

Abstract
Heat shock proteins 90 (Hsp90) are promising therapeutic targets due to their involvement in stabilizing several aberrantly expressed oncoproteins. In cancerous cells, Hsp90 expression is elevated, thereby exerting antiapoptotic effects, which is essential for the malignant transformation and tumor progression. Most of the Hsp90 inhibitors (Hsp90i) under investigation target the ATP binding site in the N-terminal domain of Hsp90. However, adverse effects, including induction of the prosurvival resistance mechanism (heat shock response or HSR) and associated dose-limiting toxicity, have so far precluded their clinical approval. In contrast, modulators that interfere with the C-terminal domain (CTD) of Hsp90 do not inflict HSR. Since the CTD dimerization of Hsp90 is essential for its chaperone activity, interfering with the dimerization process by small-molecule protein–protein interaction inhibitors is a promising strategy for anticancer drug research. We have developed a first-in-class small-molecule inhibitor (5b) targeting the Hsp90 CTD dimerization interface, based on a tripyrimidonamide scaffold through structure-based molecular design, chemical synthesis, binding mode model prediction, assessment of the biochemical affinity, and efficacy against therapy-resistant leukemia cells. 5b reduces xenotransplantation of leukemia cells in zebrafish models and induces apoptosis in BCR-ABL1+ (T315I) tyrosine kinase inhibitor-resistant leukemia cells, without inducing HSR.
Short abstract
The tripyrimidonamide 5b is a first-in-class small-molecule inhibitor targeting the Hsp90 CTD dimerization interface that shows antileukemic activity without inducing a heat shock response.
Introduction
The heat shock proteins of 90 kDa (Hsp90) are abundant, molecular chaperones that modulate the folding, stabilization, and maturation of over 400 client proteins in eukaryotes that are involved in essential processes such as signal transduction, cell cycle progression, and transcription regulation.1 In cancer cells, Hsp90 is overexpressed and involved in uncontrolled proliferation and antiapoptotic effects and, in that way, is essential for the malignant transformation and progression of several cancer types, including in acute and chronic myeloid leukemia (AML and CML).2−4 Thus, cancer cells are more dependent on Hsp90 activity than normal cells.5,6 Multiple signal transduction-promoting oncoproteins are client proteins of Hsp90, including BCR-ABL1 fusion kinase, which is a molecular hallmark of CML.7 Hence, inhibiting the activity of Hsp90 is a promising strategy for the development of anticancer therapy. Several Hsp90 inhibitors (Hsp90i) have been developed so far, for instance, targeting Hsp90 N- or C-terminal domain (NTD or CTD) or with isoform selectivity, whereas most of the inhibitors studied in clinical trials target the Hsp90 NTD ATP binding site and with a pan-inhibitory profile.5,8−20 However, adverse events including dose-limiting ocular and cardiac toxicity and poor patient stratification have precluded their clinical approval.5 Another clinical challenge with the use of Hsp90 NTD-targeting inhibitors is the induction of the prosurvival heat shock response (HSR).5,8 The HSR is a stress response mechanism mediated by heat shock factor 1 (HSF-1), which leads to the expression of other heat shock proteins (HSPs) including Hsp27, Hsp40, and Hsp70, as a rescue mechanism upon Hsp90 inhibition that eventually weakens the cytotoxic effects of Hsp90i.5,8,13−16 In addition, Hsp90 NTD-targeting inhibitors potentially inflict cytotoxicity through mechanisms that involve targets other than Hsp90 (off-target effects).6,21 The off-target effect hypothesis is also supported by the significant differences between cytotoxicity concentrations of Hsp90 NTD-targeting inhibitors vs their binding affinity to Hsp90.21 Furthermore, there are two major cytosolic isoforms of Hsp90 (Hsp90α and Hsp90β) expressed in humans. Hsp90α is an inducible isoform, overexpressed in several cancer types, whereas the Hsp90β isoform is expressed constitutively. Thus, targeting Hsp90 with isoform-specific inhibitors can afford a therapeutic window.20,22 However, the Hsp90α and Hsp90β isoforms share a high degree of similarity (Figure S21), making it challenging to develop isoform-selective inhibitors.22
Hsp90 is a flexible homodimer, and each monomer consists of three major functional domains: NTD, middle domain, and CTD. The activity of Hsp90s depends on the binding and hydrolysis of ATP at the NTD and on its dimerization via the CTD.2 The middle domain (MD) that connects the NTD and the CTD mediates the binding of clients and cochaperones. The CTD is connected to the MEEVD motif, which interacts with the subset of tetratricopeptide repeat (TPR) domain-containing cochaperones.2 To our knowledge, inhibiting Hsp90 dimer formation by targeting the CTD dimerization interface constitutes a so far unexplored mode of action (MOA) of small-molecule Hsp90i. In contrast to Hsp90i targeting the N-terminal ATP binding site, C-terminal inhibitors do not generally induce HSR.8,21,23−25 The most important classes of C-terminal inhibitors are (1) inhibitors binding to the C-terminal nucleotide binding site (e.g., novobiocin and analogues), (2) modulators of the Hsp90-CDC37 interaction (e.g., celastrol, induces HSR26), (3) modulators of the Hsp90–p23 interaction (e.g., gedunin), (4) modulators of the Hsp90–HOP interaction (e.g., LB76), (5) modulators addressing an allosteric binding site between CTD and MD, and (6) aminoxyrone (AX), the first nonpeptidic inhibitor of the C-terminal dimerization of Hsp90.21,27−31
Following a strategy recently introduced by us to identify protein–protein interaction (PPI) inhibitors,32,33 we initially identified hot spot residues in the CTD dimerization interface that accounted for most of the binding affinity34 and identified the first peptidic inhibitors shown to bind to the CTD of Hsp90.35 Furthermore, we developed AX, the first peptidomimetic Hsp90 CTD dimerization inhibitor,36 which is a promising lead candidate effective against BCR-ABL1+ TKI-resistant leukemic cells.36 Based on these experiences, here, we report the rational design, chemical synthesis, binding mode model, biochemical affinity, and biological in vitro evaluation of a first-in-class small-molecule inhibitor (5b) of Hsp90 CTD dimerization based on a tripyrimidonamide scaffold.
Results
Design of Tripyrimidonamides as CTD Hsp90 Inhibitors
Based on computational predictions and subsequent experimental validation, we identified the spatially clustered hot spot residues I688, Y689, I692, and L696 in the Hsp90 CTD interface, which are located on α-helix H5, form a functional epitope, and account for most of the protein dimerization energy.34 Furthermore, conformational analysis by 2D NMR and MD simulations revealed for the recently introduced tripyrimidonamide scaffold that it can act as a potential α-helix mimetic, mimicking side chains at positions i, i + 4 (dimeric compound) or i, i + 4, i + 8 (trimeric compound).37,38 This side chain pattern is concordant with the succession of the hot spot residues in the Hsp90 CTD interface.
Together, this provided the incentive for us to design and synthesize the tripyrimidonamide 5a, which mimics the hot spots I688, I692, and L696. In compound 5a, the side chain of V was used instead of I to avoid diastereomers. 6, which lacks the isopropyl side chain, was also designed to probe the influence of the absence of the third side chain in a tripyrimidonamide. Next, we aimed to design compounds that can also form polar interactions, as these should confer specificity of binding.34 In addition, the binding to a well-defined cleft or groove in a PPI region has been described to yield a particularly effective PPI inhibitor.34 The Y689 side chain of 7a should be accommodated in an indentation in the binding epitope of helix H4′ (Figure 1);34 we also designed the homologue 7b with a prolonged (4-hydroxy-phenyl)-ethyl side chain. Although both compounds mimic the three hot spots Y689, I692, and L696, with the longer side chain in 7b, we intended to accommodate for the apparent mismatch between the preferred side chain orientations in tripyrimidonamides and the side chain pattern of the hot spots (i, i + 3, i + 4). The side chain patterns of 5b and 7a are almost identical to that of the α-aminoxy-peptide AX, which was shown to bind to the CTD.36
Figure 1.

Rational design and synthesis of tripyrimidonamides. (a) Cryo-EM structure of the dimer of human Hsp90β (PDB ID 5FWK),39 shown in surface and cartoon representations. For one of the Hsp90 monomers, the N-terminal domain (NTD) is colored in red, the middle domain in beige, and the C-terminal domain (CTD) in blue. Above and below the protein structure, the structures of Hsp90i and their potential binding sites (see refs (40−44), color-coded according to the domains) are shown. (b) Dimeric CTD of human Hsp90β with the two monomers in blue and white. Helices H4, H4′, H5, and H5′ of the CTDs form the dimerization interface. (c) Residues forming the CTD dimerization interface in human Hsp90α are primarily located on helices H4, H4′, H5, and H5′.34 (d) Tripyrimidones can adopt conformations resembling the side chain orientation of an α-helix in i, i + 4, and i + 7 positions.38 (e) Synthesis of tripyrimidonamides: (a) COMU, DMF, r.t., 18 h; (b) NaOH, MeOH, 80 °C, 6 h; (a) 2a–2d, COMU, DMF, r.t., 18 h; (c) 6 via 5b, BBr3, DCM, −78 °C, 1 h, r.t., 1 h; (d) via 7a via 5c and 7b via 5d, H2, Pd(C), MeOH, DCM, r.t., 1 h.
Further analysis of the physicochemical properties of the CTD dimerization interface revealed a particular hydrophobic patch there (Figure 1). Interestingly, the 4-methoxy-benzyl side chain of 5b should act as a (weak) hydrogen bond acceptor for S673′ and T669′ on helix H4′ but, at the same time, decrease the side chain’s hydrophilicity for a more favorable burial in the overall hydrophobic interface. To probe this with a larger substituent, we also designed the benzyloxy derivatives 5c and 5d, which are also precursors of 7a and 7b, respectively.
Synthesis of Tripyrimidonamides
The monomeric building blocks 1 and 2a–2e were prepared according to our previously published protocol.38 Subsequently, the designed tripyrimidonamides 5a–5d were synthesized using a modular approach. Briefly, a COMU-mediated amide coupling of the lithium carboxylate 2e with 5-aminopyrimidone 1 afforded the benzoyl-protected dimer 3 in 75% yield. Deprotection of the benzoyl-group by treatment of 3 with sodium hydroxide in methanol at 80 °C afforded the unprotected dimer 4 (77% yield). Additional coupling reactions of 4 with the respective lithium salts 2a–2d in the presence of COMU furnished the tripyrimidonamides 5a–5d in 39–76% yield. Compound 6 with an N-unsubstituted N-terminal pyrimidone ring was synthesized by treating the corresponding 4-methoxybenzyl-substituted derivative 5b with BBr3 in DCM (Scheme S1). Finally, the tripyrimidonamides 7a and 7b with free phenolic groups were prepared by catalytic hydrogenation of their respective O-benzyl-protected precursors 5c and 5d (40% and 87% yield).
Selection of 5b as a Lead Candidate
To evaluate the inhibition of Hsp90 dimerization, Escherichia coli BL21 (DE3) pETSH-3 cells were used to display Hsp90α on their surface (Figure 2a).35 Passenger-driven dimer formation of Hsp90α is facilitated through the motility of the β-barrel domain within the outer membrane of E. coli, as reported for other proteins.45 To demonstrate the functionality of dimerized Hsp90 on the surface of E. coli, the transcription factor p53, a natural client protein of Hsp90, was labeled with fluorescein isothiocyanate (FITC) and added to cells displaying Hsp90 on their surface. A subsequent flow cytometer analysis revealed a high green fluorescence for cells displaying Hsp90, indicating dimerized and functional Hsp90 (Figure 2b). Compounds 5a, 6, and 7a showed only weak inhibition of 3.27%, 14.65%, and 24.35%, respectively. In contrast, 5b–5d and 7b showed moderate inhibition of 39.92%, 41.83%, 55.23%, and 31.33%, respectively (Figure 2b).
Figure 2.

Selection of 5b as a lead candidate. (a) Schematic view of the Hsp90 dimerization assay using Autodisplay. (b) Flow cytometry measurements of the inhibition of dimerized Hsp90α displayed on E. coli cells.36E. coli BL21 (DE3) cells displaying Hsp90α incubated with 1 μM FITC-labeled p53 lead to a high cellular fluorescence indicating dimerization of Hsp90α. The value obtained was set as 0% inhibition. In contrast, E. coli cells without displaying Hsp90α (control cells) show no cellular fluorescence. The value obtained here was set as 100% inhibition. Preincubation of E. coli cells with surface-displayed Hsp90α with 50 μM of the respective substance leads to a lowered cellular fluorescence intensity indicating a lowered binding affinity of FITC-labeled p53 to surface-displayed Hsp90α. These values were set in relation to obtain the relative inhibition of dimerization. (c) Apparent KD values of the purified CTD of Hsp90α and the respective substance measured via the MST method. A constant amount of 50 nM labeled CTD of Hsp90 was used, and three independent measurements were performed. The resulting mean values were determined and used in the KD fit formula. (d) Cellular viability assessment of a leukemic cell line (K562) measured by incubating with the indicated inhibitors for 72 h, followed by a viability measurement using an ATP-based Celltiter Glo assay. (e) Selection of 5b as a lead candidate on the basis of high inhibition of Hsp90α dimerization, low apparent KD, and low IC50 (μM) in a tested leukemic cell line.
