

Abstract
Three new diterpene alkaloids, (+)-8-epiagelasine T (1), (+)-10-epiagelasine B (2), and (+)-12-hydroxyagelasidine C (3), along with three known compounds, (+)-ent-agelasine F (4), (+)-agelasine B (5), and (+)-agelasidine C (6), were isolated from the sponge Agelas citrina, collected on the coasts of the Yucatán Peninsula (Mexico). Their chemical structures were elucidated by 1D and 2D NMR spectroscopy, HRESIMS techniques, and a comparison with literature data. Although the synthesis of (+)-ent-agelasine F (4) has been previously reported, this is the first time that it was isolated as a natural product. The evaluation of the antimicrobial activity against the Gram-positive pathogens Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus faecalis showed that all of them were active, with (+)-10-epiagelasine B (2) being the most active compound with an MIC in the range of 1–8 µg/mL. On the other hand, the Gram-negative pathogenes Acinetobacter baumannii, Pseudomonas aeruginosa, and Klebsiella pneumoniae were also evaluated, and only (+)-agelasine B (5) showed a moderate antibacterial activity with a MIC value of 16 μg/mL.
Keywords: diterpene alkaloids, agelasines, agelasidines, marine sponge, Agelas citrina, antibacterial
1. Introduction
One of the most common sponges in tropical and subtropical areas around the world are marine sponges of the genus Agelas (class Demospongiae, order Agelasida, family Agelasidae) [1]. Covering the period from 1971 to November 2021, 355 compounds have been isolated from Agelas sponges with interesting biological activities [2]. Many of these metabolites are of mixed biogenetic origins, as illustrated by alkaloids (especially pyrrole alkaloids and terpenoid alkaloids) [3,4], glycosphingolipids, sterols, and carotenoids that display diverse biological activities and unique structural features [5,6,7].
In particular, the nitrogenated diterpenes that include hypotaurocyamines (agelasidines) and N9-adeninium alkaloids (agelasines) represent an important class of compounds isolated from this genus and they show interesting biological activities, including antibacterial, cytotoxic, antifouling, antifungal, and inhibitory effects on Na+/K+-ATPase [8].
As part of our ongoing efforts to find new bioactive natural compounds from marine organisms collected off the coast of the Yucatán Peninsula (Mexico) [9,10,11,12], specimens of the sponge A. citrina were investigated due to the antibacterial activity displayed by their organic extracts. Former studies of the sponge A. citrina reported the presence of diterpene alkaloids [13] and pyrrole—imidazole alkaloids [14]. Our preliminary chemical investigation of this sponge led to the isolation of the major known component (−)-agelasine B, which showed significant antibacterial activity against two Gram-positive bacteria S. aureus strains [10]. Further bioactivity-guided fractionation of the CH2Cl2 and aqueous methanolic fractions led to the isolation of three new compounds (1, 2, and 3), as well as three known compounds (4, 5, and 6). All isolated compounds (1–6) were evaluated for their antibacterial activity. Herein, we describe the isolation, structure elucidation, and antibacterial activity of 1–6.
2. Results and Discussion
2.1. Isolation and Identification of Agelasines
Specimens of the sponge A. citrina collected on Cozumel Island of the Yucatán Penisula (Mexico) were extracted several times with CH3OH/CH2Cl2 to produce an organic extract, which was partitioned following our standard partitioning procedure [15] into several fractions of differing polarities. The CH2Cl2 and aqueous methanolic fractions were submitted to Solid Phase Extraction (SPE) with RP-18 cartridges and finally to RP-HPLC to produce pure compounds 1–6 (Figure 1).
Figure 1.

The structures of compounds 1–6 isolated from Agelas citrina.
Compound 1 was obtained as a yellow oil [α]25D = +12 (c 0.36, MeOH). The molecular formula of 1 was established based on the [M]+ peak at m/z 440.3375, observed in its (+)-HRESIMS (calculated for C26H42N5O, m/z 440.3384, Δ = 2.04 ppm).
The 1H and 13C NMR spectral data, along with an HSQC spectrum of 1 in DMSO-d6 (Table 1), were indicative of an agelasine-type diterpene alkaloid compound. The adenine moiety was identified by the characteristic signals of two methines at δH/δC 8.46 (1H, s, H-2′)/155.4 (C-2′) and 9.51 (1H, s, H-8′)/140.8 (C-8′); a N-methyl group at δH/δC 3.88 (3H, s)/31.1; and three non-protonated sp2 carbons at δC 152.4 (C-6′), 149.0 (C-4′), and 109.2 (C-5′). The diterpene portion of 1 was assigned from the 1H and 13C NMR signals of the bicyclic ring corresponding to four diasterotopic methylenes observed at δH/δC 1.58 (1H, m, H-1a)-0.89 (1H, m, H-1b)/39.2 (C-1), 1.53 (1H, m, H-2a)-1.19 (1H, m, H-2b)/19.8 (C-2), 1.33 (1H, m, H-3a)-1.10 (1H, m, H-3b)/41.3 (C-3), and 1.70 (1H, dt, J = 11.8, 3.0 Hz, H-7a)-1.30 (1H, m, C-7b)/43.7 (C-7); one homotopic methylene at δH/δC 1.34 (2H, m, H-6)/17.7 C-6; two methines at δH/δC 0.81 (1H, m, H-5)/55.3 (C-5) and 0.98 (1H, m, H-9)/59.9 (C-9); four methyl groups at δH/δC 1.00 (3H, s, H3-17)/23.6 (C-17), 0.84 (3H, s, H3-18)/33.0 (C-18), 0.75 (3H, s, H3-19)/21.1 (C-19), and 0.74 (3H, s, H3-20)/15.0 (C-20); two quaternary carbons at δC 32.9 (C-4) and 38.5 (C-10); and one non-protonated carbon linked to oxygen at δC 72.1 (C-8). Moreover, the 1H and 13C NMR signals corresponding to three methylenes at δH/δC 1.55 (1H, m, H-11a)-1.27 (1H, m, H-11b)/22.7 (C-11), 2.22 (1H, td, J = 12.2, 6.1 Hz, H-12a)-2.09 (1H, td, J = 12.2, 4.9 Hz, H-12b)/42.2 (C-12), and 5.13 (2H, d, J = 7.3 Hz, H-15)/46.6 (C-15); a vinyl methyl at δH/δC 1.79 (3H, s, H3-16)/16.5 (C-16); an olefinic methine at δH/δC 5.44 (1H, t, J = 7.0 Hz, H-14)/114.6 (CH); and a quaternary sp2 carbon at δC 147.0 (C-13) confirmed the presence of the 3-methylpentenyl chain.
