

Abstract
Iron is an essential nutrient that forms cofactors required for the activity of hundreds of cellular proteins. However, iron can be toxic and must be precisely managed. Poly r(C) binding protein 1 (PCBP1) is an essential, multifunctional protein that binds both iron and nucleic acids, regulating the fate of both. As an iron chaperone, PCBP1 binds cytosolic iron and delivers it to iron enzymes for activation and to ferritin for storage. Mice deleted for PCBP1 in the liver exhibit dysregulated iron balance, with lower levels of liver iron stores and iron enzymes, but higher levels of chemically-reactive iron. Unchaperoned iron triggers the formation of reactive oxygen species, leading to lipid peroxidation and ferroptotic cell death. Hepatic PCBP1 deletion produces chronic liver disease in mice, with steatosis, triglyceride accumulation, and elevated plasma ALT levels. Human and mouse models of fatty liver disease are associated with mitochondrial dysfunction. Here we show that, although deletion of PCBP1 does not affect mitochondrial iron balance, it does affect mitochondrial function. PCBP1 deletion affected mitochondrial morphology and reduced levels of respiratory complexes II and IV, oxygen consumption, and ATP production. Depletion of mitochondrial lipids cardiolipin and coenzyme Q, along with reduction of mitochondrial oxygen consumption, were the first manifestations of mitochondrial dysfunction. Although dietary supplementation with vitamin E ameliorated the liver disease in mice with hepatic PCBP1 deletion, supplementation with coenzyme Q was required to fully restore mitochondrial lipids and function. In conclusion, our studies indicate that mitochondrial function can be restored in livers subjected to ongoing oxidative damage from unchaperoned iron by supplementation with coenzyme Q, a mitochondrial lipid essential for respiration that also functions as a lipophilic radical-trapping agent.
Keywords: PCBP1, Steatosis, NASH, NAFLD, Oxidative stress, Ferroptosis, Cardiolipin, Coenzyme Q
Virtually all organisms rely on iron, as it is required for the formation of the essential cofactors heme, iron-sulfur clusters, and mono- and dinuclear iron centers [1]. Heme and iron-sulfur clusters are synthesized via multi-step, enzymatic reactions [2,3], while mono- and dinuclear iron centers are formed via direct insertion of ferrous iron [4]. Heme is synthesized exclusively in the mitochondria, iron-sulfur clusters are assembled in both the mitochondria and cytosol, while mono-and diiron centers (non-heme iron centers) are formed in mitochondria, the cytosol, and possibly the nucleus. The cellular iron for each of these cofactors originates in the kinetically-exchangeable, chemically-reactive, low-molecular-weight pool of iron in the cytosol called the labile iron pool (LIP). Iron within the LIP is largely in its reduced, ferrous form, Fe(II). While its capacity to exist in multiple oxidation states makes it crucial for electron transfer reactions, this capacity also accounts for its potential toxicity, as Fe(II) can catalyze the formation of damaging lipid and oxygen radicals [5].
The cell is protected from uncontrolled, iron-mediated oxidation by ligands that coordinate the LIP, specifically the tripeptide thiol glutathione and the iron chaperone poly rC binding protein 1 (PCBP1). Reduced glutathione (GSH) coordinates Fe(II) through its free sulfhydryl and, at normal cytosolic concentrations of both, this complex of iron and GSH makes up the bulk of the LIP [6]. PCBP1 (and its paralog PCBP2) are multifunctional adaptor proteins that can bind single-stranded DNA/RNA and iron, altering the fate of each ligand by mediating additional protein-protein interactions. The PCBP1 chaperone coordinates iron in the form of Fe-GSH complexes [7]. It can mediate the transfer of iron to the iron storage protein, ferritin [8], and to non-heme iron enzymes, such as prolyl hydroxylase [9] and deoxyhypusine hydroxylase [10]. PCBP1 with bound Fe-GSH can also deliver iron to the [2Fe–2S] chaperone complex of Glrx3-BolA2 [11]; this is accomplished by forming an intermediate complex with BolA2. By coordinating cytosolic iron, PCBP1 also ensures the proper sensing of cellular iron levels.
Animal models of PCBP1 deficiency have revealed additional roles for the iron chaperone in cellular iron homeostasis. Mice constitutively deleted for PCBP1 die in early embryogenesis, although the critical function of PCBP1 in early mammalian development has not been explored [12]. PCBP1 is necessary for efficient erythropoiesis in mice, as inducible depletion of PCBP1 is associated with microcytic anemia similar to that of iron deficiency. The anemia has been traced to a failure to efficiently transfer iron to ferritin in terminal stages of erythroid differentiation, revealing an important role for ferritin and the iron chaperone in erythroid iron utilization [13]. The liver is the primary site of iron storage in mammals and is also the major organ regulating body iron balance through the synthesis and secretion of the iron-regulatory hormone, hepcidin [14,15]. Hepatocytes within the liver are highly metabolically active and require large amounts of iron for the synthesis of heme and iron-sulfur clusters that support the TCA cycle and respiration within mitochondria as well as biosynthetic and detoxification processes throughout the cell. Mice specifically deleted for PCBP1 in hepatocytes and cholangiocytes exhibit dysregulated iron homeostasis, with low levels of liver iron, low levels of ferritin iron storage, and reduced activity of some iron-dependent enzymes [16]. These mice also develop chronic liver disease with steatosis, ballooning degeneration, elevated plasma transaminases and the accumulation of di- and triglycerides in the liver. The origins of the liver disease were traced to the enhanced redox activity of unchaperoned iron, which produced oxidative stress, lipid peroxidation, steatosis and cell death [16]. Whether iron chaperones or other delivery systems are required for delivery of iron to hepatic mitochondria has not been determined.
There is abundant evidence that mitochondrial dysfunction plays a role in the pathophysiology of human non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) [17–19]. Mitochondria are proposed to be both a target and a source of reactive oxygen species (ROS) implicated in the pathogenesis of these diseases. In mitochondria, enhanced fatty acid oxidation and flux through the TCA cycle in the setting of respiratory chain deficiency generates ROS in NAFLD and NASH. These ROS oxidize fatty acyl chains to generate lipid hydroperoxides and peroxyl radicals that propagate and release highly reactive aldehydic derivatives, which damage hepatocytes. In this study, we investigated the consequences of liver-specific PCBP1 deletion and unchaperoned iron accumulation on mitochondrial function and development of steatosis. Although PCBP1 chaperone activity was not required for the delivery of iron to mitochondria, unchaperoned cytosolic iron promoted depletion of mitochondrial lipids and mitochondrial dysfunction.
