

Abstract
A series of 25 new benzothiazole–urea–quinoline hybrid compounds were synthesized successfully via a three-step synthetic sequence involving an amidation coupling reaction as a critical step. The structures of the synthesized compounds were confirmed by routine spectroscopic tools (1H and 13C NMR and IR) and by mass spectrometry (HRMS). In vitro evaluation of these hybrid compounds for their antitubercular inhibitory activity against the Mycobacterium tuberculosis H37Rv pMSp12::GPF bioreporter strain was undertaken. Of the 25 tested compounds, 17 exhibited promising anti-TB activities of less than 62.5 µM (MIC90). Specifically, 13 compounds (6b, 6g, 6i–j, 6l, 6o–p, 6r–t, and 6x–y) showed promising activity with MIC90 values in the range of 1–10 µM, while compound 6u, being the most active, exhibited sub-micromolar activity (0.968 µM) in the CAS assay. In addition, minimal cytotoxicity against the HepG2 cell line (cell viability above 75%) in 11 of the 17 compounds, at their respective MIC90 concentrations, was observed, with 6u exhibiting 100% cell viability. The hybridization of the quinoline, urea, and benzothiazole scaffolds demonstrated a synergistic relationship because the activities of resultant hybrids were vastly improved compared to the individual entities. In silico ADME predictions showed that the majority of these compounds have drug-like properties and are less likely to potentially cause cardiotoxicity (QPlogHERG > −5). The results obtained in this study indicate that the majority of the synthesized compounds could serve as valuable starting points for future optimizations as new antimycobacterial agents.
Keywords: quinoline–urea–benzothiazole hybrids, antitubercular activity, minimum inhibitory concentration, HepG2 cell line, cytotoxicity, in silico ADME properties
1. Introduction
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb) and it contributes to the high mortality rate, driven by infectious diseases, in most sub-Saharan African countries [1]. In 2019, the World Health Organization (WHO) reported over 10 million new TB cases and 1.2 million deaths. [2]. Furthermore, nearly one-third of the global population is estimated to be latently infected with TB. Although latent TB is not transmittable, at least 5–10% of people with latent TB will see it manifest into active TB at some point in their lifetime [2]. TB treatment with what are commonly referred to as first-line drugs usually lasts six months, but this may, in some cases, extend up to nine. Failure to complete this treatment course increases the risk of developing drug-resistant TB. Studies have shown that the administration of substandard drugs and/or sub-optimal drug doses also contributes to the manifestation and spread of drug-resistant TB [3]. There are three reported types of drug-resistant TB, namely, multidrug-resistant (MDR), extensively drug-resistant (XDR), and totally drug-resistant (TDR) TB [4]. MDR-TB is caused by a strain that is resistant to two at least of the first-line anti-TB drugs (i.e., isoniazid and rifampicin). In contrast, XDR-TB is resistant to several anti-TB drugs, any fluoroquinolone, and at least one injectable drug, while TDR-TB is a generic term for strains virtually resistant to all TB drugs [4,5]. Drug resistance is governed mainly by several mechanisms associated with (i) the thick, waxy, and hydrophobic cell wall envelope [6,7], (ii) the presence of drug modifying enzymes [8,9], and (iii) chromosomal or gene mutations [10,11], among others.
Several anti-TB drugs are at various phases of development, approval, and use, and three of the most recent breakthroughs in anti-TB research resulted in the discovery of SQ-109, UPAR-174, and bedaquiline (or TMC-207) (Figure 1).
Figure 1.

Breakthrough anti-TB compounds in various stages of development, approval, and use.
SQ-109, a member of the 1,2-ethylenediamine-based anti-TB drugs, is currently undergoing phase II clinical trials and is active against MDR pulmonary TB [12,13,14]. SQ-109 is made up of a triage of moieties, namely, adamantane head, geranyl tail and 1,2-ethylenediamine linker, which are critically important for the drug’s anti-TB activity [14]. This drug functions by inhibiting the mycobacterial membrane protein large 3 (MmpL3), from the cytoplasm, which is involved in cell wall synthesis [15,16]. Other studies have reported that this drug also inhibits Mtb’s respiration by blocking menaquinone biosynthesis [17] and also acts by collapsing the pH gradient and cell wall membrane potential [18]. The major disadvantage of this compound, in terms of medicinal chemistry parameters, is its high lipophilicity (clog P = 6.82) [19]. Highly lipophilic compounds tend to have rapid metabolic turnover, low solubility and poor absorption [20]. In addition, these compounds tend to bind to off-target cells/tissues, leading to an increased risk of toxicity [21].
UPAR-174, a tricyclic 2-aminothiazole compound still in early development, possesses good anti-TB activity [22]. This compound is believed to exert its activity via potent efflux inhibitory properties similar to verapamil [22]. It also reduces the rate of drug efflux in both sensitive and drug-resistant Mtb [22]. This compound is highly lipophilic (clogP = 6.60) and has been reported to penetrate macrophages to facilitate the killing of Mtb when administered with isoniazid and rifampicin in small doses [23].
Bedaquiline is a diarylquinoline-based compound whose approval was accelerated by WHO and the US Food and Drug Administration (US-FDA). It is used with isoniazid, rifampicin, and pyrazinamide in a highly effective combination therapy used to treat MDR-TB [24]. It targets the atpE-encoded subunit c of the membrane-bound mycobacterial ATP synthase enzyme [25]. ATP synthase is responsible for transporting ions across the cell membrane and generating power for ATP synthesis. The quinoline core in this drug is critical for its activity. As effective as bedaquiline is, it also has several shortcomings. Firstly, its very high lipophilicity (clogP = 7.25) has been shown to result in its deposition in fatty tissues in the body [26]. This lipophilicity has also been directly linked to the inhibition of the cardiac potassium heERG channels, resulting in QTc interval prolongation, and hepatic toxicity [27]. Secondly, the recent reported emerging resistance toward bedaquiline among MDR-TB and XDR-TB clinical isolates is worrisome [28]. Drug resistance is by far the most significant challenge in TB chemotherapy. The mechanism of resistance to bedaquiline is thought to involve a direct mutation within the atpE gene.
All three anti-TB drugs discussed above are highly lipophilic and various studies have shown that lipophilic compounds tend to exhibit high anti-TB activity due to their abilities to penetrate the hydrophobic cell wall. However, newer compounds of similar activity but with lower lipophilicity and improved solubility are desirable. Previously, we reported several 4-amino-7-chloroquinoline-based hybrid compounds that showed promising anti-TB activities, low lipophilicity, and improved solubility profiles [29,30]. In a continuation of our efforts to discover anti-infective agents, we report here on the synthesis, in vitro anti-TB activity, cytotoxicity studies, and in silico ADME profiling of new hybrid compounds. These compounds contain the chain length varying diamine functionality of SQ-109, the quinoline nucleus found in bedaquiline/TMC 207, and the aminothiazole (in our case, benzothiazole) found in UPAR-174 (Figure 2). The diamine and benzothiazole are linked via the urea moiety. Benzothiazoles and their derivatives are known to possess many important biological properties, such as antifungal [31], antibacterial [32], anticancer [33,34], and antituberculosis activities [35,36,37,38], among others.
