

Abstract
The continual development of potent immunosuppressive regimens has led to decreased incidence of acute rejection and improvement of short-term kidney allograft survival. In contrast to acute rejection, glomerular diseases of the kidney allograft are being encountered more frequently and are emerging as leading causes of late kidney allograft failure.
While data on the pathogeneses of glomerular diseases in the kidney allograft are sparse, cumulative evidence suggests that post-transplant glomerular diseases may be the result of inherited predispositions and immunologic triggers. Whereas studying immunologic signals and performing genome-wide association studies are ideal approaches to tackle glomerular diseases in the kidney allograft, such studies are challenging due to the lack of adequately powered cohorts.
In this review, we will focus on the most commonly encountered recurrent and de novo glomerular diseases in the kidney allograft. We will address the important advances made in understanding the immunopathology and genetic susceptibility of glomerular diseases in the native kidney and how to benefit from such knowledge to further our knowledge of post-transplant glomerular diseases.
Defining genomic and immune predictors for glomerular diseases in the kidney allograft would support novel donor-recipient matching strategies and development of targeted therapies to ultimately improve long-term kidney allograft survival.
Keywords: Kidney transplantation, Glomerular diseases, Immunopathology, Genomic
INTRODUCTION
Kidney transplantation, which is considered the ultimate treatment of renal failure 1, has seen a dramatic improvement in allograft survival over the past two decades 2. This success has been attributed mainly to enhancements in one-year kidney allograft survival 3. Long-term allograft survival has improved as well, though at a much slower rate as compared to one-year survival 4. Despite these advancements, 10-year death-censored allograft failure rates are 26% and 18% for deceased and living donor kidneys, respectively 2. As a result, allograft failure constitutes the 4th most common cause of kidney failure in the United States 5.
Several mechanisms involved in long-term allograft loss have been identified. These include alloimmune factors such as human leukocyte antigen (HLA) mismatch (in particular HLA class-II mismatches) 6, which are associated with the development of de novo donor-specific antibodies (DSA) that increase the likelihood of allograft rejection 7. The advent of rigorous HLA matching of donor-recipient pairs 8, together with the improvement in induction and maintenance immunosuppression therapies, has been crucial in decreasing the incidence of rejection and prolonging allograft survival 9. This has led to a significant drop in the rates of acute rejection in the first year post-transplantation from 50-60% in the 1980s to 10% in the current era 10.
However, as allograft survival has improved, non-alloimmune factors have surfaced as important contributors to long-term allograft failure. Glomerular diseases in specific are gaining attention in recent years 11. Data from worldwide registries indicates that glomerular diseases constitute one of the main causes of native kidney failure in kidney transplant recipients. In the United States, 27% of kidney transplant recipients have glomerular diseases as the underlying cause of kidney failure 12. Nevertheless, these numbers are thought to underrepresent the true prevalence of native kidney glomerular diseases in transplant recipients given that many patients do not undergo native kidney biopsies prior to transplantation. Additionally, glomerular diseases are the second most common cause of allograft failure in kidney transplant recipients 11,13. The incidence of glomerular diseases increases the further the patient is out from transplant, with one report citing the cumulative incidence of glomerular diseases to be as high as 42% at 10 years post-transplantation 14.
Glomerular diseases in the kidney allograft can present in three forms: donor-derived, recurrent or de novo. Recurrent glomerular diseases are the most commonly encountered glomerular diseases post-transplantation 15 and appears to impact allograft survival to at least the same degree as an episode of acute rejection 16. Importantly, different primary glomerular diseases have different rates of recurrence; for example, C3 glomerulonephritis recurs in greater than 90% of recipients 17, IgA nephropathy (IgAN) and membranous nephropathy (MN) recurs in up to 50% of recipients 14,18, and primary focal segmental glomerulosclerosis (FSGS) recurs in up to 32% of recipients 19. This variability is presumed to be due to an interplay of recipient and donor genetic susceptibilities with non-genetic factors such as immunologic triggers. Compared to recurrent glomerular diseases, the prevalence, pathogenesis, and impact of de novo glomerular diseases, is even less understood.
Our focus is to review the current state of knowledge on recurrent and de novo glomerular diseases in the allograft, with a particular focus on recurrent primary IgAN, MN and FSGS , as well de novo collapsing glomerulopathy and immune complex-mediated glomerulonephritis (ICMGN). We delve into each of them individually with a special emphasis on the application of precision medicine approaches to further our understanding of these glomerular diseases.
IGA NEPHROPATHY (IGAN)
Primary IgAN has an estimated incidence of 2.5 cases per 100,000 adults per year 20. Common clinical presentation includes micro-hematuria, sub-nephrotic proteinuria, and systemic hypertension 21. Adults often present with a smoldering clinical course, hypertension, and slow progression of chronic kidney disease 21. The disease progresses to renal failure in 40% of cases over 20 years.
Established clinical risk factors for worse renal outcome, as in other kidney diseases, are proteinuria, lower glomerular filtration rate (GFR) at diagnosis and hypertension 22. Conservative therapy with high-dose renin angiotensin system blockade and blood pressure control is a well-established first-line treatment in most individuals affected by IgAN, since it effectively reduces proteinuria and slows the rate of renal function decline 23. The use of immunosuppressants is reserved for patients at high risk for rapid progression of renal disease 24.
Immunopathology
IgAN has been linked to certain bacterial and viral infections 25 and is believed to be the result of a multi-hit pathogenic model 26. The first hit is represented by a defects in glycosylation on the hinge region of IgA1, which leads to the secretion of polymeric galactose-deficient IgA1 (Gd-IgA1) by plasma cells 27. Poorly glycosylated IgA1 molecules constitute a new epitope that triggers the secretion of IgG autoantibodies (second hit) to form large immune complexes in the circulation (third hit) 28. These immune complexes typically deposit in the mesangium and sometimes in the adjacent subendothelial space (forth hit) and induce activation of the alternative and lectin pathways of complement cascade 29. Histologically, such deposits can be detected by electron microscopy as granular electron dense deposits and by immunofluorescence as dominant typically polyclonal IgA staining and less intense C3 staining, with or without IgG staining, and typically without C1 staining or intense C4d staining 30.
Based on the amount and location of the deposits, the magnitude of local complement activation, and the strength of the host immune response, light microscopy assessment may reveal a spectrum of pathological lesions, ranging from no significant proliferation, to isolated mesangial hypercellularity, endocapillary leukocyte infiltration, and occasionally cellular crescents. Glomerulosclerosis, fibrous crescents, and tubulointerstitial scarring are seen in the late stages of the disease as manifestation of progressive kidney disease or healed acute lesions. Histologic classification systems, such as Oxford IgAN classification, were built based on the proposed histo-pathogenic model to assess prognosis 31. A composite prognostic score was later created using several clinical, biochemical and histopathological predictors and found to effectively predict 5-year risk of renal failure in patients with IgAN 32.
Genetic susceptibility
IgAN prevalence vary based on ancestry and geography. The disease is most prevalent in East Asians, has intermediate prevalence in Europeans, and is relatively rare in individuals of African ancestry 33. Although familial clustering of IgAN cases have suggested an inherited mechanism of disease 34, family-based studies have not identified specific causal genes to date. The latter raises the possibility that IgAN may have an oligogenic or polygenic architecture.
Strong associations between IgAN and multiple HLA antigens have been demonstrated 35–38. Genome-wide association studies (GWAS) 35–39 have identified HLA and non-HLA susceptibility loci, which have helped elucidate the multi-hit pathogenesis of IgAN. Such loci implicate defects in antigen presentation, complement cascades, intestinal IgA production, and innate immunity against mucosal pathogens.
The largest IgAN GWAS, leveraging upon multi-ethnic cohorts and a relatively large sample size, generated a genetic risk score (GRS), expressed as the sum of each of the 15 IgAN HLA and non-HLA risk alleles weighted on their effect sizes (IgAN 15-SNP GRS) 39. This GRS explained 6% of disease risk in European populations and 8% of disease risk in East Asian populations and correlated with lower age at disease onset and with helminth diversity across the globe. The latter association has given rise to the hypothesis that IgAN genetic risk may be the consequence of a protective adaptation against local pathogens.
So far, only a small fraction of IgAN heritability is explained by known susceptibility variants. This expresses the need for larger and more powerful multi-ethnic GWAS. The use of a genetic risk scores has the potential to stratify index cases and their family members and guide the clinician towards a better definition of disease prognosis.
Post-transplant IgAN
The morphologic manifestation of recurrent IgAN in the kidney allograft is similar to that encountered in the native kidney (Figure 1). However, in contrast to numerous recent insights on the pathogenesis, genetics, and prognosis of the disease in the native kidney, there is limited knowledge about risk factors and prognosis of recurrent IgAN post-transplantation.
Figure 1: Pathogenesis of recurrent IgAN.
