

Abstract
Chimeric antigen receptor (CAR) T cell therapy has emerged as a revolutionary treatment option for highly aggressive B cell malignancies. Clinical trials of CD19 CAR T cells for the management of relapsed and/or refractory non-Hodgkin lymphoma (NHL) have shown markedly improved survival and response rates. The goal of this review is to evaluate whether the results from these clinical trials are reflective of real-world practices through the analysis of published literature of the commercially available CAR T cell products. We have found that despite the significantly different patient characteristics, the adverse events and response rates of real-world patients were similar to those of the clinical trials. Of interest, several groups excluded from the clinical trials, such as patients with HIV infection, chronic viral hepatitis, and secondary CNS (central nervous system) lymphoma, had case reports of promising outcomes.
Keywords: lymphoma, Non-Hodgkin lymphoma, cellular therapy, chimeric antigen receptor, CAR T cell therapy
INTRODUCTION
Curative treatment options for aggressive relapsed or refractory (r/r) B cell non-Hodgkin lymphoma (NHL) are limited despite the rapid development of new immunotherapy and antineoplastic drugs. A promising therapy, chimeric antigen receptor (CAR) T cells, has shown durable response rates in clinical trials for various types of malignancies, including NHL.[1–4] Currently, there are four commercially available CAR T cell therapies for B cell NHL. Axicabtagene ciloleucel (axi-cel), tisagenlecleucel (tisa-cel), and lisocabtagene maraleucel (liso-cel) were approved by the US Food and Drug Administration (FDA) in 2017, 2018, and 2021, respectively, for r/r large B cell lymphoma (LBCL) after two lines of systemic therapy, whereas brexucabtagene autoleucel (BA) was approved for r/r mantle cell lymphoma in 2020. Axi-cel is also approved for r/r follicular lymphoma after two lines of systemic therapy (2021). This comprehensive review of commercially available CAR T cell products in LBCL compares the pivotal clinical trials against real-world outcomes, including several patient groups that were initially excluded from clinical trials.
CHIMERIC ANTIGEN RECEPTOR
All of the currently available commercial products target the B cell–specific surface marker CD19 but differ slightly in manufacturing and processing. Axi-cel contains an extracellular single chained variable fragment (scFv) CD19 fused with a CD28 costimulatory domain followed by a CD3ζ signaling domain.[2] Tisa-cel and liso-cel have a similar extracellular CD19 scFv attached to a 4-1BB intracellular costimulatory domain followed by CD3ζ signaling domain.[1,3] Liso-cel has a unique manufacturing process. After leukapheresis, CD4 and CD8 T cells are separated, independently transduced, expanded, and administered to patients at equal target concentrations, providing consistent product doses of liso-cel for each patient.[1]
BRIDGING THERAPY
Processing and manufacturing of individualized CAR T cells requires two or more weeks after leukapheresis.[5,6] During this period, the oncologist may choose to administer bridging therapy (BT) to reduce tumor burden in patients with symptomatic or bulky disease.[1,3,7,8] Currently, BT is given at the discretion of the physician and therapy options include corticosteroids, radiation therapy, systemic chemotherapy, and targeted therapy. Studies have demonstrated that BT is associated with worse overall survival (OS) and progression-free survival (PFS); however, this may be due to the fact that patients who received BT were more likely to have higher-risk baseline characteristics such as worse performance status (PS), bulky disease, or elevated serum lactate dehydrogenase levels.[7,8]
CONDITIONING REGIMEN
Prior to infusion of the CAR T cells, patients undergo lymphodepletion conditioning, often with fludarabine and cyclophosphamide (flu/cy), to improve engraftment, persistence, and efficacy of the transferred T cells.[9] The mechanism by which lymphodepletion promotes these functions has yet to be fully elucidated; however, it is believed that lymphodepletion conditioning provides a favorable immunologic environment through several mechanisms including downregulation of suppressive regulatory cells, such as T-regulatory and myeloid-derived suppressor cells, and altering the cytokine milieu to support CAR T-cell proliferation.[10,11] Hirayama et al[10] showed that a favorable cytokine profile may play a greater benefit in PFS rather than the intensity of the lymphodepletion chemotherapy. Although high-intensity flu/cy was associated with a higher probability of PFS than low-intensity, multivariate analysis showed that there was no difference between low-intensity flu/cy and high-intensity flu/cy with unfavorable cytokine profile. Patients who received high-dose flu/cy conditioning with favorable cytokine profile had the highest probability of PFS.
ADVERSE EFFECTS
The most common adverse effects (AEs) of CAR T cell therapy are cytokine release syndrome (CRS), neurotoxicity, and hematologic toxicities. Many of these toxicities are manageable and non–life-threatening. Multiple grading systems for toxicities have been applied over the past few years. These include the Lee et al system,[12] the University of Pennsylvania grading scale,[3] Common Terminology Criteria for Adverse Events (CTCAE; version 4.03, US National Cancer Institute), the American Society for Transplantation and Cellular Therapy (ASTCT) consensus grading,[13] and several others[14,15] (Table 1). Owing to the heterogeneity in the grading systems used in the early clinical trials, it is challenging to compare the safety profiles across the prospective studies. As CAR T cell products became more widely used, there was a need for a universal grading system for the early identification and management of CAR T cell toxicities. The ASTCT consensus became the most common grading system of CRS and neurotoxicity associated with immune effector cell therapy. In an effort to standardize the treatment of CAR T cell–related toxicities, experts experienced in treating patients with CAR T cell therapy formed a consortium named the CAR T cell therapy-associated TOXicity (CARTOX) Working Group and created a consensus guideline on the management of CAR T cell toxicities.[14] It is important to recognize that the grading criteria and management approaches have evolved and will continue to evolve as we learn more about the clinical application of cellular therapy.
Table 1.
