

SUMMARY
The protein TRIM5α has multiple roles in antiretroviral defense, but the mechanisms underlying TRIM5α action are unclear. Here, we employ APEX2-based proteomics to identify TRIM5α-interacting partners. Our proteomics results connect TRIM5 to other proteins with actions in antiviral defense. Additionally, they link TRIM5 to mitophagy, an autophagy-based mode of mitochondrial quality control that is compromised in several human diseases. We find that TRIM5 is required for Parkin-dependent and -independent mitophagy pathways where TRIM5 recruits upstream autophagy regulators to damaged mitochondria. Expression of a TRIM5 mutant lacking ubiquitin ligase activity is unable to rescue mitophagy in TRIM5 knockout cells. Cells lacking TRIM5 show reduced mitochondrial function under basal conditions and are more susceptible to immune activation and death in response to mitochondrial damage than are wild-type cells. Taken together, our studies identify a homeostatic role for a protein previously recognized exclusively for its antiviral actions.
In brief
The protein TRIM5α is well known for its roles in antiretroviral defense. Saha et al. show that TRIM5α also has key homeostatic functions. They report that TRIM5α helps to maintain mitochondrial quality control by enabling the autophagy-dependent removal of damaged mitochondria (mitophagy).
Graphical abstract
INTRODUCTION
The tripartite motif containing protein 5 isoform α (TRIM5α; TRIM5) has evolved as a cell-intrinsic antiviral factor capable of restricting diverse RNA viruses (Chiramel et al., 2019; Ganser-Pornillos and Pornillos, 2019) and endogenous retroelements (Volkmann et al., 2020). The best-studied antiviral activity of TRIM5 is its capacity to potently block retroviral infection at a stage after the entry of the virus but prior to the integration of viral DNA into the host cell genome (Ganser-Pornillos and Pornillos, 2019). This mode of retroviral restriction correlates with the ability of TRIM5 to recognize the assembled retroviral capsid (Strem-lau et al., 2006). While TRIM5 from rhesus macaques (RhTRIM5) efficiently recognizes the HIV-1 capsid in a manner that can block infection, human TRIM5’s (HuTRIM5) ability to recognize HIV-1 is substantially reduced, and consequently, HuTRIM5 has historically been considered ineffectual against HIV-1. Nevertheless, epidemiological studies have demonstrated that different alleles of TRIM5 are associated with altered risk of HIV-1 infection and disease progression in people (Cloherty et al., 2021). Since HuTRIM5 does not carry out capsid-specific restriction of HIV-1, the human genetics studies imply that additional TRIM5-dependent mechanisms may be important for determining HIV-1 outcomes in humans.
Recent studies have identified three novel TRIM5 actions whereby HuTRIM5 can impact the outcome of HIV-1 infection in vitro. First, activation of human cells with type I interferon enables HuTRIM5 to restrict HIV-1 (Jimenez-Guardeno et al., 2019; OhAinle et al., 2018). The enhanced potency of HuTRIM5 is linked to increased interactions between TRIM5 and the immunoproteasome (Jimenez-Guardeno et al., 2019). Second, TRIM5 can activate antiviral signaling pathways leading to the production of cytokines and the generation of an antiviral state (Merindol et al., 2018; Pertel et al., 2011; Saha et al., 2020). The signaling function of TRIM5 at least partially depends on TRIM5’s interaction with the TGF-β activated kinase 1 (TAK1) protein complex (Pertel et al., 2011). Finally, TRIM5 interacts with multiple proteins having key roles in autophagy (Mandell et al., 2014; Ribeiro et al., 2016). Autophagy is a homeostatic mechanism of cellular waste management in which cytoplasmic contents (e.g., damaged mitochondria) are sequestered in a vesicle termed an autophagosome that ultimately fuses with lysosomes for elimination (Deretic, 2021). TRIM5’s connections to the autophagy pathway underlie the relative resistance to HIV-1 infection seen in human Langerhans cells (Ribeiro et al., 2016). Since autophagy has a plethora of functions in controlling cell survival, cellular metabolism, and innate immune responses (Deretic, 2021), it is likely that TRIM5 could contribute to these functions in a way that impacts the outcome of viral infection.
Clearly, an understanding of TRIM5’s protein-protein interactions has been crucial to understanding its numerous functions, yet questions about TRIM5 mechanisms remain elusive. The premise of this study is that identifying TRIM5-interacting proteins (the TRIM5 interactome) will provide fundamental knowledge required for addressing these questions. To this end, we used an APEX2-based proximity ligation system to study TRIM5 protein-protein interactions, because this approach allows for the direct labeling of TRIM5-proximal proteins in intact, living cells, leading to a high degree of coverage and labeling specificity (Rhee et al., 2013). Using this approach, we identified more than 300 proteins with diverse functions as putative TRIM5 interactors. We noted an unexpected enrichment of mitochondrial proteins in the TRIM5 interactome, which led us to identify a role for TRIM5 in the autophagy-based elimination of dysfunctional mitochondria (mitophagy). TRIM5 acted as an assembly scaffold, linking markers of damaged mitochondria with upstream autophagy regulators at the site where autophagosome assembly initiates. TRIM5-dependent mitophagy was crucial for preventing inflammation and cell death triggered by mitochondrial damage. In addition to identifying a key role for TRIM5 in mitophagy, our definition of the TRIM5 interactome will enable future discoveries of TRIM5 functions and mechanisms in and beyond TRIM5 actions in antiviral defense.
RESULTS
Proteomic definition of the TRIM5 interactome
We chose to use the APEX2 system to perform an unbiased identification of TRIM5-interacting partners (Trinkle-Mulcahy, 2019). APEX2 is an engineered form of soybean ascorbate peroxidase that catalyzes the ligation of biotin to electron-rich amino-acid residues within a radius of less than 20 nm from APEX2 or to APEX2-fusion proteins in response to hydrogen peroxide. Bio-tinylated proteins or protein complexes can then be isolated from cell lysates with streptavidin-coated beads and identified by liquid chromatography-tandem mass spectrometry (LC-MS/ MS) (Figure 1A) (Lam et al., 2015; Rhee et al., 2013). We designed our screen to allow us to detect TRIM5-proximal proteins under homeostatic conditions and to detect potential changes in the TRIM5 interactome stimulated by TRIM5 binding to assembled retroviral cores that are released into the cytoplasm following fusion of the viral envelope with host cell membranes shortly after infection (Figure 1B). For these experiments, we chose to use the well-established model of HIV-1 restriction by RhTRIM5 since rhesus is much better at recognizing and binding HIV-1 viral cores than is HuTRIM5 (Ganser-Pornillos and Pornillos, 2019; Selyutina et al., 2020).
Figure 1.

Proximity biotinylation screen for TRIM5-proximal proteins
(A) Schematic of APEX2-based labeling, purification, and identification of proteins in the near vicinity (within 20 nm) of TRIM5. HEK293T cells stably expressing rhesus (Rh) TRIM5-APEX2-V5 or APEX2-V5 alone were cultured in the presence of biotin-phenol for 30 min and then pulsed with H2O2 for 1 min to induce covalent biotinylation of nearby proteins. Biotinylated proteins were then purified on streptavidin-coated beads under stringent conditions and subjected to data-independent analysis (DIA) mass spectrometry. B, biotin; S, streptavidin.
(B) Diagram of the comparisons performed for analysis of mass-spectrometry data. Biotinylation reactions and cell lysis were performed 3 h after synchronized infection of cells with VSV-G pseudotyped HIV-1 (HIV) or mock infection. Three independent biological replicates were analyzed per treatment. Data for the comparisons indicated by the arrows can be found in Tables S1, S2, and S3.
(c) Intensity measurements of selected immune-related proteins identified by mass spectrometry. Data shown are from HIV-infected samples.
(D) Gene set enrichment analysis of immune-related reactome pathways over-represented in HIV-infected TRIM5-APEX2 datasets relative to those from APEX2 alone. Data: mean + SEM; p values determined by t test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We generated HEK293T cells stably expressing either HuTRIM5 or RhTRIM5 fused at its C terminus to V5-tagged APEX2 (TRIM5-APEX2). As a negative control for our proteomics experiments, we also generated HEK293T cells stably expressing APEX2-V5 (APEX2; Figure S1A). These cell lines displayed the expected susceptibility to infection with vesicular stomatitis virus G protein (VSV-G)-pseudotyped HIV-1, in which the RhTRIM5-APEX2 robustly restricted HIV-1 infection relative to both the HuTRIM5-APEX2 and APEX2-alone cell lines (Figures S1B and S1C). We also confirmed that over-expression of TRIM5-APEX2 fusions could activate nuclear factor kB (NF-kB) and AP1 transcription factors (Figures S1D and S1E), in accordance with published reports (Fletcher et al., 2018; Pertel et al., 2011). The findings from these control experiments demonstrate that the TRIM5-APEX2 fusion proteins retained important biological activities of untagged TRIM5.
The results of our data-independent acquisition mass spectroscopic analysis are summarized in Tables S1, S2, and S3 and can be accessed at https://simplifi.protifi.com/. Principal-component analysis demonstrated that the RhTRIM5-APEX2 samples (both HIV infected and mock) grouped together and differed from the APEX2-alone samples (Figure S1F). We identified 356 proteins that were more than 1.6-fold enriched in the TRIM5-APEX2 samples (p < 0.05; Tables S1, S2, and S3). Encouragingly, among these proteins, we identified two previously known TRIM5 interactors that are involved in TRIM5-dependent antiviral signaling: tumor growth factor beta (TGF-β)-activated kinase binding protein 1/TAB1 and ubiquitin-conjugating enzyme E2 N/UBC13 (Figure 1C). Consistent with TRIM5’s actions in innate immune signaling, we identified multiple proteins that have roles in regulating signaling pathways that result in NF-kB or IRF-3 activation (Figure 1C; Tables S1). In fact, gene set enrichment analysis of the proteins enriched in the TRIM5 in-teractome identified several immune-related pathways to be over-represented (Figure 1D). These results were consistent regardless of the presence of HIV-1 (Tables S1, S2, and S3), and we did not detect robust effects of HIV infection on the TRIM5 in-teractome (Figure S1G). Coimmunoprecipitation analysis of selected putative TRIM5 interactors identified in our screen show that a high percentage of those tested were in protein complexes with hemagglutinin (HA)-tagged HuTRIM5 in HeLa cells under homeostatic conditions (Figures S1H, 2E, 2F, and 2H). Of 13 “hits” tested, 9 showed positive interactions with TRIM5-HA in these experiments (Figure S1I). Together, these data help validate the findings from our APEX2-based identification of TRIM5-interacting proteins.