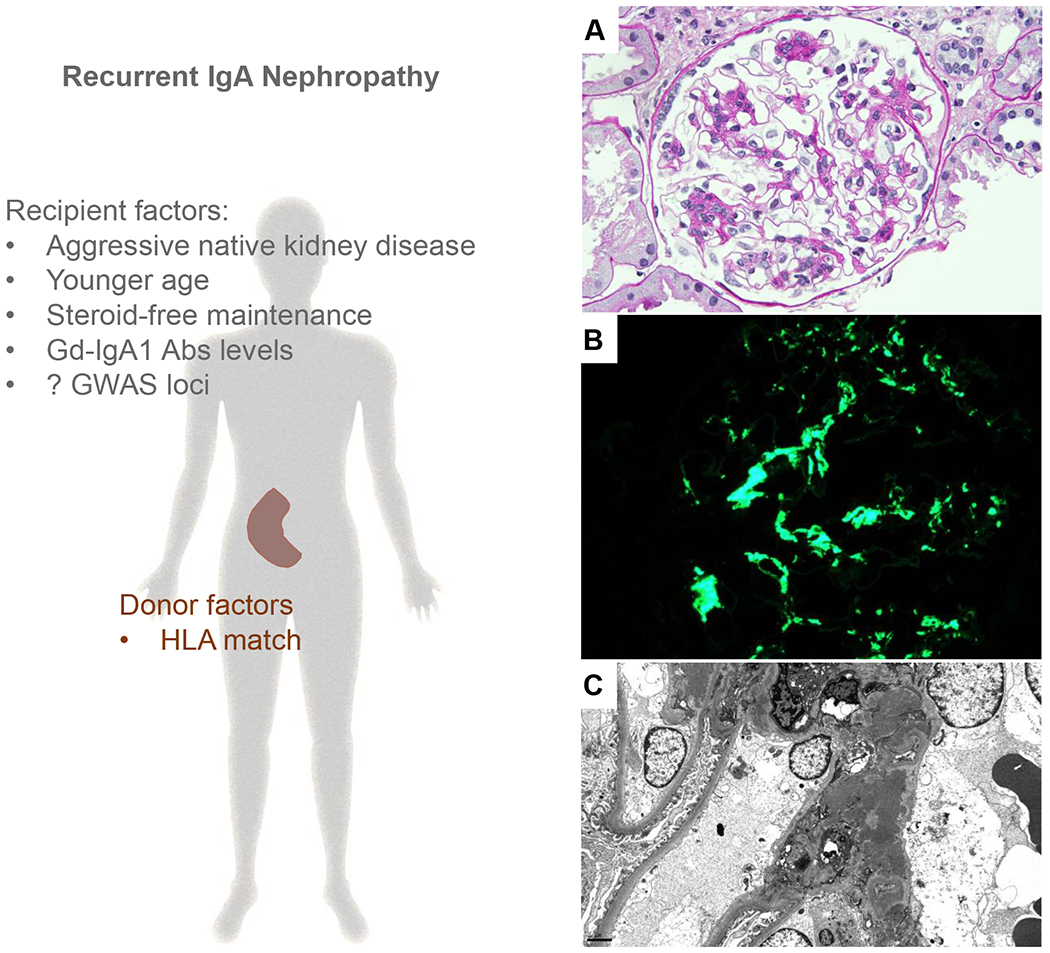
Left panel: Previous published reports have found that variables broadly related to recipients’ immune system status and donor inherited factors can predict recurrent IgAN. The genomic studies in the native kidney also suggest that recipient inherited factors can contribute to recurrent IgAN. Right panel: Representative photomicrographs of recurrent IgAN (A) A glomerulus showing mesangial expansion and proliferation (periodic acid–Schiff, original magnification ×400) (B) This was associated with global granular to confluent staining for IgA in the mesangium (immunofluorescence, original magnification, ×400) and (C) granular and often confluent mesangial electron dense deposits (electron microscopy, original magnification ×5,000).
Abbreviations: Abs, antibodies; HLA, human leukocyte antigen; Gd IgA1, Galactose-deficient IgA1; GWAS, genome wide association studies.
IgAN recurrence seems to negatively impact graft survival 15,40 and it is believed to be the third leading cause of graft loss at 10 years post-transplant after chronic allograft nephropathy and death with functioning graft in patients affected by IgAN in the native kidney 41. It has also been estimated that there is a 30% average risk of IgAN recurrence after transplantation, with a highly variable prevalence that ranges between 9 and 53% among different series 15,42,43. These wide discrepancies are mainly attributable to differences in biopsy indications and the length of post-transplant follow-up. Therefore, short follow-up time and lack of protocol biopsies can miss late cases of IgAN recurrence and IgAN recurrence in patients with slow and progressive deterioration of kidney function that does not trigger a kidney allograft biopsy.
Clinical factors associated with risk of IgAN recurrence after transplant are still not well defined. To date, risk factors of IgAN recurrence have only been studied in single-center retrospective observational studies or registry data. Single center studies suffer from small sample size, while studies based on registry data, although with higher statistical power, often lack complete clinical data and are more subject to selection and omitted variable bias.
Among recipients’ related factors, younger age 15,44 and crescents in the native kidney biopsy 45 are associated with higher risk of recurrent disease in the kidney allograft. A few studies suggested a protective role of steroid maintenance in preventing recurrent disease 46,47. Finally, elevated early post-transplant serum IgA, pre-transplant Gd-IgA1 antibodies, IgA-IgG complexes, and lower serum IgA-soluble CD89 complexes have been associated with higher risk of IgAN recurrence 48,49.
With regard to donor factors, while a few studies have reported that living donation was associated with higher risk of IgAN recurrence over time 50,51, higher recurrence rates have been consistently observed in zero HLA-mismatched grafts compared with ≥1 HLA-mismatched allografts 52,53.
In summary, it appears that recurrent IgAN is associated with recipient immune factors, donor-inherited factors, and probably recipient-inherited factors (as suggested from GWAS studies for IgAN in the native kidney) (Figure 1). To date, the role donor and recipient HLA subtypes have not been thoroughly examined in the context of recurrent IgAN. Similarly, the importance of IgAN risk variants at non-HLA loci studied. Genetic studies addressing these questions have a potential to provide better insights into the pathobiology of IgAN recurrence. The ultimate goals of such efforts would be to guide the clinician towards improving donor-recipient matching and develop targeted approaches to prevent IgAN recurrence and improve allograft survival.
MEMBRANOUS NEPHROPATHY (MN)
Primary Membranous nephropathy (MN) is a major cause of nephrotic syndrome in adults of European ancestry 54 and has an estimated annual incidence of 1.2 cases in 100,000 adults 20. While it can present at any age, the incidence of nephrotic syndrome reaches a peak between 30 and 50 years of age 55. The typical clinical phenotype is that of a nephrotic syndrome, often characterized by slow progression of edema over weeks or months. Microhematuria occurs in 30% of cases and hypertension in 10% 56. MN is characterized by progressive disease course with deterioration in renal function in 30% of cases, of which 50% can reach renal failure in 10-15 years, especially if left untreated 55. Treatment of MN includes supportive therapy for the management of MN-related complications (such as proteinuria, edema, deep vein thrombosis, hyperlipidemia, hypertension) and immunosuppressive therapy, which includes B-cell therapy (e.g. rituximab), calcineurin inhibitors, and steroids sometime in combination with alkylating agents 56. However, it is still debated who would benefit from immunosuppressive therapy, the timeline for its initiation, or the type of specific regimen to employ.
Immunopathology
MN is often primary (80%) 56 and the majority of primary MN cases are associated with circulating pathogenic autoantibodies against phospholipase A2 receptor (PLA2R, detected in 60-70% of MN cases), or thrombospondin type-1 domain- containing 7A (THSD7A, detected in 5% of anti-PLA2R negative cases) 57. Although specific triggers are still not completely known, it is plausible that dysregulation of B-cell activation and/or regulatory T cell function may contribute to MN 58,59.
In case of PLA2R-associated MN, immune dysregulation and/or loss of self-tolerance may lead to production of IgG (largely IgG4) autoantibodies that presumably bind in situ with PLA2R antigen, a transmembrane protein expressed in the podocytes of normal human glomeruli. The prognostic and predictive role of anti-PLA2R antibodies suggests that its dosage could be used to guide management of MN patients 60.
Antibody-antigen interaction would form IgG-PLA2R immune complexes that are shed into the subepithelial space of the glomerular capillaries. While these immune complexes may be capable of activating the alternative complement pathway 61, the sub-epithelial localization of the immune complexes allows them to evade the immune system, thus avoiding more widespread activation of inflammatory response. The immune complexes can be identified by electron microscopy as subepithelial electron dense deposits, and by immunofluorescence typically as dominant and polyclonal IgG (mostly IgG4) granular deposits along glomerular capillaries that co-localize with PLA2R and are often associated with less intense C3 staining 54,62. Injured podocytes produce new extracellular matrix around the immune deposits, leading to glomerular capillary thickening, often accompanied by “spikes” that are best seen in light microscopy slides using Jones' methenamine silver stain. Later in the course of the disease, these deposits undergo resorption, which manifests as decreased immunofluorescence intensity, lighter appearance of the deposits by electron microscopy.
More recently, additional MN target antigens have been discovered in PLA2R-negative and THSD7A-negative MN patients, including exostosin 1 and 2, neural epidermal growth factor-like 1 (NELL-1), Sema3B, and PCDH7 63, most of which are associated with circulating antibodies. However, their role in MN pathogenesis and their possible correlation with secondary forms of MN need further investigation.
Genetic susceptibility
Whereas only rare familial cases of MN have been reported to date 64, most insights into the genetic architecture of MN came from GWAS. The first GWAS involving patients of European ancestry identified 2 genetic susceptibility loci with large effects on disease risk: HLA-DQA1 on chromosome 6p21 (top SNP: rs2187668 tagging the HLA-DQA1*05:01 risk allele) and a locus located on chromosome 2q24 encoding the PLA2R1 gene (top SNP: rs4664308) 65. These findings, which were subsequently confirmed in independent European and East Asian cohorts 66,67, suggested that a genetic predisposition was likely due to a permissive HLA haplotype in combination with a risk variant altering the immunogenicity of PLA2R.
More recently, a much larger multi-ethnic GWAS performed on 12,820 individuals of East Asian and European ancestries confirmed the previous association of HLA loci and MN 68. Additionally, the latter study suggested ethnicity-specific associations of individual classical HLA risk alleles with MN (including the DRB1*0301 risk allele shared between Europeans and East Asians, DQA1*0501 risk allele as originally described in Europeans, and DRB1*1501 risk allele with effects specific to East Asians) and discovered two new genome-wide significant loci encompassing NFKB1 and IRF4 genes, which encode transcriptional regulators of inflammation 68.. The PLA2R locus was further fine-mapped, and a regulatory risk variant was prioritized that appears to specifically increase the PLA2R expression in glomeruli while decreasing its expression in all other tissues. Taken together, this study demonstrated that primary MN has an unusual genetic architecture, where most of the genetic risk is conveyed by a small number of risk loci with relatively large effect sizes. A genetic risk score (GRS) based on these loci explained over 30% of disease variance and was associated with anti-PLA2R positivity and higher proteinuria at disease onset. The GRS may also have a diagnostic utility when combined with serum test for anti-PLA2R antibodies. In fact, accounting for GRS information in the interpretation of anti-PLA2R ELISA test increased the overall sensitivity of a serological test and correctly re-classified 20-37% of MN cases 68.
Post-transplant MN
Similar to MN in the native kidney, recurrent MN in the kidney allograft is typically characterized by polyclonal global granular staining for IgG along the glomerular capillaries in a subepithelial distribution. However, in contrast to MN in the native kidney, recurrent MN often shows sparse electron dense deposits and lack of well developed “spikes” by light microscopy (Figure 2).
Figure 2: Pathogenesis of recurrent MN.
