

Abstract
Estrogen Related Receptors (ERRs) are key regulators of energy homeostasis and play important role in the etiology of metabolic disorders, skeletal muscle related disorders, and neurodegenerative diseases. Among the three ERR isoforms, ERRα emerged as a potential drug target for metabolic and neurodegenerative diseases. Although ERRβ/γ selective agonist chemical tools have been identified, there are no chemical tools that effectively target ERRα agonism. We successfully engineered high affinity ERRα agonism into a chemical scaffold that displays selective ERRβ/γ agonist activity (GSK4716), providing novel ERRα/β/γ pan agonists that can be used as tools to probe the physiological roles of these nuclear receptors. We identified the structural requirements to enhance selectivity toward ERRα. Molecular modeling shows that our novel modulators have favorable binding modes in the LBP of ERRα and can induce conformational changes where Phe328 that originally occupies the pocket is dislocated to accommodate the ligands in a rather small cavity. The best agonists up-regulated the expression of target genes PGC-1α and PGC-1β, which are necessary to achieve maximal mitochondrial biogenesis. Moreover, they increased the mRNA levels of PDK4, which play an important role in energy homeostasis.
Keywords: Estrogen-Related Receptors, Pan Agonists, Molecular Modeling, N-Acyl Hydrazones
1. Introduction
Estrogen Related Receptors (ERRs) are members of the nuclear hormone receptors (NRs) superfamily. The ERRs subfamily comprise three members, ERRα, ERRβ and ERRγ; and are closely related to the estrogen receptors (ERα and ERβ). ERRα and ERRβ were identified by Giguère and his co-workers in 1988 [1], and ERRγ was identified in 1999 by Chen et al. [2].
The ERRs are orphan receptors since no natural ligands have been identified for any of the three ERR isoforms. Unlike ERs, ERRs have constitutive activity and can function in absence of ligands. Although ERRs are structurally related to ERs and share sequence similarity with these receptors, they do not bind with estrogens, which are the endogenous ligands of the ERs. ERRα is expressed mainly in tissues of high energy demand such as brown adipose tissue, intestine, and skeletal muscles and functions as a sensor of energy metabolism. ERRβ is expressed in low levels in adult skeletal muscle, heart, kidney, eyes, and ears. This isoform is known to play an important role in the early development and its postnatal expression is highly restricted. ERRγ is expressed mainly in skeletal muscle, heart, pancreas, kidney, placenta, spinal cord and brain [3–5]. Within the ERR subfamily, ERRα is the most divergent of the α/β/γ trio, especially with regard to the ligand binding domain (LBD). The ligand binding pocket (LBP) of ERRα is only 100 Å3, which makes it the smallest LBP among NRs. ERRα is constitutively active because the LBP is partially filled with the amino acid residue Phe328, which allows helix 12 (H12) to adopt an active conformation that can interact with coactivators. The crystal structure of ERRα LBD in complex with coactivator PGC-1α shows that estrogen-like ligands cannot fit the small LBP and most likely will disrupt the active conformation [6]. This is probably why the majority of ERRα identified ligands are inverse agonists. Co-crystal structure of inverse agonist 1 with ERRα LBD shows that only part of this ligand fits in the LBD and the rest of the ligand sticks out from the LBP. The LBP undergoes significant conformational changes to accommodate the ligand and Phe510 (on H12) was dislocated to avoid steric clash with Phe328. Consequently, H12 was displaced to fill the coactivator groove of the AF-2 and therefore inactivate the receptor. This molecular mechanism is unique to ERRα and highlights the possibility of rationally designing drugs to modulate ERRα despite the small size of the LBP. Even though ERRα LBP is small in size and partially filled, a well-designed small-molecule ligand can be accommodated if it can promote conformational changes and cause the cavity to open up.
Several ERRα inverse agonists have been identified. Kaempferol (2), dietary flavonol found in many natural products, can reduce gene transcription by inhibiting the interaction between ERRα and DNA response elements [7]. XCT790 (3) is another ERRα inverse agonist that works by inducing ERRα ubiquitin-dependent proteasomal degradation [8,9]. This compound can inhibit the proliferation of A549 lung cancer cell [10] and HepG2 hepatocarcinoma and its multi-drug resistance (MDR) sub-line R-HepG2 by inducing mitochondrial reactive oxygen species [11] and reducing mitochondrial mass. XCT790 (3) reduces the expression of PGC-1α [12] and PGC-1β [13], thus suppressing mitochondrial biogenesis and increasing glucose uptake.
Scientists at GlaxoSmithKline suggested that ERRα may be intractable as a drug target after they failed to identify ERRα agonists using high-throughput screening and structure guided design [14]. However, natural phytoestrogens were identified by virtual screening as potential ERRα agonists. For example, in addition to functioning as ERα and ERβ agonists, genistein (4a), daidzein (4b), and biochanin (4c) are three isoflavones, which behaved as agonists for all ERRs and were shown to activate ERRα in mammalian cell transfection and mammalian two-hybrid experiments [15].
Ding et al. identified a series of pyrido[1,2-a]pyrimidin-4-ones as ERRα agonists by screening their carbonyl focused library [16]. These compounds enhanced the uptake of glucose and fatty acid in C2C12 muscle cells through the elevation of mRNA and protein levels of ERRα in downstream targets. Compound 5 has an agonistic effect on ERRα and was shown to increase the transcription of ERRα in a dose dependent manner. Moreover, compound 5 activated the receptor in 293FT cells pretreated with kaempferol (2), which is known inverse agonist of ERRα. Interestingly, 5 increased the mRNA levels of key target genes involved in oxidative metabolism such as MCAD, PDK4, and ATP5b. This compound was selective for ERRα over ERα, ERβ, and ERRβ. However, 5 moderately agonized ERRγ [16].
As part of our ongoing research program to develop chemical tools for various nuclear receptors (e.g. LXR [17–20], ROR [21], REV-ERB [22]), we became interested in developing ERRα/γ dual agonists with the aim of eventually developing small molecule agonists specific to ERRα to evaluate the function of this receptor in animal models as well as determine the potential utility of ERRα ligands in human diseases. The lack of chemical probes to this drug target has hindered our ability to study the relationship between ERRα activity and diseases states and to launch the clinical translatability of this target. GSK4716 (6) (Fig. 1) was reported as an agonist of ERRβ and ERRγ with no observed activity on ERRα [23]. In our hands, GSK4716 (6) showed very weak activity against ERRα, which prompted us to revisit this lead compound to identify ERRα/γ dual agonists. Herein we describe our probe of the structure activity relationships (SARs) of GSK4716 (6) through the chemical modification of rings A and B while maintaining the acyl hydrazone moiety to identify the structural requirements to promote selectivity toward the ERRα isoform.
Fig. 1.

Examples of ERRα modulators.
2. Results and discussion
2.1. Chemistry
The synthesis of compounds 10–46 is illustrated in Scheme 1. The acyl hydrazides (8) were prepared from their corresponding methyl esters (7) through reaction with hydrazine in methanol under reflux. All acyl hydrazides (8) were obtained in quantitative yields and their identity were confirmed by comparing their melting points and mass to literature. The acyl hydrazones (10–46) were synthesized by condensation of acyl hydrazides (8) with aldehydes (9) in ethanol in presence of catalytic amount of trifluoroacetic acid.
Scheme 1.

Synthesis of N-acyl hydrazones 10–46.
2.2. SAR study of acyl hydrazone derivatives
Synthesized compounds were screened in a cell-based (HEK293 cells) co-transfection assay using ERRE reporter construct and pcDNA3.1 ERRγ or pcDNA3.1 ERRα. Luciferase activity was measured using the Dual-Glo luciferase reporter assay system (Promega). The values indicated represent the means ± S.E. from four independently transfected wells. EC50 values for all described compounds were obtained by titration in an eleven-point dose response format. All compounds were screened against GSK4716 (6) as a reference compound for agonist activity. Compounds XCT790 (3), which is a potent and specific inverse agonist for ERRα, and 4-hydroxytamoxifen (4-OHT), which is a nonselective inverse agonist for ERRγ, were used as positive controls to identify inverse agonism activity.