The TRIM5 interactome is enriched in mitochondrial proteins
Previous studies have shown that TRIM5 primarily localizes to puncta in the cytoplasm (Campbell et al., 2007) and can also be found in the nucleus (Diaz-Griffero et al., 2011). However, Gene Ontology analysis of our mass spectroscopy data revealed an unexpected enrichment of mitochondrial proteins within the TRIM5 interactome (Figure 2A). Analysis of the mitochondrial proteins enriched in the TRIM5 interactome showed that TRIM5 was proximal to proteins designated as localizing to the outer mitochondrial membrane, the intermembrane space, the inner mitochondrial membrane, and the mitochondrial matrix in previous APEX-based proteomics studies (Figure 2B) (Hung et al., 2014, 2017; Rhee et al., 2013). While our proteomics experiments were performed with RhTRIM5, for the remainder of this study, we focused on the roles of HuTRIM5 (RhTRIM5 and HuTRIM5 are 87% identical at the amino-acid level). We used imaging and biochemical approaches to test the association between TRIM5 and mitochondria. In confocal microscopy experiments, cytoplasmic bodies of HA-tagged TRIM5 were often found to be very close to and likely in physical contact with structures positive for the outer mitochondrial membrane marker TOM20 (Figure 2C). Remarkably, published correlative light electron-cryotomography studies of TRIM5 localization also found TRIM5 structures that appeared to contact mitochondria (Carter et al., 2020), in agreement with our data. Furthermore, TRIM5 was present in preparations of purified mitochondria (Figure 2D). Protease treatment of intact mitochondria eliminated mitochondria-associated TRIM5 and TOM20 but not the inner mitochondrial membrane protein TIM23, indicating that TRIM5 primarily associated with the cytoplasmic face of the outer mitochondrial membrane. Overall, these studies indicate a previously unappreciated association between TRIM5 and mitochondria. Given the key roles of mitochondria as sources of endogenous inflammation and as orchestrators of innate antiviral signaling, we pursued further studies into what role(s) mitochondria-associated TRIM5 may play in the cell.
Figure 2.

Mitochondrial localization of TRIM5 under basal conditions
(A) Cellular localization of TRIM5-proximal proteins based on Gene Ontology analysis.
(B) Analysis of the sub-organellar localization of mitochondrial TRIM5 interactors based on Gene Ontology analysis. OMM, outer mitochondrial membrane; IMS, inner mitochondrial space; IMM, inner mitochondrial membrane.
(C) Left: maximum image projection of a HeLa cell stained to detect TRIM5-HA (green) and the mitochondrial marker TOM20 (red). A rotated 3D reconstruction of the boxed region is shown on the right. Arrows indicate areas of TRIM5 and TOM20 intersection (yellow).
(D) Protease-protection analysis of the mitochondrial localization of TRIM5. Mitochondria were purified from HEK293T cells stably expressing TRIM5-APEX2-V5 and treated or not with proteinase K in the presence or absence of detergent (Triton-X) prior to immunoblotting with the indicated antibodies. n = 3 biological replicates.
(E) Mass-spectrometry- and coimmunoprecipitation-based analysis of interactions between TRIM5 and the mitophagy receptor NIPSNAP2. Graph: mass-spectrometry results comparing the intensity of NIPSNAP2 peptides identified in streptavidin-enriched RhTRIM5-APEX2 samples relative to APEX2-alone samples following HIV infection of HEK293T cells. Immunoblots: lysates from untreated HeLa cells stably expressing TRIM5-HA (human) were immunoprecipitated with either anti-HA or -NIPSNAP2 (or isotype control) prior to immunoblotting as indicated.
(F) Mass-spectrometry and coimmunoprecipitation (coIP) analysis of interactions between TRIM5 and NIPSNAP1.
(G) Proximity ligation assay (PLA) demonstrating close spatial relationships between NIPSNAP2 and TRIM5 in HeLa cells stably expressing TRIM5-HA.
(H) Mass-spectrometry and coIP analysis of interactions between TRIM5 and the mitophagy receptor prohibitin 2.
(I) coIP analysis of the TRIM5 protein-protein interaction under basal conditions and following mitochondrial depolarization with CCCP. TRIM5-HA expressing HeLa cells were treated with CCCP or vehicle control (DMSO) for 2 h in the presence of MG132 and then subjected to lysis, immunoprecipitation with anti-HA, and immunoblotting with the indicated antibodies. N = 2 biological replicates.
(J) Immunoblot analysis of TRIM5-HA abundance in cells treated or not with CCCP for 2 h in the presence of the indicated inhibitors or DMSO vehicle control. n = 2 biological replicates. Data: mean + SEM; p values determined by t test; ***p < 0.001.
TRIM5 interacts with mitochondrial proteins associated with mitophagy and relocalizes to mitochondria-proximal ER domains in response to mitochondrial damage
Coimmunoprecipitation experiments confirmed that TRIM5 was in protein complexes with mitochondrial proteins NIPSNAP1, NIPSNAP2, prohibitin 2, and LRPPRC in HeLa cell lysates (Figures 2E, 2F, 2H, S1H, and S1I). We also confirmed TRIM5-NIPSNAP2 interactions in HeLa cells by in situ proximity ligation assay (PLA), which indicates the presence of two proteins within 16 nm of each other by the formation of fluorescent puncta (Figure 2G). Interestingly, NIPSNAP1/2, prohibitin 2 (Figures 2E–2H), and SAMM50 (Tables S1) are reported to have roles in an auto-phagy-dependent mode of mitochondrial quality control referred to as “mitophagy” (Abudu et al., 2021; Princely Abudu et al., 2019; Wei et al., 2017). Mitophagy involves the selective removal of damaged or unwanted mitochondria, and aberrant mitophagy has been connected to multiple aging-related inflammatory conditions and cancers (Chen et al., 2020). In healthy mitochondria, the NIPSNAPs and prohibitin 2 are primarily localized on the inside of the mitochondrion and away from the cytoplasm. However, mitochondrial damage increases their exposure to the cytoplasm, where they are proposed to facilitate mitophagy by interacting with autophagosome membrane-associated proteins (LC3s and GABARAPs, the mammalian orthologues of yeast Atg8 [mAtg8s]) or the autophagy adaptor p62 (Abudu et al., 2021; Princely Abudu et al., 2019; Wei et al., 2017). Given TRIM5’s previously reported interactions with mAtg8s, p62, and additional components of the autophagy machinery (Mandell et al., 2014; Ribeiro et al., 2016), we investigated whether TRIM5 might be involved in mitophagy. We found that mitochondrial depolarization using carbonyl cyanide 3-chlorophenylhydrazone (CCCP) increased the abundance of prohibitin 2 and NIPSNAP2 in TRIM5-protein complexes (Figure 2I). This experiment was performed under conditions of proteasome inhibition with the compound MG132 since we observed that mitochondrial depolarization enhanced the proteasome-dependent turnover of TRIM5 (Figure 2J). These data show that TRIM5 interactions with mitophagy “eat-me” tags increase in response to mitochondrial damage.
NIPSNAPs and prohibitin 2 are both reported to contribute to a pathway referred to as “Parkin-dependent mitophagy” (Princely Abudu et al., 2019; Wei et al., 2017). In this pathway, the ubiquitin ligase Parkin relocalizes from the cytoplasm to the surface of damaged mitochondria. Once there, Parkin catalyzes the ubiquitylation of many outer mitochondrial membrane proteins and the subsequent recruitment of ubiquitin-binding autophagy adaptors including NDP52 and optineurin (Harper et al., 2018). A classic model of Parkin-dependent mitophagy involves inducing mitochondrial depolarization with CCCP in HeLa cells expressing exogenous Parkin, as HeLa cells lack Parkin expression. We used this model to determine if CCCP treatment could impact the mitochondrial localization of TRIM5 as a first test of whether TRIM5 contributes to mitophagy. As described previously, in untreated cells, mCherry-tagged Parkin has a diffuse cytoplasmic localization, and TRIM5 primarily localizes to small cytoplasmic puncta (Figure 3A). However, 2 h after CCCP treatment, Parkin has almost completely re-localized to numerous ovoid structures in the cell (presumably mitochondria; Figure 3B). TRIM5’s localization also changes dramatically in response to CCCP treatment, forming elongated TRIM5 filaments. These filaments, which are often curved and sometimes have a branching morphology, appeared to intersect with and sometimes partially surround the Parkin-positive structures (Figures 3B, S2A, and S2B). Parkin expression enhanced the transition of TRIM5 structures from being spherical to becoming filamentous, but this process was still observed in Parkin-null cells (Figure S2B). Examination of these filaments in triple-labeling confocal microscopy experiments demonstrated that they closely associate with mitochondria (Figures 3C and S2C). Interestingly, while many mitochondria showed labeling with both Parkin and TRIM5, the mitochondrial TRIM5 does not always localize to the same site on the mitochondria as does Parkin (Figure 3C), and TRIM5-positive, Parkin-negative mitochondria are readily observed (Figure S2C). These data indicate that TRIM5 and Parkin are independently recruited to damaged mitochondria and suggest that they may play distinct roles in mitophagy.
Figure 3.

TRIM5 and autophagy proteins localize to mitochondria-proximal ER in response to mitochondrial depolarization
HeLa cells stably expressing TRIM5-HA (human) were transiently transfected with mCherry-Parkin prior to 2 h treatment with CCCP (20 μM) or vehicle control (DMSO). Cells were fixed and stained as indicated, and deconvolved confocal micrographs were acquired.
(A and B) Localization of TRIM5 and mCherry-Parkin under control conditions (DMSO) and following mitochondrial uncoupling (CCCP). Boxes indicate the location of the zoomed-in region shown to the right.
(C)onfocal analysis of the TRIM5-HA, mCherry-Parkin, and TOM20 following mitochondrial depolarization.
(D)Representative images of cells treated as above and stained to detect TRIM5-HA (green), mCherry-Parkin (red), and ER marker Calnexin (blue). The box showsthe location of the zoomed-in images at the right. Arrows indicate foci of colocalized green and blue signals on the outside of Parkin-decorated mitochondria.
Based on their morphology, we tested whether CCCP treatment re-localized TRIM5 to previously described mitochondria-associated actin filaments (Hsieh and Yang, 2019; Kruppa et al., 2018) but observed that TRIM5 filaments did not colocalize with the F-actin marker phalloidin (Figure S2D). Instead, we found substantial colocalization between TRIM5 filaments and the endoplasmic reticulum (ER) marker calnexin in CCCP-treated cells (Figures 3D and S2E). Accordingly, analysis of our mass spectrometry results identified calnexin among several ER-localized or ER-mitochondria contact site-localized proteins (Hung et al., 2017) enriched in the TRIM5 interactome (Table S4). In situ PLA confirmed that TRIM5 and calnexin were in close proximity in untreated cells (Figure S2F), and CCCP treatment substantially increased the PLA signal (Figure S2G). Taken together, our findings indicate that TRIM5 is recruited to ER that is closely associated with mitochondria following mitochondrial damage.
