

Abstract
Background
AP4 (TFAP4) encodes a basic helix-loop-helix leucine zipper (bHLH-LZ) transcription factor and is a direct target gene of the oncogenic transcription factor c-MYC. Here, we set out to determine the relevance of AP4 in human colorectal cancer (CRC) cells.
Methods
A CRISPR/Cas9 approach was employed to generate AP4-deficient CRC cell lines with inducible expression of c-MYC. Colony formation, β-gal staining, immunofluorescence, comet and homologous recombination (HR) assays and RNA-Seq analysis were used to determine the effects of AP4 inactivation. qPCR and qChIP analyses was performed to validate differentially expressed AP4 targets. Expression data from CRC cohorts was subjected to bioinformatics analyses. Immunohistochemistry was used to evaluate AP4 targets in vivo. Ap4-deficient APCmin/+ mice were analyzed to determine conservation. Immunofluorescence, chromosome and micronuclei enumeration, MTT and colony formation assays were used to determine the effects of AP4 inactivation and target gene regulation on chromosomal instability (CIN) and drug sensitivity.
Results
Inactivation of AP4 in CRC cell lines resulted in increased spontaneous and c-MYC-induced DNA damage, chromosomal instability (CIN) and cellular senescence. AP4-deficient cells displayed increased expression of the long non-coding RNA MIR22HG, which encodes miR-22-3p and was directly repressed by AP4. Furthermore, Mediator of DNA damage Checkpoint 1 (MDC1), a central component of the DNA damage response and a known target of miR-22-3p, displayed decreased expression in AP4-deficient cells. Accordingly, MDC1 was directly induced by AP4 and indirectly by AP4-mediated repression of miR-22-3p. Adenomas and organoids from Ap4-deficient APCmin/+ mice displayed conservation of these regulations. Inhibition of miR-22-3p or ectopic MDC1 expression reversed the increased senescence, DNA damage, CIN and defective HR observed in AP4-deficient CRC cells. AP4-deficiency also sensitized CRC cells to 5-FU treatment, whereas ectopic AP4 conferred resistance to 5-FU in a miR-22-3p and MDC1-dependent manner.
Conclusions
In summary, AP4, miR-22-3p and MDC1 form a conserved and coherent, regulatory feed-forward loop to promote DNA repair, which suppresses DNA damage, senescence and CIN, and contributes to 5-FU resistance. These findings explain how elevated AP4 expression contributes to development and chemo-resistance of colorectal cancer after c-MYC activation.
Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s12943-022-01581-1.
Keywords: AP4, c-MYC, MIR22HG, miR-22-3p, MDC1, DNA damage, DNA repair, Homologous recombination, Chemo-resistance, Colorectal cancer
Background
Colorectal cancer (CRC) is one of the most common human malignancies accounting for approximately 10% of worldwide cancer incidence and mortality [1]. While early-stage CRC is curable by surgery, treatment of metastatic colorectal cancer (mCRC) remains an unmet clinical need. Moreover, about 25% of CRC cases are diagnosed only at the metastatic stage. Despite the extensive molecular and functional knowledge on this disease, systemic therapy for mCRC still relies on traditional 5-fluorouracil (5-FU)-based chemotherapy regimens [2]. So far, targeted therapies and immunotherapy have shown effectiveness only in a limited subset of CRC patients. Therefore, there is an urgent need to understand the molecular mechanisms and regulations underlying CRC development and treatment resistance in order to implement novel, rationally driven, tailored therapies.
AP4 (TFAP4) is a basic helix-loop-helix leucine zipper (bHLH-LZ) transcription factor that exclusively forms homodimers, which bind to the E-box motif CAGCTG [3]. We previously identified the AP4 gene as a direct transcriptional target of c-MYC [4]. In the intestine, AP4 expression is confined to progenitor and stem cells [4–6]. Deletion of AP4 causes premature senescence and defects in mitogen-induced proliferation in mouse embryonic fibroblasts (MEFs) [5, 7]. Furthermore, AP4 expression is strongly elevated in several types of cancer [5, 8, 9]. AP4 presumably contributes to the phenotype of cancer cells by activating or repressing genes that harbor CAGCTG elements in their promoter regions, thereby controlling processes such as proliferation, metabolism, apoptosis, epithelial-mesenchymal transition (EMT) and metastasis [5]. In addition, AP4 was shown to maintain a c-MYC-induced transcriptional program in murine T cells after exposure to IL2 once c-MYC is down-regulated [10]. Recently, we showed that Ap4 is critical for adenoma initiation and growth by controlling the homeostasis of intestinal stem cells in the APCmin/+ mouse model of intestinal cancer [6]. However, the molecular mechanisms by which the c-MYC/AP4 axis promotes CRC development and progression are still largely unknown.
In the current study, we show that AP4 deletion in CRC cells results in increased DNA damage, senescence, chromosomal instability and a decrease in homologous recombination (HR). Comparative analysis of transcriptional profiles obtained from AP4-deficient CRC cells implicated MIR22HG and MDC1/Mediator of DNA Damage Checkpoint as AP4 target gene candidates that may be relevant for the increase in DNA damage observed after AP4 deletion. Indeed, AP4 directly activated MDC1 expression and repressed miR-22-3p, which targets MDC1, thereby further enhancing MDC1 expression. In addition, we could show that the AP4/miR-22-3p/MDC1 axis is important for an effective response to spontaneous and c-MYC-induced DNA damage by increasing DNA repair by HR. Therefore, deregulation of the AP4/miR-22-3p/MDC1 axis by an activated Wnt/c-MYC pathway, which is a hallmark of CRCs, may ultimately contribute to chemo-resistance.
Materials and methods
Cell culture and treatments
The CRC cell lines DLD-1 and SW480 were cultured in McCoy’ 5A medium (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS) (Invitrogen) containing 100 units/ml penicillin and 0.1 mg/ml streptomycin in 20% O2, 5% CO2 and 37 °C. Doxycyclin (DOX) was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in water (100 μg/ml stock solution). The final concentration of DOX used in cell treatment was 100 ng/ml. To maintain cell pools harboring pRTR vectors, a final concentration of 8 μg/ml puromycin was used and changed fresh medium every 2 days. pRTR vectors and pcDNA3.1 vectors were transfected with Lipofectamine 2000 (Invitrogen). MiRNA mimic, siRNAs and negative controls were transfected with Lipofectamine RNAiMAX Transfection Reagent (Invitrogen). For transfections the final concentration was 500 ng/ml for plasmids and 12.5 nM for RNAs. miR-22-3p mimics, antagomirs and controls were purchased from Qiagen (Hilden, Germany). The sequence information of miR-22-3p mimic and controls are listed in Table S1.
Generation of AP4-deficient cell lines
To generate single cell clones with complete AP4 protein expression abrogation, a CRISPR/Cas9 approach was employed. We designed three guide RNAs (listed in Table S2) targeting exon 2 of the TFAP4/AP4 gene, and cloned each of them as two complementary DNA oligonucleotides into the BbsI sites of pSp-Cas9-GFP to generate single-guide (sg) RNA expression plasmids, as described previously [11]. DLD-1 and SW480 cells were then transfected with 2 μg of each pSp-Cas9-sgRNA-GFP plasmid, or transfected with “empty” pSp-Cas9-GFP harboring no guide RNA sequence. Forty-eight hours posttransfection, GFP-positive cells were sorted into 96-well plates using a FACSARIA cell sorter (BD Biosystems, NJ, USA) and expanded as single-cell clones for 2 weeks before screening by Western blot analysis. Cells transfected with pSp-Cas9-GFP harboring no guideRNA sequence were treated in a similar manner to obtain AP4 wild type single-cell clones.
Generation of pRTR-c-MYC-VSV pools
The DLD-1 AP4 WT1/KO2 and SW480 AP4 WT1/KO1 clones were subsequently used for the generation of pRTR-c-MYC-VSV pools as described previously [5]. The pRTR vector system allows the stringent control of c-MYC expression by addition of DOX to the media. Cell pools with more than 80% of the cells expressing a fluorescent marker protein from the pRTR vector were generated by selection for 2 weeks. The percentage of GFP-positive cells in pools derived from AP4 WT and KO clones harboring pRTR-c-MYC-VSV was determined by flow cytometry (CFlow6; Accuri, Ann Arbor, MI, USA).
Sample isolation from APCmin/+ mice with deletion of Ap4
Generation of APCmin/+ mice with inactivation of Ap4 in intestinal epithelial cells (IEC), isolation of samples and derivation of organoids from IECs and adenomas from these mice has been described before [6]. Mice were kept in individually ventilated cages with a 12-hour light/dark cycle and ad libitum access to water and standard rodent diet. All animal experimentations and analyses were approved by the Government of Upper Bavaria, Germany (AZ 55.2–1-54-2532-4-2014).
Beta-galactosidase (β-gal) staining
β-gal staining was performed according to instructions provided in Senescence β-Galactosidase Staining Kit (#9860, Cell Signaling Technology, Massachusetts, USA). Briefly, cells were seeded into 6-well plate at the density of 2 × 105 cells/well. For fixation, cells were washed once with PBS and then fixed for 30 min. Next, fixed cells were stained with β-gal staining solution containing X-gal overnight at 37 °C. Cells were imaged by using a microscope (Axiovert 25, Zeiss, Jena, Germany) with Axiovision software (Version 4.8.0.0, Zeiss).
Immunofluorescence analysis
For detection by indirect immunofluorescence cells were seeded on a round glass in a 6-well plate at the density of 2 × 105 cells/well. After different treatments, cells were fixed with 4% paraformaldehyde in PBS for 20 min. Then 0.2% Triton X 100 was used to permeabilize cells for 5 min at room temperature. Then cells were blocked in 1% BSA/PBS for 1 h at room temperature. γH2AX and MDC1 were detected using the respective antibodies listed in Table S3. Cellular chromatin was stained by DAPI (Roche, Switzerland). Stained cells were covered with ProLong Gold antifade (Invitrogen). Image acquisition was performed with a confocal microscope (LSM 700, Zeiss) and the ZEN 2009 software (Zeiss). Foci quantification was performed with Image J software. Cells with over 10 foci were considered as positive. The fluorescence intensity was normalized to DAPI. For each condition at least three microscope fields with a total of 150 cells were quantified.
