

SUMMARY
Asymmetric alkene hydroamination could be a direct route to valuable chiral amines from abundant feedstocks. However, most asymmetric hydroaminations have limited synthetic value because they require a large excess of alkene, occur with modest enantioselectivity, and proceed with limited tolerance of functional groups. We report an enantioselective, intermolecular hydroamination of unactivated terminal alkenes that occurs with equimolar amounts of alkene and amine, tolerates many functional groups, and occurs in high yield, with high enantioselectivity and turnover numbers. Mechanistic studies revealed factors, including reversibility of the addition, reversible oxidation of the product amine, competing isomerization of the alkene reactant, and unfavorable replacement of sacrificial ligands in standard catalyst precursors by the chiral bisphosphine, that needed to be addressed to achieve enantioselective N–H additions to alkenes.
Graphical Abstract
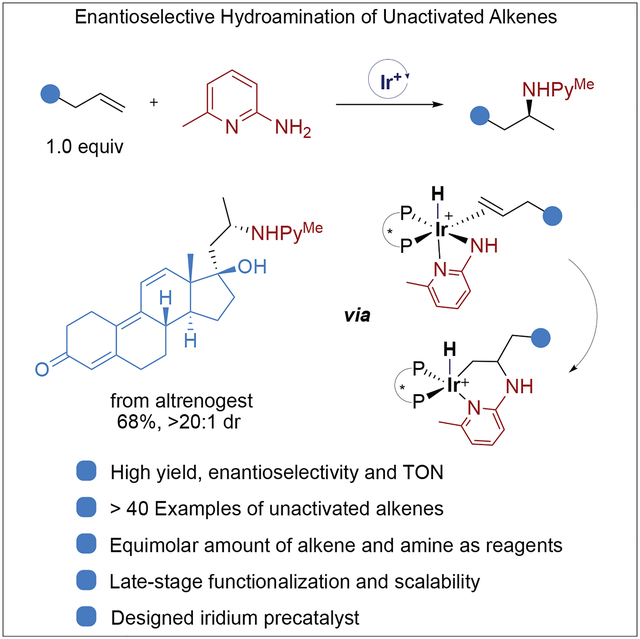
Enantioselective addition of an N–H bond to an alkene is an attractive yet challenging transformation for the synthesis of chiral amines from abundant feedstocks. To this end, we report a highly enantioselective, intermolecular hydroamination of structurally diverse unactivated terminal alkenes under mild conditions. A precatalyst crucial for this reaction was identified by revealing the undesired side reactions that occur in competition with hydroamination.
INTRODUCTION
Catalytic, asymmetric hydrofunctionalization of alkenes can provide direct access to an array of enantioenriched, chiral building blocks from simple chemical feedstocks.1–4 The hydrofunctionalization of terminal alkenes is particularly valuable for converting feedstocks to structurally diverse compounds and for the late-stage derivatization of medicinally relevant molecules, because terminal alkenes are broadly accessible and possess orthogonal reactivity to common polar functional groups, such as ketones, halides, and alcohols.5 However, hydrofunctionalizations that proceed with high Markovnikov selectivity, excellent enantioselectivity, and a high degree of generality are rare.6
Among potential enantioselective hydrofunctionalizations, the asymmetric Markovnikov hydroamination of a terminal alkene is especially valuable because it produces chiral amines bearing an α-“alkyl-methyl” stereocenter, which is found in a wide range of pharmaceuticals and agrochemicals (Figure 1A).7 These amines are commonly prepared by enantioselective reductive amination8–12 and hydrogenation of enamides and imines,10,13 enzymatic amination of alcohols,14 and the addition of an organometallic reagent to an imine bearing a chiral auxiliary.15 Although valuable, these approaches have limited compatibility with functional groups, high dependence of the yield and enantioselectivity on the structure of the substrate, and a need to install a reactive functional group into feedstock hydrocarbons before the amination reaction.
Figure 1. Amines as biologically active compounds and catalytic hydroamination of terminal alkenes.
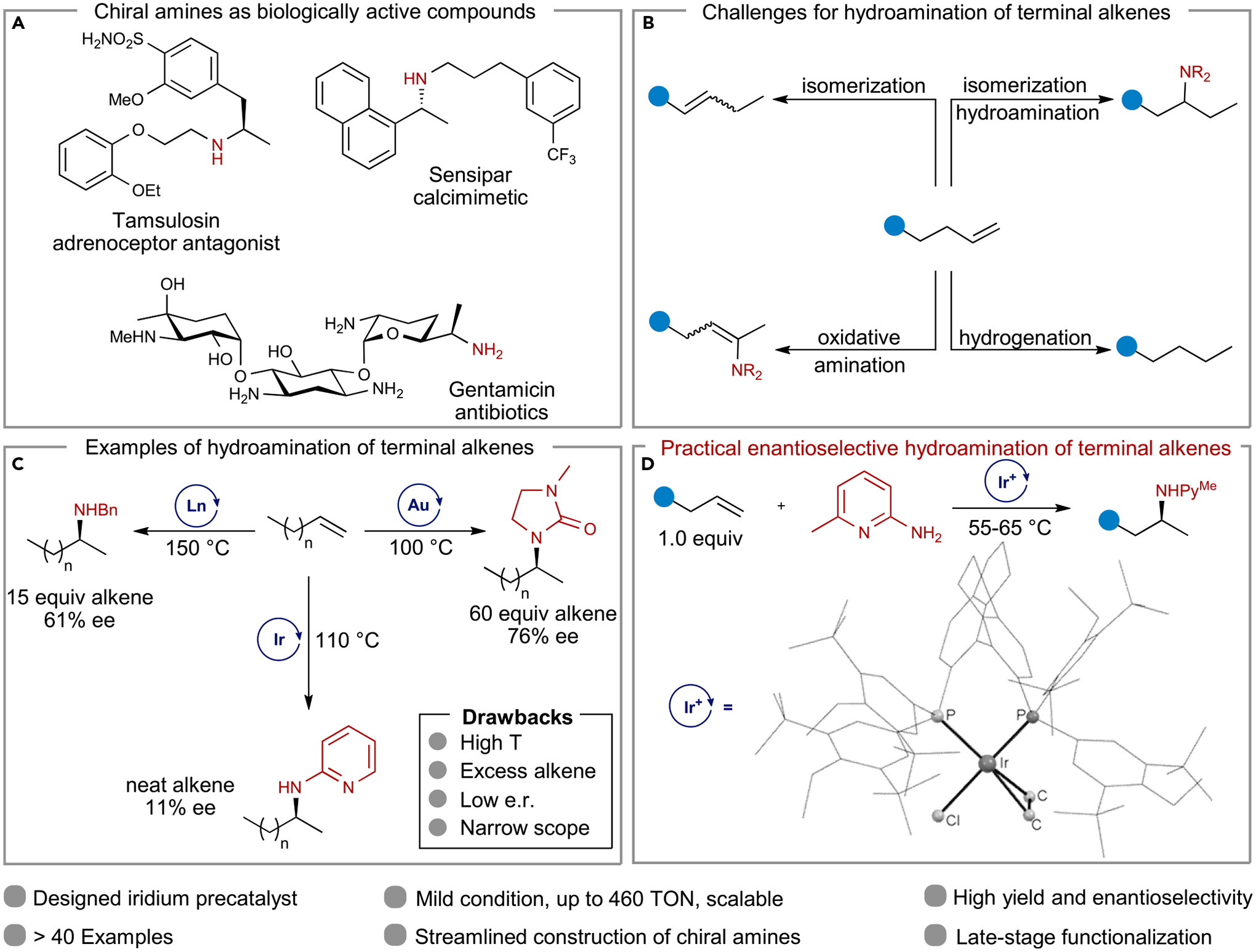
(A) Biologically active compounds containing chiral alkyl methyl amines.
