

Abstract
Heparan sulfate (HS) is a cell surface polysaccharide recently identified as a coreceptor with the ACE2 protein for the S1 spike protein on SARS-CoV-2 virus, providing a tractable new therapeutic target. Clinically used heparins demonstrate an inhibitory activity but have an anticoagulant activity and are supply-limited, necessitating alternative solutions. Here, we show that synthetic HS mimetic pixatimod (PG545), a cancer drug candidate, binds and destabilizes the SARS-CoV-2 spike protein receptor binding domain and directly inhibits its binding to ACE2, consistent with molecular modeling identification of multiple molecular contacts and overlapping pixatimod and ACE2 binding sites. Assays with multiple clinical isolates of SARS-CoV-2 virus show that pixatimod potently inhibits the infection of monkey Vero E6 cells and physiologically relevant human bronchial epithelial cells at safe therapeutic concentrations. Pixatimod also retained broad potency against variants of concern (VOC) including B.1.1.7 (Alpha), B.1.351 (Beta), B.1.617.2 (Delta), and B.1.1.529 (Omicron). Furthermore, in a K18-hACE2 mouse model, pixatimod significantly reduced SARS-CoV-2 viral titers in the upper respiratory tract and virus-induced weight loss. This demonstration of potent anti-SARS-CoV-2 activity tolerant to emerging mutations establishes proof-of-concept for targeting the HS–Spike protein–ACE2 axis with synthetic HS mimetics and provides a strong rationale for clinical investigation of pixatimod as a potential multimodal therapeutic for COVID-19.
Short abstract
Heparan sulfate (HS) has emerged as a SARS-CoV-2 coreceptor. Pixatimod (PG545), an HS mimetic, inhibits infectivity of multiple variants offering a novel therapeutic approach against COVID-19.
Introduction
The coronavirus disease 2019 (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has, according to the World Health Organisation, recently surpassed 410 million confirmed cases and 5.8 million deaths worldwide. Although vaccines against COVID-19 are currently being developed and deployed, given the severe pathophysiology induced by SARS-CoV-2,1 there is a clear need for therapeutic alternatives to alleviate and stop the COVID-19 epidemic that complement vaccination campaigns. Heparan sulfate (HS) is a highly sulfated glycosaminoglycan found on the surface of most mammalian cells which is used by many viruses as an entry receptor or coreceptor,2 including coronaviruses.3 Various compounds that mimic cellular HS such as clinically used heparins have been investigated and have been shown to block infectivity and cell-to-cell spread in a multitude of different viruses, including SARS-associated coronavirus strain HSR1.4 The glycosylated spike (S) protein of SARS-CoV-2 mediates host cell infection via binding to a receptor protein, angiotensin-converting enzyme 2 (ACE2).5 An analysis of the sequence and experimentally determined structures of the S protein reveals that the receptor binding domain (RBD) of the S1 subunit contains an HS binding site. Recent studies have clearly demonstrated binding of heparin and HS to S1 RBD,6−9 including induction of significant conformational change in the S1 RBD structure,6 and studies have also revealed that HS is a coreceptor with ACE2 for SARS-CoV-2.10 Collectively these data strongly suggest that blocking these interactions with heparins and HS mimetics has potential as an effective strategy for COVID-19 therapy. Although heparins have major potential for repurposing for such applications, limitations in the global supply of natural product heparins will greatly restrict its availability,11 highlighting an urgent need to find synthetic alternatives.
Pixatimod (PG545) is a clinical-stage HS mimetic (based on a chemically 2,3,6-O-sulfated glucose tetrasaccharide moiety with a cholestanol aglycone tail; Figure 1A) with potent anticancer12,13 and anti-inflammatory properties.14 However, significant antiviral and virucidal activity for pixatimod has also been reported against a number of viruses that use HS as an entry receptor with EC50’s ranging from 0.06 to 14 μg/mL. This includes HSV-2,15 HIV,16 RSV,17 Ross River, Barmah Forest, and Chikungunya viruses18 as well as Dengue virus.19In vivo efficacy has been confirmed in a prophylactic mouse HSV-2 genital infection model,15 a prophylactic Ross River virus mouse model,18 and a therapeutic Dengue virus mouse model.19 Pixatimod has been evaluated in a Phase Ia clinical trial in patients with advanced solid tumors where it demonstrated a tolerable safety profile and some evidence of disease control.13 It has been safely administered to over 80 cancer patients in Phase I studies as a monotherapy or in combination with nivolumab (NCT02042781 and ACTRN12617001573347), prompting us to examine its antiviral activity against SARS-CoV-2.
Figure 1.

Molecular dynamics modeling defines direct interactions of pixatimod with S1 RBD: (A) Structure of pixatimod. (B) Model (pose-a) showing interactions of pixatimod on the Coulombic surface of the RBD domain of spike protein. The sulfated tetrasaccharide partially occupies the HS/heparin binding site I. (C) Model (pose-b) showing interactions of pixatimod with the RBD domain of spike protein wherein the sulfated tetrasaccharide partially occupies the HS/heparin binding site III. The RBD is rendered using the Coulombic surface whereas the ligand is shown as a stick. (D) The lipophilic tail of pixatimod in both models wraps around the hydrophobic residues, thereby creating a steric clash with the helix of ACE2 protein. (E) The RBD is shown as a ribbon colored from the N- to C-terminal (blue to red). The residues of RBD (shown as a ball and stick) responsible for binding to ACE2 (shown in a light blue ribbon) are labeled. Pixatimod uses similar residues from the receptor binding motif on the RBD and thereby inhibits binding of ACE2. The Coulombic surface was rendered using UCSF Chimera coloring defaults: ε = 4r, thresholds ±10 kcal/mol. Red corresponds to negative charges, white to neutral, and blue to positive charges. Hydrogens are not shown for clarity.
Here, we provide evidence of a direct and destabilizing interaction of pixatimod with the S1 spike protein RBD, supported by molecular modeling data. Pixatimod was able to inhibit the interaction of S1-RBD with ACE2 and also A549 and Vero cells which are known to express the ACE2 receptor, indicating a direct mechanism of action. We established that pixatimod is a potent inhibitor of attachment and invasion of Vero cells and human bronchial epithelial cells by multiple clinical isolates of live SARS-CoV-2 virus, and pixatimod reduces its cytopathic effect at concentrations within the known therapeutic range of this drug. Importantly, pixatimod also retained potency against emerging variants of concern. Finally, we showed that pre-exposure prophylactic treatment resulted in a significant reduction of SARS-CoV-2 viral titers and virus-induced weight loss in the K18-hACE2 mouse model of severe infection and disease. Our data demonstrate that synthetic HS mimetics can target the HS–Spike protein–ACE2 axis to inhibit SARS-CoV-2 infection. They provide strong support for clinical investigation of the potential of pixatimod as a novel therapeutic intervention for prophylaxis and treatment of COVID-19 and have implications for wider applications against other HS-binding viruses and emerging global viral threats.
Results and Discussion
Modeling of Pixatimod–Spike Protein Interactions
We initially used molecular docking to map the potential binding sites of pixatimod (Figure 1A) on the S1 RBD surface of monomeric spike. Results of clustering analyses on the docked poses indicate two prominent clusters involving previously reported heparin binding sites I and III.8 The representative poses (referred to as pose-a and pose-b) of the clusters are shown in Figure 1B,C. The sulfates in the tetrasaccharide in pose-a form ionic interactions and hydrogen bonds with Arg346, Asn354, Arg355, Lys356, and Arg466 at heparin binding site I. The side-chains of residues Arg403, Arg408, Gln409, Lys417, Gln493, Gln498, Asn501, and Tyr505 of site III form ionic interactions and hydrogen bonds with the sulfates of pixatimod in pose-b. The cholestanol in both poses interacts with Tyr449, Leu452, Ser494, and Phe490 (Figure 1D). A total of 24 unique residues of RBD are known to interact with ACE2 based on the X-ray structures. Interestingly, a few of these residues (Lys417, Tyr449, Phe490, Gln498, Asn501, and Tyr505) are also predicted to be involved in binding to pixatimod (Figure 1E).
To investigate which of pose-a or pose-b is the more stable conformation, MD simulations of RBD and pixatimod were carried out in combination with relative free energy calculations using the molecular mechanics–generalized Born (GB) equation (MM/GBSA; ref (20)). The evaluation of pixatimod interactions with S1 RBD at heparin binding site I resulted in an MM/GBSA interaction energy of −21.8 kcal/mol (Table S1). This is less than what is obtained for pose-b (−36.3 kcal/mol). We found that, in both cases, van der Waals (vdw) forces contribute significantly compared to electrostatics toward the total free energy of binding. Heparin binding site III prefers heparin dp <6 which explains the stability of the sulfated tetrasaccharide of pixatimod (pose-b). Based on our previous modeling studies of heparin oligosaccharides, site I has a preference for heparin octasaccharides or longer and therefore helps explain the predicted lower affinity of pixatimod for this site.
We analyzed the per-residue (Figure S1) and pairwise energy decomposition (Figures S2 and S3) between S1 RBD and pixatimod in the complex. Amino acids making significant interactions with pixatimod were identified on the basis of their individual contributions to the total interaction energy, considering only the residues that contribute less than −1.0 kcal/mol. The decomposition approach was helpful for locating residues of the RBD domain such as Arg346, Asn354, Lys356, and Lys444 that transiently interact to form hydrogen bonds or ionic interactions with the sulfated tetrasaccharide moiety of pixatimod (Figures S1 and S2). The free energy of binding for residues Arg346, Lys444, and Tyr449 is favorable (less than −5 kcal/mol). The cholestanol residue also formed stabilizing interactions with Tyr449, Asn450, Leu452, and Gln493. Unlike heparin octasaccharides, pixatimod does not form interactions with Arg355, Arg357, Asn360, and Arg466 which explains the lower stability of pose-a of pixatimod at site I.
The favorable interactions of pixatimod pose-b with RBD heparin binding site III were examined and include residues Arg403, Glu406, Arg408, Gln409, Lys417, Tyr449, Leu452, Phe490, Gln493, Ser494, Tyr495, Gln498, Asn501, Gly502, and Tyr505. The strength of the interactions of these residues is in the range from −1.0 to −20 kcal/mol (Figures S1 and S3). The residues Asp405–Glu406 poorly contribute to the pixatimod–RBD interaction. Overall, our modeling data strongly support the notion of direct binding of pixatimod via multiple amino acid contacts in S1 RBD at heparin binding site III, predicting interference with binding to ACE2 (see the Supporting Information, Video S1).
At the time of writing this work, cryoelectron microscopy structural and molecular modeling studies have revealed the presence of a cryptic pocket in the SARS-CoV-2 spike trimer required for the binding of free fatty acid linoleic acid, vitamins, retinoids, and steroids.21,22 Molecules like linoleate and dexamethasone stabilize the locked spike conformation and thus are unlikely to interact with ACE2. It is possible that the cholestanol moiety of pixatimod could bind to the cryptic pocket in one of the monomers of the trimer whereas the tetrasaccharide might interact at heparin binding site III of the neighboring S1 RBD; however, to study this possibility is beyond the scope of this work in the absence of a structure of the open conformation of spike trimer.
Pixatimod Interacts with Spike Protein
Spectroscopic studies employing circular dichroism (CD) were undertaken to investigate the direct binding of pixatimod to the Spike protein RBD (Figure 2A,B), the region which interacts with the ACE2 receptor on human cells. CD spectroscopy in the far UV region (λ = 190–260 nm) detects conformational changes that occur in the protein secondary structure in solution and can infer binding by an added ligand. Such secondary structural changes can be quantified using spectral deconvolution. The mammalian-expressed S1-RBD (mS1 RBD) underwent a conformational change in the presence of either pixatimod or heparin as a comparator sulfated molecule, known to bind the RBD,6−10 consisting of an increased α-helical content for pixatimod, in contrast to the decrease that is observed for heparin (Figure 2C). Furthermore, a decrease in turn structure is seen for pixatimod, while for heparin this is less prominent (Figure 2C); minimal changes in beta-sheet structures are observed (Figure 2C). A pronounced secondary structure change is also observed upon the binding of pixatimod to an Escherichia coli-expressed RBD (EcS1 RBD), although differences can be seen between the secondary structures, potentially due to the lack of glycosylation on the ecS1 RBD and/or the need to refold the bacterial RBD variant, which has been produced via inclusion bodies.
Figure 2.

