z.; Popova A.; Herreros B.; He H. Measuring Nanometer Distances in Nucleic Acids Using a Sequence-Independent Nitroxide Probe. Nat. Protoc. 2007, 2, 2354–2365. 10.1038/nprot.2007.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin P. Z.; Butcher S. E.; Feigon J.; Hubbell W. L. Quantitative Analysis of the Isolated GAAA Tetraloop/Receptor Interaction in Solution: A Site-Directed Spin Labeling Study. Biochemistry 2001, 40, 6929–6936. 10.1021/bi010294g. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Xu C.-X.; Di Felice R.; Sponer J.; Islam B.; Stadlbauer P.; Ding Y.; Mao L.; Mao Z.-W.; Qin P. Z. Conformations of Human Telomeric G-Quadruplex Studied Using a Nucleotide-Independent Nitroxide Label. Biochemistry 2016, 55, 360–372. 10.1021/acs.biochem.5b01189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquiaqui J. M.; Sherman E. M.; Ionescu S. A.; Ye J. D.; Fanucci G. E. Characterizing the Dynamics of the Leader-Linker Interaction in the Glycine Riboswitch with Site-Directed Spin Labeling. Biochemistry 2014, 53, 3526–3528. 10.1021/bi500404b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez Reyes C.; Tangprasertchai N. S.; Yogesha S. D.; Nguyen R. H.; Zhang X.; Rajan R.; Qin P. Z. Nucleic Acid-Dependent Conformational Changes in CRISPR–Cas9 Revealed by Site-Directed Spin Labeling. Cell Biochem. Biophys. 2017, 75, 203–210. 10.1007/s12013-016-0738-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova A. M.; Kálai T.; Hideg K.; Qin P. Z. Site-Specific DNA Structural and Dynamic Features Revealed by Nucleotide-Independent Nitroxide Probes. Biochemistry 2009, 48, 8540–8550. 10.1021/bi900860w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Tung C.-S.; Sowa G. Z.; Hatmal M. M.; Haworth I. S.; Qin P. Z. Global Structure of a Three-Way Junction in a Phi29 Packaging RNA Dimer Determined Using Site-Directed Spin Labeling. J. Am. Chem. Soc. 2012, 134, 2644–2652. 10.1021/ja2093647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Dantas Machado A. C.; Ding Y.; Chen Y.; Lu Y.; Duan Y.; Tham K. W.; Chen L.; Rohs R.; Qin P. Z. Conformations of P53 Response Elements in Solution Deduced Using Site-Directed Spin Labeling and Monte Carlo Sampling. Nucleic Acids Res. 2014, 42, 2789–2797. 10.1093/nar/gkt1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant G. P. G.; Boyd N.; Herschlag D.; Qin P. Z. Motions of the Substrate Recognition Duplex in a Group I Intron Assessed by Site-Directed Spin Labeling. J. Am. Chem. Soc. 2009, 131, 3136–3137. 10.1021/ja808217s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P.; Shi X.; Sigurdsson S. T.; Herschlag D.; Qin P. Z. A Single-Stranded Junction Modulates Nanosecond Motional Ordering of the Substrate Recognition Duplex of a Group I Ribozyme. ChemBioChem 2013, 14, 1720–1723. 10.1002/cbic.201300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y.; Zhang X.; Tham K. W.; Qin P. Z. Experimental Mapping of DNA Duplex Shape Enabled by Global Lineshape Analyses of a Nucleotide-Independent Nitroxide Probe. Nucleic Acids Res. 2014, 42, e140–e140. 10.1093/nar/gku695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangprasertchai N. S.; Di Felice R.; Zhang X.; Slaymaker I. M.; Vazquez Reyes C.; Jiang W.; Rohs R.; Qin P. Z. CRISPR-Cas9Mediated DNA Unwinding Detected Using Site-Directed Spin Labeling. ACS Chem. Biol. 2017, 12, 1489–1493. 10.1021/acschembio.6b01137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards T. E.; Sigurdsson S. T. Site-Specific Incorporation of Nitroxide Spin-Labels into 2′-Positions of Nucleic Acids. Nat. Protoc. 2007, 2, 1954–1962. 10.1038/nprot.2007.273. [DOI] [PubMed] [Google Scholar]
- Saha S.; Jagtap A. P.; Sigurdsson S. T. Site-Directed Spin Labeling of RNA by Postsynthetic Modification of 2′-Amino Groups. Methods Enzymol. 2015, 563, 397–414. 10.1016/bs.mie.2015.07.017. [DOI] [PubMed] [Google Scholar]
- Edwards T. E.; Okonogi T. M.; Robinson B. H.; Sigurdsson S. T. Site-Specific Incorporation of Nitroxide Spin-Labels into Internal Sites of the TAR RNA; Structure-Dependent Dynamics of RNA by EPR Spectroscopy. J. Am. Chem. Soc. 2001, 123, 1527–1528. 10.1021/ja005649i. [DOI] [PubMed] [Google Scholar]
- Edwards T. E.; Sigurdsson S. T. Electron Paramagnetic Resonance Dynamic Signatures of TAR RNA–Small Molecule Complexes Provide Insight into RNA Structure and Recognition. Biochemistry 2002, 41, 14843–14847. 10.1021/bi026299a. [DOI] [PubMed] [Google Scholar]
- Edwards T. E.; Okonogi T. M.; Sigurdsson S. T. Investigation of RNA-Protein and RNA-Metal Ion Interactions by Electron Paramagnetic Resonance Spectroscopy: The HIV TAR-Tat Motif. Chem. Biol. 2002, 9, 699–706. 10.1016/S1074-5521(02)00150-3. [DOI] [PubMed] [Google Scholar]
- Schiemann O.; Weber A.; Edwards T. E.; Prisner T. F.; Sigurdsson S. T. Nanometer Distance Measurements on RNA Using PELDOR. J. Am. Chem. Soc. 2003, 125, 3434–3435. 10.1021/ja0274610. [DOI] [PubMed] [Google Scholar]
- Edwards T. E.; Sigurdsson S. T. EPR Spectroscopic Analysis of TAR RNA–Metal Ion Interactions. Biochem. Biophys. Res. Commun. 2003, 303, 721–725. 10.1016/S0006-291X(03)00411-X. [DOI] [PubMed] [Google Scholar]
- Edwards T. E.; Robinson B. H.; Sigurdsson S. T. Identification of Amino Acids That Promote Specific and Rigid TAR RNA-Tat Protein Complex Formation. Chem. Biol. 2005, 12, 329–337. 10.1016/j.chembiol.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Edwards T. E.; Sigurdsson S. T. EPR Spectroscopic Analysis of U7 Hammerhead Ribozyme Dynamics during Metal Ion Induced Folding. Biochemistry 2005, 44, 12870–12878. 10.1021/bi050549g. [DOI] [PubMed] [Google Scholar]
- Helmling C.; Bessi I.; Wacker A.; Schnorr K. A.; Jonker H. R. A.; Richter C.; Wagner D.; Kreibich M.; Schwalbe H. Noncovalent Spin Labeling of Riboswitch RNAs To Obtain Long-Range Structural NMR Restraints. ACS Chem. Biol. 2014, 9, 1330–1339. 10.1021/cb500050t. [DOI] [PubMed] [Google Scholar]
- Kaminker I.; Bye M.; Mendelman N.; Gislason K.; Sigurdsson S. T.; Goldfarb D. Distance Measurements between Manganese(II) and Nitroxide Spin-Labels by DEER Determine a Binding Site of Mn2+ in the HP92 Loop of Ribosomal RNA. Phys. Chem. Chem. Phys. 2015, 17, 15098–15102. 10.1039/C5CP01624J. [DOI] [PubMed] [Google Scholar]
- Schnorr K. A.; Gophane D. B.; Helmling C.; Cetiner E.; Pasemann K.; Fürtig B.; Wacker A.; Qureshi N. S.; Gränz M.; Barthelmes D.; et al. Impact of Spin Label Rigidity on Extent and Accuracy of Distance Information from PRE Data. J. Biomol. NMR 2017, 68, 53–63. 10.1007/s10858-017-0114-9. [DOI] [PubMed] [Google Scholar]
- Kim N. K.; Murali A.; DeRose V. J. A Distance Ruler for RNA Using EPR and Site-Directed Spin Labeling. Chem. Biol. 2004, 11, 939–948. 10.1016/j.chembiol.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Shevelev G. Y.; Krumkacheva O. A.; Lomzov A. A.; Kuzhelev A. A.; Trukhin D. V.; Rogozhnikova O. Y.; Tormyshev V. M.; Pyshnyi D. V.; Fedin M. V.; Bagryanskaya E. G. Triarylmethyl Labels: Toward Improving the Accuracy of EPR Nanoscale Distance Measurements in DNAs. J. Phys. Chem. B 2015, 119, 13641–13648. 10.1021/acs.jpcb.5b03026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaender M.; Sicoli G.; Fontecave T.; Mathis G.; Saint-Pierre C.; Boulard Y.; Gambarelli S.; Gasparutto D. Site-Specific Insertion of Nitroxide-Spin Labels into DNA Probes by Click Chemistry for Structural Analyses by ELDOR Spectroscopy. Nucleic Acids Symp. Ser. 2008, 52, 147–148. 10.1093/nass/nrn075. [DOI] [PubMed] [Google Scholar]
- Hardwick J. S.; Haugland M. M.; El-Sagheer A. H.; Ptchelkine D.; Beierlein F. R.; Lane A. N.; Brown T.; Lovett J. E.; Anderson E. A. 2’-Alkynyl Spin-Labelling Is a Minimally Perturbing Tool for DNA Structural Analysis. Nucleic Acids Res. 2020, 48, 2830–2840. 10.1093/nar/gkaa086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebald L. M.; DeMott C. M.; Ranganathan S.; Asare Okai P. N.; Glazunova A.; Chen A.; Shekhtman A.; Royzen M. Cu(II)-Based Paramagnetic Probe to Study RNA-Protein Interactions by NMR. Inorg. Chem. 2017, 56, 3773–3780. 10.1021/acs.inorgchem.6b02286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebald L. M.; DeMott C. M.; Ranganathan S.; Asare-Okai P. N.; Glazunova A.; Chen A.; Shekhtman A.; Royzen M. Cobalt-Based Paramagnetic Probe to Study RNA-Protein Interactions by NMR. J. Inorg. Biochem. 2017, 170, 202–208. 10.1016/j.jinorgbio.2017.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner L.; Javadi-Zarnaghi F.; Höbartner C. Site-Specific Labeling of RNA at Internal Ribose Hydroxyl Groups: Terbium-Assisted Deoxyribozymes at Work. J. Am. Chem. Soc. 2014, 136, 8131–8137. 10.1021/ja503864v. [DOI] [PubMed] [Google Scholar]
- Shevelev G. Y.; Krumkacheva O. A.; Lomzov A. A.; Kuzhelev A. A.; Rogozhnikova O. Y.; Troitskaya T. I.; Tormyshev V. M.; Fedin M. V.; Pyshnyi D. V.; et al. Physiological-Temperature Distance Measurement in Nucleic Acid Using Triarylmethyl-Based Spin Labels and Pulsed Dipolar EPR Spectroscopy. J. Am. Chem. Soc. 2014, 136, 9874–9877. 10.1021/ja505122n. [DOI] [PubMed] [Google Scholar]
- Shevelev G. Y.; Gulyak E. L.; Lomzov A. A.; Kuzhelev A. A.; Krumkacheva O. A.; Kupryushkin M. S.; Tormyshev V. M.; Fedin M. V.; Bagryanskaya E. G.; Pyshnyi D. V. A Versatile Approach to Attachment of Triarylmethyl Labels to DNA for Nanoscale Structural EPR Studies at Physiological Temperatures. J. Phys. Chem. B 2018, 122, 137–143. 10.1021/acs.jpcb.7b10689. [DOI] [PubMed] [Google Scholar]
- Caron M.; Dugas H. Specific Spin-Labeling of Transfer Ribonucleic Acid Molecules. Nucleic Acids Res. 1976, 3, 19–34. 10.1093/nar/3.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pscheidt R. H.; Wells B. D. Different Conformations of the 3′ Termini of Initiator and Elongator Transfer Ribonucleic Acids. J. Biol. Chem. 1986, 261, 7253–7256. 10.1016/S0021-9258(17)38383-7. [DOI] [PubMed] [Google Scholar]
- Luoma G. A.; Herring F. G.; Marshall A. G. Flexibility of End-Labeled Polymers from Electron Spin Resonance Line-Shape Analysis: 3′ Terminus of Transfer Ribonucleic Acid and 5S Ribonucleic Acid. Biochemistry 1982, 21, 6591–6598. 10.1021/bi00268a042. [DOI] [PubMed] [Google Scholar]
- Hara H.; Horiuchi T.; Saneyoshi M.; Nishimura S. 4-Thiouridine-Specific Spin-Labeling of E. Coli Transfer RNA. Biochem. Biophys. Res. Commun. 1970, 38, 305–311. 10.1016/0006-291X(70)90713-8. [DOI] [PubMed] [Google Scholar]
- Ramos A.; Varani G. A New Method to Detect Long-Range Protein-RNA Contacts: NMR Detection of Electron-Proton Relaxation Induced by Nitroxide Spin-Labeled RNA. J. Am. Chem. Soc. 1998, 120, 10992–10993. 10.1021/ja982496e. [DOI] [Google Scholar]