(B) Major challenges for the enantioselective hydroamination of terminal alkenes.
(C) Representative examples of enantioselective hydroamination of terminal alkenes and their limitations.
(D) This work: practical enantioselective hydroamination of terminal alkenes.
Although the addition of an N–H bond to an alkene has long been envisioned as a route to chiral amines from abundant alkene feedstocks, intermolecular additions of an amine to unactivated alkenes that occur in high enantiomeric excess are rare, and very few have been reported to occur with unactivated terminal alkenes with high enantiomeric excess (e.e.).16–20 Despite decades of effort toward the development of efficient catalytic hydroaminations, current methods suffer from competing side reactions, such as the isomerization,21 oxidative amination,22 and hydrogenation of the alkenes (Figure 1B).22 Consequently, they often occur only with alkenes that lack additional functional groups, and they occur at relatively high temperatures, in moderate yields, and with modest enantioselectivity (Figure 1C). Moreover, no asymmetric hydroamination has been reported in which the amine adds to the alkene lacking a directing group with nearly equal amounts of the alkene and amine, although this stoichiometry is needed for the reaction to be practical. Emerging, alternative strategies, such as formal hydroamination form linear amines exclusively, precluding the formation of α-chiral amines from terminal alkenes.23–27 We report a system for the catalytic hydroamination to form chiral amines bearing an α-“alkyl-methyl” stereocenter by an operationally simple, highly enantioselective hydroamination of structurally diverse, unactivated terminal alkenes under mild conditions (Figure 1D). Suppression of a series of side reactions and promotion of the N–H addition process were essential to achieving the high efficiency and high stereoselectivity of this hydroamination.
RESULTS AND DISCUSSION
Development of the reaction and mechanistic investigation
We recently reported that the combination of a cationic iridium catalyst, {[(R)-TMS-SYNPHOS]Ir(COD)}NTf2, 6,6′-bis(bis(3,5-bis(trimethylsilyl)phenyl)phosphaneyl)-2,2′,3,3′-tetrahydro-5,5′-bibenzo[b][1,4]dioxine; COD, cyclooctadiene; NTf2, bis(trifluoromethylsulfonyl)imide and a carefully designed amine, 2-amino-6-methylpyridine, enabled the direct asymmetric addition of an N–H bond across unactivated internal alkenes.16 Although the components of this catalyst and the accompanying reagent significantly enhanced the rate of hydroamination over that of prior hydroaminations, excess alkene (i.e., 10 equivalents) was required for the hydroamination to occur with high selectivity over alkene isomerization, and the compatibility of this reaction with functional groups was limited by the harsh reaction conditions (120°C). Therefore, we sought to address these limitations by developing a highly active system that would catalyze enantioselective hydroamination of unconjugated, unstrained terminal alkenes with a 1:1 ratio of alkene and amine under mild conditions.
At the outset of our study, we selected allylbenzene (2a) as the model alkene because of its propensity to undergo isomerization to form a thermodynamically stable, conjugated alkene, β-methylstyrene. This alkene would be a stringent test of a new system to resist isomerization. In the presence of {[(R)-TMS-SYNPHOS]Ir(COD)}NTf2 (Ir-0) as the catalyst, the equimolar reaction of allylbenzene and 6-methyl-2-aminopyridine (1a) at 120°C afforded the desired amine (2b) in only 42% yield with a modest 71:29 enantiomeric ratio (e.r.) after 35 h, along with β-methylstyrene (2c) in 58% yield (Figure 2A, entry 1).
Figure 2. Reaction development and mechanistic investigation.