Later, the binding affinity of the compounds was determined with microscale thermophoresis (MST) measurements using the NT-647-labeled recombinant CTD of the Hsp90α protein.36 A nonlinear regression curve was fitted with the KD formula, and as expected, substances showing weak inhibition have high dissociation constants (6, 249 μM; 7a, 286 μM; Figure 2c,e). The lowest KD value was observed for 5b (3.42 ± 1.0 μM) and the second lowest for 5d (11.74 ± 1.0 μM) (Figure 2c,e). These findings are paralleled by the in vitro cytotoxicity assessment of compounds 5a–5d, 6, 7a, and 7b, which also revealed 5b as the most promising candidate (with low IC50: 1.3 ± 0.3 μM) in a BCR-ABL1+ tested leukemia cell line K562 (Figure 2d,e).
Based on the inhibition of Hsp90α CTD dimerization, the low apparent KD value for the Hsp90α CTD, and the potent antileukemic activity, 5b was selected for further detailed affinity and efficacy assessments.
5b Binds Specifically to CTD of Hsp90α and Blocks Its Cochaperone Function
One of the major limitations of NTD-targeting inhibitors is their off-target activity.6,21 Hence, it is important that the selected hit 5b has a high degree of selectivity against its target, the CTD of Hsp90. To assess the selectivity of 5b, biochemical cell-free and cellular assays were performed. First, we evaluated the affinity of 5b against Hsp90 in a cell-free assay, where 5b protected recombinant (full-length) Hsp90α protein in a dose-dependent fashion from degradation against thermolysin enzyme digestion, an assay commonly used to quantify drug affinity-responsive target stability (DARTS)23,46 (Figure 3a). Next, we performed the cell-free thermal shift assay47 to determine the potential binding affinity of 5b to recombinant Hsp90α CTD and NTD protein. 5b specifically destabilized the CTD of Hsp90α protein Hsp90 CTD (ΔTm: −8.45 ± 0.55 °C), whereas reference CTD-targeting Hsp90i coumermycin A1 (CA1) stabilized Hsp90α CTD protein (Figure 3b, Figure S22 and Table S1). NTD-targeting Hsp90i tanespimycin (TM) and PUH-71 served as a positive (Hsp90α NTD) or negative (Hsp90α CTD) control in this assay. The thermostabilizing effect of 5b to its target (total Hsp90) was also assessed in a cellular setup, termed the cellular thermal shift assay (CETSA),47−49 a biophysical method based on the ligand-induced thermal stabilization of the protein to directly probe the target engagement in the living cells (Table S1 and Figure S23). The protein quantification for CETSA was performed using a digital Western blotter for a sensitive and quantitative evaluation of the ligand-protected intracellular Hsp90, whereas TM and PU-H71 served as controls. Next, the thermal stability of intracellular Hsp90 in an increasing concentration of 5b (at a fixed temperature) was determined, a method termed isothermal dose–response fingerprint ITDRFCETSA.485b induced the thermal stability of Hsp90 in a dose-dependent fashion, confirming its intracellular and specific target engagement (Figure 3c, Table S1).
Figure 3.

Specificity of 5b against Hsp90 CTD and its cochaperone function. (a) Recombinant (full-length) Hsp90α (1 μg) was incubated with 5b at indicated concentrations, followed by digestion with thermolysin. Treated protein samples were electrophoresed (SDS-PAGE) and immunoblotted with anti-Hsp90α for detecting the protection of Hsp90α protein by 5b (the upper band is protected from proteolysis). (b) A cell-free thermal shift assay was performed by incubating recombinant Hsp90α CTD protein with 5b at an increasing temperature (up to 95 °C). The melting temperature (Tm) without inhibitors (DMSO) was used as a control. (c) Dose-dependent intracellular (K562 cells) thermal stabilization (CETSAITDRF) of Hsp90 after 5b incubation (24 h) at its increasing concentration (1.25–5 μM). (d) 5b inhibits the Hsp90α chaperone function, comparable to TM and GM, in the cell-free luciferase refolding assay, where the incubation of the inhibitors prevented the rabbit reticulocyte lysate (a source of Hsp90)-assisted refolding of denatured luciferase. (e) Incubation of 5b blocked the binding of Hsp90 CTD-interacting cochaperone (PPID) in TR-FRET measurements. (f) 5b did not reduce the amount of Hsp90-bound FITC-labeled GM and, therefore, does not compete for the GM binding pocket of full-length Hsp90α. Unlabeled GM, GP, PUH71, and TM served as positive controls and NB and CA1 as negative controls.
Next, to assess the ability of 5b to inhibit the Hsp90 chaperone function, a cell-free luciferase-refolding assay50−52 was performed using rabbit reticulocyte lysates as a source of Hsp90. Exposure of 5b decreased the luciferase refolding capacity in a dose-dependent manner by blocking the chaperone function of Hsp90 (Figure 3d). The known Hsp90 NTD inhibitors geldanamycin (GM) and TM served as positive controls. In addition, to assess the specific effect of 5b in obstructing Hsp90 CTD-interacting cochaperones, a time-resolved fluorescence resonance energy transfer (TR-FRET) assay was conducted.535b blocked the binding of PPID (or cyclophilin D, an Hsp90 CTD-interacting chaperone) to recombinant Hsp90α or Hsp90β CTD protein comparable to the CA1 treatment, whereas PU-H71, TM, and GM served as negative controls (Figure 3e, Table S1). To rule out the possible interaction of 5b with the NTD of Hsp90α, a fluorescence polarization (FP) competitive assay was carried out using FITC-labeled GM50 (Figure 3f, Table S1). As expected, 5b did not show any interaction with the NTD of Hsp90, whereas unlabeled Hsp90 NTD-targeting inhibitors GM, ganetespib (GP), TM, and PU-H71 served as positive controls.
Binding Mode Prediction of 5b at Hsp90α
To provide structural insights into how 5b binds to the CTD of human Hsp90, we performed 40 independent molecular dynamics (MD) simulations of free diffusion of 5b in the presence of truncated monomeric Hsp90α (aa 294–699) using the Amber 18 suite of molecular simulation programs54 and the ff14SB55 and a modified GAFF38,56 force field for protein and ligand. Initially, we generated 40 individual starting configurations by randomly placing 5b and the CTD structure, leaving at least 10 Å between atoms in 5b and the CTD structure. After minimization, thermalization, and density adaptation, we performed MD simulations of 500 ns length, in which the 5b molecule diffused freely. To counter the high flexibility of the C-terminal helix interface, we introduced positional restraints on the backbone atoms, adjusting the reference coordinates every 100 ns to allow for moderate protein movements.
From the trajectories, first, we extracted all frames where 5b is bound to Hsp90 (no-fit RMSD of 5b ≤1.5 Å to the previous frame after superimposing Hsp90). Mapping the probability density of occurrence of 5b onto the surface of Hsp90 (Figure 4a) revealed two main binding regions: one in the C-terminal helix interface (Figure 4a, green), where binding occurred in 10 out of the 40 replicas, and another in a cleft between the CTD and middle domain (Figure 4a, blue), where binding occurred in 6 out of 40 replicas. In the latter case, an area of high density with the shape of 5b is observed (Figure 4a, light blue), which resulted from a single trajectory. This indicates that the ligand was kinetically trapped in this one case, although the position is thermodynamically not favorable. By contrast, the densities in the C-terminal interface are more ambiguously shaped, indicating that, while binding there is favorable, the ligand can still explore multiple binding modes, which are also seen to interchange.
Figure 4.

MD simulations of 5b diffusion and effective binding energy calculations to predict the binding mode. (a) The relative densities of the bound poses of 5b after 500 ns are mapped on the Hsp90α monomer fragment used in the simulations (PDB ID 3q6m). The missing NTD is shown in red, based on the Hsp90β structure (PDB ID 5fwk). Particularly high densities are observed in the region between H4 and H5 (green circle). A second, less preferred site is in the cleft between the CTD and middle domain (blue circles). (b) Effective binding energy calculations over a single trajectory that resulted in 5b binding in the C-terminal helix interface as a function of the center-of-mass distance between 5b and H4 and the simulation time (see the color scale). The dashed line at 12.1 Å corresponds to the H4–H5′ distance in the crystal structure of PDB ID 3q6m. (c) Possible binding mode of 5b in the helix interface, where 5b mimics H5′. (d) Blow-up of the possible binding mode of 5b showing how its side chains mimic side chains of H5′.
To further study these binding modes, we clustered the bound frames of 5b mapped on the protein surface with respect to their RMSD after superimposing Hsp90. Among the binding modes were several that form interactions to the C-terminal helix interface, with 5b positioned such that it mimics interactions formed by H5′ in the dimer (Figure 4c,d). To corroborate that this binding mode is favorable, we computed the effective binding free energies by the MM-GB/SA approach for the trajectory that led to it. The first transient interactions with the protein already resulted in effective energies down to ∼−30 kcal mol–1. The effective energies decreased further to ∼−45 kcal mol–1 once the ligand was bound in the C-terminal interface, thereby forming interactions with Hsp90 that remained stable even when the trajectory was extended to 1 μs, indicating that such poses are particularly favorable (Figure 4b). Regarding the magnitude of the effective energies, note that configurational entropy contributions were not considered, since estimating such contributions by a normal-mode analysis may introduce additional uncertainties.57,58
Overall, the probability density of bound 5b poses, the proportion of replicas, and the results of the MM-GB/SA computations indicate that 5b preferentially binds to the C-terminal helix interface, where it can adopt poses that mimic H5′.
Comparison to 5b Binding at Hsp90β
We then set out to study whether there is an isoform specificity for the binding of 5b because the helical interface regions differ in three positions: α, S641; β, P633; α, S658; β, A650; α, A685; β, S677 (Figure S21). Using the same setup as before, we performed MD simulations of free ligand diffusion around Hsp90β. The probability density of bound 5b again revealed that the C-terminal helix interface is the most preferred region, followed by the cleft between the CTD and middle domain (Figure S24). Notably, no high density in this cleft was then found, in contrast to Hsp90α, confirming that the observation there resulted from kinetic trapping. Hence, despite the few sequence variations in the C-terminal helix interface between Hsp90α and Hsp90β, the same preferred binding region of 5b was found in both cases.
5b Interferes with Hsp90α CTD Dimers and Disrupts Intracellular Hsp90 Multiprotein Complexes
CTD dimerization of Hsp90 is necessary for its function.59,60 To study the effect of 5b exposure on the dissociation of Hsp90 dimers in a cell-free assay, we used Hsp90α CTD protein after incubation with amine-reactive cross-linker BS3, as previously described.50,53 A dose-dependent reduction of Hsp90α CTD dimers along with an increase in the high-order oligomeric species was noticed upon incubation with 5b (Figure 5a). Next, we performed small-angle X-ray scattering (SAXS) with the Hsp90α CTD protein, which was coupled to a size exclusion chromatography column (SEC-SAXS) at the ESRF beamline BM29 in Grenoble.61,62 In the absence of 5b, a clear dimeric profile of the Hsp90α CTD protein was visible on the chromatogram, with an additionally minor tetrameric species (Figure S25). We used the program CHROMIXS63 to merge the frames containing the dimer from this SEC-SAXS profile. Buffer frames were then subtracted using PRIMUS.64 From the SAXS data, a radius of gyration (Rg) of 3.23 nm was calculated, which describes the average particle dimension in solution. The ab initio model fit from DAMMIF65 shows a χ2 of 1.127, indicating good agreement with the experimental data (Figure 5b and Table S2). The corresponding dimeric envelope is highlighted in Figure 5c, superimposed with the calculated dimeric model of Hsp90α CTD. Further, we tested the effect of 5b on the Hsp90 CTD dimer using SAXS (Figure 5d). Due to the low solubility of 5b, we needed to measure the sample as an ensemble of species in solution. First, we tested Hsp90α CTD without 5b on Xeuss 2.0 with the Q-Xoom system and observed an increase of Rg to 3.40 nm. This increase is likely due to the small amount of tetramer in solution in the ensemble. Theoretically, the Rg of a tetrameric Hsp90α CTD species is 3.78 nm, using CRYSOL, indicating that, even in batch mode SAXS measurements, the Hsp90α CTD protein is predominantly in a dimeric state. We added 5b with an equimolar concentration to Hsp90α CTD protein, and the Rg value slightly increased from 3.40 to 3.46 nm. However, with an increasing concentration of 5b to 1 mM, the Rg value increased to 4.11 nm. Compared to the theoretical Rg value of 3.78 nm for the tetramer, we observed that 5b induces oligomerization of Hsp90α CTD to species even larger than the tetrameric form (Figure 5e). It is unclear, however, if the oligomers are formed from Hsp90α CTD monomers or dimers, as the single species could not be resolved in the ensemble measurement.