1. Materials and methods
1.1. For detailed material and methods, see supplementary information
1.1.1. Animal studies
PCBP1fl/fl female mice were crossed with Alb-Cre PCBP1 fl/fl males (Alb-Cre mice B6. Cg-Tg (Alb-cre) 21Mgn/J, #003574; The Jackson Laboratory, Bar Harbor, ME). Male offspring were weaned onto purified diets (Envigo TD.80,396, Indianapolis, IN) supplemented with 50 ppm iron as ferric citrate or to standard, natural-ingredient diet (NIH-31). Vitamin E was added to purified diet at 500 IU/Kg. CoQ10 (LiQsorb, Tishcon 9306-L) was added to drinking water at 0.5 mg/ml. Mice had ad libitum access to diets and double-distilled water and were euthanized after 16–18 days on the iron-defined diet (age 5–6 weeks). Littermates lacking the Alb-Cre transgene served as controls. All animal study protocols were reviewed and approved by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Animal Care and Use Committee and performed in compliance with National Institutes of Health (NIH) guidelines for the humane care of animals.
1.2. Metabolite and lipid analyses: see supplementary methods
1.2.1. Assays
Citrate synthase activity (Biovision) and ATP levels (Abcam) were measured in dounce-homogenized liver tissue using kits according to the manufacturers’ instructions.
1.2.2. Mitochondrial isolation
Mitochondria from liver tissue was isolated [20] with the following modifications. Freshly isolated liver was minced and dounce-homogenized in 10 vol of ice-cold isolation buffer (100 mM Hepes, pH 7.3, 200 mM sucrose, 2 mM sodium citrate, protease inhibitors cocktail (Roche), 1 mM DTT). Aconitase activity was measured using the coupled aconitase-isocitrate dehydrogenase method [10]. Mitochondrial non-heme iron assay was performed as described [21].
1.2.3. Mitosox assay
Primary hepatocytes were isolated as described [16], plated on collagen-coated 2 ml optical culture plates and cultured overnight at 37 °C. Hepatocytes were washed (10 mM HEPES pH 7.3, 121 mM NaCl, 5 mM NaHCO3, 10 mM glucose, 4.8 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1.4 mM CaCl2) and then treated with 2.5uM Mitosox, 200 nM of Mitotracker and with 1 μg/mL Hoechst dye and incubated at 37 °C for 15 min. Cells were washed with buffer twice and fluorescence analyzed by confocal microscopy. The fluorescence signal was quantified using Image J. Mitochondrial ROS was indicated by Mitosox (red signal) after normalizing for mitochondrial content (Mitotracker, green signal).
1.2.4. Mitostress assay
Mitochondrial oxygen consumption rate (OCR) was measured in primary hepatocytes using the XF96 Extracellular Flux analyzer (Seahorse Bioscience) as described by the manufacturer. After the OCR measurements, cells were stained using 1 μg/mL Hoechst dye and incubated for 20 min to determine cell numbers. Results were analyzed by the WAVE desktop software (Agilent) and normalized to the number of cells, determined after the assay.
1.2.5. Western blots, fluorescence immunohistochemistry, other assays
Assays were performed as previously described [16]. Antibodies, dilutions, and sources are presented in Supplementary Table S1.
1.2.6. Quantification and statical analysis
Fluorescent images were processed and analyzed using Fiji, ImageJ (version 2.0.0-rc-49/1.51d; NIH, Bethesda, MD). Data were analyzed with Prism software (version 8; GraphPad Software Inc., San Diego, CA). Data are reported as means ± SD. Outliers were identified using the integrated ROUT method and excluded. Differences between two groups were analyzed by unpaired Student t-tests with Welsh corrections. Multiple comparisons were analyzed using the False Discovery Rate with two-stage step-up method of Benjamini, Krieger, and Yekutieli and Q = 5%. Differences among groups were determined by two-way analysis of variance (ANOVA), followed by Bonferroni’s post-hoc analysis.
2. Results
2.1. PCBP1 deletion reduces total cellular iron but not mitochondrial iron
Deletion of PCBP1 in hepatocytes and cholangiocytes led to hepatic iron dysregulation with overall reduction in hepatic iron levels but increases in unchaperoned iron, leading to oxidative damage to lipids and hepatic steatosis [16]. PCBP1-deleted livers exhibited lower levels of ferritin, lower levels of xanthine oxidase (a [2Fe–2S] enzyme) and prolyl hydroxylase (a non-heme iron enzyme) activities, and 50% lower levels of non-heme iron (Fig. 1A, left). Mitochondria serve as a major hub for iron cofactor synthesis and iron enzyme activity. It is the site for heme biosynthesis and the major site for iron-sulfur cluster biogenesis and usage [22]. We investigated whether deletion of PCBP1 affected mitochondrial iron by isolating mitochondria from fresh liver tissue obtained from wild type (fl/fl) and PCBP1-deleted (Δhep) mice fed for 16–18 days on a defined-iron, purified diet containing 50 ppm iron (normal iron diet). Mitochondrial non-heme iron levels did not differ between wild type and Δhep mice (Fig. 1A, right). Mitochondrial aconitase is an abundant [4Fe–4S]-cluster containing enzyme that catalyzes the conversion of citrate to isocitrate via a cis-aconitate intermediate. We measured mitochondrial aconitase activity in these isolated mitochondria and found that PCBP1 deletion did not decrease activity, but instead increased it by 46% (Fig. 1B). We confirmed mitochondrial isolation efficiency by immunoblot analysis using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as cytoplasmic marker and voltage-dependent anion channel (VDAC) as mitochondrial marker (Fig. 1C). Lipoic acid synthase is a mitochondrial Fe–S enzyme required for the lipoylation of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase. Anti-lipoic acid immunoblotting indicated that lipoic acid synthase activity was also unaffected (Fig. 1D). These results suggest that PCBP1 is not required for iron distribution to the mitochondria and that cytosolic iron depletion was not associated with mitochondrial iron deficiency.
Fig. 1. Mitochondrial iron and iron-sulfur clusters are unaffected in PCBP1Δhep livers.