- Duss O.; Diarra dit Konté N.; Allain F. H. T. Cut and Paste RNA for Nuclear Magnetic Resonance, Paramagnetic Resonance Enhancement, and Electron Paramagnetic Resonance Structural Studies. Methods Enzymol. 2015, 565, 537–562. 10.1016/bs.mie.2015.05.029. [DOI] [PubMed] [Google Scholar]
- Duss O.; Yulikov M.; Jeschke G.; Allain F. H. T. EPR-Aided Approach for Solution Structure Determination of Large RNAs or Protein-RNA Complexes. Nat. Commun. 2014, 5, 3669. 10.1038/ncomms4669. [DOI] [PubMed] [Google Scholar]
- Duss O.; Michel E.; Yulikov M.; Schubert M.; Jeschke G.; Allain F. H. T. Structural Basis of the Non-Coding RNA RsmZ Acting as a Protein Sponge. Nature 2014, 509, 588–592. 10.1038/nature13271. [DOI] [PubMed] [Google Scholar]
- Cai S.; Zhu L.; Zhang Z.; Chen Y. Determination of the Three-Dimensional Structure of the Mrf2-DNA Complex Using Paramagnetic Spin Labeling. Biochemistry 2007, 46, 4943–4950. 10.1021/bi061738h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin P. Z.; Hideg K.; Feigon J.; Hubbell W. L. Monitoring RNA Base Structure and Dynamics Using Site-Directed Spin Labeling. Biochemistry 2003, 42, 6772–6783. 10.1021/bi027222p. [DOI] [PubMed] [Google Scholar]
- Sprinzl M.; Krämer E.; Stehlik D. On the Structure of Phenylalanine TRNA from Yeast. Eur. J. Biochem. 1974, 49, 595–605. 10.1111/j.1432-1033.1974.tb03863.x. [DOI] [PubMed] [Google Scholar]
- McIntosh A. R.; Caron M.; Dugas H. A Specific Spin Labeling of the Anticodon of E.Coli TRNAGlu. Biochem. Biophys. Res. Commun. 1973, 55, 1356–1363. 10.1016/S0006-291X(73)80043-9. [DOI] [PubMed] [Google Scholar]
- Cedergren R. J.; Beauchemin N.; Toupin J. Incorporation of Acyl Groups into the Anticodon of Escherichia Coli Glutamic Acid Transfer Ribonucleic Acid. Biochemistry 1973, 12, 4566–4570. 10.1021/bi00747a003. [DOI] [PubMed] [Google Scholar]
- Lebars I.; Vileno B.; Bourbigot S.; Turek P.; Wolff P.; Kieffer B. A Fully Enzymatic Method for Site-Directed Spin Labeling of Long RNA. Nucleic Acids Res. 2014, 42, e117 10.1093/nar/gku553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vileno B.; Lebars I.Site-Specific Spin Labeling of RNA for NMR and EPR Structural Studies. In RNA Spectroscopy: Methods and Protocols; Arluison V., Wien F., Eds.; Springer US: New York, 2020; pp 217–235. [DOI] [PubMed] [Google Scholar]
- MacMillan A. M.; Verdine G. L. Synthesis of Functionally Tethered Oligodeoxynucleotides by the Convertible Nucleoside Approach. J. Org. Chem. 1990, 55, 5931–5933. 10.1021/jo00311a005. [DOI] [Google Scholar]
- Okamoto A.; Inasaki T.; Saito I. Nitroxide-Labeled Guanine as an ESR Spin Probe for Structural Study of DNA. Bioorg. Med. Chem. Lett. 2004, 14, 3415–3418. 10.1016/j.bmcl.2004.04.076. [DOI] [PubMed] [Google Scholar]
- Sicoli G.; Wachowius F.; Bennati M.; Höbartner C. Probing Secondary Structures of Spin-Labeled RNA by Pulsed EPR Spectroscopy. Angew. Chem., Int. Ed. 2010, 49, 6443–6447. 10.1002/anie.201000713. [DOI] [PubMed] [Google Scholar]
- Halbmair K.; Seikowski J.; Tkach I.; Höbartner C.; Sezer D.; Bennati M. High-Resolution Measurement of Long-Range Distances in RNA: Pulse EPR Spectroscopy with TEMPO-Labeled Nucleotides. Chem. Sci. 2016, 7, 3172–3180. 10.1039/C5SC04631A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budil D. E.; Kolaczkowski S. V.; Perry A.; Varaprasad C.; Johnson F.; Strauss P. R. Dynamics and Ordering in a Spin-Labeled Oligonucleotide Observed by 220 GHz Electron Paramagnetic Resonance. Biophys. J. 2000, 78, 430–438. 10.1016/S0006-3495(00)76605-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowski S. V.; Perry A.; McKenzie A.; Johnson F.; Budil D. E.; Strauss P. R. A Spin-Labeled Abasic DNA Substrate for AP Endonuclease. Biochem. Biophys. Res. Commun. 2001, 288, 722–726. 10.1006/bbrc.2001.5827. [DOI] [PubMed] [Google Scholar]
- Sicoli G.; Mathis G.; Delalande O.; Boulard Y.; Gasparutto D.; Gambarelli S. Double Electron-Electron Resonance (DEER): A Convenient Method to Probe DNA Conformational Changes. Angew. Chem., Int. Ed. 2008, 47, 735–737. 10.1002/anie.200704133. [DOI] [PubMed] [Google Scholar]
- Sicoli G.; Mathis G.; Aci-Sèche S.; Saint-Pierre C.; Boulard Y.; Gasparutto D.; Gambarelli S. Lesion-Induced DNA Weak Structural Changes Detected by Pulsed EPR Spectroscopy Combined with Site-Directed Spin Labelling. Nucleic Acids Res. 2009, 37, 3165–3176. 10.1093/nar/gkp165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner L.; Seikowski J.; Wawrzyniak K.; Ochmann A.; Höbartner C. Synthesis of Spin-Labeled Riboswitch RNAs Using Convertible Nucleosides and DNA-Catalyzed RNA Ligation. Bioorg. Med. Chem. 2013, 21, 6171–6180. 10.1016/j.bmc.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Wawrzyniak-Turek K.; Höbartner C. Deoxyribozyme-Mediated Ligation for Incorporating EPR Spin Labels and Reporter Groups into RNA. Methods Enzymol. 2014, 549, 85–104. 10.1016/B978-0-12-801122-5.00004-0. [DOI] [PubMed] [Google Scholar]
- Ding P.; Wunnicke D.; Steinhoff H.-J.; Seela F. Site-Directed Spin-Labeling of DNA by the Azide-Alkyne “click” Reaction: Nanometer Distance Measurements on 7-Deaza-2′-Deoxyadenosine and 2′-Deoxyuridine Nitroxide Conjugates Spatially Separated or Linked to a “dA-dT” Base Pair. Chem. - Eur. J. 2010, 16, 14385–14396. 10.1002/chem.201001572. [DOI] [PubMed] [Google Scholar]
- Flaender M.; Sicoli G.; Aci-Seche S.; Reignier T.; Maurel V.; Saint-Pierre C.; Boulard Y.; Gambarelli S.; Gasparutto D. A Triple Spin-Labeling Strategy Coupled with DEER Analysis to Detect DNA Modifications and Enzymatic Repair. ChemBioChem 2011, 12, 2560–2563. 10.1002/cbic.201100550. [DOI] [PubMed] [Google Scholar]
- Jakobsen U.; Shelke S. A.; Vogel S.; Sigurdsson S. T. Site-Directed Spin-Labeling of Nucleic Acids by Click Chemistry: Detection of Abasic Sites in Duplex DNA by EPR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 10424–10428. 10.1021/ja102797k. [DOI] [PubMed] [Google Scholar]
- Kerzhner M.; Abdullin D.; Więcek J.; Matsuoka H.; Hagelueken G.; Schiemann O.; Famulok M. Post-Synthetic Spin-Labeling of RNA through Click Chemistry for PELDOR Measurements. Chem. - Eur. J. 2016, 22, 12113–12121. 10.1002/chem.201601897. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Kathiresan V.; Chen Y.; Hu Y.; Jiang W.; Bai G.; Liu G.; Qin P. Z.; Fang X. Posttranscriptional Site-Directed Spin Labeling of Large RNAs with an Unnatural Base Pair System under Non-Denaturing Conditions. Chem. Sci. 2020, 11, 9655–9664. 10.1039/D0SC01717E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuebben C.; Blume S.; Abdullin D.; Brajtenbach D.; Haege F.; Kath-Schorr S.; Schiemann O. Site-Directed Spin Labeling of RNA with a Gem-Diethylisoindoline Spin Label: PELDOR, Relaxation, and Reduction Stability. Molecules 2019, 24, 4482. 10.3390/molecules24244482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuebben C.; Vicino M. F.; Mueller M.; Schiemann O. Do the P1 and P2 Hairpins of the Guanidine-II Riboswitch Interact?. Nucleic Acids Res. 2020, 48, 10518–10526. 10.1093/nar/gkaa703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerzhner M.; Matsuoka H.; Wuebben C.; Famulok M.; Schiemann O. High-Yield Spin Labeling of Long RNAs for Electron Paramagnetic Resonance Spectroscopy. Biochemistry 2018, 57, 2923–2931. 10.1021/acs.biochem.8b00040. [DOI] [PubMed] [Google Scholar]
- Babaylova E. S.; Ivanov A. V.; Malygin A. A.; Vorobjeva M. A.; Venyaminova A. G.; Polienko Y. F.; Kirilyuk I. A.; Krumkacheva O. A.; Fedin M. V.; Karpova G. G.; et al. A Versatile Approach for Site-Directed Spin Labeling and Structural EPR Studies of RNAs. Org. Biomol. Chem. 2014, 12, 3129–3136. 10.1039/c3ob42154f. [DOI] [PubMed] [Google Scholar]
- Babaylova E. S.; Malygin A. A.; Lomzov A. A.; Pyshnyi D. V.; Yulikov M.; Jeschke G.; Krumkacheva O. A.; Fedin M. V.; Karpova G. G.; Bagryanskaya E. G. Complementary-Addressed Site-Directed Spin Labeling of Long Natural RNAs. Nucleic Acids Res. 2016, 44, 7935–7943. 10.1093/nar/gkw516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malygin A. A.; Graifer D. M.; Meschaninova M. I.; Venyaminova A. G.; Krumkacheva O. A.; Fedin M. V.; Karpova G. G.; Bagryanskaya E. G. Doubly Spin-Labeled RNA as an EPR Reporter for Studying Multicomponent Supramolecular Assemblies. Biophys. J. 2015, 109, 2637–2643. 10.1016/j.bpj.2015.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malygin A. A.; Krumkacheva O. A.; Graifer D. M.; Timofeev I. O.; Ochkasova A. S.; Meschaninova M. I.; Venyaminova A. G.; Fedin M. V.; Bowman M.; Karpova G. G.; et al. Exploring the Interactions of Short RNAs with the Human 40S Ribosomal Subunit near the MRNA Entry Site by EPR Spectroscopy. Nucleic Acids Res. 2019, 47, 11850–11860. 10.1093/nar/gkz1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham S. U.; Lippard S. J. Long-Range Distance Constraints in Platinated Nucleotides: Structure Determination of the 5′ Orientational Isomer of Cis-[Pt(NH3)(4-AminoTEMPO){d(GpG)}]+ from Combined Paramagnetic and Diamagnetic NMR Constraints with Molecular Modeling. J. Am. Chem. Soc. 1995, 117, 10702–10712. 10.1021/ja00148a012. [DOI] [Google Scholar]
- Dunham S. U.; Dunham S. U.; Turner C. J.; Lippard S. J. Solution Structure of a DNA Duplex Containing a Nitroxide Spin-Labeled Platinum d(GpG) Intrastrand Cross-Link Refined with NMR-Derived Long-Range Electron–Proton Distance Restraints. J. Am. Chem. Soc. 1998, 120, 5395–5406. 10.1021/ja9742592. [DOI] [Google Scholar]
- Piton N.; Mu Y.; Stock G.; Prisner T. F.; Schiemann O.; Engels J. W. Base-Specific Spin-Labeling of RNA for Structure Determination. Nucleic Acids Res. 2007, 35, 3128–3143. 10.1093/nar/gkm169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto A.; Taiji T.; Tainaka K.; Saito I. Oligonucleotides Containing 7-Vinyl-7-Deazaguanine as a Facile Strategy for Expanding the Functional Diversity of DNA. Bioorg. Med. Chem. Lett. 2002, 12, 1895–1896. 10.1016/S0960-894X(02)00334-7. [DOI] [PubMed] [Google Scholar]
- Eggert F.; Kath-Schorr S. A Cyclopropene-Modified Nucleotide for Site-Specific RNA Labeling Using Genetic Alphabet Expansion Transcription. Chem. Commun. 2016, 52, 7284–7287. 10.1039/C6CC02321E. [DOI] [PubMed] [Google Scholar]
- Domnick C.; Hagelueken G.; Eggert F.; Schiemann O.; Kath-Schorr S. Posttranscriptional Spin Labeling of RNA by Tetrazine-Based Cycloaddition. Org. Biomol. Chem. 2019, 17, 1805–1808. 10.1039/C8OB02597E. [DOI] [PubMed] [Google Scholar]