Figure 5.

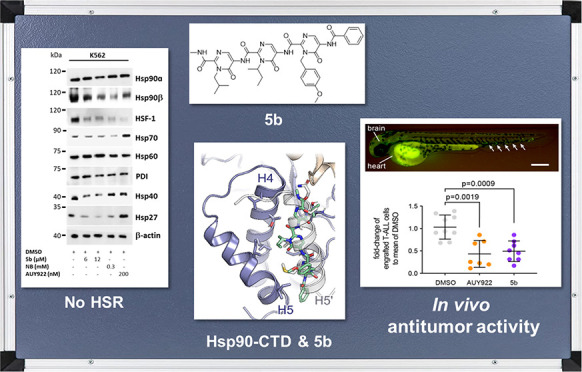
Effect of 5b on Hsp90 oligomeric species and CTD-mediated dimerization. (a) Recombinant Hsp90α CTD was incubated with 63 μM BS3 cross-linker with (at the indicated concentration) or without 5b, followed by immunoblotting with the anti-Hsp90 (AC88) antibody. (b) The scattering data of Hsp90α CTD is shown in black dots, with gray error bars. The ab initio DAMMIF model fit is shown as a red line. The intensity is displayed as a function of momentum transfer s. (c) The volumetric envelope, calculated from the scattering data using DAMMIF,65 is shown as a blue surface. The monomers of the predicted Hsp90 CTD dimer model are shown in green and cyan. Superimposing was performed using SUPCOMB.65 (d) The radius of gyration (Rg) of the different Hsp90α CTD protein samples was calculated using the Guinier approximation.66 The theoretical Rg of the tetramer was calculated using CRYSOL based on the structure PDB ID 3q6m.67 (e) Native Hsp90 complexes in K-562 (24 h administration of 5b) were identified by running blue native (BN) gels followed by immunoblotting analysis. The cytotoxic concentration of 5b resulted in the potent disruption of Hsp90α, Hsp90β, Hsp40, and Hsp27 complexes and monomers/dimers. AUY922 exposure elevated the expression of HSR associated protein complexes and monomers/dimers (Hsp40 and Hsp27), whereas Hsp60 served as loading controls.
In a cellular context, Hsp90 acts in multiprotein complexes.23 Therefore, interfering with Hsp90 function may lead to the disruption of these complexes. In a cellular assay, Western blotting was performed under reducing (+dithiothreitol or +DTT) and nonreducing (−DTT) conditions after 5b incubation of the K562 cells.36 Similarly to AX,365b inhibited the formation of Hsp90 higher-order multimeric species, in contrast to novobiocin (NB) but in concordance with AUY922 (Figure S26). Next, to study the effect of 5b exposure on Hsp90 native multiprotein complexes, blue native (BN) PAGE analysis was carried out with K562 cell lysates after 5b incubation.23,36 At cytotoxic concentrations of 5b, Hsp90α and Hsp90β multiprotein complexes were disrupted, including monomers/dimers of Hsp40 and Hsp27; Hsp60 (primarily in mitochondria) multiprotein complexes, serving as a loading control, were not affected (Figure 5e). The extent of Hsp90α or β complex/monomer/dimer disruption by 5b was comparable to the controls (NB and AUY922). Moreover, the expression of detected Hsp90α monomeric/dimeric species was prominently lower than the Hsp90β monomeric/dimeric species in the blue native PAGE analysis, which makes it difficult to conclude whether 5b had any intracellular isoform selectivity, especially in disrupting Hsp90 dimerization.
Taken together, these results confirm that 5b interferes with the Hsp90α CTD dimerization, induces oligomerization, and disrupts intracellular Hsp90 multiprotein complexes.
Basic Physicochemical Properties and Microsomal Stability of 5b
Next, we assessed the aqueous solubility, chemical stability, and in vitro metabolic stability of 5b (Supporting Information). The thermodynamic solubility of 5b was determined in phosphate-buffered saline (PBS, 25 °C, pH 7.4) after 4 and 24 h of incubation. Ondansetron was used as a reference compound with a high solubility of 95 μM. The thermodynamic solubility of 5b was low, ranging from 5 μM after 4 h to 8 μM after 24 h (n = 2). To study the chemical stability of 5b at physiological pH, the compound was dissolved in a mixture of Tween20/ethanol/phosphate buffer pH 7.5 (7/3/90) and monitored over 24 h. After 24 h, almost no decomposition was detected (0.7% drug decomposition, n = 2). The chemical stability of 5b at acidic pH was determined by dissolving 5b in a mixture of Tween20/ethanol/phosphate buffer pH 2 (7/3/90) and monitoring over 24 h. After 24 h, only slight decomposition was detected (1.3% drug decomposition, n = 2).
A metabolic stability screening of 5b in human liver microsomes revealed 91% stability after a 40 min incubation at 37 °C. Propanolol, a reference drug with medium to high metabolic stability, showed 74% of the parent compound remaining and therefore demonstrated slightly decreased stability compared with 5b. Calculated results for the intrinsic clearance suggest that 5b [6 μL/(min mg)] is a low-clearance compound with an estimated long half-life (n = 2).
5b Is Effective against Resistant Leukemia Cells and in the Zebrafish Xenotransplantation Model
Elevated Hsp90 expression is reported in several resistant leukemia subtypes such as BCR-ABL1+ CML/BCP-ALL, FLT3-ITD-driven AML, and Ph-like BCP-ALL.68−71 In addition, client proteins of Hsp90 include several kinases (e.g., AKT, BCR-ABL1, BRAF, EGFR2, HER2, and JAK1/2), growth and steroid receptors, and apoptotic factors (e.g., BCL-2 and mutant p53), which are often aberrantly regulated in several malignancies.2,72 Therefore, we determined the efficacy of 5b on therapy-resistant cell lines obtained from different leukemia subtypes (B-ALL, T-ALL, CML, and AML), including imatinib (IM) and multi-TKI-resistant and bortezomib (BTZ)-resistant leukemic cell lines.36 Average IC50 values of 5b in these cell lines were determined using an ATP-based viability assay and were plotted in a clustered heat map, indicating the superior efficacy against BCR-ABL1+ and AML leukemic cell lines when compared to T-ALL cell lines (Figure 6a).
Figure 6.

5b is effective against human leukemic cell lines without inducing any HSR. (a) Comparative cytotoxicity assessment of different subgroups of leukemic cell lines (K562, KCL22, SUPB15, HL60, MOLT4, DND41, TALL1, and HPBALL), imatinib-resistant cell lines (K562-IMr, KCL22-IMr, SUPB15-IMr), and the bortezomib-resistant cell line (HL60-BTZr) after 72 h of exposure to 5b. The IC50 data was plotted as a clustered heat map, followed by unsupervised hierarchical clustering. The vertical axis of the dendrogram exemplifies the dissimilarity between clusters, whereas the color of the individual cell is related to its position along a log IC50 (μM) gradient. (b) The treatment of K562 cells with 5b and respective controls (AUY922 and NB) for 48 h resulted in the downregulation of BCR-ABL1+ and subsequent downstream signaling pathways including phosphorylated and unphosphorylated Stat5a, Crkl, Akt, S6 (mTOR), and cMyc. (c) K562 cells were treated with the indicated (cytotoxic) concentration of 5b, NB, and AUY922 for 48 h, and later, protein lysates were subjected to immunoblot analysis. As expected, 5b and NB did not induce expression of Hsp70, Hsp40, and Hsp27, whereas AUY922 led to HSR induction. Hsp60 (primarily present in mitochondria) and PDI (endoplasmic reticulum) served as a control. (d) (upper) description of the experimental rationale; (middle) representative image of a xenotransplanted zebrafish embryo at 32 hpf [scale bar, 250 μm; note that human T-ALL cells (green) were distributed in the yolk, brain, and hematopoietic tissue (arrows)]; (lower) fold-change of labeled cells normalized to the average percentage of labeled cells in the DMSO-treated group. Each dot represents three embryos pooled as one biological sample. Data are mean ± standard deviation. The p-values were calculated with the Mann–Whitney test.
As previously performed with AX,36 we next determined the destabilizing effect of 5b on BCR-ABL1 expression and associated downstream signaling. In K562 cells, 48 h of exposure of 5b downregulated the phospho-BCR-ABL1 and total-BCR-ABL1 levels as well as the related downstream signaling pathways, as evaluated by an immunoblot analysis (Figure 6b). 5b additionally reduced the expression of client proteins associated with Hsp90 chaperone activity, involving Akt, Stat5, and c-Myc (Figure 6b). In contrast to AUY922, the exposure of 5b on the K562 cells did not induce the expression of Hsp70, Hsp40, and Hsp27 involved in HSR (Figure 6c). Exposure of 5b to the leukemic cell lines (K562, KCL22, and HL60) inhibited their proliferation (Figure S27) and induced apoptosis in a caspase 3/7 enzyme-dependent assay, with an induction of an approximately 2- to 8-fold increase of apoptotic cells, in accordance to the reference Hsp90 CTD-targeting inhibitor, novobiocin (NB) (Figure S28). In addition, 5b facilitated early differentiation measured by the expression of differentiation markers involving CD14 vs CD11b in HL60 cells and CD133 vs CD11b in Mutz-2 cells (Figure S29). In this line, a 48 h exposure of 5b to K562 cells significantly reduced the colony-forming capacity (Figure S30). To further evaluate the efficacy of 5b on leukemic cells, we used the zebrafish xenotransplantation model73 (Figure 6d). MOLT-4 cells were transplanted into zebrafish embryos at 32 h postfertilization (hpf). At this stage, the adaptive immune system is not yet developed; therefore, human cells can be tolerated by the host. The transparency of zebrafish embryos also enabled us to monitor the distribution of human cells, which were stained with a vital fluorescent dye. Xenotransplanted embryos were treated with AUY922 (500 nM) and 5b (500 nM) for 48 h, whereas DMSO was used as a negative control. No toxicity of drugs at the given concentration was noticed on the development of xenotransplanted embryos. We then determined the number of MOLT4 cells in each treated group using flow cytometry. Compared to the DMSO-treated group, the number of transplanted cells was significantly reduced in AUY922- and 5b-treated embryos.
To conclude, this data confirms the antileukemic effect of 5b without inducing HSR.
5b Acts on TKI-Resistant BCR-ABL1+ Leukemic Cells
The integration of specific tyrosine kinase inhibitors (TKI) such as imatinib (Gleevec) into polychemotherapy treatment protocols has significantly improved the response rate in BCR-ABL1+ leukemia patients (initial remission went from 35% to 88%).74 However, stable remission cannot be sustained in many cases as the leukemic cells pursue several escape mechanisms against TKI treatment; one of them is the occurrence of mutations in the ABL1 kinase domain. In particular, in the BCR-ABL1T315I mutant, only ponatinib (TKI) is effective,75 albeit with severe cardiovascular side effects.76 As Hsp90 facilitates the correct folding of several oncogenic newly synthesized or denatured proteins, among them BCR-ABL1,77−79 targeting Hsp90 with small-molecule inhibitors would destabilize BCR-ABL1 and can serve as a therapeutic target.5,12 Therefore, we next tested the efficiency of 5b in a murine BA/F3 cell line stably expressing clinically relevant mutants (BCR-ABL1T315I, BCR-ABL1E255K, and BCR-ABL1M351T) with prominent TKI resistance profiles.36 As expected, similar to K562 cells, exposure of 5b significantly reduced proliferation (Figure 7a) and induced apoptosis (Figure 7b) at 6 and 12 μM, comparable to NB (at 0.3 mM) in BA/F3 cells expressing BCR-ABL1T315I, BCR-ABL1E255K, or BCR-ABL1M351T mutants. Additionally, after the application of 5b, BCR-ABL1 oncoprotein was destabilized, and downstream signaling pathways (Akt and Stat5) were blocked with increasing concentrations of 5b, comparable to the human leukemic cell lines (Figure 7c). Furthermore, 24 h of exposure of 5b on BCR-ABL1T315I, BCR-ABL1E255K, or BCR-ABL1M351T mutant-expressing BA/F3 cells significantly inhibited the colony formation ability (Figure 7d). In addition, in our generated human BCR-ABL1+ IM-resistant cell line models (K562-IMr and KCL22-IMr),365b did not differentiate in inducing apoptosis between IM-resistant vs IM-sensitive clones, proposing a superior effect of 5b in human IM-resistant BCR-ABL1-positive cells (Figure 7e and Figure S31). Encouraged by these results, we next tested 5b on three primary CMLCD34+ IM-resistant patient samples in the range of cytotoxic concentrations (5–10 μM) obtained from leukemic cell lines. Similar to BCR-ABL1+ leukemic cell lines, the exposure of 5b reduced the proliferation, induced apoptosis, and reduced the colony-forming ability of CMLCD34+ IM-resistant patient cells and also revealed a potent combinatorial inhibitory effect when used in combination with IM (Figure 7f–h).