Table 1.
1H NMR (500 MHz) and 13C NMR (125 MHz) data for 8-epiagelasine T (1), 10-epiagelasine B (2), and agelasine B (5).
| Post. | 1 a | 2 b | 5 b | |||
|---|---|---|---|---|---|---|
|
δH, Mult, (J in Hz) |
δC, Type |
δH, Mult, (J in Hz) |
δC, Type |
δH, Mult, (J in Hz) |
δC, Type | |
| 1 | 1.58, m 0.89, m |
39.2, CH2 | 1.64 m 1.33, m |
19.7, CH2 | 1.38, m 1.15, m |
18.2, CH2 |
| 2 | 1.53, m | 19.8, CH2 | 1.98, m | 22.9, CH2 | 1.49, m | 26.8, CH2 |
| 1.19, m | 1.91, m | 1.29, m | ||||
| 3 | 1.33, m | 41.3, CH2 | 5.30, brs | 120.7, CH | 5.16, brs | 120.5, CH |
| 1.10, m | ||||||
| 4 | 32.9, C | 136.2, C | 144.3, C | |||
| 5 | 0.81, m | 55.3, CH | 40.5, C | 38.3, C | ||
| 6 | 1.34, m | 17.7, CH2 | 1.80, m 1.71, m |
33.7, CH2 | 1.70, d, (12.6) 1.14 m |
36.7, CH2 |
| 7 | 1.70, dt, (11.8, 3.0) 1.30, m |
43.7, CH2 | 1.64, m 1.42, m |
29.2, CH2 | 1.43 m | 27.4, CH2 |
| 8 | 72.1, C | 1.31, m | 33.5, CH | 1.41, m | 36.3, CH | |
| 9 | 0.98, m | 59.9, CH | 39.1, C | 38.9, C | ||
| 10 | 38.5, C | 2.28, brd, (12.9) | 42.0, CH | 1.29 m | 46.4, CH | |
| 11 | 1.55, m | 22.7, CH2 | 1.35, m | 30.0, CH2 | 1.60 m | 32.5, CH2 |
| 1.27, m | 1.25, m | 1.45 m | ||||
| 12 | 2.22, td, (12.2, 6.1) | 42.2, CH2 | 2.03, dt, (11.8, 5.6) | 35.1, CH2 | 2.00 m | 33.0, CH2 |
| 2.09, td, (12.2, 4.9) | 1.97, m | 1.92 m | ||||
| 13 | 147.0, C | 149.2, C | 148.8, C | |||
| 14 | 5.44, t, (7.0) | 114.6, CH | 5.45, t, (6.4) | 115.0, CH | 5.43, t, (5.6) | 115.0, CH |
| 15 | 5.13, d, (7.3) | 46.6, CH2 | 5.25, d, (6.5) | 48.6, CH2 | 5.34, d, (6.0) | 48.9, CH2 |
| 16 | 1.79, s | 16.5, CH3 | 1.86, s | 17.2, CH3 | 1.85, s | 17.2, CH3 |
| 17 | 1.00, s | 23.6, CH3 | 1.07, d, (7.4) | 18.7, CH3 | 0.78, d, (5.9) | 15.9, CH3 |
| 18 | 0.84, s | 33.0, CH3 | 1.62, s | 21.7, CH3 | 1.56, s | 18.0, CH3 |
| 19 | 0.75, s | 21.1, CH3 | 0.86, s | 17.2, CH3 | 0.98, s | 20.0, CH3 |
| 20 | 0.74, s | 15.0, CH3 | 0.94, s | 15.6, CH3 | 0.70, s | 18.3, CH3 |
| 2′ | 8.46, s | 155.4, CH | 8.54, s | 155.8, CH | 8.52, s | 155.2, CH |
| 4′ | 149.0, C | 149.7, C | 149.6, C | |||
| 5′ | 109.2, C | 110.0, C | 110.5, C | |||
| 6′ | 152.4, C | 151.8, C | 151.7, C | |||
| 8′ | 9.51, s | 140.8, CH | 9.87, s | 142.2, C | 10.06, s | 142.3, CH |
| 9′-N-Me | 3.88, s | 31.1, CH3 | 4.06, s | 31.9, CH3 | 4.07, s | 32.2, CH3 |
| NH2 | 7.92, s | |||||
a In DMSO-d6. b In CDCl3.
The analysis of COSY and HMBC spectra was able to confirm the planar structure of 1 (Figure 2). The 1D NMR spectral data of 1 were similar to those for agelasine T, a previously reported agelasine isolated from an unidentified Agelas collected in Okinawa [16]. However, the difference of the carbon chemical shift of the C-17 methyl group at δC 23.6 in 1 instead of the reported value of that group at δC 30.7 in agelasine T, suggested that they differed in the spatial disposition of the C-17 methyl group, and consequently in the stereochemistry at C-8 position.
Figure 2.

Key 1H-1H COSY and HMBC correlations of 1–3.
The relative configuration was assigned by 2D and selective 1D-NOESY experiments of 1. The E-geometry of the Δ13 double bond was established by the observed NOESY correlation between the signals assigned to the methylene CH2-15 at δH 5.13 and vinyl methyl CH3-16 at δH 1.79. On the other hand, the irradiation of methyl protons (Figure S6) at the position CH3-17 (δH 1.00) showed a 2.0% NOE response to the methyl protons at the position CH3-20 (δH 0.74); the irradiation of methyl protons (Figure S7) at the position CH3-18 (δH 0.84) also displayed NOE to the methyl protons at the position CH3-20 (δH 0.74) (3.0%) (see Figure 3A). Thus, these NOE correlations suggest that these three methyl groups are located on the same face of the molecule, assigned as the β face. Furthermore, the carbon chemical shift of CH3-17 (δC 23.6) resonates similarly to that of a synthetic terpene having a bicyclic ring bearing the same spatial disposition of the hydroxy group (δC 24.2) [17].
Figure 3.

Key NOE correlations for compounds 1 (A) and 2 (B).
This analysis makes it possible to propose the structure of compound 1 as (+)-8-epiagelasine T (Figure 1).
Compound 2 was obtained as a yellow oil [α]25D = +16 (c 0.13, MeOH) and its molecular formula was established on the basis of the [M]+ peak at m/z 422.3280, observed in its HRESIMS spectrum (calculated for C26H40N5, m/z 422.3279, Δ = 0.23 ppm).