Various scoring systems used to grade cytokine release syndrome (CRS)
|
Grade
|
CRS Scoring Systems
|
CARTOX14
|
||
|
ASTCT13
|
CTCAE version 4.03
|
Lee et al12
|
||
| 1 | Temperature ≥ 38°C | Fever, with or without constitutional symptoms | Symptoms are not life-threatening and require symptomatic treatment only (fever, nausea, fatigue, headache, myalgias, malaise) |
|
| 2 | Temperature ≥ 38°C with:
|
|
Symptoms require and respond to moderate intervention:
|
|
| 3 | Temperature ≥ 38°C with:
|
|
Symptoms require and respond to aggressive intervention:
|
|
| 4 | Temperature ≥ 38°C with:
|
Life-threatening consequences; urgent intervention needed | Life-threatening symptoms:
|
|
| 5 | N/A | Death | Death | Death |
As per CTCAE version 4.03.
CRS: cytokine release syndrome; ASTCT: American Society for Transplantation and Cellular Therapy; CTCAE: Common Terminology Criteria for Adverse Events; CARTOX: CAR T-cell-therapy-associated TOXicity; IV: intravenous; FiO2: fraction of inspired oxygen; CPAP: continuous positive�irway pressure; BiPAP: bilevel positive airway pressure; N/A: not applicable.
Cytokine Release Syndrome (CRS)
Engagement of the CAR to its ligand leads to T cell activation and proliferation.[16] Activation of the CAR T cell will induce the secretion of cytokines and proinflammatory signals from activated lymphocytes and other immune cells.[17] These cytokines may lead to increased endothelial cell permeability and an exaggerated systemic inflammatory response called CRS.[18] Mild cases of CRS present with fevers, tachycardia, and myalgias and can progress to hypotension, hypoxemia, or even end-organ damage. Both the symptoms of CRS and sepsis overlap greatly, and it is important to rule out underlying infections. Patients with CRS are often started on empiric antibiotics given that it is difficult to distinguish CRS from sepsis during the early days post CAR T cell infusion.
The risk of CRS is influenced by both pretreatment factors, such as tumor burden, and treatment-related factors such as the costimulatory domain of the CAR, dose of CAR T cells infused, and lymphodepletion.[18,19]
Treatment of CRS is guided by institutional policies and is mostly supportive. For grade 1 CRS, fevers (≥ 38°C) are often managed with antipyretics and external cooling interventions, such as ice baths and/or cooling blankets.[14,20] Patients who have persistent grade 1 CRS (> 72 hours) or progress to grade 2—fever plus hypotension (manageable with intravenous [IV] fluids, not requiring vasopressors) and/or hypoxia (requiring low-flow nasal cannula ≤ 6 L/min)—are often administered tocilizumab, an interleukin (IL) 6 receptor monoclonal antibody.[21] Corticosteroids can also be considered for grade 2 or higher CRS after maximum doses of tocilizumab (8 mg/kg every 8 hours for up to 3 doses in a 24-hour period) or simultaneously with tocilizumab for patients who are high risk for severe CRS (e.g., high tumor burden, early onset CRS < 3 days, comorbidities).[14,20] Severe cases of CRS (≥ grade 3) are often transferred to the intensive care unit and may require additional supportive measures in addition to anticytokine therapy and corticosteroids such as vasopressor agents for hypotension and supplemental oxygen or intubation for hypoxemia.[14,20] The judicial use of corticosteroids may have limited effect on treatment outcomes, or persistence of CAR T cells, and should be considered for the treatment of acute CRS during CAR T cell therapy.[22–24] However, careful consideration of the dose and duration is warranted.[25]
Immune Effector Cell–Associated Neurotoxicity Syndrome (ICANS)
The second most common acute toxicity associated with CAR T cell therapy is neurotoxicity, which is termed immune effector cell–associated neurotoxicity syndrome (ICANS).[13,26] The pathogenesis of ICANS is poorly understood; however, like CRS, it is believed that cytokines play a major role in its development.[16,26,27] Patients who develop ICANS have evidence of increased endothelial activation, presumably from high concentrations of systemic cytokines, causing disruption of the blood brain barrier (BBB).[26] Paired samples of cerebrospinal fluid (CSF) and serum show significantly increased levels of tumor necrosis factor α, IL-6, IL-10, and interferon-γ and CAR constructs in the CSF.[28] It is unclear whether the elevated levels of proinflammatory cytokines in the CSF are due to the diffusion of systemic cytokines into the central nervous system (CNS) by BBB disruption or production of these cytokines by infiltrative CAR T cells in the CNS. CAR T cells can be found in the CNS after treatment, but the number of CAR T-cells do not correlate with the severity of neurotoxicity.[29] Similarly, cellular components other than T cells have been implicated in the pathogenesis of neurotoxicity.[30,31] Single-cell analyses have also identified brain mural cells expressing CD19, which may provide a rationale for observations of neurotoxicity observed with CD19-directed bispecific T cell–engaging antibodies in addition to CD19-directed CAR T cell therapy, suggesting a potential off-tumor target effect.[32]
The symptoms of ICANS range greatly from mild word-finding difficulties, aphasia, toxic encephalopathy, impaired cognitive skills, altered consciousness, or hallucinations to more devastating symptoms including seizures, motor weakness, and cerebral edema. Neurotoxicity has a median time of onset of 5–6 days with resolution typically within 4 weeks. Because ICANS can lead to significant morbidity, it is critical to perform frequent neurological assessments to allow for early intervention. Questionnaires are administered periodically to screen for deficits in various cognitive domains. Lower questionnaire scores correspond to higher ICANS severity.[13] A comprehensive neurological workup is warranted for any patient who develops altered mentation or sensorimotor deficits.[14] Neuroimaging with magnetic resonance imaging (MRI) and computerized tomography (CT) scan are used to evaluate for cerebral edema, encephalitis, leptomeningeal disease, and to rule out structural causes of encephalopathy.[30,33] Lastly, electroencephalography (EEG) is obtained to detect abnormal brain wave activity that may suggest subclinical or overt seizures.