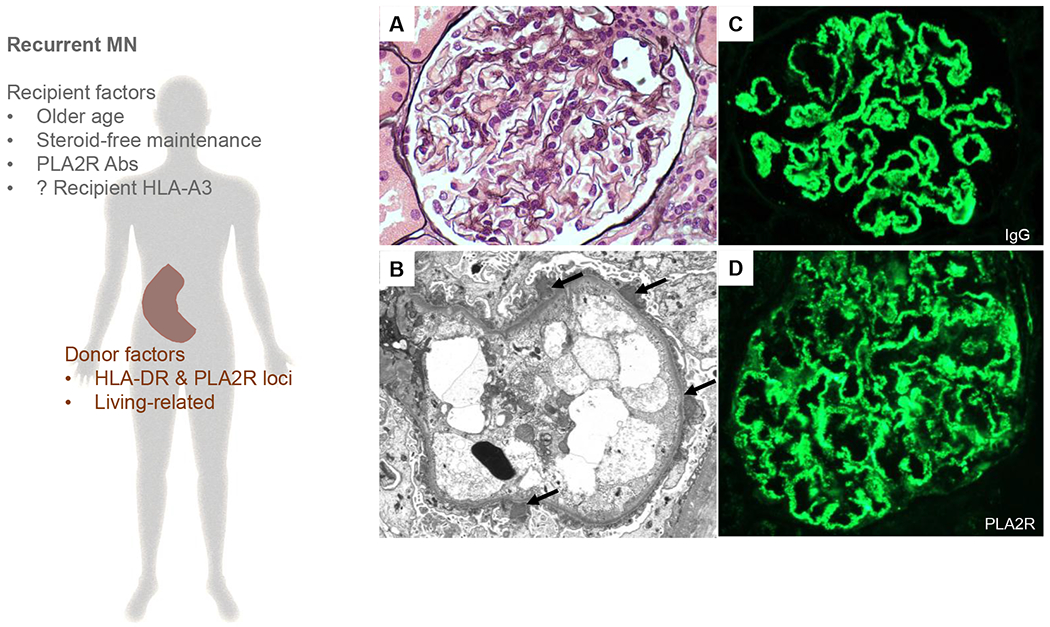
Left panel: Previous published reports have found that variables broadly related to recipients’ immune system status and donor inherited factors can predict recurrent MN. The association between recipient HLA-A3 and recurrent MN is trickier and need to be confirmed in other cohorts given the different distribution of HLA-A3 amongst different populations worldwide. Right panel: Representative photomicrographs of recurrent MN (A) A normocellular glomerulus showing minimally thickened glomerular capillaries without prominent “spikes” (Jones’ methenamine silver, original magnification ×600). This was associated with (B) scattered subepithelial deposits without significant glomerular basement membrane reaction (arrows, electron microscopy, original magnification ×6,000) (C) granular global staining for IgG along glomerular basement membranes in a subepithelial distribution (immunofluorescence, original magnification, ×400) and (D) granular global staining for PLA2R in the same distribution (immunofluorescence, original magnification, ×400). Right lower panel: Potential algorithm for future studies to improve our understanding of recurrent MN.
Abbreviations: HLA, human leukocyte antigen; PLA2R, phospholipase A2 receptor.
Despite the recent advancements in the pathogenesis of primary MN in the native kidney, our knowledge of post-transplant MN in patients with native renal failure secondary to primary MN is still limited. The role of MN recurrence on allograft survival is still a matter of debate. Most 15,69–72 but not all 73 observational studies have shown detrimental effects of MN recurrence on allograft survival. The prevalence of MN recurrence in the allograft ranges from 7 to 44% of cases, with higher rates and earlier occurrence encountered in centers performing protocol biopsies 14,73.
Recipient factors that have been anecdotally reported to be associated with recurrent MN include higher proteinuria at the time of transplantation, shorter time on waitlist, older age at transplant and a steroid-free immunosuppressive regimen 18,69,74. The last two may be linked to the status of recipient’s immune system. Small studies have also suggested that anti-PLA2R antibodies can predict recurrent MN 75,76.
To date, a few studies have investigated the role of HLA risk alleles of native MN in the recipient and MN recurrence. In a small study of 19 kidney transplant recipients with renal failure secondary to primary MN, HLA-DR3 was more frequent in patients with recurrent MN compared with those without recurrence (40% versus 21.4%) 77. Furthermore, Quintana et al reported the presence of the known native MN risk allele HLA DQA1*0501 in 6 of 7 recipients with recurrent MN 76. However, these findings were not be confirmed in a larger multicenter study, which instead suggested that recipient HLA-A3 antigen was associated with recurrent MN 74.
With regard to donor factors, a small retrospective observational study reported higher donor HLA-A3 prevalence in kidney transplants with recurrent MN compared with non-recurrent MN 78, but this could not be confirmed in a larger study 74.
Donors’ and recipients’ genetic risk for native MN has been explored in a recent study of 105 recipients with renal failure secondary to primary MN and their respective donors 79. Interestingly, only donor risk alleles in HLA-DRB1/DQA1 and PLA2R1 significantly predicted the risk of MN recurrence in the recipients independent from other clinical predictors. No effect of recipient’s HLA-DR or PLA2R1 risk alleles was found. This study, which was the first to test donor and recipient genetic risk for MN in the transplant setting, points to the importance of donor-inherited factors in recurrent MN. No donor or recipient GWAS of recurrent MN exist at present.
In summary, because of small sample size in single-center observational studies, and current lack of complete data on disease recurrence in renal transplant registries, the existing data on recipient and donor-related risk factors for recurrent MN are extremely limited. Still, current evidence suggests that recipient immune factors and donor inherited factors may represent important contributors to the risk of recurrent MN (Figure 2). Together, this stresses the need for well-designed multicenter studies with adequate power to systematically assess specific immune, genomic, and clinical factors in multivariable survival models. Future studies may widen the exploration of donors’ and recipients’ genetic risk factors, with a broader analysis of genetic effects, including the newly discovered NFKB1 and IRF4 loci.
PRIMARY FOCAL SEGMENTAL GLOMERULOSCLEROSIS (FSGS)
Focal segmental glomerulosclerosis (FSGS) represents a non-specific histologic manifestation of an injury causing podocyte depletion, which eventually leads to obliteration of the glomerular tuft, adhesion of the affected areas with Bowman’s capsule, and eventually hyaline deposition. FSGS can be primary (idiopathic), secondary or genetic. When a specific etiology cannot be defined, FSGS is considered primary (idiopathic) and it is believed to be the consequence of the effect of a circulating factor on the integrity of the glomerular filtration barrier 80. However, our increased knowledge of the role of inherited and immune factors in primary FSGS suggests that the above terminologies are not ideal.
Primary FSGS is usually more common in adolescents and young adults and in African Americans 20,81. Clinical features typically include nephrotic syndrome, manifested as nephrotic range proteinuria (>3.5 g/g urine protein/creatinine), reduced serum albumin (<3.5 g/dL) and edema 82. Treatment of primary FSGS relies on the use of immunosuppressants, including corticosteroids as first-line treatment, and calcineurin inhibitors 83. Recently, B cell depleting agents such as rituximab have shown encouraging results 84.
Immunopathology
It is believed that the pathogenesis of primary FSGS likely involves circulating factors 85. This has been suggested by evidence of the therapeutic effect of immunoadsorption and plasmapheresis on reduction of proteinuria. Moreover, the serum of FSGS patients increases the permeability to albumin in glomeruli in vitro, and when injected into rats, it induces foot process effacement and proteinuria 86. Several candidate circulating factors responsible for primary FSGS have been proposed, although none of them have been confirmed so far. Among these are anti-CD40 antibody 87, and soluble urokinase-type plasminogen activator receptor (suPAR) 88, and most recently, anti-nephrin antibodies (Weins A, presented at ASN Kidney Week 2020).
Since podocytes are terminally differentiated cells that cannot regenerate through cell division, podocyte injury from potential circulating factors can cause extensive progressive podocyte depletion. In an attempt to repair the damage, podocytes stretch to cover the denuded areas. This can be observed by electron microscopy as near complete foot process effacement (≥ 80% of total glomerular capillary surface areas) 82,89,90. Therefore, from a pathophysiologic point of view, primary FSGS can be seen as diffuse podocytopathy with near complete foot process effacement affecting all the glomeruli.
Persistent injury to the podocytes subsequently causes segmental adherence of the injured tuft to the Bowman’s capsule, permitted by the effect of the glomerular capillary hydrostatic pressure 80, followed by entrapment of plasma proteins (including macromolecules) with/without hyaline accumulation and foam cells that can be reflected as segmental trapping of IgM and C3 staining within the areas of hyalinosis by immunofluorescence. Alongside these findings, FSGS may show different morphologic features by light microscopy, including not-otherwise-specified, perihilar, cellular, tip, and collapsing variants 91.
Genetic susceptibility
In steroid-resistant nephrotic syndrome, sequencing panels identified mutations in up to 30% of patients < 25 year-old 92 and 12% of adults 93. However, even when a genetic mutation is identified, the pattern of inheritance may be complex and disease penetrance may be variable 94. These findings suggest that there may be additional genetic and/or environmental factors necessary for the manifestation of the disease. Whereas GWAS can be a powerful approach for the detection of such common susceptibility variants across the entire genome, GWAS approaches have been challenging in the setting of primary FSGS given the low incidence of the disease, the limited availability of a histological diagnosis, and the difficulties in its classification into primary or secondary.
GWAS studies have discovered associations between steroid-sensitive nephrotic syndrome and HLA loci 95, and more recently with common variants in NPHS1-KIRREL2 (a podocyte gene) and TNFSF15 (involved in other immune-mediated diseases, such as primary biliary cirrhosis) 96 supporting the notion that steroid-sensitive nephrotic syndrome is a polygenic disease, similar to systemic autoimmune disorders, where immune dysregulation seems to play a preponderant pathogenic role.
Post-transplant FSGS
Recurrence of primary FSGS in the kidney allograft often manifests very early after transplantation supporting the role of recipients’ circulating factors in the pathogenesis of recurrent FSGS 80. Early on, the morphologic appearance of recurrent FSGS would be reminiscent to that of minimal change disease while segmental sclerotic lesions develop later in the course of transplantation (Figure 4).
Figure 4: Potential pathways for de novo collapsing glomerulopathy.