Pixatimod interacts with SARS-CoV-2 S1-RBD and inhibits binding to cells and the ACE2 receptor. (A) Circular dichroism spectra (190–260 nm) of SARS-CoV-2 m(His)S1-RBD alone (black), or with pixatimod (blue) or heparin (red). The vertical dotted line indicates 193 nm. (B) The same spectra expanded between 200 and 240 nm. Vertical dotted lines indicate 222 and 208 nm. (C) Secondary structure content analyzed using BeStSel for SARS-CoV-2 m(His)S1-RBD (analyses using BeStSel were performed on smoothed data between 200 and 260 nm). (D) Differential scanning fluorimetry of binding of pixatimod (blue, 10 μg) or heparin (red, 10 μg) to mS1-RBD (1 μg, black line, protein-only control). *Tm values for RBD alone (48.4 °C, SD = 0.3) and in the presence of pixatimod (39.3 °C, SD = 1) were statistically different, t(4) = 15.25, p = 0.0001. (E) Effects of pixatimod and unfractionated porcine mucosal heparin on binding of mammalian expressed mS1-RBD-monomeric Fc to A549 human lung epithelial cells. Data were normalized to the control with no addition of mammalian expressed mS1-RBD-mFc (n = 3 ± CV). (F) Competitive ELISA assay using biotinylated human ACE2 protein immobilized on streptavidin coated plates to measure the inhibition of binding of mS1-RBD in the presence of various concentrations of inhibitor compounds. Pixatimod (IC50, 10.1 μg/mL) and porcine mucosal heparin (IC50, 24.6 μg/mL). n = 3, ±SD; representative example shown.
We explored the effects of pixatimod on protein stability using differential scanning fluorimetry (DSF) in which the thermal denaturation of a protein is monitored in the presence of a hydrophobic fluorescent dye (see the Materials and Methods section). Binding of pixatimod induced a notably large reduction in melting temperature (ΔTm) of 9.1 °C (Figure 2D; p = 0.0001), indicating major destabilization of the mammalian expressed S1-RBD (mS1 RBD) protein. In contrast, heparin at an equivalent dose only partially destabilized the RBD protein, evidenced by a small side peak shifted by ∼5–6 °C indicating populations of RBD in a bound and unbound state (Figure 2D).
The observed changes demonstrate that the SARS-CoV-2 S1 RBD interacts with pixatimod in aqueous conditions of physiological relevance. Notably, the conformational changes and destabilization observed were distinct for pixatimod compared to heparin, suggesting distinct interactions (Figure 2). Consistent with the modeling results, these data confirm direct interactions of pixatimod with S1 RBD, resulting in induction of a conformational change, consistent with the notion that HS mimetics such as pixatimod have the potential to interfere with the interaction of S1-RBD with ACE2.
Pixatimod Inhibits S1-RBD Cell Binding
We next evaluated inhibition of binding of mammalian expressed mS1-RBD-monomeric Fc to A549 human lung epithelial cells [which are known to express both HS proteoglycans (HSPGs) and the ACE2 protein receptor required for SARS-CoV-2 attachment and cell invasion]. Fixed cells were exposed to mS1-RBD-Fc for 1 h in the presence or absence of additional compounds with subsequent washing and detection by ELISA for the Fc tag. Inhibition of binding was observed for both pixatimod and heparin as a comparator compound with 78% and 52% inhibition achieved at 100 μg/mL, respectively. Data were normalized to the control with no addition of mS1-RBD. Comparable data were obtained for E. coli-expressed EcS1-RBD binding to monkey Vero cells and also demonstrated a clear dose–response relationship (Figure S5). These data confirm that pixatimod can interfere with the binding of S1-RBD (both glycosylated and nonglycosylated) to cells containing HSPGs and ACE2 protein receptors.
Pixatimod Directly Inhibits S1-RBD Binding to ACE2
To further evaluate the mechanism of action of pixatimod, its direct effects on the interaction of S1-RBD with the ACE2 protein receptor were measured using a competitive ELISA assay. Inhibition of binding of mS1-RBD preincubated with various concentrations of inhibitor compounds was measured by detection with an anti-RBD antibody. A dose response was observed with pixatimod showing an IC50 of 10.1 μg/mL (Figure 2F). In comparison, heparin also demonstrated inhibitory activity but with lower potency (24.6 μg/mL; Figure 2F). Importantly, these data confirm a direct mechanism of action of pixatimod via inhibition of S1-RBD binding to the ACE2 protein receptor, as previously noted for heparin.23
Pixatimod Inhibits SARS-CoV-2 Infection
The effect of pixatimod on SARS-CoV-2 infection of Vero E6 cells was examined using a standard plaque reduction neutralization assay. Pixatimod was preincubated with the SARS-CoV-2 clinical isolate from Victoria, Australia (VIC01), for 1 h before infecting the cells. Significant decreases were observed in the number of plaque forming units (PFU) after pixatimod treatment of SARS-CoV-2 (Figure 3A). An analysis of multiple dose response curves yielded an EC50 for pixatimod in the range 2.4–13.8 μg/mL (mean 8.1 μg/mL; n = 3 assays) (Table 1). In comparison, an EC50 of ∼10 μg/mL has been observed for unfractionated heparin (average Mw ∼ 12 000 Da) with a SARS-CoV-2 Italy UniSR1/2020 isolate8 and 25–41 μg/mL for the SARS-CoV-2 VIC01 isolate.23 Comparisons on a weight rather than molar basis are considered reasonable since unfractionated heparin chains are much longer (on average 30–40 monosaccharide units) and thus contain multiple binding sites for spike protein (compared to 4 units for pixatimod sulfated oligosaccharide with Mw 2075 Da).
Figure 3.

Pixatimod inhibits infection of Vero E6 and human bronchial epithelial cells with different SARS-CoV-2 virus isolates. Live virus infectivity assays were performed as described in the Materials and Methods section for 3 different SARS-CoV-2 isolates (representative data shown). (A) Plaque reduction neutralization assay of Victoria isolate (VIC01) (EC50 8.1 μg/mL; n = 3, ±SD). (B) Plaque reduction assay of DE isolate, EC50 2.7 μg/mL; n = 3, ±SD. (C) Cytopathic assay of Queensland isolates, EC50 13.2 (QLD02) and 0.9 (QLD935 with D614G mutation) μg/mL n = 6, ±SEM. Representative examples are shown in each case. Results of pixatimod inhibition of SARS-CoV-2 infectivity are expressed as percent plaque reduction (A), plaque number as a percent of control (B), or percent inhibition from the cytopathic effect (C). Panel B also shows cytotoxicity data for Vero cells for a calculation of the CC50 value (>236 μg/mL). In panel C, data is also shown for octyl β-maltotetraoside tridecasulfate (Octyl-βMTTS; Figure S2), an analogue of pixatimod which lacks the steroid side-chain. (D) Plaque assay to measure the inhibitory effect of pixatimod on viral shedding in BCi-NS1.1 human bronchial epithelial cells grown in an air liquid interface (ALI). ALI cultures were infected for 2 h with SARS-CoV-2 (VIC01, MOI = 0.2) previously preincubated for 1 h at 37 °C with 0.4, 4, and 40 μg/mL pixatimod or HBSS for untreated. After 72 h, an apical rinse was performed with 200 μL of HBSS, and 100 μL of the wash was used in a plaque assay in Vero E6 cells. Values are expressed as plaque forming unit (PFU)/mL, n = 2, ±SD. (E) Cytotoxicity data for BCi-NS1.1 human bronchial epithelial cells. The CC50 value for pixatimod is >300 μg/mL. Values correspond to the mean of n = 3 independent experiments with technical duplicates, ±SEM.
Table 1. Anti-SARS-CoV-2 Activities of Pixatimod in Vero E6 Cells.
| SARS-CoV-2 isolate | assay method | EC50a (μg/mL) | CC50 (μg/mL) | selectivity indexa |
|---|---|---|---|---|
| VIC01 isolate | plaque reduction | 8.1 ± 3.1 (2.4–13.8)a | >236d ≫300e |
29 (>17 to >98a) |
| DE-Gbg20 isolate | plaque reduction | 2.7b (0.8–11.6c) | >87 | |
| cytopathic effect | 2.7b (0.8–11.6c) | >20 to >295 | ||
| QLD02 isolate | cytopathic effect | 13.2 (8.0–21.6) | >17 | |
| QLD935 isolate | cytopathic effect | 0.9 (0.4–1.9) | >200 |
Mean values, individual assay result ranges, and resulting selectivity index ranges in brackets.
Mean EC50 ± SE based on the data from three independent virus plaque reduction assays (PRNT50 values).
EC50 computed by the Reed and Muench formula based on the cytopathic effect assay. Range indicates upper (complete protection of cells) and lower (partial protection) limits of EC50 estimation.
To establish that these antiviral effects were relevant for wider clinical viral isolates, assays were conducted with the isolate DE-Gbg20 from Sweden in a plaque reduction assay. Pixatimod inhibited infectivity of the DE-Gbg20 isolate with an EC50 value of 2.7 μg/mL (Figure 3B), similar to that found in experiments with the VIC01 isolate. An analysis of pixatimod cytotoxicity for Vero cells using a tetrazolium-based assay revealed that pixatimod decreased by 50% (CC50) the viability of Vero cells at a concentration >236 μg/mL, i.e., well above the EC50 values observed in the plaque reduction assay (Figure 3B; Table 1). Selectivity index (SI) values for pixatimod ranged from >17 to >98 for these assays.
In addition to the plaque reduction assays, pixatimod inhibition of SARS-CoV-2 infectivity was assessed using assays that measured the cytopathic effects of the virus as an end point. Using the Swedish DE-Gbg20 isolate, and two Australian isolates from Queensland (QLD02 and QLD935), the EC50 values for pixatimod inhibition of SARS-CoV-2 infectivity were determined to be 0.8–11.6, 13.2, and 0.9 μg/mL, respectively (Table 1), values comparable with those observed for the plaque reduction assays (Table 1). We also noted that a pixatimod analogue octyl β-maltotetraoside tridecasulfate (without the steroid side-chain) (Figure S6) lacked efficacy for both QLD02 and QLD935 isolates (Figure 3C), demonstrating the importance of the steroid side-chain for activity and supporting the notion of the sterol moiety promoting RBD switching position and enhancing availability of the heparin binding site. Notably, both DE-Gbg20 and QLD935 isolates contain the D614G mutation of the spike protein commonly present in recent isolates (Table S2).24 The QLD935 isolate exhibited lower cytopathicity, which could partially contribute to the observed lower EC50 for pixatimod against this isolate.
To provide evidence of antiviral properties of pixatimod in a more physiologically relevant model for infection of human cells, we used a bronchial airway epithelial in vitro system of SARS-CoV-2 infection and replication. The hTERT transformed bronchial epithelial cell line BCi-NS1.1 differentiates into airway multiciliated cells when grown in Transwells at the air–liquid interface (ALI), cultures which show robust infection with SARS-CoV-2 when harvested 72 h after inoculation.25,26 To determine if pixatimod has inhibitory effects on SARS-CoV-2 infection and replication in this model, the virus was preincubated with 0.4, 4, or 40 μg/mL pixatimod (or HBSS for untreated) for 1 h at 37 °C before the inoculum was added to the apical side of the cells. We examined infectious virus in the apical wash from BCI ALI cultures by plaque assay on Vero E6 cells. Viral shedding of ALI cultures infected with SARS-CoV-2 in the presence of pixatimod was essentially abolished in comparison to the cells infected with the untreated virus as a control (Figure 3D), especially at the lower doses of 0.4 and 4 μg/mL pixatimod (Figure 3D). Thus, the inhibitory potency of pixatimod against infection of these bronchial cells is apparently higher than for Vero cells. We investigated the possibility that this might be related to cytotoxicity, but this was excluded by demonstration of a high CC50 for these cells (>300 μg/mL) (Figure 3E). Overall, these data in a physiologically relevant human bronchial cell model show that pixatimod is a potent inhibitor of SARS-CoV-2 in terms of lowering both viral shedding in the apical compartment and viral load in the cell layer.