The similarity of 1H and 13C NMR spectral in CDCl3 of 2 to those of 1 suggested that 2 was also an agelasine-type alkaloid with an N9-methyladenine unit.
The adenine moiety was identified by the characteristic signals of two methines at δH/δC 8.54 (1H, s, H-2′)/155.8 (C-2′) and 9.87 (1H, s, H-8′)/142.2 (C-8′), an N-methyl group at δH/δC 4.06 (3H, s)/31.9, and three non-protonated sp2 carbons at δC 151.8 (C-6′), 149.7 (C-4′) and 110.0 (C-5′). The bicyclic ring for the diterpene portion of 2 was assigned from the 1H and 13C NMR signals corresponding to four diasterotopic methylenes observed at δH/δC 1.64 (1H, m, H-1a)-1.33 (1H, m, H-1b)/19.7 (C-1), 1.98 (1H, m, H-2a)-1.91 (1H, m, H-2b)/22.9 (C-2), 1.80 (1H, m, H-6a)-1.71 (1H, m, H-6b)/33.7 (C-6), and 1.64 (1H, m, H-7a)-1.42 (1H, m, H-7b)/29.2 (C-7); an olefinic methine at δH/δC 5.30 (1H, brs, H-3)/120.7 (C-3); two aliphatic methines at δH/δC 1.31 (1H, m, H-8)/33.5 (C-8) and 2.28 (1H, brd, J = 12.9 Hz, H-10)/42.0 (C-10); a vinyl methyl at δH/δC 1.62 (3H, s, H3-18)/21.7 (C-18); three methyl groups at δH/δC 1.07 (3H, d, J = 7.4 Hz, H3-17)/18.7 (C-17), 0.86 (3H, s, H3-19)/17.2 (C-19), and 0.94 (3H, s, H3-20)/15.6 (C-20); two quaternary carbons at δC 40.5 (C-5) and 39.1 (C-9); and a quaternary sp2 carbon at δC 136.2 (C-4). Additionally, the 1H and 13C NMR signals corresponding to three methylenes at δH/δC 1.35 (1H, m, H-11a)-1.25 (1H, m, H-11b)/30.0 (C-11), 2.03 (1H, dt, J = 11.8, 5.6 Hz, H-12a)-1.97 (1H, m, H-12b)/35.1 (C-12), and 5.25 (2H, d, J = 6.5 Hz, H-15)/48.6 (C-15); a vinyl methyl at δH/δC 1.86 (3H, s, H3-16)/17.2 (C-16); an olefinic methine at δH/δC 5.45 (1H, t, J = 6.4 Hz, H-14)/115.0 (C-14); and a quaternary sp2 carbon at δC 149.2 (C-13) confirmed the presence of the 3-methylpentenyl chain.
The constitution of 2 was established from the analysis of the 2D NMR spectral data (Figure 2). Two spin systems corresponding to the bicyclic system were deduced from the COSY experiment of 2: the first spin system was assigned to the H-1, H-2, H-3, and H-10 protons, and the second spin system to the H-6, H-7, H-8, and H3-17 protons (Figure 2). Key HMBC correlations from H3-19 (δH 0.86) to C-4 (δC 136.2), C-5 (δC 40.5)/C-6 (δC 33.7) and C-10 (δC 42.0); from H3-17 (δH 1.07) to C-8 (δC 33.5), C-9 (δC 39.1) and C-7 (δC 29.2); and from H3-20 (δH 0.94) to C-8 (δC 33.5), C-9 (δC 39.1) and C-10 (δC 42.0) allowed us to connect the two spin systems. Finally, the HMBC correlation from H3-20 (δH 0.94) to C-11 (δC 30.0) showed the link between the bicyclic ring and the 3-methylpentenyl chain (Figure 2). All these data indicate the presence of a clerodane skeleton in 2 as in agelasine B (5), also isolated from this sponge.
The relative configuration of 2 was deduced from NOESY data and confirmed by a IPAP-HSQMBC experiment. The E-configuration of the Δ13 double bond of 2, as in agelasine B (5), was deduced from NOE correlations observed in the NOESY experiment between the signals assigned to the methylene CH2-15 at δH 5.25 and vinyl methyl CH3-16 at δH 1.86, and between the methylene CH2-12 at δH 2.03 and ofefinic proton CH-14 at δH 5.45.
On the other hand, the strong NOE correlations from H3-17 (δH 1.07) to H3-19 (δH 0.86) and H3-20 (δH 0.94) in the NOESY experiment of 2 indicate that these methyl groups (H3-17, H3-19, and H3-20) are oriented on the same face of the molecule, assigned as β-face.
The drastic change in the chemical shift of H-10 in 2 at δH 2.28 (1H, brd, J = 12.9 Hz) in relation to that in agelasine B (5) at δH 1.29 (1H, m) (Table 1), and considering that agelasine B (5) has the same spatial disposition for the three methyl groups H3-17, H3-19, and H3-20, suggested they must differ in the remaining stereogenic center at C-10. Similar chemical shifts and the same HMBC and NOESY correlations were observed in 2 when the NMR spectra were run in C6D6 (Table S1). Therefore, these results suggest a cis-clerodane disposition for compound 2, instead of the trans-clerodane disposition for agelasine B (5).
Curiously, when the NMR spectra of 2 was repeated this time in CDCl3, an important displacement of the carbon and proton chemical shifts was observed in the NMR spectra after 24 h, probably due to the acidic character of the deuterated solvent. Thus, the carbon and proton chemical were assigned again by 2D-NMR experiments (see Table S1 and Figures S23–S31). The low value of the 3JCH between H-10 and C-19 of 3.42 Hz measured in an IPAP-HSQMBC experiment of this sample (Figure S31) agrees with the proposed cis-clerodane disposition for 2. Unfortunately, the 3JCH between H-10 and C-19 in agelasine B (5) could not be determined due to the insufficient amount. In order to obtain a direct proof for the cis- vs. trans-ring connections in decaline-like systems using the IPAP-HSQMBC experiment, we measured the corresponding values in the available synthetic model compounds 7 and 8 (Figure 4) [18]. In this way, the corresponding value of the cis-decalin-like system in 7 (3JC7H5 = 2.9 Hz) resulted to be similar to that of 2 (Figure S32). As expected, the corresponding value of the trans-decalin-like system (3JC7H5 = 8.42 Hz) was much larger (Figure S33).
Figure 4.

Structure of model compounds 7 and 8 used for the measurement of 3JCH coupling constants.