Figure 2.

Components making up the desired quinoline–urea–benzothiazole hybrid compounds.
In terms of anti-TB activity, benzothiazoles have been found to be effective in combatting MDR-TB [39]. They are thought to act by inhibiting the production of an enzyme used in the synthesis of biotin, an essential vitamin for the survival of Mtb [39,40]. Furthermore, this scaffold has also been shown to target another enzyme known as decaprenylphosphoryl-β-D-ribose 2′-epimerase, which is responsible for the synthesis of decaprenylphosphoryl arabinose [41,42,43]. Decaprenylphosphoryl arabinose plays a significant role in forming cell-wall arabinoses and inhibiting it leads to mycobacterial cell lysis and ultimately cell death. Other studies on benzothiazole-containing compounds, especially the 6-substituted benzothiazoles, resulted in efficacious compounds in vitro and in vivo [40]. A benzothiazole moiety is generally used in tandem with urea, and this combination has shown promising anti-TB activity [44]. Urea was selected as an active linker to covalently link both the benzothiazole and 4-aminoquinoline moieties and further enhance the anti-TB activity of the designed hybrid compounds. It is also anticipated that most of these designed hybrid compounds will be less lipophilic than SQ-109, UPAR-174, and bedaquiline, and will possess multimodal activity against Mtb.
2. Results and Discussion
2.1. Chemistry
The synthesis of target quinoline–urea–benzothiazole hybrids (6a–y) followed a three-step synthetic protocol outlined in Scheme 1.
Scheme 1.

Reagents and conditions: (i) heat at 80 °C, 1 h (without stirring), then at 135 °C, 4 h (with stirring); (ii) CDI, dry DCM, reflux at 45 °C, 20 h; (iii) dry ACN, reflux at 85 °C, 24 h.
Briefly, the first step involved the synthesis of the intermediate 4-aminoquinoline diamines (2a–e and 3: Routes A and B, respectively), in excellent yields (76–98%), from the reaction of 4,7-dichloroquinoline (1) and various excess diamines following a previously reported procedure [29]. In parallel and separately, various 2-amino-6-substituted benzothiazoles (4a–g) were converted to benzothiazole-1H-imidazol-1-carboxamide intermediates (5a–g), also in excellent yields (74–97%), using the 1,1′carbonyldiimidazoles (CDIs) in refluxing dry DCM according to modified methods reported in the literature [44]. It is worth noting that the CDI, in addition to facilitating the amide coupling reaction, also provides the urea functionality required in the target hybrids. Furthermore, the CDI must be in excess to avoid two benzothiazoles coupling to a single CDI moiety, which would be undesirable. The last step in the synthesis was the coupling of the 4-aminoquinoline diamines with benzothiazole-1H-imidazol-1-carboxamide intermediates (5a–g) in refluxing ACN to yield the desired compounds (6a–y) in excellent yields (Table 1). All the synthesized target compounds were confirmed by 1H and 13C NMR and by HRMS (see the Supplementary Materials).
Table 1.
Yields and melting points of target hybrid compounds 6a–y.
| Compound | Diamine Linker | X | m.p. (°C) | Yield (%) |
|---|---|---|---|---|
| 6a |

|
H | 298–302 | 88.52 |
| 6b | CF3 | 343–345 | 84.81 | |
| 6c | F | 340–342 | 86.11 | |
| 6d | NO2 | 339–341 | 78.75 | |
| 6e |
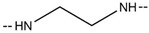
|
CF3 | 249–251 | 64.90 |
| 6f | Cl | 244–245 | 83.23 | |
| 6g | Br | 240–242 | 78.41 | |
| 6h | F | 230 –232 | 75.23 | |
| 6i |
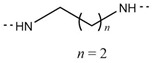
|
CF3 | 198–200 | 83.42 |
| 6j | Cl | 286–288 | 76.82 | |
| 6k | CH3 | 234–239 | 55.94 | |
| 6l | H | 285–288 | 59.31 | |
| 6m | Br | 254–257 | 60.20 | |
| 6n | F | 241–243 | 78.50 | |
| 6o |
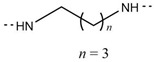
|
Cl | 189–191 | 83.04 |
| 6p | Br | 192–194 | 82.34 | |
| 6q | F | 256–259 | 92.05 | |
| 6r |
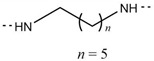
|
Cl | 239–241 | 79.72 |
| 6s | Br | 192–194 | 82.32 | |
| 6t | F | 256–259 | 92.03 | |
| 6u |
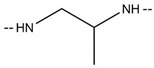
|
CF3 | 243–245 | 89.01 |
| 6v | F | 228–230 | 87.52 | |
| 6w | Br | 246–248 | 89.99 | |
| 6x | CH3 | 229–231 | 78.81 | |
| 6y | Cl | 301–304 | 81.19 |
2.2. In Vitro Antitubercular Activity Studies
The synthesized new hybrid compounds (6a–y) were evaluated for their potential in vitro antitubercular activity against the H37Rv strain over seven days of incubation in two slightly different media (7H9/CAS/Glu/Tx and 7H9/ADC/Glu/Tw) (Table 2). The two media shared the standard mycobacterial broth base (Middlebrook 7H9) and glucose (Glu) supplement. The only difference was that the former contained casitone (CAS) and tyloxapol (Tx) and the latter albumin–dextrose–catalase (ADC) and Tween-80 (Tx). ADC is considered a very useful indicator of potential protein binding (binding to serum) since it contains albumin (bovine albumin fraction V), while CAS is a source of amino acids for the bacteria [45]. Both Tx and Tw are useful surfactants in preventing mycobacterial clumping/agglomerating in liquid assays. The lowest concentration of a compound resulting in no visible mycobacterial growth is scored as the MIC90 (minimum inhibitory concentration required to inhibit 90% of the Mtb growth). Rifampicin (RIF) was used as a reference or positive control in the assays.
Table 2.