- Domnick C.; Eggert F.; Wuebben C.; Bornewasser L.; Hagelueken G.; Schiemann O.; Kath-Schorr S. EPR Distance Measurements on Long Non-Coding RNAs Empowered by Genetic Alphabet Expansion Transcription. Angew. Chem., Int. Ed. 2020, 59, 7891–7896. 10.1002/anie.201916447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Täubert S.; Zhang Y. H.; Martinez M. M.; Siepel F.; Wöltjen E.; Leonov A.; Griesinger C. Lanthanide Tagging of Oligonucleotides to Nucleobase for Paramagnetic NMR. ChemBioChem 2020, 21, 3333–3337. 10.1002/cbic.202000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannwarth W.; Schmidt D. Oligonucleotides Containing Spin-Labeled 2′-Deoxycytidine and 5-Methyl-2′-Deoxycytidine as Probes for Structural Motifs of DNA. Bioorg. Med. Chem. Lett. 1994, 4, 977–980. 10.1016/S0960-894X(01)80666-1. [DOI] [Google Scholar]
- Giordano C.; Fratini F.; Attanasio D.; Cellai L. Preparation of Spin-Labeled-2-Amino-DA, DA, DC and 5-Methyl-DC Phosphoramidites for the Automatic Synthesis of EPR Active Oligonucleotides. Synthesis (Stuttg). 2001, 2001, 565–572. 10.1055/s-2001-12355. [DOI] [Google Scholar]
- Weinrich T.; Jaumann E. A.; Scheffer U.; Prisner T. F.; Göbel M. W. A Cytidine Phosphoramidite with Protected Nitroxide Spin Label: Synthesis of a Full-Length TAR RNA and Investigation by In-Line Probing and EPR Spectroscopy. Chem. - Eur. J. 2018, 24, 6202–6207. 10.1002/chem.201800167. [DOI] [PubMed] [Google Scholar]
- Cekan P.; Sigurdsson S. T. Identification of Single-Base Mismatches in Duplex DNA by EPR Spectroscopy. J. Am. Chem. Soc. 2009, 131, 18054–18056. 10.1021/ja905623k. [DOI] [PubMed] [Google Scholar]
- Gophane D. B.; Sigurdsson S. T. TEMPO-Derived Spin Labels Linked to the Nucleobases Adenine and Cytosine for Probing Local Structural Perturbations in DNA by EPR Spectroscopy. Beilstein J. Org. Chem. 2015, 11, 219–227. 10.3762/bjoc.11.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duh J.-L.; Bobst A. M. Sequence-Specific Spin Labeling of Oligothymidylates by Phosphotriester Chemistry. Helv. Chim. Acta 1991, 74, 739–747. 10.1002/hlca.19910740407. [DOI] [Google Scholar]
- Strobel O. K.; Kryak D. D.; Bobst E. V.; Bobst A. M. Preparation and Characterization of Spin-Labeled Oligonucleotides for DNA Hybridization. Bioconjugate Chem. 1991, 2, 89–95. 10.1021/bc00008a003. [DOI] [PubMed] [Google Scholar]
- Liang Z.; Freed J. H.; Keyes R. S.; Bobst A. M. An Electron Spin Resonance Study of DNA Dynamics Using the Slowly Relaxing Local Structure Model. J. Phys. Chem. B 2000, 104, 5372–5381. 10.1021/jp994219f. [DOI] [Google Scholar]
- Kao S.-C.; Bobst A. M. Local Base Dynamics and Local Structural Features in RNA and DNA Duplexes. Biochemistry 1985, 24, 5465–5469. 10.1021/bi00341a028. [DOI] [PubMed] [Google Scholar]
- Spaltenstein A.; Robinson B. H.; Hopkins P. B. A Rigid and Nonperturbing Probe for Duplex DNA Motion. J. Am. Chem. Soc. 1988, 110, 1299–1301. 10.1021/ja00212a053. [DOI] [Google Scholar]
- Strube T.; Schiemann O.; MacMillan F.; Prisner T.; Engels J. W. A New Facile Method for Spin-Labeling of Oligonucleotides. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 1271–1274. 10.1081/NCN-100002534. [DOI] [PubMed] [Google Scholar]
- Krstić I.; Hänsel R.; Romainczyk O.; Engels J. W.; Dötsch V.; Prisner T. F. Long-Range Distance Measurements on Nucleic Acids in Cells by Pulsed EPR Spectroscopy. Angew. Chem., Int. Ed. 2011, 50, 5070–5074. 10.1002/anie.201100886. [DOI] [PubMed] [Google Scholar]
- Krstić I.; Frolow O.; Sezer D.; Endeward B.; Weigand J. E.; Suess B.; Engels J. W.; Prisner T. F. PELDOR Spectroscopy Reveals Preorganization of the Neomycin-Responsive Riboswitch Tertiary Structure. J. Am. Chem. Soc. 2010, 132, 1454–1455. 10.1021/ja9077914. [DOI] [PubMed] [Google Scholar]
- Fischhaber P. L.; Reese A. W.; Nguyen T.; Kirchner J. J.; Hustedt E. J.; Robinson B. H.; Hopkins P. B. Synthesis of Duplex DNA Containing a Spin Labeled Analog of 2′ Deoxycytidine. Nucleosides Nucleotides 1997, 16, 365–377. 10.1080/07328319708001356. [DOI] [Google Scholar]
- Schiemann O.; Piton N.; Mu Y.; Stock G.; Engels J. W.; Prisner T. F. A PELDOR-Based Nanometer Distance Ruler for Oligonucleotides. J. Am. Chem. Soc. 2004, 126, 5722–5729. 10.1021/ja0393877. [DOI] [PubMed] [Google Scholar]
- Schiemann O.; Piton N.; Plackmeyer J.; Bode B. E.; Prisner T. F.; Engels J. W. Spin Labeling of Oligonucleotides with the Nitroxide TPA and Use of PELDOR, a Pulse EPR Method, to Measure Intramolecular Distances. Nat. Protoc. 2007, 2, 904–923. 10.1038/nprot.2007.97. [DOI] [PubMed] [Google Scholar]
- Gannett P. M.; Darian E.; Powell J.; Johnson E. M. II; Mundoma C.; Greenbaum N. L.; Ramsey C. M.; Dalal N. S.; Budil D. E. Probing Triplex Formation by EPR Spectroscopy Using a Newly Synthesized Spin Label for Oligonucleotides. Nucleic Acids Res. 2002, 30, 5328–5337. 10.1093/nar/gkf634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Azarkh M.; Exner T. E.; Hartig J. S.; Drescher M. Human Telomeric Quadruplex Conformations Studied by Pulsed EPR. Angew. Chem., Int. Ed. 2009, 48, 9728–9730. 10.1002/anie.200902146. [DOI] [PubMed] [Google Scholar]
- Gophane D. B.; Sigurdsson S. T. Hydrogen-Bonding Controlled Rigidity of an Isoindoline-Derived Nitroxide Spin Label for Nucleic Acids. Chem. Commun. 2013, 49, 999–1001. 10.1039/C2CC36389E. [DOI] [PubMed] [Google Scholar]
- Gophane D. B.; Endeward B.; Prisner T. F.; Sigurdsson S. T. Conformationally Restricted Isoindoline-Derived Spin Labels in Duplex DNA: Distances and Rotational Flexibility by Pulsed Electron–Electron Double Resonance Spectroscopy. Chem. - Eur. J. 2014, 20, 15913–15919. 10.1002/chem.201403726. [DOI] [PubMed] [Google Scholar]
- Erlenbach N.; Endeward B.; Schöps P.; Gophane D. B.; Sigurdsson S. T.; Prisner T. F. Flexibilities of Isoindoline-Derived Spin Labels for Nucleic Acids by Orientation Selective PELDOR. Phys. Chem. Chem. Phys. 2016, 18, 16196–16201. 10.1039/C6CP02475K. [DOI] [PubMed] [Google Scholar]
- Gophane D. B.; Endeward B.; Prisner T. F.; Sigurdsson S. T. A Semi-Rigid Isoindoline-Derived Nitroxide Spin Label for RNA. Org. Biomol. Chem. 2018, 16, 816–824. 10.1039/C7OB02870A. [DOI] [PubMed] [Google Scholar]
- Collauto A.; von Bülow S.; Gophane D. B.; Saha S.; Stelzl L. S.; Hummer G.; Sigurdsson S. T.; Prisner T. F. Compaction of RNA Duplexes in the Cell. Angew. Chem., Int. Ed. 2020, 59, 23025–23029. 10.1002/anie.202009800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. R.; Alley S. C.; Reese A. W.; Solomon M. S.; McCallister W. V.; Mailer C.; Robinson B. H.; Hopkins P. B. A Probe for Sequence-Dependent Nucleic Acid Dynamics. J. Am. Chem. Soc. 1995, 117, 9377–9378. 10.1021/ja00141a040. [DOI] [Google Scholar]
- Miller T. R.; Hopkins P. B. Toward the Synthesis of a Second-Generation Nitroxide Spin Probe for DNA Dynamics Studies. Bioorg. Med. Chem. Lett. 1994, 4, 981–986. 10.1016/S0960-894X(01)80667-3. [DOI] [Google Scholar]
- Okonogi T. M.; Reese A. W.; Alley S. C.; Hopkins P. B.; Robinson B. H. Flexibility of Duplex DNA on the Submicrosecond Timescale. Biophys. J. 1999, 77, 3256–3276. 10.1016/S0006-3495(99)77157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonogi T. M.; Alley S. C.; Reese A. W.; Hopkins P. B.; Robinson B. H. Sequence-Dependent Dynamics of Duplex DNA: The Applicability of a Dinucleotide Model. Biophys. J. 2002, 83, 3446–3459. 10.1016/S0006-3495(02)75344-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhate N.; Cekan P.; Massey A. P.; Sigurdsson S. T. A Nucleoside That Contains a Rigid Nitroxide Spin Label: A Fluorophore in Disguise. Angew. Chem., Int. Ed. 2007, 46, 2655–2658. 10.1002/anie.200603993. [DOI] [PubMed] [Google Scholar]
- Cekan P.; Smith A. L.; Barhate N.; Robinson B. H.; Sigurdsson S. T. Rigid Spin-Labeled Nucleoside Ç: A Nonperturbing EPR Probe of Nucleic Acid Conformation. Nucleic Acids Res. 2008, 36, 5946–5954. 10.1093/nar/gkn562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards T. E.; Cekan P.; Reginsson G. W.; Shelke S. A.; Ferré-D’Amaré A. R.; Schiemann O.; Sigurdsson S. T. Crystal Structure of a DNA Containing the Planar, Phenoxazine-Derived Bi-Functional Spectroscopic Probe Ç. Nucleic Acids Res. 2011, 39, 4419–4426. 10.1093/nar/gkr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höbartner C.; Sicoli G.; Wachowius F.; Gophane D. B.; Sigurdsson S. T. Synthesis and Characterization of RNA Containing a Rigid and Nonperturbing Cytidine-Derived Spin Label. J. Org. Chem. 2012, 77, 7749–7754. 10.1021/jo301227w. [DOI] [PubMed] [Google Scholar]
- Juliusson H. Y.; Segler A.-L. J.; Sigurdsson S. T. Benzoyl-Protected Hydroxylamines for Improved Chemical Synthesis of Oligonucleotides Containing Nitroxide Spin Labels. Eur. J. Org. Chem. 2019, 2019, 3799–3805. 10.1002/ejoc.201900553. [DOI] [Google Scholar]
- Cekan P.; Jonsson E. ö; Sigurdsson S. T. Folding of the Cocaine Aptamer Studied by EPR and Fluorescence Spectroscopies Using the Bifunctional Spectroscopic Probe Ç. Nucleic Acids Res. 2009, 37, 3990–3995. 10.1093/nar/gkp277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann O.; Cekan P.; Margraf D.; Prisner T. F.; Sigurdsson S. T. Relative Orientation of Rigid Nitroxides by PELDOR: Beyond Distance Measurements in Nucleic Acids. Angew. Chem., Int. Ed. 2009, 48, 3292–3295. 10.1002/anie.200805152. [DOI] [PubMed] [Google Scholar]
- Smith A. L.; Cekan P.; Brewood G. P.; Okonogi T. M.; Alemayehu S.; Hustedt E. J.; Benight A. S.; Sigurdsson S. T.; Robinson B. H. Conformational Equilibria of Bulged Sites in Duplex DNA Studied by EPR Spectroscopy. J. Phys. Chem. B 2009, 113, 2664–2675. 10.1021/jp808260b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko A.; Denysenkov V.; Margraf D.; Cekan P.; Schiemann O.; Sigurdsson S. T.; Prisner T. F. Conformational Flexibility of DNA. J. Am. Chem. Soc. 2011, 133, 13375–13379. 10.1021/ja201244u. [DOI] [PubMed] [Google Scholar]
- Cekan P.; Sigurdsson S. T. Conformation and Dynamics of Nucleotides in Bulges and Symmetric Internal Loops in Duplex DNA Studied by EPR and Fluorescence Spectroscopies. Biochem. Biophys. Res. Commun. 2012, 420, 656–661. 10.1016/j.bbrc.2012.03.059. [DOI] [PubMed] [Google Scholar]
- Grytz C. M.; Marko A.; Cekan P.; Sigurdsson S. T.; Prisner T. F. Flexibility and Conformation of the Cocaine Aptamer Studied by PELDOR. Phys. Chem. Chem. Phys. 2016, 18, 2993–3002. 10.1039/C5CP06158J. [DOI] [PubMed] [Google Scholar]
- Grytz C. M.; Kazemi S.; Marko A.; Cekan P.; Güntert P.; Sigurdsson S. T.; Prisner T. F. Determination of Helix Orientations in a Flexible DNA by Multi-Frequency EPR Spectroscopy. Phys. Chem. Chem. Phys. 2017, 19, 29801–29811. 10.1039/C7CP04997H. [DOI] [PubMed] [Google Scholar]