On the other hand, the selective 1D-NOESY correlations from H3-17 (δH 1.10) to H3-20 (δH 0.87) (0.30%), H3-18 (δH 1.62) (0.36%) and H-10 (δH 1.25) (0.23%) (Figure S28); from H3-19 (δH 1.15) to H3-20 (δH 0.87) (0.5%) (Figure S29); and from H3-20 (δH 0.87) to H3-17 (δH 1.10) (0.25%), H3-19 (δH 1.15) (0.32%), and H-10 (δH 1.25) (0.30% (Figure S30), indicating that H-10, H3-17, H3-19, and H3-20 protons are in the same face of the molecule, confirmed the cis-clerodane disposition for compound 2 (Figure 3B). Thus, we named this new agelasine as 10-epiagelasine B (2).
The molecular formula of 3, also obtained as a yellow oil [α]25D = + 2 (c 0.2, MeOH), was established from its (+)-HRESIMS, which displayed a [M + H]+ adduct at m/z 440.2950 (calculated for C23H42N3SO3, m/z 440.2942, Δ = 2.04 ppm) and from its 13C NMR spectrum.
The 1H-NMR and 13C NMR spectral data of 3 in CDCl3 (Table 2) were indicative of an agelasidine-type diterpene alkaloid compound. The hypotauro-cyamine moiety (-SO2-CH2CH2NHC(NH)NH3+) was identified by the characteristic guanidine carbon signal at δC 157.5 (C-3′) and two methylenes at δH/δC 3.28 (2H, brs, H-1′)/0.3 (C-1′) and 3.70 (2H, brs, H-2′)/34.5 (C-2′). The diterpene portion of 3 was assigned by the 1H and 13C NMR signals of the monocyclic ring corresponding to two homotopic methylenes observed at δH/δC 1.44 (2H, m, H-1)/27.2 (C-1) and 1.95 (2H, m, H-2)/25.7 (C-2); an olefinic methine at δH/δC 5.40 (1H, brs, H-3)/124.4 (C-3); an aliphatic methine at δH/δC 1.71 (1H, m, H-6)/33.3 (C-6); a vinyl methyl at δH/δC 1.60 (3H, brs, H3-18)/19.3 (C-18); two methyl groups at δH/δC 0.84 (3H, s, H-19)/16.0 (C-19) and 0.85 (3H, d, J = 6.7 Hz, H-20)/21.2 (C-20); a quaternary carbon at δC 40.5 (C-5); and a quaternary sp2 carbon at δC 139.7 (C-4). Furthermore, the 1H and 13C NMR signals corresponding to four methylenes at δH/δC 1.43 (2H, m, H-7)/35.3 (C-7), 1.95 (1H, m, H-8a)-1.48 (1H, m, H-8b)/34.6 (C-8), 2.27 (2H, t, J = 6.5 Hz, H-11)/34.0 (C-11), and 3.84 (1H, dd, J = 14.5, 8.0 Hz, H-15a)-3.80 (1H, dd, J = 14.5, 8.0 Hz, H-15b)/53.8 (C-15); two vinyl methyls at δH/δC 1.71 (3H, brs, H-16)/13.1 (C-16) and 1.61 (1H, brs, H-17)/16.6 (C-17); two olefinic methines at δH/δC 5.06 (1H, t, J = 6.5 Hz, H-10)/118.6 (C-10) and 5.52 (1H, t, J = 8.0 Hz, H-14)/110.0 (C-14); and two quaternary sp2 carbons at δC 140.1 (C-9) and 149.2 (C-13) confirmed the presence of the 3,7-dimethyl-nona-3,7-dienyl chain.
Table 2.
1H NMR (500 MHz) and 13C (125 MHz) NMR data for 12-hydroxyagelasidine C (3) in CDCl3.
| Position | 3 | |
|---|---|---|
| δH, Mult (J in Hz) | δC, Type | |
| 1 | 1.44, m | 27.2, CH2 |
| 2 | 1.95, m | 25.7, CH2 |
| 3 | 5.40, brs | 124.4, CH |
| 4 | 139.7, C | |
| 5 | 40.5, C | |
| 6 | 1.71, m | 33.3, CH |
| 7 | 1.43, m | 35.3, CH2 |
| 8 | 1.95, m; 1.48, m | 34.6, CH2 |
| 9 | 140.1, C | |
| 10 | 5.06, t, (6.5) | 118.6, CH |
| 11 | 2.27, t, (6.5) | 34.0, CH2 |
| 12 | 4.09, t, (6.5) | 76.3, CH |
| 13 | 149.2, C | |
| 14 | 5.52, t, (8.0) | 110.0, CH |
| 15 | 3.84, dd, (14.5, 8.0); 3.80, dd, (14.5, 8.0) | 53.8, CH2 |
| 16 | 1.71, brs | 13.1, CH3 |
| 17 | 1.61, brs | 16.6, CH3 |
| 18 | 1.60, brs | 19.3, CH3 |
| 19 | 0.84, s | 16.0, CH3 |
| 20 | 0.85, d, (6.7) | 21.2, CH3 |
| 1′ | 3.28, brs | 50.3, CH2 |
| 2′ | 3.70, brs | 34.5, CH2 |
| 3′ | 157.5, C | |
The 1D NMR spectral data of 3 were similar to those of (+)-agelasidine C, isolated from Agelas nakamurai collected in Okinawa [19]. The presence of an additional hydroxyl group in 3 in relation to (+)-agelasidine C was suggested by the difference of an oxygen (16 Da) between their molecular formula. This was confirmed by the presence of an additional oxymethine at δH/δC 4.09 (1H, t, J = 6.5 Hz, H-12)/76.3 (C-12) in the 1H and 13C NMR spectra of 3, instead of the corresponding methylene group present in (+)-agelasidine C. The HMBC correlations between the vinyl methyl protons at δH 1.71 (3H, s, H3-16) and the oxymethine carbon at δC 76.3 (C-12), the quaternary sp2 carbon at δC 149.2 (C-13) and the olefinic methine carbon at δC 110.0 (C-14), located the hydroxyl group at C-12 position (Figure 2). The analysis of COSY and HMBC spectra was able to confirm the constitution of 3 (Figure 2). These data indicate that 3 is a new agelasidine, which was named 12-hydroxyagelasidine C.