In vitro 90% Mtb inhibition in 7H0/CAS/Glu/Tx and 7H9/ADC/Glu/Tw with clogP values of the target compounds 6a–y.
| Compound | a 7H9/CAS/GLU/Tx | b 7H9/ADC/GLU/Tw | c clogP |
|---|---|---|---|
| 7 Days (µM) | 7 Days (µM) | ||
| 6a | 21.001 | >125 | 4.32 |
| 6b | 4.943 | 6.854 | 5.22 |
| 6c | >125 | >125 | 4.44 |
| 6d | >125 | >125 | 4.10 |
| 6e | ND | ND | 6.35 |
| 6f | 125 | >125 | 6.10 |
| 6g | 8.89 | 14.898 | 6.25 |
| 6h | 62.5 | 62.5 | 5.53 |
| 6i | 7.812 | 12.837 | 6.72 |
| 6j | 4.389 | 11.748 | 6.48 |
| 6k | 31.25 | 62.33 | 6.25 |
| 6l | 9.628 | 9.447 | 5.75 |
| 6m | 15.924 | 16.863 | 6.63 |
| 6n | >125 | 125 | 5.91 |
| 6o | 7.455 | 23.529 | 6.49 |
| 6p | 7.812 | 15.609 | 6.64 |
| 6q | >125 | >125 | 5.92 |
| 6r | 7.597 | 14.617 | 7.54 |
| 6s | 8.76 | 20.954 | 7.69 |
| 6t | 6.974 | 31.25 | 6.97 |
| 6u | 0.968 | 5.732 | 6.66 |
| 6v | >125 | >125 | 5.84 |
| 6w | 8.191 | 14.001 | 6.56 |
| 6x | 7.219 | 10.35 | 6.19 |
| 6y | 2.331 | 8.455 | 6.41 |
| 2e | >125 | >125 | 4.07 |
| 4b | >125 | >125 | 1.97 |
| 5b | >125 | >125 | 0.474 |
| Urea | >125 | >125 | −1.66 |
| 2e:4b:Urea (1:1:1) | >125 | >125 | - |
| RIF | 0.03 | 0.001 | 3.71 |
a Protein-poor Mtb media. b Protein-rich Mtb media; ND = not determined. c Calculated using PerkinElmer ChemDraw Professional V18.0.0.231 [46].
From the data, it is evident that activity is dependent on the structural features the compounds possess (i.e., the type of the diamine and its chain length, as well as the substituent on the benzothiazole moiety). In the hydrazine sub-series (6a–d), the CF3-containing compound 6b was the only one showing excellent activity in both media. In contrast, 6a (which had a H on the benzothiazole ring) only showed moderate activity (21.0 µM) in the CAS media. The F- and NO2-substituted compounds in this sub-series were devoid of activity, as seen by MIC90 values >125 µM in both media. In the ethylenediamine sub-series (6e–h), the Br-containing compound 6g was the most active, displaying an excellent activity ranging from 8.89–14.90 µM in both media, while the F-substituted compound 6h showed moderate activity of 62.5 µM in both media. In the propyldiamine sub-series (6i–n), generally, all the tested compounds showed excellent activity (<20 µM) in both media, with the Cl-substituted 6j being the most active in the CAS assay (4.39 µM), while in the ADC assay the unsubstituted compound 6l (9.45 µM) was the most active. Similar to the hydrazine series, the F-containing compound 6n was devoid of any activity. In the butyldiamine sub-series (6o–q) (MIC90 in the range of 7.81–15.61 µM), the Br-containing compound (6p: 7.81 and 7.05 µM) was the most active, while the F-containing compound (6q) was not active at all. Interestingly, the F-substituted compound 6t, in the hexyldiamine sub-series (6r–t), was the most active compound in comparison with the other sub-series tested. The last sub-series, the propane-1,2-diamine sub-series (6u–y), yielded the most active compounds (<10 µM) of them all. The CF3 substituted 6u was the most active of all compounds tested with a sub-micromolar activity of (0.97 µM) in the CAS assay and 5.73 µM in the ADC assay. The fact that the activity decreased drastically (i.e., a six-fold decrease), as evidenced by the increasing MIC90 values, in the ADC media, could indicate potential protein binding to the albumin protein. The reference or positive control drug (RIF) was more active than all the tested compounds in both media.
In terms of intermolecular comparison across the six sub-series (Table 2), the propane-1,2-diamine sub-series provided the most active compound (6u). Notably, all the tested CF3-containing 6b, 6i, and 6u appear to be the consistently active compounds in both media in their respected sub-series. This observation is not surprising because various studies have confirmed the essential biological role attributed to the CF3 substituent by virtue of containing fluorides [47]. The CF3-containing propane-1,2-diamine compound 6u could serve as an interesting starting point in the next round of structure–activity relationship (SAR) studies to discover the next generation of these designed hybrids. Lipophilicity did not appear to play any prominent role in the activities of these compounds since there was no observable trend in this property based on Mtb activities.
In assessing whether the hybridization strategy offered any advantage or not, the individual intermediates (2e, 4b (including 5b) and urea) making up compound 6t were tested separately and in combination (weight/weight), and their activities were compared to that of 6t. All intermediates making up compound 6t and their combination were found to be largely inactive (>125 µM) compared to the hybrid, which had MIC90 values ranging from 6.97–31.25 µM. Secondly, the combination was also not active, conclusively suggesting that the hybridization was advantageous compared to the individual entities (and their combination) making up the hybrid. Furthermore, this could also indicate that the intermediates making up the hybrid have a synergistic relationship, as seen by the improved activity when hybridized.
2.3. In Vitro Cytotoxicity Studies
In vitro cytotoxicity of the synthesized compounds was determined using the MTT assay. This cell viability assay quantifies the enzymatic conversion of the yellow tetrazolium salt into purple formazan crystals by healthy, living mammalian cells. The formazan crystals produced are directly proportional to the number of viable cells [48]. The synthesized compounds showed variable cytotoxic responses for HepG2 cells at the concentrations tested (0–200 µM), with cell viability ranging from 10% to 100% (Figure 3). The ISO norm requires >75% cell viability for biomedical products [49]. In this study, an IC20 where HepG2 cells retained 80% viability was applied for the compound to be deemed safe at the MIC90 obtained in the CAS assay; only those with MIC90 values <60 µM were tested for cytotoxicity. For compound 6a, part of the hydrazine sub-series containing only H on the benzothiazole ring, the IC20 of 15.14 µM in HepG2 cells was lower than its MIC90 (21.0 µM), where HepG2 cells at this concentration retained 75% cell viability in accordance with the ISO norm.
Figure 3.

The dose–response curves for HepG2 cells exposed to 0–200 µM concentrations of the selected number of synthesized compounds. The 20% inhibitory concentration (IC20) and cell viability at the 90% Mtb inhibitory concentration for CAS (MIC90) were extrapolated from the dose–response curve.
Compound 6g in the ethylenediamine sub-series was cytotoxic to HepG2 cells; HepG2 cell viability was 48% at the MIC90 for 6g, and an IC20 of 7.47 µM was calculated. In the propyldiamine sub-series, only 6i exhibited a dose-dependent toxic response, with 6j–6m showing a sharp drop in cell viability at concentrations >25 µM. The IC20s of HepG2 cells for this series were all above the respective MIC90s. Moreover, cell viability at the MIC90 ranged from 86% to 100% in accordance with the ISO norm for biomedical products, and 6l yielded the best safety profile over a more comprehensive concentration range, with an IC20 of 104.7 µM and 100% cell viability at the MIC90. Both 6o (MIC90 7.43 µM) and 6p (MIC90 7.81 µM) from the butyldiamine sub-series were cytotoxic to HepG2 cells and did not conform to the ISO norm, retaining only 30% and 56% cell viability at the respective MIC90s. Although the IC20s for 6o and 6p were both 2.13 µM in HepG2 cells, it is interesting to note a protective effect in the Br-containing 6p at the MIC90. It is also interesting to note that the F-substituted compound 6t in the hexyldiamine sub-series exhibited the highest cell viability at the MIC90 (92%) compared to 6r (72%) and 6s (76%). Compound 6u (CF3 substituted), the most active of all the synthesized compounds of the propane-1,2-diamine sub-series, yielded 100% cell viability at its MIC90 (0.97 µM), but 6w was extremely cytotoxic, with only 19% cell viability at its MIC90. Both 6x and 6y conformed to the ISO standard, with MIC90 HepG2 cell viability levels of 96% and 78%, respectively.