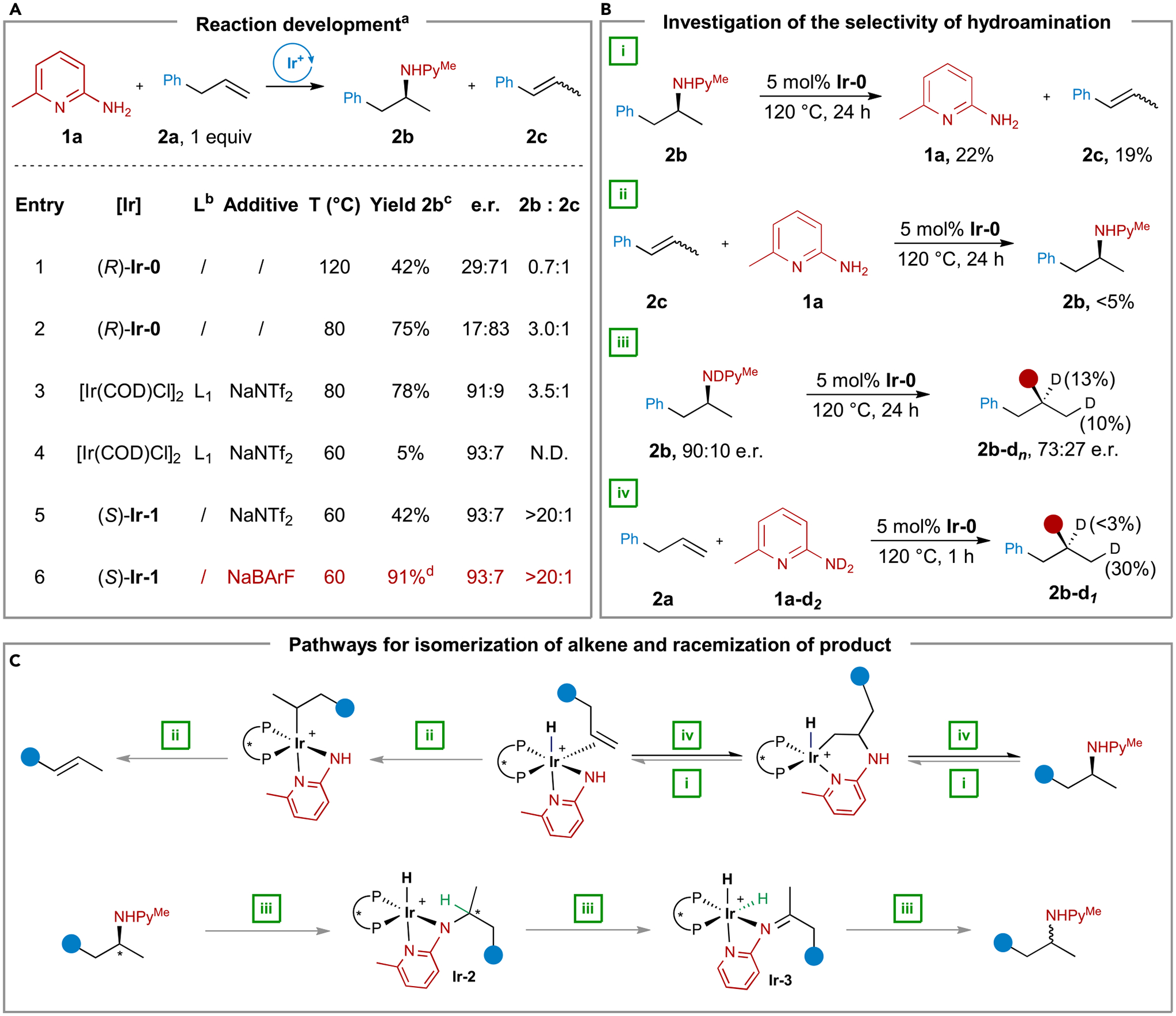
(A) Development of the reaction condition for the hydroamination of allylbenzene. aConditions: 1a (0.1 mmol), 2a (0.1 mmol), [Ir] (5 mol % by Ir), ligand (5 mol %), additive (6 mol %), dioxane (200 μL). bL1 = (S)-DTBM-SEGPHOS. cDetermined by 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as the internal standard. dIsolated yield.
(B) Investigation on the selectivity of the hydroamination of allylbenzene.
(C) Pathways for the isomerization of the alkene and the racemization of the product of hydroamination.
To understand the low selectivity toward hydroamination, we monitored the progress of this reaction by 1H NMR (nuclear magnetic resonance) spectroscopy and found that the ratio of amine 2b to β-methylstyrene 2c decreased throughout the reaction. Moreover, this ratio decreased further, even after >95% conversion of allylbenzene was observed (see supplemental information for details). This result and the knowledge that hydroamination is close to ergoneutral28 led us to consider that retrohydroamination (Figure 2C, pathway i), the reverse of hydroamination, could be occurring. To probe whether retrohydroamination is occurring, we subjected amine 2b to Ir-0 at 120°C for 24 h and observed the formation of β-methylstyrene 2c and the starting amine 1a in 19% and 22% yield, respectively (Figure 2B; reaction i). Furthermore, when a mixture of cis- and trans-2c was heated at 120°C in the presence of Ir-0 and amine 1a for 24 h, we did not observe the formation of either allylbenzene or amine 2b in significant amounts (<5%) (Figure 2B; reaction ii). These results implied that amine product 2b either underwent retrohydroamination to form β-methylstyrene directly or underwent retrohydroamination to form allylbenzene, which isomerized to β-methylstyrene irreversibly (Figure 2C, pathway ii). Therefore, a catalyst that promotes the N–H addition at sufficiently low temperatures is required for the hydroamination to be favored over retrohydroamination and isomerization of the alkene.
To probe the origin of the low enantioselectivity, we monitored the change in the e.r. of N-deuterated amine 2b when it was subjected to Ir-0 at 120°C. We observed a decrease of the e.r. from 90:10 to 73:27 after 24 h, a 13% decrease in the intensity of the 1H NMR signal for the proton at the α-position of amine 2b (Figure 2B; reaction iii), and a 2H NMR signal corresponding to a deuterium at this α-position, indicating that the N–D deuterium exchanges with the α-C–H proton (see supplemental information for more details).
In contrast, the reaction of allylbenzene with N,N-dideuterio-6-methyl-2-aminopyridine (1a-d2) and Ir-0 at 120°C for 1 h led to less than 3% incorporation of deuterium at the α-position of amine 2b (Figure 2B; reaction iv), as determined by 2H NMR spectroscopy and comparison of integrations in the 1H NMR spectra of 2b from reaction with 1a and 1a-d2. These results imply that deuterium at the α-position of amine 2b observed after heating N-deuterated amine 2b with catalyst Ir-0 did not result from the reversibility of the hydroamination process that forms amine 2b (Figure 2C, pathway iv). Instead, it was most likely incorporated by a separate pathway (Figure 2C, pathway iii) that also leads to racemization and that occurs by oxidative addition of the N–D bond of amine 2b to form intermediate Ir-2, subsequent β-hydrogen elimination of Ir-2 to generate intermediate Ir-3, site exchange between the hydride and the deuteride, and addition of deuteride to the imine to form the amine 2b with incorporation of deuterium at the α-position. This pathway, together with the retrohydroamination of amine 2b, would then account for the erosion of the enantiopurity of amine 2b, and a catalyst that undergoes the competing β-hydrogen elimination more slowly is needed to suppress the racemization of the enantioenriched amine product.21
As a consequence of these detrimental processes, a highly chemo- and enantioselective hydroamination of terminal alkenes without the use of an excess amount of alkene would require a catalytic system that promotes the pathway for hydroamination (Figure 2C, pathway iv) over the three undesired pathways for retrohydroamination (Figure 2C, pathway i), alkene isomerization (Figure 2C, pathway ii), and reversible dehydrogenation of the product amine to form the imine (Figure 2C, pathway iii). To identify such a system, we first attempted to conduct the reaction at temperatures lower than 120°C. We observed that the ratio between amine 2b and β-methylstyrene 2c increased from 0.7 to 3.0 and the e.r. of product 2b increased from 71:29 to 83:17 when the reaction was conducted at 80°C (Figure 2A, entry 2). The ratio between 2b and 2c and the e.r. further increased to 3.5 and 91:9 by replacing Ir-0 with a combination of [Ir(COD)Cl]2, (S)-DTBM-SEGPHOS ((S)-L1, (S)-(+)-5,5′-Bis[di(3,5-di-tert-butyl-4-methoxyphenyl)phosphino]-4,4′-bi-1,3-benzodioxole) and NaNTf2 (Figure 2A, entry 3). We were unable to increase the selectivity of the reaction further by conducting the reaction at temperatures lower than 80°C because 2b was formed in only 5% yield when the reaction was conducted at 60°C, due to low conversion (Figure 2A, entry 4). Therefore, the development of a more active catalyst that could promote the reaction below 80°C was required to enhance the selectivity of the reaction further.