Pixatimod Inhibits SARS-CoV-2 Infection in Vivo
The potential efficacy of pixatimod in vivo was explored using the K18-hACE2 transgenic mouse model. These mice develop a robust respiratory disease resembling severe COVID-1927 but also present with a fulminant brain infection that is associated with mortality,28 with the neuroepithelium and neurons shown to be infected.29 Although human disease is not generally associated with fulminant brain infections, infection of both neuroendothelial cells and neurons resulting in Alzheimer’s-like neuropathology has recently been described in the brains of COVID-19 patients.30−32 A single prophylactic treatment of 16 mg/kg of pixatimod was given to K18-hACE2 mice 1 day before intranasal infection with SARS-CoV-2 (QLD02 isolate). Virus was inoculated in 50 μL via the intranasal route into the lungs.27 Two control groups were included, K18-hACE2 mice that were treated with saline and infected with virus and C57BL/6J mice given pixatimod but no virus. Mice were weighed daily, and all mice were euthanized on day 5, when weight loss in the control group approached 20%, the ethically defined end point. As reported previously in other preclinical models of infectious disease,18,19 pixatimod led to an initial body weight loss of ≈8% over ∼2 days; notably, this transient effect is a known feature in mice that is not observed in humans. Weight loss was thus presented relative to day 1 postinfection in order to focus on the infection-induced weight loss (Figure 4A), which generally occurs after day 3;33 comparison data relative to d-1 are provided in the Supporting Information for reference (Figure S7). The mean weight loss for saline-treated K18-hACE2 mice was significantly higher than for pixatimod-treated K18-ACE2 mice (p = 0.043, repeat measures ANOVA including data from days 4 and 5), with the latter showing no significant infection-associated weight loss (Figure 4A).
Figure 4.

Pixatimod inhibits SARS-CoV-2 infection in K18-hACE2 transgenic mice. (A) Mean percentage weight change relative to day 1 postinfection (QLD02 isolate). Statistics by repeat measures ANOVA for days 4 and 5. n = 4 mice per group, ±SE. Mice were euthanized on day 5. (B) Mean tissue titers on day 5 postinfection. Statistics by Kolmogorov–Smirnov test (nasal turbinates) and t test (brain). n = 4 mice per group, ±SE.
Tissue viral loads were assessed on day 5 postinfection, with pixatimod-treated animals showing a significant 3.1 log10-fold reduction in viral titers in nasal turbinates (p = 0.037, Kolmogorov–Smirnov test, nonparametric data distribution) and a 4.6 log10-fold reduction in viral titers in the brain, which approached significance (p = 0.0588, t test, parametric data distribution). However, we noted that viral titers in the lungs were not significantly affected by pixatimod treatment (Figure 4B). Nevertheless, in view of the robust nature of this model, with rapid effects such as pronounced weight loss and high level of lethality, these data indicate a potent prophylactic anti-SARS-CoV-2 effect of a single dose of pixatimod.
Pixatimod Inhibits SARS-CoV-2 Infection of Multiple New Variants
Multiple new variants of concern (VOC) of SARS-CoV-2 have continued to emerge and represent a significant public health challenge since some have displayed higher infectivity rates and reduced susceptibility to existing vaccines. To explore whether pixatimod retains potency against such variants, we tested the inhibitory activity of pixatimod against three SARS-CoV-2 VOC—the B.1.1.7 (Alpha or Kent) variant, the B.1.351 (Beta or South African) variant, and the B.1.617.2 (Delta or Indian) variant—in a microinhibitory assay. The amount of pixatimod required to inhibit 50% of viral foci (mini-plaques) in Vero E6 cells was similar for each of the variants of concern tested. Relative potency against all three of the variants was very comparable to the earlier Victoria (Wuhan-like virus) isolate, with EC50 values for Victoria isolate, B.1.1.7, B.1.351, and B.1.617.2 variants being 3.6, 6.5, 2.1, and 1.0 μg/mL, respectively (Figure 5A). Analysis indicated no statistically significant differences in EC50 values for inhibition by pixatimod by any of the three variants tested (as shown by a nonparametric ANOVA, Kruskal–Wallis rank sum test; P > 0.05, so a post hoc analysis was not run).
Figure 5.
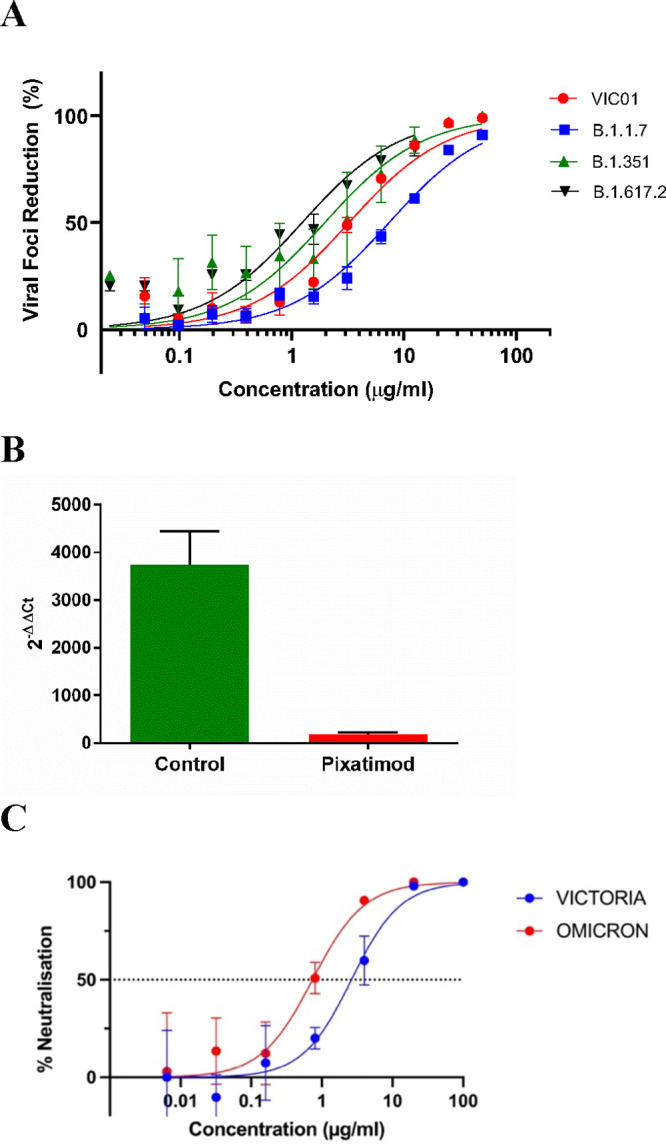
Pixatimod retains potency against multiple SARS-CoV-2 virus variants of concern. Live virus infectivity assays were performed as described in the Materials and Methods section for multiple different SARS-CoV-2 isolates (representative data shown). (A) Microinhibitory assay to assess the % viral foci reduction caused by increasing concentrations of pixatimod when incubated with live Wuhan-like SARS-CoV-2 (VIC01), B.1.1.7 Alpha variant, B.1.351 Beta variant, or B.1.617.2 Delta variant, in vitro with Vero E6 cells. Data represent 3 independent experiments per variant with n = 2 technical replicates, ±SD, except for the B.1.617.2 Delta variant where only 2 independent experiments were performed with n = 2 technical replicates. The EC50 values for VIC01 (Victoria isolate), B.1.1.7 variant, B.1.351 variant, and B.1.617.2 variant were 3.6 ± 1.8, 6.5 ± 0.8, 2.1 ± 1.7, and 1.0 ± 0.2 μg/mL, respectively. (B) Inhibitory effect of pixatimod (4 μg/mL) on the B.1.1.7 (Alpha) SARS-CoV-2 variant used to infect BCi-NS1.1 bronchial epithelial cells (ALI). Viral load in the cells was determined by an RT-PCR analysis and compared to a 2 h infected (untreated) control (n = 2 with technical duplicates, ±SD). (C) Data for SARS-CoV2 microneutralization assays for Omicron (B.1.1.529) and Vic01 variants treated with pixatimod (Oxford assay). IC50 values obtained were 0.74 ± 0.28 and 2.63 ± 0.93 μg/mL for Omicron and Vic01, respectively (n = 3, ±SD).
Next, we confirmed that pixatimod retained activity against infection of human bronchial epithelial cells by the B.1.1.7 Alpha SARS-CoV-2 variant. Cells were treated 16 h preinfection and 24 h postinfection with 4 μg/mL pixatimod, and cells were harvested 72 h postinfection for a viral RNA measurement by an RT-PCR analysis. Cells exposed to infection media only were used as a control. In the absence of any treatment, we observed an approximately 4000-fold increase in viral load at 72 h postinfection compared to the 2 h infected control. However, the viral load was significantly reduced by approximately 20-fold in the presence of pixatimod (Figure 5B). Overall, these data in a physiologically relevant human bronchial cell model show that pixatimod retains potency as an inhibitor of infection of human cells by the Alpha variant of SARS-CoV-2.
We also confirmed the ability of pixatimod to neutralize the recent Omicron (B.1.1.529) variant of SARS-CoV2, in comparison to Vic01 in microneutralization assays. IC50 values obtained were 0.74 ± 0.28 and 2.63 ± 0.93 μg/mL for Omicron and Vic01, respectively (n = 3, ±SD) (Figure 5C). Repeat assays gave similar results (Figure S8), and combined data gave averaged IC50 values of 1.13 ± 0.55 and 2.32 ± 0.44 μg/mL for pixatimod and Vic01, respectively (n = 2 assays with triplicates, ±SD). These data confirmed that pixatimod fully retained potency against the Omicron variant.
Finally, we also undertook modeling to explore potential differences in the interactions of pixatimod with VOC harboring mutations in the RBD (Figure S9). Molecular docking of pixatimod with the variant spike RBD with N501Y, as found in Alpha and Beta variants, was initially carried out. We obtained two clusters of docked poses: pose-a and pose-b similar to the wild-type S1 RBD. However, the pose-b was less populated compared to the wild-type S1 RBD. The interactions, free energies, and dynamics of pose-a with the mutant S1 RBD were similar to those of pose-a with wild-type RBD (data not shown). The mutation N501Y is relevant for pose-b wherein some repulsive effects may contribute to reducing the total ΔGbind favorable energetic contribution compared to the WT strain (Table S1). Unlike the wild-type RBD-pose-b of pixatimod, the free energy based on both residue-wise and pairwise decomposition suggests that Arg403, Arg408, and Lys417 scarcely contribute to ΔGbind (Figure S10). The largest contributions (range from −1 to −6 kcal/mol) to the ΔGbind came from the residues Tyr449, Gln498, Tyr501, Gly502, Val503, Gly504, and Tyr505 (Figure S10). These results suggested the possibility that N501Y mutants may bind slightly more weakly to pixatimod, though mutations in other residues (such as E484K in the Beta variant) could also influence binding. The Delta variant lacks the N501Y mutation but does have other mutations in the RBD (such as L452R and T478K) which could potentially influence binding to pixatimod. The mutation L452R, through its side-chain, will contribute to hydrophobic interactions, similar to the wild-type RBD. However, the residue T478 is not involved in binding with pixatimod. The K417N mutation present in Beta and Delta (detected in a very few sequences) variants is likely to retain hydrogen bond interactions with the pixatimod tetrasaccharide. However, any such differences in binding for all three VOC must be very minor compared to the original Wuhan-like isolate and each other, since the experimental EC50 values for pixatimod inhibition are statistically equivalent. These data clearly indicate that pixatimod is highly tolerant to mutations in multiple VOC.
Discussion and Conclusions
The current COVID-19 pandemic illustrates the critical need to develop both effective vaccines and therapeutics for emerging viruses; established antiviral agents appear to have limited utility against SARS-CoV-2. Owing to their involvement as coreceptors for cell attachment by many viruses, HS represents an ideal broad-spectrum antiviral target.2 Binding of a viral protein to cell-surface HS is often the first step in a cascade of interactions that is required for viral entry and the initiation of infection.34 As HS and heparin contain the same saccharide building blocks, and HS-binding proteins also interact with heparin, this drug is gaining attention beyond its anticoagulant properties in COVID-19 treatment.34 Here, we demonstrate a direct mechanism of action of pixatimod on attenuating S1-RBD binding to ACE2. These data are supported by recent studies on heparin using native mass spectrometry35 and also reveal for the first time the ability of HS mimetics to inhibit S1-RBD binding to the ACE2 receptor.
