

Abstract
Although epithelial-mesenchymal transition (EMT) is a common feature of fibrotic lung disease, its role in fibrogenesis is controversial. Recently, aberrant basaloid cells were identified in fibrotic lung tissue as a novel epithelial cell type displaying a partial EMT phenotype. The developmental origin of these cells remains unknown. To elucidate the role of EMT in the development of aberrant basaloid cells from the bronchial epithelium, we mapped EMT-induced transcriptional changes at the population and single-cell levels. Human bronchial epithelial cells grown as submerged or air-liquid interface (ALI) cultures with or without EMT induction were analyzed by bulk and single-cell RNA-Sequencing. Comparison of submerged and ALI cultures revealed differential expression of 8,247 protein coding (PC) and 1,621 long noncoding RNA (lncRNA) genes and revealed epithelial cell-type-specific lncRNAs. Similarly, EMT induction in ALI cultures resulted in robust transcriptional reprogramming of 6,020 PC and 907 lncRNA genes. Although there was no evidence for fibroblast/myofibroblast conversion following EMT induction, cells displayed a partial EMT gene signature and an aberrant basaloid-like cell phenotype. The substantial transcriptional differences between submerged and ALI cultures highlight that care must be taken when interpreting data from submerged cultures. This work supports that lung epithelial EMT does not generate fibroblasts/myofibroblasts and confirms ALI cultures provide a physiologically relevant system to study aberrant basaloid-like cells and mechanisms of EMT. We provide a catalog of PC and lncRNA genes and an interactive browser (https://bronc-epi-in-vitro.cells.ucsc.edu/) of single-cell RNA-Seq data for further exploration of potential roles in the lung epithelium in health and lung disease.
Keywords: aberrant basaloid cells, EMT, epithelial-mesenchymal plasticity (EMP), fibrosis, ILD
INTRODUCTION
Epithelial-mesenchymal transition (EMT) is a dynamic process in which epithelial cells transdifferentiate into mesenchymal cells (1, 2). First described in embryogenesis, EMT has been shown in wound healing, cancer, and tissue fibrosis (3). Evidence for EMT has been observed in lung diseases where fibrosis has been implicated in lung pathobiology, which include asthmatic airway remodeling, interstitial lung disease (ILD), and chronic obstructive pulmonary disease (COPD) (4–10). Moreover, TGF-β, a potent EMT activator, and TGF-β signaling pathways are elevated in these lung diseases (11–13). However, the role of EMT in lung fibrosis is still a matter of debate (14). A central issue is the origin of the myofibroblasts that accumulate in fibrotic tissues and drive remodeling (15). It was originally proposed that myofibroblasts were produced through EMT (16); however, lineage-tracing experiments suggest that this is unlikely (17). Recently, single-cell RNA-Sequencing (scRNA-Seq) of ILD lung tissue revealed a novel population of “aberrant basaloid” cells that display EMT characteristics but still maintain key epithelial features (18). Such partial EMT phenotypes have been previously noted in ILD (19–23) and may represent an intermediate state termed epithelial-mesenchymal plasticity (EMP) (24). Whether these cells play an active role in ILD pathogenesis and/or pathobiology remains unclear. Thus, a deeper understanding of the molecular mechanisms controlling EMT in the lung is clearly required.
Long noncoding RNAs (lncRNAs) are powerful regulators of cellular identity, function, and EMT (25–33). LncRNAs do not encode proteins; rather, many produce functional RNA transcripts, and they are frequently expressed in a more tissue- or disease-specific manner than protein coding (PC) genes (34–36), which makes them attractive biomarkers and therapeutic targets. The catalog of lncRNAs is rapidly expanding, and more than 96,000 have been mapped to the human genome, significantly outnumbering protein coding genes (37). However, very little is known regarding the expression and/or function of lncRNAs in either the healthy lung or in regulating lung epithelial EMT or EMP (38, 39). To explore the transcriptional landscape of the differentiating lung epithelium, we used bulk RNA-Seq and scRNA-Seq to analyze primary human bronchial epithelial cells (HBECs) grown in submerged cultures or at air-liquid interface (ALI). Bulk RNA-Seq analysis revealed that epithelial differentiation was associated with differential expression of 8,247 PC and 1,621 lncRNA genes. Next, EMT was induced in ALI cultures to evaluate the impact of EMT on the differentiated epithelium, which demonstrated differential expression of 6,020 PC and 907 lncRNA genes. This EMT-associated gene signature shared a significant overlap with multiple lung diseases, including ILD. Interestingly, scRNA-Seq analysis revealed no evidence of fibroblast/myofibroblast conversion. Rather, we observed a gene signature consistent with EMP, in which epithelial cells acquired mesenchymal markers yet maintained much of their epithelial identity. These cells closely resembled aberrant basaloid cells observed in ILD (18), which suggests a common origin. We also identified EMT-associated lncRNAs, and those specifically expressed by aberrant basaloid cells, such as the lncRNA CASC15, which we confirmed in ILD lung tissue. Thus, this work provides additional evidence that HBECs do not convert into myofibroblasts and validates ALI cultures as a physiologically relevant and tractable system to study aberrant basaloid-like cells and mechanisms of EMT.
METHODS
Cells and Tissues
All primary human bronchial epithelial cells (HBECs) used in this study were purchased from Lonza (Supplemental Table S1; see https://doi.org/10.6084/m9.figshare.19093763). Primary human tissues were obtained from patients with idiopathic pulmonary fibrosis (IPF) undergoing lung transplantation at University Hospital Leuven, Belgium, and from healthy donor lungs that were not suitable for transplantation. Human lungs were collected following local hospital ethical committee approval (ML6385) and written informed patient consent. According to Belgian legislation, declined donor lungs can be used for research purposes (Supplemental Table S1).
Bronchial Air-Liquid Interface Cultures for Bulk RNA Sequencing
HBECs were expanded and differentiated according to the manufacturer’s protocols (Clonetics B-ALI air-liquid interface cultures). Briefly, 1 × 106 HBECs were seeded into a T-75 flask containing 25 mL of prewarmed B-ALI growth medium and were grown as submerged cultures. For ALI cultures, 80% confluent submerged cultures were harvested and seeded (50,000 cells per 24-well insert) into the apical chamber of inserts—coated with type-I rat tail collagen solution (30 μg/mL), incubated for 2 h, and washed with 1× PBS to remove excess collagen—in 100 μL of B-ALI growth medium; the basal chambers of the inserts were cell-free and immersed in 500 μL of B-ALI growth medium. After 3 days, B-ALI growth medium from the apical and basal chambers was removed and 500-μL B-ALI differentiation medium was added to the basal chamber only (airlift). On airlifting, medium was replaced every other day with 500-μL B-ALI differentiation medium only in the basal chamber until the cultures reached full differentiation (day 25).
Bronchial Air-Liquid Interface Cultures for Single-Cell RNA Sequencing
HBECs were expanded and differentiated according to the manufacturer’s protocols (PneumaCult-ALI, Stem Cell Technologies; Cat. No. 05001). Briefly, 1 × 106 HBECs were seeded into T-25 flasks containing 5 mL of Rho Kinase (ROCK)-inhibitor supplemented with PneumaCult-ALI Complete Base Medium. When 80% confluence was achieved, cells were harvested and seeded (50,000 cells per 24-well insert) into the apical chamber of inserts (precoated with collagen solution 30 μg/mL) in 100 μL of ROCK-inhibitor supplemented PneumaCult-ALI Complete Base Medium; the basal chambers of the inserts were cell-free and immersed in 500 μL of medium. After 3 days, the medium from the apical and basal chambers was removed and 500-μL PneumaCult-ALI Complete Maintenance Medium was added to the basal chamber only (airlift). On airlifting, medium was replaced every other day with 500-μL PneumaCult-ALI Complete Maintenance Medium only to the basal chamber until the cultures reached full differentiation (day 25).
EMT Induction in Air-Liquid Interface Cultures
On day 25, the medium of the basal chambers of fully differentiated ALI cultures was supplemented with 1× of StemXVivo EMT Inducing Media (R&D; CCM017) or PBS. Medium was replaced every other day with 500-μL complete medium containing 1× of StemXVivo EMT Inducing Media or PBS only to basal chambers. After treatment, samples were collected on either day 0, day 1, day 5, day 7, or day 14.
TEER Measurement
The transepithelial electrical resistance (TEER) was measured in differentiating ALI cultures using an EVOM2 epithelial volt-ohm meter (World Precision Instruments). Resistance readings were measured and quantified starting from day 7 after airlift to 21–28 days to confirm development and maintenance of tight junctions. Briefly, medium was aspirated and replaced with 500 µL in the basolateral and 100 µL in the apical compartments. Cultures were equilibrated in the incubator for 30 min before measurement of TEER. Apical medium was then aspirated, and basolateral medium was replenished to restore ALI. TEER of blank—insert and medium without cells—was subtracted from measured TEER and Ω·cm2 calculated by multiplying by the insert area.
Single-Cell Suspension of Cell Cultures
The ALI cultures were harvested using 0.05% trypsin/EDTA (Fisher Scientific, Cat. No. 25–300-054), washed, and pelleted (250 g, 5 min, room temperature). Whereas submerged cultures were harvested and pelleted according to the manufacturer’s protocols (PneumaCult-ALI, Stem Cell Technologies; Cat. No. 05001). The pelleted cells were suspended in 3–4 mL dispase I/DNase I solution [38 U dispase I, 20 mL 1× PBS, and 20 μL DNase I (100 mg/mL)] for 15 min at 37°C. The cell suspension was then filtered using 30-µm MACS SmartStrainers (Miltenyi, Cat. No. 130-098-458) to obtain single-cell suspension.
RNA Extraction, Sequencing, and Analysis
Total RNA was extracted using RLT buffer supplemented with β-mercaptoethanol (Qiagen) according to the manufacturer’s instructions. Isolated RNA was quantified by spectrophotometry, and RNA concentrations were normalized. PolyA enriched RNA was sequenced on Illumina platform generating paired-end reads of 150 bps. Fragments were trimmed using Trim Galore software (https://github.com/FelixKrueger/TrimGalore) and reads with quality <20 were filtered out. Fragments were quasi-mapped to the human transcriptome hg38 obtained from Gencode v25 using salmon (40). Differential expression analysis was performed using DESeq2 package in R (41). A custom-curated reference transcriptome was compiled by merging the Gencode v25 and NONCODE v5 catalogs. NONCODE transcripts overlapping with Gencode transcripts by ≥90% were filtered out using bedtools intersect (https://bedtools.readthedocs.io/en/latest/content/tools/intersect.html).
For downstream analysis, the cut-off for differentially expressed (DE) genes was set at false discovery rate (FDR) ≤ 0.05, |log2FC| ≥ 2, transcripts per million (TPM) ≥ 1. DE genes were then used for different analyses and plotting using R, PCA using ggplots2 (42), Volcano plots using EnhancedVolcano (43), GO analysis using g:Profiler (44), and heatmaps using pheatmaps (45).
cDNA Synthesis and Quantitative RT-PCR
Isolated RNA was quantified by spectrophotometry, and RNA concentrations were normalized. cDNA was synthesized using SuperScript III Reverse Transcriptase (ThermoFisher Scientific) according to the manufacturer’s instructions. Resulting cDNA was analyzed by SYBR Green (KAPA SYBR Fast, KAPABiosystems) using gene-specific primers. Primer sequences are listed in Supplemental Table S2 (see https://doi.org/10.6084/m9.figshare.19093730). All reactions were performed in triplicates using ViiA7 Real-Time PCR instrument (Thermo Fischer Scientific). Data were log2 transformed before heatmap generation.
Variance Analysis
We analyzed the PCA bulk-cell RNA-Seq data variability using the within-cluster sum of squares and silhouette plots. The within-cluster sum of squares quantifies the data variability of a cluster as the sum of all pairwise (Euclidean) distances squared, divided by twice the number of points in that cluster, that is,
where ni is the number of observations in cluster i and xi, yi are the (x, y-coordinates of the samples in i. In our study, we analyzed two PCA clusters with n1 = n2 = n = 4, making directly comparable across clusters. To estimate and compare the cluster silhouettes, we utilized the silhouette() function of the cluster R package. The technique provides a measure of how well each sample is classified, i.e., how close each sample in cluster is to the samples of other clusters. Typically, the silhouette coefficient of a sample ranges in [0,1] with large numbers indicating that is well classified in . Mathematically, the coefficient has the form:
where is the average distance between si and all other data within i whereas is the average distance of si to all samples belonging to the closest cluster j.
Ingenuity Pathway Analysis
Differentially expressed genes filtered on the adjusted P values and fold-changes were used for the pathway analysis with Ingenuity Pathway Analysis (IPA; QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis). The ILD, asthma, and COPD gene lists were derived from the associated molecules of the corresponding disease in IPA software and manually curated to remove chemical compounds and other molecules. These resulting genes lists were then supplemented with differentially expressed genes obtained from published lung scRNA-Seq from donors with IPF and COPD (18).
NetAct and RACIPE Analysis
Transcription factor activity was determined from bulk gene expression data using the NetAct method. NetAct integrates transcription factor (TF)-target data from multiple literature-based resources (46–49) with the context-specific gene expression data. In this method, the enriched TFs are identified by the gene set enrichment analysis (50) using literature-based TF-target database and the activity of the selected TFs’ is calculated from the gene expression of their targets. The context-specific network was inferred by aggregating interactions between enriched TFs. An interaction between TFs was included in the inferred network if the activities of the transcription factors are highly correlated (Pearson’s correlation > 0.9) and the interaction is supported by the database. Any TF in the network, which had only outgoing interactions, was removed from the network. This inferred network was simulated using random circuit perturbations (RACIPE) to verify whether the simulations resemble the activities or not. Correlation cutoff for selection of interactions was adjusted for high agreement between simulated and inferred activities. The final network interactions file was loaded into Cytoscape for visualization (51).
EMT Quantification Analysis
EMT score was calculated using the method proposed by Tan et al. (52). In this method, two-sample Kolmogorov–Smirnov test score for difference between the estimated empirical cumulative distribution function for epithelial and mesenchymal gene sets is used as EMT score. The EMT score varies between −1 and 1 where a negative score for a sample implies that it exhibits a more epithelial phenotype, whereas a positive score reflects a more mesenchymal phenotype. We used their cell lines gene set consisting of 218 genes (170 epithelial and 48 mesenchymal genes) to compute the EMT scores.