Comet assay
The comet assay was conducted according to instructions provided in Comet Assay Kit (3-well slides, ab238544, Abcam, USA). After detachment by trypsin cells were suspended in ice-cold PBS at 1 × 105 cells/ml. Suspended cells were combined with pre-heated comet agarose (37 °C) at a 1/10 ratio (v/v) and mixed by pipetting. 70–80 μl cell-agarose mixture was added to comet agarose base layer and incubated at 4 °C for 15 min in the dark. Then the slides with agarose were immersed into 25 ml ice-cold lysis buffer for 60 min at 4 °C in the dark. Lysis buffer was replaced with ice-cold alkaline solution and incubated for 30 min at 4 °C in the dark. Slides were immersed with ice-cold alkaline electrophoresis solution for 5 min and then subjected to electrophoresis in cold alkaline electrophoresis solution (electrophoresis condition: 1 V/cm for 10–15 min). Subsequently, slides were immersed twice with pre-chilled water for 2 min. Finally, slides were immersed into 70% ice-cold Ethanol for 1 min and placed in the dark for drying. Signals were evaluated using a confocal microscope (LSM 700, Zeiss) with a FITC filter. For data analysis and DNA tail moment determination the ImageJ software in conjunction with the OpenComet plugin [12] was used.
RNA isolation and quantitative real-time polymerase chain reaction (qPCR) analysis
Total RNA of cells was isolated and purified by using High Pure RNA Isolation Kit (Roche) based on the protocol provided by the manufacturer. Total RNA from ApcMin/+ adenomas was isolated using the RNAeasy Kit (QIAGEN). 5 adenomas per mouse were used for each sample. For each sample, 1 μg RNA was used to generate cDNA via using Verso cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). For quantitative real-time polymerase chain reaction (qPCR), a Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA) was used to perform the reaction in LightCycler 480 (Roche) system. Gene expression was normalized to GAPDH or β-actin with the ΔΔCt method [13]. Experiments were performed in triplicates. Sequence information of the primers is provided in Table S4.
Western blot analysis
Cells were lysed in RIPA lysis buffer (50 mM Tris/HCl, pH 8.0, 250 mM NaCl, 1% NP40, 0.5% [w/v] sodium deoxycholate, 0.1% SDS) containing mini protease inhibitors (Roche) and PhosSTOP Phosphatase Inhibitor Cocktail Tablets (Roche). Cell lysates were sonicated for 5 seconds in each sample and centrifuged at 13,000 rpm for 20 min at 4 °C. Supernatants containing proteins were collected and quantified by Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). For each sample, total 40 μg protein was loaded and separated by 10% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis. Immobilon PVDF membranes (Millipore, Burlington, MA, USA) were used for transferring after electrophoresis using standard protocols (Bio-Rad Laboratories, Hercules, CA). ECL (Millipore) system was used and imaged through LI-COR Odyssey FC imaging system (Bad Homburg, Germany). Antibodies are provided in Table S3.
Immunohistochemical analysis
FFPE tissue was cut into 2 μm sections on a microtome and de-paraffinized. After antigen retrieval slides were incubated with primary antibody (the primary antibodies used are listed in Table S3) over night at 4 °C and washed with Tris-HCL (Tris hydrochloride) buffer (pH 7.5) followed by a secondary antibody. Antibodies were detected with the Vectastain Elite ABC (avidin-biotin complex) kit (Vector) using DAB (3,3′-diaminobenzidine) (Vector Laboratories and Dako) for brown stainings or AEC (3-Amino-9-ethylcarbazole) (Thermo Fisher Scientific) for magenta stainings. The slides were counterstained with hematoxylin (Vector Laboratories) and mounted with Roti®-Histokitt II (Carl Roth, Germany). Images were captured on an Axioplan2 imaging microscope (Carl Zeiss) equipped with an AxioCamHRc Camera (Carl Zeiss) or slides were scanned with a Vectra Polaris imaging system (PerkinElmer, Hopkinton, MA, USA) and quantified by ImageJ software.
Chromatin immunoprecipitation
Chromatin immunoprecipitation in DLD-1 pRTR-AP4-VSV cells was performed according to instruction provided in the iDeal ChIP-qPCR kit (Diagenode, Belgium). The sequence information of qChIP primers is provided in Table S5.
Micronucleus assessment
Cells were washed twice with HBSS and then fixed in 4% PFA at room temperature for 30 min. After fixation, cells were incubated with 1 μg/ml 4′6-diamidino-2-phenylindole (DAPI, Sigma) dissolved in HBSS for 5 min. Then cells were covered with ProLong™ Gold Antifade (Thermo Fisher). In order to reveal cell margins filamentous actin (F-actin) was stained with phalloidin (Alexa Fluor™ 488 Phalloidin, Thermo Fisher) at 1:50 dilution in 1% BSA. Micronuclei were microscopically determined as rounded chromatin fragments located adjacent to nuclei with a diameter not exceeding one third of the diameter of the neighboring nucleus. Microscopic analysis was performed by using a LSM 700 system (Zeiss) equipped with 63x Plan-Apochromat oil-immersion lens and ZEN 2009 software (Zeiss). For quantification, three different microscope fields were selected, and in each field, a total 50–100 cells were counted.
Mitotic spreads and chromosome enumeration
1 × 107 cells were seeded to a T-75 flask to grow 24–48 h until they reached a confluence of 90%. Cells were treated with Colcemid at a final concentration of 0.02 μg/ml for 4 h. After incubation, cells were detached by 0.5 ml 0.05% trypsin and collected in a 15 ml falcon at 160 g for 10 min. The cell pellet was resuspended in 0.5 ml of the remaining supernatant and incubated in 5 ml 0.075 M KCl solution for 30 min at room temperature. After centrifugation the pellet was resuspended in methanol-glacial acetic acid (3:1) solution. This was repeated 3 times. A small volume (5–10 μl) of cell suspension was dropped on a slide vertically using a Pasteur pipette and air dried. Chromosomes were stained by 1 μg/ml DAPI and embedded in ProLong™ Gold Antifade (Thermo Fisher). Chromosome spreading was evaluated and documented using LSM 700 system (Zeiss) equipped with 63x Plan-Apochromat oil-immersion lens. For quantification, a total 50 cells were counted in each condition.
MTT assay
Cell viability was measured using a MTT assay. Briefly, cells were seeded in 96-well plates at 3 × 103 cells/well. Before treatment with 5-FU, cells were transfected with indicated siRNAs, miRNA mimic and vectors. After 48 h, cells were treated with different doses of 5-FU for 48 h. Then 10 μl MTT solution was added per well at the concentration of 0.5 μg/μl for 4 h. The resulting Formazan was dissolved in DMSO. After agitation of the plate the absorbance was measured at 570 nm by a Varioscan system (Thermo Fisher).
Colony formation assay
Cells were seeded into 6-well plates and transfected with indicated siRNAs, miRNA mimic and vectors for 24 h. Next, 10 μM of 5-FU was added into medium and cells were continuously cultivated for another 48 h. After finishing treatments, cells were resuspended and seeded into 12-well plates at a density of 1000 cells/well without 5-FU treatment. Colony formation was determined after 3 weeks. Colonies were recorded by a digital camera (Nikon, Japan) and enumerated by using Image J software.
Apoptosis detection with Annexin V
Apoptosis was determined by flow cytometry after staining with Annexin V-FITC (apoptotic cell marker) and PI (necrotic cell marker) with the Annexin V-FITC/PI staining kit (556,570; BD Pharmingen, San Diego, CA, USA) according to manufacturer’s instructions. In brief, cells were harvested by addition of 0.05% trypsin (EDTA free) and washed 3 times with 1 x HBSS (Gibco, USA). Then cells were resuspended in 1 x binding buffer (0.01 M HEPES/NaOH [pH 7.4], 0.14 M NaCl, 2.5 mM CaCl2) at a density of 1 x 106 cells/ml. 100 μl of cell suspension (1 x 105 cells) was incubated with 5 μl of FITC Annexin V and 5 μl of PI for 15 min at room temperature in the dark. Before flow cytometry (CFlow6; Accuri, Ann Arbor, MI), another 400 μl of 1 x binding buffer was added to each tube and the samples were analyzed within 1 hour. Apoptotic cells were determined using the BD Accuri C6 Plus software template (BD Biosciences) with FL1-H (Annexin V-FITC) and FL3-H (PI).
Assessment of proliferation by real-time impedance measurement
Cell proliferation was evaluated using impedance measurements (Xcelligence RTCA DP, Roche). Cells were seeded at a density of 3 x 103 cells per E-plate well and subjected to the indicated treatments. Impedance was recorded every 60 min for a period of up to 120 h. A dimension-less parameter named cell-index was used to represent the electric impedance. The calculations were performed by the RTCA software integrated in the Xcelligence system. To validate impedance measurements, cells were also seeded into 96-well plates in triplicates and counted at the end time point using a Neubauer-chamber.
Assessment of nascent RNA
To monitor de novo RNA synthesis the amount of nascent RNA was determined using the Click-iT™ Nascent RNA Capture Kit (C10365, Thermo Fisher) followed by qPCR analysis. Briefly, cells were seeded into a 12-well plate and labeled by 0.2 mM 5-ethynyl uridine (EU) for 12 h. Labeled RNA was isolated from cells by using the High Pure RNA Isolation Kit (Roche). 500 ng of labeled RNA was used for click reaction with biotin-azide, and then the reaction system was incubated with streptavidin T1 beads and washed 5 times with the provided wash-buffers. Bead-coupled RNAs were used for cDNA synthesis with the Verso cDNA Synthesis Kit (Thermo Fischer). cDNA was subjected to qPCR analysis.
3′-UTR dual reporter assay
The full length human MDC1 3’-UTR was PCR-amplified from cDNA obtained from DLD-1 cells. The PCR product was cloned into pGL3-control-MCS. To delete the miR-22-3p seed-matching sequence (SMS) in the MDC1 3′-UTR a QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA, USA) was used according to the manufacturer’s instructions. The miR-22-3p complementary sequence (miR-22-3p antisense) was cloned as complementary DNA oligonucleotides into pGL3-control-MCS. All plasmids were verified by Sanger sequencing. The oligonucleotides used for cloning and mutagenesis are listed in Table S6. For the dual reporter assays, DLD-1 cells were seeded into a 12-well plate at 3 × 104 cells/well and cultivated overnight. Transfections were performed using HiPerFect Transfection Reagent (Qiagen), 100 ng of indicated reporter vectors, and 20 ng Renilla plasmid as normalization control. After 48 h incubation with the indicated treatments, luciferase activity was measured with a Dual Luciferase Reporter assay kit (Promega) according to manufacturer’s instructions using an Orion II Microplate Luminometer (Berthold, Germany) and the Simplicity software package.
