

Abstract
Aim
We investigated the mechanisms by which N1-(β-d-ribofuranosyl)-5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), an activator of AMP-activated protein kinase (AMPK), decreases lung injury and mortality when administered to mice post exposure to bromine gas (Br2).
Methods
We exposed male C57BL/6 mice and heme oxygenase-1 (HO-1)-deficient (HO-1−/−) and corresponding wild-type (WT) littermate mice to Br2 (600 ppm for 45 or 30 min, respectively) in environmental chambers and returned them to room air. AICAR was administered 6 h post exposure (10 mg·kg−1, intraperitoneal). We assessed survival, indices of lung injury, high mobility group box 1 (HMGB1) in the plasma, HO-1 levels in lung tissues and phosphorylation of AMPK and its upstream liver kinase B1 (LKB1). Rat alveolar type II epithelial (L2) cells and human club-like epithelial (H441) cells were also exposed to Br2 (100 ppm for 10 min). After 24 h we measured apoptosis and necrosis, AMPK and LKB1 phosphorylation, and HO-1 expression.
Results
There was a marked downregulation of phosphorylated AMPK and LKB1 in lung tissues and in L2 and H441 cells post exposure. AICAR increased survival in C57BL/6 but not in HO-1−/− mice. In WT mice, AICAR decreased lung injury and restored phosphorylated AMPK and phosphorylated LKB1 to control levels and increased HO-1 levels in both lung tissues and cells exposed to Br2. Treatment of L2 and H441 cells with small interfering RNAs against nuclear factor erythroid 2-related factor 2 or HO-1 abrogated the protective effects of AICAR.
Conclusions
Our data indicate that the primary mechanism for the protective action of AICAR in toxic gas injury is the upregulation of lung HO-1 levels.
Introduction
World production of toxic gases, such as the halogens Cl2 and Br2 and the halogenated compound phosgene (COCl2), exceeds millions of tons per year. The three main producing countries are the USA, the UK and Israel. Current industrial uses for Cl2 and Br2 include pulp bleaching, waste sanitation, organic compound and pharmaceutical manufacturing, drinking water treatment and maintenance of pathogen-free swimming pools [1–3]. Bromine is the most common organohalide found in nature and is rapidly replacing Cl2 in industrial applications. The Dead Sea alone is estimated to contain nearly a billion tons of bromine. Cl2 and Br2 are produced in central facilities and transported to manufacturing plants by rail or road.
In general, Cl2 and Br2 have similar reactivities with biological compounds and result in similar types of injury [1]. There have been numerous industrial and transportation accidents during the last 10 years, resulting in the release of these toxic gases into the atmosphere with disastrous consequences. Chemical accidents due to inadvertent release of Br2 have occurred in the Netherlands, Germany, Belgium, Israel, the USA, Russia and many other countries. A major accident occurred during Br2 transport by train in the city of Chelyabinsk, Russia (population 1.1 million), resulting in 42 people being hospitalised and over 200 seeking medical attention. In another train accident, a freight train collided with three rail cars in Dimona, Israel, one of which was carrying liquid bromine. There have been numerous incidents worldwide in which trucks carrying Br2 have overturned, spilling their contents into the atmosphere. In an industrial accident, 25000 people were exposed to Br2 vapours during the transfer of Br2 between containers in a factory in Geneva, Switzerland, in 1988.
Exposure to Cl2 or Br2 may lead to skin disorders [4], bronchospasm, acute lung injury (ALI), peribronchiolar abscess, delay in healing [5] and death from cardiorespiratory failure [1, 2]. A study conducted on human volunteers demonstrated that exposure to as little as 0.9 ppm Br2 for 5 min resulted in cough, headache and irritation of the eyes, nose and upper respiratory tract [6]. Human exposure to as little as 10 ppm of Br2 for a very short time is harmful when inhaled, causing severe burns and serious inflammation throughout the respiratory tract, which is frequently followed by pneumonia [2]. Patients exposed to Br2 vapours have required endotracheal intubation and ventilation for 5 days, and hospitalisation for 41 days [7], and have developed pneumonitis and pulmonary fibrosis [8].
Treatment following halogen gas exposure is mainly supportive and includes humidified oxygen administration, β2 agonists for bronchospasms and antibiotics for potential infections [1, 9], as well as supported ventilation with or without intubation and positive-pressure ventilation when patients develop ALI and adult respiratory distress syndrome (ARDS). Corticosteroids and sodium bicarbonate (NaHCO3) have been shown to alleviate ARDS following Cl2 exposure in animal studies, but their efficacy in improving respiratory function after human exposure has not been established [10, 11]. Therefore, animal models of halogen gas inhalation provide us with a unique opportunity to elucidate the basic mechanisms of direct lung injury-induced ARDS and to test novel therapeutic agents to reduce morbidity and mortality.
A number of previous studies have shown that in many human diseases, including inflammation and ARDS, there is a significant decrease of AMP-activated protein kinase (AMPK) and its upstream activator liver kinase B1 (LKB1) in lung tissue, and that activation of AMPK decreases lung injury [12, 13]. However, whether AMPK plays a role in the development of oxidant-induced lung injury has not been elucidated. Herein, we exposed mice, rat and human lung epithelial cells to Br2 gas (in concentrations likely to be found in the vicinity of industrial accidents) and measured the level of AMPK at various times post exposure. Because our findings indicated that there was a significant decrease of phosphorylated AMPK (pAMPK) in both lung tissues and cells, we tested the hypothesis that post Br2 administration of the AMPK activator N1-(β-d-ribofuranosyl)-5-aminoimidazole-4-carboxamide (AICAR) will decrease lung and cell injury and mortality, and we examined the mechanisms involved. Based on previous findings documenting the presence of free haem in the plasma of patients exposed to Cl2 [14], as well as in the lungs and plasma of mice exposed to either Cl2 or Br2 [14–16], protective effects of AMPK activators may be due to the upregulation of heme oxygenase-1 (HO-1), which breaks down free haem, released into the circulation following turnover of red blood cells, to carbon monoxide (CO) and biliverdin, two anti-inflammatory and antioxidant molecules [17]. Consequently, we hypothesised that this would result in the upregulation of HO-1 in lung tissue, decrease lung injury and improve mortality. We also performed experiments with rat and human lung epithelial cells exposed to Br2 and treated with small interfering RNAs (siRNAs) (or their scrambled controls) against the HO-1 gene, and showed that HO-1 is necessary for the protective effects of AICAR in limiting apoptosis, necrosis and cell death.
Methods
Animals
Adult male C57BL/6 mice (20–25 g) were bought from Charles River, non-Frederick/National Cancer Institute. HO-1-deficient (HO-1−/−) mice and their WT littermates (mixed C57BL/6 and FVB backgrounds) were provided by A. Agarwal. All mice used in the study were males. All mice were under a 12-h dim light/12-h dark cycle with access to a standard diet and tap water ad libitum. A euthanasia protocol based on intraperitoneal (i.p.) injections of ketamine and xylazine was used in the study for mice to minimise pain and distress. All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Alabama at Birmingham.
Exposure to Br2
Two mice were exposed to Br2 gas at 600 ppm between 06:00 and 12:00 in cylindrical glass chambers, as previously described [18, 19]. The duration of exposure was 45 min for C57BL/6 mice and 30 min for HO-1−/− and their WT littermate mice. Control mice breathed air for the same experimental period as Br2-exposed mice. Immediately following exposure, the mice were returned to their cages in the vivarium where they breathed room air.
Cell culture and exposure to Br2
Rat alveolar type II-like epithelial (L2) cells were provided to us by Dr Judith Creighton, previously at the University of Alabama at Birmingham. Their origin was characterised and verified using the presence of human and rat genes using PCR, immunohistochemistry and Western blots of appropriate markers (supplementary figure S1). NCI-H441 (H441) (ATCC HTB-174), a human club-like cell line, was purchased from ATCC. L2 and H441 cells were cultured as described (supplementary material: cell culture), plated on T25 flasks and allowed to grow to confluence. 24 h prior to exposure, cells were lifted from the flasks using trypsin-EDTA (Atlanta Biologicals) and seeded in six-well plates coated with collagen (0.1 mg·mL−1; Sigma-Aldrich) at 5×104 cells·cm−2 and incubated overnight at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were exposed to Br2 gas at 100 ppm in a cylindrical glass chamber for 10 min, as previously described [19, 20]. Immediately after exposure, cells were placed in a humidified incubator at 37°C vented with 95% air and 5% CO2 until used for the various experiments. Control cells remained in the same incubator for similar periods of time. AICAR or vehicle was added into the cell culture medium 6 h after Br2 exposure at a final concentration of 800 μM.
AICAR preparation
AICAR (Tocris Biosciences) was dissolved in sterile Earle’s balanced salt solution (EBSS) just prior to its use. At 6 h post Br2 or air exposure, mice were given a single i.p. injection of 100 μL of either EBSS (vehicle control) or AICAR in EBSS (10 mg per kg of body weight). The mice used for survival studies were monitored at least every 12 h over the next 10 days. As per IACUC protocols, mice that lost >30% of their initial body weight were euthanised.