The relative configuration of the monocyclic ring system in 3 was confirmed by a comparison of its 13C NMR spectral data with that of (+)-agelasidine C [19] and other agelasidines [20] having the same relative configuration. The E-configuration of the Δ9 and Δ13 double bonds was established from the NOESY correlation between the signals assigned to the methylene CH2-11 at δH 2.27 and the vinyl methyl CH3-17 at δH 1.61, and between the methylene CH2-15 at δH 3.84–3.80 and the vinyl methyl CH3-16 at δH 1.71, respectively. Absolute configuration at C-12 in 3 was determined by the Mosher’s method using the MTPA esters [21]. The 12-hydroxyagelasidine C (3) was treated with R-(−)- and S-(+)-α-methoxy-α-(trifluoromethyl) phenyl acetic acid (MTPA-OH) to afford the S- and R-MTPA esters. The analysis of the 1H NMR and 1H−1H COSY led to the assignment of both esters’ chemical shifts in proximity of C-12. The ΔδSR values between R- and S-MTPA esters of 3 at 12-OH were negative for H-11/10/17 and positive for H-14/15/18 (Figure 5), which suggested the absolute configuration of C-12 as R.
Figure 5.
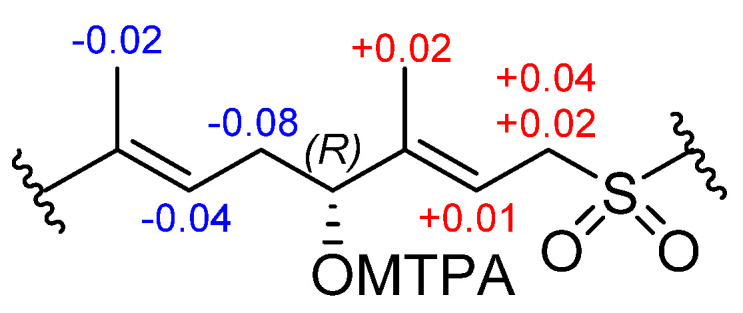
ΔδSR values (ppm) for the MTPA derivatives of 3 in CDCl3.
Compounds 4–6 (Figure 1) were identified as the known (+)-ent-agelasine F (4) [22], (+)-agelasine B (5) [8] and (+)-agelasidine C (6) [19], by comparing their NMR data with that reported in the literature. Although (+)-ent-agelasine F (4) have been previously synthetized, this is the first time that is reported as a natural product.
2.2. Antibacterial Activity of Agelasines
The antibacterial evaluation of 1–6 against the Gram-negative pathogens A. baumannii ATCC 17978, K. pneumoniae ATCC 700603, and P. aeruginosa ATCC 27823 displayed that only (+)-agelasine B (5) showed a moderate antibacterial activity with an MIC value of 16 μg/mL. In contrast, all of them exhibited antibacterial activity against Gram-positive pathogens (Table 3). These results are in accordance with previous reports, where (+)-ent-agelasine F (4), (+)-agelasine B (5) [8], and (+)-agelasidine C (6) [19] showed antimicrobial activity against Gram-positive bacteria.
Table 3.
Antibacterial activity (MIC, μg/mL) of 1–6 against Gram-positive pathogens.
| Compound | S. aureus ATCC 29213 |
S. aureus USA300LAC |
S. pneumoniae ATCC 49619 | S. pneumoniae 549 CHUAC | E. faecalis ATCC 29212 | E. faecalis 256 CHUAC | E. faecium 214 CHUAC |
|---|---|---|---|---|---|---|---|
| (+)-8-epiagelasine T (1) | 16 | 16 | 16 | 32 | 32 | ≥64 | 32 |
| (+)-10-epiagelasine B (2) | 1 | 2 | 4 | 8 | 4 | 4 | 4 |
| (+)-12-hydroxyagelasidine C (3) | 8 | 8 | 16 | - | 16 | 32 | 8 |
| (+)-ent-agelasine F (4) | 4 | 4 | 4 | - | 8 | 8 | 8 |
| (+)-agelasine B (5) | 2 | 2 | 4 | 16 | 8 | 8 | 4 |
| (+)-agelasidine C (6) | 8 | 8 | 4 | - | 8 | 8 | 8 |
Interestingly, the higher activity shown by (+)-10-epiagelasine B (2), having a cis-clerodane, than its isomer (+)-agelasine B (5), bearing a trans-clerodane, in Gram-positive pathogens, was indicative of the impact of decalin stereochemistry in the detected activity.
3. Materials and Methods
3.1. General Experimental Chemical Procedures
Optical rotations were measured on a JASCO DIP-1000 polarimeter, with an Na (589 nm) lamp and filter. 1H, 13C, and 2D NMR spectra were recorded on a Bruker spectrometer of 950 MHz, equipped with a 5 mm Cryo Probe (NEO console); a Bruker spectrometer (800 MHz for 1H and 200 MHz for 13C) equipped with a 5 mm cryo probe and an NEO console; a Bruker (800 MHz for 1H and 200 MHz for 13C) spectrometer equipped with a 3 mm cryo probe and a NEO console; a Bruker spectrometer (700 MHz for 1H and 175 MHz for 13C) equipped with a 5 mm cryo probe and an Avance III console; and a Bruker Avance 500 spectrometer (500 MHz for 1H and 125 MHz for 13C) equipped with a 5 mm cryo probe, using DMSO-d6 and CDCl3 as solvents. Chemical shifts are reported in δ scale relative to DMSO-d6 (δ 2.50 ppm for 1H NMR, δ 39.51 ppm for 13C NMR) and CDCl3 (δ 7.26 ppm for 1H NMR, δ 77.0 ppm for 13C NMR).
HRESIMS experiments were performed on the Applied Biosystems QSTAR Elite system or a Thermo MAT95XP spectrometer. HPLC separations were performed on the Agilent 1100 liquid chromatography system equipped with a solvent degasser, quaternary pump, and diode array detector (Agilent Technologies, Waldbronn, Germany) using a semipreparative reversed phase column Luna C18 (5 μ, 100 Å, 250 × 10 mm).
3.2. Animal Material
The sponge A. citrina was collected by hand and traditional SCUBA-diving off Cozumel Island, Quintana Roo (20°41′00.00″ N/87°01′32.66″ W) at depths ranging from 10 to 15 m in March 2017, and frozen immediately after collection. A voucher specimen CZE56 was deposited in the Phylum Porifera Gerardo Green National Collection of the Institute of Marine Sciences and Limnology (ICMyL) at the National Autonomous University of Mexico (UNAM), Mexico City.