Zuercher et al. [23] reported that the hydroxyl group or analogous hydrogen bond donor (HBD) group on ring A is required to obtain ERRγ activity. Recently, Lin et al. [24] showed that removing or repositioning this hydroxyl group led to significant decrease or even complete loss of ERRγ activity. 4-Isopropyl and 4-diethylamino groups were the optimal substituents at ring B to maintain ERRγ activity.
We started probing the SAR of GSK4716 (6) by removing both substituents at both rings A and B, which led to complete loss of activity against both ERRα and ERRγ (i.e. 10). Then, we investigated the SAR of ring B while ring A was unsubstituted phenyl. ERRγ activity was regained upon introduction of 4-dimethylamino (i.e. 11) or 4-chloro (i.e. 12), but no ERRα activity was observed. Compound 12 was > 8-fold more potent than 11. While hydroxyl group at the ortho-position of ring B was not tolerated (i.e. 13) and the compound was inactive against both ERRα and ERRγ, compound 14 with hydroxyl group at the para-position showed weak activity against both isoforms and was more selective toward ERRγ (> 3-fold) (Table 1).
Table 1.
In vitro ERRα and ERRγ agonistic activities of 10–24.
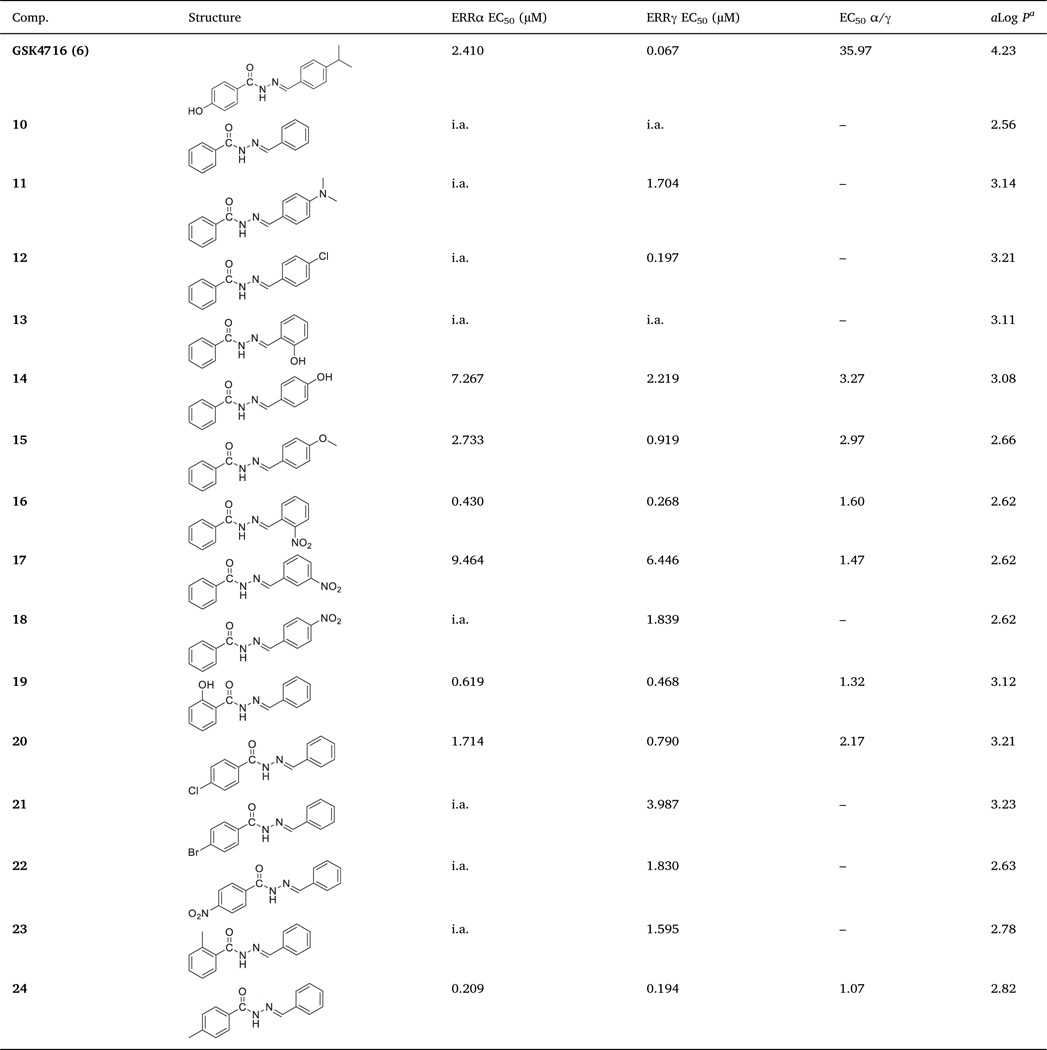
|
ALOGPS 2.1, molecular property prediction [26]. i.a. = inactive
A methoxy group at para-position of ring B (i.e. 15) enhanced the activity toward both ERRα and ERRγ but remained more selective to-ward ERRγ (≈3-fold). Compound 15 was ≈ 2.66-fold more potent toward ERRα than 14. Introduction of nitro group at the ortho-position of ring B (i.e. 16) increased the potency toward ERRα > 6-fold if compared to 15 and ≈17-fold if compared to 14. Compound 16 remained more selective toward ERRγ over ERRα (1.6-fold). Moving the nitro group from ortho -position to meta- (i.e. 17) or para-position (i.e. 18) led to the loss of ERRα activity and significant decrease in ERRγ activity (Table 1). Docking of compound 16 in both ERRα and ERRγ resulted in a similar binding pose in both receptors. The ligand is involved in hydrophobic interactions with surrounding residues in the ligand binding pocket (LBP). The aromatic ring B makes π-π interactions with Tyr326 in ERRγ and π-π interactions with Phe328 in ERRα. The nitro group in this binding pose is oriented towards the inside of the cavity of ERRα, and involved in cation- π interactions with Phe328 which correspond to Ala272 in ERRγ (Fig. 2).
Fig. 2.

Computational docking of 16 in ERRα (A) and ERRγ (B). Hydrogen bonds are shown as green dashed lines and hydrophobic interactions are shown as purple dashed lines. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We then begin investigating the SAR of ring A while ring B remained fixed as an unsubstituted phenyl. When we introduced the hydroxyl group at ortho-position, we obtained compound 19 that was active toward both ERRα (EC50 = 0.619 μM) and ERRγ (EC50 = 0.468 μM) with significant enhancement in selectivity toward ERRα (EC50 α/γ = 1.32). Evans et al. [25] showed that compound DY162, which is an analogue of 19 with hydroxyl group at para-position of ring A, was ERRγ agonist with no activity toward ERRα.
Substituting ring A with chlorine at para-position led to identification of compound 20, which was weakly active against ERRα (EC50 = 1.714 μM) and moderately potent against ERRγ (EC50 = 0.790 μM). Bromine (i.e. 21) or nitro (i.e. 22) groups were not tolerated at the same position. While both 21 and 22 maintained moderate ERRγ activity, they completely lost ERRα activity. Methyl substitution at ortho-position (i.e. 23) showed a similar pattern with moderate activity toward ERRγ (EC50 = 1.595 μM) and no activity toward ERRα. Surprisingly, methyl substituent at para-position (i.e. 24) enhanced the potency toward both ERRα (EC50 = 0.209 μM) and ERRγ (EC50 = 0.194 μM) significantly. Compound 24 was equipotent toward both isoforms (EC50 α/γ = 1.07) (Table 1 and Fig. 3).
Fig. 3.

A cell-based co-transfection assay using ERR responsive luciferase reporter for GSK4716 (6) and 24.