Heparin has been shown to inhibit binding of SARS-CoV-2 spike protein to a human cell line36 and to inhibit entry into human cells of pseudovirus carrying the SARS-CoV-2 spike protein.10,37 However, the question of whether therapeutic doses of heparins are effective for COVID-19 patients as an antiviral treatment awaits the outcome of prospective clinical trials;38 bleeding complications are possible,39 though non-anticoagulant heparin or HS preparations could be deployed that reduce cell binding and infectivity without a risk of causing bleeding.9,10 However, HS mimetics offer additional advantages in comparison to heparin beyond simply reducing anticoagulant activity,9 most notably their ready availability at scale via synthetic chemistry production that addresses the well-known fragility of the heparin supply chain.11 As a clinical-stage HS mimetic, pixatimod provides better control over structure, molecular weight diversity (a single molecular entity), sulfation, purity, and stability. Herein, we reveal a direct interaction of the clinical candidate pixatimod with the S1 spike protein RBD, supported by molecular modeling data. Notably, pixatimod induced much greater destabilization of the S1-RBD structure than heparin, observed by DSF; we speculate that this may be related to its different modes of interactions with the RBD protein surface, especially with regard to its unique cholestanol tail moiety. Pixatimod also inhibited the interaction of S1 RBD with Vero cells which express the ACE2 receptor. Moreover, infectivity assays of two types (plaque reduction and cytopathic effect, Table 1) confirmed that pixatimod is a potent inhibitor of infection of Vero E6 and human cells by various strains of SARS-CoV-2 infection (including new variants of concern), at EC50 concentrations ranging from 0.9 to 13.2 μg/mL which are well within its known therapeutic range. Interestingly, we noted that the lipophilic steroid side-chain of pixatimod was critical for its potency and is predicted from modeling to interact with S1-RBD. This unique feature, making it an unusual amphiphilic HS mimetic, has also been shown to confer virucidal activity against Herpes Simplex virus by disruption of the viral lipid envelope.15 The ability of pixatimod to potently inhibit all SARS-CoV2 strains tested, exemplified here by the Alpha, Beta, Delta, and Omicron VOC strains, is supported by our molecular modeling studies which indicate only minor likely effects on binding to RBD due to mutations observed. However, It should be noted that, besides the mutations in the RBD, the variants possess a number of other mutations/deletions in other domains on the spike protein. Nevertheless, it is clear from our data that pixatimod retains excellent and broad potency for inhibition of cell infection by all of the major VOC to date, indicating high tolerance to the complex sets of mutations that they display in their spike protein RBD. This aspect, along with chemical stability, confers significant potential advantages for pixatimod over other therapeutics targeting inhibition of viral adhesion/invasion, such as neutralizing antibodies.
Pixatimod has additional potential advantages, not least its mild anticoagulant activity12 and the fact that it has been administered i.v. to over 80 cancer patients, being well tolerated with predictable pharmacokinetics (PK) and no reports of heparin-induced thrombocytopenia.12 Further, pixatimod cytotoxicity in vitro is low; we observed a CC50 value of >300 μg/mL and >236 μg/mL in human bronchial epithelial and Vero cells, respectively, consistent with previous cytotoxicity data on human cells40 (see also a cytotoxicity data summary in the Supporting Information). Importantly, the maximum plasma concentration (Cmax) of pixatimod following a single treatment of 100 mg in cancer patients is 29.5 μg/mL with a Cmin of 2.7 μg/mL measured 1 week following treatment,12 indicating that an equivalent dosing regimen should be sufficient to achieve antiviral activity in human subjects. The low anticoagulant activity of pixatimod is an advantage since it could be used as a direct antiviral agent in combination therapies with heparins, which are being used to treat coagulopathies observed in COVID-19 patients.39 It was also encouraging that pixatimod inhibition of multiple clinical isolates of SARS-CoV-2 was noted, including a number of emerged VOC including Delta and Omicron, demonstrating potential for widespread effectiveness. The presence of multiple binding sites for pixatimod in the Spike protein would suggest robustness against mutations that may continue to arise in the pandemic and/or in the following coronavirus outbreaks. While widespread isolates with D614G spike mutants appear to be 2–3 fold more sensitive to the antiviral activity of pixatimod, caution needs to be taken in interpreting the data of the cytopathicity assay used to determine this activity as G614 isolates (at least QLD935) exhibited lower cytopathicity than D614 isolates.
Since SARS-CoV-2 is predominantly a respiratory infection, our data from human bronchial cells are directly supportive of high potency of pixatimod against infection of clinically relevant human cells. Further, the potential efficacy in humans is supported by our initial data from the established K18-hACE2 transgenic mouse model which demonstrated the ability of a single prophylactic dose to rescue the pronounced weight loss phenotype observed. Given that the half-life of pixatimod in mice is 38 h,40 compared to 141 h in humans,13 future studies should adopt repeat-dose schedules to identify the optimal schedule in models of SARS-CoV-2 infection and to establish potential postexposure therapeutic efficacy. We speculate that significant therapeutic benefits might be gained in combination with antiviral drugs with complementary mechanisms of action such as Paxlovid (nirmatrelvir/ritonavir) and molnupiravir. Prophylactic treatments are also of potential high value for COVID19.42 Although oral availability of pixatimod is poor, we also suggest that direct intranasal delivery of pixatimod as a pre-exposure prophylactic strategy would be worth investigation,23 in view of recent positive data on the delivery of nebulized heparin for treatment of ARDS,43 and also COVID-19 treatment.38
Why pixatimod should have such a significant effect on viral titers in the nasal turbinates (and approaching significance in the brain), but not lungs in the K18-hACE2 model, is unclear but may reflect very high lung (supraphysiological) K18-driven hACE2 receptor expression in the lungs in this model44 and/or pixatimod pharmacokinetics.40 Nevertheless the results are encouraging, especially as recent data suggest that viral loads in the upper respiratory tract are increased for the Omicron variant.45 In addition, the initial infection in humans occurs in the upper respiratory tract, with ARDS developing only after infection has over time spread down into the lungs.46 This progression is not recapitulated in the mouse model where fulminant lung infection requires direct inoculation of virus into the lungs via the intranasal route. Infection of the brain may occur via the olfactory epithelium,47 so the reduced infection of the brain may reflect the reduced infection of nasal turbinates. Although brain infection in COVID-19 in humans is now well documented, the fulminant brain infection seen in this mouse model does not seem to be a feature of human disease. Nevertheless, brain infection may emerge as an important contributor to long COVID.48
It is notable that there are multiple potential mechanisms of action of pixatimod against SARS-CoV-2 (summarized in Figure 6), including direct inhibition of HS–Spike–ACE2 interactions via complex and multiple interactions with amino acids in RBD. Notably, the latter contrasts with the more restricted epitopes typical of antibodies which are observed to be subject to mutational escape potentially requiring vaccine redesign or use of multivalent strategies.49 In this respect, we speculate that HS mimetics like pixatimod may prove important therapeutic tools for initial responses to escape variants. Beyond this, pixatimod also has immunomodulatory effects which may alleviate some of the immunopathologies associated with moderate–severe COVID-19 disease. Pixatimod also inhibits the proinflammatory enzyme heparanase12 and has been demonstrated to suppress IL-6 in inflammatory (pancreatitis) and viral (Ross River virus) animal models.14,18 Moreover, it blocks the heparanase-dependent invasion of macrophages into tumors in mouse cancer models40,41 which may be relevant to invasion of monocytes and macrophages into the lungs associated with severe COVID-19 disease.50 The vaccinia virus has recently been shown to rely on host heparanase to degrade HS in order to spread to distant sites,51 revealing a role for heparanase in the progression of disease that may also apply for SARS-CoV-2 in COVID-19. Notably, increased plasma heparanase activity is associated with COVID-19.52 Thus, multiple additional beneficial effects of pixatimod might be anticipated from its heparanase inhibitory properties.
Figure 6.

Proposed multimodal mechanisms of pixatimod activity against SARS-CoV-2 and other viruses. The principal mode of action demonstrated here is that pixatimod acts as a decoy receptor [1], blocking S1-RBD binding to HS coreceptors and inhibiting viral attachment to host cells, thus blocking viral infection. Additional potential modes of action include: [2] virucidal activity of pixatimod, dependent upon the cholestanol moiety,15 which may lead to degradation and permanent inactivation of SARS-CoV-2 virus particles; [3] suppression of IL-6 secretion by antigen presenting cells, primarily macrophages;14 and [4] blocking viral escape from host cells by inhibiting heparanase which otherwise promotes viral escape by cleaving HS receptors.51,52
In conclusion, based on the data presented here, pixatimod has potent antiviral activity against a number of clinical strains of SARS-CoV-2 at therapeutically relevant concentrations both in vitro and in vivo and with physiologically relevant human bronchial cells. Importantly, potency is retained against examples of recently emerged Alpha, Beta, Delta, and Omicron VOC of SARS-CoV-2, indicating that pixatimod is highly tolerant to emerging mutations. These activities are in addition to the known heparanase-inhibitory and immunomodulatory properties of pixatimod which may further support the host response to COVID-19 infection. Collectively, this provides a strong rationale to justify entry of pixatimod into clinical trials for COVID-19. Furthermore, we have demonstrated the first proof-of-concept for employing HS mimetics against SARS-CoV-2 with implications for wider future applications of this class of broad-spectrum antivirals, potentially against SARS-CoV-2 vaccine escape variants, other HS-binding viruses, and also those that may emerge as future global threats.
Materials and Methods
Computational Methods
The crystal structures of the RBD-ACE2 complex (PDB ID: 6LZG; ref (53)) and the mutant N501Y RBD-ACE2 complex (PDB ID: 7MJN; ref (54)) were retrieved from the RCSB Protein Data Bank. Structures were stripped of water molecules, ACE2 chains, and any cofactors and/or ligands present. UCSF Chimera 1.15 was used to edit the structure and for visualization. In the absence of the known binding site for pixatimod on wild-type and mutant RBD, a “blind” docking was carried out using the GlycoTorch Vina docking program.55 All sulfate and hydroxyl groups and glycosidic torsion angles of the ligand were treated as flexible. The box size was sufficient to cover the entire RBD molecule. A blind docking run was performed by setting an energy range of 12, an exhaustiveness of 80, chi_cutoff = 1, chi_coeff = 2, and the number of modes = 100. The docking simulations were repeated twice. To look for common binding modes and pixatimod–protein interactions, we performed a cluster analysis using UCSF Chimera 1.15 on multiple docking solutions obtained from blind docking, looking for poses that could give similar interactions with the protein. The docked poses from the most populated clusters were subjected to molecular dynamics (MD) simulations and relative free energies of binding calculations. All of the MD simulations were carried out using the pmemd.cuda module56 of the AMBER 16 molecular dynamics package, and the analyses were performed using the cpptraj module of AmberTools16.57 Simulation systems were set up by placing the spike RBD domain at the center of the octahedral simulation box (with an extension of at least 12 Å from each side). Pixatimod was randomly placed in the box. This was followed by an addition of TIP3P water58 and Na+ ions for neutralizing the charge of the system. Proteins were parametrized using the Amber99SB-ildn force field59 whereas Glycam-06 (version j)60 and Lipid1461 force fields were used for the sulfated tetrasaccharide and cholestanol moieties of pixatimod, respectively. Periodic boundary conditions were applied, and the time step was set to 2 fs. The electrostatic energy was calculated with the particle mesh Ewald (PME) method. SHAKE constraints were applied on the bonds involving hydrogen. A cutoff of 12 Å was applied to the Lennard-Jones and direct space electrostatic interactions with a uniform density approximation included to correct for the long-range van der Waals interactions.
The system was first minimized without electrostatics for 500 steps and then with a restraint of 25 kcal/(mol Å2) applied on the protein and pixatimod. This minimization was followed by a 100 ps MD simulation with 25 kcal/(mol Å2) positional restraints applied on the protein and ligand, and the temperature was slowly increased from 0 to 300 K. Then, this was followed by 500 steps of steepest descent cycles, followed by 500 steps of conjugate gradient minimization and 50 ps equilibrations with a restraint force constant of 5 kcal/(mol Å2) in the protein and ligand, followed by a final 2 ns equilibration without restraints to equilibrate the density. The first few steps were all carried out at constant volume followed by at least a 150 ns production MD simulation at 300 K (Langevin dynamics, collision frequency: 5 ps–1) with 1 atm constant pressure. Two replicas of the MD simulations were performed for all systems, and the results presented here are the average of two runs. Trajectories were collected, and data analyses such as RMSD, RMSF, and free energy of binding were performed on the last 12 500 frames. The binding free energy and per-residue and pairwise residue contributions62 were calculated using the molecular mechanics–generalized Born (GB) equation (MM/GBSA) procedure implemented in AmberTools16. The details of this method have been extensively reviewed.63 The polar solvation energy contribution was calculated by using GBOBC II (igb = 5).64 The values of the implicit solvent dielectric constant and the solute dielectric constant for GB calculations were set to 80 and 1, respectively. The solvent probe radius was set to 1.4 Å as the default. The entropy calculation is computationally expensive and therefore not performed for the purposes of this study.