Figure 7.

5b as a potent inhibitor against the TKI-resistant BCR-ABL1 mutant and a primary patient sample. (a) BA/F3 cells expressing BCR-ABL1 mutants (T315I, M351T, and E255K) were treated with the indicated concentration of 5b, and later, viable cells were counted after every 24 h interval for 3 days. (b) Apoptosis induction of the same cells after 48 h of incubation of the compounds at the indicated concentration, determined by a caspase 3/7 dependent Glo assay. (c) Exposure of 5b to these cells destabilizes BCR-ABL1 and, subsequently, its associated downstream signaling pathways. (d) BA/F3 BCR-ABL1 mutant-expressing cells were seeded in methylcellulose-based semisolid medium after 24 h of treatment with 5b. Colonies were counted after 14 days. (e) K562-IMr cells were treated with the indicated concentration of 5b for 48 h, later dually stained with annexin V/PI, and subsequently measured by FACS. (f) Primary CMLCD34+ patient cells were treated with the indicated concentration of 5b or IM alone or in combination, and later, viable cells were counted after every 24 h interval for 6 days. (g) Apoptosis induction in primary CMLCD34+ patient cells after exposure of 5b or IB alone or in combination. (g) Primary CMLCD34+ patient cells were seeded in methylcellulose medium after 24 h of treatment with 5b, IM alone, or both in combination. Colonies were counted after 14 days. Columns depict the mean of three independent experiments (n = 3).
Discussion
The CTD of Hsp90 contains several binding areas: the C-terminal ATP binding site, the MEEVD motif at the end of the CTD, the region at the border between the MD and the CTD (located ∼60 Å away from the NTD ATP binding site, which has been indicated to host a druggable allosteric binding site), and the primary dimerization interface of Hsp90.28,80,81 The C-terminal nucleotide binding site is only available upon occupation of the N-terminal ATP binding pocket and binds purine and pyrimidine nucleotides, while the N-terminal ATP binding site (NTD) is more specific for adenine derivatives.82 The MEEVD motif binds the TRP-domain of cochaperones such as Hop and immunophilins, which is formed by a four-helix bundle and is crucial for Hsp90 dimerization.2 For the mitochondrial Hsp90 paralog TRAP1, small-molecule inhibitors were rationally found that target the allosteric site,30 and found Hsp90 activators were indicated to also act via this site.29 Protein–protein interactions in the interface of the four-helix bundle maintain the dimeric state of Hsp90.59
In this study, we present the development of a first-in-class small-molecule inhibitor of Hsp90, 5b, which was rationally designed to target the C-terminal dimerization interface. Based on our prior hotspot prediction34 and the prototype compound AX,36 we successfully performed scaffold-hopping from the aminoxy peptide-backbone toward more druglike tripyrimidones. 5b mimics α-helical side-chains that form hotspot residues located on H5 in the dimerization interface. A 3-fold difference in the KD between 5b and 5d was reported, which is likely because of the larger 4-benzyloxyphenylethyl substituent compared to 4-methoxybenzyl (Figure 1e) that sterically interferes when binding to the H4/H5 interface. In contrast to the 4-methoxy-benzyl side chain of 5b, the 4-benzyloxyphenylethyl substituent of 5d cannot mimic the Y689′ hot spot, and the 4-methoxy-benzyl side chain of 5b does mimic it and should also act as a (weak) hydrogen bond acceptor for S673′ and T669′. To independently predict the binding site and mode of 5b, we performed extensive MD simulations, in which the inhibitor was allowed to diffuse freely around an MD-CTD construct of Hsp90α, similar to our analysis on AX binding36 and related studies.83−85 The results revealed the C-terminal dimerization interface as the most likely binding region of 5b, which was confirmed by effective binding energy computations, corroborating the hypothesis underlying the design of 5b. Following the high sequence-identity in the interface region, similar results were obtained for Hsp90β, suggesting that 5b does not exhibit isoform specificity. Furthermore, in the TR-FRET assay by taking Hsp90α and β CTD recombinant proteins, 5b did not display any Hsp90 isoform selectivity in blocking the binding of a CTD interacting chaperone (PPID).
Next, the selective binding of 5b to Hsp90 was validated in a variety of biochemical and cellular assays, including DARTS, thermal, and isothermal shift assays, whereas intracellular Hsp90 engagement and disruption of Hsp90 multiprotein complexes were established via CETSA and ITDRFCETSA assays and immunoblotting under reducing conditions (±DTT) and nondenaturing (BN-PAGE) conditions. Moreover, a cross-linker and autodisplay dimerization assay, as well as SEC-SAXS measurements, repeatedly confirmed the destabilization of Hsp90α CTD dimers upon 5b incubation, whereas no unspecific binding of 5b was reported on the Hsp90α NTD protein in an FP-based competitive assay. However, during SAXS measurements, which were performed on an ensemble of species in solution, we were unable to determine whether 5b-induced oligomers originated from Hsp90α CTD monomeric or dimeric species. In addition, 5b blocks the chaperone function of Hsp90, as determined by the TR-FRET assay and in the cell-free luciferase refolding assay. Markedly, even though there are differences in the inhibitory concentrations against tested leukemia cells between 5b (IC50 in a sub-micromolar range) and reference Hsp90 NTD control inhibitors (IC50 in sub-nanomolar range), a comparative selectivity profile (in cell-free or cell-based biochemical assays) toward Hsp90 was observed between 5b and Hsp90 NTD reference inhibitors. This data indicates that the conventional Hsp90 NTD inhibitors induce cellular inhibitory effects through their off-target activity, in addition to targeting Hsp90.6,21
Moreover, 5b exhibited potent in vitro anticancer activity against a broad spectrum of therapy-resistant leukemia cell lines (including TKI and proteasome inhibitor-resistant) and primary TKI-resistant (BCR-ABL1+) leukemia patient cells. 5b significantly reduced the leukemia burden in the zebrafish xenotransplantation model and induced apoptosis in TKI-resistant BCR-ABL1T315I, BCR-ABL1E255K, or BCR-ABL1M351T mutant cells by destabilizing the BCR-ABL1 expression and, thereby, hampering related downstream signaling cascades without HSR induction. This data collectively established 5b as a first-in-class small-molecule inhibitor that targets the C-terminal dimerization interface.
Conclusion
Through structure-based molecular design, chemical synthesis, a molecular simulations-based prediction of the binding mode, and an evaluation of biochemical affinity, we have developed the first low-molecular-weight compound interfering with the Hsp90 CTD dimerization. The C-terminal Hsp90 inhibitor 5b contains a tripyrimidonamide scaffold and is active against therapy-resistant leukemia cells as well as in a zebrafish xenotransplantation model without exhibiting the prosurvival resistance mechanism HSR.
Methods
Chemical Synthesis
See the Supporting Information for general methods, synthetic protocols, compound characterization, and spectral data (Figures S1–S20).
Expression and Purification of Recombinant Hsp90
Human Hsp90α CTD (563–732 amino acids) recombinant protein purification was performed as previously described.34 Human Hsp90α NTD (amino acids 9–236; Addgene 22481) protein was expressed in E. coli BL21-DE3 cells.86 BL21-DE3 expression strains were grown overnight and used to inoculate LB medium at 37 °C supplemented with 100 μg/mL ampicillin to an OD600 = 0.5–0.8, followed by overnight induction of protein expression with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 25 °C. After induction, cells were harvested by centrifugation at 5000g and lysed using B-PER bacterial protein extraction reagent (ThermoFisher Scientific, Wesel, Germany). GST-tagged Hsp90 CTD and NTD proteins were affinity purified using spin columns (ThermoFisher Scientific) and eluted using glutathione elution buffer. Protein aliquots were made and supplemented with 5% glycerol and stored at −80 °C.
Microscale Thermophoresis (MST)
The labeling of the CTD of Hsp90 (Hsp90 CTD) has been described before.34,36 For a determination of the apparent KD value of Hsp90 CTD and the respective inhibitor, 10 μL of 50 nM labeled Hsp90 CTD in PBS containing 0.5% BSA and 0.1% Tween-20 was mixed with 10 μL of the respective inhibitor in different concentrations ranging from 15 nM to 500 μM and incubated for 15 min at room temperature (RT) in the dark. Thermophoresis of each mixture was induced at 1475 ± 15 nm and measured using a Monolith NT.115 instrument (NanoTemper Technologies GmbH, München, Germany).87 The measurement was repeated three times independently for each sample, and each measurement was performed at 25 °C for 25 s at 70% LED power and 40% infrared laser power. The resulting fluorescence values were plotted against the concentration of the titrant, and the apparent KD was calculated using the KD fit formula Y = E + (A – E)/2 × (T + x + KD – sqrt((T + x + KD)2 – 4 × T × x)) by GraphPad Prism software (GraphPad Software, Inc. La Jolla, CA). T, the concentration value for the labeled Hsp90 CTD, was set to 50 nM.
Autodisplay Dimerization Assay
E. coli strain BL21 (DE3) [B, F–, dcm, ompT, lon, hsdS (rB–mB−), gal, λ(DE3)] pETSH-3 Hsp90 encoding CtxB signal peptide, AIDA-I autotransporter, and Hsp90 was used for the surface display of Hsp90.35,36E. coli strain BL21 (DE3) [B, F–, dcm, ompT, lon, hsdS (rB–mB−), gal, λ(DE3)] pETSH-3 SDH08 encoding CtxB signal peptide, AIDA-I autotransporter, and sorbitol dehydrogenase was used as a control.45 Recombinant bacteria were routinely grown at 37 °C in lysogeny broth (LB) containing carbenicillin (100 μg/mL). Cells were grown to the mid-log phase (OD600 = 0.5), and the protein expression was induced by adding 1 mM IPTG for 16 h at 30 °C. Cells were then washed three times and suspended in PBS to an OD600 = 0.35 in a final volume of 100 μL. Prior to p53-FITC incubation at a final concentration of 1 μM for 1 h at RT in the dark, cells were incubated for 15 min either with or without inhibitor. Subsequently, cells were washed three times with PBS containing 0.1% Tween-20 to avoid unspecific binding. The cellular fluorescence was measured with a FACS Aria III instrument (BD, Heidelberg, Germany) using 488 nm as the excitation wavelength and 530 nm for detection, and a mean fluorescence value of at least 50 000 events for each measurement was determined.
Drug Affinity Responsive Target Stability (DARTS)
A DARTS assay was performed to assess protease protection of Hsp90 protein from thermolysin digestion after incubation of Hsp90i, as described previously.23,36 Briefly, 1 μg of recombinant Hsp90α was incubated on ice for 15 min with 25, 50, and 100 μM 5b, NB, AUY922, and DMSO. After incubation, the samples were treated with thermolysin (1:50 of protein) for 5 min. The reaction was halted by the addition of 50 mM EDTA and later examined by immunoblot analysis.
Luciferase Refolding Assay
A luciferase refolding assay was performed using recombinant firefly luciferase from Photinus pyralis (Sigma-Aldrich, St. Louis, MO; 10 × 1010 units/mg), which was diluted (1:100) in denaturation buffer (25 mM Tricine, pH 7.8, 8 mM MgSO4, 0.1 mM EDTA, 1% Triton X-100, 10% glycerol, and 10 mg/mL BSA) at 38 °C for 8 min.50−52,88 Rabbit reticulocyte lysate (Promega, Madison, WI) was diluted 1:1 by the addition of cold mix buffer (100 mM Tris, pH 7.7, 75 mM Mg(OAc)2, 375 mM KCl, and 15 mM ATP), creatine phosphate (10 mM), and creatine phosphokinase (16 U/mL) and was preincubated at 30 °C with the respective inhibitors and controls for 1 h. Afterward, 1 μL of denatured luciferase or active luciferase (as a control) was added to 20 μL of a rabbit reticulocyte mixture. As a control, denatured or active luciferase was incubated without reticulocyte lysate in buffer containing 20 mM Tris, pH 7.5, 150 mM NaCl, 1% hemoglobin, and 4% BSA. At desired time points, 1.5 μL samples were removed and added to 40 μL of assay buffer (25 mM Tricine, pH 7.8, 8 mM MgSO4, 0.1 mM EDTA, 33 μM DTT, 0.5 mM ATP, and 0.5 mM luciferin), and the luminescence was read using a Spark microplate reader (Tecan). Percent luciferase refolding was determined using luminescence of DMSO at 120 min as 100% and comparing all samples to this value.