Single-Cell Hashing and Sequencing
To enable sample multiplexing, 1–2 million filtered single cells were suspended in 100-μL staining buffer (2% BSA/0.01% Tween in PBS) and blocked with 10-μL Fc blocking reagent (FcX, BioLegend) for 10 min at 4°C, then incubated with 0.5 µg of a unique cell hashing antibody (BioLegend TotalSeq-A anti-human Hashtag, Cat. No. A0251-A0256) for 20 min at 4°C. After that, cells were washed three times with 1 mL 1× PBS + 0.04% BSA and pelleted at 4°C for 5 min at 350 g. The pellet was resuspended in 1× PBS + 0.04% BSA and cell viability was calculated using Countess II FL (ThermoFisher). Labeled cells were pooled together (40,000 cells in total ∼3,500 cells of each hashtagged sample) and loaded onto one lane of a 10× Chromium Controller Chip. Single-cell capture, barcoding, and library preparation were performed using the 10× Chromium platform (53), version 3.1 chemistry, and according to the manufacturer’s protocol (Cat. No. CG00052) with modifications for generating the hashtag library (54). cDNA and libraries were checked for quality on Agilent 4200 Tapestation, quantified by KAPA qPCR, and pooled using a ratio of 95% gene expression library and 5% hashtag library before sequencing; each gene expression-hashtag library pair was sequenced at 50% of an Illumina NovaSeq 6000 S2 flow cell lane, targeting 20,000 barcoded cells with an average sequencing depth of 50,000 reads per cell.
Illumina base call files for all libraries were converted to FASTQs using bcl2fastq v2.20.0.422 (Illumina) and FASTQ files associated with the gene expression libraries were aligned to the GRCh38.93 reference genome [10× Genomics GRCh38 reference 3.0.0 (including all transcribed unitary pseudogenes)] using the version 3.1.0 Cell Ranger count pipeline (10× Genomics). FASTQ files representing the hashtag libraries were processed into hashtag-count matrices using CITE-Seq-Count (version 1.4.3) (https://zenodo.org/badge/latestdoi/99617772).
Single-Cell RNA-Seq Data Processing and Analysis
The single-cell RNA-Seq data were generated in two ALI culture libraries, AW20003 and AW21001, and one submerged culture library, SC2100310. AW20003 contained the data of donor ID 34 (11-yr-old Caucasian male). Hashtag-oligos (HTOs) 5 and 6 labelled the day 0 cells (D0), HTOs 3 and 4 the day 1 (D1) cells and HTOs 1 and 2 the day 5 (D5) cells. AW21001 combined the data of donor IDs 27 (60-yr-old black male) and 54 (19-yr-old black female) in different HTOs. HTOs 1 and 4 labelled the day 0 cells of each donor respectively. Similarly, HTOs 2 and 5 and HTOs 3 and 6 held the data of day 1 and day 5 cells. SC2100310 combined the data of all three donors with HTO 1 marking the cells of donor IDs 27, HTO 2 the cells of donor ID 34 and HTO 3 the cells of donor ID 54.
Chromium 10× data processing.
The Illumina single-cell RNA sequencing base call files of each of the two libraries were demultiplexed by cellranger mkfastq that generated the raw fastq file which was processed with the standard cellranger-3.1.1 pipeline. The reads were aligned to the Ensembl human genome GRCh38 (https://uswest.ensembl.org/Homo_sapiens/Info/Index) with STAR for each of the 6,794,880 Gel bead-in Emulsions (GEMs). The reads were confidently assigned into the exonic, intronic, and intergenic categories according to the default cellranger protocol. Those compatible with the exons of an annotated transcript having a single gene annotation were considered for unique molecular identified (UMI) counting. For barcode calling the algorithm first identified the high RNA content cells based on the total UMI count for each barcode and then applied the EmptyDrops background model to filter out empty droplets and extract the final set of 15,929 (AW20003), 23,677 (AW21001), and 19,389 (SC2100310) barcodes coming from nonempty GEMs.
HTO demultiplexing.
The hashtag demultiplexing of the six pooled HTO-distinct ALI samples per library and the three HTOs in the submerged sample was performed with Seurat’s HTODemux pipeline (55). Briefly, the raw UMI counts of the 15,929, 23,677, and 19,389 barcodes, respectively, were normalized by the Centered Log Ratio transform and subsequently separated into k = 7 (in ALI) and k = 4 (in submerged) clusters by the k-medoids algorithm. As expected, k-1 clusters were enriched for expression of a particular HTO. For each HTO, the cluster with the lowest average was considered as the negative group and its data was modeled with a negative binomial distribution whose qth quantile was used to classify each cell into HTO-assigned or HTO-unassigned (negative). We tested a series of quantile values in [0.95, 0.995] and selected q = 0.99, exhibiting a plateau in the number of cells assigned to each HTO at perplexity p = 100 (default Seurat parameter). Cells assigned to more than one HTOs were annotated as doublets and filtered out. In summary, of the starting 15,929 cells of AW20003, 1608 (10.0%) were predicted as doublets and 2,146 (13.5%) as negatives leading to 12,175 HTO-specific singlets with comparable HTO rates. Similarly, AW21001’s 23,677 starting cell set featured 2,769 (11.7%) predicted doublets, 2,993 (12.5%) negatives and 17,915 HTO-specific cells, whereas among SC2100310’s 19,389 cells we estimated 2,210 (11.4%) doublets and 2,922 (15%) negatives.
Quality control.
Quality control analysis was performed iteratively using data from each donor separately. We followed Seurat’s standard preprocessing workflow from data normalization to cell clustering and investigated whether the low-quality cells, if any, tend to cluster together (see Seurat clustering). The main quality control pipeline was performed on the clustered data in these steps:
1st step: We generated violin plots of the number of UMIs, the number of expressed genes and the percentage of reads mapped to the mitochondrial (MT) genome per cell (y-axis) against the cluster IDs (x-axis) to determine appropriate cutoffs for the low-quality cells. The dynamic range of the number of UMIs was [1k, 100k] whereas for the number of expressed genes it was [0.5k, 10k]. We remove from further analysis whole clusters with more than 95% of cells exhibiting less than 2,000 expressed genes and less than 4,800 UMIs. In addition, we removed whole clusters with more than 18% of reads mapped to the MT genome. For the rest of the clusters, we simply applied the above cutoffs to remove the low-quality cells. Not to discard meaningful data, we checked that the low-quality clusters did not highly and uniformly express any marker gene (18, 56, 57). In ALI, we filtered 2,736 cells from donor’s 34 data (22.5%), 2,925 from donor’s 27 data (37%) and 3,053 cells from donor’s 54 data (29%), whereas in the submerged library, the numbers of low-quality cells were 367 (6.5%), 306 (6.5%), and 166 (4.3%), respectively.
2nd step: We utilized Scrublet (58) and DoubletFinder (59) to predict potential doublets (see Doublet estimation). We flagged the cells whose doublet score was higher than the 95 quantiles of the doublet score distribution of each method or those predicted as doublets by both methods independently of the score. In ALI, we removed 261 cells from AW20003, 121 from AW21001’s donor 27 and 216 cells from AW21001’s donor 54. In submerged, the respective numbers per donor were 58 (donor 34), 48 (donor 27), and 33 (donor 54).
3rd step: We utilized DecondX (60) to estimate and remove the ambient RNA contamination in individual cells (see Ambient RNA decontamination). Due to noticeable differences in the quality control metrics, DecontX was applied to the data of each donor separately in both the ALI and the submerged cultures. The decontaminated UMI matrices were subjected to a second round of Seurat normalization, clustering, and filtering of low-quality cells. In ALI, we kept 8,221 high-quality cells from AW20003, 4,404 from AW21001 donor 27, and 7,029 from AW21001 donor 54 for further analysis. In submerged, we kept 5,129 (donor 34), 4,227 (donor 27), and 3,626 (donor 54) high-quality cells. DecontX was also run in the merged data set and obtained very similar results.
Seurat clustering.
The singlet expression profiles of each donor were SCT-normalized using log-transform and variance stabilization transformation (vst) (61). We derived the top 3,000 variable features for downstream analysis. The raw counts were fitted in a regularized negative binomial regression model with the sequencing depth included as a covariate for library size adjustment. The model’s Pearson’s residuals were used to calculate the scaled expression profiles. The scaled data of the 3,000 variable features were fitted in the principal components analysis (PCA) model for linear dimensionality reduction. We visualized the PC loadings and the associated heatmaps of gene expression to explore the primary sources of heterogeneity and determine the optimal number of PCs. We kept the first 100 PCs from which the Uniform Manifold Approximation and Projection (UMAP) representation was subsequently retrieved. We constructed a shared nearest neighbor graph by calculating the neighborhood overlap (Jaccard index) between every cell and its 20 nearest neighbors obtained from the cell Euclidean distances. We clustered the data with the Leiden method (62), which detects well-connected communities in a network by maximizing the difference between the actual number of edges in a community and the expected number of edges. The clustering resolution parameter was set to 1.
Doublet estimation.
We flagged potential doublets from the HTO data (see HTO demultiplexing) and subsequently from the raw UMI matrices of each donor using Scrublet (58) and DoubletFinder (59). Both methods simulated multiplets from the observed transcriptomes of clean data (HTO-doublets removed and completion of 1st step QC) and combined them with the real scRNA-Seq data to predict the outcome. Scrublet utilized a nearest neighbor algorithm to estimate the local density of the simulated doublets and assigned a doublet score to each observed cell i indicating i’s likelihood to be a doublet. DoubletFinder integrated the artificial doublets into the observed data at a user-defined proportion, pN, and defined each cell’s neighborhood in gene expression space, pK. A range of and value combinations were iteratively tested (parameter sweep) and the proportion of artificial nearest neighbors was estimated for each observed cell, indicating as before each cell’s likelihood to be a doublet.
Ambient RNA decontamination.
Ambient RNA is the pool of mRNA molecules released in the cell suspension likely from stressed or apoptotic cells. It is incorporated into the droplets resulting in cross contamination of transcripts between different cell populations. We estimated and removed contamination in individual cells by DecontX’s Bayesian model (60) using the celda R package (63). Briefly, DecontX fits to the raw UMIs of each cell a mixture of two multinomial distributions, i.e., one that models the native transcript counts from the cell’s actual population and one that models the contaminating counts. The cells are subjected to Seurat clustering and separated into k = 1, …, K populations. Similar to a Bayesian hierarchical model, the probabilities of gene g being expressed in population k and gene g contamination population k′ are explicitly defined, as well as the proportion of counts derived from the native expression distribution for each cell. Each transcript count has a hidden state that denotes transcripts t membership to the native or the contamination expression distribution. The joint posterior distribution is approximated with Variational Inference deep learning that deconvolutes the two sources of variation and subtracts the ambient profiles from the original raw counts of each cell.
Data integration.
The data from the three donors were integrated with Seurat’s SCTransform workflow (see Seurat clustering) that adjusted for the percentage of MT mapping. In ALI, donors 34 and 27/54 come from different libraries and as such the donor integration strategy also accounted for the batch effects due to the library preparation and the time of the experiment. In submerged, the data of the three donors were separated and subsequently integrated to account for the gender differences.
The SCT-transformation was applied to the data of each donor and the subsequent integration was performed with integration anchors among the data of the three donors in ALI and the submerged cultures separately. The anchors, representing pairwise correspondences between individual cells assumed to originate from the same biological state, were used to adjust the donor-specific data sets and minimize library-associated technical effects.
The data clustering analysis was carried out as in Seurat clustering. The integrated data set was fed to Seurat’s pipeline and the clusters were estimated with the resolution parameter set to 1, returning 22 clusters in ALI and 13 in submerged. In the former data set, we also separated the data by day, D0, D1, and D5, and we repeated the same procedure to generate day-specific clusters with resolution parameter set to 1. We found 15 clusters for D0 and D13 in each of D1 and D5 data, implying a higher resolution in our day-specific estimates.
Differential expression analysis.
We identified cluster biomarkers from the day-specific integrated and clustered data using Seurat’s differential expression model under the Likelihood Ratio test. The model adjusted for the percentage of MT mapping. In ALI, we compared the expression of ∼20,000 expressed genes between cluster versus all other day-specific clusters, where kd = 1, …, Kd are the Seurat clusters estimated from day’s d, integrated data, , integrated data (see Data integration). We noticed that the day-specific data offered higher cluster resolution and allowed us to identify important markers and associated cell types with greater accuracy. The differentially expressed genes were selected at and . We estimated 9,512 differentially expressed genes among all D0 clusters, 9,742 among all D1 clusters, and 10,466 among all D5 clusters. In submerged, using the same cutoffs, we estimated 7,266 differentially expressed genes.
Cell type estimation.
We estimated cell types from the day-specific data starting from a set of ∼3,800 literature-retrieved canonical markers of healthy individuals and patients with IPF (18, 56, 57) The list was reduced into gene signatures of markers found to be highly differentially expressed in our analysis (FDR ≤ 1e − 5). We followed Garnett’s pipeline (53) to establish rules of expression that, if needed, merged, and essentially labeled the Seurat clusters according to the marker expression patterns. In D0, we estimated seven cell types, i.e., Basal, Basal Cycling, Basal Supranasal, Multiciliated, Deuterosomal, Goblet, and Secretory cells. D1 and D5 exhibited three subtypes transitional basaloid-like and two subtypes of aberrant basaloid-like cells, respectively, as well as Multiciliated, Deuterosomal, Goblet, and Secretory cells. The submerged data consisted of two Basal subtypes, Basal 1 with 6,922 (53.3%) cells and Basal 2 with 2,938 (22.6%) cells, and a relatively large cycling basal population of 3,122 (24%) cells.
Next, we applied Seurat’s differential expression model to identify biomarkers for each day-specific and submerged cell type at and (see Differential expression analysis). For each day, we collected the top 500 genes of each cell type’s markers and refined the cell type annotation with a novel iterative hierarchical clustering strategy. In each iteration the cells of each cell type were subjected to adaptive hierarchical clustering using the dynamicTreeCut R package (64) that searched for subclusters of minimum 20 cells. The iteration stopped when no splits occurred across subsequent iterations. This procedure split the D0 secretory cells into Secretory type 1 and Secretory type 2. The cell type labels from the day-specific analysis were transferred to the integrated data to complete the annotation.
