

Abstract
Our understanding of the degradation of organic matter will benefit from a greater appreciation for the genes encoding enzymes involved in the hydrolysis of biopolymers such as chitin, one of the most abundant polymers in nature. To isolate representative and abundant chitinase genes from uncultivated marine bacteria, we constructed libraries of genomic DNA isolated from coastal and estuarine waters. The libraries were screened for genes encoding proteins that hydrolyze a fluorogenic analogue of chitin, 4-methylumbelliferyl β-d-N,N′-diacetylchitobioside (MUF-diNAG). The abundance of clones capable of MUF-diNAG hydrolysis was higher in the library constructed with DNA from the estuary than in that constructed with DNA from coastal waters, although the abundance of positive clones was also dependent on the method used to screen the library. Plaque assays revealed nine MUF-diNAG-positive clones of 75,000 screened for the estuarine sample and two clones of 750,000 for the coastal sample. A microtiter plate assay revealed approximately 1 positive clone for every 500 clones screened in the coastal library. The number of clones detected with the plaque assay was consistent with estimates of the portion of culturable bacteria that degrade chitin. Our results suggest that culture-dependent methods do not greatly underestimate the portion of marine bacterial communities capable of chitin degradation.
Chitin, a (1→4)-β-linked homopolymer of N-acetyl-d-glucosamine, is an abundant structural polysaccharide produced by many marine organisms. It is a constituent of the exoskeletons of zooplankton and invertebrate larvae (10), the cell walls of some chlorophytes (18), and the extracellular material of some diatoms (3, 28) and prymnesiophytes (5). The first step in chitin degradation, which is primarily done by microbes (10), is the hydrolysis of the glycosidic bonds between N-acetyl-d-glucosamine residues by chitinases (EC 3.2.1.14). The capacity to degrade chitin is widespread among taxonomic groups of prokaryotes including the gliding bacteria, vibrios, Photobacterium spp., enteric bacteria, actinomycetes, bacilli, clostridia (11), and archaea (12). Bacteria employ several proteins, including chitin-binding proteins (17, 31), to degrade chitin, but the hydrolysis by chitinase is the key step in the solubilization and mineralization of chitin.
The capacity to degrade chitin would seem to be an important attribute of marine bacteria given the presumed high input of detrital chitin into the sea (14). Chitinolytic bacteria are typically detected by either the production of clearing zones on agar containing chitin or hydrolysis of a fluorogenic substrate analogue of chitin. The assay for clearing zones suggests that ca. 10% of culturable bacteria degrade chitin (4, 20, 25), while the portion of strains hydrolyzing the analogue of chitin has been estimated to be as high as 90% (16). It is not clear which assay produces the more accurate estimate since both assays have drawbacks; the production of clearing zones requires export and diffusion of the chitinase into the surrounding media, while hydrolysis of the analogue may simply reflect the capacity to degrade small oligomers (2). Furthermore, whether either culture-based assay reflects the true portion of chitin degraders in natural bacterial assemblages is unclear, since only a small fraction (<1%) of the bacteria in seawater can be cultured (6, 7, 15) and those bacteria in culture are not thought to be representative of uncultured, natural bacteria (13, 32).
Molecular methods are needed to study chitin degraders without the isolation of bacteria into pure cultures. Methods that use nucleic acid probes and PCR primers cannot be designed solely with cultured bacteria because the nucleotide sequences of chitinase genes from cultured bacteria so far characterized are very different (33), suggesting that chitinases from uncultured bacteria may differ greatly from those in cultured bacteria. Although it is possible that conservation within groups of chitinases may become clear as more cultured bacteria are examined, information for cultured bacteria probably will not be sufficient to design “universal” PCR primers to retrieve chitinase genes from uncultured bacteria. One alternative approach that does not rely on conserved nucleotide sequences is to use genomic libraries to retrieve genes from natural bacterial communities without cultivation (24, 30, 36).
In this study we used genomic DNA libraries to retrieve chitinase genes from environmental DNA (26). A high-efficiency lambda phage cloning vector was used to produce libraries that were screened with a fluorogenic analogue of chitin to identify chitinase genes. We found that the frequencies of active clones identified by screening the libraries by plaque assay were consistent with culture-based estimates of the portion of marine bacteria that degrade chitin.
MATERIALS AND METHODS
Sample collection and preparation of DNA.