TRIM5 contributes to Parkin-dependent mitophagy
Previous studies have indicated that mitophagosome biogenesis originates from ER-mitochondria contact sites in both yeast and mammalian cells (Bockler and Westermann, 2014; Gelmetti et al., 2017; Yang et al., 2020; Zachari et al., 2019). One of the earliest steps in autophagosome/mitophagosome formation is the generation of phosphatidyl inositol 3-phosphate (PI3P). DFCP1 is a PI3P-binding protein that marks specialized ER domains known as “omegasomes” that serve as autophagy initiation sites. Following CCCP treatment, TRIM5 and GFP-DFCP1 colocalized with each other on the outside of Parkin-decorated mitochondria (Figure 4A). Mitochondria-associated TRIM5 also colocalizes with endogenous FIP200 and ATG13, both key early players in autophagy regulation, in CCCP-treated cells (Figures 4B and 4C). Typically, TRIM5 and autophagy factors colocalized at discrete points on the mitochondria rather than completely surrounding them (Figures S3A and S3B). Importantly, colocalized ATG13 and TRIM5 were observed to be associated with mitochondria in CCCP-treated HeLa cells even in the absence of Parkin expression (Figure S3C), suggesting that ATG13 can be recruited to damaged mitochondria independently of Parkin expression in cells that over-express TRIM5.
Figure 4.

TRIM5 is required for mitophagy in response to mitochondrial depolarization
(A) HeLa cells stably expressing TRIM5-HA (blue) were transiently transfected with both mCherry-Parkin (red) and eGFP-DFCP1 (green) prior to 2 h CCCP treatment and confocal microscopy. Arrows indicate regions of TRIM5/DFCP1 colocalization that are associated with Parkin-labeled mitochondria.
(B and C) Representative confocal images of cells treated as above and stained to detect TRIM5-HA (green), mCherry-Parkin (red), and the autophagy factors FIP200 (B) or ATG13 (C) (blue). Arrows indicate foci of colocalized green and blue signals on the outside of Parkin-decorated mitochondria.
(D) Immunoblot-based analysis of mitochondrial-protein abundance in lysates from WT and TRIM5 knockout Huh7 cells under basal conditions and following 24 h treatment with CCCP. The abundance of these proteins from three independent experiments are plotted relative to actin.
(E and F) High-content-imaging-based analysis of the abundance of mitochondrial nucleoids in WT and TRIM5 KO Huh7 cells treated with CCCP (10 μM) or oli-gomycin and antimycin (O/A; 10 and 5 μM, respectively) for 24 h prior to fixation and staining with an antibody that preferentially recognized mitochondrial DNA (red) and nuclear staining (blue). Left: representative images showing automatically defined nuclear and cell boundaries (blue and green rings, respectively) and mitochondrial nucleoids (aqua mask). The number of mitochondria per cell was determined for 2,000 cells per experiment. n = 6 biological replicates. Scale bar, 10 mm. Data: mean + SEM; p values determined by ANOVA; *p < 0.05, ***p < 0.001, ****p < 0.0001; †, not significant.
Since TRIM5 localized to apparent mitophagosome-formation sites, we tested its role in mitophagy using the Parkin-expressing HeLa cell model. As expected, treatment of HeLa cells stably over-expressing YFP-Parkin with CCCP or the mitochondrial-respiration inhibitors oligomycin/antimycin (O/A) induced mitophagy as measured by increased degradation of the mitochondrial proteins COXII and VDAC1. TRIM5 knockout partially reversed this effect (Figures S3D and S3E). TRIM5 knockout also reduced the formation of the lipidated form of LC3 (LC3-II) in response to CCCP (Figure S3F), indicating that TRIM5 contributes to mitophagy. High content imaging experiments showed that TRIM5 knockout did not affect the kinetics of CCCP-dependent re-localization of Parkin to mitochondria (Figures S3G and S3H) or CCCP-induced formation of ubiquitin puncta (Figures S3I and S3J), suggesting that TRIM5 contributes to mitophagy either by acting downstream of Parkin-mediated ubiquitination or by participating in a complementary pathway.
If the latter of these two models is correct, then the relative importance of TRIM5-dependent mitophagy might be higher in an experimental system in which the expression of TRIM5 more closely matched the expression of Parkin. Unlike HeLa cells, the hepatocellular adenocarcinoma cell line Huh7 expresses endogenous Parkin, and so we tested the role of TRIM5 in mitophagy using these cells. CCCP treatment of wild-type (WT) Huh7 cells reduced the levels of mitochondrial proteins COXII, VDAC1, and TOM20 by ~50%. This effect was almost completely lost in TRIM5 knockout Huh7 cells at multiple time points (Figures 4D and S3K). Expression of HuTRIM5-APEX2 restored the ability of CCCP to induce COXII degradation in TRIM5 knockout Huh7 cells (Figure S3L). Additionally, high content imaging revealed that CCCP or O/A treatment strongly reduced the number of mitochondrial nucleoids per cell in WT Huh7 cells but not in TRIM5 knockout Huh7 cells (Figures 4E and 4F). These results show that TRIM5 is an important contributor to Parkin-dependent mitophagy pathways in two different cell lines.
TRIM5 bridges damaged mitochondria with upstream autophagy regulators
We next sought to define the mechanism underlying TRIM5 action in CCCP-induced mitophagy. Confocal microscopy indicated that cells lacking TRIM5 have a defect in autophagosomal targeting of mitochondria. In these experiments, we measured the total intensity of a fixable mitochondrial stain within three-dimensional structures demarcated by the autophagosome marker LC3B. We found that LC3B structures from WT Huh7 cells contained significantly more mitochondria-derived material than did TRIM5 knockout Huh7 cells (Figure 5A). TRIM5, along with several other TRIMs, interacts with multiple autophagy proteins including ULK1 (Mandell et al., 2020). ULK1 along with its binding partners ATG101, ATG13, and FIP200 are the most upstream components of the core autophagy machinery. Our data showed that TRIM5 colocalized with ATG13 and FIP200 on mitochondrial surfaces after CCCP treatment (Figures 4B, 4C, and S3A–S3C), suggesting that TRIM5 may act in mitophagy by recruiting these proteins to damaged mitochondria. We tested how TRIM5 knockout would impact the abundance of autophagy proteins that copurify with mitochondria in CCCPtreated or untreated cells (Figures 5B and 5C). In WT Huh7 cells, CCCP treatment increased the abundance of ATG13, FIP200, ULK1, and LC3-II in mitochondrial fractions. Remarkably, we observed little to no enrichment of any of these proteins in mitochondrial fractions harvested from CCCP-treated TRIM5 knockout Huh7 cells. In contrast, TRIM5 knockout did not prevent the mitochondrial accumulation of either PINK1 or Parkin in CCCP-treated Huh7 cells (Figure S4A). CCCP treatment did not impact the abundance of endogenous TRIM5 associated with mitochondria (Figure S4B), indicating that while TRIM5 recruits autophagy machinery to mitochondria in response to mitochondria damage, TRIM5’s interactions with mitochondria are not impacted. Taken together, our data show that TRIM5 is required for the recruitment of upstream autophagy regulators to damaged mitochondria in cells expressing physiologically relevant levels of Parkin.
Figure 5.

TRIM5 recruits upstream autophagy regulatory machinery to damaged mitochondria
(A) Left: confocal microscopic analysis of WT and TRIM5 KO Huh7 cells 6 h after CCCP treatment stained to detect LC3B and mitochondria. 3D reconstructions of deconvolved images were generated, and the intensity of mitochondrial signal within LC3-positive structures was quantified relative to the volume of the LC3 structure and plotted as shown on the right. n > 700 LC3 positive structures. Data: median and quartiles shown; ****, p < 0.0001 by Mann-Whitney.
(B and C) Recruitment of autophagy machinery to mitochondria following CCCP treatment. Lysates from mitochondria purified from WT or TRIM5 Huh7 cells treated with CCCP (20 mM) or vehicle control (DMSO) for 6 h were subjected to immunoblotting with the indicated antibodies. Plots show the abundance of the indicated proteins in the mitochondrial fractions relative to TOM20. n = 3 biological replicates.
(D–G) Immunoblot analysis of the effect of TRIM5 knockout on ivermectin-stimulated degradation of COXII, VDAC1, and TOM20 in HeLa cells. Cells were treated or not with ivermectin (15 μM) for 24 h prior to lysis and immunoblotting. The abundance of the proteins indicated in the graphs was determined relative to actin. n = 3 biological replicates. Data: mean + SEM by ANOVA, *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, †, not significant.
TRIM5 acts independently of Parkin in mitophagy
While Parkin is required for the best-studied mitophagy pathways, Parkin-independent mitophagic pathways exist, and there are an increasing number of examples in which Parkin-independent mitophagy is an essential contributor to normal physiological function (Villa et al., 2018). We tested whether TRIM5 might be important for mitophagic responses to the iron chelator deferiprone (DFP) (Allen et al., 2013) or the antihelminthic drug ivermectin (IVM) (Zachari et al., 2019), as both of these compounds induce Parkin-independent mitophagy. While DFP triggered substantial degradation of mitochondrial proteins in HeLa cells, this effect was unchanged in TRIM5 knockout cells (Figure S4C). In contrast, we found that TRIM5 was required for IVM-induced degradation of COXII and VDAC1 in HeLa cells (Figures 5D–5G). We saw similar results when we performed this experiment in WT and TRIM5 knockout Huh7 cells (Figure S4D). Expression of TRIM5APEX2, but not APEX2 alone, restored the ability of IVM to promote COX-II degradation in TRIM5 knockout HeLa cells (Figure S4E).
Our data indicate that TRIM5 plays a similar role in IVM-induced mitophagy to what we described above for mitophagy induced by the mitochondrial uncoupling agent CCCP. As was seen with CCCP, IVM treatment enhanced the proteasomal degradation of TRIM5 (Figure S5A). Confocal imaging experiments revealed that IVM treatment transitioned TRIM5 structures from being round to being more elongated (Figure S5B). These structures closely associated with mitochondria in IVM-treated HeLa cells and colocalized with ATG13 (Figure S5C). Mitochondrial-fractionation experiments revealed that TRIM5 was required for the recruitment of autophagy factors to mitochondria after IVM treatment. Whereas the proteins optineurin, NDP52, ATG13, FIP200, ULK1, ATG9, and LC3B were strongly enriched in mitochondrial fractions by 4 h after IVM treatment, we observed no recruitment of these proteins to mitochondria in TRIM5 knockout cells (Figures 6A and 6B). Interestingly, IVM treatment does not recruit TRIM5 to the mitochondria from the cytosol (Figure 6C). Instead, IVM treatment increased the interactions between HA-tagged TRIM5 and GFP-ATG13 in HeLa cells (Figures 6D, 6E, and S5D) and increased the colocalization between TRIM5-HA and endogenous ATG13 as measured by high content imaging (Figure S5E). Furthermore, IVM treatment increased the interactions between TRIM5-HA and endogenous FIP200, prohibitin 2, and NIPSNAP2 (Figure 6F). These findings raised the possibility of TRIM5 acting as an assembly platform for mitophagy eat-me signals and the upstream autophagy regulatory machinery. In agreement, we found that expression of TRIM5-APEX2 increased interactions between GFP-ATG13 and endogenous prohibitin 2 or NIPSNAP2 in HEK293T cells relative to what is seen in cells expressing APEX2 alone. This effect was most pronounced under conditions of mitochondrial damage (Figure 6G).