Molecular modeling of 24 in the active site of ERRγ show that ring B is making π-π interactions with Tyr326 and the NH makes a hydrogen bond with the carbonyl oxygen of the same residue (Fig. 4B). In ERRα, the predicted binding pose is a slightly tilted from the binding pose in ERRγ and the compound is predicted to make π-π interactions with Phe328 on helix 3 (H3) (Fig. 4A).
Fig. 4.

Computational docking of 24 in ERRα (A) and ERRγ (B). Hydrogen bonds are shown as green dashes lines and hydrophobic interactions are shown as purple dashed lines. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To further probe the SAR of this scaffold, we decided to assess the effect of having two different substituents at both rings A and B on the activity and selectivity toward ERRα and ERRγ (Table 2). When we introduced methyl group in ortho-position of ring A in 11, we obtained compound 25. Like 11, compound 25 was inactive against ERRα and the potency toward ERRγ dropped > 4-fold. Introducing chlorine to the para-position of 12 gave compound 26, which was inactive against both ERR isoforms. Interestingly, introducing hydroxyl group to the ortho-position of either ring A (i.e. 19, Table 1) or ring B (i.e. 13, Table 1) showed a very distinct pharmacological behavior. While 19 was moderately potent against both isoforms with slightly higher selectivity toward ERRγ, 13 was completely inactive against both isoforms. This further supports the importance of having a hydrogen bond donor (HBD) on ring A, especially in absence of any substituents at ring B, for the compound to have affinity toward ERRs. Introducing two hydroxyl groups to the ortho-position of both rings gave compound 27, which was more potent toward ERRα (EC50 = 0.378 μM) than ERRγ (EC50 = 1.645 μM). Compound 27 is > 4-fold more selective toward ERRα over ERRγ (EC50 α/γ = 0.23) (Table 2). This compound is expected to have poor pharmacokinetic properties because it is susceptible to metabolic clearance via glucuronidation or sulfonation of the two hydroxyl groups.
Table 2.
In vitro ERRα and ERRγ agonistic activities of 25–38.
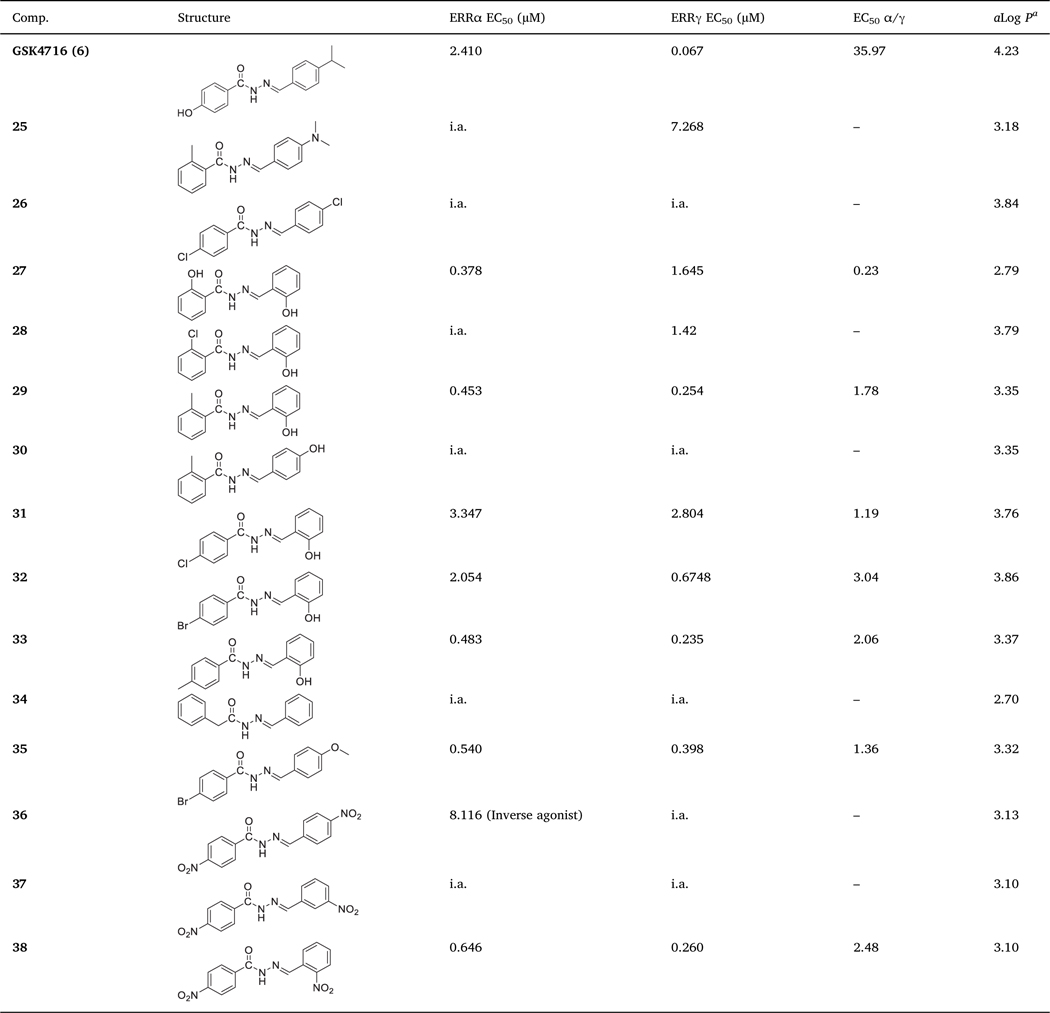
|
In ERRγ, 27 is predicted to make π-π interactions with Tyr326 that lies on the β-sheets region and hydrogen bonding interactions with Glu247. However, docking this compound in ERRα resulted in a docking pose where the molecule is slightly tilted to interact with Phe328 (Fig. 5). It is possible that this interaction contributes to selectivity of this compound towards ERRα. The hydroxyl group on ring A is predicted to make hydrogen bonding interactions with Glu331 in ERRα. The amide group is making hydrogen bonding interaction with the backbone of Ty326 in ERRγ and Phe328 in ERRα (Fig. 5).
Fig. 5.

Computational docking of 27 in ERRα (A) and ERRγ (B). Hydrogen bonds are shown as green dashed lines and hydrophobic interactions are shown as purple dashed lines. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The activity toward ERRα was lost completely when we replaced the hydroxyl group on ring A of 27 with chlorine atom in 28. Compound 28 preserved activity toward ERRγ. On the contrary, 29 with methyl group in ortho-position of ring A was active toward both ERRα (EC50 = 0.453 μM) and ERRγ (EC50 = 0.254 μM). Ring B of 29 is predicted to make π-π interactions with Tyr326 in ERRγ. In ERRα, the same group is predicted to make π-π interactions with two aromatic side chains; Phe328 and Phe382. Moreover, the NH group of 29 is predicted to make hydrogen bonding interactions with the backbone of Tyr328 in ERRγ and Phe382 in ERRα (Fig. 6).
Fig. 6.

Computational docking of 29 in ERRα (A) and ERRγ (B). Hydrogen bonds are shown as green dashed lines and hydrophobic interactions are shown as purple dashed lines. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Moving hydroxyl group on ring B of 29 from ortho-position to para-position gave compound 30, which was inactive toward both isoforms. Introducing chlorine or bromine in para-position of ring A while maintaining hydroxyl group in ortho-position of ring B, gave compounds 31 and 32, respectively. These two compounds showed weak activity toward ERRα and ERRγ but were more selective toward ERRγ. Compound 33 with methyl group in the para-position of ring A showed good activity toward both ERRα (EC50 = 0.483 μM) and ERRγ (EC50 = 0.235 μM) (Table 2). Attempt to replace ring A with benzyl group gave compound 34 that was completely inactive against both isoforms. Compound 34 has no affinity toward ERRs most probably due to the extra flexibility gained by having methylene linker between ring A and the carbonyl group.