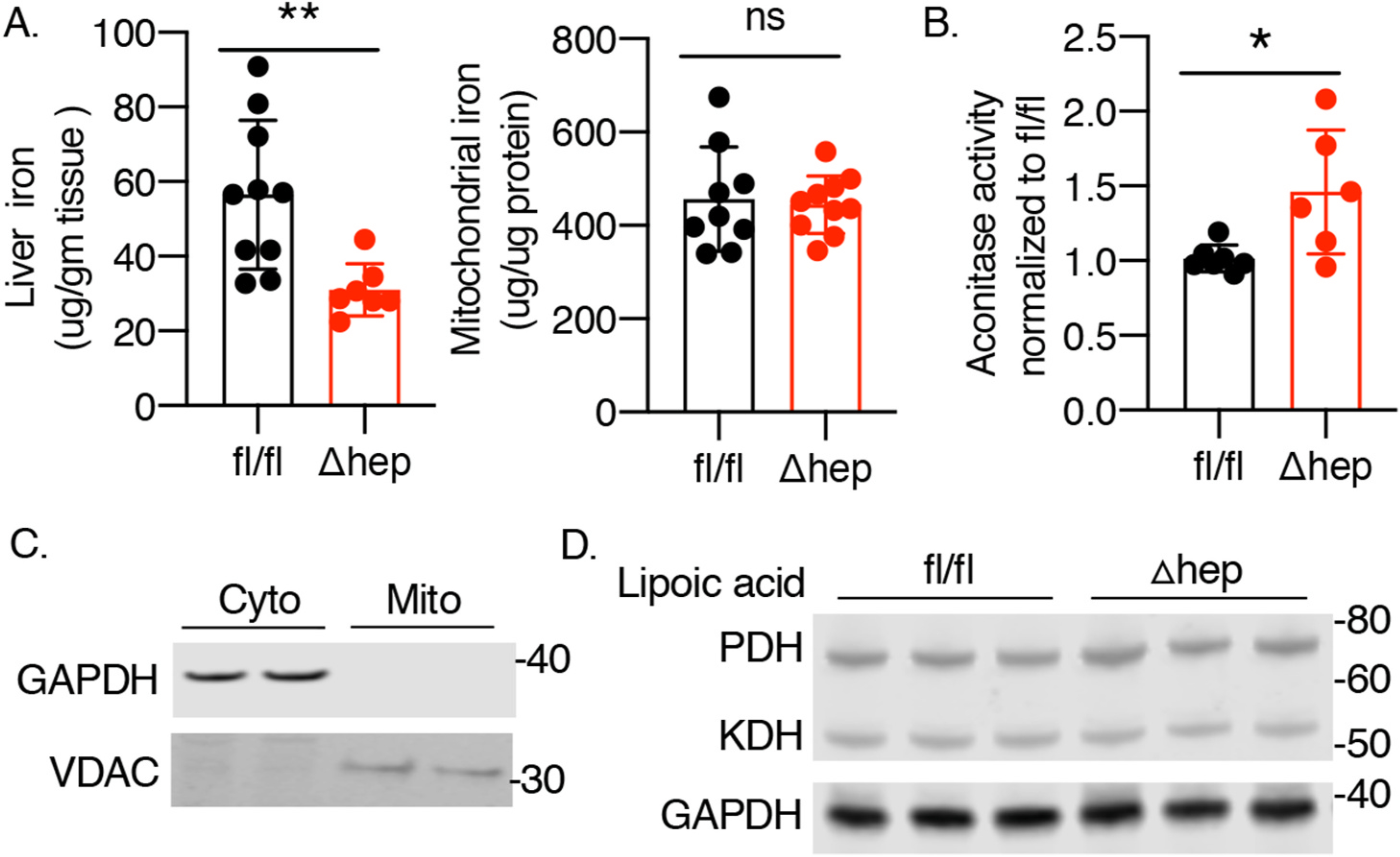
Male littermates of fl/fl and Δhep mice were weaned onto iron-defined diets (50 ppm, normal iron) for 16–18 days prior to analysis. Mitochondria were isolated from fresh liver samples. A. Non heme iron levels. B. Mitochondrial aconitase activity. C. Mitochondrial isolation efficiency. Efficiency was confirmed by Western blotting for cytoplasmic (GAPDH) and mitochondrial proteins (VDAC). D. Lipoic acid synthase activity. Liver extracts were analyzed by Western blotting using anti-lipoic acid antibodies. N = 6–12. Data represent mean ± SD. *P<0.05. Abbreviation: ns, not significant.
2.2. PCBP1 deletion leads to mitochondrial dysfunction
Mitochondrial dysfunction is frequently associated with fatty liver disease in humans and models of hepatic steatosis in mice. We analyzed mitochondrial function in PCBP1 fl/fl and Δhep mice. Primary hepatocytes isolated from these mice were stained with mitotracker green; morphological analysis demonstrated that, while wild type hepatocytes exhibited small, spherical mitochondria, mitochondria from Δhep mice appeared elongated and hyperfused (Fig. 2A), suggesting that these mitochondria were under stress [23]. Mitofusin 2 (Mfn2) is a mitochondrial membrane protein involved in dynamic alterations of mitochondrial morphology and the transfer of lipids between the endoplasmic reticulum and mitochondrial membranes. Levels of Mfn2 are depressed in patients with NASH and in mouse models of steatosis [19,24]. We measured the levels of Mfn2 protein in livers from PCBP1 fl/fl and Δhep mice and found a 25% reduction in Mfn2 in Δhep mice (Fig. 2B). These data suggested that Δhep mice exhibited mitochondrial dysfunction. To further investigate the effect of PCBP1 deletion on mitochondrial function, we measured the levels of respiratory complexes in PCBP1 fl/fl and Δhep livers. PCBP1 deletion decreased levels of complex II and IV by 25% and 33%, respectively (Fig. 2C) and ATP levels were decreased 27% in Δhep mice (Fig. 2D). Next, we examined oxygen consumption in hepatocytes isolated from these mice and found that PCBP1-deleted hepatocytes exhibited lower levels of basal and maximal oxygen consumption (Fig. 2E and Supplemental Fig. S2). These results showed that PCBP1 is required to maintain normal mitochondrial function in mice.
Fig. 2. PCBP1Δhep mice livers exhibit mitochondrial dysfunction.