Figure 6.

Mechanisms of TRIM5 in Parkin-independent and -dependent mitophagy
(A and B) The impact of TRIM5 knockout on the ivermectin-induced recruitment of the indicated autophagy-related proteins to mitochondria. Intact mitochondria were isolated from WT or TRIM5 knockout HeLa cells that were treated with ivermectin for 2 or 4 h or with vehicle control (DMSO) for 4 h. n = 3 biological replicates.
(C) The abundance of TRIM5-HA in mitochondrial and cytosolic fractions following 4 h treatment with ivermectin (IVM) or DMSO control. All samples were treated with proteasome inhibitor MG132. n = 3 biological replicates.
(D and E) coIP analysis of interactions between TRIM5-HA and GFP-ATG13 in HeLa cells treated or not with IVM. The relative abundance of immunoprecipitated (IPed) TRIM5 relative to IPed GFP-ATG13 is plotted in (E). n = 3 biological replicates.
(F) coIP analysis of interactions between TRIM5-HA and the indicated endogenous proteins from lysates of HeLa cells treated or not with IVM in the presence ofMG132. n = 2 biological replicates.
(G) coIP analysis of interactions between the indicated endogenous proteins with GFP-ATG13 in cells treated or not with mitochondrial damaging agents andexpressing TRIM5-APEX2 or APEX2 alone. n = 2 biological replicates.
(H–K) The ability of WT or E11R mutant TRIM5 to rescue CCCP- (H and I) or IVM-triggered (J and K) mitophagy in TRIM5-deficient Huh7 cells. The relative loss of COXII in CCCP- or IVM-treated cells relative to untreated cells is expressed as a percentage and referred to as “COXII degradation.” Plots (I and K) show the results of three independent experiments. Data: mean + SEM analyzed using two-way ANOVA or Student’s t test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, †, not significant.
TRIM5 ubiquitin ligase activity is required for its mitophagy functions
We tested whether TRIM5’s enzymatic activity was important for its actions in mitophagy stimulated by CCCP or IVM by adding back GFP-tagged WT or ubiquitination-disabled TRIM5 into TRIM5 knockout cells. For these experiments, we selected an E11R mutant since this mutation prevents TRIM5’s interactions with E2 conjugating enzymes and dramatically reduces TRIM5 auto-ubiquitination but retains other important biological functions of TRIM5 including its ability to carry out retroviral restriction (Fletcher et al., 2018). In TRIM5 knockout cells expressing GFP alone, CCCP or IVM treatment had a minimal effect (~10% reduction) on the abundance of the mitochondrial protein COXII (Figures 6H–6K and S5F). When WT GFP-TRIM5 is expressed in these cells, both CCCP and IVM treatments reduce the expression of COXII by more than 50%. In contrast, expression of E11R mutant GFP-TRIM5 does not restore the ability of either mitophagy-inducing agent to promote COXII degradation (Figures 6H–6K and S5F). Taken together, these results suggest that TRIM5’s enzymatic activity as a ubiquitin ligase is essential to its functions in mitophagy.
TRIM5-mediated mitophagy protects against mitochondrial damage and excessive inflammation
We next carried out metabolic-profiling experiments to determine if TRIM5 was required for the maintenance of mitochondrial health. We found that WT Huh7 cells had a higher oxygen consumption rate (OCR) and had a higher capacity to carry out mitochondrial ATP synthesis than did TRIM5 knockout cells (Figures 7A–7D). In these experiments, we used IVM as a positive control for inducing mitochondrial damage (Zachari et al., 2019). IVM treatment of WT Huh7 cells phenocopied what was seen in TRIM5 knockout Huh7 cells with or without IVM treatment. This result indicates that TRIM5 deficiency results in mitochondrial impairment even in the absence of exogenous mitochondrial damage. Together, these results hint that TRIM5 may also contribute to basal mitophagy.
Figure 7.

TRIM5 protects cells from excessive inflammation and cell death in response to mitochondrial damage
(A–D) Seahorse analysis of oxygen consumption rate (OCR) in WT and TRIM5 knockout Huh7 cells in response to IVM (1.5 μM) or DMSO alone. n = 3 biological replicates.
(E)Immunoblot analysis of immune- and inflammation-related proteins in WT or TRIM5 knockout Huh7 cells following a 24 h treatment with 15 μM IVM or DMSO control. Plots show the abundance of the indicated protein relative to a loading control (actin). n = 3 biological replicates.
(F)The impact of IVM treatment (20 μM; 24 h) on the viability of WT and TRIM5 knockout Huh7 cells. The intensity of propidium-iodide labeling, indicative of plasma-membrane disruption, was determined in single cells by flow cytometry.
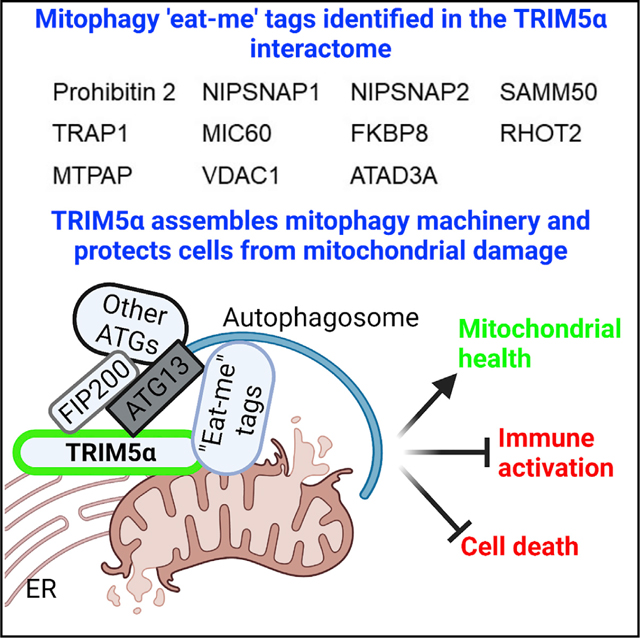
(G)Schematic illustration of TRIM5 action in mitophagy. Left: a portion of the cellular pool of TRIM5 associates with mitochondria under basal conditions.Following mitochondrial damage, TRIM5 links mitophagy eat-me tags (e.g., NIPSNAP2) with FIP200, ATG13, and other autophagy proteins (ATGs) at ER-mito-chondria contact sites. Data: mean + SEM; p values determined by ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001, †, not significant.
If not removed from cells by mitophagy, damaged mitochondria can be important endogenous sources of immune-activating damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) and can lead to cell death. We tested whether TRIM5’s actions in mitophagy are connected to its actions in regulating innate immunity and in antiviral defense. We found that 24 h treatment of WT Huh7 cells with IVM modestly increased the protein levels of active phosphorylated IRF3, the interferon-stimulated genes OAS2 and IFIT1, and the inactive form of the pro-inflammatory cytokine interleukin (IL)-1β (Figure 7E). Importantly, the impact of IVM treatment on the expression of these immune-related proteins was substantially more pronounced in TRIM5 knockout Huh7 cells, suggesting that the defect in mitophagy in these cells is connected with enhanced inflammation. We also saw that TRIM5 knockout Huh7 cells were more susceptible to death following exposure to IVM than were WT Huh7 cells (Figures 7F and S6A). Lastly, we tested whether TRIM5’s activities as a retroviral restriction factor were connected to its roles in mitophagy. While HuTRIM5 is generally incapable of recognizing the capsid of HIV-1, a single amino-acid substitution in the HIV capsid (P90A) renders the virus sensitive to HuTRIM5based restriction (Kim et al., 2019; Selyutina et al., 2020). Furthermore, the P90A capsid (CA) virus can stimulate TRIM5-based antiviral signaling (Saha et al., 2020). However, infection of YFP-Parkin expressing HeLa cells with VSV-G pseudotyped HIV-1 P90A CA did not decrease the abundance of mitochondrial proteins, nor did it increase the abundance of autophagy marker LC3-II (Figure S6B). We also tested whether pre-treatment of cells with mitochondrial damaging agents CCCP or IVM could impact the ability of RhTRIM5 to potently restrict WT HIV-1, but we did not observe any effect (Figure S6C). Taken together, our results show that TRIM5-dependent mitophagy is required to prevent excessive inflammation and cell death yet appears to be disconnected from TRIM5’s actions in retroviral restriction.
DISCUSSION
In this study, we identified proteins proximal to or interacting with the HIV restriction factor TRIM5α and used these data to identify a role for TRIM5 in maintaining mitochondrial health through the autophagic removal of damaged mitochondria, a process termed mitophagy. In the absence of TRIM5, overall mitochondrial health is reduced, and cellular sensitivity to some mitochondrial damaging agents such as IVM increase, resulting in increased inflammation and cell death. In contrast to previous studies that have focused on TRIM5’s specialized activities in protecting cells against viral infection, our study establishes that TRIM5 also has important homeostatic roles beyond antiviral defense that may be of relevance to the ever-increasing number of pathological conditions associated with aberrant mitophagy.
The first indication that TRIM5 could be linked to mitochondria came from our finding that mitochondrial proteins were unexpectedly over-represented in the TRIM5 interactome (Tables S1, S2, S3, and S4). Among these mitochondrial TRIM5 interactors were multiple proteins that were previously identified as mitophagy eat-me tags. Under conditions of mitochondrial stress, these proteins are presented on the cytoplasmic face of the outer mitochondrial membrane where they can flag damaged mitochondria for autophagic elimination through interactions with cytosolic autophagy proteins such as LC3/GABARAP, p62, and ALFY (Abudu et al., 2021; Princely Abudu et al., 2019; Wei et al., 2017). Our data show that up-stream autophagy factors such as ATG13 also interact with mitophagy eat-me tags. TRIM5 mediates these interactions in response to mitochondrial damage (Figure 7G).
TRIM5 is a ubiquitin ligase, and its enzymatic activity is required for some, but not all, of its previously reported actions in antiviral defense (Fletcher et al., 2018). Our data indicate that a TRIM5 mutant that lacks ubiquitin ligase activity is unable to rescue mitophagy in TRIM5 knockout cells. An important remaining question is to determine what proteins are ubiquitylated by TRIM5 in response to mitochondrial damage and how this post-translational modification contributes to TRIM5-dependent mitophagy.
While multiple distinct mechanisms for mitophagy exist in the literature (Zachari and Ktistakis, 2020), historically these mechanisms have been segregated based on their dependency on the ubiquitin ligase Parkin. Our data showed an involvement of TRIM5 in both Parkin-dependent mitophagy experimentally stimulated by treatment of cells with the compounds CCCP or O/A and in Parkin-independent mitophagy stimulated by IVM. However, it is important to note that our data show that TRIM5 is not required for all mitophagic pathways, as TRIM5 knockout did not impair mitophagy driven by iron depletion caused by deferiprone (DFP) treatment. Since TRIM5 is a part of the large TRIM family of proteins consisting of 70 members in the human genome, it is possible that other TRIM(s) contribute to mitophagic pathways induced by DFP or other stimuli.