- Gränz M.; Erlenbach N.; Spindler P.; Gophane D. B.; Stelzl L. S.; Sigurdsson S. T.; Prisner T. F. Dynamics of Nucleic Acids at Room Temperature Revealed by Pulsed EPR Spectroscopy. Angew. Chem., Int. Ed. 2018, 57, 10540–10543. 10.1002/anie.201803682. [DOI] [PubMed] [Google Scholar]
- Bowen A. M.; Erlenbach N.; van Os P.; Stelzl L. S.; Sigurdsson S. T.; Prisner T. F. Orientation Selective 2D-SIFTER Experiments at X-Band Frequencies. Appl. Magn. Reson. 2018, 49, 1355–1368. 10.1007/s00723-018-1057-3. [DOI] [Google Scholar]
- Hetzke T.; Vogel M.; Gophane D. B.; Weigand J. E.; Suess B.; Sigurdsson S. T.; Prisner T. F. Influence of Mg2+ on the Conformational Flexibility of a Tetracycline Aptamer. RNA 2019, 25, 158–167. 10.1261/rna.068684.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetiner E. C.; Jonker H. R. A.; Helmling C.; Gophane D. B.; Grünewald C.; Sigurdsson S. T.; Schwalbe H. Paramagnetic-Iterative Relaxation Matrix Approach: Extracting PRE-Restraints from NOESY Spectra for 3D Structure Elucidation of Biomolecules. J. Biomol. NMR 2019, 73, 699–712. 10.1007/s10858-019-00282-0. [DOI] [PubMed] [Google Scholar]
- Juliusson H. Y.; Sigurdsson S. T. Reduction Resistant and Rigid Nitroxide Spin-Labels for DNA and RNA. J. Org. Chem. 2020, 85, 4036–4046. 10.1021/acs.joc.9b02988. [DOI] [PubMed] [Google Scholar]
- Iwahara J.; Anderson D. E.; Murphy E. C.; Clore G. M. EDTA-Derivatized Deoxythymidine as a Tool for Rapid Determination of Protein Binding Polarity to DNA by Intermolecular Paramagnetic Relaxation Enhancement. J. Am. Chem. Soc. 2003, 125, 6634–6635. 10.1021/ja034488q. [DOI] [PubMed] [Google Scholar]
- Iwahara J.; Clore G. M. Detecting Transient Intermediates in Macromolecular Binding by Paramagnetic NMR. Nature 2006, 440, 1227–1230. 10.1038/nature04673. [DOI] [PubMed] [Google Scholar]
- Lawless M. J.; Sarver J. L.; Saxena S. Nucleotide-Independent Copper(II)-Based Distance Measurements in DNA by Pulsed ESR Spectroscopy. Angew. Chem., Int. Ed. 2017, 56, 2115–2117. 10.1002/anie.201611197. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Lawless M. J.; Brubaker H. J.; Singewald K.; Kurpiewski M. R.; Jen-Jacobson L.; Saxena S. Cu2+-Based Distance Measurements by Pulsed EPR Provide Distance Constraints for DNA Backbone Conformations in Solution. Nucleic Acids Res. 2020, 48, e49 10.1093/nar/gkaa133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.; Casto J.; Bogetti X.; Arora C.; Wang J.; Saxena S. Orientation and Dynamics of Cu2+ Based DNA Labels from Force Field Parameterized MD Elucidates the Relationship between EPR Distance Constraints and DNA Backbone Distances. Phys. Chem. Chem. Phys. 2020, 22, 26707–26719. 10.1039/D0CP05016D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhard D. M.; Meyer A.; Berndhäuser A.; Schiemann O.; Clever G. H. Di-Copper(II) DNA G-Quadruplexes as EPR Distance Rulers. Chem. Commun. 2018, 54, 7455–7458. 10.1039/C8CC04053B. [DOI] [PubMed] [Google Scholar]
- Stratmann L. M.; Kutin Y.; Kasanmascheff M.; Clever G. H. Precise Distance Measurements in DNA G-Quadruplex Dimers and Sandwich Complexes by Pulsed Dipolar EPR Spectroscopy. Angew. Chem., Int. Ed. 2021, 60, 4939–4947. 10.1002/anie.202008618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.-X.; Zhang X.; Zhou Y.-W.; Wang H.; Cao Q.; Shen Y.; Ji L.-N.; Mao Z.-W.; Qin P. Z. A Nitroxide-Tagged Platinum(II) Complex Enables the Identification of a DNA G-Quadruplex Binding Mode. Chem. - Eur. J. 2016, 22, 3405–3413. 10.1002/chem.201504960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N. U.; Venot A.; Umemoto K.; Leffler H.; Prestegard J. H. Distance Mapping of Protein-Binding Sites Using Spin-Labeled Oligosaccharide Ligands. Protein Sci. 2001, 10, 2393–2400. 10.1110/ps.17401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Meng L.; Moremen K. W.; Prestegard J. H. Nuclear Magnetic Resonance Structural Characterization of Substrates Bound to the α-2,6-Sialyltransferase, ST6Gal-I. Biochemistry 2009, 48, 11211–11219. 10.1021/bi9015154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T.; Kamiya Y.; Choo Y.-M.; Yamamoto S.; Kato K. Terminal Spin Labeling of a High-Mannose-Type Oligosaccharide for Quantitative NMR Analysis of Its Dynamic Conformation. Chem. Lett. 2013, 42, 544–546. 10.1246/cl.130040. [DOI] [Google Scholar]
- Yamamoto S.; Yamaguchi T.; Erdélyi M.; Griesinger C.; Kato K. Paramagnetic Lanthanide Tagging for NMR Conformational Analyses of N-Linked Oligosaccharides. Chem. - Eur. J. 2011, 17, 9280–9282. 10.1002/chem.201100856. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T.; Sakae Y.; Zhang Y.; Yamamoto S.; Okamoto Y.; Kato K. Exploration of Conformational Spaces of High-Mannose-Type Oligosaccharides by an NMR-Validated Simulation. Angew. Chem., Int. Ed. 2014, 53, 10941–10944. 10.1002/anie.201406145. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Kajino M.; Yanaka S.; Zhu T.; Yagi H.; Satoh T.; Yamaguchi T.; Kato K. Conformational Analysis of a High-Mannose-Type Oligosaccharide Displaying Glucosyl Determinant Recognised by Molecular Chaperones Using NMR-Validated Molecular Dynamics Simulation. ChemBioChem 2017, 18, 396–401. 10.1002/cbic.201600595. [DOI] [PubMed] [Google Scholar]
- Mallagaray A.; Canales A.; Domínguez G.; Jiménez-Barbero J.; Pérez-Castells J. A Rigid Lanthanide Binding Tag for NMR Structural Analysis of Carbohydrates. Chem. Commun. 2011, 47, 7179–7181. 10.1039/c1cc11860a. [DOI] [PubMed] [Google Scholar]
- Erdélyi M.; D’Auvergne E.; Navarro-Vázquez A.; Leonov A.; Griesinger C. Dynamics of the Glycosidic Bond: Conformational Space of Lactose. Chem. - Eur. J. 2011, 17, 9368–9376. 10.1002/chem.201100854. [DOI] [PubMed] [Google Scholar]
- Yamamoto S.; Zhang Y.; Yamaguchi T.; Kameda T.; Kato K. Lanthanide-Assisted NMR Evaluation of a Dynamic Ensemble of Oligosaccharide Conformations. Chem. Commun. 2012, 48, 4752–4754. 10.1039/c2cc30353a. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yamamoto S.; Yamaguchi T.; Kato K. Application of Paramagnetic NMR-Validated Molecular Dynamics Simulation to the Analysis of a Conformational Ensemble of a Branched Oligosaccharide. Molecules 2012, 17, 6658–6671. 10.3390/molecules17066658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canales A.; Mallagaray A.; Pérez-Castells J.; Boos I.; Unverzagt C.; André S.; Gabius H. J.; Cañada F. J.; Jiménez-Barbero J. Breaking Pseudo-Symmetry in Multiantennary Complex N-Glycans Using Lanthanide-Binding Tags and NMR Pseudo-Contact Shifts. Angew. Chem., Int. Ed. 2013, 52, 13789–13793. 10.1002/anie.201307845. [DOI] [PubMed] [Google Scholar]
- Canales A.; Boos I.; Perkams L.; Karst L.; Luber T.; Karagiannis T.; Domínguez G.; Cañada F. J.; Pérez-Castells J.; Häussinger D.; et al. Breaking the Limits in Analyzing Carbohydrate Recognition by NMR Spectroscopy: Resolving Branch-Selective Interaction of a Tetra-Antennary N-Glycan with Lectins. Angew. Chem., Int. Ed. 2017, 56, 14987–14991. 10.1002/anie.201709130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández de Toro B.; Peng W.; Thompson A. J.; Domínguez G.; Cañada F. J.; Pérez-Castells J.; Paulson J. C.; Jiménez-Barbero J.; Canales Á. Avenues to Characterize the Interactions of Extended N-Glycans with Proteins by NMR Spectroscopy: The Influenza Hemagglutinin Case. Angew. Chem., Int. Ed. 2018, 57, 15051–15055. 10.1002/anie.201807162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canales Á.; Mallagaray Á.; Berbís M. Á.; Navarro-Vázquez A.; Domínguez G.; Cañada F. J.; André S.; Gabius H.-J.; Pérez-Castells J.; Jiménez-Barbero J. Lanthanide-Chelating Carbohydrate Conjugates Are Useful Tools to Characterize Carbohydrate Conformation in Solution and Sensitive Sensors to Detect Carbohydrate-Protein Interactions. J. Am. Chem. Soc. 2014, 136, 8011–8017. 10.1021/ja502406x. [DOI] [PubMed] [Google Scholar]
- Mallagaray A.; Domínguez G.; Peters T.; Pérez-Castells J. A Rigid Lanthanide Binding Tag to Aid NMR Studies of a 70 KDa Homodimeric Coat Protein of Human Norovirus. Chem. Commun. 2016, 52, 601–604. 10.1039/C5CC05827A. [DOI] [PubMed] [Google Scholar]
- Moure M. J.; Eletsky A.; Gao Q.; Morris L. C.; Yang J.-Y.; Chapla D.; Zhao Y.; Zong C.; Amster I. J.; Moremen K. W.; et al. Paramagnetic Tag for Glycosylation Sites in Glycoproteins: Structural Constraints on Heparan Sulfate Binding to Robo1. ACS Chem. Biol. 2018, 13, 2560–2567. 10.1021/acschembio.8b00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilder J.; Ubbink M. Formation of Transient Protein Complexes. Curr. Opin. Struct. Biol. 2013, 23, 911–918. 10.1016/j.sbi.2013.07.009. [DOI] [PubMed] [Google Scholar]
- Ma L.; Jørgensen A. M. M.; Sørensen G. O.; Ulstrup J.; Led J. J. Elucidation of the Paramagnetic R1 Relaxation of Heteronuclei and Protons in Cu(II) Plastocyanin from Anabaena Variabilis. J. Am. Chem. Soc. 2000, 122, 9473–9485. 10.1021/ja001368z. [DOI] [Google Scholar]
- Liang B.; Bushweller J. H.; Tamm L. K. Site-Directed Parallel Spin-Labeling and Paramagnetic Relaxation Enhancement in Structure Determination of Membrane Proteins by Solution NMR Spectroscopy. J. Am. Chem. Soc. 2006, 128, 4389–4397. 10.1021/ja0574825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matei E.; Gronenborn A. M. 19F Paramagnetic Relaxation Enhancement: A Valuable Tool for Distance Measurements in Proteins. Angew. Chem., Int. Ed. 2016, 55, 150–154. 10.1002/anie.201508464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara J.; Schwieters C. D.; Clore G. M. Ensemble Approach for NMR Structure Refinement against 1H Paramagnetic Relaxation Enhancement Data Arising from a Flexible Paramagnetic Group Attached to a Macromolecule. J. Am. Chem. Soc. 2004, 126, 5879–5896. 10.1021/ja031580d. [DOI] [PubMed] [Google Scholar]
- Kosen P. A. Spin Labeling of Proteins. Methods Enzymol. 1989, 177, 86–121. 10.1016/0076-6879(89)77007-5. [DOI] [PubMed] [Google Scholar]
- Flügge F.; Peters T. Complete Assignment of Ala, Ile, Leu, Met and Val Methyl Groups of Human Blood Group A and B Glycosyltransferases Using Lanthanide-Induced Pseudocontact Shifts and Methyl–Methyl NOESY. J. Biomol. NMR 2018, 70, 245–259. 10.1007/s10858-018-0183-4. [DOI] [PubMed] [Google Scholar]
- John M.; Otting G. Strategies for Measurements of Pseudocontact Shifts in Protein NMR Spectroscopy. ChemPhysChem 2007, 8, 2309–2313. 10.1002/cphc.200700510. [DOI] [PubMed] [Google Scholar]
- Bahramzadeh A.; Huber T.; Otting G. Three-Dimensional Protein Structure Determination Using Pseudocontact Shifts of Backbone Amide Protons Generated by Double-Histidine Co2+-Binding Motifs at Multiple Sites. Biochemistry 2019, 58, 3243–3250. 10.1021/acs.biochem.9b00404. [DOI] [PubMed] [Google Scholar]