Blood gas measurements
Mice were anaesthetised with 2% isoflurane mixed with compressed air at 24 h post Br2 or air exposures. A midline incision was made through the skin, subcutaneous tissue and peritoneum. Bowels were then gently displaced laterally to allow visualisation of the inferior vena cava and abdominal aorta. A 25 G needle attached to a 1 mL syringe containing 25 μL heparin was inserted into the abdominal aorta and at least 500 μL of arterial blood was drawn. pH, HCO3−, arterial O2 saturation (SaO2), arterial O2 tension (PaO2) and arterial CO2 tension (PaCO2) were measured using an OPTI Medical CCA-TS Blood Gas and Electrolyte Analyzer with glucose cassettes (product no. BP7558; OPTI Medical). The remaining blood samples were centrifuged at 10000·g for 10 min. The upper plasma fraction was aliquoted, frozen and stored at −80°C until used as described below.
Measurement of plasma high mobility group box 1
High mobility group box 1 (HMGB1) levels were measured in the plasma samples using a mouse HMGB1 ELISA kit (product no. NBP2–62767; Novus Biologicals) following the manufacturer’s instructions.
Histological analysis and lung wet/dry ratios
The lungs were removed and the right lung was fixed in 10% formalin for 24 h and dehydrated in 70% ethanol before being embedded in paraffin. Paraffin-embedded tissues were cut into 5 μm sections, deparaffinised and rehydrated using CitriSolv (d-limonene-based solvent) and isopropanol, sequentially. The sections were stained with haematoxylin and eosin. The left lung was weighed, placed in an oven at 80°C for 72 h, and then weighed again to determine the dry weight.
Myeloperoxidase staining of the mouse lung
After being deparaffinised and rehydrated, the sections were quenched with 0.3% H2O2 then stained with the anti-myeloperoxidase (MPO) antibody (product no. ab9535; Abcam), using the HRP-DAB Cell and Tissue Staining Kit (product no. CTS005; R&D Systems) according to the manufacturer’s instructions. The MPO-stained slides were then evaluated by scoring for the presence of neutrophils within the alveolar and interstitial spaces.
Lung injury scoring
Lung injury was scored using five criteria: 1) neutrophils in the alveolar space; 2) neutrophils in the interstitial space; 3) development of hyaline membranes; 4) proteinaceous debris filling the airspaces; and 5) alveolar septal thickening. Lung injury scoring was performed using the guidelines described by the official American Thoracic Society workshop report on features and measurements of experimental ALI in animals [21]. Two trained personnel analysed the slides individually, using double blinding of specimens to avoid bias.
Measurements of plasma protein and cells in bronchoalveolar lavage fluid
Mice were anaesthetised with an i.p. injection of ketamine and xylazine (100 and 10 mg per kg of body weight, respectively). An incision was made at the neck to expose the trachea, and a 3-mm endotracheal cannula was inserted. The lungs were lavaged with 1 mL of PBS; ~0.7–0.9 mL was recovered in all groups. Recovered aliquots of lavage fluid were kept on ice and centrifuged immediately at 3000·g for 10 min to pellet the cells. Supernatants were removed and stored on ice, and protein concentrations were measured with a BCA Protein Assay Kit (product no. 23225; Thermo Fisher Scientific). Cells were re-suspended in 100 μL of PBS and counted using a Neubauer Haemocytometer Counting Chamber. Cells were then placed on slides using a Cellspin (Tharmac) and stained using a two-stain set consisting of eosin Y and a solution of thiazine dyes (Quik-Stain; Siemens). Differential counts (specifically macrophages, neutrophils and lymphocytes) were then performed on slides via light microscopy.
Western blot analysis
Mice were divided into four groups: Air control, Air+AICAR, Br2 and Br2+AICAR. 24 h after Br2 exposure, mice were anaesthetised and killed, their chest cavities opened and their lungs perfused with PBS until they were clear of blood. Their lungs were then removed. L2 and H441 cells were also exposed to Br2 (100 ppm for 10 min) or rHMGB1 (10 μg·mL−1) and collected 24 h after intervention. Lung tissue and cell samples were homogenised in a lysis buffer (product no. 89901; Thermo Fisher Scientific) containing protease/phosphatase inhibitors. The samples were then sonicated for 10 s on ice in 1.5-mL Eppendorf tubes using an ultrasonic liquid processor and centrifuged at 14000·g for 20 min at 4°C. Cleared supernatants were used to measure the protein concentration using a bicinchoninic acid assay. Equal amounts of protein (50–80 μg) from different lysates were electrophoresed on 4–20% Tris-HCl Criterion precast gels (product no. 567–1093; Bio-Rad Laboratories). Proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories) and immunostained with the following antibodies: Total AMPK (product no. 2532S; Cell Signaling Technology); T172-Phospho-AMPK (product no. 2535S; Cell Signaling Technology); total LKB1 (product no. 3047S; Cell Signaling Technology); S482-Phospho-LKB1 (product no. 3482S; Cell Signaling Technology); HMGB1 (product no. H9539; Sigma-Aldrich), nuclear factor erythroid 2-related factor 2 (Nrf2) (product no. sc722; Santa Cruz Biotechnology); HO-1 (product no. ADI-SPA-896F; Enzo Life Sciences); α-tubulin (product no. T5168; Sigma-Aldrich), E-cadherin (product no. 610182; BD Biosciences); E-selectin (product no. sc-137203; Santa Cruz Biotechnology) and pro-surfactant protein C (product no. WRAB-9337; Seven Hills Bioreagents). Bands were detected by chemiluminescent horseradish peroxidase substrate. Protein loading was normalised by reprobing the membranes with an antibody specific to β-actin (product no. sc-47778; Santa Cruz Biotechnology).
Flow cytometry analysis
L2 and H441 cells were grown in DMEM and RPMI 1640 media, respectively, supplemented with 10% FBS and 1% antibiotics in a six-well plate until 70% confluence. Cells were knocked down for either HO-1 or Nrf2 using either control scrambled RNA or siRNA against HO-1 and Nrf2, respectively, for 24 h before Br2 exposure. Cells were exposed to air or Br2 (100 ppm for 10 min), and AICAR was added into the cell culture medium 6 h after Br2 exposure at a final concentration of 800 μM for the Br2+AICAR group. Cells were collected after 24 h of exposure. Half of the cells were used to assess the HO-1 protein level by immunoblotting and half were stained using the Annexin V FITC Assay Kit (product no. 600300; Cayman Chemicals Company), according to the manufacturer’s instructions. The stained cells were then analysed for the presence of early apoptotic, late apoptotic/necrotic and necrotic cells using flow cytometry as described previously [15, 22].
Co-immunoprecipitation
For immunoprecipitations, mice lungs were rinsed with Hanks’ balanced salt solution ( product no. 14175095; Gibco) before the addition of ice-cold lysis buffer. Initial extracts were pre-cleared with normal IgG and Protein A/G plus Sepharose (product no. ab193262; Abcam). LKB1 or HMGB1 antibody conjugated sepharose were added to the tissue extract and rocked overnight. Total immunoprecipitated proteins were recovered by re-suspending the Protein A/G Sepharose pellet in 3% SDS and heating briefly at 95°C. Protein A/G Sepharose was then pelleted by centrifugation, and the supernatant containing SDS-soluble material was subjected to methanol-chloroform phase separation to remove non-protein contaminants. The purified protein pellet was washed once in methanol. The protein pellet was re-suspended in 2× SDS sample buffer and heated briefly. Aliquots were frozen at −80°C or used immediately for SDS-PAGE/Western blot analysis.
Statistical analysis
Figures were generated and statistics performed using GraphPad Prism version 8 for Windows (GraphPad Software). The mean±sem was calculated in all experiments, and statistical significance was determined by either one-way or two-way ANOVA. For one-way ANOVA analyses, Tukey’s multiple comparisons test was employed, while for two-way ANOVA analyses, Bonferroni post hoc tests were used. Overall survival was analysed by the Kaplan-Meier method. Differences in survival were tested for statistical significance by the log-rank test. A value of p<0.05 was considered significant.
Results
Phosphorylated AMPK levels are reduced in murine lung tissue and L2 cells after Br2 exposure
Western blots of peripheral lung tissue from mice exposed to Br2 600 ppm for 45 min and returned to room air for 6 h and 24 h revealed a significant decrease of pAMPK as normalised to the levels of total AMPK (figure 1a, b). Injection of AICAR i.p. at 6 h post exposure returned pAMPK/AMPK to normal levels at 24 h post exposure (figure 1a, b). Similarly, exposure of L2 cells to Br2 (100 ppm for 10 min) resulted in a significant decrease of pAMPK/AMPK at 24 h post exposure (figure 1c, d). Addition of AICAR in the medium at 6 h post exposure returned pAMPK/AMPK to control levels at 24 h (figure 1e, f). AICAR did not have any effect on the pAMPK/AMPK level of air-breathing mice (figure 1a, b) or on non-exposed L2 cells (figure 1e, f).
FIGURE 1.