Thermal Shift Assay
CTD or NTD of r-Hsp90α (5 μM) protein and inhibitors at the indicated concentrations were mixed together in the assay buffer (1× PBS, pH = 7.5) and were incubated for 2 h. Then, 6× SYPRO orange dye (Sigma-Aldrich) was added to the mixture (20 μL), which binds to the exposed hydrophobic residues of unfolded protein (during thermal exposure) thereby showing an increase in fluorescence.47,89 96-well polymerase chain reaction (PCR) plates and a PCR system (BioRad, CFX Connect real-time system) were used to heat the samples from room temperature to 95 °C in increments of 0.5 °C for 10 s, with the excitation wavelength at 470 nm and emission wavelength at 570 nm. For a determination of protein melting temperature values (Tm), the melting curve for each data set was analyzed by GraphPad Prism 8.0.2 and fitted with the sigmoidal Boltzmann fit. Melting temperatures without the inhibitors were used as a control.
An isothermal shift assay was performed following a procedure similar to that for the thermal shift assay. However, increasing indicated concentrations of the inhibitors (98 nM to 200 μM) were used and were incubated with either C-terminal or N-terminal Hsp90 (5 μM) in the assay buffer (1× PBS, pH = 7.5) for 2 h. The samples were heated to 46 °C after the addition of 6× SYPRO orange dye (Sigma-Aldrich). Percent unfolding was calculated by normalizing to 100% unfolding and comparing to the vehicle control (DMSO).
Cellular Thermal Shift Assay (CETSA)
A CETSA assay was performed as described previously,47−49 with minor modifications. K562 cells were incubated with the indicated inhibitors (or DMSO) for 24 h. Cells were harvested by centrifugation (400g for 5 min at RT) and washed three times with PBS. The pellets were dissolved in PBS and later equally divided into 200 μL PCR tubes. Solutions were heated at the indicated temperature gradient for 3 min (T-Gradient Cycler, Biometra). Aliquots were then snap-frozen in liquid nitrogen and thawed at 25 °C in a thermal cycler (GeneAMP PCR System2700, Applied Biosystems) three times, followed by centrifugation at 10 000g for 20 min at 4 °C. The supernatants were harvested, and protein levels were measured by a quantitative simple western immunoassay (JESS, BioTechne, Minneapolis, MN). Protein levels represented by the area under the curve of the electropherograms were normalized to the lowest temperature set as 0% degradation. ΔTm values for compounds were determined by plotting normalized data using a sigmoid dose curve and nonlinear regression (GraphPad Prism 8.0.2).
Isothermal Dose-Dependent Fingerprint (ITDRF) CETSA
Based on the previously determined IC50 values, K562 cells were treated with the indicated dilution range of inhibitor for 24 h. Then, cells were harvested by centrifugation (400g for 5 min at RT) and washed three times with PBS. The pellets were dissolved in 200 μL of PBS and transferred into PCR tubes (Eppendorf). Results of prior CETSA runs were utilized to determine the optimal melting temperature of 54.1 °C for Hsp90. Samples were heated once at 54.1 °C for 3 min (T-Gradient Cycler, Biometra) and then snap-frozen in liquid nitrogen and thawed at 25 °C in a thermal cycler (GeneAMP PCR System2700, Applied Biosystems) three times, followed by centrifugation at 10 000g for 20 min at 4 °C. The supernatants were harvested, and protein levels were measured by a quantitative simple western immunoassay (JESS, BioTechne, Minneapolis MN). Protein levels are calculated by the area under the curve of the electropherograms.
Simple Western Immunoassay
Fluorescent (5×) master mix, DTT, and biotinylated ladder were prepared following the manufacturer’s instructions (BioTechne). Lysates were diluted with 0.1× sample buffer and mixed 5:1 with fluorescent 5× master mix to obtain a target sample concentration of 0.40 μg/μL per well. Samples were then denatured for 5 min at 95 °C in a PCR cycler (GeneAMP PCR System2700, Applied Biosystems). The assay plate was loaded following the manufacturer’s instructions and centrifuged for 5 min at 1000g at RT. The immunoassay was performed using a 12–230 kDaA separation module with 25 cartridges (SM-W004, BioTechne). Lysates were separated for 25 min at 375 V, blocked for 5 min with antibody diluent 2, and incubated for 30 min with primary antibody and for 30 min with secondary antibody, subsequently. Primary antibody multiplex mix consisted of 1:100 anti-Hsp90 (4877S, Cell Signaling Technology, Danvers, MA) and 1:50 anti-β-actin (MAB8929, R&D) diluted in antibody diluent 2. Signals were detected using a JESS antirabbit detection module (DM-001, BioTechne) multiplexed with an antimouse secondary NIR antibody (043-821, BioTechne).
Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET)
An evaluation of the Hsp90 binding affinity to PPID (cyclophilin D) was performed using the Hsp90 CTD TR-FRET assay kit (50289, BPS Bioscience, San Diego, CA). Terbium (Tb)-labeled donor and dye-labeled acceptor were diluted 100-fold in (1×) Hsp90 assay buffer 2. A 10 μL portion of diluted Tb-labeled donor and 10 μL of diluted dye-labeled acceptor were mixed with 4 μL of inhibitor (at the indicated concentration), 10 μL of 3 ng/μL PPID-GST-tag (BPS Bioscience), and 6 μL of 2 ng/μL biotin-labeled CTD of Hsp90 (BPS Biosciences). For the positive control, the inhibitor was substituted for DMSO, and for the negative control, PPID-GST-tag was substituted for 1× Hsp90 assay buffer. Samples were incubated for 2 h at RT protected from light and measured with a microplate-reader (SPARK10M, Tecan). Fluorescence was measured using a time-resolved reading mode with two subsequent measurements: The first measurement was performed using a 340 nm/620 nm (excitation/emission) wavelength with a lag time of 60 μs and integration time of 500 μs. The second measurement was performed using a 340 nm/665 nm (excitation/emission) wavelength with a lag time of 60 μs and integration time of 500 μs. A data analysis was performed using the TR-FRET ratio (665 nm emission/620 nm emission). The TR-FRET ratios are normalized to % activity by setting the negative control as 0% activity and the positive control as 100% activity
Fluorescence Polarization (FP) Measurements
An evaluation of the binding affinity of compounds toward the ATP pocket of Hsp90 NTD was determined by a competitive binding assay against FITC-labeled geldanamycin (GM) using the Hsp90 NTD assay kit (50293, BPS Bioscience).50 The inhibitor sample wells were filled with 15 μL of 1× Hsp90 assay buffer, 5 μL of 40 mM DTT, 5 μL of 2 mg/mL BSA, 40 μL of H2O, 5 μL of FITC-labeled GM (100 nM), and 10 μL of inhibitor (at the indicated concentration). The reaction was initiated by adding 20 μL of Hsp90 (17 ng/μL) and incubating at room temperature for 3 h with slow shaking. Background wells (master mix only), negative controls (FITC-labeled GM, buffer, and DMSO), and positive controls (FITC-labeled GM, buffer, DMSO, and Hsp90) were also included within the assay plate. Fluorescence was measured at a 470 nm excitation wavelength and 525 nm emission wavelength in a microtiter-plate reader (Infinite M1000pro by Tecan). Polarization was calculated using (III – G(I⊥)/(III + G(I⊥)) × 1000 and a g-factor of 1.187. The percentage of Hsp90-bound FITC-labeled GM was calculated using
Molecular Dynamics (MD) Simulations
The structures of Hsp90α and β (PDB IDs 3q6m and 5fwk, respectively) were prepared using Schrödinger Maestro.90 For each of the isoforms, 40 individual MD simulations were performed. The initial random placement of 5b and solvation in TIP3P water91 was done using PACKMOL,92 neutralizing the system by the addition of sodium ions. ff14SB was used as the force field for the protein and a modified GAFF version 1.5 for 5b.34,56 All simulations were carried out using the Amber18 software package.54 To treat long-range electrostatics, the particle mesh Ewald method93 was used with a cutoff of 9.0 Å for equilibration and 10.0 Å for production. The SHAKE algorithm94 and hydrogen mass repartitioning95 were used to allow for simulation steps of 2 fs in the equilibration and 4 fs in the production.
Initially, the systems were energy-minimized using the steepest descent (500 steps) and conjugate gradient (2000 steps) methods and placing positional restraints with a force constant of 5 kcal mol–1 Å–2 on all protein atoms; the restraints were reduced in a second energy minimization to a force constant of 1 kcal mol–1 Å–2 (for 2000 steps of steepest descent followed by 8000 steps of conjugate gradient) and removed in a third one (for 1000 steps of steepest descent followed by 4000 steps of conjugate gradient). Placing positional restraints with a force constant of 1 kcal mol–1 Å–2 on the backbone atoms, first, the system was heated to 100 K in 50 ps of NVT MD and further heated to 300 K in 5 ps of NPT MD. A further 65 ps of NPT MD was performed for density equilibration, still applying the backbone restraints. Afterward, over the course of a further 300 ps of NPT MD, the backbone restraints were gradually reduced to a force constant of 0.2 kcal mol–1 Å–2.
After the thermalization, 500 ns of NPT MD simulation was carried out with positional restraints with a force constant of 0.1 kcal mol–1 Å–2 on the backbone atoms. The Langevin thermostat with a collision frequency of 1 ps–1 and the Monte Carlo barostat with a pressure relaxation time of 1 ps were used. The reference coordinates for these restraints were readjusted every 100 ns to allow for moderate protein flexibility. For Hsp90α, these simulations were later extended to 1 μs. The trajectories were postprocessed and analyzed with CPPTRAJ,96 and results were visualized with PyMol.86
MM-GB/SA Computations
Effective binding energies were computed over one MD trajectory that resulted in binding of 5b in the CTD dimerization interface and led to a binding mode in which 5b mimics H5′. In the computations, the single trajectory approach was used, where complex, protein, and ligand configurations were extracted from the complex trajectory.97 After removing water molecules and counterions, gas-phase energies (van der Waals and electrostatic contributions) were evaluated on every frame sampled at an interval of 200 ps using MMPBSA.py;98 the polar contribution to the solvation free energy was calculated using the “OBC II” generalized Born model,99 together with mbondi2 radii and a dielectric constant of 1 for the solute and 80 for the solvent. The ionic strength was set to 150 mM of a 1:1 salt. The nonpolar contribution to the solvation free energy was calculated as a function of the solvent-accessible surface area using 0.0050 kcal mol–1 Å–2 as the surface tension. No absolute binding free energies can be derived from this approach since configurational entropy contributions are not considered here to reduce the uncertainty in the computations.57,58 However, the sum of gas-phase energies and solvation free energies, the effective energy, is suitable to estimate relative differences in binding free energies for differential binding poses.36
WB and Blue Native Gels
Cells were treated with the indicated concentration of the compound or vehicle (DMSO) for 48 h. An immunoblot analysis was performed by following a standard protocol using antibodies: anti-Hsp90 (4877), anti-Hsp70 (4872), anti-Hsp40 (4871), anti-Hsp27 (2402), anti-HSF-1 (4356), anti-Hsp60 (12165), anti-PDI (2446), anti-Hsp90α (8165), anti-Hsp90β (5087), anti-c-Abl (2862), antiphospho-c-Abl (2865), anti-Stat5 (9363), antiphospho-Stat5 (9351), anti-CrkL (3182), antiphospho-Crkl (3181), anti-Akt (2920), antiphospho-Akt (4060), anti-S6 ribosomal protein (2217), antiphospho-S6 ribosomal protein (4858), and anti-c-Myc (13987) from Cell Signaling Technology and anti-β-actin (Sigma-Aldrich). Blue native (BN) gels were performed following the manufacturer’s instructions (Invitrogen) and as performed previously.36 Briefly, lysates were generated from the K562 cell line after 48 h of treatment with inhibitors (at the indicated concentration) using a NativePAGE Sample Prep kit (Invitrogen) by 2–3 freezing–thawing cycles followed by centrifugation at 20 000g for 25–30 min at 4 °C.
Dimerization Assay
Hsp90 CTD dimerization was evaluated using an amine-reactive chemical cross-linker bis(sulfosuccinimidyl) suberate (BS3) (Pierce).50,53 Hsp90α CTD protein (2 μM) was diluted in Na2HPO4 (25 mM; pH 7.4) and treated with different concentrations of the inhibitor to make a final volume of 25 μL. The reaction mixture was incubated at RT for 1 h. The amine-reactive cross-linker BS3 was added to a final concentration of 63 μM, and the samples were incubated for 1 h at RT. Cross-linking was quenched by the addition of SDS sample buffer and subsequent heating for 5 min at 95 °C. Samples were run in 12% SDS-PAGE gels followed by Western blotting. Blots were probed with anti-Hsp90 (AC88, Abcam) antibody.