To achieve this goal, we first sought to understand the large decrease in catalytic activity at 60°C. The 31P NMR spectrum of the reaction catalyzed by the mixture of [Ir(COD)Cl]2, (S)-DTBM-SEGPHOS, and NaNTf2 at 60°C contained two resonances that correspond to [Ir[(S)-DTBM-SEGPHOS](COD)]NTf2 and free DTBM-SEGPHOS. This result suggests that cyclooctadiene coordinated more strongly to iridium than did (S)-DTBM-SEGPHOS and aminopyridine at 60°C, forming off-cycle intermediates that did not catalyze the desired hydroamination.
We hypothesized that initiation of the hydroamination with an iridium precursor containing a monodentate, unstrained, and volatile alkene would suppress the formation of off-cycle intermediates, leading to an increase in the rate of the catalytic reaction. Thus, we prepared [Ir[(S)-DTBM-SEGPHOS](ethylene)(Cl)] ([(S)-Ir-1]),29 which contains ethylene as the ancillary ligand in place of COD, and examined the hydroamination of allylbenzene with (S)-Ir-1 as the precatalyst. The reaction catalyzed by the combination of (S)-Ir-1 and NaNTf2 at 60°C was significantly faster than that catalyzed by the combination of an iridium precursor containing COD as the ligand, affording 2b in 42% yield with 93:7 e.r. (Figure 2A, entry 5). The yield of 2b increased to 91% when NaBArF was used instead of NaNTf2 (Figure 2A, entry 6). Side products, such as β-methylstyrene, were not observed under these conditions, indicating that the catalyst formed from (S)-Ir-1 was selective for hydroamination, even with an alkene that is prone to isomerization. In addition, the 31P NMR spectrum of the reaction catalyzed by the combination of (S)-Ir-1 and NaBArF (Sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) did not contain a resonance corresponding to free DTBM-SEGPHOS. This observation supported our hypothesis that the reaction with an iridium precursor containing a monodentate, unstrained, and volatile alkene suppressed the formation of unligated, off-cycle intermediates and led to higher catalytic activity for hydroamination. Finally, heating N-deuterated amine 2b with (S)-Ir-1 and NaBArF at 60°C for 24 h to probe for reversible hydroamination did not lead to the incorporation of deuterium into the positions α or β to the nitrogen, and only minimum erosion of the enantiomeric excess (1%) of amine 2b was observed (see supplemental information for more details). These results imply that the catalyst formed from (S)-Ir-1 leads to less racemization of the enantioenriched product via retrohydroamination (Figure 2C, pathway i) and the formation of an imine intermediate (Figure 2C, pathway iii) than the catalyst we reported previously for the hydroamination of internal alkenes.16
Investigation of the scope of the reaction
Having established suitable conditions for the enantioselective hydroamination of the model alkene, we examined the scope of hydroaminations with amine 1a catalyzed by (S)-Ir-1 (Figures 3 and 4). The scope of the asymmetric hydrogenation is remarkably broad. Both simple (2b–5b) and functionalized terminal alkenes (6b–34b) underwent hydroamination in high yields and with good to excellent enantioselectivity. The asymmetric hydroamination tolerates a wide range of functional groups, including phenols (8b, 9b), ethers (10b), internal alkenes (11b), silyl-protected and free alcohols (12b, 13b), phosphates (14b), esters (15b–17b), amides (18b), protected amines (19b–21b), ketals (22b), acetals (23b), and cyclopropanes (24b). Nitro- (25b) and haloarenes (26b), which are often sensitive to reductive and nucleophilic conditions, were also compatible with the conditions of this hydroamination. Furthermore, hydroamination of alkenes in substrates containing medicinally privileged motifs, such as trifluoromethyl (27b, 28b), furyl (29b), thienyl (30b), pyridyl (31b), indolyl (33b), and carbazolyl (34b) groups, furnished products in high yields and with excellent enantioselectivity.
Figure 3. Scope of the hydroamination of unactivated alkenes with common functional groups.