Homologous recombination assay
Homologous recombination (HR) activity was assessed as described previously [14]. Briefly, DLD-1 and SW480 cells were transfected with pDR-GFP and pCBAScel vectors (kind gifts from Maria Jasin (Memorial Sloan Kettering Cancer Center, NY, USA) using lipofectamine LTX (Invitrogen), and co-transfected with the indicated oligonucleotides or/and plasmids. To normalize for transfection efficiency, cells were co-transfected with pcDNA3.1-mCherry (RFP). After 72 h, the percentage of GFP-expressing cells among RFP-positive cells was quantified by flow cytometry (CFlow6; Accuri).
Bioinformatics analysis of online databases
Expression data from tumor samples and normal mucosa for relevant mRNAs was obtained and analyzed from TCGA-COAD downloaded from the National Cancer Institute’s Genomic Data Commons (https://gdc.cancer.gov/) and NCBI GEO (www.ncbi.nlm.nih.gov/geo) [15]. Expression data from colorectal cancer cell lines were obtained from the Cancer Cell Line Encyclopedia (CCLE) [16, 17]. PDX RNA expression data were obtained from GSE76402 [18]. Association of tumor samples with CMS categories was obtained and analyzed from the Cancer Subtyping Consortium (CRCSC) at www.synapse.org. AP4 ChIP-seq data from human COLO-320 cells and murine B cells, as well as c-MYC ChIP-seq data from LOVO cells were obtained from the Cistrome Data Browser (http://cistrome.org/db/#/). Expression and clinical data of the GSE14333 cohort was downloaded from NCBI GEO. The statistics for Kaplan-Meier survival curves was calculated by log-rank test. For binary classification of cases (high/low expression), the Survminer R-package (https://CRAN.R-project.org/package=survminer) was used to determine optimal cutoff values.
Statistics
Significant differences between two groups were calculated and analyzed by a Student’s t test (two-tailed; unpaired). For multiple group comparisons, we performed 1-way analysis of variance followed by a Tukey multiple comparisons post hoc test. For gene expression associations, Pearson’s correlation was employed. P values less than 0.05 were considered as significant differences (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Statistics were performed with Prism 8 (GraphPad Software, USA).
Results
Generation and characterization of AP4-deficient CRC cell lines
In order to generate AP4-deficient colorectal cancer cell lines, DLD-1 and SW480 cells were transfected with pSp-Cas9-GFP vectors expressing three guide RNAs targeting exon 2 of the TFAP4/AP4 gene, which encodes the DNA binding region of AP4 (Fig. 1A). Three AP4-deficient single cell derived clones were obtained for each cell line that showed a lack of AP4 protein expression (Fig. 1B and Fig. S1A). Interestingly, AP4-deficient DLD-1 and SW480 were often enlarged and flattened (Fig. 1C and Fig. S1B) and the number of cells positive for senescence-associated β-galactosidase at pH 6 (SA-β-gal) was significantly increased in AP4-deficient versus AP4-proficient DLD-1 cells. Ectopic expression of AP4 reduced the number of cells positive for SA-β-gal in AP4-deficient DLD-1 cells (Fig. 1D), confirming that the observed effect is due to loss of AP4 function. Therefore, AP4 suppresses senescence in CRC cells. In addition, the colony-forming capacity was decreased in AP4-deficient DLD-1 and SW480 cells (Fig. S1C). The frequency of apoptotic cells was increased in AP4-deficient DLD-1 and SW480 cells when compared to AP4-proficient cells. Again, ectopic expression of AP4 reversed this effect (Fig. 1E and Fig. S1E). Furthermore, the viability of AP4-deficient DLD-1 and SW480 cells was significantly lower when compared to AP4-proficient cells as determined in an MTT assay (Fig. 1F and Fig. S1F). AP4-deficient DLD-1 and SW480 cells also displayed a pronounced decrease in proliferation. Ectopic AP4 largely reverted the decreased proliferation of AP4-deficient DLD-1 and to a lesser degree that of AP4-deficient SW480 cells (Fig. 1G and Fig. S1G). The proliferation defect caused by AP4 deficiency observed here is presumably largely due the induction of senescence. Since senescence is known to result from DNA damage we evaluated the amount of spontaneous DNA damage. Indeed, untreated AP4-deficient DLD-1 cells displayed elevated levels of endogenous DNA damage as evidenced by a significant increase in γH2AX-positive foci and γH2AX protein levels when compared to AP4-proficient DLD-1 cells (Fig. 1H, I). The increase in γH2AX-positive foci and γH2AX levels in AP4-deficient cells was reversed by ectopic expression of AP4. In a comet assay, AP4-deficient DLD-1 cells showed a longer tail of unrepaired, damaged DNA when compared to AP4-proficient DLD-1 cells (Fig. 1J), suggesting that AP4-deficiency causes a defect in DNA repair. Ectopic expression of AP4 in AP4-deficient DLD-1 cells suppressed DNA damage. A similar effect of AP4-deficiency on DNA damage was observed in AP4-deficient SW480 cells (Fig. S2A-C). We have previously shown that expression of AP4 is directly induced by c-MYC in breast cancer cells, human diploid fibroblasts and SW620 CRC cells [4, 19]. In line with these findings, down-regulation of c-MYC by specific siRNAs resulted in a decrease in AP4 expression in DLD-1 and SW480 cells (Fig. 1K and Fig. S2D). Furthermore, activation of a conditional c-MYC allele by addition of DOX for up to 8 days resulted in more DNA damage in AP4-deficient than in AP4-proficient CRC cells (Fig. 1L and Fig. S2E-F). Taken together, the results indicate that AP4 suppresses spontaneous and c-MYC-induced DNA damage in CRC cells. This may be due to role of AP4 in facilitating the repair of DNA damage. Therefore, the suppression DNA of damage by AP4 may decrease the rates of senescence and apoptosis in CRC cells displaying enhanced expression of c-MYC.
Fig. 1.

AP4 inactivation induces DNA damage and senescence in CRC cells. A Scheme of targeting exon 2 (shown in red) of TFAP4/AP4 using CRISPR/Cas9. B AP4 detection by Western blot analysis. β-actin served as a loading control. C Phase contrast images of untreated cells. Scale bars: 50 μm. D Detection of senescent cells using pH 6 β-gal staining 48 h after transfection. Ectopic expression of AP4 was achieved using a pcDNA-AP4-VSV vector described in [4]. Three fields of 120 cells in total were evaluated. Scale bars: 100 μm. E Quantification of apoptotic cells by Annexin V detection 48 h after transfection. F MTT assay results obtained 72 h after seeding. G Proliferation was determined by impedance measurement in E-plates 48 h after transfection in the indicated cells (left panel). Cell numbers were determined at the last time point (right panel). H Detection of γH2AX foci 48 h after transfection. Quantification of 5 fields with 150 cells in total. Scale bars: 20 μm. I Western blot analysis 48 h after transfection. J Comet assay 48 h after transfection. Quantification of DNA tail moment in 10 fields with 150 cells in total. Scale bars: 10 μm. K Western blot analysis after 48 h after c-MYC siRNA transfection. L γH2AX foci detection after c-MYC induction by addition of DOX. Quantification of 5 fields with 150 cells in total. Scale bars: 20 μm. Results are presented as the mean + SD with (n = 3) for D-G, (n=5) for H+L and (n=10) for J with *: p < 0.05, **: p < 0.01, ***: p < 0.001
AP4 directly represses MIR22HG
Next, we comprehensively determined the effects of AP4 deletion on global mRNA expression by RNA-Seq analysis (Kaller et al., in preparation). Among the genes up-regulated after AP4 deletion in DLD-1 cells was CDKN1A/p21 (Fig. 2A), which we had previously identified as a direct AP4 target gene [4]. Notably, the long non-coding/lncRNA MIR22HG was also up-regulated in AP4-deficient CRC cells (Fig. 2A). Interestingly, the MIR22HG-derived microRNA miR-22-3p is known to induce premature senescence [20]. An AP4-dependent regulation of CDKN1A/p21 and MIR22HG was also detected in SW480 cells (Fig. S3A). The expression of miR-22-3p was also elevated in AP4-deficient CRC cells (Fig. 2B). Also the amount of newly synthesized, nascent MIR22HG mRNA was increased in AP4-deficient DLD-1 and SW480 cells (Fig. 2B and Fig. S3B), suggesting a direct effect of AP4 on the transcription of MIR22HG. Furthermore, the amount of total and nascent MIR22HG mRNA was decreased after ectopic expression of AP4 from an episomal pRTR vector in DLD-1 cells (Fig. 2C). Also after ectopic expression of c-MYC, the expression of MIR22HG and miR-22-3p and that of nascent MIR22HG was repressed in an AP4-dependent manner in DLD-1 and SW480 cells (Fig. 2D-E and Fig. S2C- E). The known AP4 targets, SNAI1 and CDKN1A/p21 were also regulated in an AP4-dependent manner after activation of ectopic c-MYC in these cells (Fig. S3F). We detected AP4 occupancy at the MIR22HG promoter by ChIP-Seq analysis (Fig. 2F; Kaller et al., in preparation). Since we also identified E-box motifs (CAGCTG), which represent the binding sites of AP4, under the ChIP-Seq peaks, AP4 presumably binds directly to the promoter of MIR22HG (Fig. 2F). These results were confirmed by qChIP analysis (Fig. 2G). The increased expression of MIR22HG in AP4-deficient CRC cells was reverted by ectopic expression of AP4, whereas a mutant AP4 lacking its basic region (AP4 ΔBR), described in [4], had no effect (Fig. 2H and Fig. S3G). Therefore, the repression of MIR22HG requires the DNA binding region of AP4 and occurs by direct binding of AP4 to the MIR22G promoter. In addition, the levels of functional miR-22-3p were repressed by ectopic AP4 in AP4-deficient SW480 cells, as determined by a luciferase assay using a miR-22-3p antisense reporter (Fig. 2I). This effect was also dependent on the DNA-binding ability of AP4. Therefore, AP4 directly represses MIR22HG and thereby leads to a functionally relevant decrease of miR-22-3p.