Coastal seawater was collected from a depth of 1 m at a station 14 km outside the entrance to the Delaware Bay estuary in September 1997. Estuarine water was collected 0.23 km inside the bay in November 1996. Plankton and particles were collected from the coastal seawater (10 liters) by filtration onto Gelman Supor filters (0.2 μm) and stored frozen at −80°C in a storage buffer (9). The estuarine sample was prefiltered through a 0.8-μm (pore-size) polycarbonate filter. Frozen samples were thawed, and the cells were lysed by using sodium dodecyl sulfate (SDS) and proteinase K. The lysate was extracted sequentially with phenol-chloroform and chloroform. RNA was removed by treatment with RNase A, and the DNA was precipitated with ethanol and further purified by using the IsoQuick Nucleic Acid Extraction Kit (ORCA Research, Inc., Bothel, Wash.) according to the manufacturer’s instructions.
Size fractionation and cloning of plankton DNA.
The genomic DNA (5 μg) was prepared for ligation by partial restriction digestion with the restriction enzyme Tsp509I (New England Biolabs, Beverly, Mass.) (3.3 U of enzyme per μg of DNA, 65°C, 15 min). The restriction fragments ranging from 2 to 10 kb were collected by ethanol precipitation from the 30% portion of a sucrose step gradient (40,000 rpm in a Beckman TLS-55 rotor for 12 h). A 400-ng portion of restriction fragments was ligated into Lambda Zap II predigested EcoRI/CIAP-treated vector (Stratagene, La Jolla, Calif.) by using T4 DNA ligase (Boehringer Mannheim, Indianapolis, Ind.) according to the manufacturer’s protocol. Recombinant lambda phage DNA was packaged by using Gigapack III packaging extract (Stratagene), and the titer and fraction of phage containing inserts were determined by plaque assay with blue-white color selection. The library was amplified according to the procedure described by the manufacturer of the cloning reagents.
Screening for chitinase genes.
Two approaches were used to identify clones that hydrolyze the analogue of chitin, 4-methylumbelliferyl β-d-N,N′-diacetylchitobioside (MUF-diNAG). The first was a plaque assay (4 × 104 plaques per 150-by-15-mm petri plate) screened by spraying MUF-diNAG (50 μM in 100 mM sodium phosphate buffer [pH 8]) onto the plaques as soon as they were 1 to 3 mm in diameter (22). The library from the coastal sample was also assayed with the fluorogenic substrate analogue of cellulose (MUF-cellobioside). Fluorescing plaques were detected by using a UV (366-nm) light source and transferred to SM buffer (100 mM NaCl, 0.8 mM MgSO4, 50 mM Tris-HCl, 0.01% gelatin). Strains of the active phage were purified with two iterations of plaque isolation.
The second approach used an excised copy of the library with each lambda phage clone represented by a phagemid. The mass excision of the library was performed by using ExAssist helper phage (Stratagene) according to the manufacturer’s protocol to produce phagemids. The assay was performed in microtiter plates with phagemids adsorbed to XLOLR cells at a multiplicity of infection of 2 × 10−5. Each well of the plate contained 150 infected cells. A microtiter plate containing XLOLR cells growing in Luria broth-tetracycline (12.5 μg/ml) served as a control. After 1 day of growth, aliquots (75 μl) of the cultures were transferred to sterile microtiter plates for the MUF-diNAG hydrolysis assay. After the substrate was added (50 μM), the plates were incubated at 37°C and examined daily with UV light. Pure strains of positive clones were obtained from fluorescing wells by two iterations of serial dilution followed by the isolation of single colonies on agar plates.
Enzymatic activities in protein extracts from positive clones.
Enzymatic activities of clones were assayed with protein extracts prepared from recombinant Escherichia coli bearing plasmids with the cloned DNA. Cells were collected by centrifugation, washed three times with Tris-buffered saline (20 mM Tris, 150 mM NaCl [pH 7.5]), resuspended in Tris-buffered saline, and sonicated. Sarcosyl (1% [wt/vol]) was added to the lysate before incubation on ice for 1 h. Particulate matter was removed from the extract by centrifugation (9,000 × g, 10 min), and the activities of the supernatant were assayed.
The capacity of the protein extracts to hydrolyze the fluorogenic substrate analogues 4-methylumbelliferyl N-acetyl-β-d-glucosaminide (MUF-NAG), MUF-diNAG, and 4-methylumbelliferyl β-d-N,N′,N"-triacetylchitotrioside (MUF-triNAG) was determined with 50 μM substrate additions and incubation at 37°C. At time intervals ranging from a few minutes to an hour, subsamples were removed and added to glycine carbonate buffer (pH 9.7) in order to measure MUF fluorescence.