Cell type annotation by CelliD.
We used CelliD to perform automatic gene signature extraction and functional annotation for each individual cell of our data set (65). CelliD is based on Multiple Correspondence Analysis (MCA) and produces a simultaneous representation of cells and genes in a low dimension space. Genes are then ranked by their distance to each individual cell, providing unbiased per-cell gene signatures.
First, we ran CelliD on the normalized D0 data. The per-cell gene rankings were calculated from the gene-to-cell Euclidean distances in the MCA space in an unbiased way, i.e., blindly of the previously identified Seurat cell clusters or cell type annotations. The gene signatures were obtained from the differentially expressed genes as before (see Cell type estimation). The enrichment of per-cell signatures was evaluated through the hypergeometric test using the top 3,000 variable features across 50 MCA dimensions.
To quantify the agreement between our annotation and CelliD’s, we used the Fisher exact test. We considered our annotation set [Basal, Basal Cycling, Basal Supranasal, Multiciliated, Deuterosomal, Goblet, Secretory] and CelliD’s set [Aberrant Basaloid, Basal, Basal_Cycling, Basal_Supra, Basal_Supranasal, Multiciliated, Multiciliated_Nasal, Deuterosomal, Fibroblast, Secretory, Secretory_Nasal, Goblet]. We generated the 2 × 2 confusion matrices between pi versus qj pairs of the same cell types and estimated the enrichment of commonly annotated cells at a = 1%. All enrichment P values were highly significant, thus reproducing our annotation with an independent method.
In a similar way, we applied CelliD on the data of D1 using the annotation set q without the transitional/aberrant basaloid cells (18). We reasoned that CelliD would be forced to associate our transitional and aberrant basaloid annotated cells to the most similar cell type of q′, thus predicting their cell type of origin. The results indicated that the transitional and aberrant basaloid cells of our study likely originated from Basal, Suprabasal, and Secretory cells.
Cell type annotation by SingleR.
We used SingleR (66) for the automatic annotation of day 1 and day 5 aberrant basaloid cells using as a reference the large collection of bulk RNA-Seq and microarray expression data from the Human Primary Cell Atlas project (67). To annotate each single cell independently, SingleR correlated each cell’s gene expression profiles with the profiles of pure cell types (66). Briefly, a Spearman’s correlation coefficient was calculated between each single cell’s and each of the reference’s expression data. The multiple, cell-specific correlation coefficients were aggregated to provide a single value per cell type. The 80th percentile of the correlation values was used to select the top, most likely cell types and to prevent misclassification. The algorithm iterated using only the cell types with correlations exceeding the 80th percentile, and at each iteration, the most likely cell type annotations were kept until only one cell type annotation remained, i.e., the one with the highest correlation to our single cell.
Raw Sequencing Data Availability and Data Visualization
All RNA-Seq (bulk and single cell) data were submitted to the Gene Expression Omnibus (GEO) database repository (https://www.ncbi.nlm.nih.gov/geo/) and can be found with the accession number GSE193684.
In addition, scRNA-Seq data were uploaded for visualization in the USCS Cell Browser (https://bronc-epi-in-vitro.cells.ucsc.edu/). This consists of three data sets, the “ALI day 0” representing the day 0 ALI cultures with PBS treatment, the “Submerged” representing the submerged cultures, and the “ALI days 0, 1, and 5” representing the integrated ALI data before and after EMT induction.
Use of Published scRNA-Seq Data Sets
UMAP plots for AC245041.2, LINC02185, and SRGAP3−AS1 were generated using Lung single-cell atlas browser. The data set was set to “grch38” gene annotation before gene search and export. UMAP plots and violin plots for CASC15, MANCR, TINCR, LINC00958, and WFDC21P were generated using IPF cell atlas browser.
Tissue Immunofluorescence Staining
ALI cultures were embedded in optimal temperature compound (OCT), cryosectioned (8 µm), and, consecutively, fixed with 4% PFA, washed with PBS, permeabilized with Triton 100× 0.01%, and treated with Fc Receptor Block (Innovex bioscience) for 40 min with Background Buster (Innovex bioscience) for 30 min. The sections were then stained with primary antibodies, diluted in PBS + 5% BSA 0.1% Saponin for 1 h at room temperature, washed, and stained with the secondary antibodies at room temperature for 30 min. Nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI; 1 µg/mL) and Phalloidin ATTO647N 1/2000 for 2 min. Tissues were mounted in Fluoromount-G mounting media.
Primary antibodies: anti-Vimentin (D21H3, Cell Signaling Technology); anti-Cytokeratin 17 (E3, NJS Bioreagents); anti-TP63 (HPA006288, Atlas Antibodies); anti-FN1 (HPA027066, Atlas Antibodies); anti-FOXJ1 (HPA005714-25UL, Sigma); anti-MUC5B (HPA008246-25UL, Sigma).
Secondary antibodies: goat anti-rabbit Alexa Fluor 488, goat anti-mouse IgG2b Alexa Fluor 568 (ThermoFisher) 1/2,000. Antibodies were validated and titrated on relevant control tissues (lung). The staining pattern was then cross referenced with existing data in the literature.
RNA in Situ Hybridization
RNA transcripts were visualized in OCT-embedded ALI sections using the QuantiGene ViewRNA ISH tissue assay kit (ThermoFisher) and “Nunc Lab-Tek II Chamber Slide System” (154534PK) submerged cultures using ViewRNA cell plus assay kit (ThermoFisher). For staining on lung tissue samples, 4-μm microtome sections of formalin-fixed, paraffin-embedded (FFPE) healthy and diseased IPF lungs were collected on slides for staining and were visualized using ViewRNA Tissue Assay Core-Fast Red Kit (ThermoFisher). Human MALAT1, NRAV1, CASC15, and TINCR ViewRNA type 1 probes were obtained from ThermoFisher. The ViewRNA assay was performed according to the manufacturer’s protocol. Probes were detected at 550 nm.
Confocal Microscopy
Images were acquired on the Leica SP8 confocal microscope and Leica SP5 (Leica Microsystems). Sequential acquisition was performed with a ×40 or ×63/1.4 NA objective. Images were analyzed with Imaris 9.7.2 software and ImageJ bundle with Java 1.8.0. ViewRNA quantification was performed with Imaris software, version 9.7.2, using the “spot” function on the channel corresponding to CAS15 signal; the threshold was based on signal “quality.”
Single Nucleotide Polymorphism Analysis
All single nucleotide polymorphisms (SNPs) both upstream and downstream (±800 kb) from the CASC15 TSS were extracted from a genome-wide association study (GWAS) catalog (68). SNPs with associations relevant to lung biology were plotted, whereas all SNP associations were tabulated.
RESULTS
Differentiation of the Lung Epithelium in Vitro Induces Differential Expression of Thousands of Genes Including 1,621 lncRNAs
Long noncoding RNAs (lncRNAs) are emerging as potential regulators of cellular identity and function and therefore have significant potential as disease biomarkers and therapeutic targets. However, little is known regarding the role of lncRNAs in lung biology. Defining the lncRNA landscape of lung epithelium is the first step to understand how they contribute to lung biology and disease pathogenesis. Primary human HBECs are frequently used as an in vitro model of the human lung epithelium that can be grown under two different culture conditions: 1) submerged cultures, thought to consist of primarily undifferentiated cells or 2) ALI cultures, which consist of multiple differentiated epithelial cell types in a pseudostratified columnar structure. To map the transcriptional landscape of lung epithelium in the aforementioned models, HBECs from eight donors (four normal and four asthma, see Supplemental Table S1) were grown under submerged and ALI culture conditions before analysis by RNA-Seq (Fig. 1A). ALI cultures demonstrated a polarized epithelium with tight junctions (Supplemental Fig. S1, A and B, see https://doi.org/10.6084/m9.figshare.19093703). Principal component analysis (PCA) of RNA-Seq data revealed higher heterogeneity between donors in ALI cultures than in submerged cultures (Fig. 1B and Supplemental Fig. S1C). Differential gene expression (DGE) analysis revealed substantial transcriptional reprogramming (10,827 genes; FDR ≤ 0.05 and |log2FC| ≥ 2) on epithelial differentiation in ALI culture (Fig. 1C and Supplemental Table S3, see https://doi.org/10.6084/m9.figshare.19093736). As expected, epithelial cell subtype marker genes, such as FOXJ1 (multiciliated), MUC5B (goblet), SCGB1A1 (club), were enriched in ALI cultures, whereas the basal cell marker TP63 was reduced (Fig. 1, C and D). Gene ontology (GO) analysis revealed significant changes associated with ciliary organization, consistent with epithelial differentiation (Supplemental Fig. S1D). Analysis of noncoding genes revealed 1,621 differentially expressed lncRNAs (FDR ≤ 0.05; |log2FC| ≥ 2) lncRNAs (e.g., ZFAS1, BLACAT1, and LINC00950), none of which have been investigated for their potential role in basal cell differentiation (Fig. 1, C and E). As previously reported, lncRNAs were, in general, less highly expressed than PC genes (Fig. 1, E and F). Despite this, PCA analysis exclusively based on lncRNA expression reproduced the higher heterogeneity between donors in ALI cultures compared with submerged cultures (Supplemental Fig. S1, E and F). To further map the lncRNA landscape of the differentiating epithelium, RNA-Seq data were aligned to a custom-curated reference transcriptome. This revealed an additional 2,069 lncRNAs transcripts (FDR ≤ 0.05; |log2FC |≥ 2), but most were substantially less abundant than those already represented in the Gencode catalog (Supplemental Fig. S1G and Supplemental Table S4, see https://doi.org/10.6084/m9.figshare.19093724). In summary, differentiation of the human airway epithelium is associated with extensive transcriptional reprogramming and establishment of a distinct lncRNA landscape.
Figure 1.
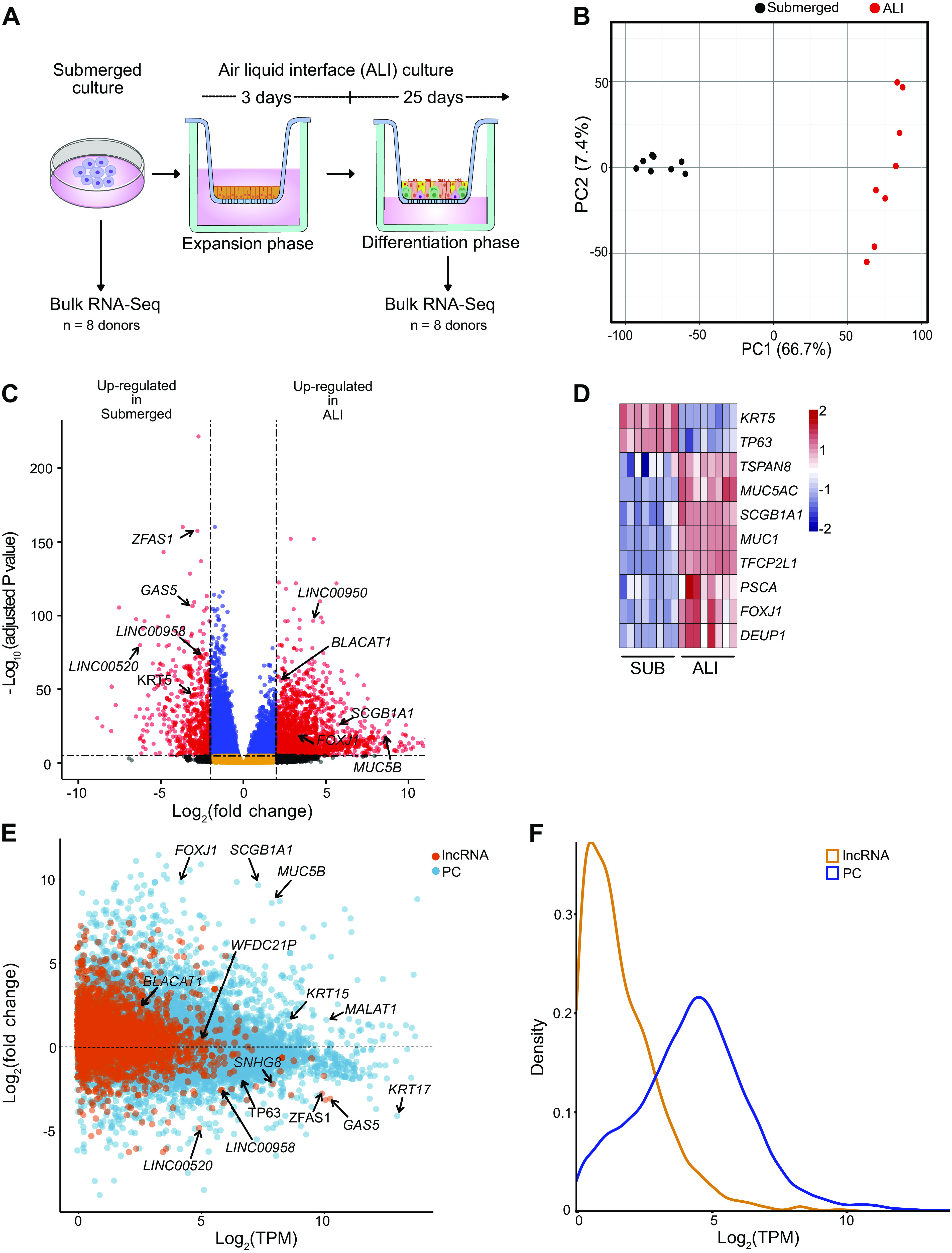
Differentiation of the lung epithelium in vitro induces differential expression of thousands of genes including 1,621 lncRNAs. A: bulk RNA-Seq experimental design. HBECs from eight donors were cultured in submerged cultures and as ALI cultures before analysis by RNA-Seq. B: unbiased clustering of all samples using principal component analysis (PCA). All genes with <1 TPM value (averaged across all samples) were removed before performing PCA, n = 8 donors. C: volcano plot of expressed genes (TPM ≥ 1) between submerged and ALI cultures, n = 8 donors. Red dots, adjusted P value <10−6 and log2 fold-change >2; blue dots, adjusted P value <10−6 and log2 fold-change <2; black dots, adjusted P value >10−6 and log2 fold-change >2; orange dots, >10−6 and log2 fold-change <2. D: lung epithelial markers, TPM values plotted as heatmap between submerged (SUB) and ALI cultures, n = 8 donors. E: MA plot of protein coding (PC) and long noncoding RNA (lncRNA) expression between submerged and ALI cultures, n = 8. F: density plot depicting expression of PC and lncRNAs. Plotted are log2 TPM values averaged across submerged and ALI cultures from all donors, n = 8 donors. ALI, air-liquid interface; HBECs, human bronchial epithelial cells; PC, protein coding.