Fig. 2.

AP4 directly represses MIR22HG. A qPCR analysis of MIR22HG and p21 in the indicated clones of DLD-1 cells. B qPCR analysis of miR-22-3p expression (left panel) and nascent MIR22HG mRNA (right panel). C qPCR analysis of MIR22HG expression after AP4 induction by DOX for the indicated duration (left panel) and nascent MIR22HG 48 h after AP4 induction by DOX (right panel). D qPCR analysis of MIR22HG (left panel) and miR-22-3p (right panel) after c-MYC induction by DOX for indicated durations. E qPCR analysis of nascent MIR22HG 48 h after c-MYC induction by DOX. F ChIP-Seq enrichment profiles for AP4-VSV associated chromatin were generated with the UCSC genome browser. The gene structure ideogram is shown below the ChIP-seq tracks. G qChIP analysis of AP4 occupancy. Cells were treated with DOX for 48 h. Chromatin was enriched by anti-AP4 or anti-rabbit-IgG antibodies. SNAI1 and 16q22 served as positive and negative controls, respectively. H qPCR analysis of MIR22HG in the indicated cells 48 h after transfections. I miR-22-3p anti-sense reporter dual-luciferase analysis 48 h after transfection. J β-gal detection at pH 6 48 h after transfection. Quantification of 3 fields with 120 cells in total. Scale bars: 50 μm. K Detection of γH2AX foci 48 h after transfection. Quantification of 3 fields with 120 cells total. Scale bars: 20 μm. In panel A-E and G-K the mean + SD (n = 3) is provided with *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001
Since miR-22-3p has been implicated in senescence of human fibroblasts and breast cancer cells before [20], we analyzed whether miR-22-3p plays a role in the cellular senescence caused by AP4-deficiency. After transfection of DLD-1 AP4 WT1 cells with a miR-22-3p mimic an increase in SA-β-gal positive cells was observed, while inhibition of miR-22-3p by a specific antagomir reduced cellular senescence in DLD-1 AP4 KO2 cells (Fig. 2J). Similar effects on senescence were detected in SW480 cells (Fig. S3H). The degree of senescence caused by ectopic miR-22-3p expression was similar to the effect of AP4 deletion, indicating that up-regulation of miR-22-3p may mediate senescence observed after deletion of AP4. Ectopic miR-22-3p also elevated the amount of DNA damage in the DLD-1 AP4 WT1 clone (Fig. 2K). Conversely, inhibition of miR-22-3p by an antagomir reduced DNA damage as evidenced by a reduction in γH2AX-positive foci in DLD-1 AP4 KO2 cells. Since miR-22-3p expression was sufficient and required for increased senescence and DNA damage in DLD-1 and SW480 cells, the up-regulation of miR-22-3p caused by AP4 inactivation in CRC cells presumably mediates a significant portion of the observed increase in DNA damage and cellular senescence.
MDC1 is a direct target of AP4
Among the known miR-22-3p targets [21], MDC1/Mediator of DNA damage Checkpoint 1 appeared to be most relevant in this context, since it is a central component of the DNA damage response [22–24]. Therefore, we asked whether down-regulation of MDC1 expression occurs via the elevated miR-22-3p expression characteristic for AP4-deficient CRC cells. Indeed, the amount of MDC1 mRNA and protein was decreased in AP4-deficient DLD-1 and SW480 cells (Fig. 3A-B and Fig. S4A-B). We confirmed that MDC1 mRNA is a direct target of miR-22-3p in DLD-1 cells in a dual reporter assay (Fig. 3C-D). Furthermore, ectopic miR-22-3p expression decreased MDC1 mRNA and protein levels in DLD-1 and SW480 cells (Fig. 3E and Fig. S4C-D). On the cellular level, more MDC1-positive and less γH2AX-positive foci were detected in the nuclei of AP4-proficient DLD-1 and SW480 cells, when compared to AP4-deficient cells (Fig. 3F and Fig. S4E). In order to determine whether the repression of MIR22HG/miR-22-3p by AP4 is relevant for the regulation of MDC1 transcript levels we performed a reporter assay (Fig. 3G). Indeed, ectopic expression of AP4 in AP4-deficient cells induced the activity of the MDC1–3′-UTR reporter in a manner dependent on the miR-22-3p SMS and the DNA binding capacity of AP4. Therefore, the repression of MIR22HG by AP4 may increase the abundance of MDC1 mRNA.
Fig. 3.

AP4-mediated repression of miR-22-3p contributes to increased MDC1 levels. A qPCR analysis of MDC1 expression. B Western blot analysis. C Scheme of the miR-22-3p seed, the seed-matching sequences and its deletion in the 3′-UTR in the MDC1 mRNA. The seed and seed-matching sequences are highlighted in red. D Dual-luciferase assay was conducted 48 h after DLD-1 cells were transfected with miR-22-3p mimic and human MDC1 3′-UTR reporter vector. The miR-22-3p anti-sense reporter served as a positive control. E qPCR (left panel) and Western blot analysis (right panel) 48 h after transfection. F MDC1 foci detection in untreated cells. Quantification of 3 fields with 120 cells in total. Scale bars: 20 μm. G Dual-luciferase assay 48 h after transfection. In panels A, D-G, the mean + SD (n = 3) is provided with *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001
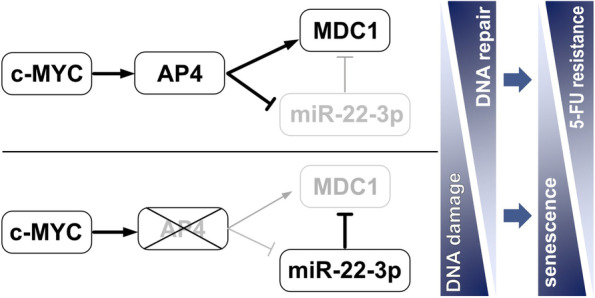
In addition, ectopic AP4 increased the amount of nascent MDC1 mRNA and AP4-deficient cells showed a decrease in nascent MDC1 mRNAs (Fig. 4A and Fig. S4F), suggesting that AP4 may also directly induce MDC1 transcription. Indeed, an E-box motif upstream of the TSS and two E-box motifs in the introns of MDC1 showed occupancy by AP4 in DLD-1 cells according to a ChIP-Seq analysis (Fig. 4B; Kaller et al., in preparation). The AP4 occupancy of these binding sites within the MDC1 promoter was confirmed by qChIP analysis (Fig. 4C). Ectopic AP4 expression resulted in increased MDC1 expression in AP4-deficient DLD-1 and SW480 cells, whereas ectopic AP4 ΔBR was unable to induce MDC1 in AP4-deficient DLD-1 and SW480 cells (Fig. 4D-E, and Fig. S4G-H). Furthermore, the expression of MDC1 protein was induced by ectopic AP4 in DLD-1 cells (Fig. 4F). c-MYC activation also induced MDC1 transcription and MDC1 protein expression in an AP4-dependent manner in DLD-1 and SW480 cells (Fig. 4G-H and Fig. S4I-J). Taken together these results show that after c-MYC activation AP4 induces MDC1 expression directly and indirectly via repressing miR-22-3p (Fig. 4I). This type of dual regulation, also termed coherent feed-forward loop, is known to confer robustness to transcriptional networks [25].
Fig. 4.

MDC1 is directly regulated by AP4. A qPCR analysis of nascent MDC1 mRNA 48 h after AP4 induction by DOX (left panel). qPCR analysis of nascent MDC1 mRNA in untreated cells (right panel). B ChIP-Seq analysis of AP4 occupancy using the UCSC genome browser. C Validation of MDC1 as AP4 direct target via qChIP assay. Cells were treated with DOX for 48 h. Chromatin was enriched by anti-AP4 or anti-rabbit-IgG antibodies. SNAI1 and 16q22 served as positive and negative controls, respectively. D-E qPCR and Western blot analysis of MDC1 in the indicated cell lines 48 h after transfection. F Western blot analysis of MDC1 after AP4 induction by DOX for the indicated durations. G qPCR analysis of nascent MDC1 mRNA after c-MYC induction by DOX for 48 h. H Western blot analysis after c-MYC induction by DOX for the indicated periods. I Model of the AP4/miR-22-3p/MDC1 coherent feed-forward loop. In panels A, C, D and G, the mean + SD (n = 3) is provided with *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001
Associations of c-MYC, AP4, MDC1 and miR-22-3p expression in primary CRCs
To determine whether the regulation of MIR22HG/miR-22-3p and MDC1 by AP4 also occurs in primary CRCs, we analyzed the expression of these genes in 15 cohorts of colorectal adenocarcinomas (COAD) provided by the TCGA consortium and GEO. Within these datasets c-MYC, AP4 and MDC1 expression was significantly increased, whereas MIR22HG expression was repressed in primary CRCs obtained from 644 patients when compared to matched normal mucosa (Fig. 5A). MIR22HG showed a significantly negative correlation with MDC1, AP4 and c-MYC expression in 471 primary CRC samples of the TCGA-COAD cohort (Fig. 5B). AP4 and c-MYC also showed a negative correlation with miR-22-3p. In addition, the expression of MDC1 and c-MYC showed a positive correlation with AP4 in CRCs. As expected, MIR22HG and its product miR-22-3p, as well as c-MYC and AP4 displayed a positive correlation in CRCs. Furthermore, c-MYC, AP4 and MDC1 showed the highest expression in CRCs belonging to the CMS2 subtype, whereas the expression of MIR22HG was lowest in CMS2 CRCs. Interestingly, CMS2 is characterized by elevated WNT and c-MYC pathway activity, as well as chromosomal instability. Since stromal cell derived mRNAs may confound the gene expression profiles of CRCs, we also performed an analysis using CRC intrinsic subtypes (CRIS). These allow tumor classification according to gene signatures obtained from patient-derived tumor xenografts in mice [18]. c-MYC, AP4 and MDC1 were mainly elevated in CRIS C-E subtypes, which are characterized by elevated EGFR, WNT signaling and TP53 mutations. Conversely, MIR22HG was decreased in CRIS C-E subtypes (Fig. 5C). Of note, MDC1, AP4 and c-MYC also displayed a negative association with MIR22HG expression in an independent patient cohort (GSE39582), as well as in expression data from CRC cell lines (CCLE) and in patient-derived xenografts (PDX) (Fig. 5D). Therefore, an inverse correlation between MIR22HG/miR-22-3p and c-MYC/AP4/MDC1 expression is also evident in primary CRCs, implying that the regulations identified here are conserved in vivo. In summary, the expression of c-MYC, AP4 and MDC1 is coordinately elevated in primary CRCs, whereas MIR22HG/miR-22-3p is repressed. This pattern is mainly associated with CMS2 and CRIS C-E subtypes, which represent CRCs with enhanced WNT signaling.