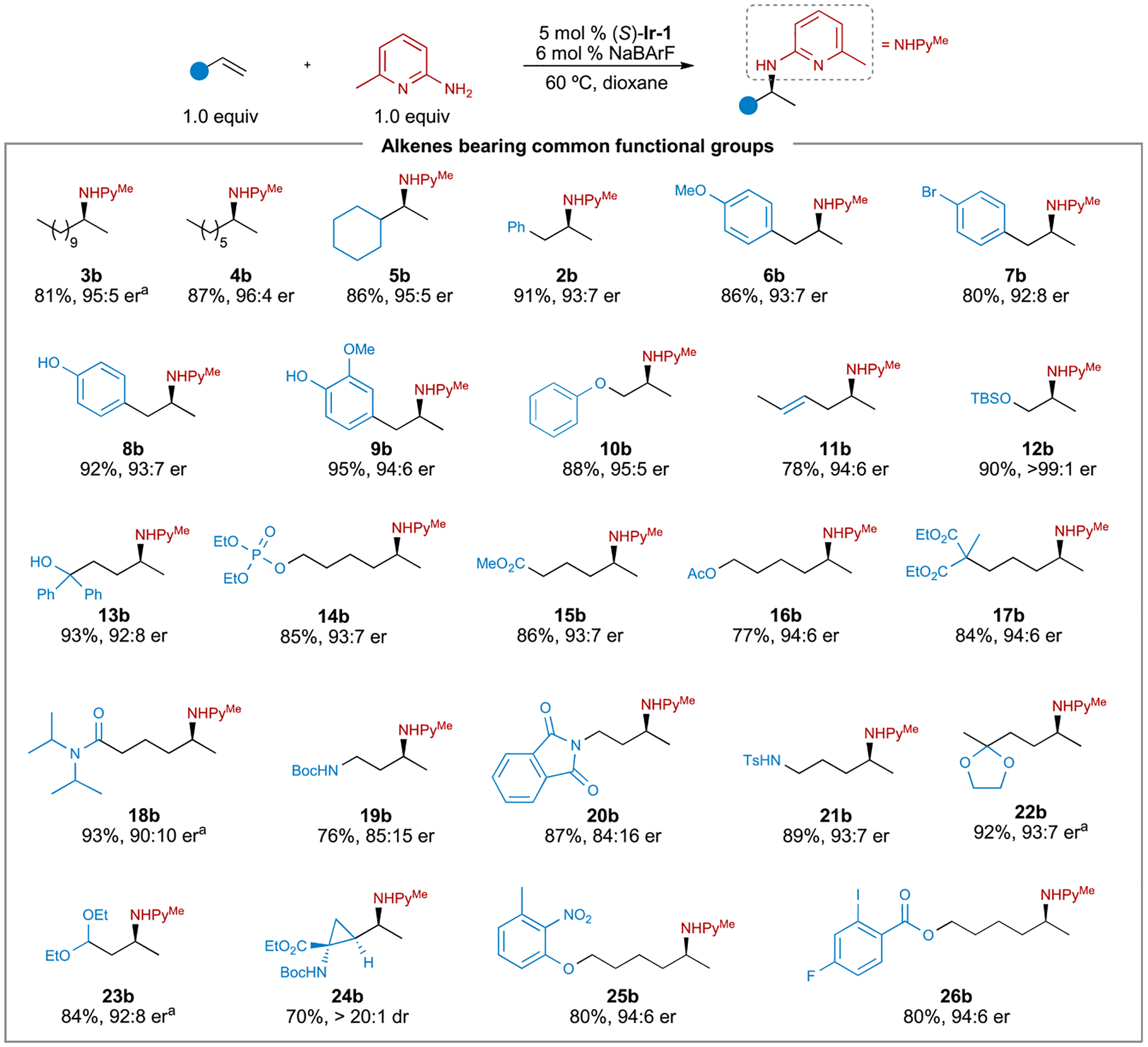
aThe reaction was performed at 55°C.
Figure 4. Scope of alkenes bearing medicinally privileged motifs and alkenes derived from natural products and pharmaceuticals.
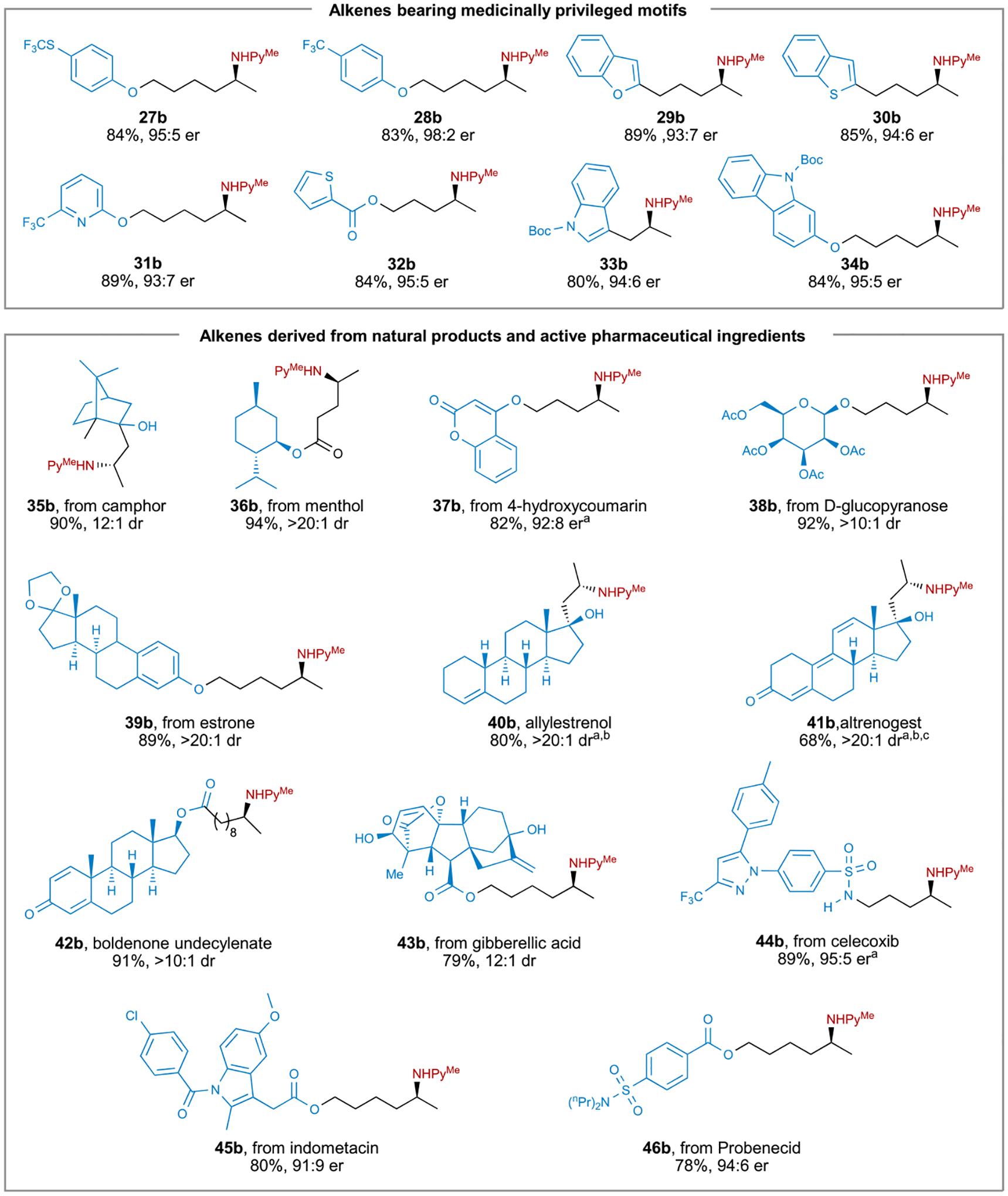
aThe reaction was performed at 65°C. bThe reaction was performed with (R)-Ir-1. cThe reaction was performed with 7.5 mol % (R)-Ir-1.