Single-Cell Analysis Reveals Epithelial Cell Type-Specific lncRNAs
To ascribe lncRNAs to specific epithelial cell types, scRNA-Seq of HBEC submerged and ALI cultures from three normal donors was performed (Fig. 2A and Supplemental Table S1). Cell identity was inferred based on expression of known marker genes, which revealed that submerged cultures consisted of two basal cell clusters and cycling-basal cells (Fig. 2, B and C), whereas ALI cultures consisted of basal, cycling-basal, suprabasal, secretory (2 clusters), goblet, multiciliated, and deuterosomal cells (Fig. 2, B–E, and Supplemental Fig. S2, A–H, see https://doi.org/10.6084/m9.figshare.19093700; Supplemental Table S5, see https://doi.org/10.6084/m9.figshare.19093754; Supplemental Table S6, see https://doi.org/10.6084/m9.figshare.19093727; and Supplemental Table S7, see https://doi.org/10.6084/m9.figshare.19093745). In addition, ALI culture annotations were further validated using CelliD (Supplemental Table S6) (18, 56, 57). As expected, we did not identify any small airway or immunological cell clusters, or any mesenchymal cells, including fibroblasts or myofibroblasts (Supplemental Table S6). As lncRNA transcripts are frequently less abundant than PC transcripts, their detection is limited with scRNA-Seq. However, we were able to identify lncRNAs enriched in different epithelial cell types in ALI cultures (Fig. 2, D and E, Supplemental Fig. S3, see https://doi.org/10.6084/m9.figshare.19093709, and Supplemental Table S7). Comparison with published scRNA-Seq data from lung (69) confirmed cell-type-specific expression of multiple lncRNAs (Supplemental Fig. S3B). Interestingly, several genes annotated as lncRNAs are known to express micropeptides such as MRLN, NBDY, and MTLN (Fig. 2D). These data indicate that ALI cultures effectively recapitulate much of the cellular complexity of the airway epithelium and allow for identification of epithelial cell-type-specific lncRNAs.
Figure 2.
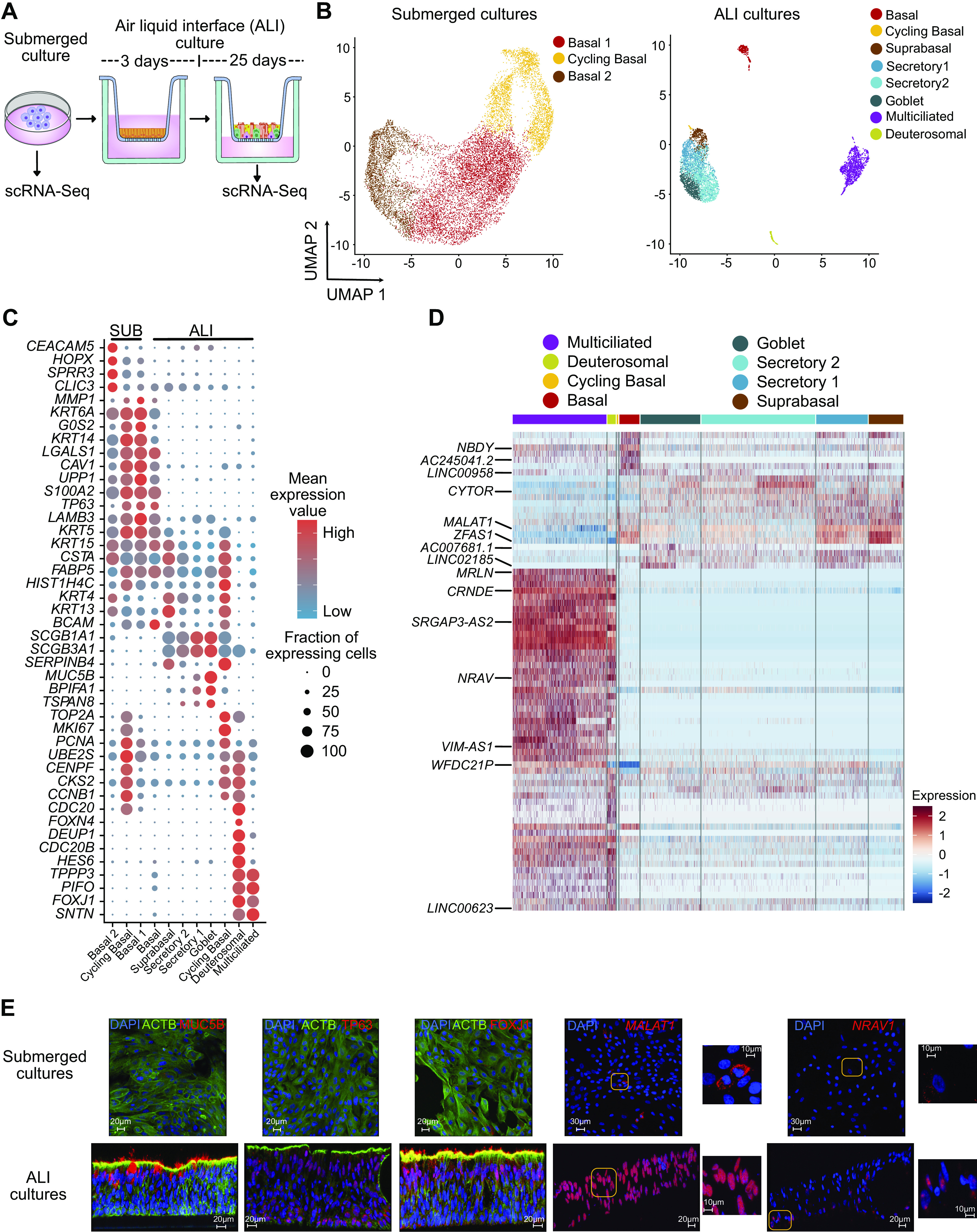
Single-cell analysis reveals epithelial cell-type-specific lncRNAs. A: schematic representation of submerged and ALI cultures. Primary human bronchial epithelial cells (HBECs) were dissociated from either submerged or ALI cultures before analysis by scRNA-Seq, n = 3 donors. B: UMAP plot of the scRNA-Seq expression data highlighting the main cell clusters observed in (left) submerged cultures and (right) ALI cultures. C: expression data highlighting selected cell-specific markers for cell clusters in submerged cultures and ALI cultures. D: heatmap depicting relative expression (normalized and scaled expression) of lncRNAs in each cluster in ALI cultures. All lncRNA names and their respective expression values are available in Supplemental Table S7. E: immunofluorescence (MUC5B, TP63, and FOXJ1) and RNA in situ hybridization (MALAT1 and NRAV) demonstrating expression of selected protein and RNA molecules in submerged and ALI cultures. Scales are depicted as micrometers; n = 2 donors, representative data from one donor is shown. ALI, air-liquid interface; lncRNA, long noncoding RNA; scRNA-Seq, single-cell RNA-sequencing.
Induction of EMT in ALI Cultures Induces a Transcriptional Reprogramming Consistent with a Partial EMT Phenotype
Although EMT is a common feature of fibrotic lung diseases, its role in fibrogenesis is controversial. Recently, aberrant basaloid cells were identified in fibrotic lung tissue as a novel epithelial cell type displaying a partial EMT phenotype, yet their developmental origin remains unknown. We sought to determine the impact of EMT on the lung epithelium and whether this contributes to the differentiation of aberrant basaloid cells. To this end, we determined transcriptome-wide changes associated with induction of EMT in ALI cultures. As both TGF-β and WNT signaling pathways have been implicated to have a significant role in fibrotic lung diseases (11, 70–76), we used a cocktail containing recombinant TGF-β1, WNT-5a, and neutralizing antibodies against E-Cadherin, sFRP-1, and Dkk-1 to induce EMT (EMT cocktail). Differentiated ALI cultures (four normal and four asthma) were treated with this EMT cocktail (referred to as EMT) before analysis by bulk RNA-Seq (Fig. 3A). PCA analysis revealed less heterogeneity between donors treated with EMT compared with PBS (Fig. 3B and Supplemental Fig. S4D, see https://doi.org/10.6084/m9.figshare.19093712). A total of 6,020 PC and 907 lncRNA genes were differentially expressed (FDR ≤ 0.05; |log2FC| ≥ 2) between PBS- and EMT-treated ALI cultures (Fig. 3C and Supplemental Table S8, see https://doi.org/10.6084/m9.figshare.19093742). This included classical markers of EMT (e.g., CDH2, CDH11, VIM, CDCK14, FGFR3, etc.; (Fig. 3, C and D and Supplemental Fig. S4A). Consistent with the known phenotypic changes associated with EMT, GO analysis revealed that transcriptional reprogramming correlated with changes in extracellular matrix, cell adhesion, and cell motility (Supplemental Fig. S4B). EMT can be incomplete, resulting in complex phenotypes (77) and Network Activity (NetAct) analysis can be used to evaluate these EMT phenotypes using bulk RNA-Seq data (78). As expected, NetAct demonstrated activity of EMT-associated transcriptional regulators such as TWIST1, NF-κB1, STAT2, CTNNB1, and SMAD3 (Fig. 3E, Supplemental Fig. S4C, and Supplemental Table S9, see https://doi.org/10.6084/m9.figshare.19093733) in EMT-induced ALI cultures. However, activity of key factors such as SNAI and ZEB family members were notably absent, suggesting a partial EMT phenotype. Networks inferred from these activities revealed a complex web of potential interactions between EMT factors (Supplemental Fig. S4C) and random circuit perturbations (RACIPE) simulations of this network resembled the activity data (79, 80). Using a scoring method that provides a quantitative measure of an EMT phenotype through analysis of transcriptional data (52), we confirmed a partial EMT phenotype of these cells (Fig. 3F). This contrasts with the more complete EMT phenotype found in TGF-β1-treated A549 cells (Supplemental Fig. S4E), which is consistent with their known EMT potential (21, 81). Furthermore, an extended 14-day EMT treatment of HBEC ALI cultures did not show substantial changes in key epithelial and mesenchymal genes (Supplemental Fig. S4F).
Figure 3.
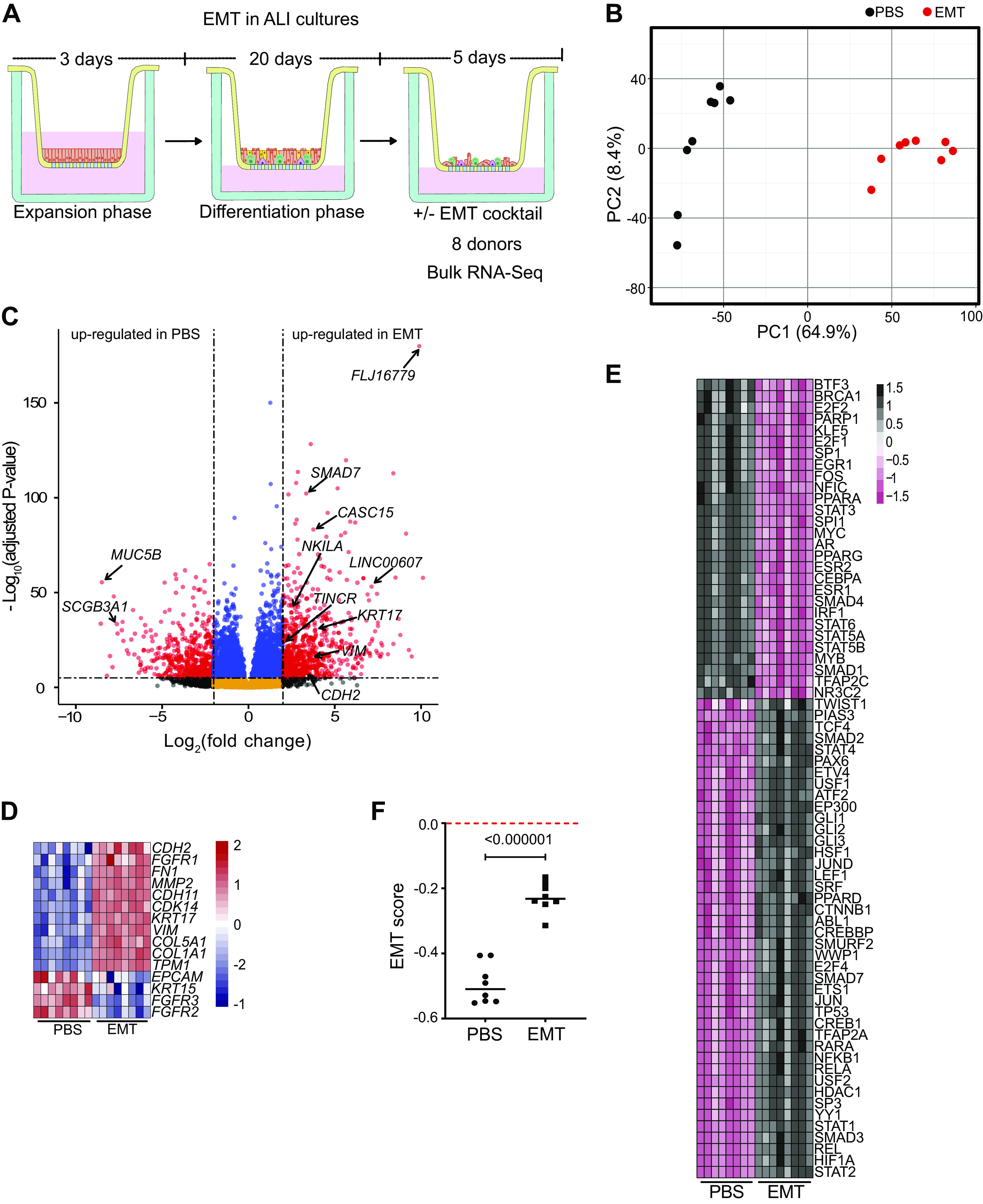
Induction of EMT in ALI cultures induces a transcriptional reprogramming consistent with a partial EMT phenotype. A: bulk RNA-Seq experimental design. Differentiated ALI cultures treated with PBS or EMT cocktail for 5 days before RNA-Seq analysis. B: unbiased clustering of all samples using principal component analysis (PCA). All genes with <1 TPM value were removed before performing PCA, n = 8 donors. C: volcano plot of expressed genes (TPM ≥ 1) between PBS- and EMT-treated ALI cultures, n = 8. Red dots, adjusted P value <10−6 and log2 fold-change >2; blue dots, adjusted P value <10−6 and log2 fold-change <2; black dots, adjusted P value >10−6 and log2 fold-change >2; orange dots, >10−6 and log2 fold-change <2. D: EMT-associated markers, TPM values plotted as heatmap between PBS- and EMT-treated ALI cultures, n = 8 donors. E: NetAct analysis depicting activity of enriched transcription factors (FDR ≤ 0.01). Presence of TWIST1 and CTNNB1 but absence of major EMT markers like SNAI and ZEB family members indicate partial EMT. F: calculated EMT score for ALI cultures treated with PBS or EMT cocktail. A negative score for EMT-treated ALI cultures demonstrates a partial EMT. ALI, air-liquid interface; EMT, epithelial-mesenchymal transition; scRNA-Seq, single-cell RNA-sequencing.