Compound 35 possess bromine in the para-position of ring A and methoxy in the para-position of ring B and showed good activity toward ERRα (EC50 = 0.540 μM) and ERRγ (EC50 = 0.398 μM) with good subtype selectivity (EC50 α/γ = 1.36). The addition of bromine to ring A enhanced the subtype selectivity > 2-fold (cf. 15; EC50 α/γ = 2.97). However, in absence of methoxy group, compound 21 was inactive toward ERRα and very weak toward ERRγ. Compound 35 demonstrates the influence of additive effect and the interplay of substituents at ring A and ring B. Therefore, it is feasible to develop isoform selective modulators for ERR subtypes by having the right combination of substituents on both rings.
Both 18 (4-NO2 on ring B) and 22 (4-NO2 on ring A) were inactive toward ERRα and weakly active toward ERRγ. Introducing the two nitro groups simultaneously at both rings in compound 36 led to a switch in activity toward ERRα from agonism to inverse agonism. Compound 36 showed a weak decrease in the basal activity of ERRα (IC50 = 8.116 μM) compared to the ERRα potent inverse agonist XCT790 (3) (IC50 = 0.175 μM). Moreover, 36 was inactive against ERRγ. Since ERRs are constitutively active receptors, ligands that decrease their constitutive activity are identified as inverse agonists. Although the observed inverse agonism activity of this compound was very weak, it demonstrates that two electron-withdrawing groups at para-position of both rings are not tolerated. The compound lost activity toward both isoforms upon moving the nitro group on ring B from para-position (i.e. 36) to meta-position (i.e. 37). The agonistic activity was retained upon moving the nitro group on ring B from meta-position in 37 to ortho-position in 38 (EC50 = 0.646 μM for ERRα, and 0.260 μM for ERRγ). Comparing 38 with nitro group in the para-position of ring A to 16 that lacks nitro group on ring A, we noticed a slight decrease in the potency of 38 against ERRα (≈1.5-fold) and insignificant change in potency against ERRγ. Moreover, subtype selectivity decreased by ≈1.5-fold (EC50 α/γ = 1.6 for 16 vs 2.48 for 38). This decrease in activity and subtype selectivity toward ERRα is most likely due to the smaller size of the ligand binding pocket of ERRα, which prefers smaller ligands.
Finally, we decided to explore different substituents in lieu of ring B while maintaining the ring A as an unsubstituted phenyl ring (Table 3). Neither small alkyl substituent (i.e. 39) nor phenylallyl analogs (i.e. 41 and 42) were tolerated and these derivatives were inactive toward both ERR isoforms. Compound 42 with thiophene ring as a bioisostere of phenyl ring in 10 showed weak agonistic effect against both ERR isoforms and good subtype selectivity (EC50 α/γ = 1.78). Despite the weak agonistic activity of 42, replacement of phenyl ring with its bioisostere (i.e. thiophene) has changed the profile of 10 from completely inactive compound to an active compound (i.e. 42) with acceptable isoform selectivity. The corresponding furan analogue (i.e. 43) was inactive toward both isoforms. The 5-methyl substituted furan analogue (i.e. 44), has weak ERRγ activity (EC50 = 2.701 μM) and no ERRα activity. The quinolin-2-ylmethylene benzo-hydrazide 45 was more potent against ERRα (EC50 = 0.487 μM) over ERRγ (EC50 = 0.836 μM). Surprisingly, the quinolin-4-ylmethylene benzohydrazide 46 showed weak inverse agonism behavior against ERRα (IC50 = 4.578 μM) in comparison to the ERRα potent inverse agonist XCT790 (3) (IC50 = 0.175 μM).
Table 3.
In vitro ERRα and ERRγ agonistic activities of 39–46.
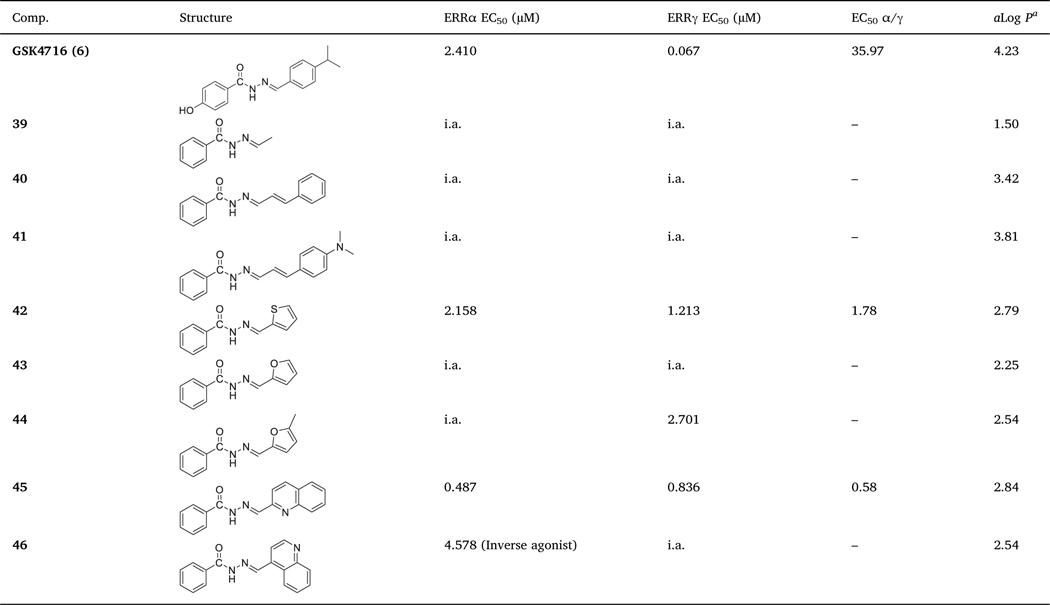
|
Our SAR data of developed agonists supported by molecular modelling show that removing the hydroxyl group at para-position of ring A in GSK4716 (6), which forms H-bond with Glu384 and avoiding using hydrogen bond donors at the same position reduced the selectivity toward ERRγ and enhanced the selectivity toward ERRα. The best functionalities at ring A were unsubstituted phenyl ring and phenyl ring substituted with Cl, Br, NO2, Me (at para-position) or OH (at ortho-position). The best functionalities at ring B were unsubstituted phenyl ring, phenyl ring substituted with OH or NO2 (at ortho-position), OMe (at para-position), and thiophene or quinoline heterocycles. Any spacer between ring A and carbonyl group or between ring B and azomethine group tend to abolish the activity.
Although ERRβ is expressed in low levels in adult tissues and its postnatal expression is highly restricted, we have tested compounds 24, 27, 29, 33, and 42 against ERRβ to address the activity of our synthesized agonists against this isoform (Table 4). All of our agonists activated ERRβ, but there was no specific pattern corresponding to how it activate the other isoforms. Compounds 24 (Fig. 3) and 33 were more active toward ERRβ than ERRα and ERRγ. Unlike 24 and 33, compound 27 was more active toward ERRα than the two other isoforms (≈2-folds higher than ERRβ and > 4-folds higher than ERRγ). Compound 29 was more active toward ERRα and ERRγ than ERRβ. The activity of 42 toward ERRβ was almost equal to ERRγ and 1.8-folds higher than ERRα.
Table 4.
In vitro ERRβ agonistic activities of 24, 27, 29, 33, and 42.
| GSK4716 (6) | 24 | 27 | 29 | 33 | 42 | |
|---|---|---|---|---|---|---|
|
| ||||||
| ERRβ EC50 (μM) | 0.280 | 0.162 | 0.772 | 0.583 | 0.123 | 1.193 |
2.3. Molecular modeling
We performed docking of selected compounds in the ligand binding pocket of ERRα and ERRγ. Molecular docking in ERRγ was performed using the ERRγ bound agonist conformation (PDBID: 2GPP) [27]. There is no reported agonist bound conformation of ERRα and molecular docking of GSK4716 (6) to the agonist pocket in apo ERRα X-ray structure (PDID: 1XB7) was not possible initially because the pocket was occupied with the side chains of amino acid residues Phe328, Arg372 and Glu331 and there was not sufficient room to accommodate the ligand [28]. The amino acid residues in both pockets are similar and the agonist GSK4716 (6) is expected to bind in similar manner in both receptors with slight differences. In order to obtain an ERRα structure suitable for docking, we performed manual modeling of ERRα bound with the agonist GSK4716 (6) followed by refinement of the ligand bound complex by performing local optimization using Schrodinger/Prime [29]. The protein-ligand complex was validated using molecular dynamics simulations for 500 ns (data will be published in due course). The refined ERRα-ligand complex was used as the receptor in the docking calculations.