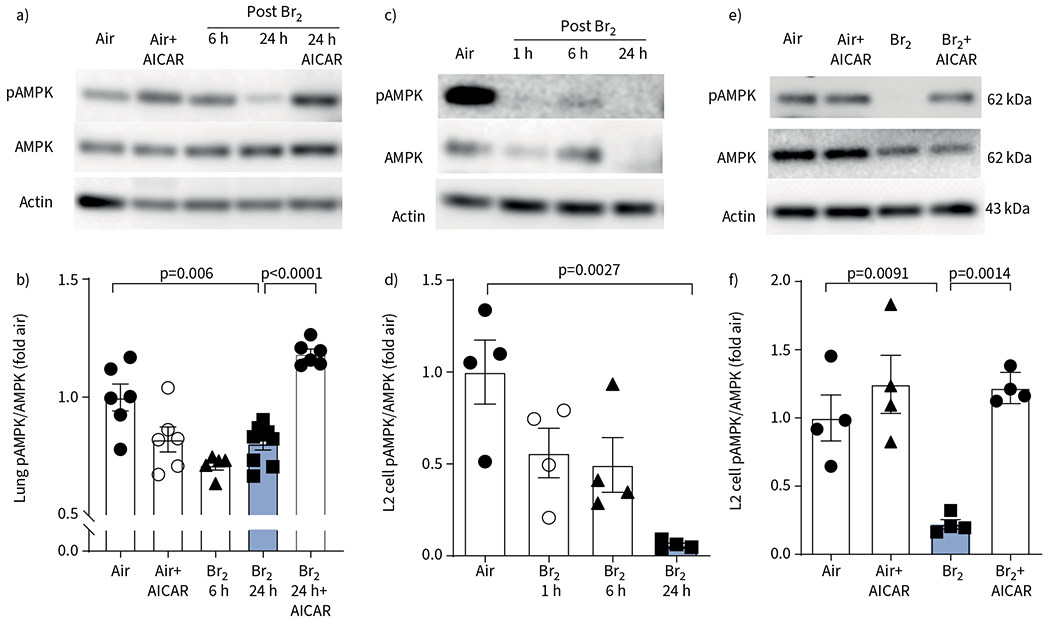
Phosphorylated AMP-activated protein kinase (pAMPK) is depleted in Br2-injured mice lung and rat alveolar type II epithelial (L2) cells. a–d) Representative Western blot images of pAMPK and total AMPK (tAMPK) levels and quantitative pAMPK to tAMPK ratio in lung tissue of mice exposed to Br2 (600 ppm for 45 min) and returned to room air for the indicated periods of time (a, b) and of rat L2 cells (100 ppm, 10 min). pAMPK level was significantly reduced in mice lung tissue and in L2 cells by 24 h post Br2 exposure (c, d). e, f) In the groups receiving AICAR 6 h post injury, pAMPK levels were significantly elevated compared to their untreated groups and approached normal (air control) values. Administration of AICAR in the absence of injury in animals and cells produced no significant difference to the air control group. Data are presented as mean±sem, n=5–7 animals per group. Significance was determined by one-way ANOVA followed by Tukey’s post hoc test. Fold air in b, d, f indicates the change relative to the air control.
AMPK activation attenuates Br2-induced lung injury, oedema and inflammation
As demonstrated by lung sections stained with haematoxylin and eosin and MPO, mice exposed to Br2 and returned to room air for 24 h exhibited significant and progressive disruption of the airway parenchyma, increased cellularity, proteinaceous debris accumulation in their alveolar spaces, increased accumulation of neutrophils in their alveolar and interstitial spaces, alveolar septal thickening in 90% of the sample area and increased lung injury score (figure 2a). Br2+AICAR-treated mice had significantly lower lung injury scores than Br2-exposed mice (figure 2b). In addition, AICAR reduced the Br2-dependent increase in bronchoalveolar lavage fluid (BALF) protein levels (figure 2c), total cell count (figure 2d), neutrophil cell count (figure 2e) and macrophage cell count (figure 2f), as well as lung wet/dry weights (figure 2g), an index of pulmonary oedema. By contrast, ACIAR had no effect on any of these variables in air-breathing mice.
FIGURE 2.
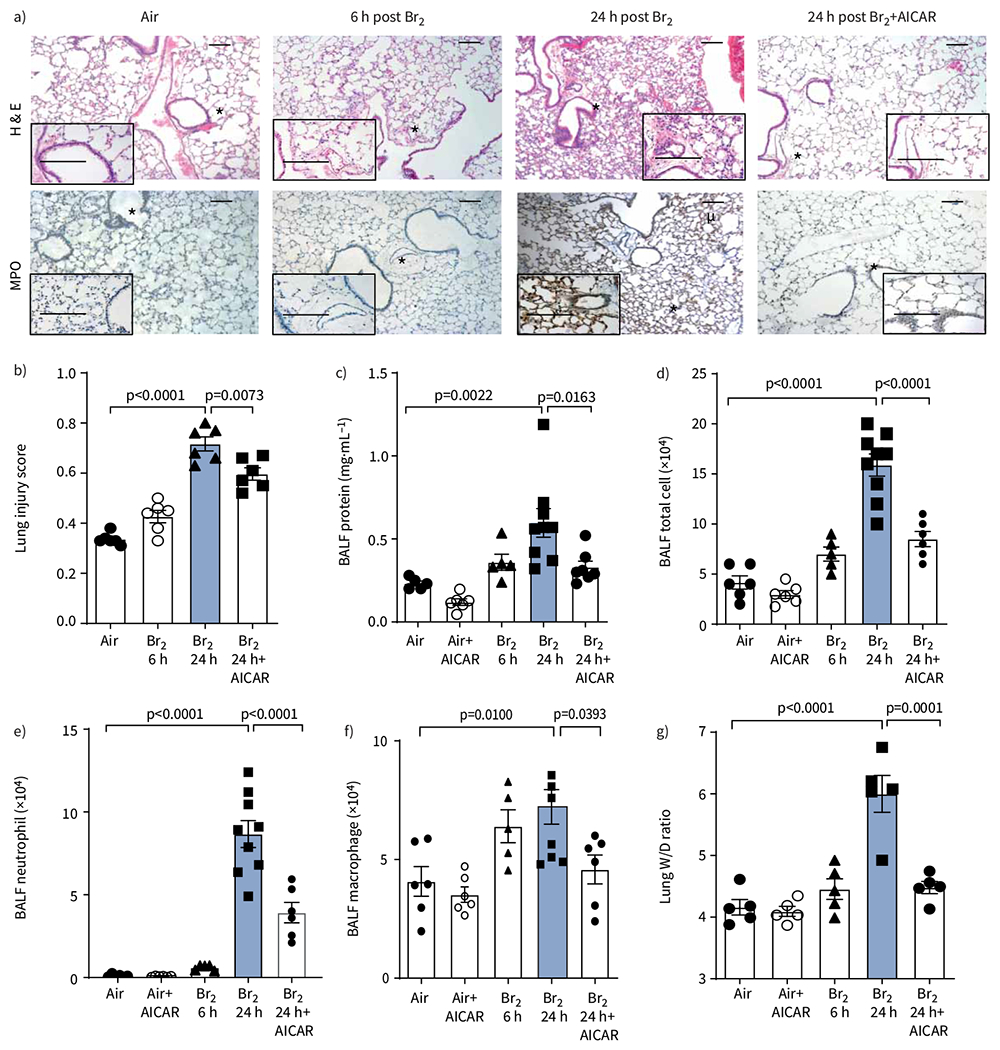
AMP-activated protein kinase (AMPK) activation attenuates lung oedema and inflammation after Br2 exposure. a) Representative images of haematoxylin and eosin (H&E) staining and myeloperoxidase (MPO) staining of peripheral lung tissue demonstrated proteinaceous and inflammatory cell infiltration into air spaces, and thickening of alveolar septae at 24 h post Br2 exposure. Lungs from Br2+AICAR-treated mice did not exhibit altered morphology or inflammatory cell infiltration and were similar in appearance to the control (air-exposed) lungs. Asterisks denote the region of lung magnified in the insert. Scale bars: 100 μm; 50 μm (inset). b) The lung injury score was significantly elevated at 24 h in the Br2+vehicle group compared to the control and AICAR-treated groups. c) Protein levels were significantly elevated in bronchoalveolar lavage fluid (BALF) at 24 h in the Br2+vehicle group compared to the control and AICAR-treated groups. d–f) Total cell, neutrophils and macrophage counts were significantly elevated in the Br2+vehicle group compared to the control and AICAR-treated groups. g) Lung oedema was evaluated by the wet/dry (W/D) ratio, which was significantly elevated at 24 h post Br2 exposure compared to the control groups, and was alleviated by AICAR treatment. Administration of AICAR in the absence of injury in animals and cells produced no significant difference to the control group. Data are presented as mean±sem, n=5–9 animals per group. Significance was determined by one-way ANOVA followed by Tukey’s post hoc test.
AMPK activation improves lung gas exchange and decreases mortality in mice exposed to Br2
In C57BL/6 mice 24 h post Br2 gas exposure, there were significant increases of PaCO2 and H+ in the plasma compared to air-exposed mice (figure 3a, b). Significant decreases of PaO2 and SaO2 were seen starting at 6 h post exposure compared to air-exposed mice (figure 3c, d). AICAR administration at 6 h post exposure returned all these variables to their corresponding air values. HCO3− levels were not altered significantly in any group (figure 3e). Most importantly, a single administration of AICAR at 6 h post Br2 improved survival significantly. None of the mice that were exposed to Br2 and returned to room air were alive 4 days post exposure; in contrast, >50% of the mice that were treated with AICAR were alive 4 days post Br2 (figure 3f). AICAR administration did not alter arterial blood gases and survival in air-exposed mice (figure 3).
FIGURE 3.