Gene Signatures from EMT-Induced ALI Cultures Overlap with Those from Different Lung Diseases
Further analysis of the RNA-Seq data revealed changes in many immune-related genes, which include cytokines, chemokines, defensins, and protease activated receptors (Fig. 4A). These changes suggest that EMT could alter innate immune lung epithelial responses to pathogens, allergens, and toxins. To determine whether these transcriptional changes displayed disease relevance, we performed an Ingenuity Pathway Analysis (IPA) using manually curated disease gene lists (Supplemental Table S10, see https://doi.org/10.6084/m9.figshare.19093751). This revealed significant enrichment in pathways for asthma, COPD, and IPF (Fig. 4B). Hepatic fibrosis was included as an unrelated disease in which EMT has also been implicated. Interestingly, most of the genes found (Fig. 4A) were associated with one or more of the lung diseases (Fig. 4B). Furthermore, there were 17 overlapping genes between EMT ALI cultures, asthma, IPF, and COPD (Fig. 4C). In conclusion, EMT in ALI cultures induces potentially pathological transcriptional changes, including those associated with multiple lung diseases.
Figure 4.
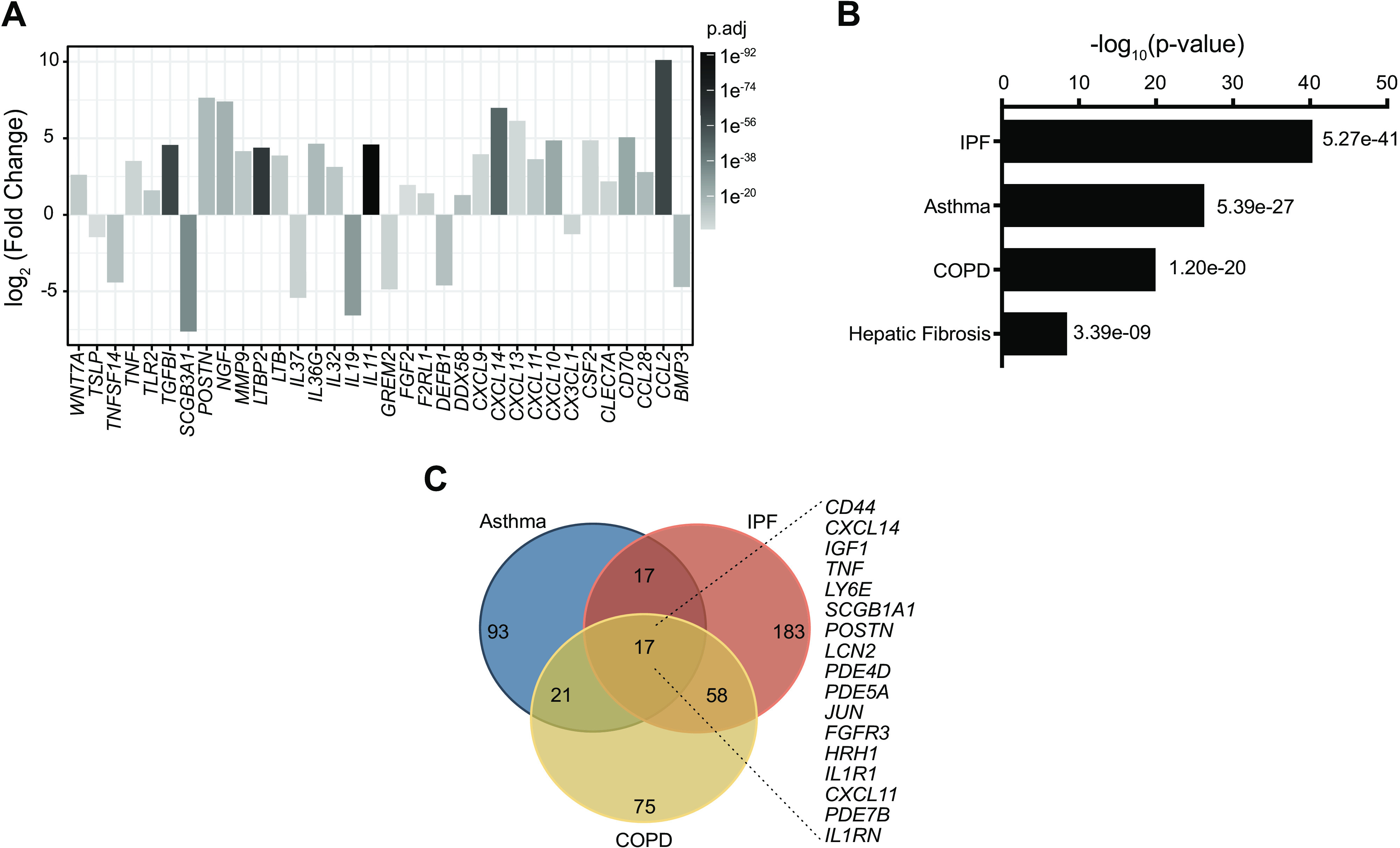
Gene signatures from EMT-induced ALI cultures overlap those from different lung diseases. A: expression or immune associated genes in RNA-Seq data from ALI cultures treated with or without EMT cocktail, identified by Gene Ontology (GO) analysis. n = 8 donors. B: Ingenuity Pathway Analysis (IPA) of genes differentially expressed between untreated (control) and EMT-treated ALI cultures, compared with custom built disease gene lists. Plotted are P values for enrichment scores. C: Venn diagram for the genes identified from IPA analysis in asthma, IPF, and COPD. Listed are differentially expressed genes between untreated (control) and EMT-treated ALI cultures that also overlap all three diseases. ALI, air-liquid interface; COPD, chronic obstructive pulmonary disease; EMT, epithelial-mesenchymal transition.
Single-Cell RNA-Seq of EMT in ALI Cultures Identifies Aberrant Basaloid-Like Cells, but No Fibroblast or Myofibroblast Conversion
To elucidate the impact of EMT at single-cell resolution, scRNA-Seq was performed on ALI cultures (n = 3) treated with PBS or EMT cocktail for 1 or 5 days (Fig. 5A). Most cells were characterized into discrete clusters corresponding to each timepoint, suggesting a progressive transcriptional reprogramming following EMT induction (Fig. 5B and Supplemental Fig. S5, A–D, see https://doi.org/10.6084/m9.figshare.19093706). Interestingly, pairwise DGE analysis across days showed that multiciliated cells were minimally impacted by the EMT treatment, both in terms of relative cell proportions and transcriptional profile (Fig. 5, C–E, Supplemental Fig. S5E, and Supplemental Table S11, see https://doi.org/10.6084/m9.figshare.19093739). In contrast, there were not significant numbers of basal, secretory, or goblet cells identified following EMT induction (Fig. 5, C and E). Instead, we identified a large population of cells that displayed both EMT and epithelial cells markers, consistent with epithelial-mesenchymal plasticity (EMP) (24) (Fig. 5, D–F, and Supplemental Table S6). To identify their likely cell type of origin, we utilized SingleR, which revealed contributions from basal, suprabasal, and secretory cells but not multiciliated or deuterosomal cells (Supplemental Fig. S5F, Supplemental Table S7, and Supplemental Table S11). Interestingly, following EMT induction, most basaloid-like cells expressed high levels of TP63, which suggests dedifferentiation to a basal cell-like phenotype (Fig. 5, D and E). Furthermore, these clusters were strikingly similar to aberrant basaloid cells recently identified in ILD lungs (18). For example, similar to aberrant basaloid cells, these basaloid-like cells coexpressed KRT17, TP63, FN1, VIM, HMGA2, CDH2, CAMK2N1, EPHB2, and OCAID2 (Fig. 5, D–F, and Supplemental Fig. S6A, see https://doi.org/10.6084/m9.figshare.19093721) (18). Therefore, we categorized day 1 clusters as “transitional aberrant basaloid-like” cells and day 5 clusters as “aberrant basaloid-like” cells. Using marker genes for fibroblasts and myofibroblasts, previously identified in scRNA-Seq analysis of human lung (18, 82, 83), we found no evidence for the presence of either fibroblasts or myofibroblast in EMT-treated ALI cultures (Supplemental Fig. S6B). This included the canonical myofibroblast marker α-SMA (ACTA2) (Supplemental Fig. S6B). Collectively, these data suggest that ALI cultures could provide a model to study the biology of aberrant basaloid-like cells in vitro, which may provide and important opportunity to reduce the need to access tissue from patients with ILD.
Figure 5.
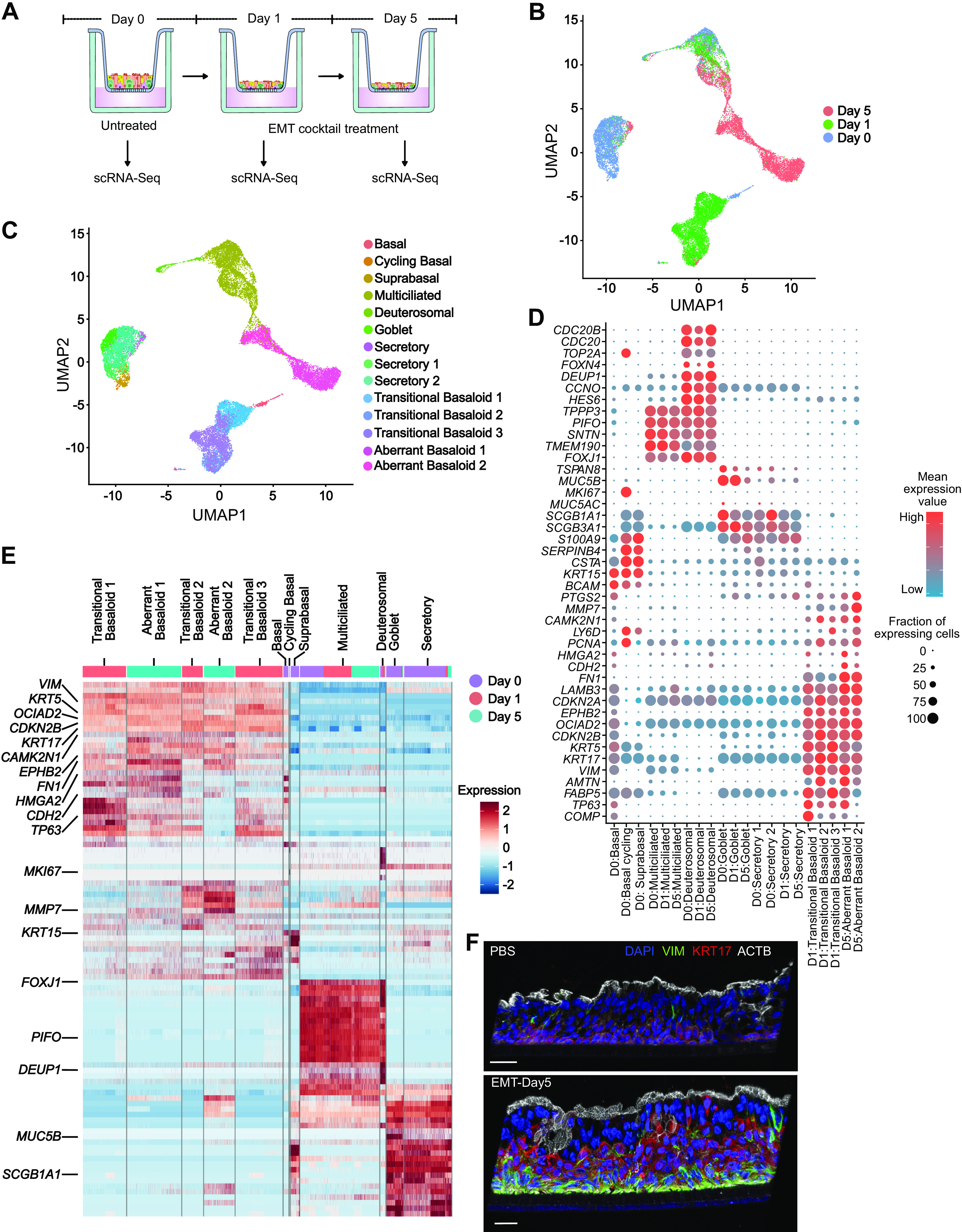
Single-cell RNA-Seq of EMT in ALI cultures identifies aberrant basaloid-like cells but no fibroblast or myofibroblast conversion. A: scRNA-Seq experimental design. Differentiated HBEC ALI cultures (25 days) were dissociated for scRNA-Seq analysis following EMT treatment for 0, 1, and 5 days, n = 3 donors. B: UMAP plot of the scRNA-Seq expression data highlighting clusters defined by timepoints of EMT treatment. C: UMAP plot depicting cluster annotations of day 0, day 1, and day 5 EMT ALI cultures. D: expression data for selected genes for each cluster. Transitional basaloid-like and aberrant basaloid-like cells express both basal and mesenchymal markers. E: heatmap depicting relative expression (normalized and scaled z-scores) of PC genes in each cluster. All PC gene names, and their respective expression values is available in Supplemental Table S7. F: immunofluorescence demonstrating colocalization of VIM and KRT17 in EMT-treated ALI cultures. Scale = 30 µm; n = 2 donors, representative data from one donor is shown. ALI, air-liquid interface; EMT, epithelial-mesenchymal transition; HBECs, human bronchial epithelial cells; PC, protein coding.