Expression of His-Tagged Recombinant SARS-CoV-2 S1 RBD in E. coli
Residues 330–583 of the SARS-CoV-2 spike protein (GenBank: MN908947) were cloned upstream of a N-terminal 6XHisTag in the pRSETA expression vector and transformed into SHuffle T7 Express Competent E. coli (NEB, UK). Protein expression was carried out in MagicMediaTM E. coli Expression Media (Invitrogen, UK) at 30 °C for 24 h, 250 rpm. The bacterial pellet was suspended in 5 mL of lysis buffer (BugBuster Protein Extraction Reagent, Merck Millipore, UK; containing DNase) and incubated at room temperature for 30 min. Protein was purified from inclusion bodies using IMAC chromatography under denaturing conditions. On-column protein refolding was performed by applying a gradient with decreasing concentrations of the denaturing agent (from 8 M urea). After extensive washing, protein was eluted using 20 mM NaH2PO4, pH 8.0, 300 mM NaCl, and 500 mM imidazole. Fractions were pooled and buffer-exchanged to phosphate-buffered saline (PBS; 140 mM NaCl, 5 mM NaH2PO4, 5 mM Na2HPO4, pH 7.4; Lonza, UK) using Sephadex G-25 media (GE Healthcare, UK). Recombinant protein (termed EcS1-RBD) was stored at −4 °C until required.
Expression of S1-RBD in Mammalian Cells
Secreted RBD-SD1 (termed mS1-RBD) was transiently produced in suspension HEK293-6E cells. A plasmid encoding RBD-SD1, residues 319–591 of SARS-CoV-2 S cloned upstream of a C-terminal HRV3C protease cleavage site, a monomeric Fc tag, and an His8x Tag were a gift from Jason S. McLellan, University of Texas at Austin. Briefly, 100 mL of HEK293-6E cells were seeded at a cell density of 0.5 × 106 cells/mL 24 h before transfection with polyethyleneimine (PEI). For transfection, 100 μg of the ACE2 plasmid and 300 μg of PEI (1:3 ratio) were incubated for 15 min at room temperature. Transfected cells were cultured for 48 h and fed with 100 mL of fresh media for an additional 48 h before harvest. mS1-RBD was purified by a HiTrap Protein G HP column (GE Healthcare, US) pre-equilibrated in PBS and eluted with 0.1 M glycine (pH 2.7). The purity of proteins was evaluated by Coomassie staining of SDS-PAGE gels, and proteins were quantified by a BCA protein assay kit (Thermo Scientific).
Secondary Structure Determination of SARS-CoV-2 S1 RBD by Circular Dichroism Spectroscopy
The circular dichroism (CD) spectra of m(His)S1-RBD (with His tag but without mFc tag; BioLegend, UK), in both the presence and absence of carbohydrate ligand, were recorded using a Chirascan V100 CD spectrometer (Applied Photophysics, UK) and Chirascan software (Applied Photophysics, UK) using a 0.2 mm path length and a quartz cuvette (Hellma, USA). All spectra were obtained scanning at 100 nm/min, with 1 nm resolution throughout the range λ = 190–260 nm and are presented as the mean of five independent scans following instrument calibration with camphorsulfonic acid. CD spectra were collected using 15 μL of a 0.5 mg/mL solution of mS1-RBD in PBS, pH 7.4. Spectra containing heparin (unfractionated porcine mucosal heparin, Celsus, USA) were collected in the same buffer at approximately comparable concentrations, since this is a polydisperse material. Collected data were analyzed, baseline corrected, and smoothed (5 neighbors) using Chirascan software prior to further processing with GraphPad Prism 7. Secondary structure predictions were calculated using the BeStSel analysis server.65
To ensure that the CD spectral change of m(His)S1-RBD-His in the presence of pixatimod did not arise from the addition of the compound alone, a difference spectrum was analyzed. The theoretical CD spectrum that resulted from the arithmetic addition of the CD spectrum of the mS1-RBD and that of pixatimod differed from the observed experimental CD spectrum of mS1-RBD mixed with compound alone. This demonstrates that the change in the CD spectrum arose from a conformational change following binding to pixatimod (Supporting Information, Figure S11). Circular dichroism experiments utilizing recombinant, E. coli expressed S1-RBD were carried out as previously described by Mycroft-West et al. (Supporting Information, Figure S4).8
Differential Scanning Fluorimetry
Differential scanning fluorimetry (DSF; ref (66)) was conducted on mammalian expressed mS1-RBD (1 μg) in PBS pH 7.6 and 1.25× Sypro Orange (Invitrogen) to a total well volume of 40 μL in 96-well qPCR plates (AB Biosystems). Unfractionated porcine mucosal heparin (Celsus) or pixatimod (10 μg) were introduced to determine the effect on the thermal stability of mS1-RBD using an AB biosystems StepOne plus qPCR machine employing the TAMRA filter setting. Melt curve experiments were performed following a 2 min initial incubation at 25 °C, with succeeding 0.5 °C increments every 30 s up to a final temperature of 90 °C. Control wells containing H2O, heparin, or pixatimod (10 μg) without mS1-RBD (1 μg) were also employed to ensure that a change in the melt curve was solely a result of protein–ligand interactions and interactions with Sypro Orange. Smoothed first derivative plots (9 neighbors, second-order polynomial, Savitxky-Golay) were constructed using Prism 8 (GraphPad). Tm values were calculated using MatLab software (R20018a, MathWorks), and ΔTm values were determined from the difference between the Tm of RBD alone or in the presence of heparin or pixatimod.
Cell Binding of S1 RBD
African green monkey Vero kidney epithelial cells and A549 human lung epithelial cells were purchased from ATCC. Cells were maintained at 50–75% confluence in DMEM supplemented with 10% fetal bovine serum, 20 mM l-glutamine, 100 U/mL penicillin-G, and 100U/mL streptomycin sulfate (all purchased from Gibco/ThermoFisher, UK). Cells were maintained at 37C, 5% CO2. For cell binding assays, cells were plated into 96-well cell culture plates at 1000 cells/well in 100 μL of maintenance medium. Cells were allowed to adhere overnight. Medium was aspirated, and wells were washed 3× with 200 μL of calcium-/magnesium-free PBS (CMF-PBS, Lonza, UK). Cells were fixed with 100 μL of 10% neutral buffered formalin (Thermofisher, UK) for 10 min at room temperature. Wells were washed 3× with 200 μL of CMF-PBS. 100 μL of CMF-PBS was added to each well, and plates were stored at 4 °C until use. Before use, wells were blocked with 200 μL of CMF-PBS + 1% BSA (Sigma-Roche, UK) for 1 h at room temperature and washed 3× with 200 μL of CMF-PBS + 0.1% Tween-20 (PBST, Sigma-Roche, UK) followed by 2× with 200 μL of CMF-PBS.
For experiments with Vero cells, EcS1-RBD (50 μg/mL), heparin, and compounds at indicated concentrations were added to each well in 25 μL of PBST + 0.1% BSA as indicated. Wells were incubated for 1 h at room temperature with rocking. Wells were washed 3× with 200 μL of PBST and 2× with 200 μL of CMF-PBS. The binding of His-tagged S1-RBD was detected with Alexa Fluor 488 anti-His tag antibody (clone J095G46, Biolegend, UK) 1:5000 in 25 μL of PBST + 0.1% BSA per well. Wells were incubated in the dark for 1 h at room temperature with rocking. Wells were washed 3× with 200 μL of PBST and 2× with 200 μL of CMF-PBS. Fluorescence was read at Ex 485:Em 535 on a Tecan Infinite M200Pro plate reader. Results are presented as normalized mean (where 0 is the fluorescence without added S1-RBD, and 1 is the fluorescence with 50 μg/mL S1-RBD) ± %CV, n = 3.
For experiments with A549 cells, mammalian expressed SARS-Cov-2 S protein RBD-monomeric Fc chimera (Biolegend, UK, 50 μg/mL) alone or with heparin (100 μg/mL) or pixatimod (100 μg/mL) was added to each well in 25 μL of PBST + 0.1% BSA as indicated. Wells were incubated for 1 h at room temperature with rocking. Wells were washed 3× with 200 μL of PBST and 2× with 200 μL of CMF-PBS. Binding of RBD was detected with HRP conjugated anti-SARS-CoV2 Spike RBD antibody (Sino Biological, UK) at 0.5 μg/mL in 25 μL of PBST + 0.1% BSA per well. Wells were incubated for 1 h at room temperature with rocking. Wells were washed 3× with 200 μL of PBST and 2× with 200 μL of CMF-PBS. Bound HRP conjugated secondary was detected with the addition of 50 μL of 1-Step Ultra TMB-ELISA reagent (ThermoScientific, UK). Wells were developed for 15 min at room temperature and the reaction was stopped with the addition of 50 μL of 2 M H2SO4. Absorbance was read at 450 nm on a Tecan Infinite M200Pro plate reader. Results are presented as normalized mean (where 0 is the absorbance without added RBD and 1 is the absorbance with 50 μg/mL RBD) ± %CV, n = 3.
Competition ELISA for S1 RBD binding to ACE2
High binding 96 well plates (Greiner) were coated with 3 μg/mL streptavidin (Fisher) in 50 mM sodium carbonate buffer pH 9.6 (50 μL/well) for 1 h at 37 °C. Plates were washed 3 times with 300 μL of PBS, 0.2% Brij35 (PBSB) and blocked with 300 μL of PBSB + 1% casein for 1 h at 37 °C. Plates were washed a further 3 times with 300 μL of PBSB prior to the addition of 100 ng/mL Biotinylated ACE2 (Sino Biological) in PBSB + 1% casein (50 μL/well) and incubated for 1 h at 37 °C. Plates were again washed 3 times with 300 μL of PBSB prior to the addition of 50 μL/well mS1-RBD (5 μg/mL) in PBSB + 1% casein, which had been preincubated for 30 min at room temperature with or without varying concentrations of heparin or pixatimod (100- 0.7 μg/mL) in separate tubes. Plates were incubated for 1 h at 37 °C to allow for mS1-RBD-ACE2 binding and were subsequently washed with 300 μL/well PBSB. Bound mS1-RBD was detected by incubation with 0.5 μg/mL Rabbit-SARS-CoV-2 (2019-nCoV) Spike RBD Antibody (Stratech) in PBSB + 1% casein (50 μL/well) for 1 h at 37 °C. Following a further 3 washes with PBSB plates were incubated for 30 min at 37 °C with horseradish peroxidase conjugated Donkey anti-Rabbit IgG diluted 1:1000, v/v in PBSB + 1% casein (Bioledgend). Plates were washed a final 5 times with 300 μL of PBSB before being developed for 10 min with 3,3′,5,5′- tetramethylbenzidine prepared according to the manufacturer’s instructions (Fisher). Reactions were stopped by the addition of 20 μL of 2 M HsSO4 and plates were read at λ = 450 nm using a Tecan Infinate M200 Pro mulit-well plate reader (Tecan Group). Control wells containing no biotinylated ACE2 were employed to ensure binding was specific.
Live SARS-CoV-2 virus assays
SARS-CoV-2 Victoria isolate (GISAID accession, EPI_ISL_406844): a plaque reduction assay was performed with the SARS-CoV-2 Victoria/01/2020 (passage 3) isolate, generously provided by The Doherty Institute, Melbourne, Australia at P1, was diluted to a concentration of 1.4 × 103 pfu/mL (70 pfu/50 μL) in minimal essential media (MEM) (Life Technologies, California, USA) containing 1% (v/v) fetal calf serum (FCS) (Life Technologies) and 25 mM HEPES buffer (Sigma) and mixed 50:50 with pixatimod dilutions, in a 96-well V-bottomed plate. The plate was incubated at 37 °C in a humidified box for 1 h to allow the virus to be exposed to pixatimod. The virus-compound mixture was transferred onto the wells of a washed 24-well plate that had been seeded with Vero E6 cells [ECACC 85020206] the previous day at 1.5 × 105 cells/well. The virus-compound mixture was left to adsorb for an hour at 37 °C, then plaque assay overlay media was applied (MEM containing 1.5% carboxymethylcellulose (Sigma, Dorset, UK), 4% (v/v) FCS and 25 mM HEPES buffer). After incubation at 37 °C in a humidified box, for 5 days, plates were fixed overnight with 20% (v/v) formalin/PBS, washed with tap water and then stained with methyl crystal violet solution (0.2% v/v) (Sigma) and plaques were counted. Compound dilutions were performed in either duplicate or quadruplicate. Compound dilutions and cells only were run in duplicate, to determine if there was any cell cytotoxicity. An internal positive control for the plaques reduction assay was run in triplicate using a sample of heat-inactivated human MERS convalescent serum known to neutralize SARS-CoV-2 (National Institute for Biological Standards and Control, UK).