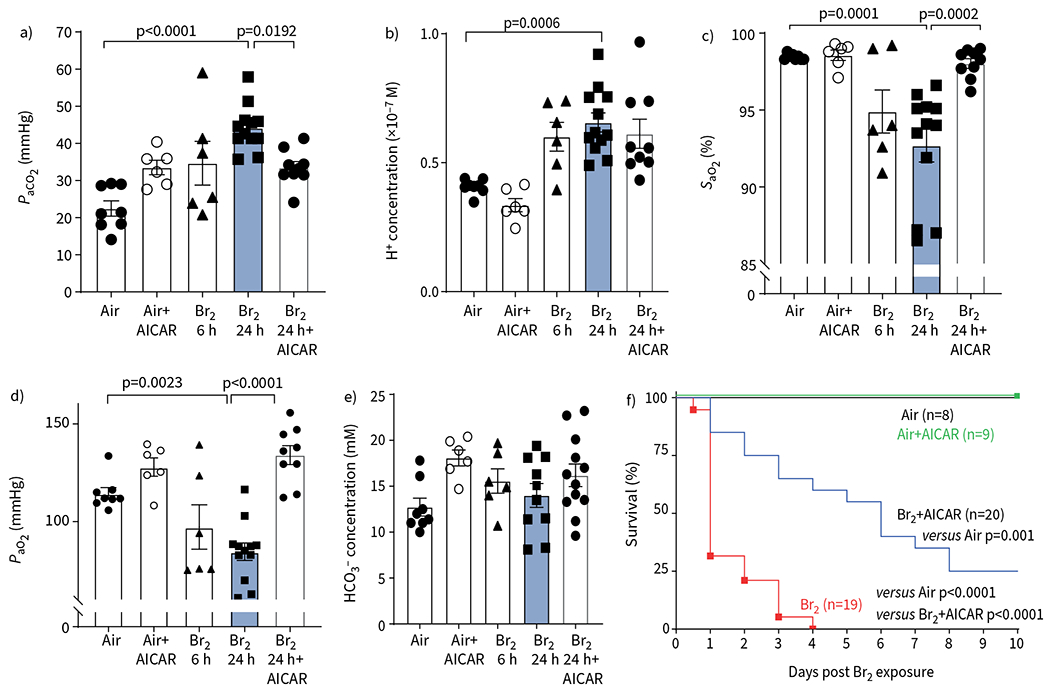
AICAR administration improves respiratory function in Br2-exposed mice. Mice were anaesthetised at 24 h post Br2 exposure (600 ppm for 45 min) and arterial blood was collected from the abdominal aorta. AICAR administration improved blood gas post Br2, including a) arterial CO2 tension (PaCO2), c) arterial O2 saturation (SaO2) and d) arterial O2 tension (PaO2). b) AICAR did not improve blood H+ significantly. e) Br2 did not cause HCO3− changes at 24 h. Data are presented as mean±sem, n=5–9 animals per group. Significance was determined by one-way ANOVA followed by Tukey’s post hoc test. f) Male and female C57BL/6 mice were exposed to Br2 (600 ppm, 45 min) and mortality was assessed over 7 days. AICAR-treated mice showed a significant improvement in survival compared to the vehicle-treated mice (n=8–20 animals per group). Administration of AICAR in the absence of injury in animals and cells produced no significant difference to the air control group. Significant changes in survival were determined by Kaplan–Meier Mantel–Cox log-rank test.
Br2 increased plasma HMGB1 and decreased pLKB1 in lung tissues of Br2-exposed mice
To understand the mechanisms by which exposure to Br2 decreased pAMPK, we measured the levels of its upstream protein regulator LKB1, which phosphorylates AMPK under normal conditions. Levels of lung pLKB1 were decreased at 24 h post Br2 exposure, while the total LKB1 level was not significantly decreased (figure 4a–c). Because HMGB1 is known to interact with and decrease LKB1 phosphorylation [23], we measured HMGB1 in plasma and its interaction with LKB1 in lung tissue. As shown in figure 4d, there was a two-fold increase in plasma HMGB1 concentrations at 24 h post Br2 exposure. Immunoprecipitation of lung tissue HMGB1 followed by Western blot with LKB1 antibody showed that the interaction between HMGB1 and LKB1 was maintained after exposure to Br2 (figure 4e, f). Finally, addition of recombinant HMGB1 in air-exposed L2 cells significantly decreased phosphorylation of both LKB1 (figure 4g, h) and pAMPK/AMPK (figure 4i, j).
FIGURE 4.
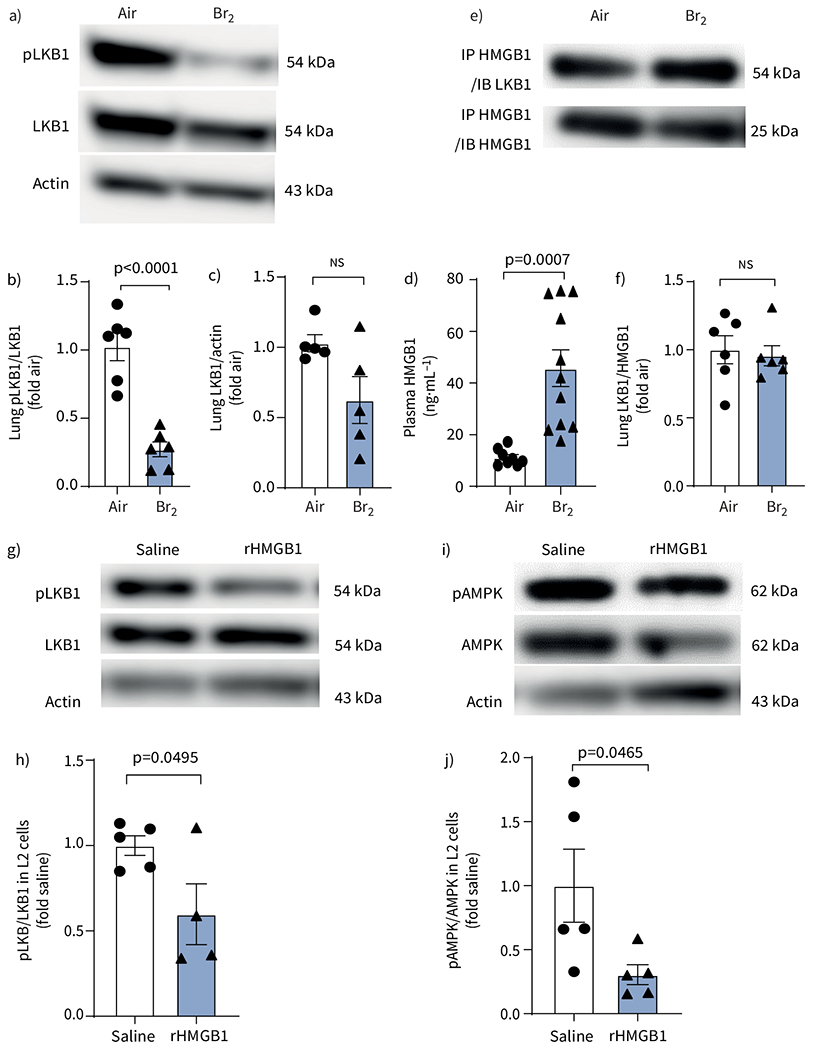
Phosphorylated liver kinase B1 (pLKB1) is decreased in lungs and high mobility group box 1 (HMGB1) is increased in plasma in Br2-exposed mice. Effect of exogenous HMGB1 on LKB1 and AMP-activated protein kinase (AMPK) signalling. a) Representative Western blot images of pLKB1 and total LKB1 levels and b, c) summarised pLKB1 to total LKB1 ratio demonstrated that pLKB1 levels (b) and total LKB1 levels (c) were significantly decreased in the lungs of mice at 24 h post Br2 exposure. d) ELISA showed that Br2-exposed mice had significantly higher levels of HMGB1. e) Representative Western blots and f) quantitative analysis from immunoprecipitation, using an antibody to HMGB1 and then pull down protein immunoblotted for LKB1, showed that LKB1 interacts with HMGB1 and forms a complex in mice lung tissue. Data are presented as mean±sem, n=5–7. Significance was determined by unpaired two-tailed t-test. g–j) The ratio of pLKB1 to total LKB1 (g, h) and phosphorylated AMPK (pAMPK) to total AMPK (i, j) in rat alveolar type II epithelial (L2) cells showed that the levels of pLKB1 and pAMPK were significantly decreased after incubation with recombinant HMGB1 (rHMGB1) protein at 10 μg·mL−1 for 24 h. Data are presented as mean±sem, n=6 independent experiments. Significance was determined by unpaired two-tailed t-test. Fold air/saline in b, c, f, h, j indicates the change relative to the air/saline control.
AICAR upregulates lung HO-1 in Br2-exposed mice
Our previous study showed that HO-1−/− mice are more susceptible to Br2, while HO-1-overexpressing mice were protected [15]. In the current study, we found significant upregulation of HO-1 in the lungs of mice exposed to Br2 that were treated with AICAR (figure 5a, b). Similar results were obtained in L2 cells exposed to Br2 and treated with AICAR (figure 5c, d). In both cases, Br2 exposure per se resulted in small, but not significant, increases in HO-1 levels, most likely brought about by increased haem [14–16].
FIGURE 5.