ICANS is managed with corticosteroids and prophylactic antiepileptic drugs. Despite high levels of IL-6 being present in the CSF of patients who develop ICANS after CAR T cell treatment, tocilizumab is ineffective at alleviating ICANS.[34] One possible explanation is that tocilizumab cannot penetrate the BBB and therefore has no action in the CNS.[26]
Given the potentially fatal sequelae of severe CRS and ICANS, additional treatment options for these CAR T cell toxicities are needed. Preclinical trials have identified several cytokines involved in the pathogenesis of CRS and ICANS. In mouse studies, inhibition of IL-1 and granulocyte-monocyte colony stimulating factor (GM-CSF) have both shown the ability to decrease the rates and severity of CRS and lethal neurotoxicity in xenografted anti-CD19 CAR T cells.[34,35] Anakinra, an anti–IL-1 receptor antibody, is under investigation for treatment of CAR T cell–related toxicities including CRS and ICANS.
Hematologic Toxicities
Like many other antineoplastic agents, CAR T cell therapy can lead to significant hematologic toxicities. The etiology of cytopenias in patients receiving anti-CD19 CAR T cell therapy for r/r NHL is complex and multifaceted. Potential bone marrow involvement of the lymphoma, prior myelotoxic therapies, lymphodepleting chemotherapy, BT, direct cytotoxicity of the CAR T cells, and CRS can all lead to myelosuppression. Prolonged cytopenias (> 28 days post cell infusion) has been reported in 20–40% of cases.[36–38] Severe thrombocytopenia and anemia due to CAR T cell toxicities are treated supportively with platelet and red blood cell transfusions, respectively. Prophylactic granulocyte–colony stimulating factor (G-CSF) can be safely administered after day 5 post CAR T cell infusion to severely neutropenic (absolute neutrophil counts < 500 μL/mL) patients to prevent infections. G-CSF given after day 5 has shown not to affect efficacy or rates of toxicities.[39] The safety of G-CSF administration prior to day 5 is not as well studied.
Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome (HLH/MAS)
Severe CRS shares many features with hemophagocytic lymphohistiocytosis (HLH) or macrophage activation syndrome (MAS), which can also be seen as an adverse event from CAR T-cell therapy.[40] HLH should be suspected when a CRS-like syndrome does not improve with the use of tocilizumab. Both entities may cause fevers, cytopenias, elevated serum ferritin, and elevated soluble IL-2 levels; however, the classic HLH criteria of hepatosplenomegaly, lymphadenopathy, and overt evidence of hemophagocytosis do not commonly occur with severe CRS. Management can be similar to CRS: anti–IL-6 antibodies and corticosteroids; despite this, some patients still face poor outcomes.
AXI-CEL
ZUMA-1 Clinical Trial
Axi-cel was the first of the commercially available CAR T cell products that gained FDA approval for r/r NHL. Through the robust results from the ZUMA-1 clinical trial, axi-cel received priority review and orphan drug designation for treatment of diffuse large B cell lymphoma (DLBCL) in 2017. The ZUMA-1 phase 2 study included 111 patients with the baseline characteristics of median age 58 years (range, 23–76), 85% with stage III–IV disease, 48% with an international prognostic index (IPI) score of 3–4, 69% with a median number of 3 or more prior therapies, and 26% with primary refractory disease.[2] Participants in the study had the following disease types: DLBCL (76%), transformed follicular lymphoma (tFL, 16%) or primary mediastinal B-cell lymphoma (PMBCL, 8%). Patients with poor PS (Eastern Cooperative Oncology Group [ECOG] scores 2–4), chronic infections with human immunodeficiency virus (HIV) or hepatitis B virus (HBV) and CNS involvement were excluded from the study. None of the patients in the ZUMA-1 clinical trial received systemic BT. Lymphodepletion conditioning was administered by using flu/cy for 3 days prior to CAR T cell infusion.
CRS of any grade (according to Lee et al[12]) occurred in 93% of the study participants, with 13% developing grade 3 or higher CRS.[2] The median time to onset of CRS was 2 days (range, 1–12) and median time to resolution, 8 days. All CRS events resolved except for one person who developed grade 5 HLH. Neurologic events occurred in 64% of patients, with 28% developing grade 3 or higher neurologic toxicities (according to CTCAE).[2] The median time to onset of neurotoxicity was 5 days (range, 1–17) and median time to resolution, 17 days. Tocilizumab was administered to 43% of patients and 27% received corticosteroids.
The response rates from the trial were astounding. The best overall response rate (ORR) was 82%, with 54% achieving a complete response (CR).[2] The median duration of response was 11.1 months. The median OS was 25.8 months and the median PFS was 5.9 months. The remarkable results from ZUMA-1 set the precedent for the FDA approval of future adoptive cellular therapy for aggressive NHLs.
Real-World Experience
Several groups have conducted multicentered postmarket studies of axi-cel for LBCL and have reported similar results to ZUMA-1 (see Table 2).[22,41] A consortium of 17 institutions in the United States, the US CAR T Consortium, performed a retrospective analysis evaluating the clinical outcomes of 298 patients treated with standard-of-care (SOC) axi-cel for r/r LBCL.[22] Patients had a median age of 60 years (range, 21–83 years). This included patients with poor PS, ECOG score 2–4 (19.5%), disease stage III–IV (82.4%), and IPI score 3–5 (54.4%). In the real world, axi-cel was used in patients with DLBCL (68.1%), PMBCL (6.4%), and tFL (25.5%). Of these, 22.8% had double- or triple-hit lymphoma, and 37.4% were double expressors; 43% would have failed to qualify for ZUMA-1 owing to their baseline characteristics: common reasons included poor PS, thrombocytopenia, recent thrombosis, and history of CNS lymphoma.
Table 2.