3.3. Extraction and Isolation
Sliced bodies of A. citrina (wet weight, 729.6 g; dry weight, 375.3 g) were exhaustively extracted with CH3OH-CH2Cl2 (1:1, 3 × 1.5 L) at 25 °C for 24 h each maceration. The combined extracts were concentrated under reduced pressure to produce 6.1 g of a crude residue. A total of 6.0 g was first partitioned between CH2Cl2/H2O (1:1 v/v) to produce aqueous and organic phases. The organic phase was concentrated under reduced pressure and partitioned between 10% aqueous CH3OH (400 mL) and hexane (2 × 400 mL). The hexane portion produced, after removing the solvent under reduced pressure, 672.2 mg of the hexane fraction (FH). The H2O content (% v/v) of the methanolic fraction was adjusted to 50% aqueous CH3OH, and the mixture was extracted with CH2Cl2 (100 mL) to afford, after removing the solvent under reduced pressure, 3.6 g of the CH2Cl2 fraction (FD) and 755.8 mg of the remaining aqueous methanolic fraction (FM) (Scheme S1).
The CH2Cl2 fraction (3.6 g) was subjected to a Solid Phase Extraction (SPE) with RP-18 (Merck KGaA) using a discontinuous gradient from H2O to CH3OH and then CH2Cl2.
The fraction eluted with H2O/CH3OH (2:1, 181.3 mg) was separated by RP-HPLC eluting, with a mobile phase consisting of 30 min of a gradient from 30% to 100% of CH3OH in H2O (v/v, each containing 0.04% trifluoroacetic acid) followed by a 10 min isocratic at 100% of CH3OH at a flow rate of 2.0 mL/min, afforded (+)-8-epilagelasine T (1) (30.0 mg; Rt = 34.9 min) and (+)-10-epiagelasine B (2) (45.0 mg; Rt = 37.0 min).
The separation of the fraction eluted with H2O/CH3OH (1:2, 391.1 mg) was performed by RP-HPLC with a mobile phase consisting of 30 min of a gradient from 40% to 100% of CH3OH in H2O (v/v, each containing 0.04% trifluoroacetic acid), followed by a 10 min isocratic at 100% of CH3OH at a flow rate of 2.0 mL/min, yielded (+)-ent-agelasine F (4) (21.3 mg; Rt = 30.1 min) and (+)-agelasine B (5) (1.5 mg; Rt = 32.5 min).
The aqueous methanolic fraction (755.8 mg) was submitted to a Solid Phase Extraction (SPE) with RP-18 (Merck KGaA) using a discontinuous gradient from H2O to CH3OH and then CH2Cl2. The separation of the fraction eluted with H2O/CH3OH (1:1, 181.5 mg) by RP-HPLC using a mobile phase consisting of 30 min of a gradient from 35% to 100% of CH3OH in H2O (v/v, each containing 0.04% trifluoroacetic acid), followed by a 10 min isocratic at 100% of CH3OH at a flow rate of 2.0 mL/min, afforded (+)-12-hidroxy agelasidine C (3) (7.0 mg; Rt = 29.8 min) and (+)-agelasidine C (6) (27.0 mg; Rt = 34.0 min).
3.4. Structural Characterization
(+)-8-epiagelasine T (1). [α]25D +12 (c 0.2, MeOH); 1H and 13C NMR see Table 1; (+)-HRESIMS m/z 440.3375 [M]+ (calcd. for C26H42N5O, 440.3384).
(+)-10-epiagelasine B (2). [α]25D +16 (c 0.2, MeOH); 1H and 13C NMR see Table 1; (+)-HRESIMS m/z 422.3280 [M]+ (calcd. for C26H40N5, 422.3279).
(+)-12-hidroxyagelasidine C (3). [α]25D +2 (c 0.2, MeOH); 1H and 13C NMR see Table 2; (+)-HRESIMS m/z 440.2950 [M + H]+ (calcd. for C23H42N3SO3, 440.2942).
(+)-ent-agelasine F (4). [α]25D +1 (c 0.2, MeOH); 1H and 13C NMR see SI; (+)-HRESIMS m/z 422.3280 [M + H]+ (calcd. for C26H40N5, 422.3279).
(+)-agelasine B (5). [α]25D +13 (c 0.2, MeOH); 1H and 13C NMR see SI; (+)-HRESIMS m/z 422.3281 [M + H]+ (calcd. for C26H40N5, 422.3284).
(+)-agelasidine C (6). [α]25D +9 (c 0.2, MeOH); 1H and 13C NMR see SI; (+)-HRESIMS m/z 424.2991 [M + H]+ (calcd. for C23H42N3SO2, 424.2993).
3.5. Preparation of the MTPA Esters
The (R)- and (S)-MTPA esters were obtained by treatment of 3 (1.5 mg, for each acid) with (R)- and (S)-MTPA (1.60 mg), DCC (1.40 mg), and DMAP (1.0 mg), in dry CH2Cl2 (1.0 mL). The reactions were stirred at room temperature for 24 h and the resulting products were purified with SPE cartridges (Hypersep C18 200 mg, MeOH/H2O 50/50, v/v) to afford the S-(−)- and R-(+)-MTPA esters 3a and 3b. The products were monitored by recording 1H NMR spectra at 500 MHz:
1H NMR data of 3a (500 MHz in CDCl3): δH 5.04 (t, 6.5, H-10), 2.22 (m, H-11), 4.94 (t, 6.5, H-12), 5.48 (t, 8.0, H-14), 3.73 (m, H-15a), 3.77 (m, H-15b), 1.58 (brs, CH3-17), 1.69 (brs, CH3-18).
1H NMR data of 3b (500 MHz in CDCl3): δH 5.08 (t, 6.5, H-10), 2.30 (m, H-11), 4.77 (t, 6.5, H-12), 5.47 (t, 8.0, H-14), 3.69 (m, H-15a), 3.65 (m, H-15b), 1.60 (brs, CH3-17), 1.67 (brs, CH3-18).
3.6. Antibacterial Activity Assays
3.6.1. Bacterial Strains and Culture Preparation
The bacterial strains used to study the antibacterial activity of the isolated compounds were the Gram-negative pathogens Acinetobacter baumannii (ATCC 17978), Pseudomonas aeruginosa (ATCC 27823), and Klebsiella pneumoniae (ATCC 700603), and the Gram-positive pathogens, including type-strains and clinical strains isolated from infections in our hospital: Staphylococcus aureus (ATCC 29213 and USA300LAC strains), Streptococcus pneumoniae (ATCC 49619 and 549 CHUAC strains), Enterococcus faecalis (ATCC 29212 and 256 CHUAC strains), and Enterococcus faecium (214 CHUAC strain).