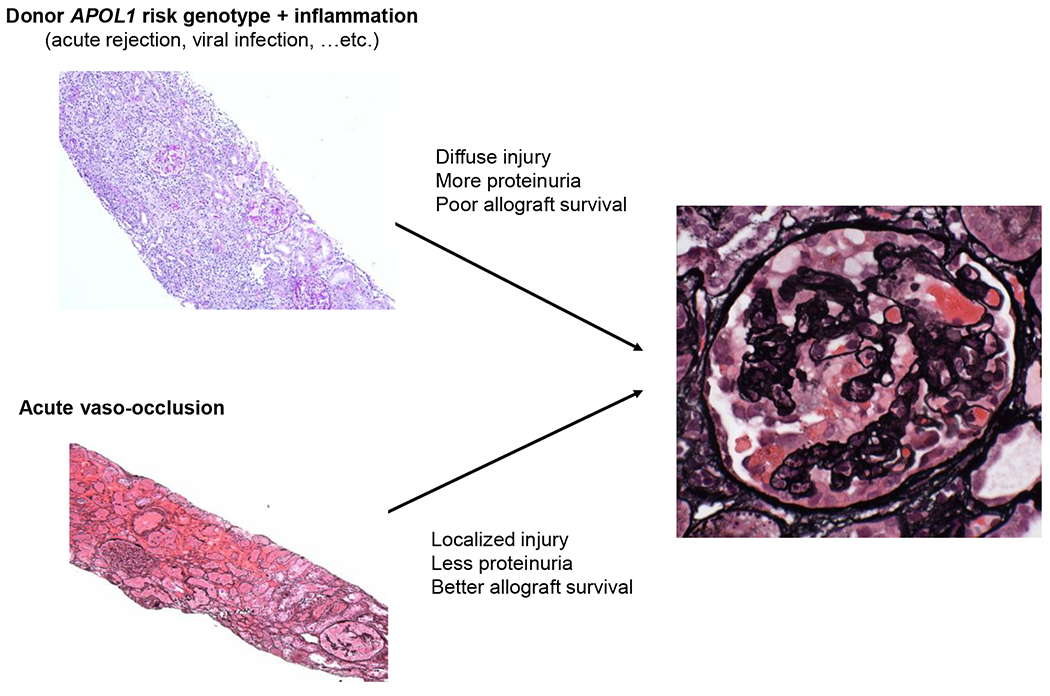
Left upper photomicrograph (periodic acid-Schiff, original magnification ×100). Left lower photomicrograph (Jones’ methenamine silver, original magnification ×100). Right photomicrograph (Jones’ methenamine silver, original magnification ×400)
Recurrent primary FSGS is expected to affect 30-40% of the patients and is associated with an increased risk of allograft loss 19,97. Primary FSGS is rare and studies aimed to assess recurrent rates, prognosis and risk factors are inevitably underpowered. While larger registry data relying on retrospectively collected data have shown lower recurrence rates 15,98 and may have missed recurrent FSGS that were not associated with allograft loss. Our knowledge of recurrent FSGS in the kidney allograft is further limited by the lack of stringent criteria to exclude cases of secondary/adaptive FSGS, which may have contaminated the studied cohorts (although using tacrolimus as a maintenance therapy may complicate the aforementioned issue).
Among recipients’ factors, European ancestry 97, lower age at diagnosis in the native kidney 98–100, and FSGS recurrence in previous allografts 101 have all been associated with higher rates of FSGS recurrence. Furthermore, pre-transplant bilateral nephrectomy was also associated with higher risk of FSGS recurrence 99, suggesting that native kidneys may absorb the potential pathogenic circulating factors 102. Recently, a panel of seven serum antibodies (CD40, PTPRO, CGB5, FAS, P2RY11, SNRPB2, and APOL2) correlated with FSGS recurrence 87.
With regard to donor factors, African American donor race has been shown to be associated with higher rates of FSGS recurrence 97. These findings may be the consequence of the detrimental effects of APOL1 risk variants in African American populations that may accelerate podocyte loss after transplantation 103. Among other donor factors, older donor age was also associated with risk of recurrence 98.
A recent multi-center study from the TANGO project analyzed 176 kidney transplant recipients affected by primary FSGS and assessed potential risk factors for disease recurrence in multivariable Cox proportional hazards models adjusted for major confounding parameters 19. This study confirmed the association of recipient White race and pre-transplant bilateral nephrectomy with higher risk of recurrence. Interestingly, older recipient age was an independent risk factor for recurrence, as well as lower BMI. However, this study did not address the role of genetic factors in disease recurrence.
In summary, little is known regarding pathogenesis and risk factors of recurrent primary FSGS (Figure 4). Our limited understanding of such risk factors in the native kidney further complicates this issue. Applying stringent criteria to diagnose primary FSGS in the native kidney and in the allograft is a good start to filter out inappropriately included cases of secondary/adaptive FSGS and assemble a pure cohort of transplant patients with recurrent and non-recurring primary FSGS. Given the increasing evidence that the immune system may play a role in disease pathogenesis, further studies should focus the attention on the interplay between donor’s and recipient’s HLA type, serologic testing, and gene expression profiles. Genomic assessment of post-transplant FSGS may discover variants associated with risk of recurrence and give better insights into donor’s and recipient’s susceptibility factors. Such studies inevitably require large sample sizes, and further emphasize the need for multi-center designs.
DE NOVO COLLAPSING GLOMERULOPATHY
Collapsing glomerulopathy, or collapsing focal segmental glomerulosclerosis, is defined histologically by the presence of at least one glomerulus with segmental wrinkling, retraction, and obliteration of glomerular capillaries, accompanied by hypertrophy and hyperplasia of overlying glomerular epithelial cells 91. Collapsing glomerulopathy is a rare disease with variable geographic distribution. It is commonly encountered in patients of African ancestry and is typically characterized by massive proteinuria and rapid progression to kidney failure 91,104.
Clinicopathologic studies have enhanced our understanding of this disease. While collapsing glomerulopathy can be idiopathic (in up to 80% of cases in some studies) 105, the list of associated triggers keeps growing over time. For example, collapsing glomerulopathy has been associated with viral infections, medication exposures (e.g. interferons and pamidronate), collagen vascular disease/autoimmunity, hemophagocytic syndrome, and acute vaso-occlusive disease 106.
Immunopathology
Animal studies have shown that developing collapsing glomerulopathy requires a genetic background and severe podocyte injury 107,108 that cause podocyte lysis and denudation of the glomerular basement membranes 108. Since the cross-talk between healthy podocytes and the endothelium is crucial for glomerular capillary integrity, the affected segment later collapses 109. In an attempt to repair the damage and seal the naked basement membranes, parietal cells seem to get activated, may detach from the parietal basement membrane, and migrate to the denuded glomerular basement membranes107,108. Ultimately, this leads to the development of characteristic features of collapsing glomerulopathy, namely proteinuria, glomerular tuft collapse, and hyperplasia of parietal cells.
In humans, collapsing glomerulopathy results from an interplay between genetic predisposition and factors inducing severe podocyte injury. Collapsing glomerulopathy is commonly encountered in subjects with recent African ancestry and more specifically in subjects with two APOL1 kidney risk variants (G1/G1, G1/G2, or rarely G2/G2), which are referred to as “APOL1 high-risk genotypes” 110. Notably, inflammation, especially when associated with elevated interferon levels, can upregulate APOL1 expression by several hundred orders of magnitude 111. Whereas APOL1 high-risk genotypes are present in 14% of the African American population, APOL1 high-risk genotypes are highly enriched in patients with collapsing glomerulopathy where they are encountered in >80% of such patients in the setting of HIV-associated nephropathy 112, COVTD-19 113,114, or interferon treatment 115.
Collapsing glomerulopathy in the kidney allograft
In contrast to collapsing glomerulopathy in the native kidney, less is known about collapsing glomerulopathy in the kidney allograft. Case series reported through 2017 were largely small and limited by incomplete clinical data (summarized in supplemental table of reference 116). Nevertheless, these studies have shown that post-transplant collapsing glomerulopathy is associated with poor allograft survival 117–119, relatively low percentage of Black recipients 118,119 and high incidence of concurrent episodes of acute vaso-occlusion 120,121 . In 2016, Shah et al. reported de novo collapsing glomerulopathy that developed in 2 patients who received their kidney allograft from a single deceased black donor who later was found to have APOL1 high-risk genotype 122, suggesting for the first time that donor APOL1 may be involved in the pathogenesis of de novo collapsing glomerulopathy.
Two years later, a single center study of 38 patients with collapsing glomerulopathy in the kidney allograft was reported 116. 47% of the recipients received allografts from African American donors, and of these, 8 (53%) had APOL1 high-risk genotypes. In these patients, concurrent acute rejection, acute vaso-occlusive disease (mainly cortical necrosis and thrombotic microangiopathy), and viremia (cytomegalovirus and rarely parvovirus or Epstein Barr virus) were present in 61%, 29%, and 13% of patients, respectively. Notably, all patients who received allografts from donors with high-risk APOL1 high-risk variants had either concurrent or prior episodes of acute rejection or viremia. Finally, multivariate analysis for allograft survival showed that a donor APOL1 high-risk genotype was a significant predictor of allograft failure, while acute vaso-occlusive disease was associated with favorable allograft survival 116.
In summary, a handful of case reports and retrospective observations suggest that donor APOL1 high-risk genotypes in the presence of allograft inflammation (such as acute rejection) can cause aggressive collapsing glomerulopathy with detrimental effects on allograft survival. In contrast, acute ischemia from acute vaso-occlusion can lead to collapsing glomerulopathy that may be independent from donor APOL1 status. Given the typically localized pattern of injury in vaso-occlusive disease, collapsing glomerulopathy in the latter setting is often associated with a favorable prognosis if the allograft can survive the vascular insults, (Figure 4). Future studies need to assess potential donor APOL1 dose effects, which may be more apparent than that occurring in the native kidney given the typical presence of a single kidney in an alloimmune environment.
DE NOVO IMMUNE COMPLEX-MEDIATED GLOMERULONEPHRITIS (ICMGN)
Establishing a diagnosis of de novo ICMGN in the kidney allograft is not always easy. Kidney transplant recipients do not consistently have a pre-transplant diagnostic native kidney biopsy. Furthermore, de novo ICMGN often occurs late in the course of transplantation. This can render early protocol biopsies of questionable diagnostic value.
While the precise prevalence of de novo ICMGN is unknown, it is estimated that at least 5% of kidney transplant patients would develop de novo MN or de novo membranoproliferative glomerulonephritis after transplantation 123. Despite this high incidence, the mechanisms of de novo ICMGN is not well understood. Because of the potent immunosuppressive therapy in kidney transplant patients, it is not surprising that early in the course of the disease, de novo ICMGN is often asymptomatic or only associated with mild renal insufficiency, sub-nephrotic proteinuria, and/or mild microhematuria 123. However, de novo ICMGN is eventually expected to have a detrimental effect on allograft survival 11,124.