Response rates of ZUMA-1 and real-world experiences of axicabtagene ciloleucel
|
|
ZUMA-12 (n
= 111 ) |
Nastoupil et al22 (n
= 298 ) |
Jacobson et al41 (n
= 122) |
Pasquini et al42 (n
= 295) |
| Overall response, % | 82 | 82 | 70 | 70 |
| Complete response, % | 54 | 64 | 50 | 52 |
Over half of the patients (53%) treated with SOC axi-cel received BT of any kind.[7] In this cohort, BT included chemotherapy with or without other therapies (54%), corticosteroids alone (23%), radiation with or without corticosteroids (12%), and targeted therapies (10%). Similar to previous studies, OS was worse in the group of patients that received BT. However, this may be a result of pretreatment factors rather than the result of the BT alone. A greater percentage of patients who received BT (30%) had worse PS than those who did not receive BT (8%). Moreover, patients who received BT (69%) were more likely to have higher IPI scores than those who did not receive BT (37%). Although there is no preferred approach for BT, radiation therapy may result in improved PFS when compared with systemic therapy in select patients.[8] In a study led by Pinnix et al,[8] BT was administered after leukapheresis to 115 patients who received axi-cel for aggressive r/r LBCL. Those who were bridged with radiation therapy had an improved median PFS of 8.9 months, compared with those bridged with systemic therapy whose median PFS was 4.7 months. The patients in the two groups did not differ in PS, disease burden, or lactate dehydrogenase. The authors did not find a difference in the OS between the radiation BT and systemic BT group. The heterogeneity in baseline characteristics and BT used makes it difficult to be definitive on the role of BT in CAR T cell therapy. Additional prospective studies are needed to better understand the role and define optimal BT strategies.
Despite differences in the baseline characteristics among patients prescribed SOC axi-cel, similar rates of toxicities were observed in comparison to ZUMA-1 (Figures 1 and 2).[22,41,42] In analyses conducted by the US CAR T Consortium, CRS of any grade occurred among 91% of patients; 7% developed grade 3 or higher CRS, and one patient died as a result of HLH.[22] Neurotoxicity occurred in 69% of patients, with grade 3 or higher occurring in 31%. One patient developed grade 5 cerebral edema. Tocilizumab and corticosteroids were given to 62% and 54% of patients for CRS, neurotoxicity, or both. In an independent analysis of the Center for International Blood and Marrow Transplant Research (CIBMTR) database, CRS was reported in 83% of patients and the incidence of grade 3 or higher CRS occurred in 11% of patients.[42] Two patients died from grade 5 CRS. Sixty-one percent of patients in this study developed neurotoxicity and one person died from grade 5 cerebral edema.
Figure 1.
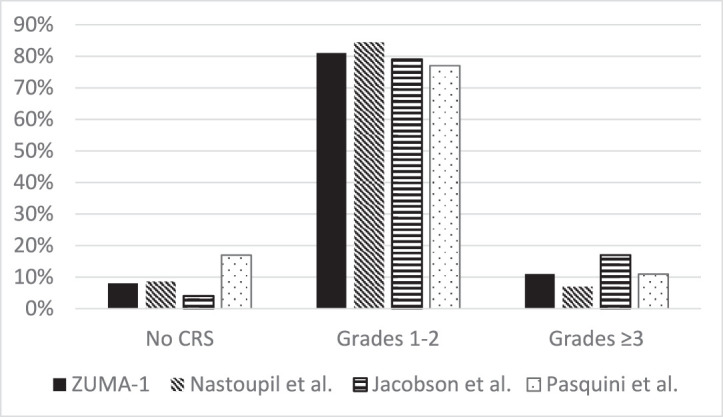
Cytokine release syndrome rates in ZUMA-1 and real-world experiences of axicabtagene ciloleucel. ZUMA-1 graded according to Lee et al12 and CTCAE version 4.03; Nastoupil et al, according to Lee et al12; Jacobson et al, according to Lee et al12; Pasquini et al, according to ASTCT.13 CRS: cytokine release syndrome; CTCAE: Common Terminology Criteria for Adverse Events; ASTCT: American Society for Transplantation and Cellular Therapy.
Figure 2.
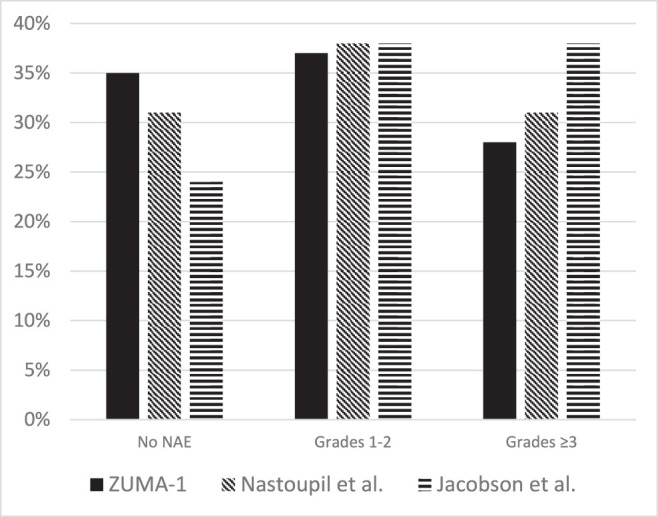
Neurologic adverse events in ZUMA-1 and real-world experiences of axicabtagene ciloleucel. ZUMA-1 graded according to CTCAE version 4.03; Nastoupil et al graded according to CTCAE version 4.03 and CARTOX14; Jacobson et al graded according to CTCAE version 4. CTCAE: Common Terminology Criteria for Adverse Events; CARTOX: CAR T-cell-therapy-associated TOXicity.
Efficacy observed with SOC axi-cel has been comparable to the ZUMA-1 trial results.[22,41,42] The US CAR T Consortium found an ORR of 82% among patients who received SOC axi-cel for LBCL with 64% achieving a CR.[22] The median PFS was 7.2 months from leukapheresis. OS and median duration of response were not yet reached at 13 months of median follow-up. In a separate multicenter analysis, Jacobson et al[41] reported an ORR of 70% and CR of 50% among 122 patients treated at seven US centers. The results from the CIBMTR registry reported 70% ORR with 52% of patients achieving a CR.[42] This is notable given the larger sample size and greater heterogeneity among SOC practices.
TISA-CEL
JULIET Clinical Trial
Prior to approval for r/r LBCL, tisa-cel was approved for use in r/r B cell precursor acute lymphoblastic leukemia (ALL), up to the age of 25 years. Following the results of JULIET, tisa-cel received FDA approval in 2018 through priority review and orphan product designation for r/r LBCL. The baseline characteristics of this study included median age of 56 years (range, 22–76), disease stage III–IV (76%), and three or more previous lines of antineoplastic therapy in 52%.[3] The JULIET trial enrolled patients with both relapsed (45%) and refractory disease (55%) with DLBCL (79%), and tFL (19%). PMBCL was excluded from this study. Similar to ZUMA-1, patients with CNS involvement and with HIV or HBV infections were excluded. Systemic BT was allowed and given to 92% of patients. BT included combinations of rituximab (54%), gemcitabine (40%), etoposide (26%), dexamethasone (25%), cisplatin (19%), cytarabine (19%), ibrutinib (9%), and lenalidomide (7%). Options for lymphodepletion chemotherapy included flu/cy (73%) or bendamustine (20%).