The hydrolysis of glycol chitin by the protein extracts was assayed by glycol chitin-SDS-polyacrylamide gel electrophoresis (33). Electrophoresis was performed with an 8% polyacrylamide gel containing 0.1% SDS and 0.01% glycol chitin. After electrophoresis, the gel was incubated at 37°C for 2 h in 100 mM sodium acetate buffer (pH 5.0) containing 1% Triton X-100. The gel was stained for 5 min in 0.01% Calcofluor White M2R in 0.5 M Tris-HCl (pH 9.0) and destained overnight in water. Equal amounts of protein (200 μg) were electrophoresed without prior heat treatment. Clearing zones produced by the hydrolytic activity of the extracts were visualized with UV light (366 nm).
RESULTS
Numbers of cloned chitinase genes recovered from libraries.
The coastal library was screened by using the fluorogenic analogues of chitin (MUF-diNAG) and cellulose (MUF-cellobiose) to detect chitinase and cellulase genes, respectively. Two MUF-diNAG-hydrolyzing clones designated pG1 and pG2 were isolated after 7.5 × 105 clones were screened by the plaque assay. Restriction digestion of clones pG1 and pG2 with a mixture of XbaI and KpnI produced identical patterns composed of 1.4- and 4-kb bands plus a 2.9-kb band representing the pBluescript KS(−) vector, indicating that these clones have identical 5.4-kb inserts (Fig. 1). Digestion with the four-base cutter Tsp509I produced identical restriction patterns (data not shown). Cellulases were not detected since no plaques fluoresced after application of MUF-cellobioside in the plaque assays.
FIG. 1.

Restriction patterns (XbaI with KpnI) of clones that hydrolyze MUF-diNAG. (A) Clone identified by screening the library by plaque assay. Lanes: 1, pG1; M, molecular weight marker. (B) Clones identified by screening the library in microtiter plates. Lanes: 1, p2F9; 2, p3A6; 3, p4H11; 4, p5C7; 5, p5F2; 6, p6D5; 7, p6E11; 8, p7C8; 9, p7D9; 10, p7F6; 11, p9D5; 12, p10D3; 13, p14E11; M, molecular weight marker.
Screening a total of 2.3 × 105 clones from the coastal library in microtiter plates with MUF-diNAG yielded 432 fluorescing wells. Clones from 14 fluorescing wells were purified by dilution series and spreading on agar plates. Thirteen wells remained MUF-diNAG positive after purification to single colonies. Restriction digestion with a mixture of XbaI and KpnI revealed that all 13 were different and were not the same as the clones isolated by the plaque assay (Fig. 1). The size of the cloned inserts, estimated by summing the restriction fragments, ranged from 1.8 to 4.2 kb.
The estuarine library was screened for chitinases by the plaque assay with MUF-diNAG. Nine clones hydrolyzing MUF-diNAG were identified after 75,000 clones were screened (see Table 2). Restriction digestion with EcoRI revealed insert sizes ranging from 5.0 to 6.1 kb (data not shown).
TABLE 2.
Results from the estuarine library
| Clone | Classification | Relative activity ofa:
|
Apparent molecular size (kDa) | ||
|---|---|---|---|---|---|
| MUF-NAG | MUF-diNAG | MUF-triNAG | |||
| pJAM2 | ND | ND | + | ND | — |
| pJAM4 | Chitobiosidase | 0 | ++ | 0 | — |
| pJAM5 | Chitobiosidase | 0 | ++ | 0 | 250 |
| pJAM6 | Exochitinase | ++ | + | + | 30 to 250b |
| pJAM9 | Exochitinase | + | + | + | 98 |
| pJAM17 | ND | ND | + | ND | — |
| pJAM19 | Endochitinase | 0 | + | + | 98 |
| pJAM24 | ND | ND | + | ND | — |
| pJAM28 | ND | ND | + | ND | — |
The hydrolytic activities of protein extracts measured with various fluorogenic N-acetylglucosamine oligomers and polyacrylamide gels containing glycol chitin as indicated are shown. Relative activities: ND, not determined; 0, background activity; +, 2- to 10-fold above background; ++, 10- to 50-fold above background; +++, 50- to 100-fold above background. —, No hydrolysis.
The clearing zone appeared as a smear from 30 to 250 kDa.
Clone phenotypes.