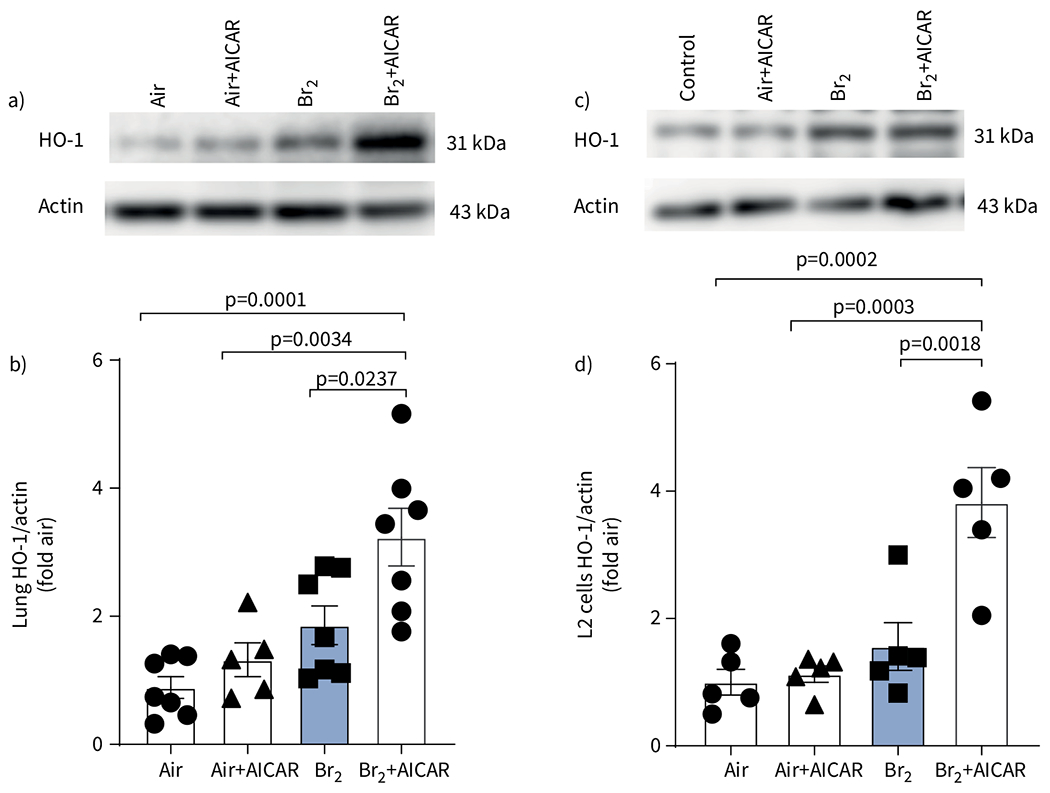
AICAR induces heme oxygenase-1 (HO-1) in mice lung tissue and rat alveolar type II epithelial (L2) cells. a) Representative Western blot images and b) quantitative graph of HO-1 in mice lung tissue showing significant upregulation of HO-1 by AICAR. c) Representative Western blot images and d) quantitative graph of HO-1 from L2 cells exposed to air or Br2 (100 ppm for 10 min) and then returned to room air 24 h later, showing significant upregulation of HO-1 by AICAR. Administration of AICAR in the absence of injury in animals and cells produced no significant difference to the air control group. Data are presented as mean±sem, n=5–6. Significance was determined by one-way ANOVA followed by Tukey’s post hoc test. Fold air in b, d indicates the change relative to the air control.
AICAR does not protect HO-1−/− mice from Br2 toxicity
To test whether the protective effects of AICAR were mediated via HO-1 upregulation, we exposed HO-1−/− mice to Br2 and treated them with AICAR. An i.p. injection of AICAR in Br2-exposed HO-1−/− mice significantly upregulated phosphorylation of AMPK and increased the pAMPK/AMPK ratio (figure 6a, b). However, there was no significant difference in survival between AICAR- and vehicle-treated HO-1−/− mice post Br2 exposure (figure 6c). In addition, AICAR did not reduce the Br2-dependent increase in BALF protein levels, total cell count or neutrophil count in HO-1−/− mice (figure 6d–f), as was the case in the WT controls (figure 2).
FIGURE 6.
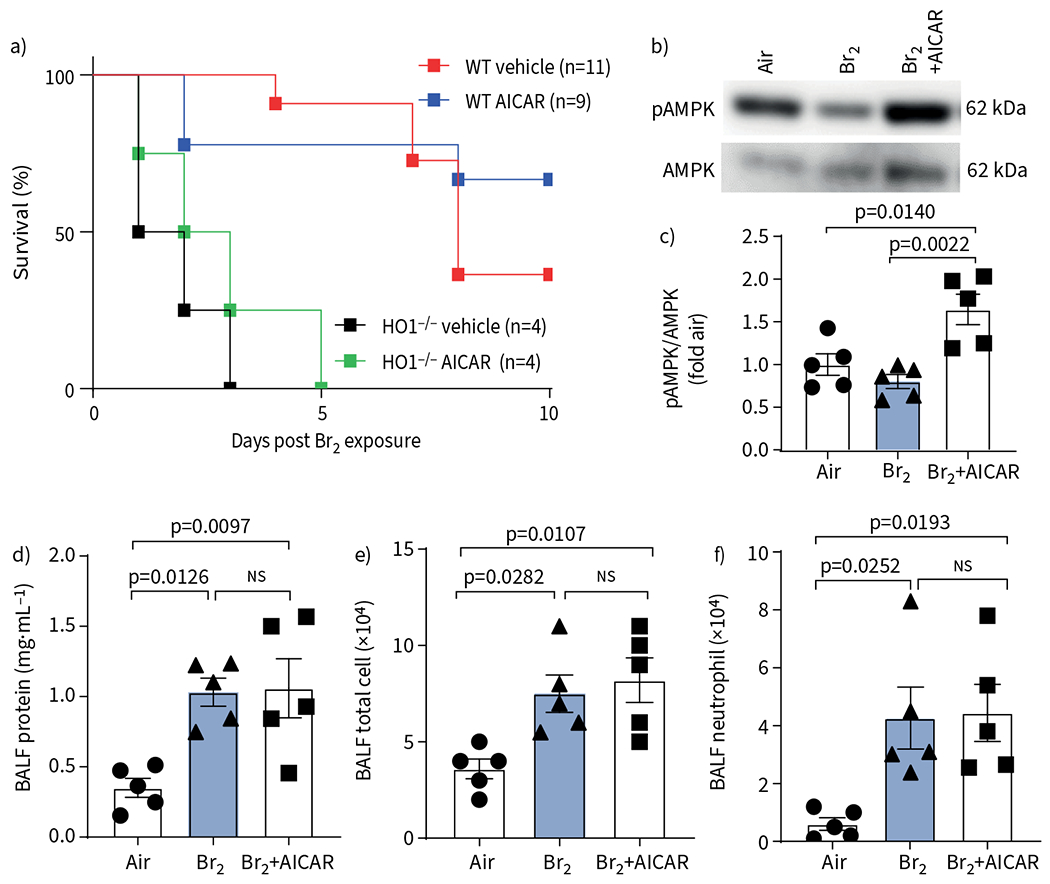
AMP-activated protein kinase (AMPK) activation does not protect HO-1−/− mice after exposure to Br2. a) Male and female HO-1−/− mice and their wild-type (WT) littermates were exposed to Br2 (600 ppm for 30 min) and mortality assessed over 10 days. AICAR treatment showed no significant protection in HO-1−/− mice. Significance was determined by Kaplan-Meier Mantel-Cox log-rank test (n=4–11 animals per group). b, c) Representative Western blot of HO-1−/− mice lung tissue showing a significant increase in AMPK and phosphorylated AMPK (pAMPK) post AICAR administration. Fold air indicates the change relative to the air control. d–f) AICAR did not reduce the levels of bronchoalveolar lavage fluid (BALF) protein (d), total cell count (e) or neutrophil count (f) in HO-1−/− mice 24 h post Br2 gas exposure. Data are presented as mean±sem, n=5. Significance was determined by one-way ANOVA followed by Tukey’s post hoc test.
AMPK activation attenuates Br2-dependent cell necrosis and apoptosis
In the next series of experiments, we treated L2 cells with siRNA or scrambled siRNAs against HO-1, exposed them to Br2 and measured cell survival using flow cytometry 24 h post Br2 exposure. There was a significant decrease in live cells with concomitant increases of early and late apoptotic cells of L2 cells treated with scrambled siRNA 24 h post Br2. The effectiveness of the siRNA against HO-1 was verified using Western blot against HO-1 (figure 7b, c). The addition of AICAR in the culture medium at 6 h post exposure significantly increased the number of live cells and decreased necrotic and late apoptotic cells in cells treated with scrambled RNA; however, no protection was seen in L2 cells treated with siRNA against HO-1 (figure 7a, d–g).
FIGURE 7.