CRS was experienced by 58% of patients, with 22% grade 3 or higher.[3] Adverse neurologic events occurred in 64% of patients, with 28% grade 3 or higher. Tocilizumab alone (14%) or tocilizumab with corticosteroids (10%) was administered to patients who developed worsening CRS or neurologic toxicities despite initial supportive measures (i.e., low-flow nasal cannula, low-dose vasopressors, antipyretics). Cytopenias of any grade that persisted past day 28 occurred in 44% of patients. Grade 3 or higher cytopenias that persisted past day 28 occurred in 32% of patients.
The best ORR was 52% and 40% of participants had a CR.3 The median OS among patients who received tisa-cel was 12 months. The median PFS among patients who had a CR was not reached. Similar to ZUMA-1, approximately 40% of patients had a durable response.
Real-World Experience
A multicenter analysis of the CIMBTR registry reported outcomes among patients who received SOC tisa-cel, including 155 with r/r NHL (see Table 3).[43] Median age was 65 years (range, 18–89), 54% were 65 years of age or older. Most had r/r disease (95%), though seven patients (5%) proceeded with tisa-cel in the setting of CR. Seventeen (11%) had double- or triple-hit features, and 27% had tFL. Median number of prior therapies was 4 (range, 0–11). Any grade CRS occurred in 45%, grade 3 or higher occurred in 4.5%. Any grade ICANS occurred in 18%, grade 3 or higher occurred in 5.1%. The response rates in the real world were comparable to those of the JULIET trial. ORR was 62% including 40% achieving a CR. Tocilizumab and corticosteroids were administered in 43% and in 10%, respectively. A separate multicenter analysis by Riedell et al[44] also found comparable response rates (ORR: 59%; CR: 44%) with low rates of severe toxicities (CRS grade ≥ 3: 1%; ICANS grade ≥ 3: 3%).
Table 3.
Response rates of JULIET and real-world experiences of tisagenlecleucel
|
|
JULIET3 (n
= 115 ) |
Pasquini et al43 (n
= 152 ) |
Riedell et al44 (n
= 79) |
| Overall response, % | 52 | 62 | 48 |
| Complete response, % | 40 | 40 | 39 |
Both flu/cy and bendamustine conditioning regimens have been used prior to infusion with tisa-cel. In a retrospective analysis of patients with r/r LBCL treated w/ tisa-cel at the University of Pennsylvania, 28 patients received bendamustine for lymphodepletion conditioning.[45] The 3-month ORR in this group was 46%, with a CR in 38% of patients. The 3-month PFS was 52%. At day 28, 11% of patients had grade 3 or higher neutropenia and 11% had grade 3 or higher thrombocytopenia. Although flu/cy is the most common regimen used for lymphodepletion conditioning, this small study suggests that bendamustine is a potential alternative for lymphodepletion chemotherapy for SOC tisa-cel.
LISO-CEL
TRANSCEND Clinical Trial
Liso-cel is the third FDA-approved autologous CD19 CAR T cell therapy for the treatment of LBCL, based on the TRANSCEND study.[1] This study was the broadest and largest among the three pivotal studies. The median age of the 269 patients enrolled was 63 years (range, 54–70), 42% were 65 years of age or older, and 10% were 75 years of age or older. [1] Four patients had an ECOG PS of 2 (1%). In addition to DLBCL not otherwise specified (51%), tFL (22%), and PMBCL (6%), the TRANSCEND study enrolled patients with grade 3B follicular lymphoma (1%) and transformed lymphoma from other indolent NHLs (7%). Secondary CNS lymphoma was not an exclusion criterion and was present in 3%. Two-third of cases were refractory to the last chemotherapy-containing regimen given. Median number of prior therapies was 3 (range, 2–4). BT (systemic therapy, radiation therapy, or both) was allowed at investigator discretion and given to 59% of patients. All patients received flu/cy lymphodepletion conditioning.
As mentioned previously, CD8+ and CD4+ T cells are independently manufactured and were administered in two sequential infusions of CD8+ followed by CD4+ CAR T cells.[1] Twenty-five patients received liso-cel infusion as outpatients. Any grade CRS occurred in 42%, and grade 3 or higher CRS occurred in 2% of patients. The median time to onset of CRS was 5 days (range, 1–14 days) and a median duration of 5 days (range, 1–17 days). Neurological events occurred in 30% of patients and grade 3 or higher occurred in 10%. The median time to onset of neurological events was 9 days (range, 1–66 days), with the median time to resolution of 11 days (range, 1–86 days). Treatment of CRS with tocilizumab only occurred in 10% of patients, tocilizumab and corticosteroids in 8% of patients, and corticosteroids only in 2% of patients. Among the 25 patients who received liso-cel in the outpatient setting, 18 patients were hospitalized for AEs, including 10 patients who were hospitalized for CRS, neurological events, or both.
In the TRANSCEND trial, treatment of r/r LBCL with liso-cel had outstanding response rates - 75% achieved an ORR, with 53% achieving a CR.1 Median OS of the study was 21.1 months and median PFS was 6.8 months. Patients with bulky disease, as measured by tumor volume, were less likely to achieve a CR.
Real-world data do not currently exist for liso-cel owing to its recent approval at the time of this publication. However, the TRANSCEND trial notably had broader eligibility criteria than JULIET or ZUMA-1, including more generous eligibility criteria such as secondary CNS involvement, ECOG PS 0–2, mild renal insufficiency, and mild depressed ejection fraction, representing important subgroups in real-world care of LBCL patients. TRANSCEND had a higher number of patients older than 65 years, 42% compared with 24% and 23% enrolled in ZUMA-1 and JULIET, respectively. This likely resulted from observations of less severe acute toxicity with liso-cel. Patients with older age and high-risk features did not have significantly different outcomes in comparison to the rest of the study population, suggesting liso-cel may have broader application in the real world. Real-world analyses are eagerly awaited.