- John M.; Park A. Y.; Dixon N. E.; Otting G. NMR Detection of Protein 15N Spins near Paramagnetic Lanthanide Ions. J. Am. Chem. Soc. 2007, 129, 462–463. 10.1021/ja066995o. [DOI] [PubMed] [Google Scholar]
- Schmitz C.; John M.; Park A. Y.; Dixon N. E.; Otting G.; Pintacuda G.; Huber T. Efficient χ-Tensor Determination and NH Assignment of Paramagnetic Proteins. J. Biomol. NMR 2006, 35, 79–87. 10.1007/s10858-006-9002-4. [DOI] [PubMed] [Google Scholar]
- John M.; Schmitz C.; Park A. Y.; Dixon N. E.; Huber T.; Otting G. Sequence-Specific and Stereospecific Assignment of Methyl Groups Using Paramagnetic Lanthanides. J. Am. Chem. Soc. 2007, 129, 13749–13757. 10.1021/ja0744753. [DOI] [PubMed] [Google Scholar]
- Skinner S. P.; Moshev M.; Hass M. A. S.; Ubbink M. PARAssign - Paramagnetic NMR Assignments of Protein Nuclei on the Basis of Pseudocontact Shifts. J. Biomol. NMR 2013, 55, 379–389. 10.1007/s10858-013-9722-1. [DOI] [PubMed] [Google Scholar]
- Lescanne M.; Skinner S. P.; Blok A.; Timmer M.; Cerofolini L.; Fragai M.; Luchinat C.; Ubbink M. Methyl Group Assignment Using Pseudocontact Shifts with PARAssign. J. Biomol. NMR 2017, 69, 183–195. 10.1007/s10858-017-0136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakomek N. A.; Walter K. F. A.; Farès C.; Lange O. F.; de Groot B. L.; Grubmüller H.; Brüschweiler R.; Munk A.; Becker S.; Meiler J.; et al. Self-Consistent Residual Dipolar Coupling Based Model-Free Analysis for the Robust Determination of Nanosecond to Microsecond Protein Dynamics. J. Biomol. NMR 2008, 41, 139–155. 10.1007/s10858-008-9244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farès C.; Lakomek N. A.; Walter K. F. A.; Frank B. T. C.; Meiler J.; Becker S.; Griesinger C. Accessing ns−μs Side Chain Dynamics in Ubiquitin with Methyl RDCs. J. Biomol. NMR 2009, 45, 23–44. 10.1007/s10858-009-9354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L.; Bax A. Modulating Protein Alignment in a Liquid-Crystalline Medium through Conservative Mutagenesis. J. Am. Chem. Soc. 2007, 129, 11326–11327. 10.1021/ja073937+. [DOI] [PubMed] [Google Scholar]
- Güntert P.Automated NMR Structure Calculation With CYANA. In Protein NMR Techniques; Downing A. K., Ed.; Humana Press: Totowa, 2004; pp 353–378. [DOI] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Marius Clore G. Using Xplor–NIH for NMR Molecular Structure Determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 48, 47–62. 10.1016/j.pnmrs.2005.10.001. [DOI] [Google Scholar]
- Schmitz C.; Vernon R.; Otting G.; Baker D.; Huber T. Protein Structure Determination from Pseudocontact Shifts Using ROSETTA. J. Mol. Biol. 2012, 416, 668–677. 10.1016/j.jmb.2011.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla K. B.; Otting G.; Huber T. Pseudocontact Shift-Driven Iterative Resampling for 3D Structure Determinations of Large Proteins. J. Mol. Biol. 2016, 428, 522–532. 10.1016/j.jmb.2016.01.007. [DOI] [PubMed] [Google Scholar]
- Pilla K. B.; Otting G.; Huber T. Protein Structure Determination by Assembling Super-Secondary Structure Motifs Using Pseudocontact Shifts. Structure 2017, 25, 559–568. 10.1016/j.str.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Yagi H.; Pilla K. B.; Maleckis A.; Graham B.; Huber T.; Otting G. Three-Dimensional Protein Fold Determination from Backbone Amide Pseudocontact Shifts Generated by Lanthanide Tags at Multiple Sites. Structure 2013, 21, 883–890. 10.1016/j.str.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Kuenze G.; Bonneau R.; Leman J. K.; Meiler J. Integrative Protein Modeling in RosettaNMR from Sparse Paramagnetic Restraints. Structure 2019, 27, 1721–1734.e5. 10.1016/j.str.2019.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick D. J.; Wang J. X.; Graham B.; Swarbrick J. D.; Mott H. R.; Nietlispach D. Integral Membrane Protein Structure Determination Using Pseudocontact Shifts. J. Biomol. NMR 2015, 61, 197–207. 10.1007/s10858-015-9899-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A.; Mott H. R.; Bostock M. J.; Kirkpatrick J. P.; Nietlispach D. Structure Determination of the Seven-Helix Transmembrane Receptor Sensory Rhodopsin II by Solution NMR Spectroscopy. Nat. Struct. Mol. Biol. 2010, 17, 768–774. 10.1038/nsmb.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B.-B.; Yang F.; Ye Y.; Wu Q.; Li C.; Huber T.; Su X.-C. 3D Structure Determination of a Protein in Living Cells Using Paramagnetic NMR Spectroscopy. Chem. Commun. 2016, 52, 10237–10240. 10.1039/C6CC05490K. [DOI] [PubMed] [Google Scholar]
- Bradley P.; Misura K. M. S.; Baker D. Toward High-Resolution de Novo Structure Prediction for Small Proteins. Science 2005, 309, 1868–1871. 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- Pilla K. B.; Leman J. K.; Otting G.; Huber T. Capturing Conformational States in Proteins Using Sparse Paramagnetic NMR Data. PLoS One 2015, 10, e0127053 10.1371/journal.pone.0127053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Del Bianco C.; Gelis I.; Katsaros N.; Luchinat C.; Parigi G.; Peana M.; Provenzani A.; Zoroddu M. A. Experimentally Exploring the Conformational Space Sampled by Domain Reorientation in Calmodulin. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 6841–6846. 10.1073/pnas.0308641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X.-N.; Sekiyama N.; Ohtani Y.; Yang F.; Miyanoiri Y.; Akagi K.; Su X.-C.; Tochio H. Conformational Space Sampled by Domain Reorientation of Linear Diubiquitin Reflected in Its Binding Mode for Target Proteins. ChemPhysChem 2021, 22, 1505–1517. 10.1002/cphc.202100187. [DOI] [PubMed] [Google Scholar]
- Nagulapalli M.; Parigi G.; Yuan J.; Gsponer J.; Deraos G.; Bamm V. V.; Harauz G.; Matsoukas J.; de Planque M. R. R.; Gerothanassis I. P.; et al. Recognition Pliability Is Coupled to Structural Heterogeneity: A Calmodulin Intrinsically Disordered Binding Region Complex. Structure 2012, 20, 522–533. 10.1016/j.str.2012.01.021. [DOI] [PubMed] [Google Scholar]
- Cerofolini L.; Fields G. B.; Fragai M.; Geraldes C. F. G. C.; Luchinat C.; Parigi G.; Ravera E.; Svergun D. I.; Teixeira J. M. C. Examination of Matrix Metalloproteinase-1 in Solution A: Preference for the Pre-Collagenolysis State. J. Biol. Chem. 2013, 288, 30659–30671. 10.1074/jbc.M113.477240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saio T.; Ogura K.; Kumeta H.; Kobashigawa Y.; Shimizu K.; Yokochi M.; Kodama K.; Yamaguchi H.; Tsujishita H.; Inagaki F. Ligand-Driven Conformational Changes of MurD Visualized by Paramagnetic NMR. Sci. Rep. 2015, 5, 16685. 10.1038/srep16685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.-N.; Loscha K. V.; Nitsche C.; Graham B.; Otting G. The Dengue Virus NS2B-NS3 Protease Retains the Closed Conformation in the Complex with BPTI. FEBS Lett. 2014, 588, 2206–2211. 10.1016/j.febslet.2014.05.018. [DOI] [PubMed] [Google Scholar]
- de la Cruz L.; Chen W.-N.; Graham B.; Otting G. Binding Mode of the Activity-Modulating C-Terminal Segment of NS2B to NS3 in the Dengue Virus NS2B-NS3 Protease. FEBS J. 2014, 281, 1517–1533. 10.1111/febs.12729. [DOI] [PubMed] [Google Scholar]
- Guan J.-Y.; Keizers P. H. J.; Liu W.-M.; Löhr F.; Skinner S. P.; Heeneman E. A.; Schwalbe H.; Ubbink M.; Siegal G. Small-Molecule Binding Sites on Proteins Established by Paramagnetic NMR Spectroscopy. J. Am. Chem. Soc. 2013, 135, 5859–5868. 10.1021/ja401323m. [DOI] [PubMed] [Google Scholar]
- Lescanne M.; Ahuja P.; Blok A.; Timmer M.; Akerud T.; Ubbink M. Methyl Group Reorientation under Ligand Binding Probed by Pseudocontact Shifts. J. Biomol. NMR 2018, 71, 275–285. 10.1007/s10858-018-0190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubbink M.; Ejdebäck M.; Karlsson B. G.; Bendall D. S. The Structure of the Complex of Plastocyanin and Cytochrome f, Determined by Paramagnetic NMR and Restrained Rigid-Body Molecular Dynamics. Structure 1998, 6, 323–335. 10.1016/S0969-2126(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Crowley P. B.; Otting G.; Schlarb-Ridley B. G.; Canters G. W.; Ubbink M. Hydrophobic Interactions in a Cyanobacterial Plastocyanin-Cytochrome f Complex. J. Am. Chem. Soc. 2001, 123, 10444–10453. 10.1021/ja0112700. [DOI] [PubMed] [Google Scholar]
- Díaz-Moreno I.; Díaz-Quintana A.; de la Rosa M. A.; Ubbink M. Structure of the Complex between Plastocyanin and Cytochrome f from the Cyanobacterium Nostoc Sp PCC 7119 as Determined by Paramagnetic NMR - The Balance between Electrostatic and Hydrophobic Interactions within the Transient Complex Determines the Relati. J. Biol. Chem. 2005, 280, 18908–18915. 10.1074/jbc.M413298200. [DOI] [PubMed] [Google Scholar]
- Pintacuda G.; Park A. Y.; Keniry M. A.; Dixon N. E.; Otting G. Lanthanide Labeling Offers Fast NMR Approach to 3D Structure Determinations of Protein-Protein Complexes. J. Am. Chem. Soc. 2006, 128, 3696–3702. 10.1021/ja057008z. [DOI] [PubMed] [Google Scholar]
- Kobashigawa Y.; Saio T.; Ushio M.; Sekiguchi M.; Yokochi M.; Ogura K.; Inagaki F. Convenient Method for Resolving Degeneracies Due to Symmetry of the Magnetic Susceptibility Tensor and Its Application to Pseudo Contact Shift-Based Protein-Protein Complex Structure Determination. J. Biomol. NMR 2012, 53, 53–63. 10.1007/s10858-012-9623-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.; Thornton J. M. Principles of Protein-Protein Interactions. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13–20. 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Las Rivas J.; Fontanillo C. Protein-Protein Interactions Essentials: Key Concepts to Building and Analyzing Interactome Networks. PLoS Comput. Biol. 2010, 6, e1000807. 10.1371/journal.pcbi.1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiruma Y.; Hass M. A. S.; Kikui Y.; Liu W.-M.; Ölmez B.; Skinner S. P.; Blok A.; Kloosterman A.; Koteishi H.; Löhr F.; et al. The Structure of the Cytochrome P450cam-Putidaredoxin Complex Determined by Paramagnetic NMR Spectroscopy and Crystallography. J. Mol. Biol. 2013, 425, 4353–4365. 10.1016/j.jmb.2013.07.006. [DOI] [PubMed] [Google Scholar]
- Tripathi S.; Li H.; Poulos T. L. Structural Basis for Effector Control and Redox Partner Recognition in Cytochrome P450. Science 2013, 340, 1227–1230. 10.1126/science.1235797. [DOI] [PubMed] [Google Scholar]