The phenotypes of 13 clones from the coastal library were characterized, and the enzymes they produced were classified by assaying protein extracts for hydrolysis of various fluorogenic N-acetylglucosamine oligomers (19). Four clones (p2F9, p3A6, p4H11, and p14E11) hydrolyzed all three N-acetylglucosamine oligomers, suggesting that they produce exochitinases (Table 1). The enzymes produced by clones p5F2, p6E11, and p10D3 were classified as chitobiosidases because they hydrolyzed only MUF-diNAG. Clone p5C7 hydrolyzed MUF-NAG and MUF-diNAG, suggesting that it might produce an exochitinase; however, it did not hydrolyze MUF-triNAG (Table 1).
TABLE 1.
Results from the coastal library
| Clone | Classification | Relative activitya of:
|
||||
|---|---|---|---|---|---|---|
| Whole cells
|
Protein extracts
|
|||||
| MUF-diNAG | MUF-NAG | MUF-diNAG | MUF-triNAG | Apparent molecular size (kDa) | ||
| p2F9 | Exochitinase | ++ | + | + | + | 98 and 250 |
| p3A6 | Exochitinase | +++ | + | 0 | + | 250 |
| p4H11 | Exochitinase | + | + | + | + | 250 |
| p5C7 | ND | ++ | + | + | 0 | 98 and 250 |
| p5F2 | Chitobiosidase | ++ | 0 | + | 0 | — |
| p6D5 | Chitobiosidase | ++ | 0 | 0 | 0 | — |
| p6E11 | Chitobiosidase | ++ | 0 | ++++ | 0 | — |
| p7C8 | Chitobiosidase | + | 0 | 0 | 0 | — |
| p7D9 | Chitobiosidase | ++ | 0 | 0 | 0 | — |
| p7F6 | Chitobiosidase | ++ | 0 | 0 | 0 | — |
| p9D5 | Chitobiosidase | ++ | 0 | 0 | 0 | — |
| p10D3 | Chitobiosidase | ++ | 0 | ++++ | 0 | — |
| p14E11 | Exochitinase | +++ | + | + | + | — |
| pG2 | Exochitinase | ND | ++++++ | +++++ | ++++ | — |
The hydrolytic activities of E. coli carrying cloned chitinase genes and protein extracts from cells measured with various fluorogenic N-acetylglucosamine oligomers and polyacrylamide gels containing glycol chitin as indicated are shown. Relative activities: 0, no activity detected above background; +, hydrolysis 1.2- to 10-fold above background; ++, hydrolysis 101- to 102-fold above background; +++, hydrolysis 103- to 104-fold above background; ++++, hydrolysis 104- to 105-fold above background; +++++, hydrolysis 105- to 106-fold above background; ++++++, hydrolysis 106- to 107-fold above background [clone bearing pBluescript KS(−) vector alone]. ND, not determined; —, no hydrolysis.
Protein extracts prepared from five clones had no activity against any of the MUF substrates, even though cultures of E. coli bearing these plasmids hydrolyzed MUF-diNAG at rates 7- to 34-fold higher than control cells possessing the pBluescript KS(−) vector (Table 1). The activity of protein extracts from all clones identified with the microtiter plates was less than that of the intact cells (Table 1) and decreased upon lysis of the cells by either sonication or lysozyme.
The estuarine library contained clones pJAM6 and pJAM9 that hydrolyzed all three chitin analogs, and the enzymes they produced were classified as exochitinases (Table 2). Clone pJAM19 was active against MUF-diNAG and MUF-triNAG but not MUF-NAG, suggesting it produced an endochitinase. Because clones pJAM4 and pJAM5 were active against only MUF-diNAG, the enzymes they produce were classified as chitobiosidases (Table 2). The relative rates of hydrolysis of the various analogues varied among the clones. Clone pJAM4 and clone pJAM5 had the highest activities (10- to 50-fold above background) against MUF-diNAG. The remaining clones hydrolyzed the various analogues at rates 2- to 10-fold above background.