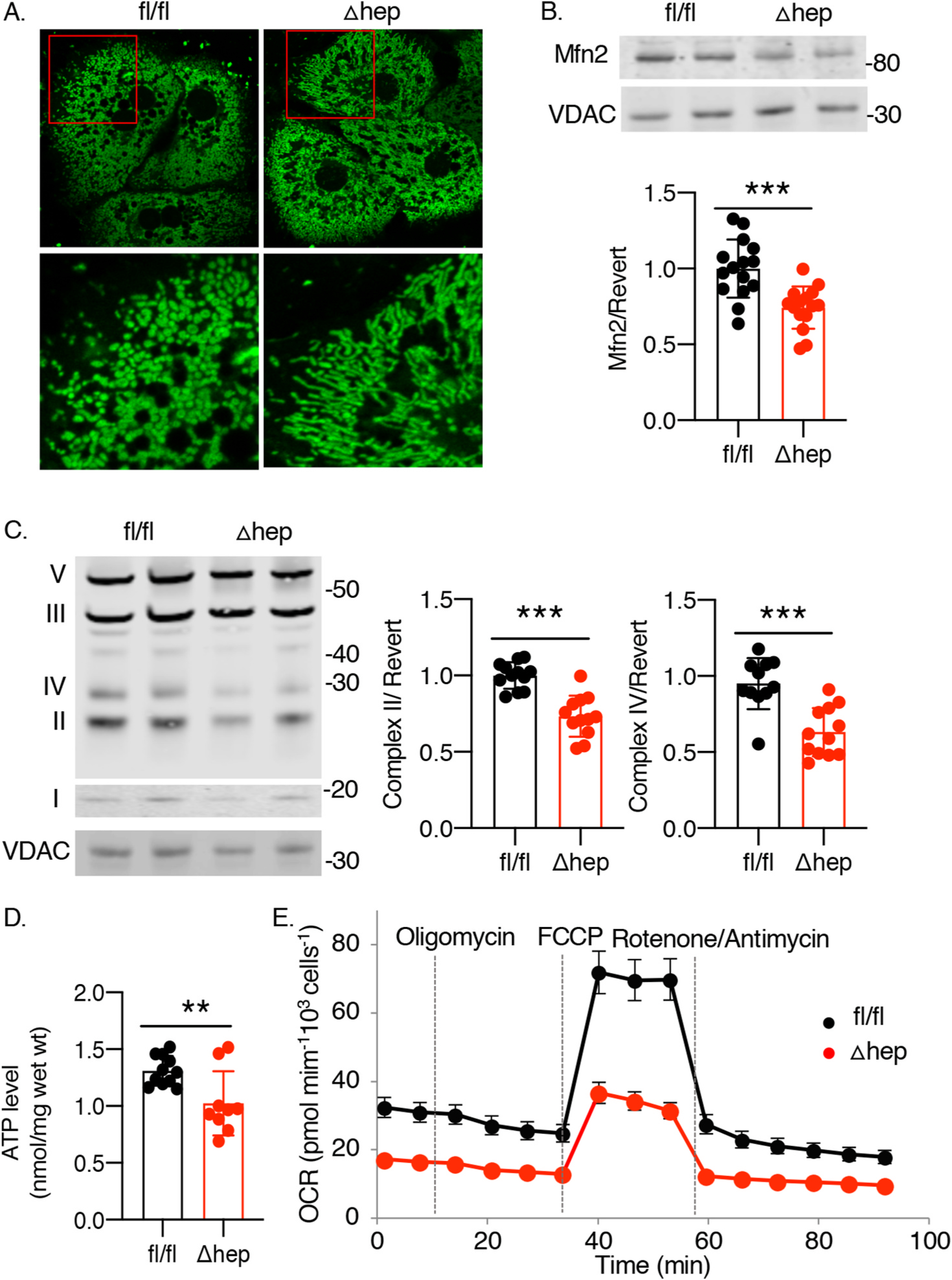
A. Elongated and hyperfused mitochondria in hepatocytes from PCBP1Δhep livers. Hepatocytes from PCBP1 fl/fl and Δhep mice were isolated and stained with mitochondrial-specific fluorescent dye Mitotracker green and subjected to live-cell confocal imaging. Inset (red) magnified in lower panels. B. Depletion of mitochondrial fusion protein mitofusin 2 in PCBP1Δhep livers. Mfn2 levels measured by quantitative Western blot with VDAC as mitochondrial loading control. C. Depletion of mitochondrial respiratory complexes C II and C IV in PCBP1Δhep livers. Western blot analysis of liver lysates probed with mixed antibodies for respiratory complexes I (NDUFB8), II (SDHB), III (UQCRC2), IV (MTCO1), and V (ATP5A). Quantification of C II and IV shown on right. D. ATP depletion in livers from PCBP1Δhep mice. Data represent mean ± SD. * indicates p < 0.05, **p < 0.01, ***, p < 0.001 as determined by unpaired t-test (B, D) or 2-way ANOVA with Bonferroni multiple comparison test (C). E. Reduced mitochondrial oxygen consumption in hepatocytes from PCBP1Δhep livers. Primary hepatocytes were isolated and mitostress test was performed to evaluate the mitochondrial oxygen consumption. Data from representative experiment are shown, see also Supplemental Fig. 2.
2.3. PCBP1 deletion induces the TCA cycle
TCA cycle flux increases in various mouse models and in humans with nutritional overload, obesity, and steatosis [25]. This increased TCA flux likely contributes to increased lipogenesis during substrate overload, causing steatosis. Persistent increases in TCA cycle flux may lead to overproduction of reducing equivalents and overload the electron transport chain, thereby leading to uncoupling and increased formation of ROS, especially superoxide anion and hydrogen peroxide [25, 26]. We performed untargeted metabolomic analyses on liver lysates from mice and found that multiple TCA metabolites were increased in Δhep mice (Fig. 3A). Overproduction of reducing equivalents in PCBP1 Δhep livers was manifest by a 40–60% reduction in NAD+/NADH ratios (Fig. 3B). Elevated levels of mitochondrial aconitase (Fig. 1B) and citrate synthase (Fig. 3C) activities, coupled with reduced levels of liver glucose (Fig. 3D) are all consistent with enhanced glycolysis and induction of the TCA cycle in livers of PCBP1 Δhep mice (Fig. 3D). These changes occurred without exposure to an obesogenic diet or nutritional overload.
Fig. 3. Induction of TCA cycle in livers of PCBP1Δhep mice.