Our identification of the TRIM5 interactome is likely to enable the elucidation of hitherto enigmatic TRIM5 mechanisms and may lead to the discovery of additional new roles for TRIM5. Although our data presented here show that the loss of TRIM5 can enhance the expression of interferon-stimulated genes in response to mitochondrial damage (presumably by disabling the cell’s ability to carry out the mitophagic removal of multiple mitochondria-associated DAMPs [Rai et al., 2021]), previous studies have shown that activation of TRIM5 by retroviral core engagement can trigger the expression of type I interferons (Fletcher et al., 2018; Merindol et al., 2018; Saha et al., 2020). While TRIM5 signaling is known to activate NF-kB and AP1 transcription factors (Pertel et al., 2011), the expression of type I interferon also requires activation of IRF-family transcription factors (e.g., IRF-3). Our proteomics data showed that several proteins known to regulate pathways upstream of IRF-3 were enriched in the TRIM5 interactome (Tables S1, S2, and S3), raising the possibility that these proteins are impacted by activated TRIM5 in a way that would trigger interferon expression. The role of these proteins in TRIM5-dependent immune activation and the establishment of an antiviral state will be a focus of future study.
In conclusion, our study has defined the interactome of TRIM5, an important antiviral restriction factor, and has uncovered a previously unanticipated homeostatic function of TRIM5 in preventing excessive inflammation and cell death in response to mitochondrial damage.
Limitations of the study
While our data demonstrated a mitophagic role for TRIM5 in multiple human cell lines, we have not tested the role of TRIM5 in carrying out mitophagy in primary cells. Additionally, we do not have in vivo data addressing the role of TRIM5-dependent mitophagy in disease. Since the mouse genome encodes seven TRIM5 homologues (Trim12a-c and Trim30a-d), developing the appropriate small-animal model to address this will be a challenge.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information or requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael Mandell (mmandell@salud.unm.edu).
Materials availability
Plasmids and cell lines generated in this study are available upon request from the lead contact.
Data and code availability
Mass Spectrometry Data is available at the MASSIVE data repository (massive.ucsd.edu) using number MSV000088006 and ProteomeExchange (http://www.proteomexchange.org) using ID number PXD028031. All datasets are publicly available as of the date of publication. This paper does not report original code. Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
HEK293T, HeLa, and Huh7 cells were obtained from the American Type Culture Collection (ATCC) and grown in Dulbecco’s modified Eagle’s medium (Life Technologies,11965126) supplemented with 10% fetal bovine serum (FBS, Life technologies, 26140–079), 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in a 5% CO2 atmosphere. HeLa cells stably expressing HA-tagged RhTRIM5α and HA-tagged HuTRIM5α were obtained from Joseph Sodroski (Harvard) and were maintained in the above media supplemented with 1 μg/mL puromycin. YFP-PARKIN stably expressing HeLa were generated by Dr. Richard J. Youle (National Institutes of Health, USA). TRIM5 knockout (KO) Huh7, HeLa and YFP-PARKIN HeLa cells were generated by transduction with lentiCRISPRv2-based lentiviruses followed by 2–4 weeks of culturing in medium containing 200 μg/mL hygromycin. Knockout lines were confirmed by immunoblot. APEX2-V5, RhTRIM5-APEX2-V5 and HuTRIM5-APEX2-V5 stable overexpression in HEK293T, and in HeLa or Huh7 TRIM5 KO lines were achieved by viral transduction followed by 14–21 days of culturing in medium containing the selective antibiotic (1 μg/mL puromycin) before expression of the target gene was confirmed by western blotting.
Generation of single-cycle lentivirus or retrovirus for stable cell line construction or infection assays
Viral particles for the generation of stable overexpressed cell lines were produced by co-transfection of pLEX_307 (a gift from David Root, Addgene plasmid # 41392) containing the target gene, psPAX2 and pMD2.G at the ratio of 1:1:1 in HEK293T cells using ProFection Mammalian Transfection System (Promega, E1200), medium was changed 16h post transfection and virus containing supernatant was harvested 48h later, clarified by centrifuging for 5 min at 1,200 rpm, 0.45 mm-filtered (Millipore, SE1M003M00), diluted with full medium at 1:1 ratio and used to transduce target cells for 48 h.
Viral particles for the generation of knockout cell lines were produced by transfecting HEK293T cells with a lentiviral vector, lenti-CRISPRv2 carrying both Cas9 enzyme and a guide RNA targeting specific gene together with the packaging plasmids psPAX2 and pMD2.G at the ratio of 10 μg, 10 μg and 10 μg/10 cm dish. We also used this approach to generate the HIV-1 CA P90A pseudoviruses. Lentiviral particles were harvested from supernatants as mentioned above and HEK293T, Huh7, and YFP-PARKIN HeLa cells were infected in the presence of polybrene for 48h in 6 cm dishes.
In this study, virus infection experiments were performed using VSV-G pseudotyped single-cycle HIV-1 produced by transfection of HEK293T cells. 24h before transfection, 2.2×106 HEK293T cells were seeded in 10 cm dishes. Two-part single cycle VSVG-pseudotyped HIV-1 (NL43 strain) was collected from the supernatants of HEK293T cells transfected with plasmids encoding VSV-G and HIV-1 lacking the Env gene (Env-, VPR-, Nef+, IRES-GFP) at 1:2 ratios. 16h post transfection, the culture media was changed, viral supernatant was harvested at 72h, clarified by centrifuging for 5 min at 1,200 rpm, passed through a 0.45 μm filter and stored at −80°C.
METHOD DETAILS
Cloning and transfection
pDest40-APEX2-V5 and pLEX_307-APEX2-V5; pDest40-RhTRIM5-APEX2-V5 and pLEX_307- RhTRIM5-APEX2-V5; pDest40-HuTRIM5-APEX2-V5 and pLEX_307-HuTRIM5-APEX2-V5 were generated using Gateway recombination cloning. First, they were PCR amplified from available cDNA clones and recombined into pDONR221 using the BP reaction (Life Technologies, 11789–020) prior to being recombined into expression plasmids by LR cloning (Life Technologies, 11791–020). Plasmid constructs were verified by DNA sequencing. The AP1 luciferase reporter plasmid was a gift from Alexander Dent (Addgene plasmid #40342; 3XAP1pGL3), the NF-κB luciferase reporter was purchased from Promega (#E8491) and the Renilla luciferase plasmid (pRL-SV40, Addgene plasmid #27163) was a gift from Ron Prywes. GFP-TRIM5 was mutated using site directed mutagenesis kit using following primers to generate GFP-TRIM5E11R: Fw, GGGGCAGGTCACCTCCCGCTTTACATTAACCAGGATTC; Rw, GAATCCTGGTTAATGTAAAGCGGGAGGTGACCTGCCCC.Plasmid transfections were performed using Lipofectamine 2000 (ThermoFisher, 11668019) or Calcium Phosphate (Promega, E1200). Samples were prepared for analysis the day after DNA transfection.
Western blotting, immunoprecipitation, and immunofluorescent labeling
SDS PAGE was carried out using pre-cast poly-acrylamide gels (Biorad) and immunoblotting was performed using standard procedures. Immunoprecipitation experiments were performed as previously described (Mandell et al., 2014). Immunoblot data was acquired using a Chemidoc MP instrument (Biorad) and quantitatively analyzed using Biorad Image Lab software. Immunofluorescent labeling of samples for high content imaging and confocal experiments, samples were fixed in 4% paraformaldehyde (Sigma) for 30 min prior to permeabilization in buffer containing 0.1% Saponin (Sigma) and 3% BSA. Following 1 h incubation in primary antibodies, AlexaFluor-conjugated secondary antibodies (Life Technologies) were used. Coverslips were mounted in ProLong Diamond anti-fade reagent (Life Technologies). Duolink in situ proximity ligation assays were performed according to the manufacturer’s instructions (Sigma).
Luciferase assays
20000 HEK293T cells were plated in 96 well plates prior to transfection with the Renilla luciferase internal control reporter plasmid pRL-TK (thymidine kinase promoter dependent Renilla luciferase), plasmids encoding firefly luciferase responsive to NF-κB or AP1 APEX2 or TRIM5-APEX2 expression plasmids as previously described (Saha et al., 2020). 40–48h after transfection, the plate was assayed using the Dual-Glo Luciferase Assay System (Promega, E2920) and read using a Microplate Luminometer (BioTek, SYNERGY HTX Multi-Mode reader). Firefly luciferase readings were normalized to Renilla luciferase readings in each well, and the data are represented as fold-change compared to control cDNA.
Infection assay using single-cycle viruses
8000 HeLa cells were seeded per well in a 96 well plate, 24h prior to virus challenge. Media containing VSV-G pseudotyped lentiviral vectors expressing GFP tagged HIV-1 (NL43, lacking Env) was added to infect cells in a total volume of 100 μL in the presence or absence of mitophagy inducers. After infection, cells were first incubated at 4°C for 1h to allow the virus to bind. Free virus was then removed by washing and cells were incubated in complete medium. 48h post infection, cells were fixed, stained with Hoechst 33342 and the fraction of transduced cells showing fluorescent protein positivity was determined by high content imaging and analysis as previously described (Saha et al., 2020).
High content imaging
High content imaging and analysis were performed using a Cellomics CellInsight CX7 scanner equipped for live-cell imaging and driven by iDEV software (Thermo Fisher Scientific). Primary objects were cells (identified based on nuclear staining with DAPI or Hoechst 33342), and regions of interest (ROI) or targets were algorithm-defined by shape/segmentation, maximum/minimum average intensity, total area and total intensity to automatically identify puncta or other profiles within valid primary objects. Transduced cells were automatically identified based on having above background fluorescent protein signal in the nucleus. All data acquisition and analysis were computer driven and independent of human operators.
APEX2-based biotin labeling and purification
Biotinylation reactions were performed as described previously (Jia et al., 2018). Briefly, HEK293T cells stably expressing APEX2-V5 or RhTRIM5-APEX2-V5 were cultured in the presence of 0.5 mM biotin-phenol (AdipoGen) for the last 30 min of the experiment. Cells were then pulsed with H2O2 (1 mM) for 1 min prior to washing the cells with a buffer that quenches the biotinylation reactions (10 mM sodium ascorbate, 10 mM sodium azide, and 5 mM Trolox in PBS) and lysis in a buffer consisting of 6M urea, 0.3 M NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM sodium ascorbate, 10 mM sodium azide, 5 mM Trolox, 1% glycerol and 25 mM TRIS-HCl (pH 7.5). Samples were then sonicated and insoluble material cleared by centrifugation. 3 mg of protein lysate per sample was then incubated with streptavidin-coated magnetic beads (Pierce) overnight. Beads were then washed with a series of buffers including: 1) IP lysis buffer (Pierce); 2) 1M KCl; 3) 50 mM Na2CO3; and 2M urea in 20 mM TRIS-HCl (pH 8.0).