To explore the possibility of late-stage functionalization of bioactive and drug-like molecules with this hydroamination process, we conducted the hydroamination of alkenes that are tethered to the cores of natural products and active pharmaceutical ingredients (Figure 4). Substrates derived from natural products containing alcohol, ester, α,β-unsaturated carbonyl, and hemiacetal groups (35b–38b) underwent hydroamination in high yields and with high diastereo- or enantioselectivity. The hydroamination of steroids with different levels of oxidation (39b–43b) also proceeded smoothly to afford the corresponding chiral amines. Moreover, alkenes derived from pharmaceuticals that contain allylic alcohols, heterocycles, aryl halides, and sulfonamides (43b–46b) underwent hydroamination in high yield and with high enantioselectivity. The absolute configuration of product 4b was determined to be S at the nitrogen-bound stereocenter by comparison of the chiral HPLC traces of the standard sample ent-4b prepared by cross-coupling.16 The absolute configurations of the other products were assigned by analogy. In all of the cases mentioned earlier, we did not observe the formation of side products from the isomerization or oxidative amination during the hydroamination, and the mass balance corresponded to the unreacted terminal alkene. The successful incorporation of amines into complex molecules containing a variety of functional groups demonstrated a high level of both chemoselectivity and stereoselectivity of our method.
Synthetic applications
The pyridyl group of the hydroamination products can be removed by a short sequence of reactions (Figure 5A). Acylation, N-methylation at the pyridine nitrogen, and nucleophilic aromatic substitution converted pyridylamines 4b, 6b, and 28b into the corresponding Boc-protected amines (4c, 6c, and 28c) in 70%–82% yield with no erosion of enantiopurity.30 The corresponding primary amine (18c) was obtained by successive hydrogenation and reduction of amine 18b in high yield and without racemization.31 To demonstrate the value of this method for the synthesis of pharmaceutically relevant chiral amines, we prepared amine ent-6c, which is a key intermediate for the synthesis of the biologically active molecules carmoterol and tamsulosin, on a 4 mmol scale (Figure 5B).32
Figure 5. Synthetic applications.
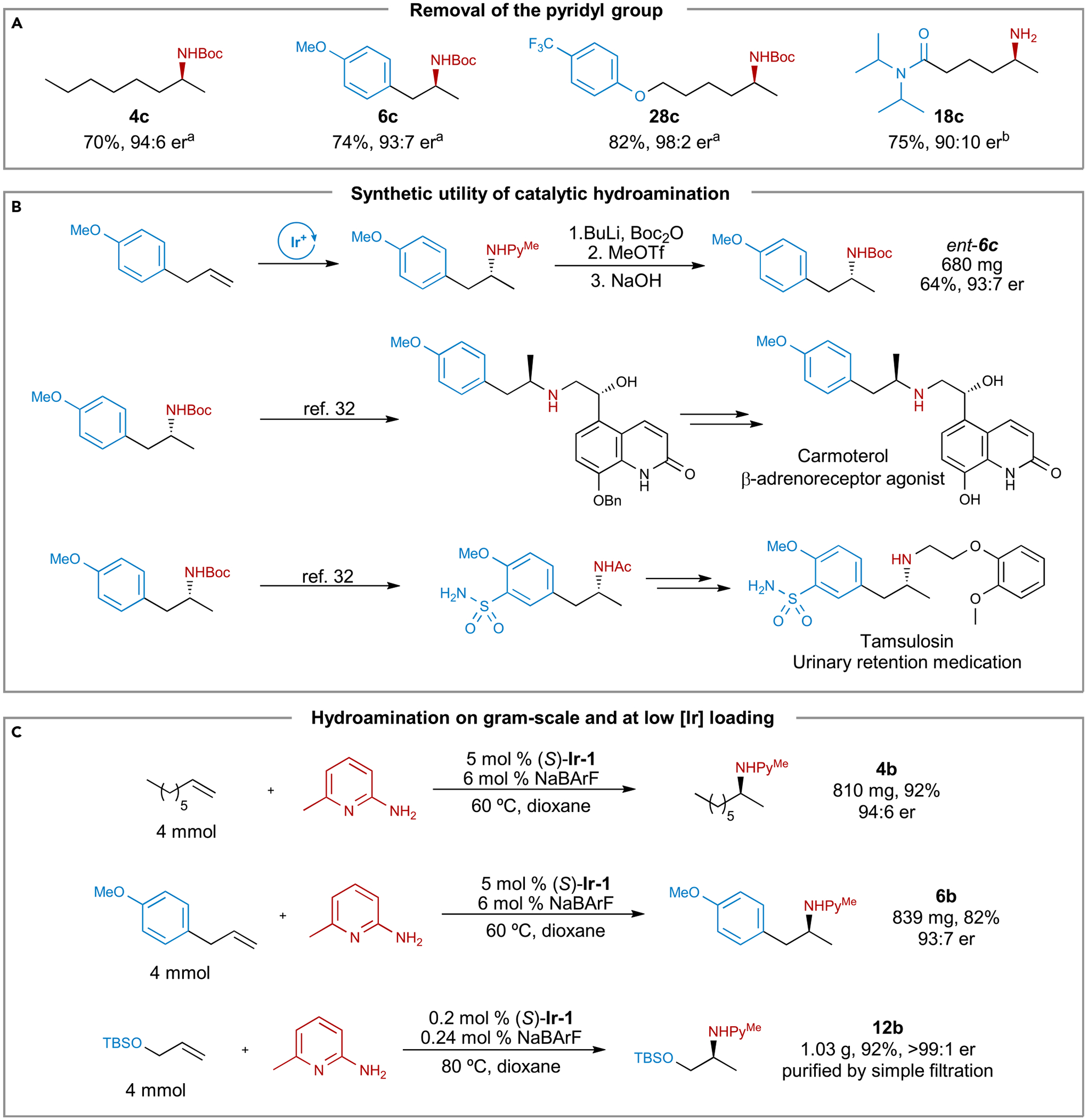
(A) Removal of the pyridyl group from the hydroamination products. aConditions: n-BuLi, Boc2O, THF; MeOTf, DCM; NaOH, EtOH, reflux. bConditions: PtO2, HCl, H2 (1 atm); NaBH4, EtOH.