- Andrałojć W.; Hiruma Y.; Liu W.-M.; Ravera E.; Nojiri M.; Parigi G.; Luchinat C.; Ubbink M. Identification of Productive and Futile Encounters in an Electron Transfer Protein Complex. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E1840–E1847. 10.1073/pnas.1616813114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelkader E. H.; Qianzhu H.; Tan Y. J.; Adams L. A.; Huber T.; Otting G. Genetic Encoding of N6-(((Trimethylsilyl)Methoxy)Carbonyl)- l -Lysine for NMR Studies of Protein-Protein and Protein-Ligand Interactions. J. Am. Chem. Soc. 2021, 143, 1133–1143. 10.1021/jacs.0c11971. [DOI] [PubMed] [Google Scholar]
- Saio T.; Ogura K.; Shimizu K.; Yokochi M.; Burke T. R.; Inagaki F. An NMR Strategy for Fragment-Based Ligand Screening Utilizing a Paramagnetic Lanthanide Probe. J. Biomol. NMR 2011, 51, 395–408. 10.1007/s10858-011-9566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John M.; Pintacuda G.; Park A. Y.; Dixon N. E.; Otting G. Structure Determination of Protein-Ligand Complexes by Transferred Paramagnetic Shifts. J. Am. Chem. Soc. 2006, 128, 12910–12916. 10.1021/ja063584z. [DOI] [PubMed] [Google Scholar]
- Chen W.-N.; Nitsche C.; Pilla K. B.; Graham B.; Huber T.; Klein C. D.; Otting G. Sensitive NMR Approach for Determining the Binding Mode of Tightly Binding Ligand Molecules to Protein Targets. J. Am. Chem. Soc. 2016, 138, 4539–4546. 10.1021/jacs.6b00416. [DOI] [PubMed] [Google Scholar]
- Anthis N. J.; Clore G. M. Visualizing Transient Dark States by NMR Spectroscopy. Q. Rev. Biophys. 2015, 48, 35–116. 10.1017/S0033583514000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzhnev D. M.; Kay L. E. Probing Invisible, Low-Populated States of Protein Molecules by Relaxation Dispersion NMR Spectroscopy: An Application to Protein Folding. Acc. Chem. Res. 2008, 41, 442–451. 10.1021/ar700189y. [DOI] [PubMed] [Google Scholar]
- Baldwin A. J.; Kay L. E. NMR Spectroscopy Brings Invisible Protein States into Focus. Nat. Chem. Biol. 2009, 5, 808. 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P.; Hansen D. F.; Stollar E.; Meirovitch E.; Kay L. E. Measurement of Bond Vector Orientations in Invisible Excited States of Proteins. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 18473–18477. 10.1073/pnas.0708296104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallurupalli P.; Hansen D. F.; Kay L. E. Structures of Invisible, Excited Protein States by Relaxation Dispersion NMR Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 11766–11771. 10.1073/pnas.0804221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichmüller C.; Skrynnikov N. R. Observation of μs Time-Scale Protein Dynamics in the Presence of Ln3+ Ions: Application to the N-Terminal Domain of Cardiac Troponin C. J. Biomol. NMR 2007, 37, 79–95. 10.1007/s10858-006-9105-y. [DOI] [PubMed] [Google Scholar]
- Sengupta I.; Nadaud P. S.; Jaroniec C. P. Protein Structure Determination with Paramagnetic Solid-State NMR Spectroscopy. Acc. Chem. Res. 2013, 46, 2117–2126. 10.1021/ar300360q. [DOI] [PubMed] [Google Scholar]
- Nadaud P. S.; Sengupta I.; Helmus J. J.; Jaroniec C. P. Evaluation of the Influence of Intermolecular Electron-Nucleus Couplings and Intrinsic Metal Binding Sites on the Measurement of 15N Longitudinal Paramagnetic Relaxation Enhancements in Proteins by Solid-State NMR. J. Biomol. NMR 2011, 51, 293–302. 10.1007/s10858-011-9536-y. [DOI] [PubMed] [Google Scholar]
- Nadaud P. S.; Helmus J. J.; Kall S. L.; Jaroniec C. P. Paramagnetic Ions Enable Tuning of Nuclear Relaxation Rates and Provide Long-Range Structural Restraints in Solid-State NMR of Proteins. J. Am. Chem. Soc. 2009, 131, 8108–8120. 10.1021/ja900224z. [DOI] [PubMed] [Google Scholar]
- Sengupta I.; Nadaud P. S.; Helmus J. J.; Schwieters C. D.; Jaroniec C. P. Protein Fold Determined by Paramagnetic Magic-Angle Spinning Solid-State NMR Spectroscopy. Nat. Chem. 2012, 4, 410–417. 10.1038/nchem.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez A.; Gaalswyk K.; Jaroniec C. P.; MacCallum J. L. High Accuracy Protein Structures from Minimal Sparse Paramagnetic Solid-State NMR Restraints. Angew. Chem., Int. Ed. 2019, 58, 6564–6568. 10.1002/anie.201811895. [DOI] [PubMed] [Google Scholar]
- Nadaud P. S.; Helmus J. J.; Sengupta I.; Jaroniec C. P. Rapid Acquisition of Multidimensional Solid-State NMR Spectra of Proteins Facilitated by Covalently Bound Paramagnetic Tags. J. Am. Chem. Soc. 2010, 132, 9561–9563. 10.1021/ja103545e. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D.; Nadaud P. S.; Shannon M. D.; Jaroniec C. P. Rapid Quantitative Measurements of Paramagnetic Relaxation Enhancements in Cu(II)-Tagged Proteins by Proton-Detected Solid-State NMR Spectroscopy. J. Phys. Chem. Lett. 2017, 8, 5871–5877. 10.1021/acs.jpclett.7b02709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadaud P. S.; Helmus J. J.; Höfer N.; Jaroniec C. P. Long-Range Structural Restraints in Spin-Labeled Proteins Probed by Solid-State Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc. 2007, 129, 7502–7503. 10.1021/ja072349t. [DOI] [PubMed] [Google Scholar]
- Bhaumik A.; Luchinat C.; Parigi G.; Ravera E.; Rinaldelli M. NMR Crystallography on Paramagnetic Systems: Solved and Open Issues. CrystEngComm 2013, 15, 8639–8656. 10.1039/c3ce41485j. [DOI] [Google Scholar]
- Wang S.; Munro R. A.; Kim S. Y.; Jung K. H.; Brown L. S.; Ladizhansky V. Paramagnetic Relaxation Enhancement Reveals Oligomerization Interface of a Membrane Protein. J. Am. Chem. Soc. 2012, 134, 16995–16998. 10.1021/ja308310z. [DOI] [PubMed] [Google Scholar]
- Milikisiyants S.; Wang S.; Munro R. A.; Donohue M.; Ward M. E.; Bolton D.; Brown L. S.; Smirnova T. I.; Ladizhansky V.; Smirnov A. I. Oligomeric Structure of Anabaena Sensory Rhodopsin in a Lipid Bilayer Environment by Combining Solid-State NMR and Long-Range DEER Constraints. J. Mol. Biol. 2017, 429, 1903–1920. 10.1016/j.jmb.2017.05.005. [DOI] [PubMed] [Google Scholar]
- Spooner P. J. R.; Veenhoff L. M.; Watts A.; Poolman B. Structural Information on a Membrane Transport Protein from Nuclear Magnetic Resonance Spectroscopy Using Sequence-Selective Nitroxide Labeling. Biochemistry 1999, 38, 9634–9639. 10.1021/bi990745l. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Marchanka A.; Carlomagno T. Structure of a Protein–RNA Complex by Solid-State NMR Spectroscopy. Angew. Chem., Int. Ed. 2020, 59, 6866–6873. 10.1002/anie.201915465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara J.; Tang C.; Marius Clore G. Practical Aspects of 1H Transverse Paramagnetic Relaxation Enhancement Measurements on Macromolecules. J. Magn. Reson. 2007, 184, 185–195. 10.1016/j.jmr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaponenko V.; Howarth J. W.; Columbus L.; Gasmi-Seabrook G.; Yuan J.; Hubbell W. L.; Rosevear P. R. Protein Global Fold Determination Using Site-Directed Spin and Isotope Labeling. Protein Sci. 2000, 9, 302–309. 10.1110/ps.9.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuita K.; Kataoka S.; Sugiki T.; Hattori Y.; Kobayashi N.; Ikegami T.; Shiozaki K.; Fujiwara T.; Kojima C. Utilization of Paramagnetic Relaxation Enhancements for High-Resolution NMR Structure Determination of a Soluble Loop-Rich Protein with Sparse NOE Distance Restraints. J. Biomol. NMR 2015, 61, 55–64. 10.1007/s10858-014-9882-7. [DOI] [PubMed] [Google Scholar]
- Schilder J.; Löhr F.; Schwalbe H.; Ubbink M. The Cytochrome c Peroxidase and Cytochrome c Encounter Complex: The Other Side of the Story. FEBS Lett. 2014, 588, 1873–1878. 10.1016/j.febslet.2014.03.055. [DOI] [PubMed] [Google Scholar]
- Olivieri C.; Subrahmanian M. V.; Xia Y.; Kim J.; Porcelli F.; Veglia G. Simultaneous Detection of Intra-and Inter-Molecular Paramagnetic Relaxation Enhancements in Protein Complexes. J. Biomol. NMR 2018, 70, 133–140. 10.1007/s10858-018-0165-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnke W.; Perez L. B.; Paris C. G.; Strauss A.; Fendrich G.; Nalin C. M. Second-Site NMR Screening with a Spin-Labeled First Ligand. J. Am. Chem. Soc. 2000, 122, 7394–7395. 10.1021/ja001241+. [DOI] [Google Scholar]
- Balogh E.; Wu D.; Zhou G.; Gochin M. NMR Second Site Screening for Structure Determination of Ligands Bound Inthe Hydrophobie Pocket of HIV-1 Gp41. J. Am. Chem. Soc. 2009, 131, 2821–2823. 10.1021/ja8094558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gochin M.; Zhou G.; Phillips A. H. Paramagnetic Relaxation Assisted Docking of a Small Indole Compound in the HIV-1 Gp41 Hydrophobic Pocket. ACS Chem. Biol. 2011, 6, 267–274. 10.1021/cb100368d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Gao J.; Li F.; Ma R.; Wei Q.; Wang A.; Wu J.; Ruan K. NMR Characterization of Weak Interactions between RhoGDI2 and Fragment Screening Hits. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 3061–3070. 10.1016/j.bbagen.2016.10.003. [DOI] [PubMed] [Google Scholar]
- Cordina N. M.; Liew C. K.; Gell D. A.; Fajer P. G.; Mackay J. P.; Brown L. J. Interdomain Orientation of Cardiac Troponin C Characterized by Paramagnetic Relaxation Enhancement NMR Reveals a Compact State. Protein Sci. 2012, 21, 1376–1387. 10.1002/pro.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthis N. J.; Clore G. M. The Length of the Calmodulin Linker Determines the Extent of Transient Interdomain Association and Target Affinity. J. Am. Chem. Soc. 2013, 135, 9648–9651. 10.1021/ja4051422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Ramelot T. A.; Ni S.; McCarrick R. M.; Kennedy M. A. Measurement of Rate Constants for Homodimer Subunit Exchange Using Double Electron-Electron Resonance and Paramagnetic Relaxation Enhancements. J. Biomol. NMR 2013, 55, 47–58. 10.1007/s10858-012-9685-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C.; Schwieters C. D.; Clore G. M. Open-to-Closed Transition in Apo Maltose-Binding Protein Observed by Paramagnetic NMR. Nature 2007, 449, 1078–1082. 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- Rovó P.; Grohe K.; Giller K.; Becker S.; Linser R. Proton Transverse Relaxation as a Sensitive Probe for Structure Determination in Solid Proteins. ChemPhysChem 2015, 16, 3791–3796. 10.1002/cphc.201500799. [DOI] [PubMed] [Google Scholar]