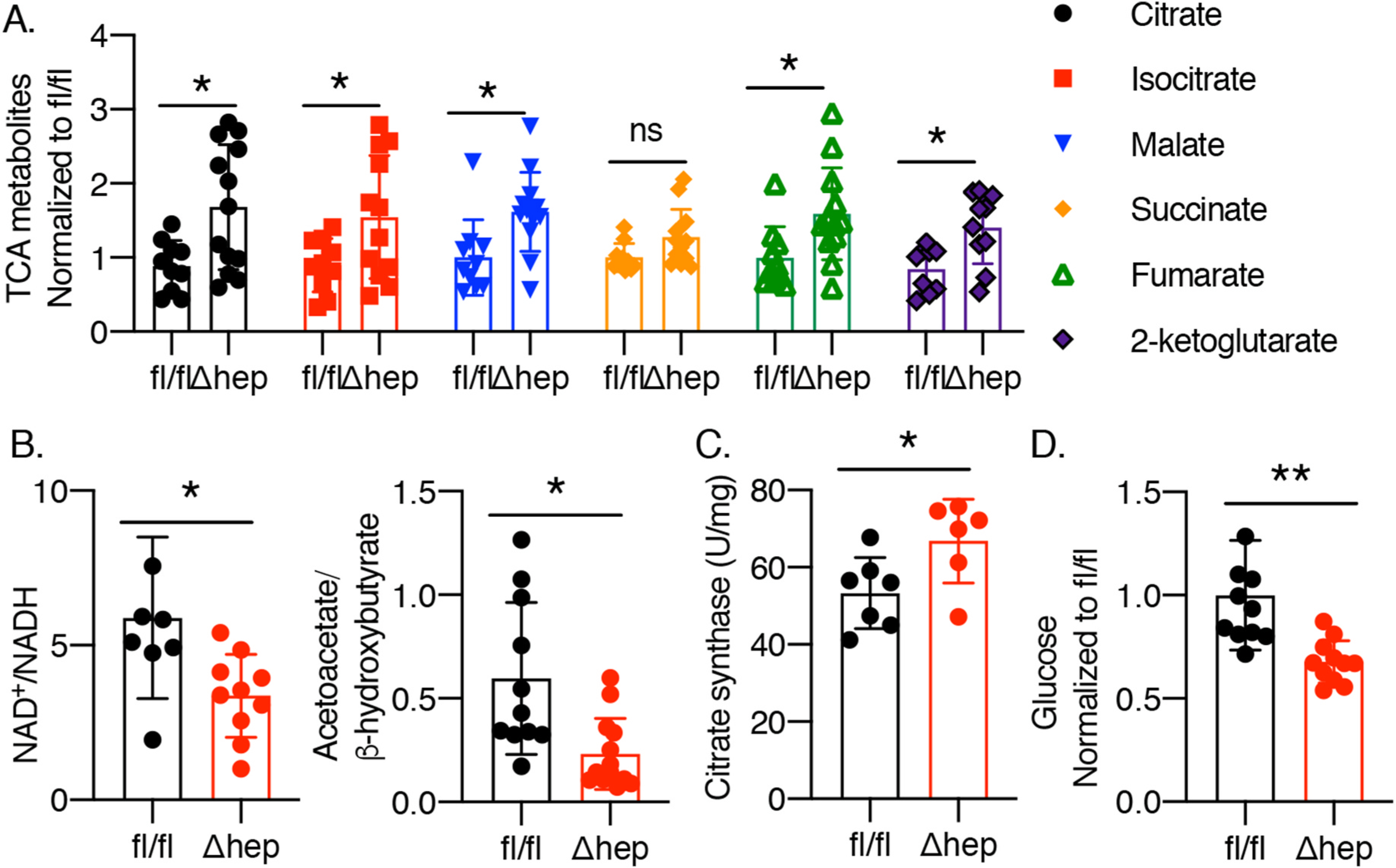
Metabolite analyses were performed on liver lysates of PCBP1fl/fl and PCBP1Δhep mice. A. Elevated TCA cycle intermediates in PCBP1Δhep livers. B. Reduction of liver NAD+/NADH and mitochondrial NAD+/NADH (acetoacetate/b-hydroxybutyrate) ratios in PCBP1Δhep livers. C. Elevated citrate synthase activity in PCBP1Δhep livers. D. Glucose depletion in PCBP1Δhep livers. Data represent mean ± SD. * indicates p < 0.05, **p < 0.01 as determined by 2-way ANOVA with Bonferroni multiple comparison test.
Deletion of PCBP1 leads to increased levels of mitochondrial ROS and decreased levels of mitochondrial lipids.
The evidence of increased flux through the TCA cycle suggested that increased production of mitochondrial ROS could be occurring in the PCBP1 Δhep livers. We assessed mitochondrial ROS in isolated hepatocytes from PCBP1 fl/fl and Δhep mice and found a 50% increase in the ROS signal from Δhep mice (Fig. 4A). We also considered that mitochondrial dysfunction could be occurring through an indirect mechanism that was secondary to the ROS generated by unchaperoned iron in the cytosol. Uncoupling protein 2 (UCP2) is a mitochondrial protein involved in the regulation of mitochondrial production of ROS and is upregulated in mice on obesogenic diets and other inflammatory stimuli [27]. UCP2 expression was increased 2.5-fold in the Δhep mice (Fig. 4B). Because lipid peroxidation was a prominent feature of the oxidative damage associated with PCBP1 deletion in liver, we examined the lipidome of these mice for changes in mitochondrial lipids. Coenzyme Q (CoQ, ubiquinone) is a redox-active mitochondrial lipid integral to the electron transport chain that also participates in extramitochondrial redox reactions [28]. Similarly, cardiolipin is a mitochondrial lipid localized to the inner membrane [29,30] where it stabilizes cristae and interacts with proteins of the electron transport chain. Its four fatty acyl chains are typically polyunsaturated, making it susceptible to peroxidation. CoQ and cardiolipin levels were depressed by 33% and 57% in the livers of Δhep mice (Fig. 4C) while acylcarnitine, a lipid carrier and marker of impaired mitochondrial lipid metabolism was increased. These data indicated that the mitochondrial dysfunction in the Δhep livers was associated with increased oxidative stress and mitochondrial lipid depletion.
Fig. 4. Stimulation of mitochondrial ROS and depletion of mitochondrial lipids in PCBP1Δhep mice.

A. Elevated ROS production in hepatocytes from PCBP1Δhep mice. Primary hepatocytes were analyzed for mitochondrial ROS using mitochondrial-specific ROS probe Mitosox. Fluorescence expressed as ratio to mitotracker green to normalize for mitochondrial content. Quantification of fluorescence intensity ratios at right. Analyses were repeated thrice; 25–40 cells were analyzed in each experiment. B. Elevated mRNA levels of mitochondrial ROS marker UCP2 in PCBP1Δhep livers. C. Depletion of Coenzyme Q and major cardiolipins in PCBP1Δhep livers. Global lipid analysis of livers was performed. Predominant cardiolipin, tetralinoleoyl (C18:2)4 cardiolipin is shown. Acylcarnitine, a lipid carrier and marker of impaired mitochondrial lipid metabolism, at right. Data represent mean ± SD. (A,B)* indicates p < 0.05, **p < 0.002 as determined by unpaired t-test. (C) * indicates q < 0.05, ** indicates q < 0.002 as determined by False Discovery Rate (FDR) determined using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%.
Oxidative stress and mitochondrial dysfunction precede the development of steatosis and liver disease in PCBP1 Δhep mice.
We have shown that PCBP1 deletion in the liver leads to both oxidative damage and mitochondrial dysfunction. Less clear, however, is whether cytosolic oxidative damage or mitochondrial dysfunction is the primary inducer of the metabolic changes leading to hepatic steatosis and liver disease. We observed that Δhep mice fed a natural ingredient diet were delayed in the development of liver disease. Younger PCBP1 Δhep mice (7–9 wks) fed a natural diet did not exhibit steatosis, elevated liver triglycerides, or elevated plasma ALT (Fig. 5A–C). In contrast, older PCBP1 Δhep mice (10 mos) exhibited each of these features on the natural diet, indicating the diet offered a temporary protective effect for the Δhep mice. This allowed us to establish a model to evaluate the primary inducer of liver disease. Young Δhep mice did not show depletion in levels of the mitochondrial respiratory complexes II and IV (Fig. 5D), Mfn2 (Fig. 5E), or ATP (Fig. 5F). However, older mice with PCBP1 deletion did exhibit decreased levels of Complex II and IV levels (Fig. 5D, Supplemental 5), Mnf2 (Fig. 5E, Supplemental 5), and ATP levels (Fig. 5F), potentially indicating that mitochondrial dysfunction was not present in the younger Δhep mice.