Immunopathology
De novo ICMGN is traditionally considered a non-alloimmune glomerular disease 125. Yet, the majority of de novo ICMGN are characterized by negative serologic work-up 126,127. Hence, it is plausible that a proportion of de novo ICMGN may occur secondary to the development of in situ immune complexes formed by ligation of recipient immunoglobulins with donor alloimmune antigens. In fact, ICMGN secondary to alloimmune response is a well-documented phenomenon. In the neonate, MN can develop following production of IgG antibodies against neutral endopeptidase in mothers who lack such antigen128. ICMGN can also be encountered following bone marrow transplantation, often in association with chronic graft versus host disease 129.
In the kidney allograft, de novo ICMGN have been detected in a non-immunosuppressed rat model of kidney transplantation (Fischer-344 kidneys to Lewis recipient) 127. In the aforementioned study, Grau et al. harvested rat kidney allografts at different time intervals and found that allografts harvested up to six weeks post-transplantation showed histopathologic findings similar to human kidney allografts with antibody-mediated rejection (AMR) while those harvested at 26 weeks showed de novo ICMGN 127.
In humans, de novo ICMGN comprise wide spectrum of glomerular diseases, including IgAN, MN, and ICMGN-not otherwise specified (ICMGN-NOS) 130,131. De novo IgAN is the least studied form of de novo ICMGN while de novo MN is probably the most characterized form of de novo ICMGN. In contrast to recurrent MN, de novo MN is typically PLA2R-negative, over expresses HLA-DR antigen in the podocytes, and occurs late in the course of transplantation 74,132,133. Whereas older studies have suggested a relation between de novo MN and HCV 134, more recent studies failed to show such an association 74. Thus, it is suspected that a proportion of de novo MN may be secondary to alloimmune response against antigens in donor podocytes 74,133
Recent studies have attempted to further characterize the group of de novo ICMGN-NOS, which includes non-IgA mesangial proliferative or membranoproliferative glomerulonephritis of unknown etiology. Giannico et al. described the clinical and histopathologic features of 28 allograft biopsies with mesangial proliferative ICMGN-NOS126. They found that 36% of these biopsies have concurrent acute T-cell mediated rejection and 25% have AMR.126 Lloyd et al. also identified 32 patients with de novo ICMGN, including 12 (37%) ICGN-NOS135. Compared to other de novo ICMGN, ICGN-NOS had higher incidence of concurrent AMR (67% vs. 5%, p<0.001) and numerically more acute T cell mediated rejection (33% vs. 10%) 135. We also found evidence to support a relation between de novo ICMGN and alloimmunity 131. Compared to recurrent glomerulonephritis (n=77), patients with de novo ICMGN (n=46) had more concurrent AMR, higher DSA at the time of diagnosis, higher number of previous solid organ transplants, and less potent induction therapy at the time of transplantation 131. However, when these cases were further studied, we found that the proportion of biopsies with AMR or DSA were highest in patients with de novo MN (63%), followed by de novo IgAN (36%) and de novo ICGN-NOS (29%) (Table 1).
Table 1.
Characteristics and potential etiologies for de novo ICMGN
| ICMGN-NOS (N=21)(a) |
IgAN (N=11)(b) |
MN (N=8)(c) |
HCV (N=4)(d) |
Infection-related (N=2)(e) |
|
|---|---|---|---|---|---|
| Post-transplant interval (years) | 4.2 (2.5, 7.3) | 4.6 (3.1, 8.1) | 3.4 (1.4, 7.8) | 3.3 (2.1, 4.3) | 1.8 and 9 |
| Induction with IL-2R | 8/20 (40%) | 3/11 (27%) | 0/7 (0%) | 4/4 (100%) | 0/2 (0%) |
| Prior solid organ transplant (%) | 6/21 (29%) | 4/11 (36%) | 3/8 (38%) | 1/4 (25%) | 0/2 (0%) |
| AMR | 2/21 (9.5%) | 2/11 (18.2%) | 4/8 (50%) | 1/4 (25%) | 0/2 (0%) |
| DSA without AMR | 4/21 (19%) | 2/11 (18.2%) | 1/8 (12.5%) | 1/4 (25%) | 0/2 (0%) |
| TCMR | 6/21 (28.5%) | 2/11 (18.2%) | 2/8 (25%) | 0/4 (0%) | 1/2 (50%) |
| ANA or other autoantibodies | 1/21 (4.8%) | 0/11 (0%) | 1/8 (12.5%) | 1/4 (25%) | 0/2 (0%) |
| HCV | 0/21 (0%) | 0/11 (0%) | 0/8 (0%) | 4/4(100%) | 0/2 (0%) |
| Infection (non-HCV) | 0/21 (0%) | 0/11 (0%) | 0/8 (0%) | 0/4 (0%) | 2/2 (100%) |
| Others | 1/11 (9%) (cirrhosis) | ||||
| No apparent etiology identified | 11/21 (52.3%) | 5/11 (45.4%) | 3/8 (37.5%) | 0/4 (0%) | 0/2 (0%) |
Abbreviations AMR, antibody-mediated rejection; DSA, circulating donor-specific antibodies; TCMR, T cell mediated rejection
CUIMC patients with de novo ICMGN (n=46, retrospectively identified between 2011-2019)
This study was approved by Columbia Institutional Review Board and published in part previously (reference 131).
In this non-published part of the study, we sought to associate de novo ICMGN with possible etiologies. One of the 21 IGMGN-NOS, was associated with positive ANA and a history of autoimmune hepatitis, and was therefore favored to be related to autoimmunity. One of the 11 patients with de novo IgAN had liver cirrhosis and was classified as IgAN secondary to liver cirrhosis.
- Data on induction therapy was not available for 2 patients (1 with GN-NOS and 1 with MN). Per protocol, all recipients with HCV receive induction therapy with IL2R
- Some patients had more than one potential etiology:
2 patients had TCMR and DSA without AMR. 1 patient had ANA and DSA without AMR.
1 patient had TCMR and DSA without AMR.
1 patient had AMR and TCMR. 1 patient had AMR, TCMR and ANA.
1 patient had HCV and DSA. 1 patient had HCV and AMR. 1 patient had HCV and ANA.
1 patient had infectious etiology and TCMR.
In summary, there is cumulative evidence to suggest that a proportion of de novo ICMGN are alloimmune in nature. Such association warrants additional investigation. Late post-transplant protocol biopsies may be needed to further characterize these glomerular diseases. Gene expression profiling, mass spectrometry, and GWAS are potential approaches that may enhance our understanding of the pathophysiology of de novo ICMGN. Until then, a practical approach in evaluating de novo ICMGN may include performing detailed serologic and histologic evaluation, including full panel immunofluorescence and even electron microscopy, to exclude traditional causes like infection (especially bacterial and HCV), autoimmune diseases, and cryoglobulinemia. In case the aforementioned causes were reasonably excluded, then the possibility of an alloimmune etiology should be considered. If the allograft biopsy meets Banff criteria for AMR, then the focus should be to treat AMR. If the biopsy does not meet diagnostic criteria for AMR, it would still possible that such ICMGN is alloimmune in nature. Physicians caring for kidney transplant patients may need to consider modify their immunosuppressive treatment (such as increase their maintenance immunosuppression) to try to slow the progression of such diseases.
CONCLUSION
Glomerular diseases in kidney allografts have a detrimental effects on allograft survival, yet our knowledge of the risk factors for these disorders is largely restricted to descriptive reports and small clinical studies. Application of precision medicine approaches to glomerular diseases of the kidney allograft is limited by inadequately powered studies. To enhance our understanding of post-transplant glomerular diseases, we need to benefit from scientific advances achieved by genetic and immunologic studies in the native kidney to better define the role of inherited and immune factors in glomerular diseases of the kidney allograft. Such an approach also has the potential to dissect intra-renal (donor) from extra-renal (recipient) genetic and immune effects, which cannot be achieved by studying these diseases in the native kidney. (Figure 5).
Figure 5: Potential algorithm for future studies to improve our understanding of glomerular diseases of the kidney allograft.
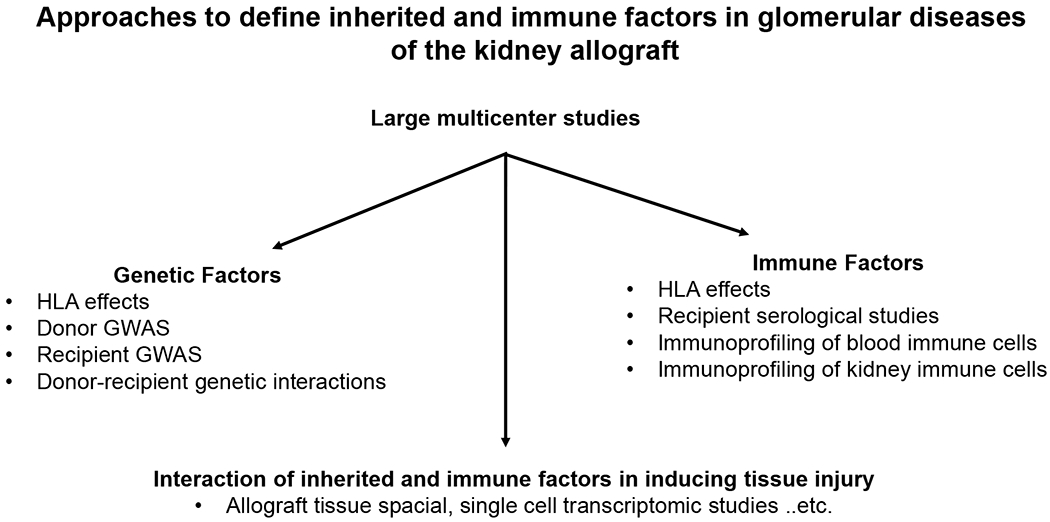
Abbreviations: HLA, human leukocyte antigen; GWAS, genome wide association studies.
Figure 3: Pathogenesis of recurrent primary FSGS.