Molecular modelling using induced fit docking of compounds 16, 24, 27, and 29 in the LBP of ERRα show that they have favorable binding modes where they make π-π interactions with Phe328, which corresponds to Ala272 in ERRγ. Moreover, the compounds tend to adopt a tilted confirmation to fit in the much smaller binding pocket of ERRα and make π-π interactions with Phe328 that originally occupies the available space in this binding pocket.
2.4. Gene expression
ERRs are the major transcriptional regulators that regulate key genes involved in energy metabolism within cells and tissues of high-energy demand. They regulate genes involved in carbohydrate and energy metabolism, lipid synthesis and fatty acid oxidation, mitochondrial activity and oxidative phosphorylation [20]. The agonistic effect of the best dual agonists (i.e. 16, 24, 27, 29, 35, 38, and 42) was validated by measuring the changes in gene expression of four ERR target genes; PGC-1α, PGC-1β, CPT1α and PDK4 (Fig. 7) in mouse myoblast cell line C2C12 at 1 μM of tested compounds. The peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) and the peroxisome proliferator-activated receptor-γ coactivator 1β (PGC-1β) regulate several metabolic pathways and their activity is of great importance to achieve maximal mitochondrial biogenesis.
Fig. 7.

Normalized mRNA expression of PGC-1α, PGC-1β, CPT1α and PDK4 with 16, 24, 35, and 42 in muscle cells.
Compounds 24 and 42 were found to increase the mRNA levels of both PGC-1α and PGC-1β, while compound 35 increased the mRNA levels of PGC-1α only. Carnitine palmitoyltransferase 1α (CPT1α) catalyzes the first step of long-chain fatty acid import into mitochondria, and its activity is believed to be rate limiting for β-oxidation of fatty acids. Compound 16 significantly increased the expression of CPT1α. The pyruvate dehydrogenase kinase isozyme 4 (PDK4) play an important role in energy homeostasis and catalyzes the conversion of pyruvate to CoA. The transcription of PDK4 is controlled by both ERRα and ERRγ in hepatic cells and muscle. Compounds 16, 24, 35, and 42 potently upregulated PDK4 transcription.
2.5. Cytotoxicity assay
Cell viability was assessed using FITC Annexin V/ Dead cell Apoptosis kit (Invitrogen) for flow cytometry according to the manufacturer’s manual. Compounds 24, 27, 29, and 42 were tested at 1 and 10 μM. No signs of toxicity were observed for active compounds (Fig. 8).
Fig. 8.

Cell viability of 24, 27, 29, and 42.
2.6. In silico ADME evaluation
In order to evaluate the drug-likeness and to to predict the absorption, distribution, metabolism, elimination and toxicity (ADMET) of some of the best identified modulators, we have calculated a set of molecular descriptors using QikProp program [30]. All evaluated compounds complied with Lipinski’s Rule of Five and showed the desired molecular weight (MW), lipophilicity (log P), number of hydrogen bond donors (HBD) and number of hydrogen bond acceptors (HBA) (Table 5). Compliance with Lipinski’s Rule of Five indicate that these molecules have drug-like properties [31]. These compounds were also found to comply with Jorgensen’s rule of 3 (LogSwat, BIPCaco-2, and number of primary metabolites) and are predicted to have oral bioavailability [32,33]. These compounds are predicted to have good aqueous solubility, cell permeability, and oral bioavailability (Table 5). The calculated apparent MDCK cell permeability, which mimic the BBB further suggests that these compounds have good cell permeability. Moreover, the estimated plasma-protein binding to human serum albumin (logKHSA) of these compounds are within the recommended range, which means that these compounds are predicted to circulate freely within the blood stream and can achieve high efficiency.
Table 5.
Calculated molecular descriptors for prediction of ADME properties for 16, 24, 27, 29, 35, and 42.
| Descriptors/Properties | aMW (Da) | bHBD | cHBA | dPSA | eLogP o/w | fLogSwat (moles/liter) |
|---|---|---|---|---|---|---|
|
| ||||||
| Range for 95% of known Drugs | 130.0 to 725.0 | 0 to 6 | 2.0 to 20.0 | 7.0 to 200.0 | −2.0 to 6.5 | −6.5 to 0.5 |
| 16 | 269.26 | 1 | 3.5 | 93.04 | 2.81 | −3.93 |
| 24 | 238.29 | 1 | 2.5 | 49.78 | 3.85 | −4.46 |
| 27 | 256.26 | 2 | 3 | 89.93 | 2.52 | −3.51 |
| 29 | 254.29 | 2 | 3.25 | 68.23 | 3.08 | −4.24 |
| 35 | 333.18 | 1 | 3.25 | 57.98 | 4.06 | −4.95 |
| 42 | 230.28 | 1 | 2.5 | 48.35 | 2.90 | −3.34 |
| Properties | g%Human oral absorption | hLog B/B | iBIPCaco-2 (nm/sec) | jMDCK | k#metab | lLogKHSA |
|
| ||||||
| Range for 95% of known Drugs | < 25% is poor | −3.0 to 1.2 | < 25 poor, >500 great | <25 poor, > 500 great | 1.0 to 8.0 | −1.5 to 1.5 |
| 16 | 88 | −1.26 | 319 | 144 | 1 | 0.13 |
| 24 | 100 | −0.34 | 2230 | 1177 | 1 | 0.34 |
| 27 | 87 | −1.25 | 353 | 160 | 2 | 0.02 |
| 29 | 100 | −0.67 | 1170 | 586 | 2 | 0.11 |
| 35 | 100 | −0.23 | 2230 | 3120 | 1 | 0.34 |
| 42 | 100 | −0.29 | 1891 | 1330 | 1 | 0.05 |
Molar weight;
Number of hydrogen bonds donated by the molecule;
Number of hydrogen bonds accepted by the molecule,
Polar surface area;
Logarithm of partitioning coefficient between n-octanol and water phases;
Logarithm of aqueous solubility;
Predicted human oral absorption on a 0–100% scale, based on a multiple linear regression model;
Logarithm of predicted blood/brain barrier partition coefficient;
Predicted apparent Caco-2 cell membrane permeability in Boehringer–Ingelheim scale;
Predicted apparent MDCK cell permeability;
Number of likely metabolic reactions;
Logarithm of predicted binding constant to human serum albumin.
3. Conclusions
We were able to modulate the estrogen-related receptors subtype selectivity by converting an ERRβ/γ specific agonist, GSK4716 (6), into ERRα/β/γ pan agonists. Molecular modeling shows that the new pan agonists have favorable binding modes in both isoforms but have different conformations in the LBP of ERRα. These ligands induce a new conformation in the cavity of ERRα where Phe328 is slightly displaced to accommodate the ligand and make π-π interactions with one of the two phenyl rings. The identified ligands increased the expression of ERR target genes, PGC-1α, PGC-1β, CPT1α and PDK4 in mouse myoblast cell line C2C12. Compounds 24, 27, 29, and 42 did not show signs of toxicity when tested at 1 and 10 μM. We identified the important structural requirements to enhance the selectivity toward ERRα and our future goal is to develop ERRα specific agonists to be used as chemical tools to validate ERRα as a putative target for treatment of neurodegenerative diseases like Alzheimer Disease.
4. Experimental
4.1. Chemistry
All starting materials were purchased from commercial suppliers and used without further purification. The purities of the final compounds were characterized by high-performance liquid chromatography (LC/MS) using a gradient elution program (Ascentis Express Peptide C18 column, acetonitrile/water 5/95/95/5, 5 min, 0.05%trifluoracetic acid) and UV detection (254 nM). The purities of final compounds were 95% or greater. NMR spectra was recorded on a Bruker NMR 400 MHz Avance III spectrometer operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR. Chemical shifts are given in part per million (ppm) relative to the deuterated solvent residual peak, coupling constants J are given in Hertz.