SEC-SAXS
We collected the SEC-SAXS data on beamline BM29 at the ESRF Grenoble.61,62 The BM29 beamline was equipped with a PILATUS 2M detector (Dectris) at a fixed distance of 2.827 m. The measurement of Hsp90 CTD (18 mg/mL) was performed at 20 °C on a Superdex 200 increase 3.2/300 column (Buffer 50 mM TRIS pH 7.5, 100 mM NaCl) with a flow rate of 0.075 mL/min, collecting one frame every two seconds. The data was scaled to absolute intensity against water. Further, we collected SAXS data on our Xeuss 2.0 Q-Xoom system from Xenocs, equipped with a PILATUS 3 R 300 K detector (Dectris) and a GENIX 3D CU ultralow divergence X-ray beam delivery system. The chosen sample to detector distance for the experiment was 0.55 m, resulting in an achievable q-range of 0.10–6 nm–1. All measurements were performed at 20 °C with protein concentrations of 9.7 and 10.8 mg/mL. Compound 5b was added and incubated for 30 min at 20 °C. Samples were injected in the low-noise flow cell (Xenocs) via an autosampler. For each sample, 18 frames with an exposure time of 10 min were collected. Data were scaled to absolute intensity against water. All programs used for data processing were part of the ATSAS software package (version 3.0.3).100 Primary data reduction was performed with the programs CHROMIXS and PRIMUS.63,64 With the Guinier approximation,66 we determine the forward scattering I(0) and the radius of gyration (Rg). The program GNOM101 was used to estimate the maximum particle dimension (Dmax) with the pair-distribution function p(r). Low-resolution ab initio models were calculated with DAMMIF.65 A superimposition of the predicted model was done with the program SUPCOMB.102
Physicochemical Properties of 5b (See the Supporting Information for More Details)
Aqueous Solubility of 5b
The aqueous thermodynamic solubility of 5b was determined in phosphate-buffered saline (PBS, pH 7.4) after 4 and 24 h of incubation time at 25 °C. Ondansetron was used as a reference compound with high solubility of 95 μM. The thermodynamic solubility of 5b was ranging from 4 μM after 4 h to 8 μM after 24 h (n = 2). For detailed information, see Bienta, Enamie Biological Services study reports.
Chemical Stability of 5b
Drug decomposition was determined by high-performance liquid chromatography (HPLC, Method 1): instrument, Knauer HPLC system in combination with a Knauer UV Detector Azura UVD 2.1L; column, KNAUER Eurospher II 100-5 C18, 150 × 4 mm; mobile phase 1, linear gradient (90–0%) of water with 0.1% trifluoroacetic acid; mobile phase 2, linear gradient (10–100%) of acetonitrile with 0.1% of trifluoroacetic acid; run time, 20 min, followed by an isocratic elution with 100% acetonitrile for 10 min; flow rate, 1 mL/min; detection, 254 nm.
5b was dissolved in a mixture of Tween20/ethanol/phosphate buffer pH 7.4 (7/3/90) and the stability monitored over a period of 24 h at 37 °C. After 24 h, almost no decomposition was detected (0.7% drug decomposition, n = 2).
The stability of 5b at acidic pH was determined by dissolving 5b in a mixture of Tween20/ethanol/phosphate buffer pH 2 (7/3/90), and the stability was monitored over a period of 24 h at 37 °C. After 24 h, only slight decomposition was detected (1.3% drug decomposition, n = 2).
In Vitro Metabolic Stability of 5b in Human Liver Microsomes
The metabolic stability screening of 5b in human liver microsomes revealed 91% stability after a 40 min incubation at 37 °C. Propanolol, a reference drug with medium to high metabolic stability showed 74% of the parent compound remaining and therefore demonstrated slightly decreased stability compared with 5b. Calculated results for the intrinsic clearance suggest that 5b (6 μL/(min mg)) is a low-clearance compound with an estimated long half-life (n = 2). For detailed information, see Bienta, Enamie Biological Services study reports.
Cell Culture
K562, KCL22, SUPB15 (BCR-ABL1+ CML/BCP-ALL), Mutz-2 (50 ng/mL SCF), HL-60 (AML), DND41, HPBALL, TALL1, and MOLT4 (T-ALL) leukemic cell lines (DSMZ, Braunschweig, Germany) were cultured in RPMI1640 GlutaMAX (ThermoFisher Scinetific) supplemented with 10–20% FCS (Sigma-Aldrich, St. Louis, MO) and penicillin/streptomycin (Invitrogen, Carlsbad, CA) and maintained at 37 °C with 5% CO2. Normal BA/F3 and BA/F3 cells expressing BCR-ABL1 mutants (T315I, M351T, and E255K) (murine pro B cell line) were cultured in RPMI1640 GlutaMAX (10% FCS) supplemented with or without IL-3 (10 ng/mL), respectively. BA/F3 cells expressing BCR-ABL1 mutants were resistant against imatinib (IM) until ∼10 μM.36 IM-resistant BCR-ABL1+ K562 (K562-IMr), KCL22 (KCL22-IMr), and SUPB15 (SUPB15-IMr) were generated by a gradual increase (1–2.5 μM) in the concentration of IM (Sigma-Aldrich, St. Louis, MO) over a period of 3 months.36,103 Bortezomib (BTZ)-resistant clones (80 nM) of HL60 (HL60-BTZr) were established following a protocol similar to that described to pick IM resistant clones. Primary patient derived CMLCD34+ blast cells were cultured in mononuclear cell medium (PromoCell, Heidelberg, Germany).
Hsp90 CTD/NTD-Targeting Reference Inhibitors
Coumermycin A1 (CA1) and novobiocin (NB) were purchased from Sigma-Aldrich, and Hsp90 NTD-targeting reference control inhibitors, geldanamycin (GM), tanespimycin (TM), and PUH-71, were purchased from Selleckchem.
Viability Assay
Cells were seeded in a white 96-well plate (Corning, NY) with an increasing concentration (50 nM to 25 μM) of inhibitors and respective controls for 48 h. Cell viability was monitored using a Celltiter Glo luminescent assay (based on the ATP quantification), following the manufacturer’s guidelines (Promega).36 IC50 values for compounds were determined by plotting raw data (normalized to controls) using a sigmoid dose curve and nonlinear regression (GraphPad Prism).
Proliferation Assay
Cell proliferation was examined after treatment with the compounds at their indicated concentration with the trypan exclusion method using an automated cell counter (Vi-CELL XR-Beckman Coulter, Fullerton, CA) after every 24 h interval.
Annexin V Staining
For evaluating apoptosis, cells treated with inhibitor for 48 h were stained with annexin V and propidium iodide (PI), following the supplier’s guidelines (Invitrogen, Carlsbad, CA), and later, the stained cells were subjected to FACS (Cytoflex, Beckman Coulter).
Caspase 3/7 Glo Assay
Cells were incubated with the respective inhibitors or control for 48 h, and later, the enzymatic activity of caspase 3/7 was examined (SPARK10M, Tecan) by using a caspase 3/7-dependent Glo assay (absorbance at 405 nm), following the manufacturer’s instructions (Promega).36
Zebrafish
A Zebrafish wild-type TE strain was maintained according to standard protocols and handled in accordance with European Union animal protection directive 2010/63/EU and the local government (Tierschutzgesetz §11, Abs. 1, Nr. 1, husbandry permit 35/9185.46/Uni TÜ). All experiments described in this study were conducted on embryos younger than 5 days postfertilization (dpf), prior to the legal onset of animal life.
Xenotransplantation in Zebrafish Embryos
A xenotransplantation experiment was performed as described previously.73 Briefly, MOLT-4 cells were labeled with a Vybrant CFDA SE cell tracer kit (Invitrogen) following the manufacturer’s instructions and were then suspended in PBS at a density of 1 × 108 cells/mL. An approximately 1 nL portion of cell suspension (around 200 cells) was injected into the perivitelline space of embryos at 32 hpf. Injected embryos were first incubated at 28 °C for 1 h. Only embryos with good engraftment were selected for treatment with DMSO (control group), AUY922 (500 nM), or 5b (500 nM) for 48 h at 35 °C. This temperature enables the maintenance of embryos with grafted cells without compromising zebrafish development. Drug-treated embryos were dissociated by passing through a 40 μm cell strainer (Greiner Bio-One) and then analyzed using a BD LSR II flow cytometer. The fold change of engrafted MOLT4 cells was calculated using the mean of DMSO-treated embryos. GraphPad Prism software (version 7) was used for graphing and statistical analysis.
Differentiation Assay
FACS measurements were performed after incubating the respective inhibitors at the indicated concentration for 48 h using fluorochrome-coupled monoclonal antibodies (mAbs) along with matched isotype controls: anti-CD11b (Bear1; Beckman Coulter), anti-CD14 (RMO52; Beckman Coulter), and anti-CD133 (AC133; MiltenyiBiotec, Gladbach, Germany).46
Colony Forming Unit (CFU) Assay
A CFU assay was performed by plating inhibitor treated cells (48 h) in methylcellulose medium (Methocult H4100, StemCell Technologies, Vancouver, BC, Canada) supplemented with 50 ng/mL SCF, 10 ng/mL IL-3, 10 ng/mL GM-CSF (Peprotech, Hamburg, Germany), and 3 U/mL erythropoietin (eBioscience, San Diego, CA).46 Colonies were counted after 14 days (n = 3).
Significance analyses of normally distributed data with variances similar between groups used a paired, two-tailed Student’s t test: *, p < 0.05; **, p < 0.005; and *** p < 0.001, unless stated otherwise.
Acknowledgments
This study was funded in part by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), 270650915 (Research Training Group GRK 2158: TP4a to H.G., TP4b to S.S., TP2c to T.K., and TP 2d to S.B.). S.B., H.G., and T.K. are supported by Forschungskommission and DSO-Netzwerkverbundes, HHU Düsseldorf. S.B. additionally acknowledges the financial support by KinderKrebsForschung e.V. J.H. and A.B. were supported by the TransOnc priority program of the German Cancer Aid within grant 70112951 (ENABLE). J.H. was supported by ERC Stg 85222 “PreventALL” and ERA PerMED 2018 “GEPARD”. T.K. and H.G. were supported by funds from the Strategischer Forschungsfonds of HHU. The Center for Structural studies is funded by the DFG (Grant 417919780 and INST 208/761-1 FUGG to S.S.). We are grateful for computational support by the “Zentrum für Informations und Medientechnologie” at the Heinrich-Heine-Universität Düsseldorf and the computing time provided by the John von Neumann Institute for Computing (NIC) to H.G. on the supercomputer JUWELS at Jülich Supercomputing Centre (JSC) (user IDs: HKF7, VSK33). A.B. acknowledges the financial support of Katharina-Hardt Foundation and Löwenstern e.V. for providing a Simple Western Instrument (JESS). We thank Anton Popov for the great support at BM29 as well as the whole ESRF Outstation Grenoble Team.
Glossary
Abbreviations
- TLC
thin-layer chromatography
- MOA
mode of action
- MD
molecular dynamics
- MM-GB/SA
molecular mechanics generalized Born surface area
- NTD
N-terminal domain
- MD
middle domain
- CTD
C-terminal domain
- PPIs
protein–protein interactions
- HSR
heat shock response
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.2c00013.
Author Contributions
▲ S.B. and L.S. contributed equally to this work. H.G. and T.K. share senior authorship. Conceptualization: H.G., T.K., S.B., and L.S. Development and application of methodology: D.B., N.D., M.V., B.F., H.P., B.L., V.W., F.L., M.G., J.L., B.B., D.D., N.A., Ba.B., J.S., J.S.-D., F.K.H., J.J., J.H., A.B., S.B., H.G., and T.K. Analysis of data: D.B., B.F., J.L., H.G., S.B., N.D., M.V., V.W., T.K., and L.S. S.S. and J.R. performed and analyzed the SAXS experiment. Manuscript writing: S.B., H.G., T.K., D.B., and V.W. The study was supervised by H.G., T.K., and S.B.
The authors declare no competing financial interest.