Fig. 5.

Correlations between c-MYC, AP4, MDC1 and miR-22-3p expression in primary CRCs. A Relative expression of the indicated mRNAs detected in expression profiles from patient-derived normal mucosa and tumor tissue deposited in the TCGA-COAD and GEO databases. B Scatter plots of pair-wise comparisons of mRNA expression of the indicated genes in TCGA-COAD samples. C Heat maps of associations between c-MYC, AP4, MIR22HG and MDC1 expression and CMS/CRIS subtypes in the indicated patient cohorts. D RNAs were ranked in descending order according to Pearson correlation coefficient r with MIR22HG expression for the indicated datasets. The positions of MDC1, c-MYC and AP4 expression in the ranked lists are indicated
Regulation of Mir22hg/MDC1 by Ap4 in mice
Next, we analyzed whether the regulation of miR-22-3p and MDC1 by AP4 is conserved in adenomas from ApcMin/+ mice, which represent a model for inherited colorectal cancer (familial adenomatous polyposis (FAP) [26]. ApcMin/+ mice harbor an inactivating mutation in one Apc allele. Upon spontaneous loss of the second Apc allele, these mice develop multiple intestinal adenomas. In RNA-Seq results obtained in our previous study [6] Mir22hg expression was significantly increased in intestinal organoids with deletion of Ap4 (Fig. 6A), which is in line with the results obtained in AP4-deficient CRC cells. Ap4 occupancy of murine Mir22hg (Fig. 6B), which coincides with the presence of two E-box motifs, was detected in published ChIP-Seq results [27]. Mdc1 expression was significantly decreased in Ap4-deficient intestinal organoids (Fig. 6C). In addition, an Ap4 E-Box binding motif displaying occupancy by Ap4 was detected in the Mdc1 promoter region (Fig. 6D). Furthermore, a decrease in Mdc1 mRNA expression was found in adenomas from ApcMin/+ mice with intestinal epithelial cell (IEC)-specific deletion of Ap4 when compared to Ap4-wild-type ApcMin/+ mice (Fig. 6E). Taken together, these results imply that the regulation of MIR22HG and MDC1 by AP4 is conserved in mice. In addition, a decrease in Mdc1 protein expression and in the number of Mdc1-positive cells was detected by immunohistochemistry in Ap4∆IEC adenomas of 120 days old ApcMin/+ mice when compared to Ap4fl/fl adenomas (Fig. 6F). Conversely, an increase in γH2AX-positive cells was detected in Ap4∆IEC adenomas (Fig. 6G), indicating higher levels of endogenous DNA damage in ApcMin/+/Ap4ΔIEC adenomas. Furthermore, Ap4ΔIEC adenomas displayed an increase of cells positive for the senescence marker p16/INK4A when compared to Ap4fl/fl adenomas (Fig. 6H). As expected, p16-positive cells were negative for the proliferation marker Ki67. Taken together, these results show that the regulation of DNA repair and senescence by AP4 via its targets MIR22HG and MDC1 is conserved in mice and occurs in vivo.
Fig. 6.

Regulation of MIR22HG and MDC1 by AP4 is conserved in mice. A Differential expression of Mir22hg in Ap4-deficient intestinal organoids. B+D ChIP-Seq enrichment profiles for Ap4-associated chromatin were generated with the UCSC genome browser. The data was obtained from GSM2132681. The structures of the B Mir22hg and the D Mdc1genes are shown below the ChIP-Seq tracks. C Differential expression of Mdc1 in Ap4-deficient intestinal organoids. E qPCR analysis of adenomas from three 120 days old ApcMin/+ mice (five adenomas per mouse) per genotype. F Left panel: Immunohistochemical detection of Mdc1 in adenomas of 120 days old ApcMin/+ mice with the indicated genotype. Counterstaining with hematoxylin. Scale bar = 100 μm. Right panel: Quantification of Mdc1 relative intensity and Mdc1-positive cells in percent (%) in adenomas from 120 days old ApcMin/+ mice in a total of 10 adenomas per genotype. G Left panel: Immunohistochemical detection of γH2AX in adenomas of 120 days old ApcMin/+ mice with the indicated genotype. Right panel: Quantification of γH2AX-positive cells in percent (%) in adenomas from 120 days old ApcMin/+, Ap4fl/fl and ApcMin/+, Ap4∆IEC mice in a total of 14 or 13 adenomas, respectively. H Left panel: Immunohistochemical detection of p16 in adenomas of 120 days old ApcMin/+ mice with the indicated genotype. Right panel: Quantification of p16-positive cells in percent (%) in adenomas from 120 days old ApcMin/+Ap4fl/fl and ApcMin/+, Ap4∆IEC mice in 26 or 22 adenomas, respectively. In panels F-H scale bars represent 100 μm. *:p < 0.05, **:p < 0.01, ***:p < 0.001
MDC1 mediates effects of AP4 on DNA damage
Next, we determined the functional relevance of MDC1 in CRC lines with varying AP4 status. When the expression of MDC1 was decreased using siRNAs in AP4-proficient DLD-1 and SW480 cells we detected an increase of cells with nuclear γH2AX foci (Fig. 7A and Fig. S5A). However, when MDC1 was ectopically expressed in AP4-deficient DLD-1 and SW480 cells the frequency of nuclei with detectable γH2AX foci decreased (Fig. 7B and Fig. S5B). Therefore, MDC1 is required to suppress DNA damage in AP4 expressing cells and sufficient to rescue a DNA damage repair defect in AP4-deficient CRC cells. Silencing of MDC1 in AP4-proficient DLD-1 and SW480 cells resulted in increased cellular senescence as evidenced by increased SA-β-gal staining (Fig. 7C and Fig. S5C). Conversely, ectopic expression of MDC1 suppressed senescence in AP4-deficient DLD-1 and SW480 cells (Fig. 7C and Fig. S5C). Therefore, the presence of unrepaired DNA damage resulting from the modulation of MDC1 levels by AP4 correlated with the amount of cell undergoing senescence. In addition, the viability of AP4-proficient CRC cells was significantly lower when MDC1 was silenced by specific siRNAs (Fig. 7D and Fig. S5D), whereas ectopic MDC1 expression increased the viability of AP4-deficient CRC cells (Fig. 7E and Fig. S5E). To determine whether the effect of miR-22-3p on cellular senescence and DNA damage was mediated by targeting MDC1 and not another miR-22-3p target mRNA, we co-expressed miR-22-3p and a miR-22-3p-insensitive MDC1 mRNA in AP4-proficient DLD-1 and SW480 cells (Fig. 7F-H and Fig. S5F-H). Indeed, expression of the miR-22-3p-insensitive MDC1 largely alleviated the DNA damage and cellular senescence caused by miR-22-3p. Taken together, AP4 suppresses DNA damage by inducing MDC1, which facilitates DNA repair. As a consequence cellular senescence is decreased and viability increased in cells expressing AP4.
Fig. 7.

MDC1 mediates effects of AP4 on DNA repair. A Detection of MDC1 and γH2AX foci by immunocytochemistry 48 h after silencing MDC1. Scale bars: 20 μm. Foci quantification was performed with Image J software. Nuclei with over 10 foci were considered as positive. The fluorescence intensity was normalized to DAPI. Quantification of 3 fields with 120 cells in total. B MDC1 and γH2AX foci were detected by immunocytochemistry 48 h after ectopic expression of MDC1. Scale bars: 20 μm. Nuclei with over 10 foci were considered as positive. The fluorescence intensity was normalized to DAPI. Quantification of 3 fields with 120 cells in total. C β-gal staining 48 h after silencing or ectopic expression of MDC1, respectively. Quantification of 3 fields with 120 cells in total. Scale bars: 50 μm. MTT assay results 48 h after D silencing MDC1 or E ectopic expression of MDC1. Detection of F γH2AX foci by immunocytochemistry, G β-gal staining and H comet assay in DLD-1 AP4 WT1 cells 48 h after transfection. MDC1-HA was rendered non-responsive to miR-22-3p by deletion of the MDC1 3'-UTR [28]. Quantification of DNA tail moment in 10 fields with 150 cells in total. The mean + SD is provided with A-E (n = 3), F-G (n = 5) and H (n = 10) with **: p < 0.01, ***: p < 0.001, ****: p < 0.0001
MDC1 mediates effects of AP4 on CIN and HR
Since DNA damage, which is not repaired before cells enter mitosis, may result in chromosomal instability (CIN) [29, 30], we determined the effect of AP4 inactivation on CIN. Micronuclei are acentric fragments or chromosomes that failed to integrate into daughter nuclei in mitosis and can be used as a proxy for CIN [31]. Indeed, the inactivation of AP4 in DLD-1 or SW480 cells resulted in an increased frequency of micronuclei (Fig. 8A and Fig. S6A). The frequency of micronuclei was also increased following siRNA-mediated down-regulation of MDC1 and transfection of a miR-22-3p mimic in AP4-proficient DLD-1 and SW480 cells (Fig. 8B and Fig. S6B). Ectopic expression of MDC1 or miR-22-3p-specific antagomirs reduced the number of micronuclei in AP4-deficient DLD-1 or SW480 cells (Fig. 8B and Fig. S6B). In AP4-deficient DLD-1 cells the overall frequency of aberrant chromosome gains was significantly higher when compared to AP4-proficient DLD-1 cells (Fig. 8C). Furthermore, the aberration of chromosome numbers paralleled the formation of micronuclei in AP4-deficient CRC cells. In summary, the increased CIN observed in AP4-deficient CRC cells is, at least in part, due to a decrease of MDC1 and an increase of miR-22-3p expression. The effects of unrepaired DNA damage on CIN occurs mainly during the G2/M transition. During this phase homologous recombination (HR) is the main pathway for repair of dsDNA breaks (DSB). Since MDC1 plays an important role in the HR [32], we asked whether AP4 regulates HR via repressing miR-22-3p and inducing MDC1. To this end, we employed a previously established HR assay [14] to evaluate HR-mediated repair of a DSB induced by the I-SceI restriction enzyme (Fig. 8D). Notably, HR was repressed in AP4-deficient DLD-1 and SW480 cells (Fig. 8E and Fig. S6C), whereas ectopic AP4 enhanced HR in DLD-1 and SW480 cells (Fig. 8F and Fig. S6D). Furthermore, HR was repressed by ectopic MDC1 siRNA or miR-22-3p mimic in AP4-proficient DLD-1 and SW480 cells (Fig. 8G and Fig. S6E). In addition, ectopic expression of MDC1 or miR-22-3p antagomir restored HR in AP4-deficient DLD-1 and SW480 cells (Fig. 8H and Fig. S6F). Finally, a miR-22-3p-mediated decrease in HR was reversed by expression of miR-22-3p-insensitive MDC1 in AP4-proficient DLD-1 and SW480 cells (Fig. 8I and Fig. S6G), implying that MDC1 is the relevant miR-22-3p target mediating the inhibitory effect of miR-22-3p on HR. Taken together, elevated AP4 expression enhances HR activity via repressing miR-22-3p and inducing MDC1 and thereby contributes to genomic integrity. The increase of CIN on AP4-deficient cells may therefore be a consequence of insufficient HR activity.