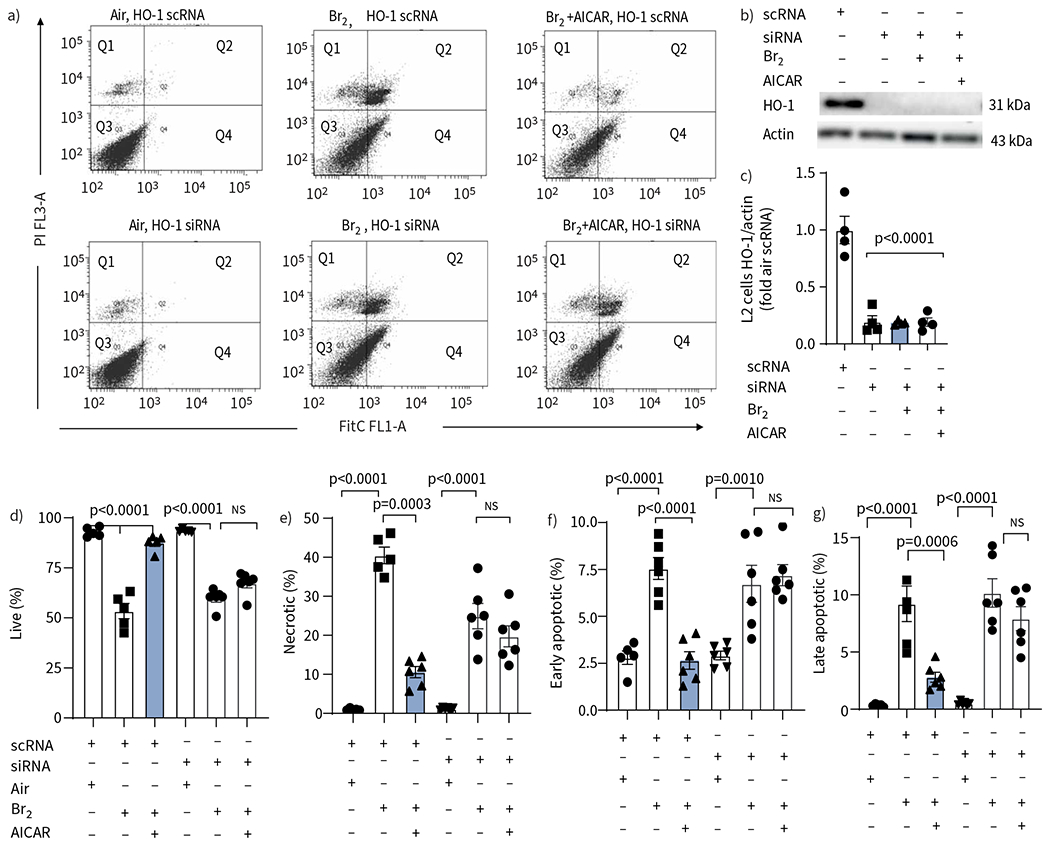
AICAR reduces apoptosis and necrosis following exposure of rat alveolar type II epithelial (L2) cells to Br2 but not in those treated with small interfering RNA (siRNA) against heme oxygenase-1 (HO-1). L2 cells were treated with scrambled RNA (scRNA) or siRNA against HO-1 and then divided into three groups: Air, Br2 and Br2+AICAR. a) Representative flow cytometry analysis of Annexin V FITC-stained cells collected 24 h post Br2 exposure showed a significant decrease in live cells (quadrant (Q) 3), an increase in late apoptotic/necrotic (Q2) cells and an increase in necrotic (Q1) cells. Q4 showed early apoptotic cells. b, c) Administration of a siRNA against HO-1 significantly reduced the levels of HO-1 protein 24 h post Br2 and in non-exposed L2 cells. AICAR did not increase the protein levels of HO-1 in siRNA-treated cells. The control scRNA did not affect the levels of HO-1. d–g) Quantification of flow cytometry data. In AICAR-treated L2 cells that received the control scRNA, all indicators of cell injury were reduced compared to the vehicle-treated Br2 groups. Administration of the siRNA against HO-1 did not increase the injury after Br2 exposure in vehicle-treated cells, but prevented the protective effects of AICAR seen in the scRNA groups. Data are presented as individual data points and mean±sem. Experiments were conducted in triplicate. Significance was determined by one-way ANOVA followed by Tukey’s post hoc test.
To further characterise the pathway involved in the protective effects of AICAR, we knocked down Nrf2, an important transcriptional regulator of HO-1, in L2 cells using siRNA. The absence of Nrf2 prevented the protective effects of AICAR in an almost identical manner to the absence of HO-1 (figure 8a–d). The knockdown of Nrf2 and HO-1 in L2 cells treated with Nrf2 siRNA was verified and quantified using Western blots (figure 8i, j). Finally, the importance of HO-1 in the beneficial effects of AICAR was also assessed in the human lung epithelial cell line H441. As shown in figure 8e–h, AICAR significantly reduced the levels of late apoptotic cells post Br2 exposure in cells treated with scrambled RNA but not in cells treated with the siRNA against HO-1. The knockdown of HO-1 in H441 cells treated with HO-1 siRNA was verified and quantified through Western blots (figure 8k, l). Representative fluorescence-activated cell sorting scatter dot plots from these experiments can be found in supplementary figure S2a.
FIGURE 8.
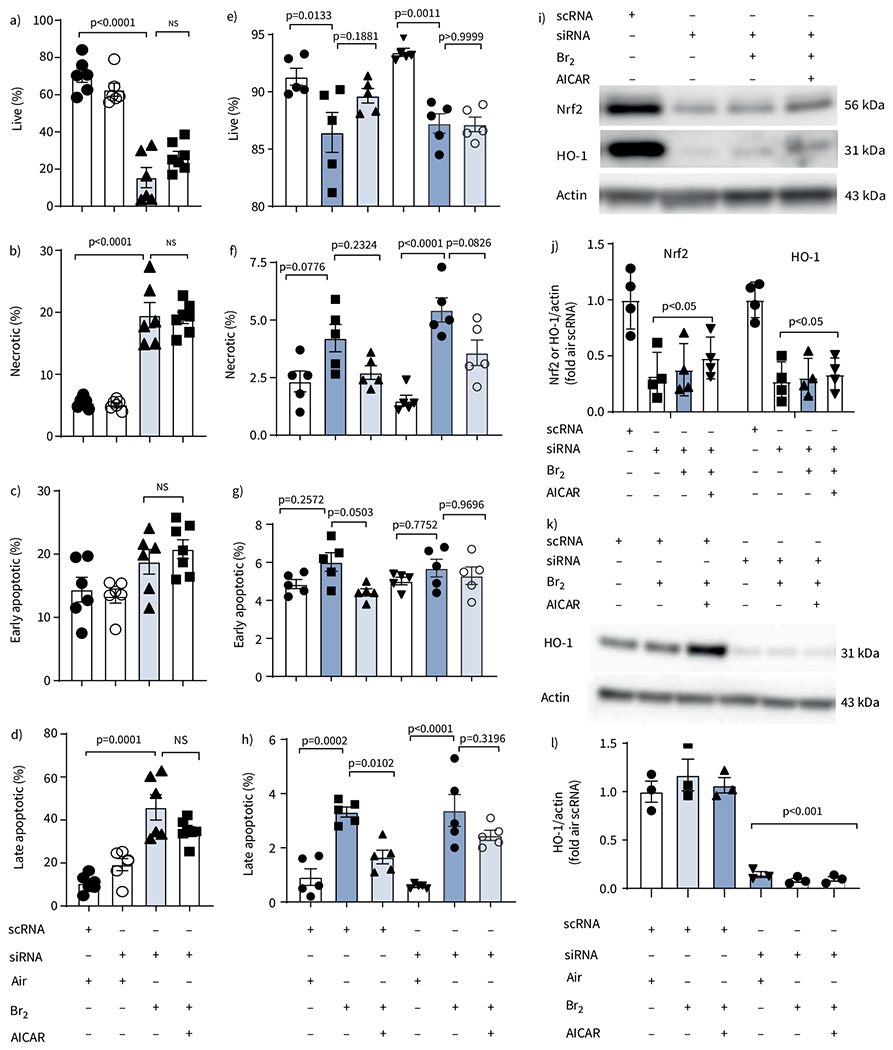
AICAR reduces apoptosis and necrosis following Br2 injury in vitro through heme oxygenase-1 (HO-1) in rat alevolar type II epithelial (L2) cells and human club-like epithelial (H441) cells but not in cells treated with small interfering RNA (siRNA) against nuclear factor erythroid 2-related factor 2 (Nrf2). a–d) Quantification of flow cytometry data from L2 cells treated with scrambled RNA (scRNA) or siRNA against Nrf2. In AICAR-treated cells that received scRNA, all indicators of cell injury were reduced compared to the vehicle-treated Br2 groups. Administration of the siRNA against Nrf2 did not increase the injury after Br2 exposure in vehicle-treated cells, but prevented the protective effects of AICAR seen in the scRNA groups. Data are presented as individual points and mean±sem. Experiments were replicated in triplicate. Significance was determined by one-way ANOVA followed by Tukey’s post hoc test. e–h) Quantification of flow cytometry data from H441 cells treated with scRNA or siRNA against HO-1 and then divided further into three groups: Air, Br2 and Br2+AICAR. In AICAR-treated cells that received the control scRNA, all indicators of cell injury were reduced compared to the vehicle-treated Br2 groups that received the scRNA. Administration of the siRNA against HO-1 did not increase the injury after Br2 exposure in vehicle-treated cells, but prevented the protective effects of AICAR seen in the scRNA groups. Data are presented as individual data points and mean±sem. Experiments repeated in triplicate. Significance was determined by one-way ANOVA followed by Tukey’s post hoc test. i, j) Representative Western blot against Nrf2 and HO-1 after Nrf2 siRNA administration and its quantification in L2 cells. Data are presented as mean±sem, n=4. Significance determined by one-way ANOVA followed by Tukey’s post hoc test. k, l) Representative Western blot against HO-1 after HO-1 siRNA administration and its quantification in H441 cells. Data are presented as mean±sem, n=4. Significance determined by one-way ANOVA followed by Tukey’s post hoc test.
Discussion
The most important findings of our study are as follows. 1) Exposure of mice to Br2 gas for 30 min in concentrations likely to be encountered in the vicinity of industrial accidents resulted in delayed lung injury, many of the symptoms of which resemble those of human ARDS. Within 10 days post exposure, 50% of the mice had died, similar to human ARDS mortality [24]. 2) Exposure to Br2 resulted in significant downregulation of the pAMPK/AMPK level in lung tissues at 6 h and 24 h post exposure. 3) A single i.p. injection of AICAR at 6 h post Br2 restored the pAMPK/AMPK to control levels, decreased the severity of lung injury and improved mortality, but had no effect on these variables in air-exposed mice. 4) In HO-1−/− mice, as well as in rat and human lung epithelial cells, in which HO-1 levels were abolished by treatment with siRNAs against HO-1 or Nrf2, AICAR administration failed to show any protective effects in spite of the restoration of pAMPK/AMPK to control levels. Taken as a whole, these in vivo and in vitro data show that the protective effects of AICAR were not related to restoration of pAMPK/AMPK but rather to the upregulation of HO-1 (supplementary figure S3).