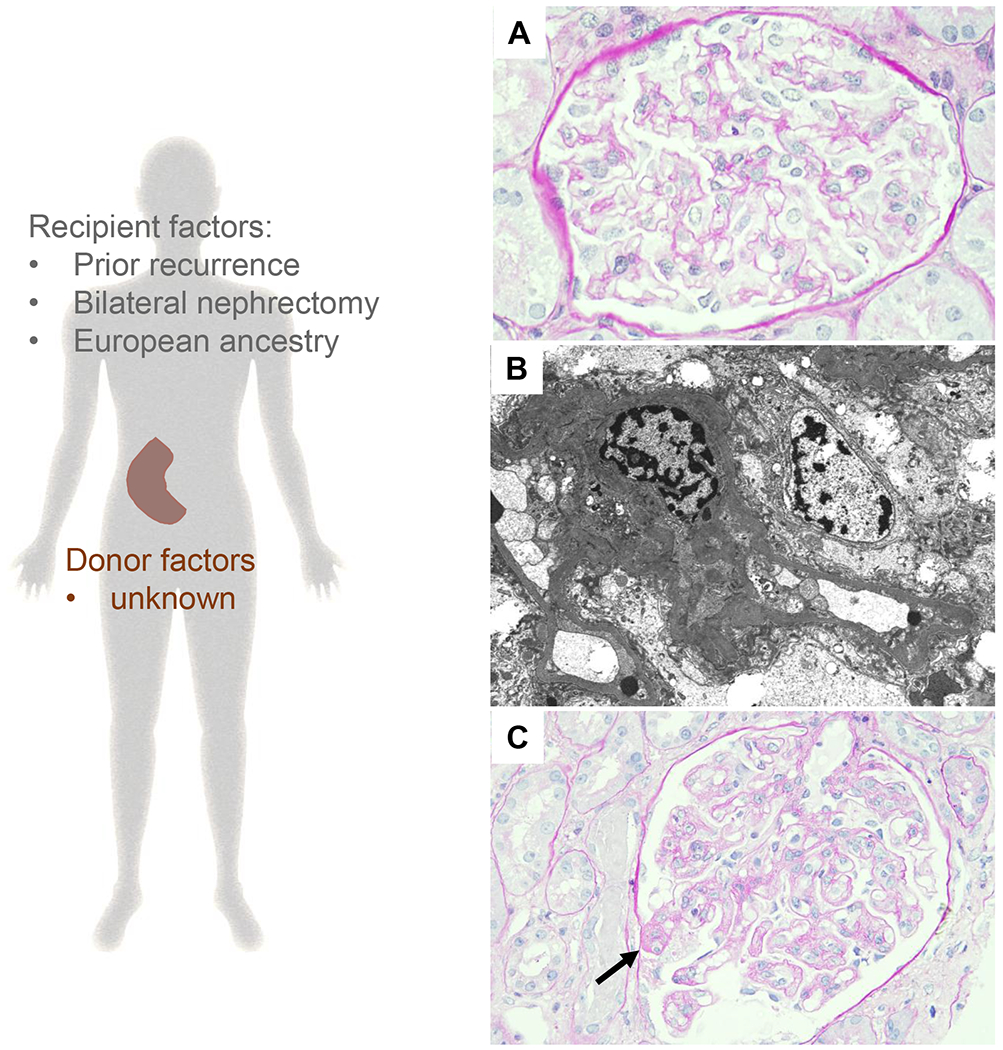
Left panel: Previous published reports have found that variables broadly related to recipients’ immune system status may predict recurrent FSGS. The association with inherited factors is even less defined at the moment. Right panel: Representative photomicrographs of recurrent primary FSGS (A) A normocellular glomerulus showing mild prominence of the podocytes on day 12 after transplantation in a patient presented with full nephrotic syndrome (periodic acid–Schiff, original magnification ×600) (B) This was associated with complete foot process effacement (electron microscopy, original magnification ×6,000) (C) Several segmental sclerotic lesion was noted in the follow-up biopsy, which was performed 3 months post-transplant (arrows, representative image, periodic acid–Schiff, original magnification ×600).
Acknowledgment
IB is supported from 2020 Mendez National Institute of Transplantation Foundation Research Grant. KK is supported by R01-DK105124, RC2-DK116690, and UH3-DK114926 from the NIH/NIDDK. FZ is supported by a transplant fellowship from the UnitedHealth Group.
Abbreviations
- APOL1
apolipoprotein L1
- AMR
antibody-mediated rejection
- DSA
circulating donor-specific antibodies
- FSGS
primary focal segmental glomerulosclerosis
- Gd IgA1
Galactose-deficient IgA1
- GFR
glomerular filtration rate
- GRS
genetic risk score
- GWAS
genome wide association studies
- HLA
human leukocyte antigen
- ICMGN
immune complex-mediated glomerulonephritis
- ICMGN-NOS
immune complex-mediated glomerulonephritis not otherwise specified
- IgAN
IgA nephropathy
- MN
membranous nephropathy
- PLA2R
phospholipase A2 receptor
- SNP
single nucleotide polymorphism
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med. 1999;341(23):1725–1730. [DOI] [PubMed] [Google Scholar]
- 2.Hart A, Smith JM, Skeans MA, et al. OPTN/SRTR 2016 Annual Data Report: Kidney. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2018; 18 Suppl 1(Suppl 1):18–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hariharan S, Johnson CP, Bresnahan BA, Taranto SE, McIntosh MJ, Stablein D. Improved graft survival after renal transplantation in the United States, 1988 to 1996. The New England journal of medicine. 2000;342(9):605–612. [DOI] [PubMed] [Google Scholar]
- 4.Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant. 2011;11(3):450–462. [DOI] [PubMed] [Google Scholar]
- 5.Saran R, Robinson B, Abbott KC, et al. US Renal Data System 2017 Annual Data Report: Epidemiology of Kidney Disease in the United States. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2018;71(3 Suppl 1):A7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi X, Lv J, Han W, et al. What is the impact of human leukocyte antigen mismatching on graft survival and mortality in renal transplantation? A meta-analysis of 23 cohort studies involving 486,608 recipients. BMC Nephrol. 2018;19(1):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiebe C, Gibson IW, Blydt-Hansen TD, et al. Evolution and Clinical Pathologic Correlations of De Novo Donor-Specific HLA Antibody Post Kidney Transplant. Am J Transplant. 2012;12(5):1157–1167. [DOI] [PubMed] [Google Scholar]
- 8.Milner J, Melcher ML, Lee B, et al. HLA Matching Trumps Donor Age: Donor-Recipient Pairing Characteristics That Impact Long-Term Success in Living Donor Kidney Transplantation in the Era of Paired Kidney Exchange. Transplant Direct. 2016;2(7):e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pallardó Mateu LM, Sancho Calabuig A, Capdevila Plaza L, Franco Esteve A. Acute rejection and late renal transplant failure: risk factors and prognosis. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2004;19 Suppl 3:iii38–42. [DOI] [PubMed] [Google Scholar]
- 10.Hart A, Smith JM, Skeans MA, et al. OPTN/SRTR 2015 Annual Data Report: Kidney. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2017;17 Suppl 1(Suppl 1):21–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Zoghby ZM, Stegall MD, Lager DJ, et al. Identifying specific causes of kidney allograft loss. Am J Transplant. 2009;9(3):527–535. [DOI] [PubMed] [Google Scholar]
- 12.Saran R, Robinson B, Abbott KC, et al. US Renal Data System 2019 Annual Data Report: Epidemiology of Kidney Disease in the United States. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2020;75(1 Suppl 1):A6–a7. [DOI] [PubMed] [Google Scholar]
- 13.Briganti EM, Russ GR, McNeil JJ, Atkins RC, Chadban SJ. Risk of renal allograft loss from recurrent glomerulonephritis. N Engl J Med. 2002;347(2):103–109. [DOI] [PubMed] [Google Scholar]
- 14.Cosio FG, Cattran DC. Recent advances in our understanding of recurrent primary glomerulonephritis after kidney transplantation. Kidney Int. 2017;91(2):304–314. [DOI] [PubMed] [Google Scholar]
- 15.Allen PJ, Chadban SJ, Craig JC, et al. Recurrent glomerulonephritis after kidney transplantation: risk factors and allograft outcomes. Kidney Int. 2017;92(2):461–469. [DOI] [PubMed] [Google Scholar]
- 16.Singh T, Astor BC, Zhong W, Mandelbrot DA, Maursetter L, Panzer SE. The association of acute rejection vs recurrent glomerular disease with graft outcomes after kidney transplantation. Clinical transplantation. 2019;33(12):e13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regunathan-Shenk R, Avasare RS, Ahn W, et al. Kidney Transplantation in C3 Glomerulopathy: A Case Series. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2019;73(3):316–323. [DOI] [PubMed] [Google Scholar]
- 18.Grupper A, Cornell LD, Fervenza FC, Beck LH Jr., Lorenz E, Cosio FG. Recurrent Membranous Nephropathy After Kidney Transplantation: Treatment and Long-Term Implications. Transplantation. 2016;100(12):2710–2716. [DOI] [PubMed] [Google Scholar]
- 19.Uffing A, Perez-Saez MJ, Mazzali M, et al. Recurrence of FSGS after Kidney Transplantation in Adults. Clin J Am Soc Nephrol. 2020;15(2):247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrol Dial Transplant. 2011;26(2):414–430. [DOI] [PubMed] [Google Scholar]
- 21.Haas M, Rahman MH, Cohn RA, Fathallah-Shaykh S, Ansari A, Bartosh SM. IgA nephropathy in children and adults: comparison of histologic features and clinical outcomes. Nephrol Dial Transplant. 2008;23(8):2537–2545. [DOI] [PubMed] [Google Scholar]
- 22.Barbour SJ, Reich HN. Risk stratification of patients with IgA nephropathy. Am J Kidney Dis. 2012;59(6):865–873. [DOI] [PubMed] [Google Scholar]
- 23.Praga M, Gutierrez E, Gonzalez E, Morales E, Hernandez E. Treatment of IgA nephropathy with ACE inhibitors: a randomized and controlled trial. J Am Soc Nephrol. 2003;14(6):1578–1583. [DOI] [PubMed] [Google Scholar]
- 24.Manno C, Torres DD, Rossini M, Pesce F, Schena FP. Randomized controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgA nephropathy. Nephrol Dial Transplant. 2009;24(12):3694–3701. [DOI] [PubMed] [Google Scholar]
- 25.Rollino C, Vischini G, Coppo R. IgA nephropathy and infections. J Nephrol. 2016;29(4):463–468. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki H, Kiryluk K, Novak J, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22(10):1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magistroni R, D’Agati VD, Appel GB, Kiryluk K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015;88(5):974–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki H Biomarkers for IgA nephropathy on the basis of multi-hit pathogenesis. Clin Exp Nephrol. 2019;23(1):26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Medjeral-Thomas NR, O’Shaughnessy MM. Complement in IgA Nephropathy: The Role of Complement in the Pathogenesis, Diagnosis, and Future Management of IgA Nephropathy. Adv Chronic Kidney Dis. 2020;27(2):111–119. [DOI] [PubMed] [Google Scholar]
- 30.Drachenberg CB, Papadimitriou JC, Chandra P, et al. Epidemiology and Pathophysiology of Glomerular C4d Staining in Native Kidney Biopsies. Kidney Int Rep. 2019;4(11):1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trimarchi H, Barratt J, Cattran DC, et al. Oxford Classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017;91(5):1014–1021. [DOI] [PubMed] [Google Scholar]
- 32.Barbour SJ, Coppo R, Zhang H, et al. Evaluating a New International Risk-Prediction Tool in IgA Nephropathy. JAMA Intern Med. 2019;179(7):942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levy M, Berger J. Worldwide perspective of IgA nephropathy. Am J Kidney Dis. 1988;12(5):340–347. [DOI] [PubMed] [Google Scholar]
- 34.Julian BA, Quiggins PA, Thompson JS, Woodford SY, Gleason K, Wyatt RJ. Familial IgA nephropathy. Evidence of an inherited mechanism of disease. N Engl J Med. 1985;312(4):202–208. [DOI] [PubMed] [Google Scholar]
- 35.Feehally J, Farrall M, Boland A, et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol. 2010;21(10):1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gharavi AG, Kiryluk K, Choi M, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu XQ, Li M, Zhang H, et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet. 2011;44(2):178–182. [DOI] [PubMed] [Google Scholar]
- 38.Li M, Foo JN, Wang JQ, et al. Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nat Commun. 2015;6:7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiryluk K, Li Y, Scolari F, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46(11):1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang SH, Kennard AL, Walters GD. Recurrent glomerulonephritis following renal transplantation and impact on graft survival. BMC Nephrol. 2018;19(1):344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moroni G, Belingheri M, Frontini G, Tamborini F, Messa P. Immunoglobulin A Nephropathy. Recurrence After Renal Transplantation. Front Immunol. 2019;10:1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Odum J, Peh CA, Clarkson AR, et al. Recurrent mesangial IgA nephritis following renal transplantation. Nephrol Dial Transplant. 1994;9(3):309–312. [PubMed] [Google Scholar]