4.1.1. General procedure for the preparation of hydrazides 8
Carboxylic acids 1a-g (5 mmol) were heated under reflux in methanol (5 mL) for 2 h in presence of conc. H2SO4 (catalytic amount) with continuous stirring. The reaction was monitored by TLC till the acids were fully converted to the corresponding esters [Eluent: EtOAc/Hexanes (1:4)]. The reaction mixture was allowed to cool down to room temperature and hydrazine monohydrate 80% (20 mmol, 0.96 mL) was added slowly in an ice bath. The reaction was then warmed to room temperature and heated under reflux for another 1–2 h and followed by TLC till formation of hydrazide. The reaction mixture was kept at refrigerator till the product precipitated. All hydrazides were isolated in quantitative yields and in a pure form. Their melting points were in full agreement with the literature melting points of the same compounds.
4.1.2. General procedure for the preparation of hydrazones 10–46
A mixture of the appropriate hydrazides 8 (1 mmol) and the appropriate aldehyde 4a-j (1.1 mmol) was refluxed in ethanol until the condensation was complete (monitored by TLC, 1–3 h). The reaction was allowed to cool to room temperature and the formed precipitate was filtered and washed with cold ethanol. The purity of most compounds was ≈98% and in few cases the obtained solid was recrystallized from ethanol to give the desired product. All acyl hydraones reported here are known compounds their identity were confirmed by comparing their melting points and mass to literature (See supporting information). 1H and 13C NMR spectra of most active compounds are described here.
4.1.2.1. (E)-N’-(2-Nitrobenzylidene)benzohydrazide (16) [34].
Pale yellow crystals, mp 222–224 °C, 89% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 8.88 (s, 1H), 8.14 (d, J = 7.3 Hz, 1H), 8.08 (d, J = 8.1 Hz, 1H), 7.83 (d, J = 7.2 Hz, 2H), 7.83 (t, J = 7.3 Hz, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.63 – 7.50 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 163.3, 148.3, 142.9, 133.7, 133.0, 132.0, 130.7, 128.8, 128.5, 127.9, 127.7, 124.7. LC/MS m/z: 270 [M+H]+.
4.1.2.2. (E)-N’-Benzylidene-4-methylbenzohydrazide (24) [35].
White needles, mp 235–236 °C, 90% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.78 (s, 1H), 8.47 (s, 1H), 7.84 (d, J = 7.7 Hz, 2H), 7.73 (d, J = 6.3 Hz, 2H), 7.50 – 7.41 (m, 3H), 7.33 (d, J = 7.9 Hz, 2H), 2.38 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 162.9, 147.5, 141.8, 134.4, 130.5, 129.0, 128.8, 127.6, 127.0, 21.0. LC/MS m/z: 239 [M+H]+.
4.1.2.3. (E)-2-Hydroxy-N’-(2-hydroxybenzylidene)benzohydrazide (27) [36].
White microcrystals, mp 265–267 °C, 85% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.03 (s, 1H), 11.78 (s, 1H), 11.20 (s, 1H), 8.69 (s, 1H), 7.90 (d, J = 7.1 Hz, 1H), 7.57 (d, J = 6.9 Hz, 1H), 7.46 (t, J = 7.7 Hz, 1H), 7.32 (t, J = 8.2 Hz, 1H), 7.05 – 6.72 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 164.5, 159.0, 157.50, 149.0, 134.0, 131.6, 129.5, 128.6, 119.4, 119.0, 118.6, 117.3, 116.5, 115.6. LC/MS m/z: 257 [M+H]+.
4.1.2.4. (E)-N’-(2-Hydroxybenzylidene)-2-methylbenzohydrazide (29) [37].
White crystals, mp 163–165 °C, 50% yield. 1H NMR (400 MHz, DMSO, two rotamers 4:1) δ 11.99 (s, 0.8H), 11.87 (s, 0.2H), 11.22 (s, 0.8H), 9.84 (s, 0.2H),8.50 (s, 0.8H), 8.27 (s, 0.2H), 7.53 – 7.26 (m, 6H), 6.95 – 6.76 (m, 2H), 2.40 (s, 2.4H), 2.25 (s, 0.6H); 13C NMR (101 MHz, DMSO) δ 164.9, 157.4, 147.9, 136.1, 134.6, 131.4, 130.7, 130.2, 129.5, 127.6, 126.7, 125.7, 125.5, 119.4, 118.6, 116.4, 19.4. LC/MS m/z: 255 [M+H]+.
4.1.2.5. (E)-N’-(2-Hydroxybenzylidene)-4-methylbenzohydrazide (33) [38].
Yellow solid, mp 199–201 °C, 83% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 11.30 (s, 1H), 8.60 (s, 1H), 7.82 (d, J = 7.9 Hz, 2H), 7.50 (d, J = 7.5 Hz, 1H), 7.35–7.25 (m, 3H), 6.89 (t, J = 8.1 Hz, 2H), 3.30 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 162.6, 157.5, 148.1, 142.1, 131.3, 129.9, 129.6, 129.1, 127.7, 119.3, 118.7, 116.4, 21.1. LC/MS m/z: 255 [M+H]+.
4.1.2.6. (E)-4-Bromo-N’-(4-methoxybenzylidene)benzohydrazide (35) [39].
White microcrystals, mp 129–131 °C, 89% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.78 (s, 1H), 8.39 (s, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.71 (d, J = 8.0 Hz, 2H), 7.68 (d, J = 8.3 Hz, 2H), 7.02 (d, J = 8.3 Hz, 2H), 3.81 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 162.0, 160.9, 148.0, 132.6, 131.5, 129.7, 128.8, 126.8, 125.4, 114.4, 55.3. LC/MS m/z: 333 [M+H+].
4.1.2.7. (E)-4-Nitro-N’-(2-nitrobenzylidene)benzohydrazide (38) [40].
White solid, mp 260–262 °C, 91% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.47 (s, 1H), 8.90 (s, 1H), 8.38 (d, J = 8.6 Hz, 2H), 8.28 – 8.05 (m, 4H), 7.84 (t, J = 7.6 Hz, 1H), 7.71 (t, J = 7.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.7, 149.4, 148.3, 144.2, 138.6, 133.8, 131.0, 129.3, 128.5, 128.1, 124.7, 123.7. LC/MS m/z: 315 [M+H]+.
4.1.2.8. (E)-N’-(Thiophen-2-ylmethylene)benzohydrazide (42) [37].
White crystals, mp 207–209 °C, 91% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.77 (s, 1H), 8.64 (s, 1H), 7.86 (d, J = 7.5 Hz, 2H), 7.79 (d, J = 7.1 Hz, 1H), 7.57 – 7.38 (m, 4H), 7.11 (dd, J = 5.0, 3.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 166.3, 143.4, 133.8, 131.4, 129.4, 128.9, 128.7, 128.3, 128.0, 127.4. LC/MS m/z: 231 [M+H]+.
4.2. Molecular modeling
Docking in ERRα and ERRγ was performed using the induced-fit docking module in Schrodinger to represent ligand-induced conformational changes [41–43]. Each ligand was initially docked using a softened potential (van der Waals radii scaling) of the ligand and the active site side chains. The scaling factors to soften the potentials of the receptors and ligands were set to 0.5 and the maximum number of poses was set to 20. The side chains of the residue within 5.0 A of the ligand are refined along with the docked ligand with Prime using a continuum solvation based molecular mechanics model, via rotamer-based library optimizations of the amino acids side chain conformations [29]. The ligand is re-docked, using the standard precision algorithm implemented in Glide into the induced-fit receptor structures and reminimized, and the final poses are rank ordered by the scoring function given by a linear combination of the Prime energy and GlideScore referred to as the IFDScore [42,44]. Images were made using Discovery Studio [45].