Sample preparation for LC-MS/MS
Protein samples on magnetic beads were washed four times with 200ul of 50 mM Triethyl ammonium bicarbonate (TEAB) with a twenty-minute shake time at 4C in between each wash. Roughly 2.5 ug of trypsin was added to the bead and TEAB mixture and the samples were digested over night at 800 rpm shake speed. After overnight digestion the supernatant was removed and the beads were washed once with enough 50 mM ammonium bicarbonate to cover. After 20 min at a gentle shake the wash is removed and combined with the initial supernatant. The peptide extracts are reduced in volume by vacuum centrifugation and a small portion of the extract is used for fluorometric peptide quantification (Thermo scientific Pierce). One microgram of sample based on the fluorometric peptide assay was loaded for each LC-MS analysis.
Liquid chromatography-tandem mass spectrometry
Peptides were desalted and trapped on a Thermo PepMap trap and separated on an Easy-spray 100 μm × 25 cm C18 column using a Dionex Ultimate 3,000 nUPLC at 200 nL/min. Solvent A = 0.1% formic acid, Solvent B = 100% Acetonitrile 0.1% formic acid. Gradient conditions = 2% B to 50% B over 60 min, followed by a 50%–99% B in 6 min and then held for 3 min than 99% B to 2%B in 2 min and total run time of 90 min using Thermo Scientific Fusion Lumos mass spectrometer. The samples were run in DIA mode; mass spectra were acquired using a collision energy of 35, resolution of 30 K, maximum inject time of 54 ms and a AGC target of 50K, using staggered isolation windows of 12 Da in the m/z range 400–1,000 m/z.
Data analysis
DIA data was analyzed using Spectronaut 15 (Biognosys Schlieren, Switzerland) using the directDIA workflow with the default settings. Briefly, protein sequences were downloaded from Uniprot (Human Proteome UP000005640), Human immunodeficiency virus type 1 (HIV-1) group M subtype B (isolate BRU/LAI; UP000007692), Tripartite motif-containing protein 5 from Macaca mulatta (Q0PF16), and common laboratory contaminant sequences from https://thegpm.org/crap/. Trypsin/P specific was set for the enzyme allowing two missed cleavages. Fixed Modifications were set for Carbamidomethyl, and variable modification were set to Acetyl (Protein N-term) and Oxidation. For DIA search identification, PSM and Protein Group FDR was set at 1%. A minimum of 2 peptides per protein group were required for quantification. A report was exported from Spectronaut using the reporting feature and imported into SimpliFi https://simplifi.protifi.com/ for QC and statistical analysis (Protifi, Farmingdale NY). Proteins ‘hits’ were defined as being enriched by 1.7-fold in the TRIM5-APEX2 samples (p < 0.05). Proteins known to be endogenously biotinylated were excluded from consideration. Gene set enrichment analysis of hits was performed using Reactome Pathway Browser (=https://reactome.org/PathwayBrowser/#TOOL=AT). Mitochondrial proteins were identified using the MitoMiner4.0 database (https://mitominer.mrc-mbu.cam.ac.uk/release-4.0/begin.do), and gene ontology component analysis was performed using the Panther database (http://www.pantherdb.org/).
Data availability
Mass Spectrometry Data is available at the MASSIVE data repository (massive.ucsd.edu) using number MSV000088006 and ProteomeExchange (http://www.proteomexchange.org) using ID number PXD028031.
Mitochondrial isolation experiments
Subcellular fractionation was performed with a QProteome mitochondria isolation kit (Qiagen) according to the instruction manual. In brief, 107 HeLa and Huh7 cells were re-suspended in 1 mL of lysis buffer, incubated for 10 min at 4°C and centrifuged at 1,000x g for 10 min. The supernatant was transferred into a separate tube as cytosolic fraction, while the pellet was re-suspended in 1.5 mL of ice-cold disruption buffer, rapidly passed through 26 g needle 10–15 times to disrupt cells and centrifuged at 1,000x g for 10 min, 4°C. The supernatant was then re-centrifuged at 6,000x g for 10 min, 4°C. The pellet obtained after centrifugation comprised the mitochondrial fraction. For proteinase K (PK) digestion, mitochondria were re-suspended in Mitochondrial buffer (MB) (210 mM mannitol, 70 mM sucrose, 10 mM HEPES, 1 mM EDTA, pH 7.5) with 50 mg/mL of PK and incubated 30 min at RT. For Triton X-100 digestion, mitochondria were re-suspended in digestion buffer (10 mM sucrose, 0.1 mM EGTA/ Tris and 10 mM Tris/HCl, pH 7.4) with Triton X-100. Both reactions were stopped by addition of 5 mM phenylmethylsulfonyl fluoride (Sigma). For the analysis of integral membrane proteins, the mitochondrial fraction was re-suspended in MB buffer or MB buffer containing freshly prepared 0.1 M Na2CO3 (pH 11.5) and incubated on ice for 30 min. The insoluble membrane fraction was centrifuged at 16000x g for 15 min.
Confocal microscopy
Sub-airy unit (0.6AU) pinhole confocal microscopy with a Zeiss LSM800 or a Leica TCS-SP8 microscope was performed followed by computational image restoration with Huygens Essential (Scientific Volume Imaging, Hilversum, Netherlands) utilizing a constrained maximum likelihood estimation algorithm. All 3D images were acquired with a 63X/1.4NA plan apochromat oil immersion objective and sampled at ideal Nyquist sampling rates in x, y, and z planes. Voxel lateral and axial dimensions were determined by utilizing an online Nyquist calculator (https://svi.nl/NyquistCalculator) allowing for sub-diffraction limited resolution following image restoration. All images were rendered on a high performance CUDA-GPA enabled workstation and 3D renders were generated for volume-object analysis with Huygens Object Analyzer software. Sphericity values for TRIM5 structures was obtained using the “RoughSphericity” feature while the intensity of mitochondrial stain signal within LC3-positive volumes was determined using the “CoSum” feature in the Huygens software.
Metabolic profiling
We used the Seahorse XF Cell Mito Stress Test Kit (Agilent) according to the manufacturer’s protocol in a Seahorse XFe96 Analyzer to measure oxygen consumption and extracellular acidification rates in Huh7 cells treated or not with 1.5 μM IVM. After initial optimization experiments, we used kit components at the following concentrations: oligomycin, 1.5 μm; FCCP, 1 mm; rotenone, 0.5 μm; antimycin A, 0.5 μm. Analysis was performed using Seahorse Analytics software (Agilent).
Cell death analysis
WT and TRIM5 knockout Huh7 cells were treated or not with 20 μM IVM for 24 h prior to measurement of cell viability by measuring propidium iodide (PI) labeling or using the alamarBlue cell viability assay (Invitrogen). For PI experiments, cells were harvested and treated with 1 μg/mL PI and 10 μg/mL Hoechst 33342 prior to flow cytometry using an Attune NxT flow cytometer (Thermo). PI staining intensity was measured in single cells as determined by forward and side scatter and by Hoechst positivity. 20k total cells were analyzed per treatment. For the alamarBlue assays, reagent was added to cells 20 h prior to determining absorbance at 570 nm and 600 nm using a SYNERGY HTX Multi-Mode Reader (BioTek) according to the manufacturer’s protocol.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are expressed as means ± SEM (n > 3). Where not otherwise indicated, data were analyzed with unpaired two-tailed t-tests or ANOVA with Bonferroni post hoc analysis. Data sets showing non-normal distributions were analyzed using Mann-Whitney or Kruskal Wallis tests. Statistical significance is defined as *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| V5-Tag (D3H8Q) Rabbit mAb | Cell Signaling Technology | #13202; RRID: AB_2687461 |
| ABIN1/TNFAIP3 | Cell Signaling Technology | #4664; RRID: AB_10547137 |
| TBK1/NAK (D1B4) Rabbit mAb | Cell Signaling Technology | #3504; RRID: AB_2255663 |
| UbcH5C (D60E2) Rabbit mAb | Cell Signaling Technology | #4330; RRID: AB_10544697 |
| UBE2N (D2A1) Rabbit mAb | Cell Signaling Technology | #6999; RRID: AB_10828936 |
| Atg13 (E1Y9V) Rabbit mAb | Cell Signaling Technology | #13468; RRID: AB_2797419 |
| FIP200 (D10D11) Rabbit mAb | Cell Signaling Technology | #12436; RRID: AB_2797913 |
| LC3A/B Antibody | Cell Signaling Technology | #4108; RRID: AB_2137703 |
| TRIM5α (D6Z8L) Rabbit mAb | Cell Signaling Technology | #14326; RRID: AB_2798451 |
| Atg9A (D4O9D) Rabbit mAb | Cell Signaling Technology | #13509; RRID: AB_2798241 |
| IRF-3 (D83B9) Rabbit mAb | Cell Signaling Technology | #4302; RRID: AB_1904036 |
| Phospho-IRF-3 (Ser396) (4D4G) Rabbit mAb | Cell Signaling Technology | #4947; RRID: AB_823547 |
| IL-1β (D3U3E) Rabbit mAb | Cell Signaling Technology | #12703; RRID: AB_2737350 |
| OAS2 Antibody | Cell Signaling Technology | #54155 |
| IFIT1 (D2X9Z) Rabbit mAb | Cell Signaling Technology | #14769; RRID: AB_2783869 |
| Anti-LRP130 Antibody (F-7) | Santa Cruz Biotechnology | sc-166178; RRID: AB_2137453 |
| Anti-ETEA Antibody (F-7) | Santa Cruz Biotechnology | sc-374098; RRID: AB_10918565 |
| Anti-Rad23B Antibody (F-8) | Santa Cruz Biotechnology | sc-390019 |
| Anti-Calnexin Antibody (AF18) | Santa Cruz Biotechnology | sc-23954; RRID: AB_626783 |
| Anti-Calumenin Antibody (F-8) | Santa Cruz Biotechnology | sc-271357; RRID: AB_10610089 |