2.4. In Silico ADME Profiling of the Synthesized Compounds Using QikProp
To further validate the potential albumin protein binding observed in the 7H9/ADC/Glu/Tw media data above and to understand the possible effects of the high lipophilicity (clogP > 4), all the synthesized compounds were profiled in silico using a Schrodinger QikProp tool [50] for their ADME properties (Table 3). QikProp is a very useful tool for comparing any new compound’s potential ADME properties against known drugs (>95%) used to develop the tool. Studies have shown that the primary reasons for the high attrition rate of drug candidates during clinical trials are poor ADME and unfavourable drug-likeness properties [51,52]. Prior to the prediction, all the structures of the synthesized compounds were neutralized, and the program was run on the normal mode. Generally, all the synthesized compounds showed high predicted human oral absorption percentage (>80%) and the majority fulfilled Lipinski’s drug-like properties by having two (compound 6s only) or fewer violations (compounds 6p, 6r, 6t, and 6u—one violation each) of the rule of five (less than four violations) [53]. The predicted brain–blood partition coefficient (QPlogBB) was also within the acceptable range. Surprisingly, the predicted binding to human serum albumin (QPlogKhsa) was within the acceptable range, contrary to the experimentally observed potential protein binding in the ADC Mtb assay. Encouragingly, all the synthesized compounds were predicted to have IC50 values for the blockage of HERG K+ channels (QlopHERG) greater than −5. This is indicative of compounds that are devoid of any potential to inhibit the cardiac potassium HERG K+ channels and less likely to cause any cardiotoxicity.
Table 3.
In silico ADME and drug-likeness properties of the selected compounds.
| Compound | % Human Oral Absorption a | QPlogHERG b | QPlogBB c | QPlogKhsa d | Ro5 e |
|---|---|---|---|---|---|
| 6a | 91 | −5.3 | −0.594 | −0.093 | 0 |
| 6b | 100 | −5.235 | −0.351 | 0.142 | 0 |
| 6c | 93 | −5.169 | −0.487 | −0.056 | 0 |
| 6d | 71 | −5.211 | −1.744 | −0.117 | 0 |
| 6e | 100 | −5.661 | −0.371 | 0.363 | 0 |
| 6f | 100 | −5.094 | −0.378 | 0.184 | 0 |
| 6g | 100 | −5.119 | −0.369 | 0.205 | 0 |
| 6h | 100 | −5.062 | −0.425 | 0.116 | 0 |
| 6i | 100 | −5.748 | −0.648 | 0.458 | 0 |
| 6j | 100 | −5.695 | −0.48 | 0.313 | 0 |
| 6k | 100 | −5.854 | −0.865 | 0.382 | 0 |
| 6l | 100 | −5.809 | −0.634 | 0.203 | 0 |
| 6m | 100 | −5.725 | −0.477 | 0.337 | 0 |
| 6n | 100 | −5.676 | −0.529 | 0.243 | 0 |
| 6o | 100 | −5.884 | −0.831 | 0.433 | 0 |
| 6p | 91 | −5.904 | −0.822 | 0.456 | 1 |
| 6q | 100 | −5.851 | −0.843 | 0.427 | 0 |
| 6r | 100 | −6.077 | −0.863 | 0.637 | 1 |
| 6s | 83 | −6.212 | −0.904 | 0.695 | 2 |
| 6t | 96 | −6.049 | −0.908 | 0.563 | 1 |
| 6u | 95 | −5.776 | −0.319 | 0.511 | 1 |
| 6v | 100 | −5.736 | −0.459 | 0.303 | 0 |
| 6w | 100 | −5.777 | −0.402 | 0.396 | 0 |
| 6x | 100 | −5.749 | −0.593 | 0.414 | 0 |
| 6y | 100 | −5.753 | −0.409 | 0.374 | 0 |
a % Human Oral Absorption: Predicted human absorption on 0 to 100% scale (recommended: >80% is good and <25% is poor). b QPlog HERG: Predicted IC50 values for blockage of HERG K+ channels (below −5 is a concern). c QPlogBB: Predicted brain–blood partition coefficient (recommended: −3.0–1.2 range). d QPlogKhsa: Prediction of binding to human serum albumin (recommended: −1.5–1.5 range). e Ro5: Number of violations of Lipinski’s rule (recommended: a maximum of four violations).
3. Materials and Methods
3.1. Chemistry
All chemical reagents were purchased from Sigma-Aldrich (Modderfontein South Africa), Merck (Modderfontein, South Africa), Alfa Aesar (Kyalami, South Africa), or Thembane chemicals (Johannesburg, South Africa) and were used without further purification. Throughout the study, 1.1′Carbonyldiimidazole was kept in a desiccator. All solvents were purchased from Sigma-Aldrich and were HPLC or analytical grade. Deionized water was used throughout the study. Nuclear magnetic resonance (NMR) spectroscopic studies were performed using a Bruker Fourier 400 MHz and a Varian Gemini 400 spectrometer at 400 MHz and 100 MHz for the 1H NMR and 13C NMR nuclei, respectively, and all chemical shifts were referenced using solvent signals. Tetramethylsilane (TMS) was used as the internal standard, with chemical shifts reported in ppm and coupling constants (J) reported in Hertz (Hz). Reference peaks for deuterated chloroform (CDCl3) were 7.26 ppm for 1H NMR and 77.2 ppm for 13C NMR; for dimethyl sulfoxide (DMSO-d6), 2.50 ppm for 1H NMR and 39.5 ppm for 13C NMR; and for deuterated methanol (Meth-d), 4.87 ppm and 3.31 ppm for 1H NMR and 49.1 ppm for 13C NMR. Melting point studies were conducted on Stuart Scientific melting point apparatus SMP3. Fourier-transform infrared (FT-IR) spectroscopy studies were performed using a PerkinElmer FT-IR spectrometer. High-resolution mass spectrometry (HRMS) data, used to predict the present elements and to confirm the molecular weights of the synthesized compounds, were obtained with Waters Synapt G2, ESI probe, ESI Pos, Cone Voltage 15 V HRMS.
3.2. General Procedure for the Synthesis of Hybrid Compounds 6a–y
N-(Benzothiazole-2-yl)-1H-imidazole-1-carboxamides (5a–g) (1.1 mmol) with the respective N-(7-Chloro-4-quinolinyl)-diamines (2a–e and 3) (1.3 mmol) in HPLC grade acetonitrile (20 mL) were refluxed in an oil bath at 85 °C for 24 h. After reaction completion, the reaction mixture was cooled to room temperature and quenched with 1 M HCl solution (30 mL). The precipitated product was filtered and washed thoroughly with cold deionized water and air dried to obtain the desired target compounds (6a–y) in good to excellent yields and at an acceptable purity.