Supplementary Material
References
- Sanchez J.; Carter T. R.; Cohen M. S.; Blagg B. S. Old and New Approaches to Target the Hsp90 Chaperone. Current cancer drug targets 2020, 20, 253. 10.2174/1568009619666191202101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopf F. H.; Biebl M. M.; Buchner J. The HSP90 chaperone machinery. Nature reviews. Molecular cell biology 2017, 18 (6), 345–360. 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- Flandrin P.; Guyotat D.; Duval A.; Cornillon J.; Tavernier E.; Nadal N.; Campos L. Significance of heat-shock protein (HSP) 90 expression in acute myeloid leukemia cells. Cell Stress Chaperones 2008, 13 (3), 357–64. 10.1007/s12192-008-0035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Shen A.; Zhang C.; Song Z.; Ai J.; Liu H.; Sun L.; Ding J.; Geng M.; Zhang A. Development of Heat Shock Protein (Hsp90) Inhibitors To Combat Resistance to Tyrosine Kinase Inhibitors through Hsp90-Kinase Interactions. Journal of medicinal chemistry 2016, 59 (12), 5563–86. 10.1021/acs.jmedchem.5b01106. [DOI] [PubMed] [Google Scholar]
- Butler L. M.; Ferraldeschi R.; Armstrong H. K.; Centenera M. M.; Workman P. Maximizing the therapeutic potential of HSP90 inhibitors. Molecular cancer research: MCR 2015, 13 (11), 1445–51. 10.1158/1541-7786.MCR-15-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Koay Y. C.; McAlpine S. R. How Selective are Hsp90 Inhibitors for Cancer Cells over Normal Cells?. ChemMedChem. 2017, 12 (5), 353–357. 10.1002/cmdc.201600595. [DOI] [PubMed] [Google Scholar]
- Wu J.; Liu T.; Rios Z.; Mei Q.; Lin X.; Cao S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38 (3), 226–256. 10.1016/j.tips.2016.11.009. [DOI] [PubMed] [Google Scholar]
- Wang Y.; McAlpine S. R. Heat-shock protein 90 inhibitors: will they ever succeed as chemotherapeutics?. Future Med. Chem. 2015, 7 (2), 87–90. 10.4155/fmc.14.154. [DOI] [PubMed] [Google Scholar]
- Hsu H. S.; Lin J. H.; Huang W. C.; Hsu T. W.; Su K.; Chiou S. H.; Tsai Y. T.; Hung S. C. Chemoresistance of lung cancer stemlike cells depends on activation of Hsp27. Cancer 2011, 117 (7), 1516–28. 10.1002/cncr.25599. [DOI] [PubMed] [Google Scholar]
- Gandhi N.; Wild A. T.; Chettiar S. T.; Aziz K.; Kato Y.; Gajula R. P.; Williams R. D.; Cades J. A.; Annadanam A.; Song D.; et al. Novel Hsp90 inhibitor NVP-AUY922 radiosensitizes prostate cancer cells. Cancer biology & therapy 2013, 14 (4), 347–56. 10.4161/cbt.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi S.; Saura C.; Henderson C.; Lin N. U.; Mahtani R.; Goddard J.; Rodenas E.; Hudis C.; O’Shaughnessy J.; Baselga J. A multicenter trial evaluating retaspimycin HCL (IPI-504) plus trastuzumab in patients with advanced or metastatic HER2-positive breast cancer. Breast cancer research and treatment 2013, 139 (1), 107–13. 10.1007/s10549-013-2510-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Shen A.; Zhang C.; Song Z.; Ai J.; Liu H.; Sun L.; Ding J.; Geng M.; Zhang A. Development of Heat Shock Protein (Hsp90) Inhibitors To Combat Resistance to Tyrosine Kinase Inhibitors through Hsp90-Kinase Interactions. J. Med. Chem. 2016, 59, 5563. 10.1021/acs.jmedchem.5b01106. [DOI] [PubMed] [Google Scholar]
- Bagatell R.; Paine-Murrieta G. D.; Taylor C. W.; Pulcini E. J.; Akinaga S.; Benjamin I. J.; Whitesell L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin. Cancer Res. 2000, 6 (8), 3312–3318. [PubMed] [Google Scholar]
- McCollum A. K.; Teneyck C. J.; Sauer B. M.; Toft D. O.; Erlichman C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer research 2006, 66 (22), 10967–75. 10.1158/0008-5472.CAN-06-1629. [DOI] [PubMed] [Google Scholar]
- Guo F.; Rocha K.; Bali P.; Pranpat M.; Fiskus W.; Boyapalle S.; Kumaraswamy S.; Balasis M.; Greedy B.; Armitage E. S.; et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer research 2005, 65 (22), 10536–44. 10.1158/0008-5472.CAN-05-1799. [DOI] [PubMed] [Google Scholar]
- Maloney A.; Clarke P. A.; Naaby-Hansen S.; Stein R.; Koopman J. O.; Akpan A.; Yang A.; Zvelebil M.; Cramer R.; Stimson L.; et al. Gene and protein expression profiling of human ovarian cancer cells treated with the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer research 2007, 67 (7), 3239–53. 10.1158/0008-5472.CAN-06-2968. [DOI] [PubMed] [Google Scholar]
- Hall J. A.; Forsberg L. K.; Blagg B. S. Alternative approaches to Hsp90 modulation for the treatment of cancer. Future Med. Chem. 2014, 6 (14), 1587–605. 10.4155/fmc.14.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee M.; Hatial I.; Keegan B. M.; Blagg B. S. J. Assay design and development strategies for finding Hsp90 inhibitors and their role in human diseases. Pharmacol Ther 2021, 221, 107747. 10.1016/j.pharmthera.2020.107747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren J. 3rd.; Blagg B. S. J. The Right Tool for the Job: An Overview of Hsp90 Inhibitors. Adv. Exp. Med. Biol. 2020, 1243, 135–146. 10.1007/978-3-030-40204-4_9. [DOI] [PubMed] [Google Scholar]
- Li L.; Wang L.; You Q. D.; Xu X. L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. Journal of medicinal chemistry 2020, 63 (5), 1798–1822. 10.1021/acs.jmedchem.9b00940. [DOI] [PubMed] [Google Scholar]
- Wang Y.; McAlpine S. R. N-terminal and C-terminal modulation of Hsp90 produce dissimilar phenotypes. Chem. Commun. 2015, 51 (8), 1410–3. 10.1039/C4CC07284G. [DOI] [PubMed] [Google Scholar]
- Khandelwal A.; Kent C. N.; Balch M.; Peng S.; Mishra S. J.; Deng J.; Day V. W.; Liu W.; Subramanian C.; Cohen M.; et al. Structure-guided design of an Hsp90beta N-terminal isoform-selective inhibitor. Nat. Commun. 2018, 9 (1), 425. 10.1038/s41467-017-02013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskew J. D.; Sadikot T.; Morales P.; Duren A.; Dunwiddie I.; Swink M.; Zhang X.; Hembruff S.; Donnelly A.; Rajewski R. A.; et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 2011, 11, 468. 10.1186/1471-2407-11-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koay Y. C.; McConnell J. R.; Wang Y.; Kim S. J.; Buckton L. K.; Mansour F.; McAlpine S. R. Chemically accessible hsp90 inhibitor that does not induce a heat shock response. ACS medicinal chemistry letters 2014, 5 (7), 771–6. 10.1021/ml500114p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers R. P.; Alexander L. D.; Johnson V. A.; Lin C. C.; Savage J.; Corral R.; Moss J.; Slugocki T. S.; Singh E. K.; Davis M. R.; et al. Design and synthesis of Hsp90 inhibitors: exploring the SAR of Sansalvamide A derivatives. Bioorganic & medicinal chemistry 2010, 18 (18), 6822–56. 10.1016/j.bmc.2010.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide S. D.; Bosman J. D.; Mbadugha B. N.; Kawahara T. L.; Matsumoto G.; Kim S.; Gu W.; Devlin J. P.; Silverman R. B.; Morimoto R. I. Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 2004, 279 (53), 56053–60. 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- Strocchia M.; Terracciano S.; Chini M. G.; Vassallo A.; Vaccaro M. C.; Dal Piaz F.; Leone A.; Riccio R.; Bruno I.; Bifulco G. Targeting the Hsp90 C-terminal domain by the chemically accessible dihydropyrimidinone scaffold. Chem. Commun. 2015, 51 (18), 3850–3. 10.1039/C4CC10074C. [DOI] [PubMed] [Google Scholar]
- Bickel D.; Gohlke H. C-terminal modulators of heat shock protein of 90 kDa (HSP90): State of development and modes of action. Bioorganic & medicinal chemistry 2019, 27 (21), 115080. 10.1016/j.bmc.2019.115080. [DOI] [PubMed] [Google Scholar]
- Sattin S.; Tao J.; Vettoretti G.; Moroni E.; Pennati M.; Lopergolo A.; Morelli L.; Bugatti A.; Zuehlke A.; Moses M.; et al. Activation of Hsp90 Enzymatic Activity and Conformational Dynamics through Rationally Designed Allosteric Ligands. Chemistry 2015, 21 (39), 13598–608. 10.1002/chem.201502211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Martin C.; Moroni E.; Ferraro M.; Laquatra C.; Cannino G.; Masgras I.; Negro A.; Quadrelli P.; Rasola A.; Colombo G. Rational Design of Allosteric and Selective Inhibitors of the Molecular Chaperone TRAP1. Cell Rep 2020, 31 (3), 107531. 10.1016/j.celrep.2020.107531. [DOI] [PubMed] [Google Scholar]
- Mak O. W.; Sharma N.; Reynisson J.; Leung I. K. H. Discovery of novel Hsp90 C-terminal domain inhibitors that disrupt co-chaperone binding. Bioorg. Med. Chem. Lett. 2021, 38, 127857. 10.1016/j.bmcl.2021.127857. [DOI] [PubMed] [Google Scholar]
- Metz A.; Pfleger C.; Kopitz H.; Pfeiffer-Marek S.; Baringhaus K. H.; Gohlke H. Hot spots and transient pockets: predicting the determinants of small-molecule binding to a protein-protein interface. J. Chem. Inf Model 2012, 52 (1), 120–33. 10.1021/ci200322s. [DOI] [PubMed] [Google Scholar]
- Metz A.; Schanda J.; Grez M.; Wichmann C.; Gohlke H. From determinants of RUNX1/ETO tetramerization to small-molecule protein-protein interaction inhibitors targeting acute myeloid leukemia. J. Chem. Inf Model 2013, 53 (9), 2197–202. 10.1021/ci400332e. [DOI] [PubMed] [Google Scholar]
- Ciglia E.; Vergin J.; Reimann S.; Smits S. H.; Schmitt L.; Groth G.; Gohlke H. Resolving hot spots in the C-terminal dimerization domain that determine the stability of the molecular chaperone Hsp90. PloS one 2014, 9 (4), e96031 10.1371/journal.pone.0096031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bopp B.; Ciglia E.; Ouald-Chaib A.; Groth G.; Gohlke H.; Jose J. Design and biological testing of peptidic dimerization inhibitors of human Hsp90 that target the C-terminal domain. Biochimica et biophysica acta 2016, 1860 (6), 1043–55. 10.1016/j.bbagen.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Bhatia S.; Diedrich D.; Frieg B.; Ahlert H.; Stein S.; Bopp B.; Lang F.; Zang T.; Kroger T.; Ernst T.; et al. Targeting HSP90 dimerization via the C terminus is effective in imatinib-resistant CML and lacks the heat shock response. Blood 2018, 132 (3), 307–320. 10.1182/blood-2017-10-810986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger V.; Ciglia E.; Thoma R.; Vasylyeva V.; Frieg B.; de Sousa Amadeu N.; Kurz T.; Janiak C.; Gohlke H.; Hansen F. K. alpha-Aminoxy Peptoids: A Unique Peptoid Backbone with a Preference for cis-Amide Bonds. Chemistry 2017, 23 (15), 3699–3707. 10.1002/chem.201605100. [DOI] [PubMed] [Google Scholar]
- Spanier L.; Ciglia E.; Hansen F. K.; Kuna K.; Frank W.; Gohlke H.; Kurz T. Design, synthesis, and conformational analysis of trispyrimidonamides as alpha-helix mimetics. Journal of organic chemistry 2014, 79 (4), 1582–93. 10.1021/jo402353z. [DOI] [PubMed] [Google Scholar]
- Verba K. A.; Wang R. Y.; Arakawa A.; Liu Y.; Shirouzu M.; Yokoyama S.; Agard D. A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352 (6293), 1542–7. 10.1126/science.aaf5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu M. G.; Chadli A.; Bouhouche I.; Catelli M.; Neckers L. M. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem. 2000, 275 (47), 37181–37186. 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- Eccles S. A.; Massey A.; Raynaud F. I.; Sharp S. Y.; Box G.; Valenti M.; Patterson L.; Brandon A. D.; Gowan S.; Boxall F.; et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer research 2008, 68 (8), 2850–2860. 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- Yin Z. Y.; Henry E. C.; Gasiewicz T. A. (−)-Epigallocatechin-3-gallate Is a Novel Hsp90 Inhibitor. Biochemistry-Us 2009, 48 (2), 336–345. 10.1021/bi801637q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren K.; Zhang H.; Brekken J.; Huser N.; Powell R. E.; Timple N.; Busch D. J.; Neely L.; Sensintaffar J. L.; Yang Y. C.; et al. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol. Cancer Ther 2009, 8 (4), 921–9. 10.1158/1535-7163.MCT-08-0758. [DOI] [PubMed] [Google Scholar]
- Pacey S.; Gore M.; Chao D.; Banerji U.; Larkin J.; Sarker S.; Owen K.; Asad Y.; Raynaud F.; Walton M.; et al. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest New Drugs 2012, 30 (1), 341–9. 10.1007/s10637-010-9493-4. [DOI] [PubMed] [Google Scholar]
- Jose J.; von Schwichow S. Autodisplay of active sorbitol dehydrogenase (SDH) yields a whole cell biocatalyst for the synthesis of rare sugars. Chembiochem 2004, 5 (4), 491–9. 10.1002/cbic.200300774. [DOI] [PubMed] [Google Scholar]
- Bhatia S.; Reister S.; Mahotka C.; Meisel R.; Borkhardt A.; Grinstein E. Control of AC133/CD133 and impact on human hematopoietic progenitor cells through nucleolin. Leukemia 2015, 29 (11), 2208–20. 10.1038/leu.2015.146. [DOI] [PubMed] [Google Scholar]
- Wang L.; Jiang J.; Zhang L.; Zhang Q.; Zhou J.; Li L.; Xu X.; You Q. Discovery and Optimization of Small Molecules Targeting the Protein-Protein Interaction of Heat Shock Protein 90 (Hsp90) and Cell Division Cycle 37 as Orally Active Inhibitors for the Treatment of Colorectal Cancer. Journal of medicinal chemistry 2020, 63 (3), 1281–1297. 10.1021/acs.jmedchem.9b01659. [DOI] [PubMed] [Google Scholar]
- Liang C.; Hao H.; Wu X.; Li Z.; Zhu J.; Lu C.; Shen Y. Design and synthesis of N-(5-chloro-2,4-dihydroxybenzoyl)-(R)-1,2,3,4-tetrahydroisoquinoline-3-carboxamid es as novel Hsp90 inhibitors. European journal of medicinal chemistry 2016, 121, 272–282. 10.1016/j.ejmech.2016.05.033. [DOI] [PubMed] [Google Scholar]
- Jafari R.; Almqvist H.; Axelsson H.; Ignatushchenko M.; Lundback T.; Nordlund P.; Martinez Molina D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc 2014, 9 (9), 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- Goode K. M.; Petrov D. P.; Vickman R. E.; Crist S. A.; Pascuzzi P. E.; Ratliff T. L.; Davisson V. J.; Hazbun T. R. Targeting the Hsp90 C-terminal domain to induce allosteric inhibition and selective client downregulation. Biochimica et biophysica acta. General subjects 2017, 1861 (8), 1992–2006. 10.1016/j.bbagen.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell J. R.; Alexander L. A.; McAlpine S. R. A heat shock protein 90 inhibitor that modulates the immunophilins and regulates hormone receptors without inducing the heat shock response. Bioorg. Med. Chem. Lett. 2014, 24 (2), 661–6. 10.1016/j.bmcl.2013.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galam L.; Hadden M. K.; Ma Z.; Ye Q. Z.; Yun B. G.; Blagg B. S.; Matts R. L. High-throughput assay for the identification of Hsp90 inhibitors based on Hsp90-dependent refolding of firefly luciferase. Bioorganic & medicinal chemistry 2007, 15 (5), 1939–46. 10.1016/j.bmc.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan R. K.; Mok D.; Ward B. K.; Ratajczak T. Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: evidence that coumarin antibiotics disrupt Hsp90 dimerization. J. Biol. Chem. 2006, 281 (11), 7161–71. 10.1074/jbc.M512406200. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. III; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Gilson M. K.; et al. AMBER 2018; University of California: San Francisco, 2018.