| Anti-NIPSNAP1 Antibody (H-9) | Santa Cruz Biotechnology | sc-515197 |
| Anti-Tim23 Antibody (H-8) | Santa Cruz Biotechnology | sc-514463 |
| Anti-Tom20 Antibody (F-10) | Santa Cruz Biotechnology | sc-17764; RRID: AB_628381 |
| Anti-Optineurin Antibody (C-2) | Santa Cruz Biotechnology | sc-166576; RRID: AB_2156554 |
| Anti-CALCOCO2 Antibody (F-6) | Santa Cruz Biotechnology | sc-376540; RRID: AB_11150487 |
| Anti-PINK1 Antibody (C-3) | Santa Cruz Biotechnology | sc-518052; RRID: AB_2861352 |
| Anti-Parkin Antibody (PRK8) | Santa Cruz Biotechnology | sc-32282; RRID: AB_628104 |
| Anti-Actin Antibody (2Q1055) | Santa Cruz Biotechnology | sc-58673; RRID: AB_2223345 |
| Anti-HA tag antibody - ChIP Grade | Abcam | ab9110; RRID: AB_2223345 |
| Anti-HA tag antibody | Abcam | ab134028 |
| Anti-mCherry antibody | Abcam | ab183628; RRID: AB_2223345 |
| Anti-GFP antibody | Abcam | ab290; RRID: AB_303395 |
| Anti-MTCO2 antibody [12C4F12] | Abcam | ab110258; RRID: AB_10887758 |
| Anti-VDAC1/Porin antibody | Abcam | ab15895; RRID: AB_2214787 |
| NCOA4 Antibody (1F11) | Novus Biologicals | H00008031-M05; RRID: AB_894201 |
| GBAS Antibody (OTI1B8) | Novus Biologicals | NBP2-45730 |
| Prohibitin 2 Antibody | Novus Biologicals | NBP2-13754 |
| N4BP1 Antibody | Novus Biologicals | NBP2-37688 |
| Anti-LC3 pAb (Polyclonal Antibody) | MBL International | PM036; RRID: AB_2274121 |
| Anti-Multi Ubiquitin mAb (Monoclonal Antibody) | MBL International | D058-3; RRID: AB_592937 |
| RB1CC1 Polyclonal antibody | Proteintech | 17250-1-AP; RRID: AB_10666428 |
| anti-DNA mouse monoclonal, AC-30-10, lyophilized, purified | Progen | 61014; RRID: AB_2750935 |
| Rabbit Anti-LC3B | Sigma Aldrich | L7543; RRID: AB_796155 |
| IRDye® 680LT Goat anti-Mouse IgG Secondary Antibody | LI-COR Biosciences | 925-68020; RRID: AB_2687826 |
| IRDye 800CW Goat anti-Mouse IgG, 0.1 mg | LI-COR Biosciences | 925-32210; RRID: AB_2687825 |
| Goat Anti-Mouse IgG (H+L)-HRP Conjugate | BIO-RAD | 1721011; RRID: AB_2617113 |
| Goat Anti-Rabbit IgG (H+L)-HRP Conjugate | BIO-RAD | 1721019; RRID: AB_11125143 |
| Mouse TrueBlot® ULTRA: Anti-Mouse Ig HRP, Rat Monoclonal eB144 | Rockland | 18-8817-30; RRID: AB_2610849 |
| Clean-Blot™ IP Detection Reagent (HRP) | ThermoFisher | 21230; RRID: AB_2864363 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | ThermoFisher | A32723; RRID: AB_2633275 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher | A-11034; RRID: AB_2576217 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | ThermoFisher | A-11004; RRID: AB_2534072 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | ThermoFisher | A-11011; RRID: AB_143157 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 | ThermoFisher | A32728; RRID: AB_2633277 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 | ThermoFisher | A32733; RRID: AB_2633282 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| NEB 5-alpha Competent E.coli (High Efficiency) | New England Biolabs | C2987 |
| XL10-Gold Ultracompetent cells | Agilent | 210518 |
|
| ||
| Chemicals, peptides, and recombinant protein | ||
|
| ||
| Biotinyl tyramide (biotin-phenol) | AdipoGen LIFE SCIENCES | CDX-B0270-M100 |
| sodium ascorbate | Sigma Aldrich | A7631 |
| sodium azide | Sigma Aldrich | S2002 |
| Trolox | Sigma Aldrich | 238813 |
| Tetracycline hydrochloride | Sigma Aldrich | T3383 |
| Puromycin dihydrochloride | Sigma Aldrich | P9620 |
| Oligomycin | Sigma Aldrich | 495455 |
| Antimycin A | Sigma Aldrich | A8674 |
| Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) | Sigma Aldrich | C2759 |
| Ivermectin | Sigma Aldrich | I8898 |
| 2-mercaptoethanol | Sigma Aldrich | M3148 |
| Phenylmethanesulfonyl fluoride solution | Sigma Aldrich | 93482 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich | 11836170001 |
| PHOSSTOP | Sigma Aldrich | 4906837001 |
| Saponin | Sigma Aldrich | 84510 |
| Tween 20 | Sigma Aldrich | P1379 |
| Proteinase K | Sigma Aldrich | P2308 |
| Dynabeads Protein G | ThermoFisher | 10003D |
| Streptavidin Magnetic Beads | ThermoFisher | 88816 |
| IP lysis buffer | ThermoFisher | 87788 |
| RIPA lysis buffer | ThermoFisher | 89901 |
| Opti-MEM Reduced Serum Medium | ThermoFisher | 31985070 |
| Lipofectamine 2000 Reagent | ThermoFisher | 11668019 |
| AlexaFluor647-labeled phalloidin | ThermoFisher | A22287 |
| Restore Plus Western Blot Stripping Buffer | ThermoFisher | 46430 |
| Hoechst 33342 | ThermoFisher | H3570 |
| LR Clonase II Plus Enzyme Mix | ThermoFisher | 11791100 |
| BP Clonase II Plus Enzyme Mix | ThermoFisher | 11789100 |
| 10x Tris/Glycine/SDS buffer | Bio-Rad | 1610732 |
| 2x Laemmli Buffer | Bio-Rad | 1610737 |
| 4x Laemmli Buffer | Bio-Rad | 1610747 |
| Clarity ECL | Bio-Rad | 1705061 |
| Glycine | Bio-Rad | 1610718 |
| Tris Base | Bio-Rad | 1610719 |
| Sodium Chloride | VWR | BDH928625KG |
| Sodium Phosphate Dibasic | VWR | 97061472 |
| Potassium Chloride | VWR | EMPX14051 |
| Potassium Phosphate Monobasic | VWR | EMDPX15651 |
| Kanamycin Sulfate | VWR | 97061-600 |
| Ampicillin Sodium Salt | VWR | IC19014805 |
| Dimethyl Sulfoxide(DMSO) | VWR | EMMX14586 |
| Ethanol | VWR | 89125172 |
| Methanol | VWR | BDH20291GLP |
| 2-Propanol (Isopropyl Alcohol) | VWR | BDH20271GLP |
| Triton X-100 | VWR | EM9410 |
| Paraformaldehyde | VWR | JTS8987 |
| Bafilomycin A1 | Invivogen | Tlrl-baf1 |
| Blasticidin | Invivogen | Ant-bl-1 |
| Hygromycin | Corning | 30-240-CR |
| MG132 | Selleckchem | S21619 |
| PhenoVue™ 641 Mitochondrial Stain | PerkinElmer | CP3D1 |
| Bovine Serum Albumin | Fisher Scientific | CAS 9048-46-8 |
|
| ||
| Critical commercial assays | ||
|
| ||
| ProFection Mammalian Transfection System | Promega | E1200 |
| Dual-Glo Luciferase | Promega | E2920 |
| QProteome mitochondria isolation kit | Qiagen | 37612 |
| Seahorse XFe96 FluxPaks | Agilent | 102601-100 |
| XF Cell Mito Stress Test Starter | Agilent | 103708-100 |
| Duolink® In Situ PLA Probe Anti-Rabbit PLUS | Sigma Aldrich | DUO92002 |
| Agilent QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent | 210518 |
|
| ||
| Deposited data | ||
|
| ||
| Raw MS data | MASSIVE data repository (massive.ucsd.edu) | MSV000088006 |
| Raw MS data | ProteomeExchange (http://www.proteomexchange.org) | PXD028031 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HeLa RhesusTRIM5α-HA | J. Sodroski, Harvard University | N/A |
| HeLa HumanTRIM5α-HA | J. Sodroski, Harvard University | N/A |
| HeLa TRIM5α knockout | This study | N/A |
| HeLa TRIM5α knockout-APEX2V5 stable overexpressed cells | This study | N/A |
| HeLa TRIM5α knockout-Human TRIM5α APEX2V5 stable overexpressed cells | This study | N/A |
| HeLa cells stably expressing YFP-Parkin | Richard J. Youle, NIH | N/A |
| HeLa cells stably expressing YFP-Parkin-TRIM5α knockout | This study | N/A |
| HEK APEX2-V5 | Saha et al., 2020, https://doi.org/10.1371/journal.ppat.1009017 | |
| HEK RhesusTRIM5α APEX2-V5 | Saha et al., 2020, https://doi.org/10.1371/journal.ppat.1009017 | N/A |
| HEK HumanTRIM5 α APEX2-V5 | Saha et al., 2020, https://doi.org/10.1371/journal.ppat.1009017 | N/A |
| Huh7 TRIM5 α knockout | This study | N/A |
| Huh7 TRIM5 α knockout-APEX2V5 stable overexpressed cells | This study | N/A |
| Huh7 TRIM5 α knockout-Human TRIM5α APEX2V5 stable overexpressed cells | This study | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers for GFP-TRIM5E11R sense: Fw, GGGGCAGGTCACCTCCCGCTTTACATTAACCAGGATTC | Integrated DNA Technologies | N/A |
| Primers for GFP-TRIM5E11R antisense: Rw, GAATCCTGGTTAATGTAAAGCGGGAGGTGACCTGCCCC | Integrated DNA Technologies | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLEX_307 | Addgene | 41392 |
| pDest40-APEX2-V5 | Saha et al., 2020, https://doi.org/10.1371/journal.ppat.1009017 | N/A |
| pLEX_307-APEX2-V5 | Saha et al., 2020, https://doi.org/10.1371/journal.ppat.1009017 | N/A |
| pDest40-RhTRIM5-APEX2-V5 | Saha et al., 2020, https://doi.org/10.1371/journal.ppat.1009017 | N/A |
| pLEX_307- RhTRIM5-APEX2-V5 | Saha et al., 2020, https://doi.org/10.1371/journal.ppat.1009017 | N/A |
| pDest40-HuTRIM5-APEX2-V5 | Saha et al., 2020, https://doi.org/10.1371/journal.ppat.1009017 | N/A |
| pLEX_307-HuTRIM5-APEX2-V5 | Saha et al., 2020, https://doi.org/10.1371/journal.ppat.1009017 | N/A |
| 3xAP1pGL3 (3xAP-1 in pGL3-basic) | Addgene | 40342 |
| pRL-SV40P | Addgene | 27163 |
| pGL4.33[luc2P/SRE/Hygro] | Promega | E8491 |
| psPAX2 | Addgene | 12260 |
| pMD2.G | Addgene | 12259 |
| mCherry-Parkin | Addgene | 23956 |
| YFP-Parkin | Addgene | 23955 |
| GFP-DFCP1 | Axe et al., https://doi.org/10.1083/jcb.200803137 | N/A |
| GFP-TRIM5 | J. Sodroski, Harvard University | N/A |
| GFP-TRIM5E11R | This study | N/A |
| pEGFP-C1-hAtg13 | Addgene | 22875 |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism 8 | GraphPad | N/A |
| Image Lab | BIO-RAD | N/A |
| Scaffold software | Proteome Software Inc | N/A |
| Spectronaut 15 | Biognosys | N/A |
| SimpliFi | Farmingdale | N/A |
| iDEV software | ThermoFisher | N/A |
| Huygens Object Analyzer and Colocalization | Scientific Volume Imaging | N/A |
| LASX acquisition software | Leica | N/A |
| Zen Blue Image Acquisition software | Carl Zeiss | N/A |
| Seahorse Analytics software | Agilent | N/A |
| BioRender | BioRender.com | N/A |
| Gen5 | Agilent | N/A |
Highlights.