N-(Benzo[d]thiazol-2yl)-2-(7-chloroquinolin-4-yl)hydrazine-1-carboxamide (6a). Peach powder; yield: 85.52%; m.p. 198–202 °C; IR (cm−1): 3292, 1534 (NH), 1649 (C=C), 1750 (C=O), 1350 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm); 1H NMR (400 MHz, DMSO-d6, ppm): δH 10.13 (NH, brs, 1H), 8.63 (H2 and H1′, dd, J 13.6, 8.0 Hz, 2H), 8.17 (H4′, d, J 2.1 Hz, 1H), 7.88–7.80 (H8 and H5, m, 2H), 7.58 (H6, d, J 8.1 Hz, 1H), 7.36 (H2′, ddd, J 8.2, 7.3, 1.3 Hz, 1H), 7.21 (H3′, ddd, J 8.3, 7.3, 1.2 Hz, 1H), 6.99 (H3, d, J 6.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6, ppm): δC 157.6, 144.3, 138.9, 128.0, 127.6, 127.1, 124.7, 123.1, 119.7, 114.3, 99.6. HRMS (TOF MS ES+): found M + 1: 370.0524, C17H13N5OSCl requires MH+, 370.0529.
2-(7-Chloroquinolin-4-yl)-N-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)hydrazine-1-carboxamide (6b). Yellow powder; yield: 84.81%; m.p. 243–245 °C; IR (cm−1): 3226, 1543 (NH), 1662 (C=C), 1655 (C=O), 1363 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 10.26 (NH, brs, 1H), 8.67 (H2 and H1′, dd, J 16.6, 8.0 Hz, 2H), 8.36 (H4′, s, 1H), 8.16 (H8, d, J 2.1 Hz, 1H), 7.84 (H5, dd, J 2.2, 0.8 Hz, 1H), 7.76 (H2′, d, J 8.5 Hz, 1H), 7.67 (H6, dd, J 8.5, 1.9 Hz, 1H), 7.02 (H3, d, J 6.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6, ppm): δC 157.5, 144.4, 138.9, 132.1, 128.8, 128.02, 126.4, 125.3, 124.9, 124.1, 123.45, 123.3, 121.9, 120.1, 119.7, 114.4, 99.6. HRMS (TOF MS ES+): found M + 1: 438.0402, C18H12N5OF3SCl requires MH+, 438.0403.
2-(7-Chloroquinolin-4-yl)-N-(6-fluorobenzo[d]thiazol-2-yl)hydrazine-1-carboxamide (6c). Yellow oxide powder; yield: 86.11%; m.p. 240–242 °C; IR (cm−1): 3362, 1543 (NH), 1648 (C=C), 1643 (C=O), 1377 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 10.32 (NH, brs, 1H), 8.74 (H-2, d, J 6.6 Hz 1H), 8.69 (H1′, d, J 10.9, 1H), 8.18 (H4′, s, 1H), 7.78 (H5 and H8, m, 2H), 7.59 (NH, s, 1H), 7.37 (H2′, J 6.9 Hz, 1H), 7.27 (H6, d, J 7.1 Hz, 1H), 6.73 (H3, d, J 3.2 Hz 1H); 13C NMR (101 MHz, DMSO-d6, ppm): δC 158.0, 146.7, 138.1, 131.1, 128.1, 127.4, 127.2, 127.0, 127.3, 126.2 (2 × C), 125.4, 124.1, 123.9, 120.4, 119.90, 99.9. HRMS (TOF MS ES+): found M + 1: 388.0434, C17H12N5OFSCl requires MH+, 388.0435.
2-(7-Chloroquinolin-4-yl)-N-(6-nitrobenzo[d]thiazol-2-yl)hydrazine-1-carboxamide (6d). Yellow powder; yield: 78.75%; m.p. 239–241 °C; IR (cm−1): 3222, 1534 (NH), 1658 (C=C), 1653 (C=O), 1393 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 8.79–8.58 (2 × NH, s, 2H), 8.59–8.45 (H2 and H1′, m, 2H), 8.20 (H5, dd, J 6.6 and 1.4 Hz, 1H), 8.10 (H-8, d, J 1.6 Hz, 1H), 7.69 (H4′, d, J 1.3 Hz, 1H), 7.40 (H2′ and H6, d, J 9.0 Hz, 2H), 7.12 (H3, m, 1H); 13C NMR (101 MHz, DMSO-d6, ppm): δC 172.2, 157.7, 156.1, 143.2, 142.5, 141.4, 138.8, 138.2, 131.5, 126.9, 125.9, 122.6, 119.3, 118.3, 117.0, 114.1, 98.6. HRMS (TOF MS ES−): found M-1: 413.0208, C17H10N6O3SCl requires M-H, 413.224.
1-(2-((7-Chloroquinolin-4-yl)amino)ethyl)-3-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)urea (6e). Cream white powder; yield: 64.90%; m.p. 249–251 °C; IR (cm−1): 3281, 1533 (NH), 1614 (C=C), 1758 (C=O), 1358 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 9.60 (NH, brt, J 5.5 Hz, 1H), 8.64 (H2, d, J 9.2 Hz, 1H), 8.60 (H1′, d, J 7.1 Hz, 1H), 8.16 (H4′, s, 1H), 7.99 (NH, s, 1H), 7.79 (H5 and H8, dd, J 7.1, 2.3 Hz, 2H), 7.55–7.32 (H2′ and NH, m, 2H), 7.24 (H6, m, 1H), 6.83 (H3, d, J 9.2 Hz, 1H), 3.62 (H10a, m, 1H), 3.22 (H10b, m, 1H), 199-178 (2H, M, H9); 13C NMR (101 MHz, DMSO-d6, ppm): δC 163.2, 158.3, 156.9, 143.2, 138.9, 138.4, 132.44, 128.2, 127.5, 127.0, 126.4, 123.3, 123.1, 120.3, 119.8, 119.4, 116.8, 99.1, 43.5, 38.3. HRMS (TOF MS ES+): found M + 1: 466.0719, C20H16N5OF3SCl requires MH+, 466.0716.
1-(6-Chlorobenzo[d]thiazol-2-yl)-3-(2-((7-chloroquinolin-4-yl)amino)ethyl)urea (6f). As a dark cream solid; yield: 83.23%; m.p. 244–245 °C; FT-IR (vmax/cm−1): v (C=O) 1535.72, v (NH) 2938.94; 3238.94, 3354.12, 1H NMR (400 MHz, DMSO-d6, ppm): δH 11.24–10.99 (2 × NH, 2 × brs, 2H), 9.28 (H2, d, J 8.90 Hz, 1H), 8.56 (H1′, m, 2H), 8.09 (H4′ and H8, m, 2H), 7.89 (H5, m, 1H), 7.73 (NH and H2′, m, 2H), 7.23 (H6, d, J 8.7 Hz, 1H), 6.99 (H3, d, J 9.1 Hz, 1H), 3.77 (H10, m, 2H), 3.55 (H9, m, 2H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 162.9, 156.7, 155.2, 154.9, 148.3, 144.3, 140.1, 138.7, 133.2, 127.9, 127.3, 127.0, 121.2, 120.3, 119.9, 116.5, 98.4, 44.3, 37.8. HRMS (TOF MS ES+): found M + 1: 432.0451, C19H16N5OSCl2 requires MH+, 432.0453.