Fig. 8.

MDC1 mediates effects of AP4 on chromosomal instability. A Examples and quantification of micronuclei after DAPI staining. Three fields of 120 cells in total were evaluated. Scale bars: 20 μm. B Kinetic evaluation of micronucleus formation 48 h after transfection with the indicated oligonucleotides or vector. C Representative images of mitotic chromosome spreads. Quantification of 50 spreads per genotype. Scale bars: 20 μm. D Scheme illustrating the assay used for the fluorescence based measurement of HR-mediated DSB repair. E The indicated cells were co-transfected with pDR-GFP and pCBAScel plasmids. A pcDNA-mCherry plasmid was co-transfected as a control of transfection efficiency. The percentage of cells expressing GFP was measured by flow cytometry. F-I The percentage of the indicated cells expressing GFP was measured by flow cytometry 72 h after transfection of the indicated plasmids or oligonucleotides. In A-B and E-I the mean + SD (n = 3) and in C the mean + SD (n = 50) are provided with *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001
AP4 modulates the sensitivity to chemotherapeutic drugs
Since AP4 enhanced the repair of DNA damage via regulation of miR-22-3p and MDC1, we asked whether AP4 decreases the sensitivity towards DNA-damage inducing chemotherapeutic drugs, such as Etoposide and 5-Fluoro-Uracil/5-FU. As shown before [33], the frequency and intensity of MDC1- and γH2AX-positive foci was elevated after treatment of CRC cells with Etoposide (Fig. 9A and Fig. S7A). In AP4-deficient cells more γH2AX foci were observed after Etoposide treatment when compared to AP4-proficient cells. In addition, AP4 inactivation further enhanced the Etoposide-mediated reduction of cell viability (Fig. 9B and Fig. S7B), suggesting that AP4 may suppress the adverse effects of etoposide by enhancing DNA damage repair. Indeed, silencing of MDC1 enhanced the effect of Etoposide on reducing cell viability in AP4-proficient DLD-1 and SW480 cells (Fig. 9C and Fig. S7C). Conversely, the viability of AP4-deficient cells was increased by ectopic MDC1 expression after exposure to Etoposide (Fig. 9D and Fig. S7D). Therefore, the relative decrease in viability of AP4-deficient compared to AP4-proficient cells is presumably due to the increased DNA damage caused by the decreased expression of MDC1. Interestingly, ectopic AP4 expression further increased the intensity of MDC1-positive, nuclear foci in the absence and presence of Etoposide (Fig. 9E). Interestingly, the intensity and frequency of γH2AX-positive foci was decreased in DLD-1 cells ectopically expressing AP4 and exposed to Etoposide when compared to DLD-1 cells treated with Etoposide alone (Fig. 9E). Similar results were obtained by Western blot analysis (Fig. 9F). In addition, exposure to Etoposide caused an extension of the so-called “comet tails”, indicating an increase in unrepaired DNA damage (Fig. 9G). Notably, this increase was largely suppressed by ectopic AP4 expression, whereas silencing MDC1 largely abrogated this effect of AP4. These results demonstrate that elevated AP4 expression facilitates the repair of DNA damage by inducing MDC1. By decreasing the amount of DNA damage elevated AP4 expression may therefore contribute to an increased viability and drug resistance of CRC cells.
Fig. 9.

AP4 confers resistance towards Etoposide via MDC1. A Immunofluorescence analysis 12 h after addition of 20 μM Etoposide. Quantification of 3 fields with 120 cells in total. Scale bars: 20 μm. B MTT assay was performed 60 h after treatment with 20 μM Etoposide for 12 h. MTT assay of cells transfected with C the indicated oligonucleotides or D expression plasmids for 48 h and then subjected to treatment with Etoposide for 12 h. E Immunofluorescence analysis after activation of ectopic AP4 expression by treatment with DOX for 48 h and addition of Etoposide for 12 h. Quantification of 3 fields with 120 cells in total. Scale bars: 20 μm. F Western blot analysis of cells treated as in (E). G DNA damage detection by comet assay. Forty-eight hours after activation of ectopic AP4 expression by treatment with DOX, cells were transfected with the indicated oligonucleotides for 48 h and then treated with Etoposide for 12 h. For the last 60 h fresh DOX was added. Quantification of DNA tail moment by evaluation of 10 fields with 150 cells in total, Scale bars: 10 μm. The mean + SD is provided in A-E with (n=3) and in G with (n=10) and *:p < 0.05, **:p < 0.01, ***:p < 0.001, ****:p < 0.0001
Next, we determined whether the AP4 status influences the sensitivity of CRC cell lines to 5-FU, a chemotherapeutic drug commonly used to treat advanced CRC. Determination of the IC50 dosage revealed that AP4-deficiency sensitizes DLD-1 cells to 5-FU when compared to AP4-proficient DLD-1 cells (Fig. 10A). Next, we determined whether modulation of MDC1 or miR-22-3p expression in CRC lines with varying AP4 status affects 5-FU resistance. Indeed, ectopic expression of miR-22-3p in AP4-proficient DLD-1 cells reduced the tolerance to 5-FU, whereas antagomir-mediated inactivation of miR-22-3p increased the 5-FU resistance in DLD-1 AP4 KO2 clone (Fig. 10B-C). Furthermore, silencing of MDC1 sensitized AP4-proficient DLD-1 cells to 5-FU (Fig. 10D). In addition, ectopic MDC1 expression enhanced 5-FU resistance in AP4-deficient DLD-1 cells (Fig. 10E). Furthermore, ectopic AP4 enhanced the tolerance of DLD-1 cells to 5-FU (Fig. 10F). Similar results were obtained in SW480 cells (Fig. S8A-E).
Fig. 10.

AP4 confers resistance towards 5-FU via MDC1. A The indicated cells were treated with increasing concentrations of 5-FU for 48 h. Then the IC50 was determined by an MTT assay. B-E The indicated cells were transfected with the indicated oligonucleotides for 48 h and subsequently treated with increasing concentrations of 5-FU for 48 h. Then the IC50 was determined by an MTT assay. F The DLD-1 pRTR-AP4-VSV cell pool was treated with DOX for 48 h and subsequently with increasing concentrations of 5-FU for 48 h. For the last 48 h fresh DOX was added. Then the IC50 was determined by an MTT assay. G-J Colony formation assay of the indicated cells transfected with the indicated oligonucleotides or plasmids for 48 h and then subjected to treatment with 10 μM 5-FU for 48 h and then cultured for additional 3 weeks. K and L Colony formation assay, as described in (G), of DLD-1 pRTR-AP4-VSV pools after DOX treatment for 48 h after transfection of the indicated oligonucleotides for 48 h. In panels G-L, the mean + SD is provided. *:p < 0.05, **:p < 0.01, ***:p < 0.001
Next, we performed a colony-formation assay over a period of 3 weeks after a treatment with 5-FU for 48 h (Fig. 10G-L). In this assay, ectopic miR-22-3p expression sensitized AP4-proficient DLD-1 cells to 5-FU, whereas inactivation of miR-22-3p increased 5-FU resistance of AP4-deficient DLD-1 cells (Fig. 10G-H). When MDC1 was silenced in AP4-proficient DLD-1 cells a decrease in colony numbers was observed (Fig. 10I). Conversely, ectopic MDC1 expression increased the number of colonies of AP4-deficient DLD-1 cells (Fig. 10J). Similar results were also obtained in SW480 cells (Fig. S8F-I). Furthermore, ectopic AP4 expression also increased the number of colonies of DLD-1 cells, whereas both ectopic miR-22-3p and silencing of MDC1 abrogated the effect of AP4 (Fig. 10K-L). Taken together, these results show that elevated AP4 expression mediates 5-FU resistance in CRC cell lines in a MDC1 and miR-22-3p dependent manner.
In addition, we analyzed the potential association between the response to chemotherapy and the expression of c-MYC, AP4, MIR22HG or MDC1 in primary CRCs (Fig. S9A-D). After receiving chemotherapy, patients with CRCs displaying high expression of c-MYC, AP4 or MDC1 showed a trend towards decreased relapse free survival, when compared to patients with low expression of these genes in primary CRCs. Notably, low expression of MIR22HG was significantly associated with decreased relapse free survival after chemotherapy. These results suggest, that high c-MYC, AP4, MDC1 and/or low MIR22HG/miR-22-3p expression in primary CRCs confer resistance to chemotherapy, whereas low expression of c-MYC, AP4, MDC1 and/or high MIR22HG/miR-22-3p expression sensitize to chemotherapy.