Fig. 5. Delayed development of liver disease in PCBP1Δhep mice fed natural ingredient diet.

Mice were fed on natural ingredient, standard NIH-31 diet and analyzed at 7–9 weeks (young) or 10 months (old). A. Hepatic steatosis in old, but not young mice. Liver sections stained with H&E. B. Elevated liver triglycerides in old, but not young mice. C. Elevated serum ALT in old, but not young mice. D. Depletion of respiratory complexes II and IV in old, but not young mice. E. Depletion of Mfn2 in old, but not young mice. F. Decreased levels of ATP in old, but not young mice. Data represent mean ± SD. * indicates p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns not significant, as determined by 2-way ANOVA with Bonferroni multiple comparison test.
However, further examination suggested that mitochondrial dysfunction was an early feature of the PCBP1-associated liver disease. Both young and old mice with PCBP1 deletion exhibited evidence of ongoing oxidative damage, including deposition of 4-hydroxynonenol (4-HNE), a product of lipid peroxidation, in hepatic proteins (Fig. 6A) and increased expression of NQO1 and MT-1, which are targets of Nrf2, a ROS-sensitive transcription factor (Fig. 6B) These data suggest that oxidative stress and lipid peroxidation are early features of PCBP1 disease. Genes involved in de novo cholesterol and fatty acid synthesis were also upregulated (HMGCS1, FASN, Fig. 6C). Global lipid analysis demonstrated that young Δhep mice exhibited 50% and 60% decreases in cardiolipin and CoQ, respectively, and 50% increased acylcarnitine levels (Fig. 6D). UCP2 was upregulated in both young and old Δhep mice (Fig. 6E) and primary hepatocytes isolated from young Δhep mice exhibited decreased oxygen consumption (Fig. 6F and Supplemental Fig. 6). Young Δhep hepatocytes did not, however, exhibit excess mitochondrial ROS production (Supplemental Fig. 6). These data suggested that the cytosolic oxidative stress catalyzed by unchaperoned iron in the Δhep mice was associated with mitochondrial lipid depletion and suppressed respiration at the earliest stages of the liver disease.
Fig. 6. Oxidative damage, mitochondrial lipid depletion, and reduced respiration precede development of liver disease in young PCBP1Δhep mice fed natural ingredient diet.

Mice were fed natural ingredient diet as in Fig. 5, above. A. Increased lipid peroxidation in livers of young PCBP1Δhep mice. 4-HNE, an end product of lipid peroxidation, measured by fluorescent immunohistochemistry in fixed liver tissue. B. Elevated transcripts indicating oxidative stress in young and old mice. qPCR analysis of Nrf2 targets MT1 and NQO1. C. Activation of SREBP1 and 2 target genes in livers of young and old PCBP1Δhep mice. mRNA levels of HMGCS1 and FASN measured by qPCR. D. Depletion of mitochondrial lipids cardiolipin and CoQ9 and accumulation of lipid carrier acylcarnitine in young PCBP1Δhep mice. Global lipid analysis of livers from young mice was performed. E. Elevation of UCP2 in young PCBP1Δhep mice. F. Reduced mitochondrial respiration in young PCBP1Δhep mice. Primary hepatocytes were isolated from 2 month old mice. Both basal and maximal oxygen consumption was decreased in hepatocytes isolated from PCBP1 deleted mice. Data represent mean ± SD. (A) ** indicates p < 0.002 by unpaired t-test. (B,C and E)* indicates p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns not significant, as determined by 2-way ANOVA with Bonferroni multiple comparison test. (D) *** indicates q < 0.001 as determined by False Discovery Rate determined using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%.
2.4. Antioxidant treatment prevents hepatic steatosis, oxidative damage and mitochondrial dysfunction
Our previous study showed that dietary supplementation with the lipophilic antioxidant α-tocopherol (vitamin E) partially prevented the hepatic steatosis and oxidative damage phenotypes in the PCBP1 Δhep mice. However, the steatosis, triglyceride accumulation and liver damage were not completely suppressed by vitamin E. Because mitochondrial dysfunction and mitochondrial lipid depletion are observed in the PCBP1 Δhep mouse, we explored whether antioxidant treatment with the mitochondrial-specific lipid CoQ could prevent the mitochondrial dysfunction observed in these mice. Mice naturally synthesize ubiquinone comprised of 9 isoprenoid subunits (CoQ9) while humans synthesize ubiquinone with 10 subunits (CoQ10). These lipids are functionally identical and readily separable analytically (Supplemental Fig. 7). We fed the PCBP1 fl/fl and Δhep mice the normal, defined-iron diet supplemented with either CoQ10 or vitamin E. Similar to our previous studies with vitamin E [16], supplementation of Δhep mice with CoQ10 largely prevented the lipid droplet formation (Fig. 7A) and partially suppressed liver triglyceride and serum ALT levels (Fig. 7B). Furthermore, both antioxidants decreased the expression of fatty acid synthase (FASN) and cholesterol biosynthesis genes (HMGS1) (Supplemental Fig. S7). Vitamin E suppressed the oxidative stress and lipid peroxidation that led to activation of Nrf2 target genes and 4-HNE deposition [16]. Similarly, CoQ10 treatment suppressed the induction of Nrf2 targets (Fig. 7C)) and 4-HNE (Supplemental Fig. S7) and [16], as well as the induction of UCP2 (Fig. 7D). These data support the hypothesis that oxidative stress was the driver of hepatic steatosis.
Fig. 7. Supplementation with Coenzyme Q prevents hepatic steatosis and oxidative damage in PCBP1Δhep mice.

Male littermates were weaned onto 50 ppm iron diet supplemented with either vitamin E (500 IU/gm) or CoQ10 via drinking water (0.5 mg/ml) for 16–18 days. A. Prevention of steatosis with CoQ10. H&E staining of fixed liver tissues. B. Normalization of liver triglycerides and reduction of serum ALT with CoQ10. C. Normalization of Nrf2 target genes MT-1, NQO1 by qPCR using RNA isolated from liver tissue. D. Normalization of mitochondrial stress marker UCP2. E. Normalization of ATP levels. F. Normalization of acylcarnitine and cardiolipin levels. Data represent mean ± SD. * indicates p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns not significant, as determined by 2-way ANOVA with Bonferroni multiple comparison test.