SPECIAL POPULATIONS
Secondary CNS Lymphoma
The safety and efficacy of CD19 CAR T cell therapy for the management of secondary CNS lymphoma has not yet been well characterized owing to small number of patients treated to date. The TRANSCEND study included seven patients with secondary CNS lymphoma who received liso-cel.[1] In the efficacy-evaluable analysis of the patients in TRANSCEND who obtained a PET-CT (positron emission tomography–computed tomography) prior to liso-cel infusion, three of six patients with secondary CNS lymphoma achieved a CR. Two of the seven patients with secondary CNS lymphoma developed ICANS.[1] Further studies are needed to evaluate the safety and efficacy of CD19 CAR T cell therapy for the treatment of r/r LBCL with CNS involvement given that this population was excluded from the ZUMA-1 and JULIET trials, and the small number of patients with CNS involvement in the TRANSCEND trial makes it challenging to draw strong conclusions.
Much of the safety and efficacy data in the real-world use of axi-cel for treatment of r/r LBCL with CNS involvement are described in small studies and single institutional experiences. Ghafouri et al[46] reported early responses with SOC axi-cel used in patients with secondary CNS lymphoma among four of five patients infused. Unfortunately, three of the four responders had disease progression. The median PFS was 134 days and median OS was 155 days. Two patients developed CRS, grade 1–2. Two patients developed ICANS. The first patient developed ICANS grade 3 without concurrent CRS, whereas the second patient developed ICANS grade 4 with concurrent grade 2 CRS. Both of the patients were treated with corticosteroids and the patient with grade 4 ICANS received dual antiepileptics for treatment of status epilepticus. None of the patients had long-term neurological AEs. The authors concluded the toxicity appeared comparable to that of patients without secondary CNS lymphoma, but the responses were not durable.
The US Lymphoma CAR T Consortium reported axi-cel resulted in similar response rates in patients with secondary CNS lymphoma (n = 17) when compared with patients with systemic disease only (n = 281).[22] The best ORR between the CNS and non-CNS cohorts was 75% vs 59%, respectively, and ongoing response rates at 6 months were comparable at 41% and 31%, respectively.[47] The incidence of CRS and ICANS of any grade was comparable between the patients with CNS involvement and those without CNS involvement.
Frigault et al[48] reported their single center experience with tisa-cel in secondary CNS lymphoma. Four of their eight patients who received tisa-cel achieved an objective response. Two patients who did not achieve an objective response died from disease progression. None of the patients developed ICANS and seven of eight patients developed grade 1 CRS. Based on these observations, a prospective study is underway exploring tisa-cel in primary CNS lymphoma (NCT04134117).
Chronic Viral Hepatitis
Immunosuppression and cytotoxic therapies can cause HBV reactivation from chronic or resolved HBV infections. The mechanism of HBV reactivation is not clearly understood; however, patients who receive rituximab chemotherapy, an anti-CD20 monoclonal antibody, are at the highest risk. Prior to rituximab administration, patients are started on antiviral drugs to decrease the risk of reactivation. Unsurprisingly, there have been reports of HBV reactivation during CD-19 CAR T cell treatment. Strati et al[49] published their experience with three patients with chronic viral hepatitis: two patients with chronic or resolved hepatitis B and one patient with hepatitis C virus (HCV). The patients with HBV had a low HBV DNA titer (< 10 IU/mL) and were given prophylatic antiviral therapy. Both patients developed CRS and neurotoxicity that eventually resolved and both patients achieved a CR that was ongoing for 8 and 31 months, respectively. No significant viral reactivation occurred during either patient's acute toxicities. The patient with resolved HBV infection self-discontinued her prophylaxis antiviral 13 months after axi-cel infusion and had HBV reactivation (79 million IU/mL) 3 months afterwards. Her HBV reactivation was successfully treated with re-initiation of antivirals. The last patient in the report had chronic HCV that was refractory to interferon and ribavirin therapy 25 years before presentation. At the time of evaluation, the patient's HCV RNA level was 15.1 million IU/mL and alanine aminotransferase was 70 U/L. His axi-cel treatment was complicated by grade 3 CRS and grade 3 ICANS that resolved. Interestingly, there was no significant increase in the patient's HCV RNA or liver function tests. The patient achieved a CR that was ongoing at 6 months.
HIV Infection
Infection with HIV significantly increases the likelihood of developing aggressive NHL.[50] The incidence of DLBCL is 17-fold higher in individuals infected with HIV than in those who are not infected. In addition, HIV-related lymphomas have higher rates of unfavorable subtypes, such as double- or triple-hit and primary CNS lymphoma.[42] Fortunately, recent case reports have suggested that CD19 CAR T cells have led to durable responses in AIDS-associated, high-grade B-cell lymphoma.[51] Abramson et al[51] reported on two HIV-infected patients with r/r high-grade NHL who achieved CR after treatment with axi-cel. The first patient was a 22-year-old man with HIV on antiviral retroviral therapy intermittently who developed r/r high-grade B cell lymphoma with rearrangements of MYC and BCL6. After his second relapse, the patient was evaluated for CAR T cell candidacy on the condition that he adhere to his ART. The patient's CD4 count was 52 cells/mm3, absolute lymphocyte count 450 cells/mm3, and HIV viral load count 67 copies/mL at the time of apheresis. He underwent lymphodepletion therapy with flu/cy followed by infusion of axi-cel. The patient subsequently developed grade 2 CRS and grade 3 ICANS with confusion, somnolence, and expressive aphasia. He was treated with tocilizumab, dexamethasone, and seizure prophylaxis with lacosamide. By the time of discharge, the patient's CRS and ICANS resolved. PET-CT 2 months after treatment showed a durable CR that was ongoing at 1 year after infusion. The second patient was a male person with a history of well-controlled HIV (viral load undetectable, CD4 count 127), prior HBV, cytomegalovirus, and Mycobacterium avium complex infections, and being treated for DLBCL. The patient was a poor candidate for high-dose chemotherapy. He received dose-reduced flu/cy for lymphodepletion conditioning followed by axi-cel infusion. The patient's treatment course was uncomplicated; he did not develop CRS or ICANS. He achieved a CR at day 28.