Protein extracts were assayed for the capacity to hydrolyze glycol chitin in polyacrylamide gels containing this soluble form of chitin. After electrophoresis, the gels were stained with Calcofluor to visualize areas of the gel in which chitin had been hydrolyzed. Four clones from the coastal library (p2F9, p3A6, p4H11, and p5C7) made clearing zones that were distinguishable from the control [pBluescript KS(−)] (Fig. 2). The enzymes appeared to have molecular masses of 98 and 250 kDa. However, their actual sizes cannot be measured from this analysis, as proteins do not run true to their molecular masses in this type of gel (34). Protein extracts of four clones (pJAM5, pJAM6, pJAM9, and pJAM19) from the estuarine library hydrolyzed glycol chitin after SDS-electrophoresis. Clone pJAM5 produced an active enzyme that appeared to have a molecular mass of 250 kDa. Clones pJAM9 and pJAM19 produced a clearing at ca. 98 kDa. Clone pJAM6 produced many regions of clearing between 250 and 30 kDa (data not shown).
FIG. 2.
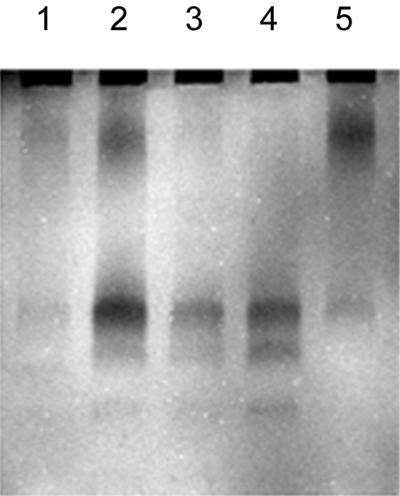
Calcofluor-stained glycol chitin gel of proteins extracted from E. coli bearing the plasmids pBluescript KS(−) (lane 1), p2F9 (lane 2), p3A6 (lane 3), p4H11 (lane 4), and p5C7 (lane 5). Regions of the gel with hydrolyzed chitin were distinguishable by their lack of fluorescence (dark bands).
DISCUSSION
The screening of genomic DNA libraries for the expression of targeted genes has been used to collect genes encoding proteins with industrial applications (26), but this approach can be used to ask ecological questions as well. In this study, we cloned genes coding for proteins that hydrolyze a fluorogenic analogue of chitin and glycol chitin from environmental DNA. Examining chitinase genes is important for understanding the ecology of chitin-degrading bacteria and chitin degradation in aquatic systems.
Screening environmental DNA libraries has its limitations. Detection of a cloned chitinase by expression requires cloning a native promoter with the chitinase gene or the alignment of the cloned gene with the reading frame of the lacZ promoter on the vector (22). In some cases, the inability to obtain an expressed clone is not clear. For example, part of the chiA gene of Vibrio harveyi was obtained by screening a plasmid library for activity (29), but a full-length chiA was never obtained in a library made in lambda phage (34). Furthermore, the fidelity with which a genomic library reflects the abundance of genes in the community can be further influenced by manipulations of the library itself. For example, clones pG1 and pG2, which have the same restriction fragment length polymorphism pattern, could represent genes from two separate genomes captured by the library; alternatively, one may be a duplicate produced when the library was amplified to create copies of the library. It is possible to screen a library without amplification, but copies of the library were necessary in order to screen with more than one substrate analogue.
In spite of the limitations, genomic DNA libraries screened for gene expression are valuable tools for ecological studies because they can provide information about enzymes from organisms without cultivation. Furthermore, they are well suited for genes such as those encoding chitinases which do not have regions of similarity needed for designing universal probes or PCR primers. In addition to recovering previously unrecognized diversity, eventually we wish to build molecular approaches to examine the frequency and expression of chitinases in natural samples and to determine the relative abundance of organisms that possess chitinases. Answering these questions will require extensive development of methods to detect genes in natural bacterial assemblages. Although a direct estimate of chitinolytic bacteria is not technically possible now, we can use our data to obtain an indirect estimate of how many bacteria degrade chitin. In addition to being ecologically interesting, the calculation is important for determining if the frequency of MUF-diNAG-positive clones we obtained is reasonable.
We used the abundance of clones hydrolyzing MUF-diNAG to estimate the portion of uncultured bacteria that are chitinolytic. The estimation was based on the relationship among insert size, genome size, and the number of clones that must be screened to find a single-copy gene in a library with a probability of 0.99 (1):
 |
1 |
where N is the number of clones that must be screened. The calculation was made by using a genome size representative of bacteria (2 × 106 kb) and an insert size of 4 kb. Because the coastal library was constructed with total plankton DNA, which typically contains about 50% bacterial DNA (21, 38), the number of clones to screen for this library was multiplied by 2.