Discussion
Here we identified a new regulatory connection that explains how AP4 preserves genomic integrity in the context of c-MYC activation (see also graphical abstract). Our results demonstrate that AP4 suppresses DNA damage, which occurs spontaneously or at an increased rate after c-MYC activation in CRC cells, by promoting the expression of MDC1. By forming a coherent feed-forward loop, in which AP4 directly induces MDC1 and represses miR-22-3p, a known inhibitor of MDC1, AP4 can exert a tight and robust regulation of MDC1 expression. Notably, MDC1 is a central effector in the DNA damage response/DDR as it serves as a molecular platform for many DNA repair proteins at the site of DNA damage and facilitates the amplification of the initial γH2AX signal [22–24]. By regulating the abundance of MDC1 protein, AP4 is therefore able to fine-tune the cellular response to DNA damage. In support of this model, ectopic AP4 suppressed DNA damage induced by Etoposide and 5-FU in a manner dependent on MDC1 and miR-22-3p. Thereby, AP4 may contribute to resistance of CRC cells towards DNA damaging substances. Since we obtained similar results in MSI/microsatellite instable (DLD-1) and MSS/microsatellite stable (SW480) CRC cell lines, our results are presumably relevant for the majority of CRCs, as these fall into either of these categories. As both cell lines harbor mutant p53 alleles, the effects of AP4 on the DDR are independent of wild-type p53. Since AP4, c-MYC and MDC1 expression are concomitantly elevated in primary CRCs along with down-regulation of MIR22HG, these findings are presumably of clinical relevance.
Our results imply that patients with CRCs that display low expression of c-MYC, AP4 and MDC1, as well as elevated miR-22-3p levels should respond better to chemotherapy than patients with CRCs that exhibit high c-MYC, AP4, MDC1 and low miR-22-3p expression. Indeed, we detected an association between poor response to chemotherapy and elevated c-MYC, AP4 and MDC1 expression, as well as low MIR22HG expression in a cohort of CRC patients. The mechanism underlying this effect is likely to be the increased DNA repair capacity of cells with elevated AP4 (and therefore high MDC1 levels) detected in this study. Therefore, further studies to validate the use of AP4 and MDC1 as predictive markers are warranted.
Furthermore, our results indicate that inhibition of AP4 sensitizes cancer cells to DNA damaging agents used for chemotherapy. In the future inhibition of AP4 function may be achieved by inhibition of AP4 homo-dimerization using small-drugs or synthetic peptides. Similar approaches have been used to interfere with c-MYC/MAX hetero-dimerization [34]. Alternatively, targeted degradation approaches, such as PROTAC/PROteolysis TArgeting Chimeras, may be used to down-regulate AP4 protein levels [35].
Interestingly, decreased expression of MIR22HG in primary CRCs is associated with poor survival of CRC patients [36]. In addition, elevated expression of AP4 protein showed a significant correlation with distant metastasis and advanced tumor grade in CRC patients [19]. Therefore, the detection of MIR22HG and/or AP4 expression may also have prognostic value for CRC.
MiR-22-3p has been characterized as a senescence-associated microRNA that functions by directly targeting and suppressing CDK6, SIRT1, Sp1 and MDC1 [20, 21]. Expression of an MDC1 variant insensitive to miR-22-3p alleviated DNA damage and senescence caused by miR-22-3p. Therefore, other miR-22-3p targets besides MDC1 are presumably not relevant in the context of c-MYC/AP4 activation and the senescence observed in AP4-deficient cells was largely due to elevated expression of miR-22-3p and the resulting decreased expression of MDC1.
Previous studies have shown that c-MYC induces G1/S transition and DNA replication by activating CDKs and also by directly interacting with replication associated proteins [37, 38]. In tumor cells c-MYC expression is often deregulated and the resulting unscheduled DNA replication causes DNA damage and genomic instability [39, 40]. Here, MDC1 expression was increased to a larger extent after c-MYC activation in AP4-proficient than in AP4-deficient CRC cells, which inversely correlated with the amount of DNA damage. Therefore, the AP4/MDC1 axis protects cells from c-MYC-induced DNA damage. In addition, repression of MDC1 by siRNA and miR-22-3p resulted in an increased frequency of micronuclei in AP4-proficient cells, indicating that the AP4-mediated repression of miR-22-3p and the resulting de-repression of MDC1 is critical for maintaining chromosomal stability. Taken together, AP4-induced MDC1 therefore limits the extent of DNA damage that cells encounter after activation of c-MYC and thereby allows the proliferation of cancer cells with deregulated c-MYC expression. Interestingly, MDC1 is also necessary for the intra-S-phase and the G2/M DNA damage checkpoints [23, 24]. Furthermore, repressing MDC1 induces apoptotic cell death following DNA damage caused by ionizing radiation [41].
In primary CRCs expression of c-MYC, AP4 and MDC1 was consistently up-regulated. Interestingly, elevated MDC1 levels have also been reported in cervical, laryngeal squamous and nasopharyngal carcinomas, which are associated with viral infections and expression of viral proteins, and therefore high levels of DNA damage [42–44]. Since up-regulation of c-MYC is a hallmark of CRCs [45], it is therefore tempting to speculate that CRCs harbor comparatively high levels of DNA damage due to elevated expression of c-MYC, which results in a requirement of MDC1 up-regulation for tumor initiation and progression. A further plausible source of DNA damage during CRC initiation and progression are presumably the products of certain bacterial strains, such as Colibactin produced by pks + E. coli, which contribute to transformation of colon epithelial cells [46].
5-FU has become the mainstay of systemic treatment of CRC since the 1990s. However, nearly 50% of patients diagnosed with metastatic CRC have a low five-year survival rate of 12% due to resistance towards 5-FU-based chemotherapy [47]. 5-FU metabolites are incorporated into RNA and DNA [48], and are subject to base excision repair (BER) or mismatch repair (MMR). DSBs generated during the repair undergo homologous recombination (HR) [49]. Accordingly, impaired HR sensitizes cancer cells to 5-FU treatment and MMR-deficient CRCs display better clinical outcomes after chemotherapy involving 5-FU [50, 51]. Interestingly, inactivation of MDC1 impaired MMR and HR [52, 53]. Here we observed, that AP4 induces HR in CRC cell lines. In addition, ectopic MDC1 expression or inhibition of miR-22-3p restored the capacity for HR in AP4-deficient CRC cells. Therefore, elevated AP4 expression promotes chemo-resistance by enhancing HR via directly and indirectly increasing the levels of MDC1. Our observation that silencing of MDC1 sensitizes AP4-proficient cells to 5-FU treatment is in line with previous studies in which MDC1 inactivation resulted in increased radio-sensitivity and sensitivity to DNA-damaging chemotherapeutics [44, 54, 55]. In addition, we found that AP4 confers 5-FU resistance via up-regulating MDC1 and thereby enhancing the repair of 5-FU-induced DNA damage. Since it has been shown that activation of c-MYC also contributes to chemotherapy resistance in CRC [56], the c-MYC/AP4/miR-22-3p/MDC1 feed-forward loop characterized here is an attractive, new molecular mechanism, which explains chemotherapy resistance in CRC with elevated c-MYC expression. As the majority of CRCs show elevated c-MYC and AP4 expression, interference with the c-MYC/AP4/miR-22-3p/MDC1 axis represents an attractive approach to sensitize CRCs to chemotherapies.
Conclusions
In summary, AP4, miR-22-3p and MDC1 form a coherent, regulatory feed-forward loop to promote DNA repair, which suppresses DNA damage, senescence and CIN, and contributes to 5-FU resistance. These findings explain how elevated AP4 expression contributes to initiation, maintenance, progression and chemo-resistance of colorectal cancer after c-MYC activation.
Supplementary Information
Acknowledgements
We are grateful to Drs. Zhenkun Lou, Stephen Jackson, Maria Jasin and Bert Vogelstein for providing vectors and cell lines, to Raffaele Conca (Dr. von Haunersches Children’s Hospital, Munich) for FACS sorting and to Ursula Götz for technical assistance.
Abbreviations
- AP4
transcription factor AP-4
- APC
Adenomatous polyposis coli
- CCLE
Cancer Cell Line Encyclopedia
- CIN
Chromosomal instability
- CMS
consensus molecular subtype
- COAD
colon adenocarcinoma
- CRC
colorectal cancer
- CRIS
CRC intrinsic subtypes
- DOX
Doxycycline
- HR
Homologous recombination
- 5-FU
5-Fluorouracil
- MDC1
Mediator of DNA damage Checkpoint 1
- Min
Multiple intestinal neoplasia
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MIR22HG
MIR22 host gene
- PDX
patient-derived xenografts
- qChIP
quantitative chromatin immunoprecipitation
- TCGA
The Cancer Genome Atlas
- TSS
transcriptional start site
- UTR
Untranslated region
Authors’ contributions
JC designed and performed experiments, analyzed results, wrote the paper; MK performed bioinformatics analyses on RNA- and Chip-Seq data and correlative expression analyses; SJ performed qPCR and IHC analysis of mouse samples; MR performed bioinformatics analysis on data from CRC patient cohorts. HH conceived and supervised the study, planned experiments and wrote the paper. All authors read and approved the final manuscript.
Funding
This work was supported by German Cancer Aid/Deutsche Krebshilfe Grant [70114235 and 70112245 to HH]. Open Access funding enabled and organized by Projekt DEAL.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Declarations
Ethics approval and consent to participate
Animal experimentations and analyses were approved by the Government of Upper Bavaria, Germany (AZ 55.2–1-54-2532-4-2014).