1-(6-Bromobenzo[d]thiazol-2-yl)-3-(2-((7-chloroquinolin-4-yl)amino)ethyl)urea (6g). Light brown solid; yield: 78.41%; m.p. 240–242 °C; FT-IR (vmax/cm−1): v (C=O) 1667.12, v (NH) 2936.09; 3340.35, 3413.64, 1H NMR (400 MHz, DMSO-d6, ppm): δH 8.49 (H2, d, J 7.8 Hz, 1H), 8.25 (H1′, dd, J 7.1, 2.1 Hz, 1H), 8.16 (H4′, d, J 1.91 Hz, 1H), 7.82 (H8, s, 1H), 7.55 (H2′ and H5, 2 × d, J 7.0 and 6.5 Hz, 2H), 7.49 (H6 and NH, m, 2H), 7.15 (NH, brs, 1H), 6.97 (NH, brs, 1H), 6.52 (H3, d, J 7.8 Hz, 1H), 3.49 (H10, m, 2H), 3.35 (H9, m, 2H); 13C NMR (101 MHz; DMSO-d6, ppm): δc 163.0, 154.9, 154.2, 153.6, 151.7, 148.3, 134.9, 134.0, 129.7, 127.3, 124.9, 123.7, 123.4, 123.1, 122.3, 118.6, 114.7, 98.6, 44.8, 37.6. HRMS (TOF MS ES+): found M + 1: 475.9948, C20H16N5OBrSCl requires MH+, 475.9947.
1-(6-Fluorobenzo[d]thiazol-2-yl)-3-(2-((7-chloroquinolin-4-yl)amino)ethyl)urea (6h). Pink solid; yield: 75.23%; m.p. 230–232 °C; FT-IR (vmax/cm−1): v (C=O) 1649.32, v (NH) 3261.71; 3354.28, 3399.65; 1H NMR (400 MHz, DMSO-d6, ppm) δH 11.05-10.55 (2 × NH, 2 × brs, 2H), 8.52 (H4′, dd, J 6.8, 1.4 Hz, 1H), 7.89 (H1′ and H2, m, 2H), 7.69 (H5 and H8, m, 2H), 7.32 (2H, H2′ and H6, m, 2H), 6.79 (H3, d, J 6.9 Hz, 1H), 3.73 (H10a, m, 1H), 3.51 (H10b, m, 1H), 3.21 (H9, m, 2H); 13C NMR (101 MHz; DMSO-d6, ppm): δc 161.9, 159.7, 157.8, 156.3, 155.0, 146.3, 143.2, 138.2, 137.5, 132.9–132.1 (C3′ coupling to F atom), 128.1, 127.3, 121.9, 119.4, 117.3, 113.9–113.0 (C2′ coupling to F atom), 108.3–107.6 (C4′ coupling to F atom), 98.3, 44.9, 32.8. HRMS (TOF MS ES+): found M + 1: 416.0740, C20H16N5OFSCl requires MH+, 416.0748.
1-(3-((7-Chloroquinolin-4-yl)amino)propyl)-3-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)urea (6i). Yellow powder; yield: 83.42%; m.p. 198–200 °C; IR (cm−1): 3255, 1532 (NH), 1691 (C=C), 152 (C=O), 1343 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 9.42 (NH, brs, 1H), 8.61 (H2, d, J 7.9 Hz, 1H), 8.55 (H1′, d, J 6.8 Hz, 1H), 8.32 (H4′, s, 1H), 7.83 (H8, s, 1H), 7.75 (H5, d, J 7.2 Hz, 1H), 7.51 (H2′ and NH, m, 1H), 7.24 (NH, t, J 7.2 Hz, 1H), 6.89 (H6, d, J 6.9 Hz, 1H), 6.89 (H3, d, J 7.8 Hz, 1H), 3.58 (H11, m, 2H), 3.20 (H9a, m, 1H and *H9b overlaps with water peak), 1.90 (H10, m, 2H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 207.1, 162.7, 153.7, 152.3, 149.5, 136.38, 134.3, 129.5, 128.3 (2 × C), 127.8 (2 × C), 125.1, 118.4, 99.5, 59.3, 44.5, 33.4. HRMS (TOF MS ES+): found M + 1: 480.0167, C21H18N5OSF3Cl requires MH+, 480.0173.
1-(6-Chlorobenzo[d]thiazol-2-yl)-3-(3-((7-chloroquinolin-4-yl)amino)propyl)urea (6j). Yellow-brown solid; yield: 76.82%; m.p. 286–288 °C; IR (cm−1): 3301, 1564 (NH), 1664 (C=C), 1618 (C=O), 1360 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 11.25 (NH, s, 1H), 9.51 (NH, brs, 1H), 8.63 (H2 and H1′, m, 2H), 8.41 (H4′, s, 1H), 8.01 (H8, s, 1H), 7.79 (H2′ and NH, m, 2H), 7.64 (H5, d, J 6.9 Hz, 1H), 7.33 (H6, d, J 6.8 Hz, 1H), 7.12 (H3, d, J 7.4 Hz, 1H), 4.12 (H11a, m, 1H and *H11b overlaps with water peak), 3.71–3.55 (H9, m, 2H), 1.33 (H10, q, J 9.1 Hz, 2H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 163.9, 158.8–158.0 (2 × C), 144.6, 138.9–138.5 (2 × C), 137.6–137.3 (2 × C), 128.3–127.9 (2 × C), 126.5–126.0 (2 × C), 124.5, 119.7–119.6 (2 × C), 117.3, 99.8, 48.6, 45.2, 18.7. HRMS (TOF MS ES+): found M + 1: 446.0607, C20H18N5OSCl2 requires MH+, 446.0609.
1-(3-((7-Chloroquinolin-4-yl)amino)propyl)-3-(6-methylbenzo[d]thiazol-2-yl)urea (6k). Faint pink powder; yield: 55.94%; m.p. 234–239 °C; IR (cm−1): 3266, 1543 (NH), 1660 (C=C), 1625 (C=O), 1373 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 9.51 (2 × NH, m, 2H), 8.81 (H2, d, J 9.0 Hz, 1H), 8.51 (H1′, d, J 7.0 Hz, 1H), 7.96 (H2′, d, J 7.0 Hz, 1H), 7.75 (H4′, d, J 2.1 Hz, 1H), 7.68 (H8, s, 1H), 7.41 (H5, d, J 8.2 Hz, 1H), 7.21 (H6, d, J 8.2 Hz, 1H), 6.85 (H3, d, J 9.0 Hz, 1H), 3.53 (H11a, m, 1H and *H11b and H9 overlaps with water peak), 2.31 (CH3, s, 3H), 1.83 (H10, m, 2H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 160.9, 157.1, 154.9, 143.2, 139.7 (2 × C), 139.4, 132.7, 132.1, 127.4 (2 × C), 126.5, 122.1 (2 × C), 119.4, 116.8, 99.8, 41.7, 37.5, 28.9, 20.1. HRMS (TOF MS ES+): found M + 1: 426.1155, C21H21N5OSCl requires MH+, 426.1155.