Next, we examined the effect of antioxidant treatment on mitochondrial function. Although both vitamin E and CoQ10 treatment rescued the decreased levels of mitochondrial proteins Mnf2 and complex II and IV (Supplemental Fig. S7), only CoQ10 rescued the depressed ATP levels of the Δhep mice (Fig. 7E). Similarly, only CoQ10 and not Vitamin E treatment normalized levels of cardiolipins and acylcarnitines (Fig. 7 F). These data suggest that although both vitamin E and Coq10 could partially suppress the oxidative damage and steatosis, only supplementation with CoQ10 could fully restore mitochondrial lipids and ATP production to wild type levels in the Δhep livers. Thus, the ongoing oxidative damage caused by unchaperoned iron in the cytosol of mice lacking PCBP1 in hepatocytes leads to the selective depletion of mitochondrial lipids and subsequent dysfunction. This dysfunction, which was corrected by restoration of CoQ levels, emphasizes the importance of this lipid for both mitochondrial function and cellular protection from ROS.
3. Discussion
Here we have examined the impact of unchaperoned cytosolic iron on mitochondrial function in the livers of mice lacking the iron chaperone PCBP1 in hepatocytes. Despite the absence of a defect in iron trafficking to mitochondria, disruption of cytosolic iron homeostasis triggered both adaptive and dysfunctional mitochondrial responses. Our data suggest a model in which deletion of PCBP1 leads to increased levels of unchaperoned cytosolic iron, producing oxidative damage and lipid peroxidation. This process also led to damage and depletion of critical mitochondrial lipids. Initially, these changes led to alterations in mitochondrial metabolism characterized by expression of uncoupling protein 2 and decreased oxygen consumption, which are likely adaptive responses. However, continued cytosolic ROS and oxidative damage ultimately led to mitochondrial dysfunction characterized by increased TCA cycle flux and mitochondrial ROS, decreased levels of mitochondrial lipids, mitofusin 2, respiratory complexes II and IV, and ATP. Here the enhanced mitochondrial ROS likely contributes to lipogenesis and hepatic steatosis.
Mitochondria are the major site of iron cofactor synthesis and utilization, yet the mechanisms by which cells direct iron to mitochondria are not clear. PCBP1 was found to be critical for the efficient delivery of iron to mitochondria in developing erythrocytes [13]. This delivery of iron occurs via an indirect mechanism in which PCBP1 first loads ferritin with iron; ferritin is then targeted to the lysosome for degradation and lysosomal iron is then transferred to mitochondria. This pathway appears not to be critical in hepatocytes, as ferritin storage was decreased in the absence of PCBP1, yet mitochondrial iron levels were unaffected. There may be an alternative cytosolic iron chaperone delivering iron to mitochondria in hepatocytes or a system for the direct transfer of iron from the endo/lysosomal compartment to mitochondria.
Despite the appropriate transfer of iron to mitochondria, loss of PCBP1 in the cytosol and the resulting unchaperoned iron has significant effects on hepatic mitochondrial function. Data presented here suggest that the initial impact of unchaperoned iron was to produce ROS and lipid peroxidation in the cytosol. Supporting this sequence of events is the activation of the ROS-sensitive transcription factor NRF2 and the accumulation of end products of lipid peroxidation in young Δhep mice prior to the onset of liver disease. ROS and lipid peroxidation likely account for the depletion of cardiolipins and CoQ in the mitochondria. Cardiolipins, specifically the tetralinoleoyl (C18:2)4 cardiolipin that is most abundant in mitochondria, are critical for maintenance of the appropriate curvature of mitochondrial inner membranes, stabilization of respiratory complexes, trapping of protons during oxidative phosphorylation, and assembly of ATP synthase [31]. However, the polyunsaturated acyl chains of cardiolipins are sensitive to oxidative damage. Peroxidation and oxidative decomposition of polyunsaturated acyl chains in extramitochondrial membranes may limit the synthesis of tetralinoleoyl cardiolipin in mitochondria and adversely affect multiple mitochondrial functions. The specific depletion of tetralinoleoyl cardiolipin observed in the PCBP1 Δhep livers contributed to the reduction in mitochondrial oxygen consumption seen in all Δhep mice as well as the loss of respiratory complexes II and IV and ATP depletion in the older Δhep mice and Δhep mice on the defined-iron diet.
A second feature of the mitochondrial dysfunction observed in the young Δhep mice is the depletion of CoQ. While CoQ is an integral part of the electron transport chain, coupling the transfer of electrons from complexes I and II to complex III, it is also involved in redox balance in membranes throughout the cell [32]. Mild depletion of CoQ in the liver can impair respiratory chain function, although the degree of depletion appears not to correlate with severity of dysfunction in mouse models of hepatic CoQ deficiency. Thus, the moderate levels of CoQ depletion observed in the PCBP1 Δhep livers likely contribute to impaired electron transport chain function and may contribute to the elevated mitochondrial ROS measured in the Δhep mice maintained on the defined-iron diet. However, an important role of CoQ in livers of the Δhep mice may originate in its extramitochondrial functions [33]. CoQ is distributed in membranes throughout the cell, including the plasma membrane. Recent studies have confirmed that it actively functions as a lipophilic antioxidant that suppresses the formation of the lipid hydroperoxides associated with ferroptosis, an iron-mediated cell death pathway. Reduced CoQ can capture lipid hydroperoxyl radicals and block their propagation through endomembranes. FSP1, the flavoenzyme catalyzing the regeneration of reduced CoQ, is critical in sustaining the antioxidant properties of CoQ and in preventing lipid oxidation and ferroptosis [34,35]. Whether total cellular CoQ levels are depleted by cellular ROS was not addressed in those studies, but our data suggest that they are and may ultimately contribute to mitochondrial dysfunction and ROS production in the livers of Δhep mice.
Although the lipophilic antioxidant effects of CoQ would appear to be similar to those of vitamin E, our data suggest they differ in important ways. While dietary supplementation with vitamin E partially reversed the steatosis and oxidative stress in livers of the Δhep mice [16], it did not restore cardiolipin or ATP to the levels of wild type mice. CoQ10 supplementation, however, did restore cardiolipin, as well as ATP, levels and reversed the depletion of mitochondrial respiratory complexes. CoQ10 also suppressed steatosis, oxidative damage, and activation of UCP2. Thus, both the antioxidant effects and the electron transport activities of CoQ may contribute to the prevention of mitochondrial dysfunction in the setting of iron-mediated oxidative stress. Clinical studies examining the effects of CoQ supplementation in patients with metabolic syndrome, non-alcoholic fatty liver disease, and non-alcoholic steatohepatitis have found some benefit to supplementation, with improvement in clinical markers of lipid peroxidation and inflammation, better blood glucose control, and reduction of liver-derived enzymes [36,37]. However, the impact of these studies is limited by the relatively small numbers of patients and short duration (<3 mos) of treatment. These studies suggest that more clinical study of Coenzyme Q is warranted.
Supplementary Material
Acknowledgements
We thank Siegfried Hekimi of McGill University, Oksana Gavrilova and Yinyan Ma of the NIDDK Mouse Metabolism Core, Shashi Shrivastav and Jeffrey Kopp of Kidney Disease Section, NIDDK and Ulrich Siebenlist from NIAD for technical assistance and helpful discussions.