(B) Synthetic utility of the catalytic enantioselective hydroamination.
(C) Hydroamination on gram-scale and at low [Ir] loading.
Our catalytic hydroamination is easily scalable and operationally simple. The hydroaminations of 1-octene and of 4-methoxyallylbenzene on a 4 mmol scale afforded the corresponding products in yields and with enantioselectivity that were comparable to those from reactions conducted on a 0.1 mmol scale (Figure 5C). In contrast to prior hydroaminations of unactivated alkenes, even those with achiral catalysts or catalysts forming products with low enantioselectivity,17–19,21,22 our current catalyst enabled the reaction to occur with a high turnover number (TON = 460). The hydroamination of silyl-protected 2-propen-1-ol reached >95% conversion with 0.2 mol % loading of (S)-Ir-1, and over 1 g of the pure chiral amine 12b was isolated by a simple filtration in 92% yield. The high TON suggests that our new iridium precursor forms a highly active yet robust catalyst.
Conclusions
The combination of high activity, broad scope, and high enantioselectivity show that the often-stated potential of hydroamination to convert commodity alkenes to chiral amines enantioselectively can be a reality. We show that recognition of competing side reactions, including the isomerization of the alkene, retrohydroamination of the product amine, and dehydrogenation of the amine to form the imine, which diminished the chemo- and enantioselectivity of this transformation, and implementation of strategies to overcome them can lead to a catalyst for the addition of an amine to a structurally diverse set of alkenes with a 1:1 ratio of the two reactants. Detailed mechanistic investigation of this Ir-catalyzed hydroamination is currently ongoing in our laboratory. We anticipate that this discovery will inspire the future development of more active and selective catalysts for the hydrofunctionalization of alkenes and that reactions with additional nitrogen-based reagents that possess the properties of our 2-aminopyridine can lead to discovery of processes to form a variety of products containing stereogenic centers alpha to nitrogen derived from simple alkene feedstocks.
EXPERIMENTAL PROCEDURES
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, John F. Hartwig (jhartwig@berkeley.edu).
Materials availability
This study did not generate new materials.
Data and code availability
All data needed to support the conclusions of this manuscript are included in the main text or supplemental information.
Supplementary Material
Highlights.
Iridium-catalytzed hydroamination with high yield, enantioselectivity, and turnover number
Reaction occurs with a 1:1 ratio of amine to alkene
Compatible with many functional groups and applicable to late-stage functionalization
Results from the design of a precatalyst based on mechanistic insight
The bigger picture.
The enantioselective hydroamination of alkenes is an atom-economic route to chiral amines with important biological activities from readily available chemical feedstocks. However, current hydroaminations of alkenes are rarely applied in synthesis because they require excess of the alkene, are incompatible with common functional groups, and occur with only modest enantioselectivity.
Through an understanding of the competing side reactions that occur during hydroaminations, we designed a system comprising a specific iridium precatalyst that effects highly enantioselective hydroaminations of structurally varied unactivated alkenes containing a wide range of functional groups with a 1:1 ratio of the amine and the alkene. This discovery and the underlying principles should facilitate the development of catalytic hydrofunctionalizations with additional types of N–H bonds and other X–H bonds to elevate the practical utility of these reactions.
ACKNOWLEDGMENTS
The enantioselective aspects of the work were supported by the National Institutes of Health under grant R35GM130387, and the catalyst development was supported by the Director, Office of Science of the US Department of Energy under contract number DE-AC02-05CH11231. We gratefully acknowledge Takasago for gifts of (S)-DTBM-SEGPHOS, H. Celik for assistance with NMR experiments (NIH S10OD024998), M. Zhang for HRMS, and N. Settineri for X-ray crystallography (NIH S10-RR027172). S.M. thanks A. Fawcett, C.S. Day, K. Choi, I.F. Yu, Y. Qiu, and B. Su for helpful discussions and suggestions of manuscript; S. Pedram for supply of substrates and ligands; and Y. Gu, R. Chen, and Y. Xie for experimental assistance during the pandemic. Y.X. thanks Bristol-Myers Squibb for a graduate fellowship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.chempr.2021.12.005.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Crudden CM, and Edwards D (2003). Catalytic asymmetric hydroboration: recent advances and applications in carbon–carbon bond-forming reactions. Eur. J. Org. Chem 2003, 4695–4712. 10.1002/ejoc.200300433. [DOI] [Google Scholar]
- 2.Guo J, Cheng Z, Chen J, Chen X, and Lu Z (2021). Iron- and cobalt-catalyzed asymmetric hydrofunctionalization of alkenes and alkynes. Acc. Chem. Res 54, 2701–2716. 10.1021/acs.accounts.1c00212. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, and Lu Z (2018). Asymmetric hydrofunctionalization of minimally functionalized alkenes via earth abundant transition metal catalysis. Org. Chem. Front 5, 260–272. 10.1039/C7QO00613F. [DOI] [Google Scholar]
- 4.Liu RY, and Buchwald SL (2020). CuH-catalyzed olefin functionalization: from hydroamination to carbonyl addition. Acc. Chem. Res 53, 1229–1243. 10.1021/acs.accounts.0c00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mlynarski SN, Schuster CH, and Morken JP (2014). Asymmetric synthesis from terminal alkenes by cascades of diboration and cross-coupling. Nature 505, 386–390. 10.1038/nature12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coombs JR, and Morken JP (2016). Catalytic enantioselective functionalization of unactivated terminal alkenes. Angew. Chem. Int. Ed. Engl 55, 2636–2649. 10.1002/anie.201507151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuentes AV, Pineda MD, and Venkata KCN (2018). Comprehension of top 200 prescribed drugs in the US as a resource for pharmacy teaching, training and practice. Pharmacy (Basel) 6, 43. 10.3390/pharmacy6020043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinhuebel D, Sun Y, Matsumura K, Sayo N, and Saito T (2009). Direct asymmetric reductive amination. J. Am. Chem. Soc 131, 11316–11317. 10.1021/ja905143m. [DOI] [PubMed] [Google Scholar]