1-(3-((7-Chloroquinolin-4-yl)amino)propyl)-3-(benzo[d]thiazol-2-yl)urea (6l). Grey powder; yield: 59.31%; m.p. 285–288 °C; 1H NMR (400 MHz, DMSO-d6, ppm): δH 9.32 (NH, brs, 1H), 8.64 (H2, d, J 8.1 Hz, 1H), 8.53 (H1′, d, J 9.1 Hz, 1H), 8.40 (H8, s, 1H), 7.82 (H4′ and H5, m, 2H), 7.69 (H3′ and H2′, m, 2H), 7.43 (H6, d, J 8.1 Hz, 1H), 6.88 (H3, d, J 8.0 Hz, 1H), 3.61 (H11, m, 2H), 3.31 (H9a, m, 1H and *H9b overlaps with water peak), 1.88 (H10, m, 2H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 154.6 (2 × C), 151.1, 150.2, 134.7 (2 × C), 127.9–127.5 (4 × C), 124.6–124.2 (3 × C), 118.9 (2 × C), 99.8, 52.5, 45.1, 23.1. HRMS (TOF MS ES+): found M + 1: 412.1002, C20H19N5OSCl requires MH+, 412.0999.
1-(6-Bromobenzo[d]thiazol-2-yl)-3-(3-((7-chloroquinolin-4-yl)amino)propyl)urea (6m). Pale yellow solid; yield: 60.20%; m.p. 254–257 °C; 1H NMR (400 MHz, DMSO-d6, ppm): δH 8.40 (H2, d, J 5.3 Hz, 1H), 8.25 (H1′, m, 1H), 8.12 (H4′ and NH, s, 2H), 7.78 (H8, d, J 1.4 Hz, 1H), 7.54 (H5, m, 1H), 7.48 (H2′, m, 1H), 7.34 (H6, t, J 5.0 Hz, 1H), 6.95 (2 × NH, 2 × brs, 2H), 6.49 (H3, d, J 5.4 Hz, 1H), 3.23 (H11 and H9, m, 4H), 1.91 (H10a, m, 1H), 1.68 (H10b, m, 1H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 162.9, 154.6, 152.3, 150.1, 148.3, 147.0, 135.2, 128.3, 126.4, 124.3, 123.8, 122.4, 118.9 (2 × C), 114.6, 99.8, 41.3, 38.6, 31.3, 27.3: HRMS (TOF MS ES+): found M + 1: 490.0110, C20H18N5OBrSCl requires MH+, 490.0104.
1-(6-Fluorobenzo[d]thiazol-2-yl)-3-(3-((7-chloroquinolin-4-yl)amino)propyl)urea (6n). Light brown solid; yield: 78.50%; m.p. 241–243 °C; 1H NMR (400 MHz, DMSO-d6, ppm): δH 8.42 (H1′, m, 1H), 8.26 (H2, d, J 8.9 Hz, 1H), 8.07 (H4′, d, J 1.7 Hz, 1H), 7.79 (H8, d, J 4.3 Hz, 1H), 7.52 (H5, d, J 10.6 Hz, 2H), 7.45 (H2′ and H6, d, J 8.6 Hz, 2H), 6.92 (NH, t, J 5.0 Hz, 1H), 6.55 (H3, d, J 8.9 Hz, 1H), 3.38 (H11a, m, 1H), 3.29 (H11b, m, 1H), 3.20 (H9, m, 2H), 1.92 (H10a, m, 1H), 1.67 (H10b, m, 1H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 163.9, 153.2, 151.1, 149.9, 148.6, 148.0, 137.9, 129.3, 127.2, 123.8, 123.3, 122.5, 119.9 (2 × C), 116.4, 98.5, 43.0, 37.9, 30.2, 26.9: HRMS (TOF MS ES+): found M + 1: 430.0896, C20H18N5OFSCl requires MH+, 430.0905.
1-(6-Chlorobenzo[d]thiazol-2-yl)-3-(4-((7-chloroquinolin-4-yl)amino)butyl)urea (6o). Yellow-brown solid; yield: 83.04%; m.p. 189.8–191.3 °C; FT-IR (vmax/cm−1): v (C=O) 1538.60v (NH) 2964.7, 3339.73, 3389.02; 1H NMR (400 MHz, CH3OH-d4, ppm): δH 8.39 (H2 and H1′, m, 2H), 7.80 (H4′ and H5, d, J 8.8 Hz, 2H), 7.69 (H8, d, J 3.3 Hz, 1H), 7.51 (H2′, d, J 7.2 Hz, 1H), 7.33 (H6, d, J 8.6 Hz, 1H), 6.87 (H3, d, J 6.5 Hz, 1H), 3.62 (H12, m, 2H), 3.36 (H9, m, 2H), 1.83 (H11, m, 2H), 1.75 (H10, m, 2H); 13C NMR (101 MHz; DMSO-d6, ppm): δc 163.1, 156.2, 154.8, 148.3, 144.6, 139.7, 138.3, 133.7, 127.5, 127.0, 126.7, 122.4, 122.1, 120.3, 116.9, 98.3, 43.6, 41.3 (*overlaps with solvent peak), 27.3, 21.2. HRMS (TOF MS ES+): found M + 1: 460.0763, C21H20N5OSCl2 requires MH+, 460.0766.
1-(6-Bromobenzo[d]thiazol-2-yl)-3-(4-((7-chloroquinolin-4-yl)amino)butyl)urea) (6p). Dark yellow solid; yield: 82.34% m.p. 192–194 °C; 1H NMR (400 MHz, DMSO-d6, ppm): δH 10.93 (NH, brs, 1H), 8.49 (H2, d, J 7.1 Hz, 1H), 8.35 (H1′, d, J 8.1 Hz, 1H), 8.19 (H4′, s, 1H), 8.19 (H8 and NH, s, 2H), 7.50–7.42 (H2′, H5 and H6, m, 3H), 6.92 (NH, brs, 1H), 6.63 (H3, d, J 7.0 Hz, 1H), 3.25 (H12 and H9, m, 4H), 1.71 (H11, m, 2H), 1.65 (H10, m, 2H); 13C NMR (101 MHz; DMSO-d6, ppm): δc 162.1, 154.3, 152.7, 151.9, 149.3, 135.3, 134.8, 129.3, 126.7, 125.9, 125.3, 124.7, 122.4, 118.1, 114.9, 99.6, 43.1, 42.2, 27.5, 26.2. HRMS (TOF MS ES+): found M + 1: 504.0263, C21H20N5OBrSCl requires MH+, 540.0260.