Solid Organ Transplant Recipients
Recipients of solid organ transplant are at increased risk of developing posttransplant lymphoproliferative disorder.[52] Given many of these patients receive long-term immune suppression to prevent allograft rejection, the feasibility of pursuing an effective autologous CAR T cell therapy is unknown, and these patients are excluded from prospective studies. A single-center experience reports the outcomes of three patients with r/r DLBCL and kidney allograft treated with SOC axi-cel.[53] Two of the three patients achieved a CR at day 30, but only one maintained that response. Two of the three developed CRS, one of these patients also developed concurrent ICANS. One of the three had no CRS or ICANS. The authors report the feasibility of discontinuing immunosuppressive therapy 2–4 weeks before leukapheresis and re-initiating 4–12 weeks post axi-cel treatment. This is a very small, single-center experience, but further study is warranted.
CONCLUSION
The real-world outcomes of CD19 CAR T cell therapy for aggressive r/r NHL result in response and survival rates that rival the remarkable results from pivotal trials. This is surprising given that many of these patients would have been excluded from the clinical trials because of their worse functional status and comorbidities. Additionally, the rates of adverse events associated with CAR T cell therapy were similar among real-world practice and clinical trials. This suggests that eligibility criteria for pivotal studies should be reexamined, as we may be excluding patients who may benefit from CAR T cell therapy. Lastly, progression after CAR T cell therapy may indicate a highly aggressive disease course and a poor prognosis. Early identification and referral for CAR T cell therapy is critical. Further research is needed to enhance our understanding of the mechanisms of resistance and identification of optimal candidates.
Funding Statement
Sources of Support: None.
Footnotes
Conflict of Interest: Kevin Tang: None. Loretta J. Nastoupil has received honoraria from ADC Therapeutics, Bayer, BMS/Celgene, Epizyme, Genentech, Gilead/Kite, Janssen, Novartis, Morphosys, Pfizer, TG Therapeutics. She has also received research support from BMS/Celgene, Epizyme, Genentech, Gilead/Kite, Janssen, Merck, Novartis, Pfizer, Takeda, TG Therapeutics.
References
- 1.Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet . 2020;396:839–852. doi: 10.1016/S0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 2.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med . 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med . 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 4.Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med . 2020;382:1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine BL, Miskin J, Wonnacott K, et al. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev . 2017;4:92–101. doi: 10.1016/j.omtm.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tyagarajan S, Spencer T, Smith J. Optimizing CAR-T cell manufacturing processes during pivotal clinical trials. Mol Ther Methods Clin Dev . 2020;16:136–144. doi: 10.1016/j.omtm.2019.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain MD, Jacobs M, Nastoupil LJ, et al. Characteristics and outcomes of patients receiving bridging therapy while awaiting manufacture of standard of care axicabtagene ciloleucel CD19 chimeric antigen receptor (CAR) T-cell therapy for relapsed/refractory large B-cell lymphoma. Blood . 2019;134(suppl 1):245. [Google Scholar]
- 8.Pinnix CC, Gunther JR, Dabaja BS, et al. Bridging therapy prior to axicabtagene ciloleucel for relapsed/refractory large B-cell lymphoma. Blood Adv . 2020;4:2871–2883. doi: 10.1182/bloodadvances.2020001837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wrzesinski C, Paulos CM, Kaiser A, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother . 2010;33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirayama AV, Gauthier J, Hay KA, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood . 2019;133:1876–1887. doi: 10.1182/blood-2018-11-887067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Innamarato P, Kodumudi K, Asby S, et al. Reactive myelopoiesis triggered by lymphodepleting chemotherapy limits the efficacy of adoptive T cell therapy. Mol Ther . 2020;28:2252–2270. doi: 10.1016/j.ymthe.2020.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood . 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector Cells. Biol Blood Marrow Transplant . 2019;25:625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PubMed] [Google Scholar]
- 14.Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy: assessment and management of toxicities. Nat Rev Clin Oncol . 2018;15:47–62. doi: 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park JH, Riviere I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med . 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xhangolli I, Dura B, Lee G, et al. Single-cell analysis of CAR-T cell activation reveals a mixed TH1/TH2 response independent of differentiation. Genomics Proteomics Bioinformatics . 2019;17:129–139. doi: 10.1016/j.gpb.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen PH, Lipschitz M, Weirather JL, et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight . 2020;5:e134612. doi: 10.1172/jci.insight.134612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med . 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 19.van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov . 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood . 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le RQ, Li L, Yuan W, et al. FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist . 2018;23:943–947. doi: 10.1634/theoncologist.2018-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nastoupil LJ, Jain MD, Feng L, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US Lymphoma CAR T Consortium. J Clin Oncol . 2020;38:3119–3128. doi: 10.1200/JCO.19.02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oluwole O, Munoz J, Vose JM, et al. Prophylactic steroid use with axicabtagene ciloleucel in patients with relapsed/refractory large b-cell lymphoma. Abstract 70. Presented at 2021 Transplantation and Cellular Therapy Meetings; Feb 8–12 2021.