- Maltsev S.; Hudson S. M.; Sahu I. D.; Liu L.; Lorigan G. A. Solid-State NMR 31P Paramagnetic Relaxation Enhancement Membrane Protein Immersion Depth Measurements. J. Phys. Chem. B 2014, 118, 4370–4377. 10.1021/jp500267y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo R. H.; Kroncke B. M.; Solomon T. L.; Columbus L. Mapping Membrane Protein Backbone Dynamics: A Comparison of Site-Directed Spin Labeling with NMR 15N-Relaxation Measurements. Biophys. J. 2014, 107, 1697–1702. 10.1016/j.bpj.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly S.; Weiner B. E.; Meiler J. Membrane Protein Structure Determination Using Paramagnetic Tags. Structure 2011, 19, 441–443. 10.1016/j.str.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Cierpicki T.; Jimenez R. H. F.; Lukasik S. M.; Ellena J. F.; Cafiso D. S.; Kadokura H.; Beckwith J.; Bushweller J. H. NMR Solution Structure of the Integral Membrane Enzyme DsbB: Functional Insights into DsbB-Catalyzed Disulfide Bond Formation. Mol. Cell 2008, 31, 896–908. 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyay M.; Ledolter K.; Kitzler M.; Sandford G.; Cobb S. L.; Konrat R. 19F NMR Spectroscopy Tagging and Paramagnetic Relaxation Enhancement-Based Conformation Analysis of Intrinsically Disordered Protein Complexes. ChemBioChem 2020, 21, 696–701. 10.1002/cbic.201900453. [DOI] [PubMed] [Google Scholar]
- Dvoretsky A.; Gaponenko V.; Rosevear P. R. Derivation of Structural Restraints Using a Thiol-Reactive Chelator. FEBS Lett. 2002, 528, 189–192. 10.1016/S0014-5793(02)03297-0. [DOI] [PubMed] [Google Scholar]
- Kamen D. E.; Cahill S. M.; Girvin M. E. Multiple Alignment of Membrane Proteins for Measuring Residual Dipolar Couplings Using Lanthanide Ions Bound to a Small Metal Chelator. J. Am. Chem. Soc. 2007, 129, 1846–1847. 10.1021/ja067089e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Chen Y.; Tabuchi A.; Cosgrove D. J.; Hong M. The Target of β-Expansin EXPB1 in Maize Cell Walls from Binding and Solid-State NMR Studies. Plant Physiol. 2016, 172, 2107–2119. 10.1104/pp.16.01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. L.; Wang X.; Xiao Y. H.; Su X. C. Resonance Assignments of Lowly Populated and Unstable Enzyme Intermediate Complex under Real-Time Conditions. ChemBioChem 2019, 20, 2738–2742. 10.1002/cbic.201900240. [DOI] [PubMed] [Google Scholar]
- Gao J.; Liang E.; Ma R.; Li F.; Liu Y.; Liu J.; Jiang L.; Li C.; Dai H.; Wu J.; et al. Fluorine Pseudocontact Shifts Used for Characterizing the Protein-Ligand Interaction Mode in the Limit of NMR Intermediate Exchange. Angew. Chem., Int. Ed. 2017, 56, 12982–12986. 10.1002/anie.201707114. [DOI] [PubMed] [Google Scholar]
- Xu D.-F.; Li B.; Gao J.; Liu Z.-J.; Niu X.-G.; Nshogoza G.; Zhang J.-H.; Wu J.-H.; Su X.-C.; He W.; et al. Ligand Proton Pseudocontact Shifts Determined from Paramagnetic Relaxation Dispersion in the Limit of NMR Intermediate Exchange. J. Phys. Chem. Lett. 2018, 9, 3361–3367. 10.1021/acs.jpclett.8b01443. [DOI] [PubMed] [Google Scholar]
- Keizers P. H. J.; Mersinli B.; Reinle W.; Donauer J.; Hiruma Y.; Hannemann F.; Overhand M.; Bernhardt R.; Ubbink M. A Solution Model of the Complex Formed by Adrenodoxin and Adrenodoxin Reductase Determined by Paramagnetic NMR Spectroscopy. Biochemistry 2010, 49, 6846–6855. 10.1021/bi100598f. [DOI] [PubMed] [Google Scholar]
- Somireddy Venkata B.; Keizers P. H. J.; Drijfhout J. W.; Ubbink M. A Focal Adhesion Kinase-Derived Peptide Binds the Src SH3 Domain in Two Orientations, As Demonstrated Using Paramagnetic Nuclear Magnetic Resonance. Biochemistry 2016, 55, 29–37. 10.1021/acs.biochem.5b00998. [DOI] [PubMed] [Google Scholar]
- Dasgupta S.; Hu X.-Y.; Keizers P. H. J.; Liu W.-M.; Luchinat C.; Nagulapalli M.; Overhand M.; Parigi G.; Sgheri L.; Ubbink M. Narrowing the Conformational Space Sampled by Two-Domain Proteins with Paramagnetic Probes in Both Domains. J. Biomol. NMR 2011, 51, 253–263. 10.1007/s10858-011-9532-2. [DOI] [PubMed] [Google Scholar]
- Ben Bdira F.; Waudby C. A.; Volkov A. N.; Schröder S. P.; AB E.; Codée J. D. C.; Overkleeft H. S.; Aerts J. M. F. G.; van Ingen H.; Ubbink M. Dynamics of Ligand Binding to a Rigid Glycosidase. Angew. Chem., Int. Ed. 2020, 59, 20508–20514. 10.1002/anie.202003236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marneth K.; van den Elst H.; Cramer-Blok A.; Codee J.; Overkleeft H. S.; Aerts J. M. F. G.; Ubbink M.; Ben Bdira F. Tuning the Transglycosylation Reaction of a GH11 Xylanase by a Delicate Enhancement of Its Thumb Flexibility. ChemBioChem. 2021, 22, 1743–1749. 10.1002/cbic.202000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner S. P.; Liu W.-M.; Hiruma Y.; Timmer M.; Blok A.; Hass M. A. S.; Ubbink M. Delicate Conformational Balance of the Redox Enzyme Cytochrome P450cam. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 9022–9027. 10.1073/pnas.1502351112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oroz J.; Kim J. H.; Chang B. J.; Zweckstetter M. Mechanistic Basis for the Recognition of a Misfolded Protein by the Molecular Chaperone Hsp90. Nat. Struct. Mol. Biol. 2017, 24, 407–413. 10.1038/nsmb.3380. [DOI] [PubMed] [Google Scholar]
- Oroz J.; Chang B. J.; Wysoczanski P.; Lee C.-T.; Pérez-Lara Á.; Chakraborty P.; Hofele R. V.; Baker J. D.; Blair L. J.; Biernat J.; et al. Structure and Pro-Toxic Mechanism of the Human Hsp90/PPIase/Tau Complex. Nat. Commun. 2018, 9, 4532. 10.1038/s41467-018-06880-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welegedara A. P.; Yang Y.; Lee M. D.; Swarbrick J. D.; Huber T.; Graham B.; Goldfarb D.; Otting G. Double-Arm Lanthanide Tags Deliver Narrow Gd3+–Gd3+ Distance Distributions in Double Electron–Electron Resonance (DEER) Measurements. Chem. - Eur. J. 2017, 23, 11694–11702. 10.1002/chem.201702521. [DOI] [PubMed] [Google Scholar]
- Pan Y. Z.; Quade B.; Brewer K. D.; Szabo M.; Swarbrick J. D.; Graham B.; Rizo J. Sequence-Specific Assignment of Methyl Groups from the Neuronal SNARE Complex Using Lanthanide-Induced Pseudocontact Shifts. J. Biomol. NMR 2016, 66, 281–293. 10.1007/s10858-016-0078-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer K. D.; Bacaj T.; Cavalli A.; Camilloni C.; Swarbrick J. D.; Liu J.; Zhou A.; Zhou P.; Barlow N.; Xu J.; et al. Dynamic Binding Mode of a Synaptotagmin-1-SNARE Complex in Solution. Nat. Struct. Mol. Biol. 2015, 22, 555–564. 10.1038/nsmb.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahawaththa M. C.; Pearce B. J. G.; Szabo M.; Graham B.; Klein C. D.; Nitsche C.; Otting G. Solution Conformations of a Linked Construct of the Zika Virus NS2B-NS3 Protease. Antiviral Res. 2017, 142, 141–147. 10.1016/j.antiviral.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Fenwick R. B.; Esteban-Martín S.; Richter B.; Lee D.; Walter K. F. A.; Milovanovic D.; Becker S.; Lakomek N. A.; Griesinger C.; Salvatella X. Weak Long-Range Correlated Motions in a Surface Patch of Ubiquitin Involved in Molecular Recognition. J. Am. Chem. Soc. 2011, 133, 10336–10339. 10.1021/ja200461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaudzems K.; Jia X.; Yagi H.; Zhulenkovs D.; Graham B.; Otting G.; Liepinsh E. Structural Basis for 5′-End-Specific Recognition of Single-Stranded DNA by the R3H Domain from Human Sμbp-2. J. Mol. Biol. 2012, 424, 42–53. 10.1016/j.jmb.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Göbl C.; Resch M.; Strickland M.; Hartlmüller C.; Viertler M.; Tjandra N.; Madl T. Increasing the Chemical-Shift Dispersion of Unstructured Proteins with a Covalent Lanthanide Shift Reagent. Angew. Chem., Int. Ed. 2016, 55, 14847–14851. 10.1002/anie.201607261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiliveri S. C.; Louis J. M.; Ghirlando R.; Baber J. L.; Bax A. Tilted, Uninterrupted, Monomeric HIV-1 Gp41 Transmembrane Helix from Residual Dipolar Couplings. J. Am. Chem. Soc. 2018, 140, 34–37. 10.1021/jacs.7b10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C. A.; Shen Y.; Ying J.; Takagi Y.; Torchia D. A.; Sellers J. R.; Bax A. Remarkable Rigidity of the Single α-Helical Domain of Myosin-VI As Revealed by NMR Spectroscopy. J. Am. Chem. Soc. 2019, 141, 9004–9017. 10.1021/jacs.9b03116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Kirkpatrick J. P.; Launay H. M. M.; de Simone A.; Häussinger D.; Dobson C. M.; Vendruscolo M.; Cabrita L. D.; Waudby C. A.; Christodoulou J. Probing the Dynamic Stalk Region of the Ribosome Using Solution NMR. Sci. Rep. 2019, 9, 13528. 10.1038/s41598-019-49190-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann K.; Joss D.; Müntener T.; Nogueira E. S.; Schäfer M.; Knörr L.; Monnard F. W.; Häussinger D. Localization of Ligands within Human Carbonic Anhydrase II Using 19F Pseudocontact Shift Analysis. Chem. Sci. 2019, 10, 5064–5072. 10.1039/C8SC05683H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland M.; Catazaro J.; Rajasekaran R.; Strub M.-P.; O’Hern C.; Bermejo G. A.; Summers M. F.; Marchant J.; Tjandra N. Long-Range RNA Structural Information via a Paramagnetically Tagged Reporter Protein. J. Am. Chem. Soc. 2019, 141, 1430–1434. 10.1021/jacs.8b11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müntener T.; Böhm R.; Atz K.; Häussinger D.; Hiller S. NMR Pseudocontact Shifts in a Symmetric Protein Homotrimer. J. Biomol. NMR 2020, 74, 413–419. 10.1007/s10858-020-00329-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucuzza S.; Güntert P.; Plückthun A.; Zerbe O. An Automated Iterative Approach for Protein Structure Refinement Using Pseudocontact Shifts. J. Biomol. NMR 2021, 75, 319–334. 10.1007/s10858-021-00376-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelkader E. H.; Yao X.-J.; Feintuch A.; Adams L. A.; Aurelio L.; Graham B.; Goldfarb D.; Otting G. Pulse EPR-Enabled Interpretation of Scarce Pseudocontact Shifts Induced by Lanthanide Binding Tags. J. Biomol. NMR 2016, 64, 39–51. 10.1007/s10858-015-0003-z. [DOI] [PubMed] [Google Scholar]
- Kaminker I.; Tkach I.; Manukovsky N.; Huber T.; Yagi H.; Otting G.; Bennati M.; Goldfarb D. W-Band Orientation Selective DEER Measurements on a Gd3+/Nitroxide Mixed-Labeled Protein Dimer with a Dual Mode Cavity. J. Magn. Reson. 2013, 227, 66–71. 10.1016/j.jmr.2012.11.028. [DOI] [PubMed] [Google Scholar]
- Yardeni E. H.; Bahrenberg T.; Stein R. A.; Mishra S.; Zomot E.; Graham B.; Tuck K. L.; Huber T.; Bibi E.; Mchaourab H. S.; et al. Probing the Solution Structure of the E. Coli Multidrug Transporter MdfA Using DEER Distance Measurements with Nitroxide and Gd(III) Spin Labels. Sci. Rep. 2019, 9, 12528. 10.1038/s41598-019-48694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalon E.; Huber T.; Hagelueken G.; Graham B.; Frydman V.; Feintuch A.; Otting G.; Goldfarb D. Gadolinium(III) Spin Labels for High-Sensitivity Distance Measurements in Transmembrane Helices. Angew. Chem., Int. Ed. 2013, 52, 11831–11834. 10.1002/anie.201305574. [DOI] [PubMed] [Google Scholar]
- Prokopiou G.; Lee M. D.; Collauto A.; Abdelkader E. H.; Bahrenberg T.; Feintuch A.; Ramirez-Cohen M.; Clayton J.; Swarbrick J. D.; Graham B.; et al. Small Gd(III) Tags for Gd(III)–Gd(III) Distance Measurements in Proteins by EPR Spectroscopy. Inorg. Chem. 2018, 57, 5048–5059. 10.1021/acs.inorgchem.8b00133. [DOI] [PubMed] [Google Scholar]