- 9.Storer RI, Carrera DE, Ni Y, and MacMillan DWC (2006). Enantioselective organocatalytic reductive amination. J. Am. Chem. Soc 128, 84–86. 10.1021/ja057222n. [DOI] [PubMed] [Google Scholar]
- 10.Tararov VI, and Börner A (2005). Approaching highly enantioselective reductive amination. Synlett 2005, 203–211. [Google Scholar]
- 11.Li C, Villa-Marcos B, and Xiao J (2009). Metal-Brønsted acid cooperative catalysis for asymmetric reductive amination. J. Am. Chem. Soc 131, 6967–6969. 10.1021/ja9021683. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, Pettman A, Basca J, and Xiao J (2010). A versatile catalyst for reductive amination by transfer hydrogenation. Angew. Chem. Int. Ed. Engl 49, 7548–7552. 10.1002/anie.201002944. [DOI] [PubMed] [Google Scholar]
- 13.Verendel JJ, Pàmies O, Diéguez M, and Andersson PG (2014). Asymmetric hydrogenation of olefins using chiral Crabtree-type catalysts: scope and limitations. Chem. Rev 114, 2130–2169. 10.1021/cr400037u. [DOI] [PubMed] [Google Scholar]
- 14.Mutti FG, Knaus T, Scrutton NS, Breuer M, and Turner NJ (2015). Conversion of alcohols to enantiopure amines through dual-enzyme hydrogen-borrowing cascades. Science 349, 1525–1529. 10.1126/science.aac9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robak MT, Herbage MA, and Ellman JA (2010). Synthesis and applications of tert-Butanesulfinamide. Chem. Rev 110, 3600–3740. 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- 16.Xi Y, Ma S, and Hartwig JF (2020). Catalytic asymmetric addition of an amine N–H bond across internal alkenes. Nature 588, 254–260. 10.1038/s41586-020-2919-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan S, Endo K, and Shibata T (2012). Ir(I)-catalyzed intermolecular regio- and enantioselective hydroamination of alkenes with heteroaromatic amines. Org. Lett 14, 780–783. 10.1021/ol203318z. [DOI] [PubMed] [Google Scholar]
- 18.Reznichenko AL, Nguyen HN, and Hultzsch KC (2010). Asymmetric intermolecular hydroamination of unactivated alkenes with simple amines. Angew. Chem. Int. Ed. Engl 49, 8984–8987. 10.1002/anie.201004570. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z, Lee SD, and Widenhoefer RA (2009). Intermolecular hydroamination of ethylene and 1-alkenes with cyclic ureas catalyzed by achiral and chiral gold(I) complexes. J. Am. Chem. Soc 131, 5372–5373. 10.1021/ja9001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanable EP, Kennemur JL, Joyce LA, Ruck RT, Schultz DM, and Hull KL (2019). Rhodium-catalyzed asymmetric hydroamination of allyl amines. J. Am. Chem. Soc 141, 739–742. 10.1021/jacs.8b09811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma S, Hill CK, Olen CL, and Hartwig JF (2021). Ruthenium-catalyzed hydroamination of unactivated terminal alkenes with stoichiometric amounts of alkene and an ammonia surrogate by sequential oxidation and reduction. J. Am. Chem. Soc 143, 359–368. 10.1021/jacs.0c11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sevov CS, Zhou J, and Hartwig JF (2012). Iridium-catalyzed intermolecular hydroamination of unactivated aliphatic alkenes with amides and sulfonamides. J. Am. Chem. Soc 134, 11960–11963. 10.1021/ja3052848. [DOI] [PubMed] [Google Scholar]
- 23.Rucker RP, Whittaker AM, Dang H, and Lalic G (2012). Synthesis of tertiary alkyl amines from terminal alkenes: copper-catalyzed amination of alkyl boranes. J. Am. Chem. Soc 134, 6571–6574. 10.1021/ja3023829. [DOI] [PubMed] [Google Scholar]
- 24.Miki Y, Hirano K, Satoh T, and Miura M (2013). Copper-catalyzed intermolecular regioselective hydroamination of styrenes with polymethylhydrosiloxane and hydroxylamines. Angew. Chem. Int. Ed. Engl 52, 10830–10834. 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]
- 25.Strom AE, and Hartwig JF (2013). One-pot anti-Markovnikov hydroamination of unactivated alkenes by hydrozirconation and amination. J. Org. Chem 78, 8909–8914. 10.1021/jo401498w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu S, Niljianskul N, and Buchwald SL (2013). Enantio- and regioselective CuH-catalyzed hydroamination of alkenes. J. Am. Chem. Soc 135, 15746–15749. 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen X, Chen X, Chen J, Sun Y, Cheng Z, and Lu Z (2020). Ligand-promoted cobalt-catalyzed radical hydroamination of alkenes. Nat. Commun 11, 783. 10.1038/s41467-020-14459-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johns AM, Sakai N, Ridder A, and Hartwig JF (2006). Direct measurement of the thermodynamics of vinylarene hydroamination. J. Am. Chem. Soc 128, 9306–9307. 10.1021/ja062773e. [DOI] [PubMed] [Google Scholar]
- 29.Ohmura T, Yagi K, Torigoe T, and Suginome M (2021). Intramolecular addition of a dimethylamino C(sp3)–H bond across C–C triple bonds using IrCl(DTBM-SEGPHOS)(ethylene) catalyst: synthesis of indoles from 2-Alkynyl-N-methylanilines. Synthesis 53, 3057–3064. [Google Scholar]
- 30.Dastbaravardeh N, Schnürch M, and Mihovilovic MD (2012). Ruthenium(0)-catalyzed sp3 C–H bond arylation of benzylic amines using arylboronates. Org. Lett 14, 1930–1933. 10.1021/ol300627p. [DOI] [PubMed] [Google Scholar]
- 31.Smout V, Peschiulli A, Verbeeck S, Mitchell EA, Herrebout W, Bultinck P, Vande Velde CML, Berthelot D, Meerpoel L, and Maes BUW (2013). Removal of the pyridine directing group from α-substituted N-(pyridin-2-yl)piperidines obtained via directed Rucatalyzed sp3 C–H functionalization. J. Org. Chem 78, 9803–9814. 10.1021/jo401521y. [DOI] [PubMed] [Google Scholar]
- 32.Wang J-W, Li Y, Nie W, Chang Z, Yu Z-A, Zhao Y-F, Lu X, and Fu Y (2021). Catalytic asymmetric reductive hydroalkylation of enamides and enecarbamates to chiral aliphatic amines. Nat. Commun 12, 1313. 10.1038/s41467-021-21600-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to support the conclusions of this manuscript are included in the main text or supplemental information.