Consent for publication
Does not apply.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Andrei P, Battuello P, Grasso G, Rovera E, Tesio N, Bardelli A. Integrated approaches for precision oncology in colorectal cancer: The more you know, the better. Semin Cancer Biol. 2021. 10.1016/j.semcancer.2021.04.007. [DOI] [PubMed]
- 2.Mauri G, Arena S, Siena S, Bardelli A, Sartore-Bianchi A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann Oncol. 2020;31:1135–1147. doi: 10.1016/j.annonc.2020.05.027. [DOI] [PubMed] [Google Scholar]
- 3.Wong MM, Joyson SM, Hermeking H, Chiu SK. Transcription Factor AP4 Mediates Cell Fate Decisions: To Divide, Age, or Die. Cancers (Basel). 2021;13:676–91. [DOI] [PMC free article] [PubMed]
- 4.Jung P, Menssen A, Mayr D, Hermeking H. AP4 encodes a c-MYC-inducible repressor of p21. Proc Natl Acad Sci U S A. 2008;105:15046–15051. doi: 10.1073/pnas.0801773105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackstadt R, Jung P, Hermeking H. AP4 directly downregulates p16 and p21 to suppress senescence and mediate transformation. Cell Death Dis. 2013;4:e775. doi: 10.1038/cddis.2013.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaeckel S, Kaller M, Jackstadt R, Gotz U, Muller S, Boos S, Horst D, Jung P, Hermeking H. Ap4 is rate limiting for intestinal tumor formation by controlling the homeostasis of intestinal stem cells. Nat Commun. 2018;9:3573. doi: 10.1038/s41467-018-06001-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackstadt R, Hermeking H. AP4 is required for mitogen- and c-MYC-induced cell cycle progression. Oncotarget. 2014;5:7316–7327. doi: 10.18632/oncotarget.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng Y, Wang L, Xu J, Zhang Q. AP4 positively regulates LAPTM4B to promote hepatocellular carcinoma growth and metastasis, while reducing chemotherapy sensitivity. Mol Oncol. 2018;12:373–390. doi: 10.1002/1878-0261.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C, Cai Q, He W, Lam TB, Lin J, Zhao Y, Chen X, Gu P, Huang H, Xue M, et al. AP4 modulated by the PI3K/AKT pathway promotes prostate cancer proliferation and metastasis of prostate cancer via upregulating L-plastin. Cell Death Dis. 2017;8:e3060. doi: 10.1038/cddis.2017.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou C, Pinto AK, Curtis JD, Persaud SP, Cella M, Lin CC, Edelson BT, Allen PM, Colonna M, Pearce EL, et al. c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat Immunol. 2014;15:884–893. doi: 10.1038/ni.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement MV. OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2014;2:457–465. doi: 10.1016/j.redox.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 14.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Cell Line Encyclopedia C, Genomics of Drug Sensitivity in Cancer C: Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015, 528:84–87. [DOI] [PMC free article] [PubMed]
- 17.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isella C, Brundu F, Bellomo SE, Galimi F, Zanella E, Porporato R, Petti C, Fiori A, Orzan F, Senetta R, et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat Commun. 2017;8:15107. doi: 10.1038/ncomms15107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackstadt R, Roh S, Neumann J, Jung P, Hoffmann R, Horst D, Berens C, Bornkamm GW, Kirchner T, Menssen A, Hermeking H. AP4 is a mediator of epithelial-mesenchymal transition and metastasis in colorectal cancer. J Exp Med. 2013;210:1331–1350. doi: 10.1084/jem.20120812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu D, Takeshita F, Hino Y, Fukunaga S, Kudo Y, Tamaki A, Matsunaga J, Takahashi RU, Takata T, Shimamoto A, et al. miR-22 represses cancer progression by inducing cellular senescence. J Cell Biol. 2011;193:409–424. doi: 10.1083/jcb.201010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JH, Park SJ, Jeong SY, Kim MJ, Jun S, Lee HS, Chang IY, Lim SC, Yoon SP, Yong J, You HJ. MicroRNA-22 Suppresses DNA Repair and Promotes Genomic Instability through Targeting of MDC1. Cancer Res. 2015;75:1298–1310. doi: 10.1158/0008-5472.CAN-14-2783. [DOI] [PubMed] [Google Scholar]
- 22.Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004;23:2674–2683. doi: 10.1038/sj.emboj.7600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- 24.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 25.Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149:515–524. doi: 10.1016/j.cell.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 27.Chou C, Verbaro DJ, Tonc E, Holmgren M, Cella M, Colonna M, Bhattacharya D, Egawa T. The Transcription Factor AP4 Mediates Resolution of Chronic Viral Infection through Amplification of Germinal Center B Cell Responses. Immunity. 2016;45:570–582. doi: 10.1016/j.immuni.2016.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu L, Luo K, Lou Z, Chen J. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci U S A. 2008;105:11200–11205. doi: 10.1073/pnas.0802885105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayashi MT, Karlseder J. DNA damage associated with mitosis and cytokinesis failure. Oncogene. 2013;32:4593–4601. doi: 10.1038/onc.2012.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asaithamby A, Hu B, Chen DJ. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc Natl Acad Sci U S A. 2011;108:8293–8298. doi: 10.1073/pnas.1016045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Ma Z, Treszezamsky A, Powell SN. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat Struct Mol Biol. 2005;12:902–909. doi: 10.1038/nsmb991. [DOI] [PubMed] [Google Scholar]
- 33.Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–543. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 34.Whitfield JR, Soucek L. The long journey to bring a Myc inhibitor to the clinic. J Cell Biol. 2021;220 (8):e202103090. 10.1083/jcb.202103090. [DOI] [PMC free article] [PubMed]
- 35.Bushweller JH. Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer. 2019;19:611–624. doi: 10.1038/s41568-019-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J, Shao T, Song M, Xie Y, Zhou J, Yin J, Ding N, Zou H, Li Y, Zhang J. MIR22HG acts as a tumor suppressor via TGFbeta/SMAD signaling and facilitates immunotherapy in colorectal cancer. Mol Cancer. 2020;19:51. doi: 10.1186/s12943-020-01174-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB. Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer. 2004;4:562–568. doi: 10.1038/nrc1393. [DOI] [PubMed] [Google Scholar]
- 38.Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR, 3rd, Menssen A, Hermeking H. Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle. 2007;6:205–217. doi: 10.4161/cc.6.2.3742. [DOI] [PubMed] [Google Scholar]
- 39.Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, Galloway DA, Gu W, Gautier J, Dalla-Favera R. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–451. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- 40.Menssen A, Epanchintsev A, Lodygin D, Rezaei N, Jung P, Verdoodt B, Diebold J, Hermeking H. c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle. 2007;6:339–352. doi: 10.4161/cc.6.3.3808. [DOI] [PubMed] [Google Scholar]
- 41.Wang Z, Zeng Q, Chen T, Liao K, Bu Y, Hong S, Hu G. Silencing NFBD1/MDC1 enhances the radiosensitivity of human nasopharyngeal cancer CNE1 cells and results in tumor growth inhibition. Cell Death Dis. 2015;6:e1849. doi: 10.1038/cddis.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuan C, Bu Y, Wang C, Yi F, Yang Z, Huang X, Cheng L, Liu G, Wang Y, Song F. NFBD1/MDC1 is a protein of oncogenic potential in human cervical cancer. Mol Cell Biochem. 2012;359:333–346. doi: 10.1007/s11010-011-1027-7. [DOI] [PubMed] [Google Scholar]
- 43.Liu X, Qiu Z, Wang Z, Zuo W, Gong Z, Liu C, Zeng Q, Qian Y, Jiang L, Li Y, et al. NFBD1/MDC1 participates in the regulation of proliferation and apoptosis in human laryngeal squamous cell carcinoma. Clin Transl Oncol. 2018;20:534–541. doi: 10.1007/s12094-017-1748-5. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Liao K, Zuo W, Liu X, Qiu Z, Gong Z, Liu C, Zeng Q, Qian Y, Jiang L, et al. Depletion of NFBD1/MDC1 Induces Apoptosis in Nasopharyngeal Carcinoma Cells Through the p53-ROS-Mitochondrial Pathway. Oncol Res. 2017;25:123–136. doi: 10.3727/096504016X14732772150226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas N: Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. [DOI] [PMC free article] [PubMed]
- 46.Iftekhar A, Berger H, Bouznad N, Heuberger J, Boccellato F, Dobrindt U, et al. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat Commun. 2021;12:1003–18. [DOI] [PMC free article] [PubMed]
- 47.Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041–1047. doi: 10.1016/s0140-6736(00)02034-1. [DOI] [PubMed] [Google Scholar]
- 48.Hagenkort A, Paulin CBJ, Desroses M, Sarno A, Wiita E, Mortusewicz O, Koolmeister T, Loseva O, Jemth AS, Almlof I, et al. dUTPase inhibition augments replication defects of 5-Fluorouracil. Oncotarget. 2017;8:23713–23726. doi: 10.18632/oncotarget.15785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wyatt MD, Wilson DM., 3rd Participation of DNA repair in the response to 5-fluorouracil. Cell Mol Life Sci. 2009;66:788–799. doi: 10.1007/s00018-008-8557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hemminki A, Mecklin JP, Jarvinen H, Aaltonen LA, Joensuu H. Microsatellite instability is a favorable prognostic indicator in patients with colorectal cancer receiving chemotherapy. Gastroenterology. 2000;119:921–928. doi: 10.1053/gast.2000.18161. [DOI] [PubMed] [Google Scholar]
- 51.Fujinaka Y, Matsuoka K, Iimori M, Tuul M, Sakasai R, Yoshinaga K, Saeki H, Morita M, Kakeji Y, Gillespie DA, et al. ATR-Chk1 signaling pathway and homologous recombinational repair protect cells from 5-fluorouracil cytotoxicity. DNA Repair (Amst) 2012;11:247–258. doi: 10.1016/j.dnarep.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 52.Merentitis D, Nguyen BD, Samartzis EP, Noske A, Brandt S, Dedes KJ. Loss of MDC1 in Endometrial Carcinoma Is Associated With Loss of MRN Complex and MMR Deficiency. Anticancer Res. 2019;39:6547–6553. doi: 10.21873/anticanres.13870. [DOI] [PubMed] [Google Scholar]
- 53.Wang Z, Zuo W, Zeng Q, Qian Y, Li Y, Liu C, Wang J, Zhong S, Bu Y, Hu G. Loss of NFBD1/MDC1 disrupts homologous recombination repair and sensitizes nasopharyngeal carcinoma cells to PARP inhibitors. J Biomed Sci. 2019;26:14. doi: 10.1186/s12929-019-0507-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peng A, Chen PL. NFBD1, like 53BP1, is an early and redundant transducer mediating Chk2 phosphorylation in response to DNA damage. J Biol Chem. 2003;278:8873–8876. doi: 10.1074/jbc.C300001200. [DOI] [PubMed] [Google Scholar]
- 55.Zeng Q, Wang Z, Liu C, Gong Z, Yang L, Jiang L, Ma Z, Qian Y, Yang Y, Kang H, et al. Knockdown of NFBD1/MDC1 enhances chemosensitivity to cisplatin or 5-fluorouracil in nasopharyngeal carcinoma CNE1 cells. Mol Cell Biochem. 2016;418:137–146. doi: 10.1007/s11010-016-2739-5. [DOI] [PubMed] [Google Scholar]
- 56.Boos SL, Loevenich LP, Vosberg S, Engleitner T, Ollinger R, Kumbrink J, Rokavec M, Michl M, Greif PA, Jung A, et al. Disease Modeling on Tumor Organoids Implicates AURKA as a Therapeutic Target in Liver Metastatic Colorectal Cancer. Cell Mol Gastroenterol Hepatol. 2021;13:517–540. doi: 10.1016/j.jcmgh.2021.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.