4.3. Cell culture
C2C12 cells, mouse myoblast cell line, were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and 1% L-Glutamine. The C2C12 cells were plated in 6 well dishes (Corning) with two and a half million cells per well and grew for 24 h until confluence is reached. The cells were then treated with either the drug in DMSO, or DMSO as the control at a concentration of 1 μM or 10 μM. All groups were tested in triplicates.
4.4. Cell viability assay
C2C12 cells were treated for 24 h with 1 μM or 10 μM of different compounds. After treatment, cells were harvested and stained for live/ dead cells using FITC Annexin V/ Dead Apoptosis Kit with FITC annexin V and PI, for Flow Cytometry (Invitrogen) according the manufacturer instruction. Cells were then analyzed by flow cytometry using BD Accuri c6 flow cytometer (BD Biosciences).
4.5. Gene expression
Cells were treated for 24 h with 10 μM of different compounds. After treatment, cells were harvested and total RNA was extracted using RNeasy Mini Kit according the manufacturer instruction. Total RNA were quantified using NanoDrop and 1 μg of total RNA was reverse transcript using qScript cDNA Synthesis Kit (QuantaBio) according the manufacturer instruction. Gene expression was assessed by qPCR using Sybr Select Master Mix (Applied Biotechnologies). All samples were run in duplicates and the analysis was completed by determining ΔΔCt values. The reference gene used was 36b4, a ribosomal protein gene.
Supplementary Material
Acknowledgments
We would like to thank Support & Development of Scientific Research Center (SDSRC) at Benha University (Egypt) for financial support for M.S. and S.G. This work was partially supported by the National Institute on Aging of the National Institutes of Health (United States) under Award Number R21AG065657 (to B.E.) and National Institute of Arthritis and Musculoskeletal and Skin Diseases (United States) under Award Number R01AR069280 (to T.B.).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2020.104079.
References
- [1].Giguère V, Yang N, Segui P, Evans RM, Identification of a new class of steroid hormone receptors, Nature 331 (1988) 91–94, 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- [2].Chen F, Zhang Q, McDonald T, Davidoff MJ, Bailey W, Bai C, Liu Q, Caskey CT, Identification of two hERR2-related novel nuclear receptors utilizing bioinformatics and inverse PCR, Gene 228 (1999) 101–109, 10.1016/S0378-1119(98)00619-2. [DOI] [PubMed] [Google Scholar]
- [3].Eudy JD, Yao S, Weston MD, Ma-Edmonds M, Talmadge CB, Cheng JJ, Kimberling WJ, Sumegi J, Isolation of a gene encoding a novel member of the nuclear receptor superfamily from the critical region of usher syndrome type IIa at 1q41, Genomics 50 (1998) 382–384, 10.1006/geno.1998.5345. [DOI] [PubMed] [Google Scholar]
- [4].Hong H, Yang L, Stallcup MR, Hormone-independent transcriptional activation and coactivator binding by novel orphan nuclear receptor ERR3, J. Biol. Chem 274 (1999) 22618–22626, 10.1074/jbc.274.32.22618. [DOI] [PubMed] [Google Scholar]
- [5].Heard DJ, Vissing H, Norby PL, Holloway J, Human ERRγ, a Third Member of the Estrogen Receptor-Related Receptor (ERR) Subfamily of Orphan Nuclear Receptors: Tissue-Specific Isoforms Are Expressed during Development and in the Adult, Mol. Endocrinol 14 (2000) 382–392, 10.1210/mend.14.3.0431. [DOI] [PubMed] [Google Scholar]
- [6].Schlaeppi J-M, Fournier B, Bitsch F, Geiser M, Schilb A, Kallen J, Riou V, Strauss A, Filipuzzi I, Graham A, Evidence for Ligand-independent Transcriptional Activation of the Human Estrogen-related Receptor α (ERRα), J. Biol. Chem 279 (2004) 49330–49337, 10.1074/jbc.m407999200. [DOI] [PubMed] [Google Scholar]
- [7].Wang J, Fang F, Huang Z, Wang Y, Wong C, Kaempferol is an estrogen-related receptor α and γ inverse agonist, FEBS Lett. 583 (2009) 643–647, 10.1016/j.febslet.2009.01.030. [DOI] [PubMed] [Google Scholar]
- [8].Stevens WC, Sapp DW, Busch BB, Zhou S, Mohan R, Martin R, Horlick RA, Ordentlich P, Identification of a Selective Inverse Agonist for the Orphan Nuclear Receptor Estrogen-Related Receptor α, J. Med. Chem 47 (2004) 5593–5596, 10.1021/jm049334f. [DOI] [PubMed] [Google Scholar]
- [9].Kersual N, Chalbos D, Vanacker J-M, Bianco S, Lanvin O, Potentiation of ICI182,780 (Fulvestrant)-induced Estrogen Receptor-α Degradation by the Estrogen Receptor-related Receptor-α Inverse Agonist XCT790, J. Biol. Chem 282 (2007) 28328–28334, 10.1074/jbc.m704295200. [DOI] [PubMed] [Google Scholar]
- [10].Wang J, Wang Y, Wong C, Oestrogen-related receptor alpha inverse agonist XCT790 arrests A549 lung cancer cell population growth by inducing mitochondrial reactive oxygen species production, Cell Prolif. 43 (2010) 103–113, 10.1111/j.1365-2184.2009.00659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wu F, Wang J, Wang Y, Kwok TT, Kong SK, Wong C, Estrogen-related receptor α (ERRα) inverse agonist XCT-790 induces cell death in chemotherapeutic resistant cancer cells, Chem. Biol. Interact 181 (2009) 236–242, 10.1016/j.cbi.2009.05.008. [DOI] [PubMed] [Google Scholar]
- [12].Chen L, Wong C, Estrogen-Related Receptor α Inverse Agonist Enhances Basal Glucose Uptake in Myotubes through Reactive Oxygen Species, Biol. Pharm. Bull 32 (2009) 1199–1203, 10.1248/bpb.32.1199. [DOI] [PubMed] [Google Scholar]
- [13].Nie Y, Wong C, Suppressing the activity of ERRα in 3T3-L1 adipocytes reduces mitochondrial biogenesis but enhances glycolysis and basal glucose uptake, J. Cell.Mol. Med 13 (2009) 3051–3060, 10.1111/j.1582-4934.2008.00382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zuercher WJ, Stein RA, Hyatt SM, Willson TM, Orband-Miller LA, Lockamy EL, McDonnell DP, Miller AB, On the Intractability of Estrogen-Related Receptor α as a Target for Activation by Small Molecules, J. Med. Chem 50 (2007) 6722–6724, 10.1021/jm7012387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Suetsugi M, Su L, Karlsberg K, Yuan Y-C, Chen S, Flavone and isoflavone phytoestrogens are agonists of estrogen-related receptors, Mol. Cancer Res 1 (2003) 981–991 http://www.ncbi.nlm.nih.gov/pubmed/14638870. [PubMed] [Google Scholar]
- [16].Peng L, Gao X, Duan L, Ren X, Wu D, Ding K, Identification of pyrido[1,2-a] pyrimidine-4-ones as new molecules improving the transcriptional functions of estrogen-related receptor α, J. Med. Chem 54 (2011) 7729–7733, 10.1021/jm200976s. [DOI] [PubMed] [Google Scholar]
- [17].Griffett K, Solt LA, El-Gendy B-E-M, Kamenecka TM, Burris TP, A liver-selective LXR inverse agonist that suppresses hepatic steatosis, ACS Chem. Biol 8 (2013) 559–567, 10.1021/cb300541g. [DOI] [PubMed] [Google Scholar]