HO-1 upregulation is induced by a variety of pathophysiological stimuli, e.g. hypoxia, inflammation, endotoxin exposure and, most importantly, free haem [17]. HO-1 has been shown to ameliorate lung injury in animal studies induced by hypoxia [25], lipopolysaccharide (LPS) [26–28], hyperoxia [29] and other forms of oxidative stress. The novel findings shown in this paper are consistent with our previous findings and those of others showing that agents that decrease free haem play an important protective effect in the development and progression of ALI in rodents and mice. For example, exposing mice to toxic gases such as Cl2, Br2 and COCl2 results in a significant increase of free haem, due to increased fragility of red blood cells [14–16, 30]. Patients accidentally exposed to Cl2 have significant levels of free haem in their plasma, 6 h post exposure [14]. Administration of hemopexin, an agent that binds to free haem with high affinity and results in a complex that is removed by the liver, decreases mortality and reverses the onset of acute and chronic lung injury in animal models of ARDS [14, 16, 31]. Mice overexpressing HO-1 are resistant and mice that lack HO-1 (HO-1−/−) are highly susceptible to lung injury caused by exposure to Br2 [15]. Also, patients with sepsis and ARDS with increased levels of haem-scavenging proteins have a better outcome and decreased mortality [32, 33].
AMPK activators also induce HO-1 that attenuates oxidative stress in the liver, in brain astrocytes in the proximal tubular cells of the kidney and in human dendritic cells [17, 34–39]. Another study has shown that AMPK stimulates HO-1 gene expression in human endothelial cells via the Nrf2/antioxidant response element signalling pathway [40, 41]. AMPK phosphorylates Nrf2 at Ser558, which promotes the nuclear accumulation of Nrf2, a necessary step for HO-1 gene activation [41]. Our data established that in vitro inhibition of Nrf2 using siRNAs blocked the upregulation of HO-1 by AICAR and negated the protective effects of AICAR on cell apoptosis and necrosis following Br2 exposure. Our data, when viewed in the prism of our current knowledge of AMPK, Nrf2 and HO-1, suggest that AICAR activates AMPK, which upregulates HO-1 through Nrf2.
AMPK and its upstream activator LKB1 are not only cellular energetic sensors but also exhibit many important beneficial effects in different human diseases including ARDS [42–44]. Decreased AMPK activity has been observed in diabetes, obesity, inflammation, lung fibrosis and several other pathophysiological conditions [45, 46]. Pharmacological activation of AMPK has been shown to be effective in preventing and reversing organ dysfunction in a variety of diseases [13]. AMPK activation in vitro decreases the production of LPS-induced inflammatory cytokines by neutrophils [47]. In vivo, AMPK activators reverse LPS-induced ALI and bleomycin-induced lung fibrosis [45, 48]. Furthermore, AMPK activators exert protective effects on lung injury, including airway remodelling in asthma and mitigation of the subsequent development of fibrosis [49, 50]. The protective effects of restoring pAMPK after injury may be due to a variety of mechanisms. AMPK activation is important to counteract inflammation. Anti-inflammatory effects of pAMPK have been observed in acute and chronic diseases, such as reducing murine colitis [51] and reversing ALI and fibrosis [23, 47]. AICAR reduces organ (liver) and cell (endothelial) injury through the inhibition of NF-κB activation and downregulation of inflammatory cytokine (e.g. tumour necrosis factor-α, interleukin-1β and interleukin-6) production [52–58].
HMGB1, an inflammatory cytokine, inhibits AMPK activity and is a biomarker for mortality in pneumonia and ARDS [12, 42, 59]. LKB1 is the major kinase that phosphorylates AMPK [60, 61] and the interaction between LKB1 and HMGB1 inhibits the phosphorylation of LKB1 and AMPK, preventing the anti-inflammatory effects of pAMPK [23]. We, therefore, hypothesised that Br2 inhalation would decrease LKB1 and AMPK phosphorylation by increasing HMGB1 in the plasma, which would result in ALI. Our data show that pLKB1, the upstream regulator of AMPK phosphorylation, was significantly decreased, while LKB1 levels remained unchanged. This finding indicates that the mechanism of AMPK phosphorylation downregulation in Br2 exposure is due to LKB1 and pLKB1 downregulation.
During an inflammatory response, damaged cells release HMGB1, a highly conserved non-histone nuclear protein, passively or actively, upon either LPS or toll like receptor 4 (TLR4) stimulation. HMGB1 inhibits AMPK activation [12, 23, 42]. HMGB1 concentration is currently regarded as an important biological marker in many inflammation-related conditions. HMGB1 interacts with LKB1, decreasing pLKB1, and inhibits AMPK activation, thereby diminishing the protective effects of AMPK [23]. Our results show that plasma of Br2-exposed mice has increased levels of HMGB1, explaining the low levels of pLKB1 found in exposed lungs.
This study has some limitations that could be addressed in further studies. First, this study does not address the dose-effect response and whether smaller doses could be as effective as the one used here. Furthermore, this study utilises a single time point of administration. It will be interesting to address the therapeutic window of AICAR after Br2 exposure. Furthermore, AICAR and AMPK have a host of different actions. For example, AMPK promotes autophagy via phosphorylation of unc-51 like autophagy activating kinase 1 (UlK1) [62, 63]. Upregulation of autophagy has been shown to decrease halogen-induced injury [64]. In this study, we chose to focus on its interaction with HO-1 through Nrf2.
In conclusion, we show that Br2 exposure causes downregulation of AMPK phosphorylation and significant lung injury and mortality. Administration of AICAR results in amelioration of the lung injury and mortality caused by Br2 exposure. Finally, our HO-1−/− data provide strong evidence that, in our model, the protective effects of AMPK activation are through the upregulation of HO-1.
Supplementary Material
Support statement:
This work was supported by the CounterACT Program, National Institutes of Health Office of the Director (NIH OD), the National Institute of Neurological Disorders and Stroke (NINDS) and the National Institute of Environmental Health Sciences (NIEHS), grant numbers 5UO1 ES026458, 3UO1 ES026458 03S1 and 5UO1 ES027697 (to S. Matalon); R21 NS090024 (to S. Matalon and A. Agarwal); NIH/NIDDK R01 DK059600 (to A. Agarwal); NIH/NIDDK P30 DK079337 (to A. Agarwal); CFAR pilot funding P30 AI027767-32 (to S. Aggarwal), CCTS pilot funding UL1TR003096 (to S. Aggarwal); NIH/NHLBI K12 HL143958 (to S. Aggarwal). Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest:
I. Ahmad has nothing to disclose. A. Molyvdas has nothing to disclose. M-Y. Jian has nothing to disclose. T. Zhou has nothing to disclose. A.M. Traylor has nothing to disclose. H. Cui has nothing to disclose. G. Liu has nothing to disclose. W. Song has nothing to disclose. A. Agarwal reports other (advisory board) from Akebia Therapeutics and Reata Pharmaceuticals, grants and other (advisory board) from Angion, and other (advisory board and stock options) from Goldilocks Therapeutics, outside the submitted work. T. Jilling has nothing to disclose. S. Aggarwal has nothing to disclose. S. Matalon has nothing to disclose.