EMT Induction Reprograms the lncRNA Landscape
Analysis of bulk RNA-Seq identified 907 differentially expressed lncRNAs (FDR ≤0.05; |log2FC| ≥ 2; Fig. 6A and Supplemental Table S8) following EMT induction in ALI cultures (Fig. 6A). A number of these lncRNAs have previously been associated with EMT (e.g., CASC15 and NKILA); however, most have yet to be studied in any system. To further identify high-confidence lncRNAs in EMT-induced ALI cultures, we performed a pseudo-bulk analysis of the scRNA-Seq data (Supplemental Table S12, see https://doi.org/10.6084/m9.figshare.19093757), which identified deferentially expressed lncRNAs (FDR ≤0.05; |log2FC| ≥ 2): 651 at Day 1 and 990 at Day 5 (Supplemental Table S12 and Supplemental Table S13, see https://doi.org/10.6084/m9.figshare.19093760), many of which were also captured in the bulk RNA-Seq . A stringent analysis of the top 100 lncRNAs (FDR ≤0.05; |log2FC| ≥ 2) between bulk (Supplemental Table S8) and scRNA-Seq (Supplemental Table S13) identified 16 lncRNAs (Fig. 6C). In addition, a cluster-based analysis revealed lncRNAs whose expression was enriched in transitional basaloid-like cells and aberrant basaloid-like cells (Supplemental Fig. S7, see https://doi.org/10.6084/m9.figshare.19093718). Of particular interest was the lncRNA CASC15, because it has been previously implicated in the regulation of EMT (84–86). Expression of CASC15 was enriched in transitional aberrant basaloid-like cells and aberrant basaloid-like cells (Fig. 6, D and E, and Supplemental Fig. S8, A and B, see https://doi.org/10.6084/m9.figshare.19093715). We then analyzed scRNA-Seq data (18) for expression of CASC15 in ILD lungs that reveled substantial expression in aberrant basaloid cells (Fig. 6F and Supplemental Fig. S8C). In addition, RNA-FISH analysis of lung tissue samples revealed higher expression of CASC15 in a donor with IPF compared with a matched non-IPF control (Fig. 6G and Supplemental Fig. S8D). Although the function of CASC15 in the lung is unknown, it is relatively highly expressed in HBECs and multiple independent GWAS implicate the CASC15 locus in lung biology (Supplemental Fig. S8, E and F, and Supplemental Table S14, see https://doi.org/10.6084/m9.figshare.19093748). Exploration of CASC15 and other lncRNAs identified could provide novel insights into the programming and function of aberrant basaloid cells in EMT.
Figure 6.
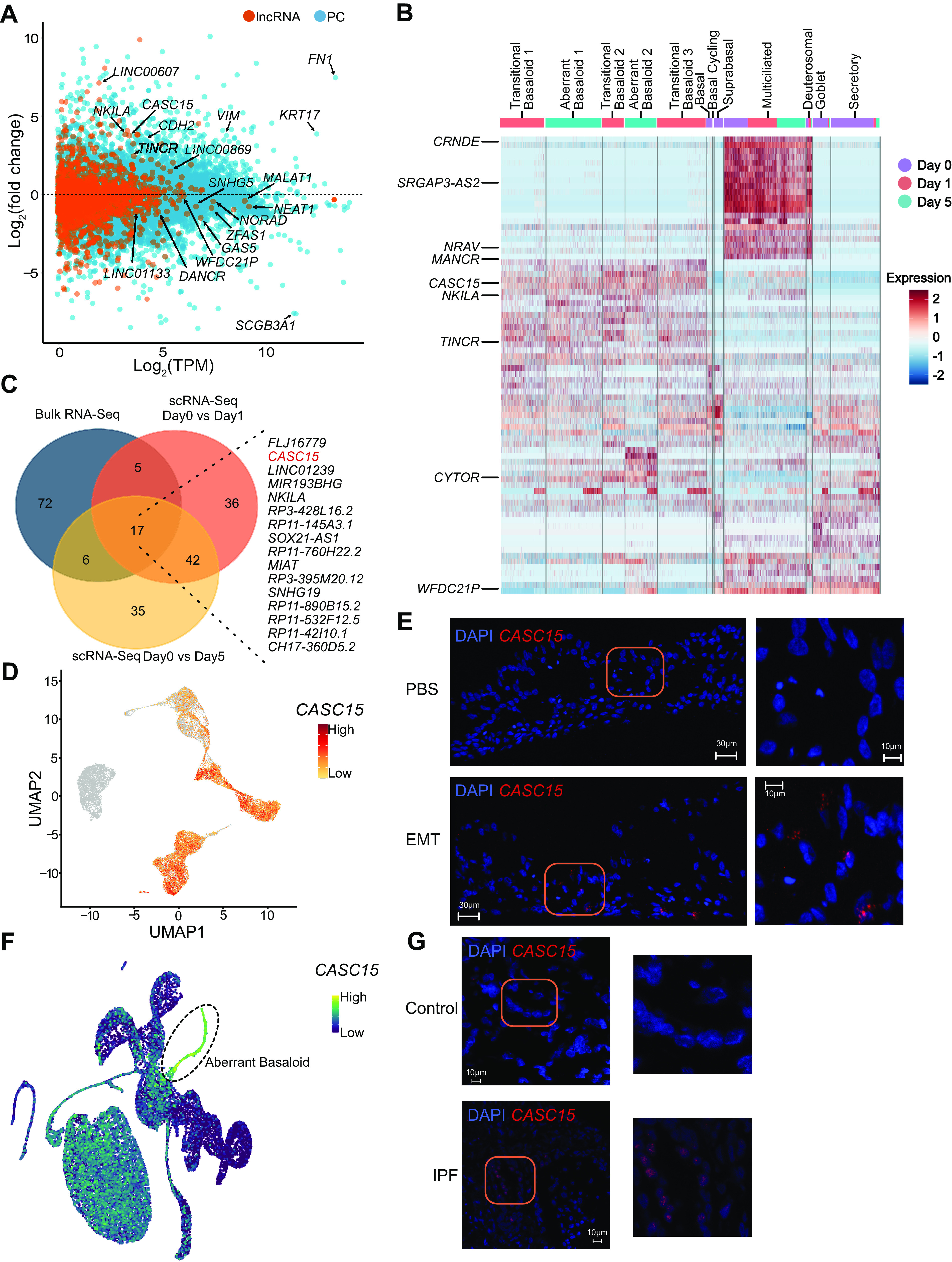
EMT induction reprograms the lncRNA landscape. A: MA plot of protein coding (PC) and long noncoding RNA (lncRNA) expression between PBS- and EMT-treated ALI cultures, n = 8. B: heatmap depicting relative expression (normalized and scaled expression) of lncRNAs in each cluster. All lncRNA names and their respective expression values are available in Supplemental Table S7. Highlighted lncRNAs CASC15, TINCR, and MANCR exclusively mark transitional basaloid-like and aberrant basaloid-like cells. C: Venn diagram of top 100 lncRNA genes (as ranked based on adjusted P value) identified from bulk RNA-Seq, and pseudo-bulk analysis of scRNA-Seq of EMT-treated ALI cultures; listed are overlapping genes. D: UMAP plot highlighting expression of CASC15 in each cluster. E: in situ hybridization showing CASC15 expression in EMT-treated ALI cultures but not in PBS-treated ALI cultures. Scale = 30 µm; n = 2, representative data from one donor is shown. F: UMAP highlighting expression of CASC15 in IPF scRNA-Seq data. G: in situ hybridization using CASC15 probes on lung tissue from donors with or without IPF; scale = 30 µm; n = 1, data shown are from one donor each. IPF sample: 64 old, male, Caucasian. non-IPF (control) sample: 66 old, male, Caucasian. Confocal imaging demonstrates absence of CASC15 expression in non-IPF lung tissue but capturing punctuated signal in IPF lung tissue. ALI, air-liquid interface; EMT, epithelial-mesenchymal transition; lncRNA, long noncoding RNA.
DISCUSSION
EMT is commonly observed in fibrotic lung disease, although its contribution to disease is unclear. Here we sought to elucidate the impact of EMT on human bronchial epithelium by mapping EMT-induced transcriptional changes at the population and single-cell level in ALI cultures.
Analysis of submerged and differentiated ALI cultures revealed a dramatic transcriptional reprogramming involving 8,247 PC and 1,621 lncRNA genes. These changes highlight the potential limitations when interpreting data from submerged cultures. The reason for these differences was partially revealed by scRNA-Seq analysis of submerged cultures compared with ALI cultures. Although ALI cultures contained a complex mixture of different epithelial cell types, submerged cultures consisted entirely of basal cells and cycling basal cells. In addition, presumed equivalent cell types in submerged and ALI cultures had noticeably distinct transcriptional profiles, which potentially reflect different medium composition or differences in oxygen levels between submerged and ALI cultures. Unexpectedly, in bulk RNA-Seq analysis, there were no observed differences between cells obtained from donors with, or without, asthma. However, because asthma is a heterogenous disease and the severity and donor endotypes were unknown (87, 88), it is possible that some, or all, of the donors used in this study had mild disease. Alternatively, disease phenotypes may be influenced by cell culture conditions. Previously, significant differences in asthmatic cell responses from donors obtained from the same source have been observed (89). Thus, it is likely that phenotypes are donor-dependent.
Experiments showed that epithelial cell differentiation was accompanied by changes in the lncRNA landscape. Although lncRNAs are generally less highly expressed than PC genes, we were able to detect a subset using scRNA-Seq, which revealed numerous lineage-specific lncRNAs, with enrichment in multiciliated cells. For example, expression of the lncRNA Negative Regulator of Antiviral Response (NRAV) was highest in multiciliated cells, which is interesting as these are target cells for respiratory viral infections. The function of most lncRNAs is completely unknown, and their study may reveal novel and important roles in airway epithelial differentiation or function. Interestingly, some lineage-specific transcripts originally annotated as lncRNAs have been shown to generate small peptides, and their function in airway epithelium is currently unknown.
Induction of EMT in ALI cultures was also associated with broad PC and lncRNA gene transcriptional changes. Although HBECs from donors with asthma have previously been described to be more sensitive to EMT induction (90), we did not find any differences based on disease status, potentially for reasons discussed above. However, pathway analysis revealed a significant overlap of the ALI-EMT gene signature with genes associated with different lung diseases that have characteristics of fibrosis implicated in their disease pathogenesis or pathobiology, which suggests some commonality in underlying mechanisms. Indeed, EMT signatures have been described in ILD, COPD, and asthma (4–10). However, only 17 genes were common between EMT induction in ALI cultures, asthma, IPF, and COPD, which likely reflects differences in the timing and extent of pathology in each of these diseases. Nevertheless, most of these genes have been associated with EMT previously. The most significant enrichment was with ILD. Although fibrosis is recognized as a larger component of lung pathology in ILD compared with asthma and COPD, this result was unexpected because ILD is generally thought to be associated with alveolar epithelium and our cultured epithelial cells are bronchial in origin. Yet recent evidence suggests that the conducting airways are involved in ILD (91, 92), and our results may suggest that common pathways are shared between epithelium in different lung compartments. Indeed, lung scRNA-Seq shows that nonalveolar cell types also display altered transcriptional profiles in ILD (75, 93, 94), and these differentially expressed genes were integrated into this studies’ pathway analysis, which may partially explain their observed enrichment.
Bioinformatic analysis of bulk RNA-Seq data revealed a partial EMT signature. Notably, NetAct reported a lack of enrichment of the key EMT mediators such as SNAI1. This is potentially due to limited SNAI1 expression by HBEC ALI cultures. In support of partial EMT, scRNA-Seq did not reveal any fibroblast or myofibroblast-like cells. Indeed, transitional basaloid-like and aberrant basaloid-like cells were either negative for most fibroblast/myofibroblast markers or expressed them at a level similar to other epithelial cell types. This underscores the prevailing hypothesis that epithelial EMT is unlikely to directly contribute to the myofibroblast population, which is replicated here in ALI cultures. Instead, we saw gene signatures consistent with partial EMT, known as epithelial-mesenchymal plasticity (EMP), in which cells coexpress both epithelial and EMT markers. EMP phenotypes have been found in ILD (19–23) and were recently described as the hallmark of aberrant basaloid cells found in IPF (18). Following EMT, ALI HBECs expressed genes associated with aberrant basaloid cells. Day 1 cells had an intermediate phenotype, which we termed transitional basaloid-like cells. One difference is that these in vitro generated aberrant basaloid-like cells maintained expression of Keratin 5. This may reflect a different cellular origin, different EMT inducing signals, or differences that occur depending on the duration of EMT treatment. However, extending EMT cultures out to 14 days did not result in additional EMT progression, which suggests that timing alone is not the issue. Indeed, recent work suggest that patient-derived aberrant basaloid cells maintain a partial EMT phenotype when cultured in vitro (95).
Following EMT induction, basal, secretory, and goblet cells were almost completely replaced by aberrant basaloid-like cells. Subclusters that were apparent within the pool of transitional and aberrant basaloid-like cells were predicted to preferentially originate from basal, suprabasal, and secretory cells. In contrast, a normal representation of multiciliated cells was clear even 5 days after EMT induction, and there was little evidence that they contributed significantly to the pool of aberrant basaloid-like cells. In addition, multiciliated cells did not appear to be directly impacted by the EMT induction and displayed a normal gene signature. However, it is unclear why multiciliated cells would be resistant to EMT induction because the scRNA-Seq data indicate that these cells express TGFβ receptor and WNT receptors, although receptor expression could fluctuate over time. Alternatively, multiciliated cells that responded to EMT may have been lost via apoptosis, or they may have converted to aberrant basaloid-like cells and lost all of their original gene signature(s), rendering it impossible to determine from which population they originated. Even though aberrant basaloid cells do not appear to contribute to the myofibroblast pool, they may still play an active role in fibrotic lung disease through altered differentiation and/or autocrine/paracrine inflammatory responses. Evidence for such a paradigm exists in kidney fibrosis where renal epithelial cells undergo partial EMT and then release paracrine signals that reshape the microenvironment to promote inflammation and fibrogenesis (24).