- 43.Ortiz F, Gelpi R, Koskinen P, et al. IgA nephropathy recurs early in the graft when assessed by protocol biopsy. Nephrol Dial Transplant. 2012;27(6):2553–2558. [DOI] [PubMed] [Google Scholar]
- 44.Nijim S, Vujjini V, Alasfar S, et al. Recurrent IgA Nephropathy After Kidney Transplantation. Transplant Proc. 2016;48(8):2689–2694. [DOI] [PubMed] [Google Scholar]
- 45.Avasare RS, Rosenstiel PE, Zaky ZS, et al. Predicting Post-Transplant Recurrence of IgA Nephropathy: The Importance of Crescents. Am J Nephrol. 2017;45(2):99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clayton P, McDonald S, Chadban S. Steroids and recurrent IgA nephropathy after kidney transplantation. Am J Transplant. 2011;11(8):1645–1649. [DOI] [PubMed] [Google Scholar]
- 47.Di Vico MC, Messina M, Fop F, Barreca A, Segoloni GP, Biancone L. Recurrent IgA nephropathy after renal transplantation and steroid withdrawal. Clin Transplant 2018;32(4):e13207. [DOI] [PubMed] [Google Scholar]
- 48.Berthoux F, Suzuki H, Mohey H, et al. Prognostic Value of Serum Biomarkers of Autoimmunity for Recurrence of IgA Nephropathy after Kidney Transplantation. J Am Soc Nephrol. 2017;28(6):1943–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berthelot L, Robert T, Vuiblet V, et al. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. 2015;88(4):815–822. [DOI] [PubMed] [Google Scholar]
- 50.Han SS, Huh W, Park SK, et al. Impact of recurrent disease and chronic allograft nephropathy on the long-term allograft outcome in patients with IgA nephropathy. Transpl Int. 2010;23(2):169–175. [DOI] [PubMed] [Google Scholar]
- 51.Andresdottir MB, Hoitsma AJ, Assmann KJ, Wetzels JF. Favorable outcome of renal transplantation in patients with IgA nephropathy. Clin Nephrol. 2001;56(4):279–288. [PubMed] [Google Scholar]
- 52.McDonald SP, Russ GR. Recurrence of IgA nephropathy among renal allograft recipients from living donors is greater among those with zero HLA mismatches. Transplantation. 2006;82(6):759–762. [DOI] [PubMed] [Google Scholar]
- 53.Andresdottir MB, Haasnoot GW, Doxiadis II, Persijn GG, Claas FH. Exclusive characteristics of graft survival and risk factors in recipients with immunoglobulin A nephropathy: a retrospective analysis of registry data. Transplantation. 2005;80(8):1012–1018. [DOI] [PubMed] [Google Scholar]
- 54.Ronco P, Debiec H. Membranous nephropathy: A fairy tale for immunopathologists, nephrologists and patients. Mol Immunol. 2015;68(1):57–62. [DOI] [PubMed] [Google Scholar]
- 55.Glassock RJ. Diagnosis and natural course of membranous nephropathy. Semin Nephrol. 2003;23(4):324–332. [DOI] [PubMed] [Google Scholar]
- 56.Couser WG. Primary Membranous Nephropathy. Clin J Am Soc Nephrol. 2017;12(6):983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tomas NM, Beck LH Jr., Meyer-Schwesinger C, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med. 2014;371(24):2277–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van de Logt AE, Fresquet M, Wetzels JF, Brenchley P. The anti-PLA2R antibody in membranous nephropathy: what we know and what remains a decade after its discovery. Kidney Int. 2019;96(6):1292–1302. [DOI] [PubMed] [Google Scholar]
- 59.Rosenzwajg M, Languille E, Debiec H, et al. B- and T-cell subpopulations in patients with severe idiopathic membranous nephropathy may predict an early response to rituximab. Kidney Int. 2017;92(1):227–237. [DOI] [PubMed] [Google Scholar]
- 60.Beck LH Jr., Fervenza FC, Beck DM, et al. Rituximab-induced depletion of anti-PLA2R autoantibodies predicts response in membranous nephropathy. J Am Soc Nephrol. 2011;22(8):1543–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Francis JM, Beck LH Jr., Salant DJ. Membranous Nephropathy: A Journey From Bench to Bedside. Am J Kidney Dis. 2016;68(1):138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beck LH Jr., Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sethi S. New ‘Antigens’ in Membranous Nephropathy. J Am Soc Nephrol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bomback AS, Gharavi AG. Can genetics risk-stratify patients with membranous nephropathy? J Am Soc Nephrol. 2013;24(8):1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stanescu HC, Arcos-Burgos M, Medlar A, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med. 2011;364(7):616–626. [DOI] [PubMed] [Google Scholar]
- 66.Sekula P, Li Y, Stanescu HC, et al. Genetic risk variants for membranous nephropathy: extension of and association with other chronic kidney disease aetiologies. Nephrol Dial Transplant. 2017;32(2):325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramachandran R, Kumar V, Kumar A, et al. PLA2R antibodies, glomerular PLA2R deposits and variations in PLA2R1 and HLA-DQA1 genes in primary membranous nephropathy in South Asians. Nephrol Dial Transplant. 2016;31(9):1486–1493. [DOI] [PubMed] [Google Scholar]
- 68.Xie J, Liu L, Mladkova N, et al. The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat Commun. 2020;11(1):1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cosyns JP, Couchoud C, Pouteil-Noble C, Squifflet JP, Pirson Y. Recurrence of membranous nephropathy after renal transplantation: probability, outcome and risk factors. Clin Nephrol. 1998;50(3):144–153. [PubMed] [Google Scholar]
- 70.Josephson MA, Spargo B, Hollandsworth D, Thistlethwaite JR. The recurrence of recurrent membranous glomerulopathy in a renal transplant recipient: case report and literature review. Am J Kidney Dis. 1994;24(5):873–878. [DOI] [PubMed] [Google Scholar]
- 71.Dabade TS, Grande JP, Norby SM, Fervenza FC, Cosio FG. Recurrent idiopathic membranous nephropathy after kidney transplantation: a surveillance biopsy study. Am J Transplant. 2008;8(6):1318–1322. [DOI] [PubMed] [Google Scholar]
- 72.El-Zoghby ZM, Grande JP, Fraile MG, Norby SM, Fervenza FC, Cosio FG. Recurrent idiopathic membranous nephropathy: early diagnosis by protocol biopsies and treatment with anti-CD20 monoclonal antibodies. Am J Transplant. 2009;9(12):2800–2807. [DOI] [PubMed] [Google Scholar]
- 73.Sprangers B, Lefkowitz GI, Cohen SD, et al. Beneficial effect of rituximab in the treatment of recurrent idiopathic membranous nephropathy after kidney transplantation. Clin J Am Soc Nephrol. 2010;5(5):790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Batal I, Vasilescu ER, Dadhania DM, et al. Association of HLA Typing and Alloimmunity With Posttransplantation Membranous Nephropathy: A Multicenter Case Series. Am J Kidney Dis. 2020;76(3):374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stahl R, Hoxha E, Fechner K. PLA2R autoantibodies and recurrent membranous nephropathy after transplantation. N Engl J Med. 2010;363(5):496–498. [DOI] [PubMed] [Google Scholar]
- 76.Quintana LF, Blasco M, Seras M, et al. Antiphospholipase A2 Receptor Antibody Levels Predict the Risk of Posttransplantation Recurrence of Membranous Nephropathy. Transplantation. 2015;99(8):1709–1714. [DOI] [PubMed] [Google Scholar]
- 77.Couchoud C, Pouteil-Noble C, Colon S, Touraine JL. Recurrence of membranous nephropathy after renal transplantation. Incidence and risk factors in 1614 patients. Transplantation. 1995;59(9):1275–1279. [PubMed] [Google Scholar]
- 78.Andresdottir MB, Wetzels JF. Increased risk of recurrence of membranous nephropathy after related donor kidney transplantation. Am J Transplant. 2012;12(1):265–266. [DOI] [PubMed] [Google Scholar]
- 79.Berchtold L, Letouze E, Alexander MP, et al. HLA-D and PLA2R1 risk alleles associate with recurrent primary membranous nephropathy in kidney transplant recipients. Kidney Int. 2020. [DOI] [PubMed] [Google Scholar]
- 80.D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365(25):2398–2411. [DOI] [PubMed] [Google Scholar]
- 81.Kitiyakara C, Kopp JB, Eggers P. Trends in the epidemiology of focal segmental glomerulosclerosis. Semin Nephrol. 2003;23(2):172–182. [DOI] [PubMed] [Google Scholar]
- 82.De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC. Differentiating Primary, Genetic, and Secondary FSGS in Adults: A Clinicopathologic Approach. J Am Soc Nephrol. 2018;29(3):759–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rovin BH, Caster DJ, Cattran DC, et al. Management and treatment of glomerular diseases (part 2): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019;95(2):281–295. [DOI] [PubMed] [Google Scholar]
- 84.Hansrivijit P, Cheungpasitporn W, Thongprayoon C, Ghahramani N. Rituximab therapy for focal segmental glomerulosclerosis and minimal change disease in adults: a systematic review and meta-analysis. BMC Nephrol. 2020;21(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rosenberg AZ, Kopp JB. Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 2017;12(3):502–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010;5(11):2115–2121. [DOI] [PubMed] [Google Scholar]