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11 (8), 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25 (9), 1157–74. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Sun H. Y.; Duan L. L.; Chen F.; Liu H.; Wang Z.; Pan P. C.; Zhu F.; Zhang J. Z. H.; Hou T. J. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20 (21), 14450–14460. 10.1039/C7CP07623A. [DOI] [PubMed] [Google Scholar]
- Gohlke H.; Case D. A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 2004, 25 (2), 238–250. 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- Pearl L. H.; Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annual review of biochemistry 2006, 75, 271–94. 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- Wayne N.; Bolon D. N. Dimerization of Hsp90 is required for in vivo function. Design and analysis of monomers and dimers. J. Biol. Chem. 2007, 282 (48), 35386–95. 10.1074/jbc.M703844200. [DOI] [PubMed] [Google Scholar]
- Pernot P.; Theveneau P.; Giraud T.; Fernandes R. N.; Nurizzo D.; Spruce D.; Surr J.; McSweeney S.; Round A.; Felisaz F.; et al. New beamline dedicated to solution scattering from biological macromolecules at the ESRF. Journal of Physics: Conference Series 2010, 247 (1), 012009. 10.1088/1742-6596/247/1/012009. [DOI] [Google Scholar]
- Pernot P.; Round A.; Barrett R.; De Maria Antolinos A.; Gobbo A.; Gordon E.; Huet J.; Kieffer J.; Lentini M.; Mattenet M.; et al. Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat 2013, 20 (4), 660–4. 10.1107/S0909049513010431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjkovich A.; Svergun D. I. CHROMIXS: automatic and interactive analysis of chromatography-coupled small angle X-ray scattering data. Bioinformatics 2018, 34, 1944. 10.1093/bioinformatics/btx846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev P. V.; Volkov V. V.; Sokolova A. V.; Koch M. H. J.; Svergun D. I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. 10.1107/S0021889803012779. [DOI] [Google Scholar]
- Franke D.; Svergun D. I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42, 342–346. 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinier A. Diffraction of x-rays of very small angles-application to the study of ultramicroscopic phenomenon. Annales de Physique 1939, 11, 161–237. 10.1051/anphys/193911120161. [DOI] [Google Scholar]
- Svergun D.; Barberato C.; Koch M. H. J. CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. 10.1107/S0021889895007047. [DOI] [Google Scholar]
- Mahalingam D.; Swords R.; Carew J. S.; Nawrocki S. T.; Bhalla K.; Giles F. J. Targeting HSP90 for cancer therapy. British journal of cancer 2009, 100 (10), 1523–9. 10.1038/sj.bjc.6605066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajapeer K. V.; Baskaran R. Hsp90 Inhibitors for the Treatment of Chronic Myeloid Leukemia. Leukemia research and treatment 2015, 2015, 757694. 10.1155/2015/757694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazenby M.; Hills R.; Burnett A. K.; Zabkiewicz J. The HSP90 inhibitor ganetespib: A potential effective agent for Acute Myeloid Leukemia in combination with cytarabine. Leukemia research 2015, 39 (6), 617–24. 10.1016/j.leukres.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata Y.; Nakamoto H.; Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Current pharmaceutical design 2013, 19 (3), 347–65. 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Xiao L.; Wang L.; Ruden D. M. Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochemical pharmacology 2012, 83 (8), 995–1004. 10.1016/j.bcp.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima T.; Krahl A. C.; Nasri M.; Xu Y.; Aghaallaei N.; Findik B.; Klimiankou M.; Ritter M.; Hartmann M. D.; Gloeckner C. J.; et al. LMO2 activation by deacetylation is indispensable for hematopoiesis and T-ALL leukemogenesis. Blood 2019, 134 (14), 1159–1175. 10.1182/blood.2019000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz K. R.; Bowman W. P.; Aledo A.; Slayton W. B.; Sather H.; Devidas M.; Wang C.; Davies S. M.; Gaynon P. S.; Trigg M.; et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2009, 27 (31), 5175–81. 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons D. L.; Pricl S.; Posocco P.; Laurini E.; Fermeglia M.; Sun H.; Talpaz M.; Donato N.; Quintas-Cardama A. Molecular dynamics reveal BCR-ABL1 polymutants as a unique mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy. Proc. Natl. Acad. Sci. U.S.A. 2014, 111 (9), 3550–5. 10.1073/pnas.1321173111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariad suspends ponatinib sales. Cancer discovery 2014, 4 ( (1), ), 6–7. 10.1158/2159-8290.CD-NB2013-163. [DOI] [PubMed] [Google Scholar]
- Blagosklonny M. V.; Toretsky J.; Neckers L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene 1995, 11 (5), 933–939. [PubMed] [Google Scholar]
- Chavany C.; Mimnaugh E.; Miller P.; Bitton R.; Nguyen P.; Trepel J.; Whitesell L.; Schnur R.; Moyer J.; Neckers L. p185erbB2 binds to GRP94 in vivo. Dissociation of the p185erbB2/GRP94 heterocomplex by benzoquinone ansamycins precedes depletion of p185erbB2. J. Biol. Chem. 1996, 271 (9), 4974–7. 10.1074/jbc.271.9.4974. [DOI] [PubMed] [Google Scholar]
- Stepanova L.; Leng X.; Parker S. B.; Harper J. W. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes & development 1996, 10 (12), 1491–502. 10.1101/gad.10.12.1491. [DOI] [PubMed] [Google Scholar]
- D’Annessa I.; Raniolo S.; Limongelli V.; Di Marino D.; Colombo G. Ligand Binding, Unbinding, and Allosteric Effects: Deciphering Small-Molecule Modulation of HSP90. J. Chem. Theory Comput 2019, 15 (11), 6368–6381. 10.1021/acs.jctc.9b00319. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin C.; Serapian S. A.; Colombo G.; Rasola A. Dynamically Shaping Chaperones. Allosteric Modulators of HSP90 Family as Regulatory Tools of Cell Metabolism in Neoplastic Progression. Front Oncol 2020, 10, 1177. 10.3389/fonc.2020.01177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soti C.; Racz A.; Csermely P. A Nucleotide-dependent molecular switch controls ATP binding at the C-terminal domain of Hsp90. N-terminal nucleotide binding unmasks a C-terminal binding pocket. J. Biol. Chem. 2002, 277 (9), 7066–75. 10.1074/jbc.M105568200. [DOI] [PubMed] [Google Scholar]
- Frieg B.; Gremer L.; Heise H.; Willbold D.; Gohlke H. Binding modes of thioflavin T and Congo red to the fibril structure of amyloid-beta(1–42). Chem. Commun. 2020, 56 (55), 7589–7592. 10.1039/D0CC01161D. [DOI] [PubMed] [Google Scholar]
- Milic D.; Dick M.; Mulnaes D.; Pfleger C.; Kinnen A.; Gohlke H.; Groth G. Recognition motif and mechanism of ripening inhibitory peptides in plant hormone receptor ETR1. Sci. Rep 2018, 8 (1), 3890. 10.1038/s41598-018-21952-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohlke H.; Hergert U.; Meyer T.; Mulnaes D.; Grieshaber M. K.; Smits S. H.; Schmitt L. Binding region of alanopine dehydrogenase predicted by unbiased molecular dynamics simulations of ligand diffusion. J. Chem. Inf Model 2013, 53 (10), 2493–8. 10.1021/ci400370y. [DOI] [PubMed] [Google Scholar]
- Fontana J.; Fulton D.; Chen Y.; Fairchild T. A.; McCabe T. J.; Fujita N.; Tsuruo T.; Sessa W. C. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ. Res. 2002, 90 (8), 866–73. 10.1161/01.RES.0000016837.26733.BE. [DOI] [PubMed] [Google Scholar]
- Seidel S. A.; Dijkman P. M.; Lea W. A.; van den Bogaart G.; Jerabek-Willemsen M.; Lazic A.; Joseph J. S.; Srinivasan P.; Baaske P.; Simeonov A.; et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 2013, 59 (3), 301–15. 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulasiraman V.; Matts R. L. Luciferase renaturation assays of chaperones and chaperone antagonists. Methods Mol. Biol. 1998, 102, 129–41. 10.1385/0-89603-520-4:129. [DOI] [PubMed] [Google Scholar]
- Lo M. C.; Aulabaugh A.; Jin G.; Cowling R.; Bard J.; Malamas M.; Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332 (1), 153–9. 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2018-1: Maestro; Schrödinger, LLC: New York, NY, 2018.
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Martinez L.; Andrade R.; Birgin E. G.; Martinez J. M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30 (13), 2157–2164. 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98 (12), 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23 (3), 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Hopkins C. W.; Le Grand S.; Walker R. C.; Roitberg A. E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput 2015, 11 (4), 1864–74. 10.1021/ct5010406. [DOI] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9 (7), 3084. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Homeyer N.; Gohlke H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inform 2012, 31 (2), 114–22. 10.1002/minf.201100135. [DOI] [PubMed] [Google Scholar]
- Miller B. R. 3rd; McGee T. D. Jr.; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput 2012, 8 (9), 3314–21. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Onufriev A.; Bashford D.; Case D. A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55 (2), 383–94. 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- Manalastas-Cantos K.; Konarev P. V.; Hajizadeh N. R.; Kikhney A. G.; Petoukhov M. V.; Molodenskiy D. S.; Panjkovich A.; Mertens H. D. T.; Gruzinov A.; Borges C. ATSAS 3.0: expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54 (1), 343. 10.1107/S1600576720013412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D. I. Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. J. Appl. Crystallogr. 1992, 25, 495–503. 10.1107/S0021889892001663. [DOI] [Google Scholar]
- Kozin M. B.; Svergun D. I. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001, 34, 33–41. 10.1107/S0021889800014126. [DOI] [Google Scholar]
- Mahon F. X.; Deininger M. W.; Schultheis B.; Chabrol J.; Reiffers J.; Goldman J. M.; Melo J. V. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood 2000, 96 (3), 1070–1079. 10.1182/blood.V96.3.1070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.