EMT in ALI cultures resulted in a reprogramming of the lncRNA landscape. Although the function of most lncRNAs is unknown, these lncRNAs could be explored as potential biomarkers or even potential therapeutic targets. The lncRNA CASC15 is particularly notable given the current understanding of its role in EMT and connection to lung biology implicated through GWAS (68, 96). However, its function in the lung has yet to be explored. In summary, this work provides additional evidence that HBECs do not convert into myofibroblasts. Furthermore, we show that ALI cultures provide a physiologically relevant and tractable system to study aberrant basaloid-like cells and mechanisms of EMP. This may result in an important step in ILD research given the limited access to ILD patient tissue, which typically reflects cells from end-stage disease. Thus, there is potential to study EMP/EMT at earlier stages of disease in this system, where the role for CASC15 can be further elucidated. Finally, we provide a catalog of airway epithelial lncRNAs and an interactive viewer for single-cell expression data hosted by USCS for further exploration for their roles in the lung during health and disease.
DATA AVAILABILITY
The RNA-Seq (bulk and single cell) data that support the findings of this study are openly available in the Gene Expression Omnibus (GEO) database repository at (https://www.ncbi.nlm.nih.gov/geo/), accession number GSE193684.
SUPPLEMENTAL DATA
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.19093763.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.19093730.
Supplemental Table S3: https://doi.org/10.6084/m9.figshare.19093736.
Supplemental Table S4: https://doi.org/10.6084/m9.figshare.19093724.
Supplemental Table S5: https://doi.org/10.6084/m9.figshare.19093754.
Supplemental Table S6: https://doi.org/10.6084/m9.figshare.19093727.
Supplemental Table S7: https://doi.org/10.6084/m9.figshare.19093745.
Supplemental Table S8: https://doi.org/10.6084/m9.figshare.19093742.
Supplemental Table S9: https://doi.org/10.6084/m9.figshare.19093733.
Supplemental Table S10: https://doi.org/10.6084/m9.figshare.19093751.
Supplemental Table S11: https://doi.org/10.6084/m9.figshare.19093739.
Supplemental Table S12: https://doi.org/10.6084/m9.figshare.19093757.
Supplemental Table S13: https://doi.org/10.6084/m9.figshare.19093760.
Supplemental Table S14: https://doi.org/10.6084/m9.figshare.19093748.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.19093703.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.19093700.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.19093709.
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.19093712.
Supplemental Fig. S5: https://doi.org/10.6084/m9.figshare.19093706.
Supplemental Fig. S6: https://doi.org/10.6084/m9.figshare.19093721.
Supplemental Fig. S7: https://doi.org/10.6084/m9.figshare.19093718.
Supplemental Fig. S8: https://doi.org/10.6084/m9.figshare.19093715.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants R01HL125897 (to J.L.K.), U19 AI142733 (to J.L.K., K.P., and A.W.), R21AI133440 (to A.W.), and R01AI141609 (to A.W.).
DISCLOSURES
N. Kaminski served as a consultant to Boehringer Ingelheim, Third Rock, Pliant, Samumed, NuMedii, Theravance, LifeMax, Three Lake Partners, Optikira, Astra Zeneca, RohBar, Veracyte, Augmanity, CSL Behring, Galapagos, Gilead, and Thyron over the past 3 years, reports Equity in Pliant and Thyron, and received a grant from Veracyte, Boehringer Ingelheim, BMS, and nonfinancial support from MiRagen and Astra Zeneca. N. Kaminski has IP on novel biomarkers and therapeutics in IPF licensed to Biotech. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
D.B.U.K., J.K., J.G., M.L., K.P., N.K., J.L.K., and A.W.conceived and designed research; D.B.U.K., J.M., T.-C.W., J.K., J.G., N.K., J.L.K., and A.W. performed experiments; D.B.U.K., E.M., M.Y., V.K., J.M., T.-C.W., J.K., J.G., M.L., K.P., N.K., J.L.K., and A.W.analyzed data; D.B.U.K., E.M., M.Y., V.K., T.-C.W., J.K., J.G., M.L., K.P., J.L.K., and A.W. interpreted results of experiments; D.B.U.K., E.M., V.K., J.M., J.K., J.G., M.L., K.P., J.L.K., and A.W. prepared figures; D.B.U.K., E.M., J.K., J.G., M.L., K.P., J.L.K., and A.W.drafted manuscript; D.B.U.K., E.M., M.Y., V.K., J.M., T.-C.W., J.K., J.G., M.L., K.P., J.L.K., and A.W. edited and revised manuscript; D.B.U.K., E.M., M.Y., V.K., J.M., T.-C.W., J.K., J.G., M.L., K.P., N.K., J.L.K., and A.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We gratefully acknowledge the contribution of Diane Luo from the Single Cell Biology service and the Genome Technologies service at The Jackson Laboratory for expert assistance with the work described in this publication. The authors thank Professors Wuyts and Vanaudenaerde from KU Leuven for providing the tissues.
REFERENCES
- 1.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15: 178–196, 2014. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20: 69–84, 2019. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 3.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol 27: 347–376, 2011. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 4.Sohal SS, Mahmood MQ, Walters EH. Clinical significance of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD): potential target for prevention of airway fibrosis and lung cancer. Clin Transl Med 3: 33, 2014. doi: 10.1186/s40169-014-0033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishioka M, Venkatesan N, Dessalle K, Mogas A, Kyoh S, Lin TY, Nair P, Baglole CJ, Eidelman DH, Ludwig MS, Hamid Q. Fibroblast-epithelial cell interactions drive epithelial-mesenchymal transition differently in cells from normal and COPD patients. Respir Res 16: 72, 2015. doi: 10.1186/s12931-015-0232-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kage H, Borok Z. EMT and interstitial lung disease: a mysterious relationship. Curr Opin Pulm Med 18: 517–523, 2012. doi: 10.1097/MCP.0b013e3283566721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji X, Li J, Xu L, Wang W, Luo M, Luo S, Ma L, Li K, Gong S, He L, Zhang Z, Yang P, Zhou Z, Xiang X, Wang CY. IL4 and IL-17A provide a Th2/Th17-polarized inflammatory milieu in favor of TGF-β1 to induce bronchial epithelial-mesenchymal transition (EMT). Int J Clin Exp Pathol 6: 1481–1492, 2013. [PMC free article] [PubMed] [Google Scholar]
- 8.Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol 12: 53–59, 2012. doi: 10.1097/ACI.0b013e32834ec6eb. [DOI] [PubMed] [Google Scholar]
- 9.Gohy ST, Hupin C, Fregimilicka C, Detry BR, Bouzin C, Gaide Chevronay H, Lecocq M, Weynand B, Ladjemi MZ, Pierreux CE, Birembaut P, Polette M, Pilette C. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur Respir J 45: 1258–1272, 2015. doi: 10.1183/09031936.00135814. [DOI] [PubMed] [Google Scholar]
- 10.Cho JH, Gelinas R, Wang K, Etheridge A, Piper MG, Batte K, Dakhallah D, Price J, Bornman D, Zhang S, Marsh C, Galas D. Systems biology of interstitial lung diseases: integration of mRNA and microRNA expression changes. BMC Med Genomics 4: 8, 2011. doi: 10.1186/1755-8794-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 11.Willis BC, Borok Z. TGF-β-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol 293: L525–L534, 2007. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 12.Verhamme FM, Bracke KR, Joos GF, Brusselle GG. Transforming growth factor-β superfamily in obstructive lung diseases. more suspects than TGF-β alone. Am J Respir Cell Mol Biol 52: 653–662, 2015. doi: 10.1165/rcmb.2014-0282RT. [DOI] [PubMed] [Google Scholar]
- 13.Halwani R, Al-Muhsen S, Al-Jahdali H, Hamid Q. Role of transforming growth factor-β in airway remodeling in asthma. Am J Respir Cell Mol Biol 44: 127–133, 2011. doi: 10.1165/rcmb.2010-0027TR. [DOI] [PubMed] [Google Scholar]
- 14.Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax 69: 760–765, 2014. doi: 10.1136/thoraxjnl-2013-204608. [DOI] [PubMed] [Google Scholar]
- 15.Jolly MK, Ward C, Eapen MS, Myers S, Hallgren O, Levine H, Sohal SS. Epithelial-mesenchymal transition, a spectrum of states: role in lung development, homeostasis, and disease. Dev Dyn 247: 346–358, 2018. doi: 10.1002/dvdy.24541. [DOI] [PubMed] [Google Scholar]
- 16.Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc 3: 377–382, 2006. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA 108: E1475–E1483, 2011. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 6: eaba1983, 2020. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaguchi M, Hirai S, Tanaka Y, Sumi T, Miyajima M, Mishina T, Yamada G, Otsuka M, Hasegawa T, Kojima T, Niki T, Watanabe A, Takahashi H, Sakuma Y. Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial-mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis. Lab Invest 97: 232–242, 2017. doi: 10.1038/labinvest.2016.135. [DOI] [PubMed] [Google Scholar]
- 20.Varma S, Mahavadi P, Sasikumar S, Cushing L, Hyland T, Rosser AE, Riccardi D, Lu J, Kalin TV, Kalinichenko VV, Guenther A, Ramirez MI, Pardo A, Selman M, Warburton D. Grainyhead-like 2 (GRHL2) distribution reveals novel pathophysiological differences between human idiopathic pulmonary fibrosis and mouse models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 306: L405–L419, 2014. doi: 10.1152/ajplung.00143.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabasa M, Duch P, Jorba I, Giménez A, Lugo R, Pavelescu I, Rodríguez-Pascual F, Molina-Molina M, Xaubet A, Pereda J, Alcaraz J. Epithelial contribution to the profibrotic stiff microenvironment and myofibroblast population in lung fibrosis. Mol Biol Cell 28: 3741–3755, 2017. doi: 10.1091/mbc.E17-01-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jonsdottir HR, Arason AJ, Palsson R, Franzdottir SR, Gudbjartsson T, Isaksson HJ, Gudmundsson G, Gudjonsson T, Magnusson MK. Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab Invest 95: 1418–1428, 2015. doi: 10.1038/labinvest.2015.114. [DOI] [PubMed] [Google Scholar]
- 23.Morbini P, Inghilleri S, Campo I, Oggionni T, Zorzetto M, Luisetti M. Incomplete expression of epithelial-mesenchymal transition markers in idiopathic pulmonary fibrosis. Pathol Res Pract 207: 559–567, 2011. doi: 10.1016/j.prp.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 21: 341–352, 2020. [Erratum in Nat Rev Mol Cell Biol 22: 834, 2021] doi: 10.1038/s41580-020-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, Wang SB, Wang YZ, Yang Y, Yang N, Zhou WP, Yang GS, Sun SH. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 25: 666–681, 2014. doi: 10.1016/j.ccr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 26.Wu H, Hu Y, Liu X, Song W, Gong P, Zhang K, Chen Z, Zhou M, Shen X, Qian Y, Fan H. LncRNA TRERNA1 function as an enhancer of SNAI1 promotes Gastric cancer metastasis by regulating epithelial-mesenchymal transition. Mol Ther Nucleic Acids 8: 291–299, 2017. doi: 10.1016/j.omtn.2017.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uthaya Kumar DB, Williams A. Long non-coding RNAs in immune regulation and their potential as therapeutic targets. Int Immunopharmacol 81: 106279, 2020. doi: 10.1016/j.intimp.2020.106279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su W, Xu M, Chen X, Chen N, Gong J, Nie L, Li L, Li X, Zhang M, Zhou Q. Long noncoding RNA ZEB1-AS1 epigenetically regulates the expressions of ZEB1 and downstream molecules in prostate cancer. Mol Cancer 16: 142, 2017. doi: 10.1186/s12943-017-0711-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu C, Lin J. Long noncoding RNA ZEB1-AS1 acts as an oncogene in osteosarcoma by epigenetically activating ZEB1. Am J Transl Res 8: 4095–4105, 2016. [PMC free article] [PubMed] [Google Scholar]
- 30.Li W, Zhang Z, Liu X, Cheng X, Zhang Y, Han X, Zhang Y, Liu S, Yang J, Xu B, He L, Sun L, Liang J, Shang Y. The FOXN3-NEAT1-SIN3A repressor complex promotes progression of hormonally responsive breast cancer. J Clin Invest 127: 3421–3440, 2017. doi: 10.1172/JCI94233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lei K, Liang X, Gao Y, Xu B, Xu Y, Li Y, Tao Y, Shi W, Liu J. Lnc-ATB contributes to gastric cancer growth through a MiR-141-3p/TGFβ2 feedback loop. Biochem Biophys Res Commun 484: 514–521, 2017. doi: 10.1016/j.bbrc.2017.01.094. [DOI] [PubMed] [Google Scholar]
- 32.Ge XS, Ma HJ, Zheng XH, Ruan HL, Liao XY, Xue WQ, Chen YB, Zhang Y, Jia WH. HOTAIR, a prognostic factor in esophageal squamous cell carcinoma, inhibits WIF-1 expression and activates Wnt pathway. Cancer Sci 104: 1675–1682, 2013. doi: 10.1111/cas.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beltran M, Puig I, Peña C, García JM, Alvarez AB, Peña R, Bonilla F, de Herreros AG. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev 22: 756–769, 2008. doi: 10.1101/gad.455708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattioli K, Volders PJ, Gerhardinger C, Lee JC, Maass PG, Melé M, Rinn JL. High-throughput functional analysis of lncRNA core promoters elucidates rules governing tissue specificity. Genome Res 29: 344–355, 2019. doi: 10.1101/gr.242222.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hessels D, Schalken JA. The use of PCA3 in the diagnosis of prostate cancer. Nat Rev Urol 6: 255–261, 2009. doi: 10.1038/nrurol.2009.40. [DOI] [PubMed] [Google Scholar]
- 36.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigó R. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22: 1775–1789, 2012. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, Li Z, Bu D, Sun N, Zhang MQ, Chen R. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res 44: D203–D208, 2016. doi: 10.1093/nar/gkv1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gokey JJ, Snowball J, Sridharan A, Speth JP, Black KE, Hariri LP, Perl AT, Xu Y, Whitsett JA. MEG3 is increased in idiopathic pulmonary fibrosis and regulates epithelial cell differentiation. JCI Insight 3: e122490, 2018. doi: 10.1172/jci.insight.122490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Omote N, Sakamoto K, Li Q, Schupp JC, Adams T, Ahangari F, Chioccioli M, DeIuliis G, Hashimoto N, Hasegawa Y, Kaminski N. Long noncoding RNA TINCR is a novel regulator of human bronchial epithelial cell differentiation state. Physiol Rep 9: e14727, 2021. doi: 10.14814/phy2.14727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419, 2017. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York; London: Springer, 2009. [Google Scholar]
- 43.Blighe K, Rana S, Lewis M. EnhancedVolcano: publication-Ready Volcano Plots with Enhanced Colouring and Labeling (Online). https://bioconductor.org/packages/devel/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html
- 44.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic acids Res 47: W191–W198, 2019. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolde R. pheatmap: Pretty Heatmaps. 2015. https://CRAN.R-project.org/package=pheatmap.