Funding sources
These studies were supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and the Office of Dietary Supplements Research Scholars Program, National Institutes of Health. J. E. C. and J. A. M. are supported by U54DK11085804, 1S10OD018210-01A1 and 1S10OD021505-01. The authors declare no conflicts of interest.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.freeradbiomed.2021.08.232.
References
- [1].Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L, Mechanisms of mammalian iron homeostasis, Biochemistry 51 (2012) 5705–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hamza I, Dailey HA, One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans, Biochim. Biophys. Acta 1823 (2012) 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lill R, Dutkiewicz R, Freibert SA, et al. , The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins, Eur. J. Cell Biol 94 (2015) 280–291. [DOI] [PubMed] [Google Scholar]
- [4].Philpott CC, Patel SJ, Protchenko O, Management versus miscues in the cytosolic labile iron pool: the varied functions of iron chaperones, Biochim. Biophys. Acta Mol. Cell Res 1867 (2020), 118830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bayir H, Anthonymuthu TS, Tyurina YY, et al. , Achieving life through death: redox biology of lipid peroxidation in ferroptosis, Cell Chem Biol 27 (2020) 387–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hider RC, Kong XL, Glutathione: a key component of the cytoplasmic labile iron pool, Biometals 24 (2011) 1179–1187. [DOI] [PubMed] [Google Scholar]
- [7].Patel SJ, Frey AG, Palenchar DJ, et al. , A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly, Nat. Chem. Biol 15 (2019) 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shi H, Bencze KZ, Stemmler TL, Philpott CC, A cytosolic iron chaperone that delivers iron to ferritin, Science 320 (2008) 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nandal A, Ruiz JC, Subramanian P, et al. , Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2, Cell Metabol. 14 (2011) 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Frey AG, Nandal A, Park JH, et al. , Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 8031–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Frey AG, Palenchar DJ, Wildemann JD, Philpott CC, Glutaredoxin A, BolA complex serves as an iron-sulfur cluster chaperone for the cytosolic cluster Assembly machinery, J. Biol. Chem 291 (2016) 22344–22356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ghanem LR, Kromer A, Silverman IM, et al. , The poly(C) binding protein Pcbp2 and its retrotransposed derivative Pcbp1 are independently essential to mouse development, Mol. Cell Biol 36 (2016) 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC, PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis, J. Clin. Invest 127 (2017) 1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang CY, Babitt JL, Liver iron sensing and body iron homeostasis, Blood 133 (2019) 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ganz T, Erythropoietic regulators of iron metabolism, Free Radic. Biol. Med 133 (2019) 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Protchenko O, Baratz E, Jadhav S, et al. , Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis, Hepatology 73 (2021) 1176–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Longo M, Meroni M, Paolini E, Macchi C, Dongiovanni P, Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): new perspectives for a fairy-tale ending? Metabolism 117 (2021), 154708. [DOI] [PubMed] [Google Scholar]
- [18].Chen Z, Tian R, She Z, Cai J, Li H, Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease, Free Radic. Biol. Med 152 (2020) 116–141. [DOI] [PubMed] [Google Scholar]
- [19].Lee J, Park JS, Roh YS, Molecular insights into the role of mitochondria in nonalcoholic fatty liver disease, Arch Pharm. Res. (Seoul) 42 (2019) 935–946. [DOI] [PubMed] [Google Scholar]
- [20].Frezza C, Cipolat S, Scorrano L, Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts, Nat. Protoc 2 (2007) 287–295. [DOI] [PubMed] [Google Scholar]
- [21].Enns CA, Ahmed R, Wang J, et al. , Increased iron loading induces Bmp6 expression in the non-parenchymal cells of the liver independent of the BMP-signaling pathway, PloS One 8 (2013), e60534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Paul BT, Manz DH, Torti FM, Torti SV, Mitochondria and Iron: current questions, Expet Rev. Hematol 10 (2017) 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yao CH, Wang R, Wang Y, Kung CP, Weber JD, Patti GJ, Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation, Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hernandez-Alvarez MI, Sebastian D, Vives S, et al. , Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease, Cell 177 (2019) 881–895 e17. [DOI] [PubMed] [Google Scholar]
- [25].Sunny NE, Bril F, Cusi K, Mitochondrial adaptation in nonalcoholic fatty liver disease: Novel mechanisms and treatment strategies, Trends Endocrinol. Metabol 28 (2017) 250–260. [DOI] [PubMed] [Google Scholar]
- [26].Satapati S, Kucejova B, Duarte JA, et al. , Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver, J. Clin. Invest 125 (2015) 4447–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim JD, Yoon NA, Jin S, Diano S, Microglial UCP2 mediates inflammation and obesity induced by high-fat feeding, Cell Metabol. 30 (2019) 952–962 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Alcazar-Fabra M, Navas P, Brea-Calvo G, Coenzyme Q biosynthesis and its role in the respiratory chain structure, Biochim. Biophys. Acta 1857 (2016) 1073–1078. [DOI] [PubMed] [Google Scholar]
- [29].Chu CT, Ji J, Dagda RK, et al. , Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells, Nat. Cell Biol 15 (2013) 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li XX, Tsoi B, Li YF, Kurihara H, He RR, Cardiolipin and its different properties in mitophagy and apoptosis, J. Histochem. Cytochem 63 (2015) 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Paradies G, Paradies V, Ruggiero FM, Petrosillo G, Role of cardiolipin in mitochondrial function and dynamics in Health and disease: molecular and pharmacological aspects, Cells 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang Y, Hekimi S, Understanding ubiquinone, Trends Cell Biol. 26 (2016) 367–378. [DOI] [PubMed] [Google Scholar]
- [33].Zheng J, Conrad M, The metabolic underpinnings of ferroptosis, Cell Metabol. 32 (2020) 920–937. [DOI] [PubMed] [Google Scholar]
- [34].Doll S, Freitas FP, Shah R, et al. , FSP1 is a glutathione-independent ferroptosis suppressor, Nature 575 (2019) 693–698. [DOI] [PubMed] [Google Scholar]
- [35].Stockwell BR, A powerful cell-protection system prevents cell death by ferroptosis, Nature 575 (2019) 597–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dludla PV, Orlando P, Silvestri S, et al. , Coenzyme Q10 supplementation improves adipokine levels and alleviates inflammation and lipid peroxidation in conditions of metabolic syndrome: a meta-analysis of randomized controlled trials, Int. J. Mol. Sci 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Farsi F, Mohammadshahi M, Alavinejad P, Rezazadeh A, Zarei M, Engali KA, Functions of coenzyme Q10 supplementation on liver enzymes, markers of systemic inflammation, and adipokines in patients affected by nonalcoholic fatty liver disease: a double-blind, placebo-controlled, randomized clinical trial, J. Am. Coll. Nutr 35 (2016) 346–353. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.