- [18].Flaveny CA, Griffett K, El-Gendy B-E-M, Kazantzis M, Sengupta M, Amelio AL, Chatterjee A, Walker J, Solt LA, Kamenecka TM, Burris TP, Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis, Cancer Cell. 28 (2015) 42–56, 10.1016/j.ccell.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].El-Gendy B-E-M, Goher SS, Hegazy LS, Arief MMH, Burris TP, Recent Advances in the Medicinal Chemistry of Liver X Receptors, J. Med. Chem 61 (2018) 10935–10956, 10.1021/acs.jmedchem.8b00045. [DOI] [PubMed] [Google Scholar]
- [20].Goher SS, Griffett K, Hegazy L, Elagawany M, Arief MMH, Avdagic A, Banerjee S, Burris TP, Elgendy B, Development of novel liver X receptor modulators based on a 1,2,4-triazole scaffold, Bioorganic Med. Chem. Lett 29 (2019) 449–453, 10.1016/j.bmcl.2018.12.025. [DOI] [PubMed] [Google Scholar]
- [21].Khan PM, El-Gendy BEDM, Kumar N, Garcia-Ordonez R, Lin L, Ruiz CH, Cameron MD, Griffin PR, Kamenecka TM, Small molecule amides as potent ROR-γ selective modulators, Bioorganic Med. Chem. Lett 23 (2013) 532–536, 10.1016/j.bmcl.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Banerjee S, Wang Y, Solt LA, Griffett K, Kazantzis M, Amador A, El-Gendy BM, Huitron-Resendiz S, Roberts AJ, Shin Y, Kamenecka TM, Burris TP, Pharmacological targeting of the mammalian clock regulates sleep architecture and emotional behaviour, Nat. Commun 5 (2014), 10.1038/ncomms6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Willson TM, Collins JL, Miller AB, Zuercher WJ, Orband-Miller LA, Chao EYH, Gaillard S, McDonnell DP, Jones DG, Shearer BG, Identification and Structure−Activity Relationship of Phenolic Acyl Hydrazones as Selective Agonists for the Estrogen-Related Orphan Nuclear Receptors ERRβ and ERRγ, J.Med. Chem 48 (2005) 3107–3109, 10.1021/jm050161j. [DOI] [PubMed] [Google Scholar]
- [24].Lin H, Doebelin C, Patouret R, Garcia-Ordonez RD, Ra Chang M, Dharmarajan V, Bayona CR, Cameron MD, Griffin PR, Kamenecka TM, Design, synthesis, and evaluation of simple phenol amides as ERRγ agonists, Bioorganic Med, Chem. Lett 28 (2018) 1313–1319, 10.1016/j.bmcl.2018.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Evans RM, Downes M, Atkins A, Yu RT, Ahmadian M, Estrogen Related Receptor Gamma (Errgamma) Enhances And Maintains Brown Fat Thermogenic Capacity, US20180207114, 2018. [Google Scholar]
- [26].Makarenko AS, Mauri A, Prokopenko VV, Tanchuk VY, Todeschini R, Ertl P, Tetko IV, Radchenko EV, Zefirov NS, Gasteiger J, Palyulin VA, Livingstone D, Virtual Computational Chemistry Laboratory – Design and Description, J. Comput. Aided. Mol. Des 19 (2005) 453–463, 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- [27].Wang L, Zuercher WJ, Consler TG, Lambert MH, Miller AB, Orband-Miller LA, McKee DD, Willson TM, Nolte RT, X-ray crystal structures of the estrogen-related receptor-γ ligand binding domain in three functional states reveal the molecular basis of small molecule regulation, J. Biol. Chem 281 (2006) 37773–37781, 10.1074/jbc.M608410200. [DOI] [PubMed] [Google Scholar]
- [28].Kallen J, Schlaeppi J-M, Bitsch F, Filipuzzi I, Schilb A, Riou V, Graham A, Strauss A, Geiser M, Fournier B, Evidence for Ligand-independent Transcriptional Activation of the Human Estrogen-related Receptor α (ERRα), J. Biol. Chem 279 (2004) 49330–49337, 10.1074/jbc.m407999200. [DOI] [PubMed] [Google Scholar]
- [29].Schrödinger Release 2020–1: Induced Fit Docking protocol; Glide, Schrödinger, LLC, New York, NY, 2020; Prime, Schrödinger, LLC, New York, NY, 2020., (n.d.). [Google Scholar]
- [30].Schrödinger Release 2020–1: QikProp, Schrödinger, LLC, New York, NY, 2020., (2020). [Google Scholar]
- [31].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev 46 (2001) 3–26, 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- [32].Jorgensen WL, Duffy EM, Prediction of drug solubility from structure, Adv. Drug Deliv. Rev 54 (2002) 355–366, 10.1016/S0169-409X(02)00008-X. [DOI] [PubMed] [Google Scholar]
- [33].Jorgensen WL, Duffy EM, Prediction of drug solubility from Monte Carlo simulations, Bioorganic Med. Chem. Lett 10 (2000) 1155–1158, 10.1016/S0960-894X(00)00172-4. [DOI] [PubMed] [Google Scholar]
- [34].Rajput AP, Rajput SS, Synthesis of benzaldehyde substituted (phenylcarbonyl) hydrazones and their formylation using Vilsmeier-Haack reaction, Int. J. PharmTech Res 1 (2009) 1605–1611. [Google Scholar]
- [35].Gillis BT, Schimmel KF, Peracetic acid oxidation of hydrazones. I. Aromatic aldehyde alkylhydrazones, J. Org. Chem 27 (1962) 413–417, 10.1021/jo01049a017. [DOI] [Google Scholar]
- [36].Patel HS, Patel SJ, Synthesis and biological activity of 2-hydroxy-N-(5-methylene-4-oxo-2-aryl-thiazolidin-3-yl)-benzamide, Phosphorus, Sulfur Silicon Relat. Elem 185 (2010) 1632–1639, 10.1080/10426500903176521. [DOI] [Google Scholar]
- [37].Ainscough EW, Brodie AM, Denny WA, Finlay GJ, Gothe SA, Ranford JD, Cytotoxicity of salicylaldehyde benzoylhydrazone analogs and their transition metal complexes: quantitative structure-activity relationships, J. Inorg. Biochem 77 (1999) 125–133, 10.1016/S0162-0134(99)00131-2. [DOI] [PubMed] [Google Scholar]
- [38].Edward JT, Gauthier M, Chubb FL, Ponka P, Synthesis of new acylhydrazones as iron-chelating compounds, J. Chem. Eng. Data 33 (1988) 538–540, 10.1021/je00054a044. [DOI] [Google Scholar]
- [39].Kumar P, Narasimhan B, Ramasamy K, Mani V, Mishra RK, Abdul Majeed AB, Synthesis, Antimicrobial, Anticancer Evaluation and QSAR Studies of 3/4-Bromo Benzohydrazide Derivatives, Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 15 (2015) 1050–1064, 10.2174/156802661511150408111252. [DOI] [PubMed] [Google Scholar]
- [40].Desai KG, Desai KR, Synthesis of some novel pharmacologically active Schiff bases using microwave method and their derivatives formazans by conventional method., Indian J. Chem. Sect. B Org. Chem. Incl, Med. Chem 44B (2005) 2097–2101. [Google Scholar]
- [41].Farid R, Day T, Friesner RA, Pearlstein RA, New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies, Bioorganic Med. Chem 14 (2006) 3160–3173, 10.1016/j.bmc.2005.12.032. [DOI] [PubMed] [Google Scholar]
- [42].Sherman W, Day T, Jacobson MP, Friesner RA, Farid R, Novel procedure for modeling ligand/receptor induced fit effects, J. Med. Chem 49 (2006) 534–553, 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- [43].Sherman W, Beard HS, Farid R, Use of an induced fit receptor structure in virtual screening, Chem. Biol. Drug Des 67 (2006) 83–84, 10.1111/j.1747-0285.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- [44].Clark AJ, Tiwary P, Borrelli K, Feng S, Miller EB, Abel R, Friesner RA, Berne BJ, Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations, J. Chem. Theory Comput 12 (2016) 2990–2998, 10.1021/acs.jctc.6b00201. [DOI] [PubMed] [Google Scholar]
- [45].BIOvIA DS Discovery studio modeling environment. San Diego, Dassault Systemes, Release, 4. 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.