Footnotes
This article has supplementary material available from erj.ersjournals.com
References
- 1.Summerhill EM, Hoyle GW, Jordt SE, et al. An Official American Thoracic Society Workshop Report: chemical inhalational disasters. Biology of lung injury, development of novel therapeutics, and medical preparedness. Ann Am Thorac Soc 2017; 14: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lam A, Vetal N, Matalon S, et al. Role of heme in bromine-induced lung injury. Ann NY Acad Sci 2016; 1374: 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlisle M, Lam A, Svendsen ER, et al. Chlorine-induced cardiopulmonary injury. Ann NY Acad Sci 2016; 1374: 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers JV, Price JA, Wendling MQ, et al. An assessment of transcriptional changes in porcine skin exposed to bromine vapor. J Biochem Mol Toxicol 2011; 25: 252–262. [DOI] [PubMed] [Google Scholar]
- 5.Schlagbauer M, Henschler D. Toxicitat von chlor und bei einmaliger und wiederholter inhalation [Toxicity of chlorine and bromine in single and repeated inhalation]. Int Arch Arbeitsmed 1967; 23: 91–98. [PubMed] [Google Scholar]
- 6.Rupp H, Henschler D. Wirkungen geringer Chlor-und bromkonzentrationen auf den menschen [Effect of low chlorine and bromine concentrations on man]. Int Arch Arbeitsmed 1967; 23: 79–90. [PubMed] [Google Scholar]
- 7.Inagaki N, Ishikawa M, Takeda M, et al. Case with bromine exposure leading to respiratory insufficiency. Chudoku Kenkyu 2005; 18: 141–147. [PubMed] [Google Scholar]
- 8.Kraut A, Lilis R. Chemical pneumonitis due to exposure to bromine compounds. Chest 1988; 94: 208–210. [DOI] [PubMed] [Google Scholar]
- 9.Sexton JD, Pronchik DJ. Chlorine inhalation: the big picture. J Toxicol Clin Toxicol 1998; 36: 87–93. [DOI] [PubMed] [Google Scholar]
- 10.Zhou T, Song WF, Shang Y, et al. Halogen inhalation-induced lung injury and acute respiratory distress syndrome. Chin Med J (Engl) 2018; 131: 1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pascuzzi TA, Storrow AB. Mass casualties from acute inhalation of chloramine gas. Mil Med 1998; 163: 102–104. [PubMed] [Google Scholar]
- 12.Wang G, Han D, Zhang Y, et al. A novel hypothesis: up-regulation of HO-1 by activation of PPARγ inhibits HMGB1-RAGE signaling pathway and ameliorates the development of ALI/ARDS. J Thorac Dis 2013; 5: 706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev 2009; 89: 1025–1078. [DOI] [PubMed] [Google Scholar]
- 14.Aggarwal S, Lazrak A, Ahmad I, et al. Reactive species generated by heme impair alveolar epithelial sodium channel function in acute respiratory distress syndrome. Redox Biol 2020; 36: 101592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aggarwal S, Lam A, Bolisetty S, et al. Heme attenuation ameliorates irritant gas inhalation-induced acute lung injury. Antioxid Redox Signal 2016; 24: 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aggarwal S, Ahmad I, Lam A, et al. Heme scavenging reduces pulmonary endoplasmic reticulum stress, fibrosis, and emphysema. JCI Insight 2018; 3: e120694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ayer A, Zarjou A, Agarwal A, et al. Heme oxygenases in cardiovascular health and disease. Physiol Rev 2016; 96: 1449–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Addis DR, Lambert JA, Ren C, et al. Vascular endothelial growth factor-121 administration mitigates halogen inhalation-induced pulmonary injury and fetal growth restriction in pregnant mice. J Am Heart Assoc 2020; 9: e013238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lazrak A, Yu Z, Doran S, et al. Upregulation of airway smooth muscle calcium-sensing receptor by low-molecular-weight hyaluronan. Am J Physiol Lung Cell Mol Physiol 2020; 318: L459–L471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazrak A, Chen L, Jurkuvenaite A, et al. Regulation of alveolar epithelial Na+ channels by ERK1/2 in chlorine-breathing mice. Am J Respir Cell Mol Biol 2012; 46: 342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matute-Bello G, Downey G, Moore BB, et al. An Official American Thoracic Society Workshop Report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 2011; 44: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Londino JD, Lazrak A, Noah JW, et al. Influenza virus M2 targets cystic fibrosis transmembrane conductance regulator for lysosomal degradation during viral infection. FASEB J 2015; 29: 2712–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tadie JM, Bae HB, Deshane JS, et al. Toll-like receptor 4 engagement inhibits adenosine 5’-monophosphate-activated protein kinase activation through a high mobility group box 1 protein-dependent mechanism. Mol Med 2012; 18: 659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000; 342: 1334–1349. [DOI] [PubMed] [Google Scholar]
- 25.Minamino T, Christou H, Hsieh CM, et al. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci USA 2001; 98: 8798–8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong Q, Yin H, Fang M, et al. Heme oxygenase-1 upregulation significantly inhibits TNF-α and Hmgb1 releasing and attenuates lipopolysaccharide-induced acute lung injury in mice. Int Immunopharmacol 2008; 8: 792–798. [DOI] [PubMed] [Google Scholar]
- 27.Otterbein LE, Kolls JK, Mantell LL, et al. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest 1999; 103: 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin H, Li X, Gong Q, et al. Heme oxygenase-1 upregulation improves lipopolysaccharide-induced acute lung injury involving suppression of macrophage migration inhibitory factor. Mol Immunol 2010; 47: 2443–2449. [DOI] [PubMed] [Google Scholar]
- 29.Jin Y, Kim HP, Chi M, et al. Deletion of caveolin-1 protects against oxidative lung injury via up-regulation of heme oxygenase-1. Am J Respir Cell Mol Biol 2008; 39: 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aggarwal S, Jilling T, Doran S, et al. Phosgene inhalation causes hemolysis and acute lung injury. Toxicol Lett 2019; 312: 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang X, Lin T, Sun G, et al. Hemopexin down-regulates LPS-induced proinflammatory cytokines from macrophages. J Leukoc Biol 2009; 86: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaver CM, Upchurch CP, Janz DR, et al. Cell-free hemoglobin: a novel mediator of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2016; 310: L532–L541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janz DR, Bastarache JA, Sills G, et al. Association between haptoglobin, hemopexin and mortality in adults with sepsis. Crit Care 2013; 17: R272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell NK, Fitzgerald HK, Fletcher JM, et al. Plant-derived polyphenols modulate human dendritic cell metabolism and immune function via AMPK-dependent induction of heme oxygenase-1. Front Immunol 2019; 10: 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee EH, Baek SY, Park JY, et al. Rifampicin activates AMPK and alleviates oxidative stress in the liver as mediated with Nrf2 signaling. Chem Biol Interact 2020; 315: 108889. [DOI] [PubMed] [Google Scholar]
- 36.Jeong YH, Park JS, Kim DH, et al. Lonchocarpine increases Nrf2/ARE-mediated antioxidant enzyme expression by modulating AMPK and MAPK signaling in brain astrocytes. Biomol Ther (Seoul) 2016; 24: 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolisetty S, Traylor A, Zarjou A, et al. Mitochondria-targeted heme oxygenase-1 decreases oxidative stress in renal epithelial cells. Am J Physiol Renal Physiol 2013; 305: F255–F264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui T, Schopfer FJ, Zhang J, et al. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J Biol Chem 2006; 281: 35686–35698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zarjou A, Bolisetty S, Joseph R, et al. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J Clin Invest 2013; 123: 4423–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matzinger M, Fischhuber K, Poloske D, et al. AMPK leads to phosphorylation of the transcription factor Nrf2, tuning transactivation of selected target genes. Redox Biol 2020; 29: 101393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joo MS, Kim WD, Lee KY, et al. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol 2016; 36: 1931–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999; 285: 248–251. [DOI] [PubMed] [Google Scholar]
- 43.Andersson U, Wang H, Palmblad K, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 2000; 192: 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Vishnubhakat JM, Bloom O, et al. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery 1999; 126: 389–392. [PubMed] [Google Scholar]
- 45.Rangarajan S, Bone NB, Zmijewska AA, et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med 2018; 24: 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaw RJ. Metformin trims fats to restore insulin sensitivity. Nat Med 2013; 19: 1570–1572. [DOI] [PubMed] [Google Scholar]
- 47.Zhao X, Zmijewski JW, Lorne E, et al. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008; 295: L497–L504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jian MY, Alexeyev MF, Wolkowicz PE, et al. Metformin-stimulated AMPK-α1 promotes microvascular repair in acute lung injury. Am J Physiol Lung Cell Mol Physiol 2013; 305: L844–L855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Z, Bone N, Jiang S, et al. AMP-activated protein kinase and glycogen synthase kinase 3β modulate the severity of sepsis-induced lung injury. Mol Med 2016; 21: 937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park CS, Bang BR, Kwon HS, et al. Metformin reduces airway inflammation and remodeling via activation of AMP-activated protein kinase. Biochem Pharmacol 2012; 84: 1660–1670. [DOI] [PubMed] [Google Scholar]
- 51.Bai A, Yong M, Ma AG, et al. Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J Pharmacol Exp Ther 2010; 333: 717–725. [DOI] [PubMed] [Google Scholar]
- 52.Yang Z, Kahn BB, Shi H, et al. Macrophage α1 AMP-activated protein kinase (α1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem 2010; 285: 19051–19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salt IP, Palmer TM. Exploiting the anti-inflammatory effects of AMP-activated protein kinase activation. Expert Opin Investig Drugs 2012; 21: 1155–1167. [DOI] [PubMed] [Google Scholar]
- 54.Kim YD, Kim YH, Cho YM, et al. Metformin ameliorates IL-6-induced hepatic insulin resistance via induction of orphan nuclear receptor small heterodimer partner (SHP) in mouse models. Diabetologia 2012; 55: 1482–1494. [DOI] [PubMed] [Google Scholar]
- 55.Cacicedo JM, Yagihashi N, Keaney JF Jr, et al. AMPK inhibits fatty acid-induced increases in NF-κB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004; 324: 1204–1209. [DOI] [PubMed] [Google Scholar]
- 56.Huang NL, Chiang SH, Hsueh CH, et al. Metformin inhibits TNF-α-induced IκB kinase phosphorylation, IκB-α degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int J Cardiol 2009; 134: 169–175. [DOI] [PubMed] [Google Scholar]
- 57.Ruderman NB, Park H, Kaushik VK, et al. AMPK as a metabolic switch in rat muscle, liver and adipose tissue after exercise. Acta Physiol Scand 2003; 178: 435–442. [DOI] [PubMed] [Google Scholar]
- 58.Ruderman NB, Cacicedo JM, Itani S, et al. Malonyl-CoA and AMP-activated protein kinase (AMPK): possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem Soc Trans 2003; 31: 202–206. [DOI] [PubMed] [Google Scholar]
- 59.Abraham E, Arcaroli J, Carmody A, et al. HMG-1 as a mediator of acute lung inflammation. J Immunol 2000; 165: 2950–2954. [DOI] [PubMed] [Google Scholar]
- 60.Willows R, Sanders MJ, Xiao B, et al. Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem J 2017; 474: 3059–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 2009; 9: 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yeganeh B, Lee J, Ermini L, et al. Autophagy is required for lung development and morphogenesis. J Clin Invest 2019; 129: 2904–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jurkuvenaite A, Benavides GA, Komarova S, et al. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic Biol Med 2015; 85: 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.