A microinhibition assay67 was used to assess the inhibitory effect of pixatimod on two variants of concern; the B.1.1.7 (Alpha) variant (passage 3), the B.1.351 (Beta) variant (passage 3) and the B.1.617.2 (Delta) variant (passage 2). Pixatimod was diluted 2-fold over a 12-step dilution range, in duplicate. A fixed concentration of VOC was added to the diluted pixatimod. Additional assay wells included virus-free and untreated virus-only controls. The plates (diluted pixatimod and virus) were then incubated for 1 h at 37 °C to allow virus to be exposed to pixatimod. The contents of the virus/pixatimod plates were then transferred into 96-well plates containing Vero-E6 cells and virus was allowed to adsorb to the cells for 1 h at 37 °C. The inocula were removed from the cell plates and a viscous overlay (1% CMC) added (pixatimod was added to the overlay media). The plates were then incubated for 24 h. The cells were then fixed using 8% formalin for >8 h and an immunostaining protocol performed on the fixed cells. Stained foci were counted using an ELISpot counter (Cellular Technology Limited (CTL)). The counted foci data were then imported into R- Bioconductor. An internal positive control, chloroquine diphosphate (50–0.02 μM), was run alongside pixatimod, on each assay plate.
A midpoint probit analysis (written in R programming language for statistical computing and graphics) was used to determine the amount (μg/mL) required to reduce SARS-CoV-2 viral plaques or viral foci by 50% (EC50) compared with the virus only control (n = 5 [24-well assay], n = 10 [96-well assay].).
SARS-CoV-2 DE-Gbg20 isolate (GISAID accession under application): Plaque reduction assay for SARS-CoV-2 clinical isolate DE-Gbg20 from Sweden was performed in a similar manner, except for the virus and the pixatimod (5-fold decreasing concentrations at a range 100–0.16 μg/mL) were diluted in DMEM supplemented with 2% heat-inactivated FCS, and 100 U of penicillin and 60 μg/mL of streptomycin (DMEM-S). The virus (100 PFU) and pixatimod (5-fold decreasing concentrations at a range 100–0.16 μg/mL) were mixed and incubated for 30 min in humidified atmosphere comprising 5% CO2 (CO2 incubator). The mixtures were then transferred to Vero cells (ATCC CCL-81) and following incubation with cells for 90 min in the CO2 incubator, the methylcellulose overlay was added. Three separate experiments each with duplicates were performed.
A cytopathic effect assay was performed with the SARS-CoV-2 DE-Gbg20 isolate and Vero cells (ATCC) plated at 2 × 104 per well in 96-well plates the day prior to the experiment. Serial 5-fold dilutions of pixatimod in DMEM supplemented with 2% heat-inactivated FCS, and 100 U of penicillin and 60 μg/mL of streptomycin (DMEM-S) were incubated with 100 TCID50 of SARS-CoV-2 isolate DE for 20 min in humidified atmosphere comprising 5% CO2 (CO2 incubator). The final concentrations of pixatimod were in a range 0.075 μg/mL to 47.2 μg/mL. The cells were rinsed once with 50 μL of DMEM-S, and then 200 μL of the virus-pixatimod mixtures were added to each well with cells (in quadruplicates). After incubation of the virus-pixatimod mixtures with cells for 3 days in the CO2 incubator, the cells were inspected under a microscope for the presence of virus induced cytopathic effect where complete protection of cells were denoted as “+” while a partial protection (~50% of cells showing no cytopathic effect) was recorded as “ ± “. The 50% end-point (EC50) was computed by the Reed and Muench method.
SARS-CoV-2 QLD02 (GISAID accession EPI_ISL_407896) and QLD935 (GISAID accession EPI_ISL_436097) clinical isolates from Australia: A cytopathic effect assay was carried out as described above for the DE-Gbg20 isolate, with 10 ffu/well and 3 days incubation. In this assay, Vero E6 cells were plated at 2 × 104 per well in 96-well plates the day prior to experiment. Serial 5-fold dilutions of pixatimod in DMEM supplemented with 2% heat-inactivated FCS, and 100 U of penicillin and 60 μg/mL of streptomycin (DMEM-S) were incubated with 10 foci forming units of SARS-CoV-2 QLD02 or QLD935 isolate and incubated for 30 min in humidified atmosphere comprising 5% CO2 (CO2 incubator). The cells were rinsed once with 50 μL of DMEM-S, and then 200 μL of the virus-pixatimod mixtures were added to each well with cells (in triplicates). After incubation of the virus-pixatimod mixtures with cells for 3 days in the CO2 incubator, the cells were fixed with 4% PFA and then stained with crystal violet. Then crystal violet was released by methanol and OD at 595 nm was measured to quantify cell viability (protection from infection). The EC50 was then calculated using GraphPad Prism.
Comparative microinhibition antiviral assays on Omicron (B1.1.529) and Vic01 variants were carried out in the Oxford lab. Compounds were 5-fold serially diluted in DMEM supplemented with 1% FBS from an initial concentration of 1 or 3 mg/mL to 64 ng/mL or 192 ng/mL respectively, forming a 10× stock. Five microlitres of 10× stock dilution was combined with 45 μL of SARS-CoV-2 virus (approximately 100 ffu) and incubated for 60 min at 37 °C. Vero E6 cells were also preincubated with compound by adding 5 μL of 10× stock dilutions to 45 μL of cells (1 × 106/mL) and incubating at 37 °C for 1 h. Following incubation, the preincubated virus was added to the preincubated cells and the virus was allowed to infect the cells for a further hour at 37 °C, 5% CO2 followed by the addition of 100 μL of compound adulterated Carboxymethyl cellulose (1.5%) to each well. The plates were incubated for a further 18 (Victoria) or 22 (Omicron) hours at 37 °C, 5% CO2. All assays were carried out in triplicate in 96 well plates. Cells were washed with 200 μL of DPBS and then fixed with paraformaldehyde 4% v/v (100 μL/well) for 30 min at room temperature. Cells were permeabilised with TritonX100 (1% in PBS) and then stained for SARS-CoV-2 nucleoprotein using a human monoclonal antibody (EY2A or FB9B). Bound antibody was detected following incubation with a goat antihuman IgG HRP conjugate (Sigma, UK) and following TrueBlue Peroxidase substrate (Insight Biotechnology, UK) addition imaged using an ELISPOT reader. The half-maximal inhibitory concentration (IC50) was defined as the concentration of compound that reduced the Foci forming unit (FFU) by 50% compared to the control wells.
Cytotoxicity assays
The assay was performed as described previously.17 Briefly, Vero cells (ATCC, 2 × 104 cells/well) were seeded in 96 well cluster plates to become nearly confluent at the day of the experiment. The cell growth medium was then removed and 100 μL of serial 5-fold dilutions of pixatimod in DMEM-S (ranging from 0.09 to 236 μg/mL) were added to cells. Following incubation of cells with pixatimod for 3 days in the CO2 incubator, 20 μL of the MTS salt containing CellTiter 96 Aqueous One Solution reagent (Promega, Madison, WI) was added and incubated for further 1–2 h at 37 °C. The absorbance was recorded at 490 nm against a background of 650 nm. Two separate experiments each in duplicates were performed and the results are expressed as percentage of absorbance value detected with pixatimod relative to control cells. For CC50 assays with human bronchial cells, submerged BCI were plated in a 96 well plate at a concentration of 1.5 × 104cells/well. After 24 h cells were exposed to pixatimod (0, 1,5,25,100,200,300μg/mL) and cell viability was measured 72 h later. Following incubation of cells with pixatimod in the CO2 incubator, 20 μL of the MTS salt containing CellTiter 96 Aqueous One Solution reagent (Promega, Madison, WI) was added and incubated for further 1 at 37 °C.
Human bronchial cell infection assays
The hTERT transformed bronchial epithelial cell line BCi-NS1.125,26 expanded in PneumaCult-Ex Plus Basal Medium supplemented with Pneumacult Ex Plus supplements, hydrocortisone, nystatin and penicillin–streptomycin. BCi-NS1.1 cells were grown at an Air Liquid Interface (ALI) in PneumaCult-ALI Basal Medium supplemented with Pneumacult ALI supplement hydrocortisone, PneumaCult ALI maintenance supplement, heparin, nystatin and penicillin–streptomycin (all from Stemcell Technologies) at 37 °C in 5% CO2. BCi-NS1.1 cell ciliation was observed by microscopy and cells were differentiated and ciliated after 4 to 5 weeks at ALI. Transepithelial electrical resistance (TER) was monitored weekly using an EVOM Voltohmmeter (World Precision Instruments) and cells with a TER ≥ 1000 WΩcm2 were used.
SARS-CoV-2 strains BetaCoV/Australia/VIC01/2020 and VUI-202012/1 (B.1.1.7 Alpha variant) were used for infection. With variant BetaCoV/Australia/VIC01/2020, 50,000 plaque-forming units (MOI = 0.1–0.2, obtained from Public Health England and propagated in Vero E6 cells for no more than two passages before use) were preincubated with 0.4, 4, and 40 μg/mL of pixatimod for 1 h at 37 °C in a total volume of 200 μL (or an equivalent volume of HBSS was added apically in the untreated controls). BCi-NS1.1 cells (4 to 8 weeks after ALI) after being washed three times with HBSS were apically infected with the preincubated virus and compounds. After 2h the virus-compound solution was removed from the apical side and washed twice each time with 200ul HBSS. The last wash was collected for plaque assays and 50 μL stored in Qiazol (Qiagen). Similarly at 72 h a further 200 μL of HBSS wash was performed and collected as at 2h. Cells were also harvested with QIAzol (QIAGEN) at 2 h after the HBSS wash and at 72 h postinfection for RNA extraction.
Plaque assay was performed to quantify extracellular virus released in the supernatant of infected ALI. Vero E6 cells were seeded at 2.5 × 105 cells/well in a 12-well plate and left for a period of 24 h in DMEM supplemented with 10% fetal bovine serum (FBS), glutamine and 50 U/mL penicillin-streptomycin at 37 °C in 5% CO2. Cells were washed once with infection medium (serum-free DMEM supplemented with 25 mM HEPES) and 100 μL from washes of infected ALI cultures added to wells in 10-fold serial dilutions. After a 1 h incubation at 37 °C in 5% CO2, infectious supernatants were removed and a 1.5 mL overlay of 1 × DMEM supplemented with 4% FBS, 25 mM HEPES and 0.6% (w/v) cellulose (Sigma) was added. Plates were incubated at 37 °C and 5% CO2 for 72 h before removing the overlay, fixing with 8% formaldehyde in PBS, and staining with 0.1% (w/v) crystal violet in a solution of 20% (v/v) ethanol.
RNA was isolated from cell lysates using standard phenol–chloroform extraction, and reverse transcribed to cDNA using a High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) following the manufacturer’s instructions. Taqman gene expression assays for N2 and N3 regions of the SARS-CoV-2 N gene were made (ThermoFisher) following CDC specifications (Division of Viral Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, Atlanta, GA, USA, 20 January 2020 copy).
Shedding of viable virions in the apical side was quantified by plaque assay and also by RT-PCR. For determination of relative changes of RNA within the cells the average Ct values of SARS-CoV-2 N gene RNA was normalized against the house keeping gene B2M and shown as fold changes compared to the 2h infected control using the 2–ΔΔCt method.
With the VUI-202012/1 (B.1.1.7) Alpha variant, BCi-NS1.1 cells (4 to 8 weeks after ALI) were treated with 4 μg/mL pixatimod at 16 h pre- and 24 h postinfection. Infection was performed apically, as described above. Cells exposed to infection media only were used as control. 72 h postinfection, cells were rinsed with HBSS and harvested for viral RNA measurement by RT-PCR analysis as previously described for the BetaCoV/Australia/VIC01/2020 isolate, and compared to a 2 h infected (untreated) control.
K18-hACE2 mouse experiments
Mouse experiments are approved by the QIMR Berghofer MRI Biosafety Committee and Animal Ethics Committee (project P3600), and are conducted in accordance with the “Australian Code for the care and use of animals for scientific purposes” as defined by the National Health and Medical Research Council of Australia. Work was conducted in a dedicated suite in a biosafety level-3 (PC3) facility at the QIMR Berghofer MRI (Australian Department of Agriculture, Water and the Environment certification Q2326 and Office of the Gene Technology Regulator certification 3445). Heterozygous K18-hACE2-transgenic mice (The Jackson Laboratory, Bar Harbour, ME, USA) were bred in-house by crossing with C57BL/6J mice (Animal Resources Center, Canning Vale, WA, Australia).68 DNA from tail tips was isolated using Extract-N-Amp Tissue PCR kit (Sigma) and PCR genotyping undertaken as described (The Jackson Laboratory. Genotyping protocols database. B6.Cg-Tg(K18-ACE2)2Prlmn/J. Stock No: 034860. Protocol 37654), except using primers Forward −5′-CTTGGTGATATGTGGGGTAGA-3′, reverse 5′ CGCTTCATCTCCCACCACTT-3′ (recommended by NIOBIOHN, Osaka, Japan).