- 24.Liu S, Deng B, Yin Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J . 2020;10:15. doi: 10.1038/s41408-020-0280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strati P, Ahmed S, Furqan F, et al. Prognostic impact of corticosteroids on efficacy of chimeric antigen receptor T-cell therapy in large B-cell lymphoma. Blood . 2019;133:2800–2802. doi: 10.1182/blood.2020008865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taraseviciute A, Tkachev V, Ponce R, et al. Chimeric antigen receptor T cell-mediated neurotoxicity in nonhuman primates. Cancer Discov . 2018;8:750–763. doi: 10.1158/2159-8290.CD-17-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taraseviciute A, Broglie L, Phelan R, et al. What is the role of hematopoietic cell transplantation (HCT) for pediatric acute lymphoblastic leukemia (ALL) in the age of chimeric antigen receptor T-cell (CART) therapy? J Pediatr Hematol Oncol . 2019;41:337–344. doi: 10.1097/MPH.0000000000001479. [DOI] [PubMed] [Google Scholar]
- 28.Hu Y, Sun J, Wu Z, et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J Hematol Oncol . 2016;9:70. doi: 10.1186/s13045-016-0299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gust J, Taraseviciute A, Turtle CJ. Neurotoxicity associated with CD19-targeted CAR-T cell therapies. CNS Drugs . 2018;32:1091–1101. doi: 10.1007/s40263-018-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santomasso BD, Park JH, Salloum D, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov . 2018;8:958–971. doi: 10.1158/2159-8290.CD-17-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng Q, Han G, Puebla-Osorio N, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med . 2020;26:1878–1887. doi: 10.1038/s41591-020-1061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parker KR, Migliorini D, Perkey E, et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell . 2020;183:126–142. doi: 10.1016/j.cell.2020.08.022. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strati P, Nastoupil LJ, Westin J, et al. Clinical and radiologic correlates of neurotoxicity after axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv . 2020;4:3943–3951. doi: 10.1182/bloodadvances.2020002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giavridis T, van der Stegen SJC, Eyquem J, et al. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med . 2018;24:731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sterner RM, Sakemura R, Cox MJ, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood . 2019;133:697–709. doi: 10.1182/blood-2018-10-881722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol . 2019;20:31–42. doi: 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cordeiro A, Bezerra ED, Hirayama AV, et al. Late events after treatment with CD19-targeted chimeric antigen receptor modified T ells. Biol Blood Marrow Transplant . 2020;26:26–33. doi: 10.1016/j.bbmt.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strati P, Varma A, Adkins S, et al. Hematopoietic recovery and immune reconstitution after axicabtagene ciloleucel in patients with large B-cell lymphoma. Haematologica . 2020. Published online Jul 30. [DOI] [PMC free article] [PubMed]
- 39.Galli E, Allain V, Di Blasi R, et al. G-CSF does not worsen toxicities and efficacy of CAR-T cells in refractory/relapsed B-cell lymphoma. Bone Marrow Transplant . 2002;55:2347–2349. doi: 10.1038/s41409-020-01006-x. [DOI] [PubMed] [Google Scholar]
- 40.Hashmi H, Bachmeier C, Chavez JC, et al. Haemophagocytic lymphohistiocytosis has variable time to onset following CD19 chimeric antigen receptor T cell therapy. Br J Haematol . 2019;187:e35–e38. doi: 10.1111/bjh.16155. [DOI] [PubMed] [Google Scholar]
- 41.Jacobson CA, Hunter BD, Redd R, et al. Axicabtagene ciloleucel in the non-trial setting: outcomes and correlates of response, resistance, and toxicity. J Clin Oncol . 2020;38:3095–3106. doi: 10.1200/JCO.19.02103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pasquini MC LF, Herrera AF, et al. Post-marketing use outcomes of an anti-CD19 chimeric antigen receptor (CAR) T cell therapy, axicabtagene ciloleucel (Axi-Cel), for the treatment of large B cell lymphoma (LBCL) in the United States (US) Blood . 2019;134(suppl 1):764. [Google Scholar]
- 43.Pasquini MC, Hu ZH, Curran K, et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv . 2020;4:5414–5424. doi: 10.1182/bloodadvances.2020003092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riedell RA, Walling C, Nastoupil LJ, et al. A multicenter retrospective analysis of outcomes and toxicities with commercial axicabtagene ciloleucel and tisagenlecleucel for relapsed/refractory aggressive B-cell lymphomas. 2020;26:S41–S42. [Google Scholar]
- 45.Svoboda J, Hatcher JB, Chong EA. Use of bendamustine for lymphodepletion before tisagenlecleucel (anti-CD19 CAR T cells) for aggressive B-cell lymphomas. Blood . 2019;134(suppl 1):1606. [Google Scholar]
- 46.Ghafouri S, Timmerman J, Larson S, et al. Axicabtagene ciloleucel CAR T-cell therapy for relapsed/refractory secondary CNS non-Hodgkin lymphoma: comparable outcomes and toxicities, but shorter remissions may warrant alternative consolidative strategies? Bone Marrow Transplant . 2021;56:974–977. doi: 10.1038/s41409-020-01099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bennani NN, Maurer M, Nastoupil LJ, et al. Experience with axicabtagene ciloleucel (Axi-cel) in patients with secondary CNS involvement: results from the US Lymphoma CAR T Consortium. Blood . 2019;134:763. [Google Scholar]
- 48.Frigault MJ, Dietrich J, Martinez-Lage M, et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood . 2019;134:860–866. doi: 10.1182/blood.2019001694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strati P, Nastoupil LJ, Fayad LE, et al. Safety of CAR T-cell therapy in patients with B-cell lymphoma and chronic hepatitis B or C virus infection. Blood . 2019;133:2800–2802. doi: 10.1182/blood.2019000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gibson TM, Morton LM, Shiels MS, et al. Risk of non-Hodgkin lymphoma subtypes in HIV-infected people during the HAART era: a population-based study. AIDS . 2014;28:2313–2318. doi: 10.1097/QAD.0000000000000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abramson JS, Irwin KE, Frigault MJ, et al. Successful anti-CD19 CAR T-cell therapy in HIV-infected patients with refractory high-grade B-cell lymphoma. Cancer . 2019;125:3692–3698. doi: 10.1002/cncr.32411. [DOI] [PubMed] [Google Scholar]
- 52.Dierickx D, Habermann TM. Post-transplantation lymphoproliferative disorders in adults. N Engl J Med . 2018;378:549–562. doi: 10.1056/NEJMra1702693. [DOI] [PubMed] [Google Scholar]
- 53.Mamlouk O, Nair R, Iyer SP, et al. Safety and efficacy of CAR T-cell therapy in kidney transplant recipients. Blood . 2021;137:2558–2562. doi: 10.1182/blood.2020008759. [DOI] [PMC free article] [PubMed] [Google Scholar]