- 46.Jiang C, Xuan Z, Zhao F, Zhang MQ. TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res 35: D137–D140, 2007. doi: 10.1093/nar/gkl1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han H, Cho J-W, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E, Lee S, Kang B, Jeong D, Kim Y, Jeon H-N, Jung H, Nam S, Chung M, Kim J-H, Lee I. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res 46: D380–D386, 2018. doi: 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Essaghir A, Toffalini F, Knoops L, Kallin A, van Helden J, Demoulin JB. Transcription factor regulation can be accurately predicted from the presence of target gene signatures in microarray gene expression data. Nucleic Acids Res 38: e120, 2010. doi: 10.1093/nar/gkq149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chi SM, Seo YK, Park YK, Yoon S, Park CY, Kim YS, Kim SY, Nam D. REGNET: mining context-specific human transcription networks using composite genomic information. BMC Genomics 15: 450, 2014. doi: 10.1186/1471-2164-15-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550, 2005. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504, 2003. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, Thiery JP. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med 6: 1279–1293, 2014. doi: 10.15252/emmm.201404208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH. Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 14049, 2017. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, Smibert P, Satija R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19: 224, 2018. doi: 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33: 495–502, 2015. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruiz García S, Deprez M, Lebrigand K, Cavard A, Paquet A, Arguel MJ, Magnone V, Truchi M, Caballero I, Leroy S, Marquette CH, Marcet B, Barbry P, Zaragosi LE. Novel dynamics of human mucociliary differentiation revealed by single-cell RNA sequencing of nasal epithelial cultures. Development 146, 2019. doi: 10.1242/dev.177428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deprez M, Zaragosi LE, Truchi M, Becavin C, Ruiz García S, Arguel MJ, Plaisant M, Magnone V, Lebrigand K, Abelanet S, Brau F, Paquet A, Pe'er D, Marquette CH, Leroy S, Barbry P. A single-cell atlas of the human healthy airways. Am J Respir Crit Care Med 202: 1636–1645, 2020. doi: 10.1164/rccm.201911-2199OC. [DOI] [PubMed] [Google Scholar]
- 58.Wolock SL, Lopez R, Klein AM. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst 8: 281–291.e9, 2019. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst 8: 329–337.e4, 2019. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang S, Corbett SE, Koga Y, Wang Z, Johnson WE, Yajima M, Campbell JD. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol 21: 57, 2020. doi: 10.1186/s13059-020-1950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20: 296, 2019. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Traag VA, Waltman L, van Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 9: 5233, 2019. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seltmann S, Stachelscheid H, Damaschun A, Jansen L, Lekschas F, Fontaine JF, Nguyen-Dobinsky TN, Leser U, Kurtz A. CELDA—an ontology for the comprehensive representation of cells in complex systems. BMC Bioinformatics 14: 228, 2013. doi: 10.1186/1471-2105-14-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 24: 719–720, 2008. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 65.Cortal A, Martignetti L, Six E, Rausell A. Gene signature extraction and cell identity recognition at the single-cell level with Cell-ID. Nat Biotechnol 39: 1095–1102, 2021. doi: 10.1038/s41587-021-00896-6. [DOI] [PubMed] [Google Scholar]
- 66.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20: 163–172, 2019. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mabbott NA, Baillie JK, Brown H, Freeman TC, Hume DA. An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genomics 14: 632, 2013. doi: 10.1186/1471-2164-14-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, Suveges D, Vrousgou O, Whetzel PL, Amode R, Guillen JA, Riat HS, Trevanion SJ, Hall P, Junkins H, Flicek P, Burdett T, Hindorff LA, Cunningham F, Parkinson H. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic acids Res 47: D1005–D1012, 2019. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schiller HB, Montoro DT, Simon LM, Rawlins EL, Meyer KB, Strunz M, Vieira Braga FA, Timens W, Koppelman GH, Budinger GRS, Burgess JK, Waghray A, van den Berge M, Theis FJ, Regev A, Kaminski N, Rajagopal J, Teichmann SA, Misharin AV, Nawijn MC. The human lung cell atlas: a high-resolution reference map of the human lung in health and disease. Am J Respir Cell Mol Biol 61: 31–41, 2019. doi: 10.1165/rcmb.2018-0416TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yue X, Shan B, Lasky JA. TGF-β: titan of lung fibrogenesis. Curr Enzym Inhib 6, 2010. doi: 10.2174/10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van Scoyk M, Randall J, Sergew A, Williams LM, Tennis M, Winn RA. Wnt signaling pathway and lung disease. Transl Res 151: 175–180, 2008. doi: 10.1016/j.trsl.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 72.Shi J, Li F, Luo M, Wei J, Liu X. Distinct roles of Wnt/β-catenin signaling in the pathogenesis of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Mediators Inflamm 2017: 3520581, 2017. doi: 10.1155/2017/3520581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saito A, Horie M, Nagase T. TGF-β signaling in lung health and disease. Int J Mol Sci 19: 2460, 2018. doi: 10.3390/ijms19082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Königshoff M, Eickelberg O. WNT signaling in lung disease: a failure or a regeneration signal? Am J Respir Cell Mol Biol 42: 21–31, 2010. doi: 10.1165/rcmb.2008-0485TR. [DOI] [PubMed] [Google Scholar]
- 75.Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, Peter L, Chung MI, Taylor CJ, Jetter C, Raju L, Roberson J, Ding G, Wood L, Sucre JMS, Richmond BW, Serezani AP, McDonnell WJ, Mallal SB, Bacchetta MJ, Loyd JE, Shaver CM, Ware LB, Bremner R, Walia R, Blackwell TS, Banovich NE, Kropski JA. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv 6: eaba1972, 2020. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baarsma HA, Konigshoff M. 'WNT-er is coming': WNT signalling in chronic lung diseases. Thorax 72: 746–759, 2017. doi: 10.1136/thoraxjnl-2016-209753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben-Jacob E, Onuchic JN, Levine H. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front Oncol 5: 155, 2015. doi: 10.3389/fonc.2015.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Su K, Kohar V, Katebi A, Gordin D, Qin Z, Karuturi K, Li S, Lu M. NetAct: A Computational Algorithm to Construct Core Transcription Factor Regulatory Network Using Gene Activity. 2021. https://github.com/lusystemsbio/NetAct [DOI] [PMC free article] [PubMed]
- 79.Kohar V, Lu M. Role of noise and parametric variation in the dynamics of gene regulatory circuits. NPJ Syst Biol Appl 4: 40, 2018. doi: 10.1038/s41540-018-0076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jia D, George JT, Tripathi SC, Kundnani DL, Lu M, Hanash SM, Onuchic JN, Jolly MK, Levine H. Testing the gene expression classification of the EMT spectrum. Phys Biol 16: 025002, 2019. doi: 10.1088/1478-3975/aaf8d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karacosta LG, Anchang B, Ignatiadis N, Kimmey SC, Benson JA, Shrager JB, Tibshirani R, Bendall SC, Plevritis SK. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat Commun 10: 5587, 2019. doi: 10.1038/s41467-019-13441-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, Berry GJ, Shrager JB, Metzger RJ, Kuo CS, Neff N, Weissman IL, Quake SR, Krasnow MA. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587: 619–625, 2020. doi: 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu X, Rowan SC, Liang J, Yao C, Huang G, Deng N, Xie T, Wu D, Wang Y, Burman A, Parimon T, Borok Z, Chen P, Parks WC, Hogaboam CM, Weigt SS, Belperio J, Stripp BR, Noble PW, Jiang D. Definition and signatures of lung fibroblast populations in development and fibrosis in mice and men (Preprint). bioRxiv, 2020. doi: 10.1101/2020.07.15.203141. [DOI]
- 84.Wu Q, Xiang S, Ma J, Hui P, Wang T, Meng W, Shi M, Wang Y. Long non-coding RNA CASC15 regulates gastric cancer cell proliferation, migration and epithelial mesenchymal transition by targeting CDKN1A and ZEB1. Mol Oncol 12: 799–813, 2018. doi: 10.1002/1878-0261.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Russell MR, Penikis A, Oldridge DA, Alvarez-Dominguez JR, McDaniel L, Diamond M, Padovan O, Raman P, Li Y, Wei JS, Zhang S, Gnanchandran J, Seeger R, Asgharzadeh S, Khan J, Diskin SJ, Maris JM, Cole KA. CASC15-S is a tumor suppressor lncRNA at the 6p22 neuroblastoma susceptibility locus. Cancer Res 75: 3155–3166, 2015. doi: 10.1158/0008-5472.CAN-14-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fernando TR, Contreras JR, Zampini M, Rodriguez-Malave NI, Alberti MO, Anguiano J, Tran TM, Palanichamy JK, Gajeton J, Ung NM, Aros CJ, Waters EV, Casero D, Basso G, Pigazzi M, Rao DS. The lncRNA CASC15 regulates SOX4 expression in RUNX1-rearranged acute leukemia. Mol Cancer 16: 126, 2017. doi: 10.1186/s12943-017-0692-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lotvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, Lemanske RF, Jr, Wardlaw AJ, Wenzel SE, Greenberger PA. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol 127: 355–360, 2011. doi: 10.1016/j.jaci.2010.11.037. [DOI] [PubMed] [Google Scholar]
- 88.Kuruvilla ME, Lee FE, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol 56: 219–233, 2019. doi: 10.1007/s12016-018-8712-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, Hammad H. Interleukin-1α controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med 209: 1505–1517, 2012. doi: 10.1084/jem.20112691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick SM, Bai TR, Knight DA. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-β1. Am J Respir Crit Care Med 180: 122–133, 2009. doi: 10.1164/rccm.200811-1730OC. [DOI] [PubMed] [Google Scholar]
- 91.Chilosi M, Poletti V, Murer B, Lestani M, Cancellieri A, Montagna L, Piccoli P, Cangi G, Semenzato G, Doglioni C. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: the role of ΔN-p63. Lab Invest 82: 1335–1345, 2002. doi: 10.1097/01.lab.0000032380.82232.67. [DOI] [PubMed] [Google Scholar]
- 92.Plantier L, Debray MP, Estellat C, Flamant M, Roy C, Bancal C, Borie R, Israël-Biet D, Mal H, Crestani B, Delclaux C. Increased volume of conducting airways in idiopathic pulmonary fibrosis is independent of disease severity: a volumetric capnography study. J Breath Res 10: 016005, 2016. doi: 10.1088/1752-7155/10/1/016005. [DOI] [PubMed] [Google Scholar]
- 93.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. . Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 199: 1517–1536, 2019. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beisang DJ, Smith K, Yang L, Benyumov A, Gilbertsen A, Herrera J, Lock E, Racila E, Forster C, Sandri BJ, Henke CA, Bitterman PB. Single-cell RNA sequencing reveals that lung mesenchymal progenitor cells in IPF exhibit pathological features early in their differentiation trajectory. Sci Rep 10: 11162, 2020. doi: 10.1038/s41598-020-66630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Khan P, Roux J, Blumer S, Fang L, Savic S, Knudsen L, Jonigk D, Kuehnel MP, Gazdhar A, Geiser T, Tamm M, Hostettler KE. In vitro culture of aberrant basal-like cells from fibrotic lung tissue (Preprint). bioRxiv, 2020. doi: 10.1101/2020.08.16.247866. [DOI]
- 96.Wyss AB, Sofer T, Lee MK, Terzikhan N, Nguyen JN, Lahousse L, et al. . Multiethnic meta-analysis identifies ancestry-specific and cross-ancestry loci for pulmonary function. Nat Commun 9: 2976, 2018. doi: 10.1038/s41467-018-05369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.19093763.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.19093730.
Supplemental Table S3: https://doi.org/10.6084/m9.figshare.19093736.
Supplemental Table S4: https://doi.org/10.6084/m9.figshare.19093724.
Supplemental Table S5: https://doi.org/10.6084/m9.figshare.19093754.
Supplemental Table S6: https://doi.org/10.6084/m9.figshare.19093727.
Supplemental Table S7: https://doi.org/10.6084/m9.figshare.19093745.
Supplemental Table S8: https://doi.org/10.6084/m9.figshare.19093742.
Supplemental Table S9: https://doi.org/10.6084/m9.figshare.19093733.
Supplemental Table S10: https://doi.org/10.6084/m9.figshare.19093751.
Supplemental Table S11: https://doi.org/10.6084/m9.figshare.19093739.
Supplemental Table S12: https://doi.org/10.6084/m9.figshare.19093757.
Supplemental Table S13: https://doi.org/10.6084/m9.figshare.19093760.
Supplemental Table S14: https://doi.org/10.6084/m9.figshare.19093748.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.19093703.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.19093700.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.19093709.
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.19093712.
Supplemental Fig. S5: https://doi.org/10.6084/m9.figshare.19093706.
Supplemental Fig. S6: https://doi.org/10.6084/m9.figshare.19093721.
Supplemental Fig. S7: https://doi.org/10.6084/m9.figshare.19093718.
Supplemental Fig. S8: https://doi.org/10.6084/m9.figshare.19093715.
Data Availability Statement
The RNA-Seq (bulk and single cell) data that support the findings of this study are openly available in the Gene Expression Omnibus (GEO) database repository at (https://www.ncbi.nlm.nih.gov/geo/), accession number GSE193684.