- 87.Delville M, Sigdel TK, Wei C, et al. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med. 2014;6(256):256ra136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei C, El Hindi S, Li J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011;17(8):952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sethi S, Zand L, Nasr SH, Glassock RJ, Fervenza FC. Focal and segmental glomerulosclerosis: clinical and kidney biopsy correlations. Clin Kidney J. 2014;7(6):531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Laurinavicius A, Hurwitz S, Rennke HG. Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int. 1999;56(6):2203–2213. [DOI] [PubMed] [Google Scholar]
- 91.D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004;43(2):368–382. [DOI] [PubMed] [Google Scholar]
- 92.Lovric S, Ashraf S, Tan W, Hildebrandt F. Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant. 2016;31(11):1802–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yao T, Udwan K, John R, et al. Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS. Clin J Am Soc Nephrol. 2019;14(2):213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barua M, Brown EJ, Charoonratana VT, Genovese G, Sun H, Pollak MR. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013;83(2):316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Debiec H, Dossier C, Letouze E, et al. Transethnic, Genome-Wide Analysis Reveals Immune-Related Risk Alleles and Phenotypic Correlates in Pediatric Steroid-Sensitive Nephrotic Syndrome. J Am Soc Nephrol. 2018;29(7):2000–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jia X, Yamamura T, Gbadegesin R, et al. Common risk variants in NPHS1 and TNFSF15 are associated with childhood steroid-sensitive nephrotic syndrome. Kidney Int. 2020;98(5):1308–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abbott KC, Sawyers ES, Oliver JD 3rd, et al. Graft loss due to recurrent focal segmental glomerulosclerosis in renal transplant recipients in the United States. Am J Kidney Dis. 2001;37(2):366–373. [DOI] [PubMed] [Google Scholar]
- 98.Francis A, Trnka P, McTaggart SJ. Long-Term Outcome of Kidney Transplantation in Recipients with Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 2016;11(11):2041–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sener A, Bella AJ, Nguan C, Luke PP, House AA. Focal segmental glomerular sclerosis in renal transplant recipients: predicting early disease recurrence may prolong allograft function. Clin Transplant. 2009;23(1):96–100. [DOI] [PubMed] [Google Scholar]
- 100.Hickson LJ, Gera M, Amer H, et al. Kidney transplantation for primary focal segmental glomerulosclerosis: outcomes and response to therapy for recurrence. Transplantation. 2009;87(8):1232–1239. [DOI] [PubMed] [Google Scholar]
- 101.Newstead CG. Recurrent disease in renal transplants. Nephrol Dial Transplant. 2003;18 Suppl 6:vi68–74. [DOI] [PubMed] [Google Scholar]
- 102.Fujisawa M, Iijima K, Ishimura T, et al. Long-term outcome of focal segmental glomerulosclerosis after Japanese pediatric renal transplantation. Pediatr Nephrol. 2002;17(3):165–168. [DOI] [PubMed] [Google Scholar]
- 103.Zanoni F, Kiryluk K. Genetic background and transplantation outcomes: insights from genome-wide association studies. Curr Opin Organ Transplant. 2020;25(1):35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Laurinavicius A, Rennke HG. Collapsing glomerulopathy--a new pattern of renal injury. Semin Diagn Pathol. 2002;19(3):106–115. [PubMed] [Google Scholar]
- 105.Nicholas Cossey L, Larsen CP, Liapis H. Collapsing glomerulopathy: a 30-year perspective and single, large center experience. Clinical Kidney Journal. 2017;10(4):443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Albaqumi M, Soos TJ, Barisoni L, Nelson PJ. Collapsing glomerulopathy. J Am Soc Nephrol. 2006;17(10):2854–2863. [DOI] [PubMed] [Google Scholar]
- 107.Smeets B, Uhlig S, Fuss A, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol. 2009;20(12):2604–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hakroush S, Cebulla A, Schaldecker T, Behr D, Mundel P, Weins A. Extensive podocyte loss triggers a rapid parietal epithelial cell response. J Am Soc Nephrol. 2014;25(5):927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111(5):707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Raghubeer S, Pillay TS, Matsha TE. Gene of the month: APOL1. J Clin Pathol. 2020. [DOI] [PubMed] [Google Scholar]
- 111.Friedman DJ, Pollak MR. APOL1 and Kidney Disease: From Genetics to Biology. Annu Rev Physiol. 2020;82:323–342. [DOI] [PubMed] [Google Scholar]
- 112.Kasembeli AN, Duarte R, Ramsay M, et al. APOL1 Risk Variants Are Strongly Associated with HIV-Associated Nephropathy in Black South Africans. J Am Soc Nephrol. 2015;26(11):2882–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kudose SBI, Santoriello D et al. Kidney Biopsy Findings in Patients with COVID-19. J Am Soc Nephrol. 2020;(In Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu H, Larsen CP, Hernandez-Arroyo CF, et al. AKI and Collapsing Glomerulopathy Associated with COVID-19 and APOL 1 High-Risk Genotype. J Am Soc Nephrol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nichols B, Jog P, Lee JH, et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2015;87(2):332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Santoriello D, Husain SA, De Serres SA, et al. Donor APOL1 high-risk genotypes are associated with increased risk and inferior prognosis of de novo collapsing glomerulopathy in renal allografts. Kidney Int. 2018;94(6):1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Meehan SM, Pascual M, Williams WW, et al. De novo collapsing glomerulopathy in renal allografts. Transplantation. 1998;65(9):1192–1197. [DOI] [PubMed] [Google Scholar]
- 118.Stokes MB, Davis CL, Alpers CE. Collapsing glomerulopathy in renal allografts: a morphological pattern with diverse clinicopathologic associations. Am J Kidney Dis. 1999;33(4):658–666. [DOI] [PubMed] [Google Scholar]
- 119.Swaminathan S, Lager DJ, Qian X, Stegall MD, Larson TS, Griffin MD. Collapsing and non-collapsing focal segmental glomerulosclerosis in kidney transplants. Nephrol Dial Transplant. 2006;21(9):2607–2614. [DOI] [PubMed] [Google Scholar]
- 120.Canaud G, Bruneval P, Noel LH, et al. Glomerular collapse associated with subtotal renal infarction in kidney transplant recipients with multiple renal arteries. Am J Kidney Dis. 2010;55(3):558–565. [DOI] [PubMed] [Google Scholar]
- 121.Nadasdy T, Allen C, Zand MS. Zonal distribution of glomerular collapse in renal allografts: possible role of vascular changes. Hum Pathol. 2002;33(4):437–441. [DOI] [PubMed] [Google Scholar]
- 122.Shah PB, Cooper JE, Lucia MS, Boils C, Larsen CP, Wiseman AC. APOL1 Polymorphisms in a Deceased Donor and Early Presentation of Collapsing Glomerulopathy and Focal Segmental Glomerulosclerosis in Two Recipients. Am J Transplant. 2016;16(6):1923–1927. [DOI] [PubMed] [Google Scholar]
- 123.Lim WH, Shingde M, Wong G. Recurrent and de novo Glomerulonephritis After Kidney Transplantation. Front Immunol. 2019;10:1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Abbas F, El Kossi M, Jin JK, Sharma A, Halawa A. De novo glomerular diseases after renal transplantation: How is it different from recurrent glomerular diseases? World J Transplant. 2017;7(6):285–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Van Loon E, Bernards J, Van Craenenbroeck AH, Naesens M. The Causes of Kidney Allograft Failure: More Than Alloimmunity. A Viewpoint Article. Transplantation. 2020;104(2):e46–e56. [DOI] [PubMed] [Google Scholar]
- 126.Giannico GA, Arnold S, Langone A, et al. Non-immunoglobulin A mesangial immune complex glomerulonephritis in kidney transplants. Hum Pathol. 2015;46(10):1521–1528. [DOI] [PubMed] [Google Scholar]
- 127.Grau V, Zeuschner P, Immenschuh S, et al. Immune Complex-Type Deposits in the Fischer-344 to Lewis Rat Model of Renal Transplantation and a Subset of Human Transplant Glomerulopathy. Transplantation. 2016;100(5):1004–1014. [DOI] [PubMed] [Google Scholar]
- 128.Debiec H, Guigonis V, Mougenot B, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med. 2002;346(26):2053–2060. [DOI] [PubMed] [Google Scholar]
- 129.Byrne-Dugan CJ, Collins AB, Lam AQ, Batal I. Membranous nephropathy as a manifestation of graft-versus-host disease: association with HLA antigen typing, phospholipase A2 receptor, and C4d. Am J Kidney Dis. 2014;64(6):987–993. [DOI] [PubMed] [Google Scholar]
- 130.Chin K-K, Charu V, O’Shaughnessy MM, Troxell ML, Cheng XS. Histologic Case Definition of an Atypical Glomerular Immune-Complex Deposition Following Kidney Transplantation. Kidney International Reports. 2020;5(5):632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Khairallah P, Kamal J, Crew RJ, et al. The Association Between Post-Kidney Transplant De Novo Glomerulonephritis and Alloimmunity. Kidney International Reports. 2020. (In Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Larsen CP, Walker PD. Phospholipase A2 receptor (PLA2R) staining is useful in the determination of de novo versus recurrent membranous glomerulopathy. Transplantation. 2013;95(10): 1259–1262. [DOI] [PubMed] [Google Scholar]
- 133.Wen J, Xie K, Zhang M, et al. HLA-DR, and not PLA2R, is expressed on the podocytes in kidney allografts in de novo membranous nephropathy. Medicine (Baltimore). 2016;95(37):e4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cruzado JM, Carrera M, Torras J, Grinyo JM. Hepatitis C virus infection and de novo glomerular lesions in renal allografts. Am J Transplant. 2001;1(2):171–178. [PubMed] [Google Scholar]
- 135.Lloyd IE, Ahmed F, Revelo MP, Khalighi MA. De novo immune complex deposition in kidney allografts: a series of 32 patients. Hum Pathol. 2018;71:109–116. [DOI] [PubMed] [Google Scholar]