We expected more MUF-diNAG-positive clones for a given library size, and thus a higher frequency of positive clones than indicated by equation 1, because bacteria typically have not one but about five chitinase genes. Thus, the expected frequency of MUF-diNAG positive clones is:
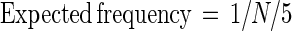 |
2 |
where N is calculated from equation 1. We assumed five chitinase genes per chitinolytic species based on observations of the enzymes produced by five chitin-degrading bacteria. Serratia marcescens (8) and Streptomyces olivaceoviridis (23) each produce five chitinases, while Streptomyces plicatus (22) and Bacillus circulans (37) produce four and six chitinases, respectively. It was assumed that the number of chitinase genes per bacterium is equal to the number of chitinases produced. The number of chitinase enzymes may be larger than the number of chitinase genes because chitinases may be modified by proteolytic cleavage to produce additional chitinases (23, 35). For example, V. harveyi appears to produce 10 chitinases by proteolytic cleavage of proteins encoded by five chitinase genes (34). The type and number of chitinases produced varies with exposure to different types of chitin (34), so estimates of the numbers of chitinases produced by various bacteria may increase as more types of chitin are tested, but probably by no more than a factor of 2.
The expected number of MUF-diNAG-positive clones is a maximum estimate because our calculation so far assumes that all genomes represented in the library contain chitinase genes. Thus, the percentage of chitin-degrading bacteria in the original water sample can be estimated by dividing the observed frequency of MUF-diNAG positive clones by the expected frequency. The estimates of the percentages of chitin degraders for the estuarine and coastal communities were 5.5 and 0.12%, respectively, based on the results from plaque assays (Table 3). In contrast, screening the coastal library as phagemids in microtiter plates produced far more MUF-diNAG-positive clones than would be expected if all of the bacteria in the community were chitin degraders. The explanation for this large number of MUF-diNAG-positive clones is not clear, but several of these clones probably produce proteins that hydrolyze the substrate analogue but are not involved in the hydrolysis of chitin.
TABLE 3.
Percentage of chitinolytic bacteria in coastal and estuarine samples inferred from the frequency of clones hydrolyzing MUF-diNAG
| Environment | Plaques or phagemids screened | Observed frequency of MUF-diNAG-positive clones | Expected frequency of MUF-diNAG-positive clonesa | Chitinolytic bacteriab (%) |
|---|---|---|---|---|
| Estuary | Plaques | 1/8,333 | 1/460 | 5.5 |
| Coastal | Plaques | 1/750,000 | 1/920 | 0.12 |
| Coastal | Phagemids | 1/533 | 1/920 | 170 |
The reciprocal of the number of clones that must be screened to find a single-copy gene (equation 1) divided by 5 (number of chitinase genes per bacterium). The numbers of clones to screen for the coastal sample were multiplied by 2 because this sample contained total plankton DNA, which typically contains only 50% bacterial DNA (22).
That is, the observed frequency of MUF-diNAG-positive clones divided by the expected frequency of MUF-diNAG-positive clones multiplied by 100.
The microtiter assay seems to detect low-level hydrolysis of MUF-diNAG not observed with the plaque assay. This greater sensitivity is likely because a microtiter well contains more cells than a plaque and because the microtiter assay uses intact cells containing excised plasmids, whereas cells in plaques have been lysed. In addition to likely higher enzyme production, enzyme activity was probably also higher in the microtiter assay with intact cells since we found that lysing MUF-diNAG-positive clones by sonification, which is analogous to viral lysis, greatly reduced the hydrolysis of MUF-diNAG. Finally, diffusion of the fluorescent signal away from plaques may be another contributing factor. Robbins et al. (22) suggested that a low signal can limit the detection of clones hydrolyzing a fluorogenic substrate and pointed out that the accumulation of fluorescence requires the production of the fluorescing compound to exceed its removal by diffusion. The microtiter wells were not subjected to this limitation and fluorescence was maintained for several days.
Given the prevalence of chitin-producing organisms in the sea, it might be anticipated that most marine bacteria are able to degrade chitin. Studies with cultures, however, suggest that relatively few marine bacteria degrade chitin, ranging from 0.4 to 19% of total cultured bacteria (4, 20, 25). Our estimates for estuarine and coastal waters were within this range. Although chitinase activity can be much higher on particles than in the surrounding seawater (27), including particle-associated bacteria in the coastal library did not greatly increase the estimate of the portion of bacteria that degrade chitin. Even though culture-based methods retrieve only a small fraction of the total bacteria, our results suggest that culture-based estimates of the percentage of chitinolytic bacteria may be correct. However, there still remains a large pool of uncultured chitin-degrading bacteria in aquatic environments, and describing their chitinases will produce a better understanding of chitin degradation in the sea.