- Cohen M. R.; Frydman V.; Milko P.; Iron M. A.; Abdelkader E. H.; Lee M. D.; Swarbrick J. D.; Raitsimring A.; Otting G.; Graham B.; et al. Overcoming Artificial Broadening in Gd3+-Gd3+ Distance Distributions Arising from Dipolar Pseudo-Secular Terms in DEER Experiments. Phys. Chem. Chem. Phys. 2016, 18, 12847–12859. 10.1039/C6CP00829A. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Yang F.; Gong Y.-J.; Bahrenberg T.; Feintuch A.; Su X.-C.; Goldfarb D. High Sensitivity In-Cell EPR Distance Measurements on Proteins Using an Optimized Gd(III) Spin Label. J. Phys. Chem. Lett. 2018, 9, 6119–6123. 10.1021/acs.jpclett.8b02663. [DOI] [PubMed] [Google Scholar]
- Dalaloyan A.; Martorana A.; Barak Y.; Gataulin D.; Reuveny E.; Howe A.; Elbaum M.; Albeck S.; Unger T.; Frydman V.; et al. Tracking Conformational Changes in Calmodulin in Vitro, in Cell Extract, and in Cells by Electron Paramagnetic Resonance Distance Measurements. ChemPhysChem 2019, 20, 1860–1868. 10.1002/cphc.201900341. [DOI] [PubMed] [Google Scholar]
- Gordon-Grossman M.; Kaminker I.; Gofman Y.; Shai Y.; Goldfarb D. W-Band Pulse EPR Distance Measurements in Peptides Using Gd3+-Dipicolinic Acid Derivatives as Spin Labels. Phys. Chem. Chem. Phys. 2011, 13, 10771–10780. 10.1039/c1cp00011j. [DOI] [PubMed] [Google Scholar]
- Petros A. M.; Neri P.; Fesik S. W. Identification of Solvent-Exposed Regions of an FK-506 Analog, Ascomycin, Bound to FKBP Using a Paramagnetic Probe. J. Biomol. NMR 1992, 2, 11–18. 10.1007/BF02192797. [DOI] [PubMed] [Google Scholar]
- Molinari H.; Esposito G.; Ragona L.; Pegna M.; Niccolai N.; Brunne R. M.; Lesk A. M.; Zetta L. Probing Protein Structure by Solvent Perturbation of NMR Spectra: The Surface Accessibility of Bovine Pancreatic Trypsin Inhibitor. Biophys. J. 1997, 73, 382–396. 10.1016/S0006-3495(97)78078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli M.; Bernini A.; Segoni C.; Molinari H.; Esposito G.; Lesk A. M.; Laschi F.; Temussi P.; Niccolai N. Tendamistat Surface Accessibility to the TEMPOL Paramagnetic Probe. J. Biomol. NMR 1999, 15, 125–133. 10.1023/A:1008319507565. [DOI] [PubMed] [Google Scholar]
- Bernini A.; Venditti V.; Spiga O.; Ciutti A.; Prischi F.; Consonni R.; Zetta L.; Arosio I.; Fusi P.; Guagliardi A.; et al. NMR Studies on the Surface Accessibility of the Archaeal Protein Sso7d by Using TEMPOL and Gd(III)(DTPA-BMA) as Paramagnetic Probes. Biophys. Chem. 2008, 137, 71–75. 10.1016/j.bpc.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Bernini A.; Spiga O.; Ciutti A.; Venditti V.; Prischi F.; Governatori M.; Bracci L.; Lelli B.; Pileri S.; Botta M.; et al. NMR Studies of BPTI Aggregation by Using Paramagnetic Relaxation Reagents. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 856–862. 10.1016/j.bbapap.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Venditti V.; Bernini A.; De Simone A.; Spiga O.; Prischi F.; Niccolai N. MD and NMR Studies of α-Bungarotoxin Surface Accessibility. Biochem. Biophys. Res. Commun. 2007, 356, 114–117. 10.1016/j.bbrc.2007.02.094. [DOI] [PubMed] [Google Scholar]
- Bernini A.; Spiga O.; Venditti V.; Prischi F.; Botta M.; Croce G.; Tong A. P.-L.; Wong W.-T.; Niccolai N. The Use of a Ditopic Gd(III) Paramagnetic Probe for Investigating α-Bungarotoxin Surface Accessibility. J. Inorg. Biochem. 2012, 112, 25–31. 10.1016/j.jinorgbio.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Almeida R. M.; Geraldes C. F. G. C.; Pauleta S. R.; Moura J. J. G. Gd(III) Chelates as NMR Probes of Protein-Protein Interactions. Case Study: Rubredoxin and Cytochrome c 3. Inorg. Chem. 2011, 50, 10600–10607. 10.1021/ic200858c. [DOI] [PubMed] [Google Scholar]
- Madl T.; Güttler T.; Görlich D.; Sattler M. Structural Analysis of Large Protein Complexes Using Solvent Paramagnetic Relaxation Enhancements. Angew. Chem., Int. Ed. 2011, 50, 3993–3997. 10.1002/anie.201007168. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Boelens R.; Bonvin A. M. J. J. HADDOCK: A Protein-Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Meyer N. H.; Tripsianes K.; Vincendeau M.; Madl T.; Kateb F.; Brack-Werner R.; Sattler M. Structural Basis for Homodimerization of the Src-Associated during Mitosis, 68-KDa Protein (Sam68) Qua1 Domain. J. Biol. Chem. 2010, 285, 28893–28901. 10.1074/jbc.M110.126185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidis L.; Schütz U.; Tripsianes K.; Madl T.; Radke J.; Rucktäschel R.; Wilmanns M.; Schliebs W.; Erdmann R.; Sattler M. Allosteric Modulation of Peroxisomal Membrane Protein Recognition by Farnesylation of the Peroxisomal Import Receptor PEX19. Nat. Commun. 2017, 8, 14635. 10.1038/ncomms14635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson H.; Jensen M. R.; Gesmar H.; Meier S.; Vinther J. M.; Keeler C.; Hodsdon M. E.; Led J. J. Specific and Nonspecific Interactions in Ultraweak Protein-Protein Associations Revealed by Solvent Paramagnetic Relaxation Enhancements. J. Am. Chem. Soc. 2014, 136, 10277–10286. 10.1021/ja503546j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F.-H.; Wang X.; Chen J.-L.; Wen X.; Sun H.; Su X.-C. Deciphering the Multisite Interactions of a Protein and Its Ligand at Atomic Resolution by Using Sensitive Paramagnetic Effects. Chem. - Eur. J. 2017, 23, 926–934. 10.1002/chem.201604393. [DOI] [PubMed] [Google Scholar]
- Swarbrick J. D.; Karas J. A.; Li J.; Velkov T. Structure of Micelle Bound Cationic Peptides by NMR Spectroscopy Using a Lanthanide Shift Reagent. Chem. Commun. 2020, 56, 2897–2900. 10.1039/C9CC09207B. [DOI] [PubMed] [Google Scholar]
- Kellner R.; Mangels C.; Schweimer K.; Prasch S. J.; Weiglmeier P. R.; Rösch P.; Schwarzinger S. SEMPRE: Spectral Editing Mediated by Paramagnetic Relaxation Enhancement. J. Am. Chem. Soc. 2009, 131, 18016–18017. 10.1021/ja905412z. [DOI] [PubMed] [Google Scholar]
- Welegedara A. P.; Adams L. A.; Huber T.; Graham B.; Otting G. Site-Specific Incorporation of Selenocysteine by Genetic Encoding as a Photocaged Unnatural Amino Acid. Bioconjugate Chem. 2018, 29, 2257–2264. 10.1021/acs.bioconjchem.8b00254. [DOI] [PubMed] [Google Scholar]
- Welegedara A. P.; Maleckis A.; Bandara R.; Mahawaththa M. C.; Dilhani Herath I.; Jiun Tan Y.; Giannoulis A.; Goldfarb D.; Otting G.; Huber T. Cell-Free Synthesis of Selenoproteins in High Yield and Purity for Selective Protein Tagging. ChemBioChem. 2021, 22, 1480–1486. 10.1002/cbic.202000785. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Feintuch A.; Collauto A.; Adams L. A.; Aurelio L.; Graham B.; Otting G.; Goldfarb D. Selective Distance Measurements Using Triple Spin Labeling with Gd3+, Mn2+, and a Nitroxide. J. Phys. Chem. Lett. 2017, 8, 5277–5282. 10.1021/acs.jpclett.7b01739. [DOI] [PubMed] [Google Scholar]
- Anthis N. J.; Doucleff M.; Clore G. M. Transient, Sparsely Populated Compact States of Apo and Calcium-Loaded Calmodulin Probed by Paramagnetic Relaxation Enhancement: Interplay of Conformational Selection and Induced Fit. J. Am. Chem. Soc. 2011, 133, 18966–18974. 10.1021/ja2082813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Calderone V.; Cerofolini L.; Fragai M.; Geraldes C. F. G. C.; Hermann P.; Luchinat C.; Parigi G.; Teixeira J. M. C. The Catalytic Domain of MMP-1 Studied through Tagged Lanthanides. FEBS Lett. 2012, 586, 557–567. 10.1016/j.febslet.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Niccolai N.; Spadaccini R.; Scarselli M.; Bernini A.; Crescenzi O.; Spiga O.; Ciutti A.; Di Maro D.; Bracci L.; Dalvit C.; et al. Probing the Surface of a Sweet Protein: NMR Study of MNEI with a Paramagnetic Probe. Protein Sci. 2001, 10, 1498–1507. 10.1110/ps.30101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccolai N.; Spiga O.; Bernini A.; Scarselli M.; Ciutti A.; Fiaschi I.; Chiellini S.; Molinari H.; Temussi P. A. NMR Studies of Protein Hydration and TEMPOL Accessibility. J. Mol. Biol. 2003, 332, 437–447. 10.1016/S0022-2836(03)00852-0. [DOI] [PubMed] [Google Scholar]
- Hartlmüller C.; Spreitzer E.; Göbl C.; Falsone F.; Madl T. NMR Characterization of Solvent Accessibility and Transient Structure in Intrinsically Disordered Proteins. J. Biomol. NMR 2019, 73, 305–317. 10.1007/s10858-019-00248-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M.; Baram A.; Luz Z. Electronic and Nuclear Relaxation in Solutions of Transition Metal Ions with Spin S= 3/2 and 5/2. Mol. Phys. 1971, 20, 67–80. 10.1080/00268977100100081. [DOI] [Google Scholar]
- Eaton D. R. The Nuclear Magnetic Resonance of Some Paramagnetic Transition Metal Acetylacetonates. J. Am. Chem. Soc. 1965, 87, 3097–3102. 10.1021/ja01092a015. [DOI] [Google Scholar]
- Alsaadi B. M.; Rossotti F. J. C.; Williams R. J. P. Hydration of Complexone Complexes of Lanthanide Cations. J. Chem. Soc., Dalton Trans. 1980, 11, 2151–2154. 10.1039/dt9800002151. [DOI] [Google Scholar]
- Fries P. H.; Belorizky E. Quantitative Interpretation of the Very Fast Electronic Relaxation of Most Ln3+ Ions in Dissolved Complexes. J. Chem. Phys. 2012, 136, 74510–74513. 10.1063/1.3685584. [DOI] [PubMed] [Google Scholar]
- Benmelouka M.; Borel A.; Moriggi L.; Helm L.; Merbach A. E. Design of Gd(III)-Based Magnetic Resonance Imaging Contrast Agents: Static and Transient Zero-Field Splitting Contributions to the Electronic Relaxation and Their Impact on Relaxivity. J. Phys. Chem. B 2007, 111, 832–840. 10.1021/jp0633289. [DOI] [PubMed] [Google Scholar]
- Wöhnert J.; Franz K. J.; Nitz M.; Imperiali B.; Schwalbe H. Protein Alignment by a Coexpressed Lanthanide-Binding Tag for the Measurement of Residual Dipolar Couplings. J. Am. Chem. Soc. 2003, 125, 13338–13339. 10.1021/ja036022d. [DOI] [PubMed] [Google Scholar]
- Martin L. J.; Hähnke M. J.; Nitz M.; Wöhnert J.; Silvaggi N. R.; Allen K. N.; Schwalbe H.; Imperiali B. Double-Lanthanide-Binding Tags: Design, Photophysical Properties, and NMR Applications. J. Am. Chem. Soc. 2007, 129, 7106–7113. 10.1021/ja070480v. [DOI] [PubMed] [Google Scholar]
- Guo F.; Friedman J. M. Charge Density-Dependent Modifications of Hydration Shell Waters by Hofmeister Ions. J. Am. Chem. Soc. 2009, 131, 11010–11018. 10.1021/ja902240j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barb A. W.; Subedi G. P. An Encodable Lanthanide Binding Tag with Reduced Size and Flexibility for Measuring Residual Dipolar Couplings and Pseudocontact Shifts in Large Proteins. J. Biomol. NMR 2016, 64, 75–85. 10.1007/s10858-015-0009-6. [DOI] [PMC free article] [PubMed] [Google Scholar]