Unbiased proteomic analysis reveals that TRIM5α associate with mitochondria
TRIM5α links upstream autophagy proteins with markers of damaged mitochondria
Mitophagy is compromised in TRIM5α knockout cells
TRIM5α knockout exacerbates the cellular impacts of mito-chondrial damage
ACKNOWLEDGMENTS
This work was supported by P20GM121176 and R01AI155746 to M.A.M. from the US National Institutes of Health. LC-MS/MS studies were supported by 1S10OD021801-01 from the NIH to B.P. We thank Dr. Wade Johnson for technical expertise at the University of New Mexico Cancer Comprehensive Center Flow Cytometry Shared Resource, which is partially supported by P30CA118100 from the NIH. We thank Dr. Ed Campbell (Loyola University Chicago) for sharing reagents, Dr. Jingyue Jia and Dr. Suresh Kumar for critically reading the manuscript, and Dr. Yiliang Zhu for advice on statistical analysis. BioRender software was used for to generate graphics.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110797.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abudu YP, Shrestha BK, Zhang W, Palara A, Brenne HB, Larsen KB, Wolfson DL, Dumitriu G, Oie CI, Ahluwalia BS, et al. (2021). SAMM50 acts with p62 in piecemeal basal- and OXPHOS-induced mitophagy of SAM and MICOS components. J. Cell Biol. 220. 10.1083/jcb.202009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GFG, Toth R, James J, and Ganley IG (2013). Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 14, 1127–1135. 10.1038/embor.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockler S, and Westermann B. (2014). Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev. Cell 28, 450–458. 10.1016/j.devcel.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Campbell EM, Dodding MP, Yap MW, Wu X, Gallois-Montbrun S, Malim MH, Stoye JP, and Hope TJ (2007). TRIM5 alpha cytoplasmic bodies are highly dynamic structures. Mol. Biol. Cell 18, 2102–2111. 10.1091/mbc.e06-12-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SD, Mamede JI, Hope TJ, and Jensen GJ (2020). Correlated cryogenic fluorescence microscopy and electron cryo-tomography shows that exogenous TRIM5a can form hexagonal lattices or autophagy aggregates in vivo. Proc. Natl. Acad. Sci. U S A 117, 29702–29711. 10.1073/pnas.1920323117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Kroemer G, and Kepp O. (2020). Mitophagy: an emerging role in aging and age-associated diseases. Front. Cell Dev. Biol. 8, 200. 10.3389/fcell.2020.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiramel AI, Meyerson NR, McNally KL, Broeckel RM, Montoya VR, Mendez-Solis O, Robertson SJ, Sturdevant GL, Lubick KJ, Nair V, et al. (2019). TRIM5a restricts flavivirus replication by targeting the viral Prote-ase for proteasomal degradation. Cell Rep. 27, 3269–3283.e6. 10.1016/j.celrep.2019.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloherty APM, Rader AG, Compeer B, and Ribeiro CMS (2021). Human TRIM5alpha: autophagy connects cell-intrinsic HIV-1 restriction and innate immune sensor functioning. Viruses 13, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. (2021). Autophagy in inflammation, infection, and immunometabolism. Immunity 54, 437–453. 10.1016/j.immuni.2021.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero F, Gallo DE, Hope TJ, and Sodroski J. (2011). Trafficking of some old world primate TRIM5α proteins through the nucleus. Retrovirology 8, 38. 10.1186/1742-4690-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher AJ, Vaysburd M, Maslen S, Zeng J, Skehel JM, Towers GJ, and James LC (2018). Trivalent RING assembly on retroviral capsids activates TRIM5 ubiquitination and innate immune signaling. Cell Host Microbe 24, 761–775.e6. 10.1016/j.chom.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, and Pornillos O. (2019). Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 17, 546–556. 10.1038/s41579-019-0225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM, and Valente EM (2017). PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13, 654–669. 10.1080/15548627.2016.1277309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Ordureau A, and Heo JM (2018). Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 19, 93–108. 10.1038/nrm.2017.129. [DOI] [PubMed] [Google Scholar]
- Hsieh CW, and Yang WY (2019). Omegasome-proximal PtdIns(4,5)P2 couples F-actin mediated mitoaggregate disassembly with autophagosome formation during mitophagy. Nat. Commun. 10, 969. 10.1038/s41467-019-08924-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung V, Lam SS, Udeshi ND, Svinkina T, Guzman G, Mootha VK, Carr SA, and Ting AY (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity bio-tinylation. eLife 6. 10.7554/elife.24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, Svinkina T, Carr SA, Mootha VK, and Ting AY (2014). Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging.Mol. Cell 55, 332–341. 10.1016/j.molcel.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, et al. (2018). Galectins control mTOR in response to endomembrane damage. Mol. Cell 70, 120–135.e8. 10.1016/j.molcel.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Guardeno JM, Apolonia L, Betancor G, and Malim MH (2019). Immunoproteasome activation enables human TRIM5alpha restriction of HIV1. Nat. Microbiol. 4, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Dauphin A, Komurlu S, McCauley SM, Yurkovetskiy L, Carbone C, Diehl WE, Strambio-De-Castillia C, Campbell EM, and Luban J. (2019). Cyclophilin A protects HIV-1 from restriction by human TRIM5alpha. Nat. Microbiol. 4, 2044–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruppa AJ, Kishi-Itakura C, Masters TA, Rorbach JE, Grice GL, Kendrick-Jones J, Nathan JA, Minczuk M, and Buss F. (2018). Myosin VI-dependent actin cages encapsulate parkin-positive damaged mitochondria. Dev. Cell 44, 484–499.e6. 10.1016/j.devcel.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, and Ting AY (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54. 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell MA, Jain A, Arko-Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Munch J, Kirchhoff F, Simonsen A, et al. (2014). TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 30, 394–409. 10.1016/j.devcel.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell MA, Saha B, and Thompson TA (2020). The tripartite nexus: autophagy, cancer, and tripartite motif-containing protein family members. Front. Pharmacol. 11, 308. 10.3389/fphar.2020.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merindol N, El-Far M, Sylla M, Masroori N, Dufour C, Li JX, Cherry P, Plourde MB, Tremblay C, and Berthoux L. (2018). HIV-1 capsids from B27/ B57+ elite controllers escape Mx2 but are targeted by TRIM5a, leading to the induction of an antiviral state. PLoS Pathog. 14, e1007398. 10.1371/journal.ppat.1007398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OhAinle M, Helms L, Vermeire J, Roesch F, Humes D, Basom R, Del-row JJ, Overbaugh J, and Emerman M. (2018). A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. eLife 7. 10.7554/elife.39823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, et al. (2011). TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365. 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Princely Abudu Y, Pankiv S, Mathai BJ, Hakon Lystad A, Bindesboll C, Brenne HB, Yoke Wui Ng M, Thiede B, Yamamoto A, Mutugi Nthiga T, et al. (2019). NIPSNAP1 and NIPSNAP2 act as “Eat Me” signals for mitophagy. Dev. Cell 49, 509–525.e12. 10.1016/j.devcel.2019.03.013. [DOI] [PubMed] [Google Scholar]
- Rai P, Janardhan KS, Meacham J, Madenspacher JH, Lin WC, Karmaus PWF, Martinez J, Li QZ, Yan M, Zeng J, et al. (2021). IRGM1 links mitochondrial quality control to autoimmunity. Nat. Immunol. 22, 312–321. 10.1038/s41590-020-00859-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, and Ting AY (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331. 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro CM, Sarrami-Forooshani R, Setiawan LC, Zijlstra-Willems EM, van Hamme JL, Tigchelaar W, van der Wel NN, Kootstra NA, Gringhuis SI, Geijtenbeek TBH, and Geijtenbeek TB (2016). Receptor usage dictates HIV-1 restriction by human TRIM5a in dendritic cell subsets. Nature 540, 448–452. 10.1038/nature20567. [DOI] [PubMed] [Google Scholar]
- Saha B, Chisholm D, Kell AM, and Mandell MA (2020). A non-canonical role for the autophagy machinery in anti-retroviral signaling mediated by TRIM5α. PLoS Pathog. 16, e1009017. 10.1371/journal.ppat.1009017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyutina A, Persaud M, Simons LM, Bulnes-Ramos A, Buffone C, Martinez-Lopez A, Scoca V, Di Nunzio F, Hiatt J, Marson A, et al. (2020). Cyclophilin A prevents HIV-1 restriction in lymphocytes by blocking human TRIM5a binding to the viral core. Cell Rep. 30, 3766–3777.e6. 10.1016/j.celrep.2020.02.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, and Sodroski J. (2006). Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5a restriction factor. Proc. Natl. Acad. Sci. U S A 103, 5514–5519. 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L. (2019). Recent advances in proximity-based labeling methods for interactome mapping. F1000Res 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa E, Marchetti S, and Ricci JE (2018). No parkin zone: mitophagy without Parkin. Trends Cell Biol. 28, 882–895. 10.1016/j.tcb.2018.07.004. [DOI] [PubMed] [Google Scholar]
- Volkmann B, Wittmann S, Lagisquet J, Deutschmann J, Eissmann K, Ross JJ, Biesinger B, and Gramberg T. (2020). Human TRIM5α senses and restricts LINE-1 elements. Proc. Natl. Acad. Sci. U S A 117, 17965–17976. 10.1073/pnas.1922366117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R Jr., Mishra P, and Levine B. (2017). Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168, 224–238.e10. 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Li C, Yang S, Xiao Y, Xiong X, Chen W, Zhao H, Zhang Q, Han Y, and Sun L. (2020). Mitochondria-associated ER membranes - the origin site of autophagy. Front. Cell Dev. Biol. 8, 595. 10.3389/fcell.2020.00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M, and Ktistakis NT (2020). Mammalian Mitophagosome Formation: a focus on the early signals and steps. Front. Cell Dev. Biol. 8, 171. 10.3389/fcell.2020.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M, Gudmundsson SR, Li Z, Manifava M, Cugliandolo F, Shah R, Smith M, Stronge J, Karanasios E, Piunti C, et al. (2019). Selective autophagy of mitochondria on a ubiquitin-endoplasmic-reticulum platform.Dev. Cell 50, 627–643.e5. 10.1016/j.devcel.2019.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass Spectrometry Data is available at the MASSIVE data repository (massive.ucsd.edu) using number MSV000088006 and ProteomeExchange (http://www.proteomexchange.org) using ID number PXD028031. All datasets are publicly available as of the date of publication. This paper does not report original code. Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.
Mass Spectrometry Data is available at the MASSIVE data repository (massive.ucsd.edu) using number MSV000088006 and ProteomeExchange (http://www.proteomexchange.org) using ID number PXD028031.