Gram-negative and Gram-positive strains were routinely grown or maintained in Luria–Bertani (LB), and in Trypticase soya broth (TSB) media, respectively, supplemented with 2% agar or the antibiotic ampicillin (30 mg/L), when needed. All strains were grown at 37 °C and stored in 10% glycerol at −80 °C.
3.6.2. Microdilution Method: Minimum Inhibitory Concentration
The minimum inhibitory concentrations (MICs) were determined by the broth microdilution method (Clinical Laboratory Standards Institute (CLSI), 2022). Briefly, the bacterial strains suspensions were cultured overnight at 37 °C in Mueller Hinton II agar plates (Becton Dickinson) and the turbidity of the bacterial suspensions was standarized at 0.5 on the McFarland scale to establish the inocula. The crude extracts of the test samples were dissolved in methanol. Two-fold serial dilutions of the extracts in Mueller Hinton II broth medium (Sigma) were carried out in 96-wells microplates, to produce a range of extract concentrations of 0.5–256 mg/L. Methanol was present at maximum concentration of 0.25% v/v in the well containing the highest concentration of compound. One well in each row contained growth media and bacterial suspension and was used as a positive growth control. Another well, containing medium only, was used as the negative control. Solvent controls of methanol and growth medium were included to determine whether the concentrations used interfered with bacterial growth. The MIC was evaluated after incubation 20–24 h to 37 °C and established as the lowest concentration of the compound at which the bacterial strains did not grow. All compounds were tested in triplicate.
As controls, MIC assays were also performed with imipenem against Gram-negative strains, and with vancomycin against Gram-positive strains (MIC values known).
4. Conclusions
In summary, three new diterpene alkaloids, (+)-8-epiagelasine T (1), (+)-10-epiagelasine B (2), and (+)-12-hydroxy agelasidine C (3), together with three known compounds, (+)-ent-agelasine F (4), (+)-agelasine B (5), and (+)-agelasidine C (6), were isolated from sponge Agelas citrina collected on Cozumel Island in the Mexican Caribbean. This is the first time that (+)-ent-agelasine F (4) is reported as a natural product. All of them display antibacterial activity against Gram-positive bacteria, (+)-10-epiagelasine B (2) being the most active one.
Acknowledgments
We gratefully acknowledge the help of colleagues, Daniel Catzim Pech, Gabriel González Mapen, Jorge Peniche Pérez, Melissa Llanes López, and Rodrigo Garcia Uribe for collecting the marine samples. We thank Patricia Gomez (ICMyL-UNAM) for the help in the taxonomic identification.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/md20050298/s1, Figures S1–S8: NMR spectra of 1 in DMSO-d6; Figure S9: (+)-HRESIMS of 1; Table S1: 1H NMR (500 MHz) and 13C NMR (125 MHz) and 1H NMR (800 MHz) and 13C NMR (200 MHz)* spectral data for 10-epiagelasine B (2) in CDCl3 and C6D6; Figures S10–S15: NMR spectra of 2 in CDCl3; Figure S16: (+)-HRESIMS of 1; Figures S17–S22: NMR spectra of 2 in C6D6; Figures S23–S30: NMR spectra of 2 in CDCl3 after 24 h; Figure S31: 2D IPAP-HSQMBC spectrum of 2; Figures S32–S33: 2D IPAP-HSQMBC of 7 and 8; Figures S34–S39: NMR spectra of 3 in CDCl3; Figure S40: (+)-HRESIMS of 3. Scheme S1: Extraction and fractionation scheme.
Author Contributions
Conceptualization, D.P.-P., J.R., A.M.F. and C.J.; Chemistry (extraction and isolation), D.P.-P., A.M.F.; NMR experiments, data analysis, and structural elucidation, J.C.F.-M., C.G., D.P.-P. and A.M.F.; Collection of samples and taxonomic identification, D.P.-P., C.G.-S., H.V.-H. and S.G.-H.; Biological assays, M.M.-G. and C.L.-M.; Resources, J.R., A.M.F. and C.J.; Writing—original draft, D.P.-P., A.M.F., J.R., A.B. and C.J.; Writing—review and editing, D.P.-P., J.R., A.B., A.M.F. and C.J. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
All datasets related to this article can be obtained from the authors.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work was supported by grants RTI2018-093634-B-C22 from the State Agency for Research (AEI) of Spain, cofunded by the FEDER Programme from the European Union (MCIN/AEI/10.13039/501100011033/FEDER) and BLUEBIOLAB (0474_BLUEBIOLAB_1_E), Programme INTERREG V A of Spain-Portugal (POCTEP). This work was supported by Projects PI17/01482 and PI20/01212 awarded to AB, all within in the National Plan for Scientific Research, Development and Technological Innovation 2017–2020 and funded by the ISCIII—General Subdirection of Assessment and Promotion of the Research-European Regional Development Fund (FEDER) “A way of making Europe”. The work was also supported by CIBERINFEC (CIBER de Enfermedades Infecciosas). The study was also funded by project IN607D 2021/12 (GAIN-Agencia Gallega de Innovación—Consellería de Economía, Emprego e Industria) awarded to AB. The study was also funded by projects GRC2018/039 from Xunta de Galicia. Dawrin Pech-Puch received his postdoctoral fellowship from the National Council of Science and Technology (CONACYT) of Mexico. This work was supported by the Max Planck Society and the DFG (Gr1211/19-1)/CAPES 418729698 project.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Manconi R., Pronzato R., Perino E. A New Species of Agelas from the Zanzibar Archipelago, Western Indian Ocean (Porifera, Demospongiae) ZooKeys. 2016;553:1–31. doi: 10.3897/zookeys.553.5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu M.-J., Li M., Ma H., Li P.-L., Li G.-Q. Secondary Metabolites from Marine Sponges of the Genus Agelas: A Comprehensive Update Insight on Structural Diversity and Bioactivity. RSC Adv. 2022;12:7789–7820. doi: 10.1039/D1RA08765G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.König G.M., Wright A.D. Two New Naturally Occurring Pyrrole Derivatives from the Tropical Marine Sponge Agelas oroides. Nat. Prod. Lett. 1994;5:141–146. doi: 10.1080/10575639408044048. [DOI] [Google Scholar]