ACKNOWLEDGMENTS
This research was funded by the U.S. Department of Energy. The collection of samples was made possible by a National Science Foundation grant.
REFERENCES
- 1.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struh K. Current protocols in molecular biology. New York, N.Y: Greene Publishing Associates/Wiley-Interscience; 1992. [Google Scholar]
- 2.Bassler B L, Yu C, Lee Y C, Roseman S. Chitin utilization by marine bacteria. Degradation and catabolism of chitin oligosaccharides by Vibrio furnissii. J Biol Chem. 1991;266:24276–24286. [PubMed] [Google Scholar]
- 3.Blackwell J, Parker K D, Rudall K M. Chitin fibers of the diatoms Thalassiosira fluviatilis and Cyclotella cryptica. J Mol Biol. 1967;28:383. doi: 10.1016/s0022-2836(67)80018-4. [DOI] [PubMed] [Google Scholar]
- 4.Brisou J, Tysset C, de Rautlin A, de la Roy Y, Curcier R, Moreau R. Etude sur la chitinolyse a milieu marin. Ann Inst Pasteur. 1964;106:469–478. [Google Scholar]
- 5.Chretiennot-Dinet M J, Giraud-Guille M M, Vaulot D, Putaux J L, Saito Y, Chanzy H. The chitinous nature of filaments ejected by Phaeocystis (Prymnesiophyceae) J Phycol. 1997;33:666–672. [Google Scholar]
- 6.Eguchi M, Ishida Y. Oligotrophic heterotrophic bacteria and in situ heterotrophic activity in pelagic seawaters. FEMS Microbiol Ecol. 1990;73:23–30. [Google Scholar]
- 7.Ferguson R, Buckley E, Palumbo A. Response of marine bacterioplankton to differential filtration and confinement. Appl Environ Microbiol. 1984;47:49–55. doi: 10.1128/aem.47.1.49-55.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuchs R L, McPherson S A, Drahos D J. Cloning of a Serratia marcescens gene encoding chitinase. Appl Environ Microbiol. 1986;51:504–509. doi: 10.1128/aem.51.3.504-509.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giovannoni S J, DeLong E F, Schmidt T M, Pace N R. Tangential flow filtration and preliminary phylogenetic analysis of marine picoplankton. Appl Environ Microbiol. 1990;56:2572–2575. doi: 10.1128/aem.56.8.2572-2575.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gooday G W. The ecology of chitin degradation. Adv Microb Ecol. 1990;11:387–430. [Google Scholar]
- 11.Gooday G W, editor. A survey of polysaccharase production: a search for phylogenetic implications. London, England: Academic Press; 1979. [Google Scholar]
- 12.Huber R, Stohr J, Hohenhaus S, Rachel R, Burggraf S, Jannasch H, Stetter K O. Thermococcus chitonophagus sp. nov., a novel, chitin-degrading, hyperthermophilic archaeum from a deep-sea hydrothermal vent environment. Arch Microbiol. 1995;164:255–264. [Google Scholar]
- 13.Hugenholtz P, Goebel B M, Pace N R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirchman, D. L., and J. White. Hydrolysis and mineralization of chitin in the Delaware Estuary. Aquat. Microb. Ecol., in press.