Female 6–8 week old K18-hACE2 mice (n = 4 per group) were treated once with 16 mg/kg of pixatimod in 200 μL of saline i.p. on the day before infection. Control K18-hACE2 mice received 200 μL of saline on the day before infection. Age and gender matched C57BL/6J mice (n = 4) were included as a drug toxicity control and received 16 mg/kg of pixatimod in 200 μL of saline i.p., but were not infected with virus.
hCoV-19/Australia/QLD02/2020 (QLD02) (GISAID Accession ID; EPI_ISL_407896) virus was used to inoculate K18-hACE2 mice (n = 4 per group) intranasally with 104 CCID50 of virus in 50 μL of medium while under light anesthesia: 3% isoflurane (Piramal Enterprises Ltd., Andhra Pradesh, India) delivered using The Stinger, Rodent Anesthesia System (Advanced Anaesthesia Specialists/Darvall, Gladesville, NSW, Australia). Mice were weighed daily and euthanized on day 5 postinfection using CO2. Tissues were fixed for histology or were weighed and then homogenized using four ceramic beads at 6500 rpm twice for 15 s (Precellys 24 Homogenizer, Bertin Instruments, Montigny-le-Bretonneux, France). After centrifugation for 10 min (9400g at 4 °C), virus titers in supernatants were determined by CCID50 assays using Vero E6 cells. All CCID50 data are per gram of wet tissue. Tissue was weighed before being homogenized.
Statistical Analysis
Experimental data are presented as means ± SD, SEM or CV as noted. Statistical analyses were performed using analysis of a two-tailed Student’s t test with GraphPad Prism (GraphPad software) unless otherwise noted. Differences were considered statistically significant if the P value was less than 0.05. Statistical analysis for the K18-hACE2 mouse work was performed using IBM SPSS Statistics for Windows, Version 19.0 (IBM Corp., Armonk, NY). The t test was used when the data were deemed parametric; i.e., the difference in variances was <4, skewness was >2, and kurtosis was <2. Otherwise the nonparametric Kolmogorov–Smirnov test was used.
Safety Statement
No unexpected, new, or significant safety hazards were associated with the reported work.
Acknowledgments
The authors would like to thank Zucero Therapeutics for provision of pixatimod and Queensland Health Forensic & Scientific Services, Queensland Department of Health, for the provision of QLD02 and QLD935 SARS-CoV-2 isolates. V. Ferro acknowledges support from the Australian Research Council (DP170104431). T. Bergström acknowledges support of the Swedish Research Council. A. A. Khromykh, and A. Suhrbier acknowledge funding support from the Australian Infectious Diseases Research Centre. Computational (and/or data visualization) resources and services used in this work were provided by the eResearch Office, Queensland University of Technology, Brisbane, Australia, and with the assistance of resources from the National Computational Infrastructure (NCI Australia), an NCRIS enabled capability supported by the Australian Government. N. S. Gandhi is supported through the Advance Queensland Industry Research Fellowship. A. Suhrbier is supported by an Investigator grant from the National Health and Medical Research Council of Australia and acknowledges philanthropic support from inter alia Clive Berghofer and Lyn Brazil, as well as contract R&D funding from Zucero. M. A. Skidmore, S. E. Guimond, C. J. Mycroft-West, and J. E. Turnbull acknowledge the support of the University of Keele. J. E. Turnbull and E. A. Yates acknowledge the support of the University of Liverpool and contract R&D funding from Zucero. Z. Yang acknowledges the Danish National Research Foundation (DNRF107) and the Lundbeck Foundation. Y.-H. Chen acknowledges the Innovation Fund Denmark. R. Karlsson acknowledges the European Commission (GlycoImaging H2020-MSCA-ITN-721297).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c01293.
Additional data and figures including binding free energies, per-residue decomposition free energy, 2D schematic, amino acid residues, pairwise decomposition free energy, circular dichroism spectra, secondary structure content, binding of EcS1-RBD to Vero cells, structures, and microneutralization assay results (PDF)
Animation of SARS-CoV-2 spike trimer overlaid with pixatimod docked pose-b (MP4)
Author Contributions
◮ S. E. Guimond, C. J. Mycroft-West, N. S. Gandhi, J. A. Tree, T. T. Le, and C. M. Spalluto contributed equally. D. Watterson, A. A. Khromykh, A. Suhrbier, M. W. Carroll, E. Trybala, T. Bergström, V. Ferro, M. A. Skidmore, and J. E. Turnbull are equal senior authors. J. E. Turnbull, V. Ferro, M. A. Skidmore, and K. Dredge conceived the project. S. E. Guimond, C. J. Mycroft-West, N. S. Gandhi, J. A. Tree, T. T. Le, C. M. Spalluto, M. V. Humbert, K. R. Buttigieg, N. Coombes, M. J. Elmore, M. Wand, K. Nyström, J. Said, Y. X. Setoh, A. A. Amarilla, N. Modhiran, J. D. J. Sng, M. Chhabra, D. Watterson, P. R. Young, D. J. Rawle, A. A. Khromykh, M. A. Lima, E. A. Yates, R. Karlsson, R. L. Miller, Y.-H. Chen, Z. Yang, D. Nguyen, S. Laidlaw, E. Trybala, M. A. Skidmore, T. M. A. Wilkinson, and A. Suhrbier designed and conducted the experiments and undertook analyses. V. Ferro, E. Hammond, K. Dredge, M. W. Carroll, J. Stewart, T. Bergström, M. A. Skidmore, and J. E. Turnbull analyzed results and prepared the manuscript.
The authors declare the following competing financial interest(s): E. Hammond and K. Dredge are employees of Zucero Therapeutics. V. Ferro, E. Hammond, and K. Dredge are inventors on pixatimod patents.
Notes
Data and Materials Availability. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supporting Information. Additional data related to this paper may be requested from the authors.
Supplementary Material
References
- Akhmerov A.; Marbán E. COVID-19 and the Heart. Circ. Res. 2020, 126, 1443–1455. 10.1161/CIRCRESAHA.120.317055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagno V.; Tseligka E. D.; Jones S. T.; Tapparel C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias?. Viruses 2019, 11, 596. 10.3390/v11070596. [DOI] [Google Scholar]
- Milewska A.; Zarebski M.; Nowak P.; Stozek K.; Potempa J.; Pyrc K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol 2014, 88, 13221–13230. 10.1128/JVI.02078-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicenzi E.; Canducci F.; Pinna D.; Mancini N.; Carletti S.; Lazzarin A.; Bordignon C.; Poli G.; Clementi M. Coronaviridae and SARS-associated coronavirus strain HSR1. Emerg Infect Dis 2004, 10, 413–418. 10.3201/eid1003.030683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Schroeder S.; Krüger N.; Herrler T.; Erichsen S.; Schiergens T. S.; Herrler G.; Wu N. H.; Nitsche A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mycroft-West C.; Su D.; Elli S.; Guimond S.; Miller G.; Turnbull J.; Yates E.; Guerrini M.; Fernig D.; Lima M.; et al. The 2019 coronavirus (SARS-CoV-2) surface protein (Spike) S1 Receptor Binding Domain undergoes conformational change upon heparin binding. bioRxiv, 2020.2002.2029.971093, 2020. https://doi.org/10.1101/2020.02.29.971093.
- Mycroft-West C. J.; Su D.; Li Y.; Guimond S. E.; Rudd T. R.; Elli S.; Miller G.; Nunes Q. M.; Procter P.; Bisio A.; et al. SARS-CoV-2 Spike S1 Receptor Binding Domain undergoes Conformational Change upon Interaction with Low Molecular Weight Heparins. bioRxiv, 2020.2004.2029.068486, 2020. https://doi.org/10.1101/2020.04.29.068486.
- Mycroft-West C. J.; Su D.; Pagani I.; Rudd T. R.; Elli S.; Guimond S. E.; Miller G.; Meneghetti M. C. Z.; Nader H. B.; Li Y.; et al. Heparin inhibits cellular invasion by SARS-CoV-2: structural dependence of the interaction of the surface protein (spike) S1 receptor binding domain with heparin. Thrombosis and Haemostasis. 2020, 120 (12), 1700–1715. 10.1055/s-0040-1721319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Chopra P.; Li X.; Wolfert M. A.; Tompkins S. M.; Boons G. J.. SARS-CoV-2 spike protein binds heparan sulfate in a length- and sequence-dependent manner. bioRxiv, 2020. https://doi.org/10.1101/2020.05.10.087288.
- Clausen T. M.; Sandoval D. R.; Spliid C. B.; Pihl J.; Perrett H. R.; Painter C. D.; Narayanan A.; Majowicz S. A.; Kwong E. M.; McVicar R. N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183 (4), 1043–1057. 10.1016/j.cell.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilanova E.; Tovar A. M. F.; Mourão P.a.S. Imminent risk of a global shortage of heparin caused by the African Swine Fever afflicting the Chinese pig herd. J. Thromb Haemost 2019, 17, 254–256. 10.1111/jth.14372. [DOI] [PubMed] [Google Scholar]
- Ferro V.; Liu L.; Johnstone K. D.; Wimmer N.; Karoli T.; Handley P.; Rowley J.; Dredge K.; Li C. P.; Hammond E.; et al. Discovery of PG545: a highly potent and simultaneous inhibitor of angiogenesis, tumor growth, and metastasis. J. Med. Chem. 2012, 55, 3804–3813. 10.1021/jm201708h. [DOI] [PubMed] [Google Scholar]
- Dredge K.; Brennan T. V.; Hammond E.; Lickliter J. D.; Lin L.; Bampton D.; Handley P.; Lankesheer F.; Morrish G.; Yang Y.; et al. A Phase I study of the novel immunomodulatory agent PG545 (pixatimod) in subjects with advanced solid tumours. Br. J. Cancer 2018, 118, 1035–1041. 10.1038/s41416-018-0006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamaysi I.; Singh P.; Nasser S.; Awad H.; Chowers Y.; Sabo E.; Hammond E.; Gralnek I.; Minkov I.; Noseda A.; et al. The Role of Heparanase in the Pathogenesis of Acute Pancreatitis: A Potential Therapeutic Target. Sci. Rep 2017, 7, 715. 10.1038/s41598-017-00715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said J. S.; Trybala E.; Görander S.; Ekblad M.; Liljeqvist J.; Jennische E.; Lange S.; Bergström T. The Cholestanol-Conjugated Sulfated Oligosaccharide PG545 Disrupts the Lipid Envelope of Herpes Simplex Virus Particles. Antimicrob. Agents Chemother. 2016, 60, 1049–1057. 10.1128/AAC.02132-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said J.; Trybala E.; Andersson E.; Johnstone K.; Liu L.; Wimmer N.; Ferro V.; Bergström T. Lipophile-conjugated sulfated oligosaccharides as novel microbicides against HIV-1. Antiviral Res. 2010, 86, 286–295. 10.1016/j.antiviral.2010.03.011. [DOI] [PubMed] [Google Scholar]
- Lundin A.; Bergström T.; Andrighetti-Fröhner C. R.; Bendrioua L.; Ferro V.; Trybala E. Potent anti-respiratory syncytial virus activity of a cholestanol-sulfated tetrasaccharide conjugate. Antiviral Res. 2012, 93, 101–109. 10.1016/j.antiviral.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Supramaniam A.; Liu X.; Ferro V.; Herrero L. J. Prophylactic Antiheparanase Activity by PG545 Is Antiviral In Vitro and Protects against Ross River Virus Disease in Mice. Antimicrob. Agents Chemother. 2018, 62 (4), 17. 10.1128/AAC.01959-17. [DOI] [Google Scholar]
- Modhiran N.; Gandhi N. S.; Wimmer N.; Cheung S.; Stacey K.; Young P. R.; Ferro V.; Watterson D. Dual targeting of dengue virus virions and NS1 protein with the heparan sulfate mimic PG545. Antiviral Res. 2019, 168, 121–127. 10.1016/j.antiviral.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Miller B. R.; McGee T. D.; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory and Computation. 2012, 8, 3314–3321. 10.1021/ct300418h. [DOI] [Google Scholar]
- Toelzer C.; Gupta K.; Yadav S. K. N.; Borucu U.; Davidson A. D.; Kavanagh Williamson M.; Shoemark D. K.; Garzoni F.; Staufer O.; Milligan R.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370, 725–730. 10.1126/science.abd3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemark D. K.; Colenso C. K.; Toelzer C.; Gupta K.; Sessions R. B.; Davidson A. D.; Berger I.; Schaffitzel C.; Spencer J.; Mulholland A. J. Molecular Simulations suggest Vitamins, Retinoids and Steroids as Ligands of the Free Fatty Acid Pocket of the SARS-CoV-2 Spike Protein. Angew. Chem., Int. Ed. Engl. 2021, 60, 7098–7110. 10.1002/anie.202015639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tree J. A.; Turnbull J. E.; Buttigieg K. R.; Elmore M. J.; Coombes N.; Hogwood J.; Yates E. A.; Gray E.; Singh D.; Wilkinson T.; et al. Unfractionated heparin inhibits live wild-type SARS-CoV-2 cell infectivity at therapeutically relevant concentrations. Br. J. Pharmacol. 2021, 178, 626–635. 10.1111/bph.15304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B.; Fischer W. M.; Gnanakaran S.; Yoon H.; Theiler J.; Abfalterer W.; Hengartner N.; Giorgi E. E.; Bhattacharya T.; Foley B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020, 182, 812–827. 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume C.; Jackson C. L.; Spalluto C. M.; Legebeke J.; Nazlamova L.; Conforti F.; et al. A novel ACE2 isoform is expressed in human respiratory epithelia and is upregulated in response to interferons and RNA respiratory virus infection. Nat. Genet. 2021, 53, 205–214. 10.1038/s41588-020-00759-x. [DOI] [PubMed] [Google Scholar]
- Walters M. S.; Gomi K.; Ashbridge B.; Moore M. A.; Arbelaez V.; Heldrich J.; et al. Generation of a human airway epithelium derived basal cell line with multipotent differentiation capacity. Respir Res. 2013, 14 (1), 135. 10.1186/1465-9921-14-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarilla A. A.; Sng J. D. J.; Parry R.; Deerain J. M.; Potter J. R.; Setoh Y. X.; Rawle D. J.; Le T. T.; Modhiran N.; Wang X.; et al. A versatile reverse genetics platform for SARS-CoV-2 and other positive-strand RNA viruses. Nat. Commun. 2021, 12, 3431. 10.1038/s41467-021-23779-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari P.; Rothan H. A.; Natekar J. P.; Stone S.; Pathak H.; Strate P. G.; Arora K.; Brinton M. A.; Kumar M. Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2Mice. Viruses. 2021, 13, 132. 10.3390/v13010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carossino M.; Montanaro P.; O’Connell A.; Kenney D.; Gertje H.; Grosz K.; Ericsson M.; Huber B. R.; Subramaniam S.; Kirkland T. A.; et al. Fatal neuroinvasion and SARS-CoV-2 tropism in K18-hACE2 mice is partially independent on hACE2 expression. bioRxiv, 2021. https://doi.org/10.1101/2021.01.13.425144.