- 4.Sauleau P., Moriou C., al Mourabit A. Metabolomics Approach to Chemical Diversity of the Mediterranean Marine Sponge Agelas oroides. Nat. Prod. Res. 2017;31:1625–1632. doi: 10.1080/14786419.2017.1285298. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H., Dong M., Chen J., Wang H., Tenney K., Crews P. Bioactive Secondary Metabolites from the Marine Sponge Genus Agelas. Mar. Drugs. 2017;15:351. doi: 10.3390/md15110351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pech-Puch D., Pérez-Povedano M., Martinez-Guitian M., Lasarte-Monterrubio C., Vázquez-Ucha J.C., Bou G., Rodríguez J., Beceiro A., Jimenez C. In Vitro and In Vivo Assessment of the Efficacy of Bromoageliferin, an Alkaloid Isolated from the Sponge Agelas dilatata, against Pseudomonas aeruginosa. Mar. Drugs. 2020;18:326. doi: 10.3390/md18060326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiménez C., Crews P. Mauritamide A and Accompanying Oroidin Alkaloids from the Sponge Agelas mauritiana. Tetrahedron Lett. 1994;35:1375–1378. doi: 10.1016/S0040-4039(00)76222-8. [DOI] [Google Scholar]
- 8.Hong L.-L., Sun J.-B., Yang F., Liu M., Tang J., Sun F., Jiao W.-H., Wang S.-P., Zhang W., Lin H.-W. New Diterpene Alkaloids from the Marine Sponge Agelas mauritiana. RSC Adv. 2017;7:23970–23976. doi: 10.1039/C7RA02547E. [DOI] [Google Scholar]
- 9.Pech-Puch D., Joseph-Nathan P., Burgueño-Tapia E., González-Salas C., Martínez-Matamoros D., Pereira D.M., Pereira R.B., Jiménez C., Rodríguez J. Absolute Configuration by Vibrational Circular Dichroism of Anti-Inflammatory Macrolide Briarane Diterpenoids from the Gorgonian Briareum asbestinum. Sci. Rep. 2021;11:496. doi: 10.1038/s41598-020-79774-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pech-Puch D., Pérez-Povedano M., Gómez P., Martínez-Guitián M., Lasarte-Monterrubio C., Vázquez-Ucha J.C., Novoa-Olmedo M.L., Guillén-Hernández S., Villegas-Hernández H., Bou G., et al. Marine Organisms from the Yucatan Peninsula (Mexico) as a Potential Natural Source of Antibacterial Compounds. Mar. Drugs. 2020;18:369. doi: 10.3390/md18070369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pech-Puch D., Berastegui-Cabrera J., Pérez-Povedano M., Villegas-Hernández H., Guillén-Hernández S., Cautain B., Reyes F., Pachón J., Gómez P., Rodríguez J., et al. Antiviral and Antiproliferative Potential of Marine Organisms from the Yucatan Peninsula, Mexico. Front. Mar. Sci. 2020;7:607. doi: 10.3389/fmars.2020.00607. [DOI] [Google Scholar]
- 12.Pech-Puch D., Rodríguez J., Cautain B., Sandoval-Castro C.A., Jiménez C. Cytotoxic Furanoditerpenes from the Sponge Spongia tubulifera Collected in the Mexican Caribbean. Mar. Drugs. 2019;17:416. doi: 10.3390/md17070416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stout E.P., Yu L.C., Molinski T.F. Antifungal Diterpene Alkaloids from the Caribbean Sponge Agelas citrina: Unified Configurational Assignments of Agelasidines and Agelasines. Eur. J. Org. Chem. 2012;2012:5131–5135. doi: 10.1002/ejoc.201200572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cychon C., Lichte E., Köck M. The Marine Sponge Agelas citrina as a Source of the New Pyrrole–Imidazole Alkaloids Citrinamines A–D and N -Methylagelongine. Beilstein J. Org. Chem. 2015;11:2029–2037. doi: 10.3762/bjoc.11.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anta C., González N., Rodríguez J., Jiménez C. A New Secosterol from the Indonesian Octocoral Pachyclavularia violacea. J. Nat. Prod. 2002;65:1357–1359. doi: 10.1021/np010592e. [DOI] [PubMed] [Google Scholar]
- 16.Kubota T., Iwai T., Takahashi-Nakaguchi A., Fromont J., Gonoi T., Kobayashi J. Agelasines O–U, New Diterpene Alkaloids with a 9-N-Methyladenine Unit from a Marine Sponge Agelas sp. Tetrahedron. 2012;68:9738–9744. doi: 10.1016/j.tet.2012.09.040. [DOI] [Google Scholar]
- 17.Bendall J., Cambie R., Moratti S., Rutledge P., Woodgate P. Synthesis of a Novel Diterpenoid Rearrangement Product. Aust. J. Chem. 1995;48:1747. doi: 10.1071/CH9951747. [DOI] [Google Scholar]
- 18.Millán R.E., Rodríguez J., Sarandeses L.A., Gómez-Bengoa E., Sestelo J.P. Indium(III)-Catalyzed Stereoselective Synthesis of Tricyclic Frameworks by Cascade Cycloisomerization Reactions of Aryl 1,5-Enynes. J. Org. Chem. 2021;86:9515–9529. doi: 10.1021/acs.joc.1c00825. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura H., Wu H., Kobayashi J., Kobayashi M., Ohizumi Y., Hirata Y. Physiologically Active Marine Natural Products from Porifera. VIII. Agelasidines. Novel Hypotaurocyamine Derivatives from the Okinawan Sea Sponge Agelas nakamurai Hoshino. J. Org. Chem. 1985;50:2494–2497. doi: 10.1021/jo00214a017. [DOI] [Google Scholar]
- 20.Abdjul D.B., Yamazaki H., Kanno S., Takahashi O., Kirikoshi R., Ukai K., Namikoshi M. Structures and Biological Evaluations of Agelasines Isolated from the Okinawan Marine Sponge Agelas nakamurai. J. Nat. Prod. 2015;78:1428–1433. doi: 10.1021/acs.jnatprod.5b00375. [DOI] [PubMed] [Google Scholar]
- 21.Seco J.M., Quiñoá E., Riguera R. The Assignment of Absolute Configuration by NMR. Chem. Rev. 2004;104:17–118. doi: 10.1021/cr000665j. [DOI] [PubMed] [Google Scholar]
- 22.Proszenyák Á., Brændvang M., Charnock C., Gundersen L.-L. The First Synthesis of Ent-Agelasine F. Tetrahedron. 2009;65:194–199. doi: 10.1016/j.tet.2008.10.081. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets related to this article can be obtained from the authors.