- 15.Kogure K, Simidu U, Taga N. Distribution of viable marine bacteria in neritic seawater around Japan. Can J Microbiol. 1980;36:318–323. doi: 10.1139/m80-052. [DOI] [PubMed] [Google Scholar]
- 16.Martinez J, Smith D C, Steward G F, Azam F. Variability in ectohydrolytic enzyme activities of pelagic marine bacteria and its significance for substrate processing in the sea. Aquat Microb Ecol. 1996;10:223–230. [Google Scholar]
- 17.Montgomery M T, Kirchman D L. Induction of chitin-binding proteins during the specific attachment of the marine bacterium Vibrio harveyi to chitin. Appl Environ Microbiol. 1994;60:4284–4288. doi: 10.1128/aem.60.12.4284-4288.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulisch M. Chitin in protistan organisms: distribution, synthesis and deposition. Eur J Protistol. 1993;29:1–18. doi: 10.1016/S0932-4739(11)80291-9. [DOI] [PubMed] [Google Scholar]
- 19.Muzzarelli R A A. Chitin. 1st ed. Oxford, England: Pergamon Press; 1977. [Google Scholar]
- 20.Okutani K, editor. Microorganisms related to mineralization of chitin in aquatic environments. Tokyo, Japan: University of Tokyo Press; 1975. [Google Scholar]
- 21.Paul J H, Jeffrey W H, DeFlaun M. Particulate DNA in subtropical oceanic and estuarine planktonic environments. Mar Biol. 1985;90:95–101. [Google Scholar]
- 22.Robbins P W, Albright C, Benfield B. Cloning and expression of a Streptomyces plicatus chitinase (chitinase-63) in Escherichia coli. J Biol Chem. 1988;263:443–447. [PubMed] [Google Scholar]
- 23.Romaguera A, Menge U, Breves R, Diekmann H. Chitinases of Streptomyces olivaceoviridis and significance of processing for multiplicity. J Bacteriol. 1992;174:3450–3454. doi: 10.1128/jb.174.11.3450-3454.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt T M, DeLong E F, Pace N R. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol. 1991;173:4371–4378. doi: 10.1128/jb.173.14.4371-4378.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seki H. Seasonal fluctuation of heterotrophic bacteria in the sea of Aburatsubo Inlet. J Oceanogr Soc Jpn. 1966;22:15–26. [Google Scholar]
- 26.Short J M. Recombinant approaches for accessing biodiversity. Nat Biotechnol. 1997;15:1322–1323. doi: 10.1038/nbt1297-1322. [DOI] [PubMed] [Google Scholar]
- 27.Smith D C, Simon M, Alldredge A L, Azam F. Intense hydrolytic enzyme activity on marine aggregates and implications for rapid particle dissolution. Nature. 1992;359:139–142. [Google Scholar]
- 28.Smucker R A, Dawson R. Products of photosynthesis by marine phytoplankton: chitin in TCA “protein” precipitates. J Exp Mar Biol Ecol. 1986;104:143–152. [Google Scholar]
- 29.Soto-Gil R W. Ph.D. dissertation. University of California, San Diego; 1988. , and San Diego State University, San Diego, Calif. [Google Scholar]
- 30.Stein J L, Marsh T L, Wu K Y, Shizuya H, DeLong E F. Characterization of uncultivated prokaryotes: isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. J Bacteriol. 1996;178:591–599. doi: 10.1128/jb.178.3.591-599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki K, Suzuki M, Taiyoji M, Nikaidou N, Watanabe T. Chitin binding protein (CBP21) in the culture supernatant of Serratia marcescens 2170. Biosci Biotechnol Biochem. 1998;62:128–135. doi: 10.1271/bbb.62.128. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki M, Rappe M, Haimberger Z, Winfield H, Adair N, Strobel J, Giovannoni S. Bacterial diversity among small-subunit rRNA gene clones and cellular isolates from the same seawater sample. Appl Environ Microbiol. 1997;63:983–989. doi: 10.1128/aem.63.3.983-989.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svitil A L, Kirchman D L. A chitin-binding domain in a marine bacterial chitinase and other microbial chitinases: implications for the ecology and evolution of 1,4-beta-glycanases. Microbiology (United Kingdom) 1998;144:1299–1308. doi: 10.1099/00221287-144-5-1299. [DOI] [PubMed] [Google Scholar]
- 34.Svitil A L, Ni Chadhain S M, Moore J A, Kirchman D L. Chitin degradation proteins produced by the marine bacterium Vibrio harveyi growing on different forms of chitin. Appl Environ Microbiol. 1997;63:408–413. doi: 10.1128/aem.63.2.408-413.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tantimavanich S, Pantuwatana S, Bhumiratana A, Panbangred W. Multiple chitinase enzymes from a single gene of Bacillus licheniformis TP-1. J Ferment Bioeng. 1998;85:259–265. [Google Scholar]
- 36.Vergin K L, Urbach E, Stein J L, DeLong E F, Lanoil B D, Giovannoni S J. Screening of a fosmid library of marine environmental genomic DNA fragments reveals four clones related to members of the order planctomycetales. Appl Environ Microbiol. 1998;64:3075–3078. doi: 10.1128/aem.64.8.3075-3078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe T, Oyanagi W, Suzuki K, Tanaka H. Chitinase system of Bacillus circulans WL-12 and importance of chitinase-A1 in chitin degradation. J Bacteriol. 1990;172:4017–4022. doi: 10.1128/jb.172.7.4017-4022.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White, R. Unpublished data.