- Shen W.-B.; Logue J.; Yang P.; Baracco L.; Elahi M.; Reece E. A.; Wang B.; Li L.; Blanchard T. G.; Han Z.; et al. SARS-CoV-2 invades cognitive centers of the brain and induces Alzheimer’s-like neuropathology. bioRxiv, 2022. https://doi.org/10.1101/2022.01.31.478476.
- Gray-Rodriguez S.; Jensen M. P.; Otero-Jimenez M.; Hanley B.; Swann O. C.; Ward P. A.; Salguero F. J.; Querido N.; Ildiko Farkas I.; Velentza-Almpani E.. Multisystem screening reveals SARS-CoV-2 in neurons of the myenteric plexus and in megakaryocytes. J. Pathol. 2022, in press. 10.1002/path.5878 [DOI] [Google Scholar]
- Wenzel J.; Lampe J.; Müller-Fielitz H.; Schuster R.; Zille M.; Müller K.; Krohn M.; Körbelin J.; Zhang L.; Özorhan U.; et al. The SARS-CoV-2 main protease M pro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24 (11), 1522–1533. 10.1038/s41593-021-00926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau G. B.; et al. Evaluation of K18-hACE2Mice as a Model of SARS-CoV-2 Infection. Am. J. Trop Med. Hyg 2020, 103, 1215–1219. 10.4269/ajtmh.20-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl U.; Li J. P. Heparin - an old drug with multiple potential targets in Covid-19 therapy. J. Thromb Haemost. 2020, 18, 2422. 10.1111/jth.14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Du Y.; Igor A.; Kaltashov I. A.. The utility of native MS for understanding the mechanism of action of repurposed therapeutics in COVID-19: heparin as a disruptor of the SARS-CoV-2 interaction with its host cell receptor. bioRxiv, 2020. https://doi.org/10.1101/2020.06.09.142794.
- Partridge L. J.; Green L. R.; Monk P. N.. Unfractionated heparin potently inhibits the binding of SARS-CoV-2 spike protein to a human cell line. bioRxiv, 2020.05.21.107870, 2020. https://doi.org/10.1101/2020.05.21.107870.
- Tandon R.; Sharp J. S.; Zhang F.; Pomin V. H.; Ashpole N. M.; Mitra D.; Jin W.; Liu H.; Sharma P.; Linhardt R. J.. Effective Inhibition of SARS-CoV-2 Entry by Heparin and Enoxaparin Derivatives. bioRxiv, 2020.06.08.140236, 2020. https://doi.org/10.1101/2020.06.08.140236.
- van Haren F. M. P.; van Loon L. M.; Steins A.; Smoot T. L.; Sas C.; Staas S.; Vilaseca A. B.; Barbera R. A.; Vidmar G.; Beccari H.. Inhaled nebulised unfractionated heparin for the treatment of hospitalised patients with COVID-19: A multicentre case series of 98 patients. Br. J, Clin. Pharm. 2022, in press. 10.1111/bcp.15212 [DOI] [Google Scholar]
- Bargellini I.; Cervelli R.; Lunardi A.; Scandiffio R.; Daviddi F.; Giorgi L.; Cicorelli A.; Crocetti L.; Cioni R. Spontaneous Bleedings in COVID-19 Patients: An Emerging Complication. Cardiovasc Intervent Radiol 2020, 43, 1095. 10.1007/s00270-020-02507-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dredge K.; Hammond E.; Handley P.; Gonda T. J.; Smith M. T.; Vincent C.; Brandt R.; Ferro V.; Bytheway I. PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. Br. J. Cancer 2011, 104, 635–642. 10.1038/bjc.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyango I.; Barash U.; Naroditsky I.; Li J. P.; Hammond E.; Ilan N.; Vlodavsky I. Heparanase cooperates with Ras to drive breast and skin tumorigenesis. Cancer Res. 2014, 74, 4504–4514. 10.1158/0008-5472.CAN-13-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zuk N.; Dechtman I.; Henn I.; Weiss L.; Afriat A.; Krasner E.; Gal Y. Potential prophylactic treatments for Covid19. Viruses 2021, 13, 1292. 10.3390/v13071292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon B.; et al. Nebulised heparin for patients with or at risk of acute respiratory distress syndrome: a multicentre, randomised, double-blind, placebo-controlled phase 3 trial. Lancet Resp. Med. 2021, 9, 360. 10.1016/S2213-2600(20)30470-7. [DOI] [Google Scholar]
- Winkler E. S.; Bailey A. L.; Kafai N. M.; Nair S.; McCune B. T.; Yu J.; Fox J. M.; Chen R. E.; Earnest J. T.; Keeler S. P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nature Immunol. 2020, 21, 1327–1335. 10.1038/s41590-020-0778-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machhi J.; et al. The Natural History, Pathobiology, and Clinical Manifestations of SARS-CoV-2 Infections. J. Neuroimmune Pharmacol 2020, 15, 359–386. 10.1007/s11481-020-09944-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui K. P. Y.; Ho J. C. W.; Cheung M. C.; Ng K.; Ching R. H. H.; Lai K.; Kam T. T.; Gu H.; Sit K.-Y.; Hsin M. K. Y.. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, in press. 10.1038/s41586-022-04479-6 [DOI] [Google Scholar]
- Ylikoski J.; Markkanen M.; Mäkitie A. Pathophysiology of the COVID-19 - entry to the CNS through the nose. Acta Otolaryngol 2020, 140, 886–889. 10.1080/00016489.2020.1773533. [DOI] [PubMed] [Google Scholar]
- Koenig P.-A.; Das H.; Liu H.; Kummerer B. M.; Gohr F. N.; Jenster L.-M.; Schiffelers L. D. J.; Tesfamariam Y. M.; Uchima M.; Wuerth J. D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371, 6530. 10.1126/science.abe6230. [DOI] [Google Scholar]
- Mehandru S.; Merad M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022, 23 (2), 194–202. 10.1038/s41590-021-01104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M.; Martin J. C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol 2020, 20, 355–362. 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna M.; Ranasinghe C.; Browne A. M.; Li J. P.; Vlodavsky I.; Parish C. R. Is host heparanase required for the rapid spread of heparan sulfate binding viruses?. Virology 2019, 529, 1–6. 10.1016/j.virol.2019.01.001. [DOI] [PubMed] [Google Scholar]
- Buijsers B.; Yanginlar C.; de Nooijer A.; Grondman I.; Maciej-Hulme M. L.; Jonkman I.; Janssen N. A. F.; Rother N.; de Graaf M.; Pickkers P.; et al. Increased plasma heparanase activity in COVID-19 patients. Front. Immunol. 2020, 11, 575047. 10.3389/fimmu.2020.575047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zhang Y.; Wu L.; Niu S.; Song C.; Zhang Z.; Lu G.; Qiao C.; Hu Y.; Yuen K. Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell. 2020, 181, 894–904. 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Mannar D.; Srivastava S. S.; Berezuk A. M.; Demers J. P.; Saville J. W.; Leopold K.; Li W.; Dimitrov D. S.; Tuttle K. S.; Zhou S.; et al. Cryo-electron microscopy structures of the N501Y SARS-CoV-2 spike protein in complex with ACE2 and 2 potent neutralizing antibodies. PLoS Biol. 2021, 19, e3001237. 10.1371/journal.pbio.3001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boittier E. D.; Burns J. M.; Gandhi N. S.; Ferro V. GlycoTorch Vina: Docking Designed and Tested for Glycosaminoglycans. J. Chem. Inf Model. 2020, 60, 6328–6343. 10.1021/acs.jcim.0c00373. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Götz A. W.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Cheatham Iii T. E.; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber biomolecular simulation programs. Journal of computational chemistry 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner K. N.; Yongye A. B.; Tschampel S. M.; González-Outeiriño J.; Daniels C. R.; Foley B. L.; Woods R. J. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej B. D.; Gould I. R.; Walker R. C. A Parameterization of Cholesterol for Mixed Lipid Bilayer Simulation within the Amber Lipid14 Force Field. J. Phys. Chem. B 2015, 119, 12424–12435. 10.1021/acs.jpcb.5b04924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohlke H.; Kiel C.; Case D. A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 2003, 330, 891–913. 10.1016/S0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 2015, 10, 449–461. 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onufriev A.; Bashford D.; Case D. A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- Micsonai A.; Wien F.; Kernya L.; Lee Y. H.; Goto Y.; Réfrégiers M.; Kardos J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E3095–E3103. 10.1073/pnas.1500851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uniewicz K. A.; Orio A.; Xu R.; Ahmed Y.; Wilkinson M.; Fernig G. D.; Yates A. E. Differential Scanning Fluorimetry Measurements of Protein Stability Changes upon Binding to Glycosaminoglycans: A Screening Test for Binding Specificity. Anal. Chem. 2010, 82 (9), 3796–3802. 10.1021/ac100188x. [DOI] [PubMed] [Google Scholar]
- Bewley K. R.; Coombes N. S.; Gagnon L.; McInroy L.; Baker N.; Shaik I.; St-Jean J. R.; St-Amant N.; Buttigieg K. R.; Humphries H. E.; et al. Quantification of SARS-CoV-2 neutralizing antibody by wild-type plaque reduction neutralization, microneutralization and pseudotyped virus neutralization assays. Nat. Protoc. 2021, 16, 3114–3140. 10.1038/s41596-021-00536-y. [DOI] [PubMed] [Google Scholar]
- Bishop C. R.; Dumenil T.; Rawle D. J.; Le T. T.; Yan K.; Tang B.; Hartel G.; Suhrbier A.. Mouse models of COVID-19 recapitulate inflammatory pathways rather than gene expression. bioRxiv, 2022/481866, 2022. https://doi.org/10.1101/2022.02.24.481866.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.