1-(6-Fluorobenzo[d]thiazol-2-yl)-3-(4-((7-chloroquinolin-4-yl)amino)butyl)urea (6q). Pale yellow solid; yield: 96.05% m.p. 256–259 °C; 1H NMR (400 MHz, DMSO-d6, ppm): δH 8.39 (H2, d, J 7.2 Hz, 1H), 8.25 (H1′, d, J 9.6 Hz, 1H), 8.15 (H4′, d, J 2.2 Hz, 1H), 7.81 (H8, d, J 3.2 Hz, 1H), 7.72 (NH, brs, 1H), 7.51 (H5, d, J 9.8 Hz, 1H), 7.42 (H2′ and H6, m, 2H), 6.81 (2 × NH, brs, 2H), 6.61 (H3, d, J 7.0 Hz, 1H), 3.36 (H12, m, 2H), 3.21 (H12, m, 2H), 1.71 (H11, m, 2H), 1.62 (H10, m, 2H); 13C NMR (101 MHz; DMSO-d6, ppm): δc 167.8, 162.1, 160.3, 157.5, 147.5, 133.9–132.9 (C3′ coupling to F atom), 125.7, 125.0, 122.7, 119.3, 114.9–114.7 (C2′ coupling to F atom), 113.9, 113.2, 109.9, 109.7–109.2 (C4′ coupling to F atom), 99.7, 44.6, 42.5, 22.5, 20.7. HRMS (TOF MS ES+): found M + 1: 444.1063, C21H20N5OFSCl requires MH+, 444.1061.
1-(6-Chlorobenzo[d]thiazol-2-yl)-3-(6-((7-chloroquinolin-4-yl)amino)hexyl)urea (6r). Yellow-brown solid; yield: 79.7%; m.p. 239–241 °C; FT-IR (vmax/cm−1): v (C=O) 1666.63, v (NH) 3076.20, 3340.97, 3412.01; 1H NMR (400 MHz, CH3OH-d4, ppm): δH 8.35 (H2, d, J 6.9 Hz, 1H), 8.19 (H1′, d, J 8.8 Hz, 1H), 7.75 (H4′ and NH, d, J 10.2 Hz, 2H), 7.56 (H5, d, J 7.2 Hz, 1H), 7.40 (H8, d, J 3.2 Hz, 1H), 7.33 (H6, d, J 7.3 Hz, 1H), 6.50 (H3, d, J 6.8 Hz, 1H), 3.27 (H14, m, 2H), 3.25 (H9, m, 2H), 1.75 (H13, m, 2H, 1.69 (H10, m, 2H), 1.56–1.47 (H11 and H12, m, 4H); 13C NMR (101 MHz; DMSO-d6, ppm): δc 156.9, 152.5, 151.7, 151.0, 149.7, 149.1, 137.8, 137.2, 128.3, 127.0, 126.8, 125.1, 122.9, 122.6, 122.1, 118.6, 98.9, 43.5, 30.6, 28.2, 27.5, 27.1, 26.9. HRMS (TOF MS ES+): found M + 1: 488.1081, C23H24N5OSCl2 requires MH+, 488.1079.
1-(6-Bromobenzo[d]thiazol-2-yl)-3-(6-((7-chloroquinolin-4-yl)amino)hexyl)urea (6s). Pale cream colour; yield: 82.32%; m.p. 192–194 °C; FT-IR (vmax/cm−1): v (C=O) 1533.51, v (NH) 2861.00, 2935.51, 3234.26; 1H NMR (400 MHz, DMSO-d6, ppm): δH 10.93, (NH, brs, 1H), 9.32 (NH, brs, NH), 8.65 (H2, d, J 8.8 Hz, 1H), 8.58 (H1′, d, J 7.6 Hz, 1H), 8.12 (H4′, s, 1H), 7.97 (H8, s, 1H), 7.75 (H5, d, J 9.3 Hz, 1H), 7.51 (H2′, m, 1H), 7.48 (H6, m, 1H), 7.21 (NH, t, J 5.0 Hz, 1H), 6.88 (H3, d, J 8.5 Hz, 1H), 3.51 (H14, m, 2H), 3.21 (H9, m, 2H), 1.71 (H13, m, 2H), 152–1.32 (H10, H11 and H12, m, 6H); 13C NMR (101 MHz; DMSO-d6, ppm): δc 161.2, 156.3, 154.2, 148.7, 144.3, 139.3, 138.2, 134.3, 129.1, 127.5, 126.5, 124.9, 122.1, 119.7, 116.3, 115.2, 98.7, 44.6, 30.9, 30.7, 27.5, 26.5, 26.0. HRMS (TOF MS ES+): found M + 1: 432.0576. C23H24N5OBrSCl requires MH+, 432.0573.
1-(6-((7-Chloroquinolin-4-yl)amino)hexyl)-3-(6-fluorobenzo[d]thiazol-2-yl)urea (6t). Yellow solid; yield: 92.0%; m.p. 256–259 °C; 1H NMR (400 MHz, DMSO-d6, ppm): δH 10.75 (NH, brs, 1H), 9.75 (NH, brs, 1H), 8.65 (H2, J 8.3 Hz, 1H), 8.58 (H1′, d, J 6.8 Hz, 1H), 8.0 (H4′, s, 1H), 7.75 (H2′, m, 1H), 7.60 (H5 and H8, m, 2H), 7.16 (H6, m, 1H), 6.81 (H3, d, J 7.8 Hz, 1H), 3.61 (H14, m, 2H), 3.36 (H9, m, 2H), 1.75 (H13, m, 2H), 1.61–1.31 (H10, H11 and H12, m, 6H); 13C NMR (101 MHz; DMSO-d6, ppm): δc 162.3, 161.9, 157.3, 155.9, 154.3, 147.6, 144.3, 139.0–138.9 (C3′ coupling to F atom), 133.2, 127.6, 126.1, 122.3, 121.4, 117.9, 114.9–114.3 (C2′ coupling to F atom), 108.6–108.2 (C4′ coupling to F atom), 98.2, 43.9, 30.1, 29.9, 28.6, 28.0, 27.6, 26.9. HRMS (TOF MS ES+): found M + 1: 472.1374, C23H24N5OFSCl requires MH+, 472.1374.
1-(1-((7-Chloroquinolin-4-yl)amino)propan-2-yl)-3-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)urea (6u). Yellow brown powder; yield: 84.81%; m.p. 243–245 °C; IR (cm−1): 3298, 1557 (NH), 1661 (C=C), 1562 (C=O), 1373 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 11.13 (NH, brs, 1H), 9.47 (NH, brs, 1H), 8.69 (H2 and H1′, d, J 7.4 Hz, 2H), 8.32 (H8, s, 1H), 7.95 (H4′, s, 1H), 7.75 (H2′, d, J 7.6 Hz, 1H), 7.63 (H5, d, J 8.7 Hz, 1H), 7.31 (H6, d, J 8.7 Hz, 1H), 7.13 (H3, d, J 7.1 Hz, 1H), 4.18 (H10, m, 1H), 3.74 (H9a, m, 1H), 3.63 (H9b, m, 1H), 1.30 (H11, d, J 5.9 Hz, 3H); 13C NMR (101 MHz, DMSO-d6, ppm): δC 163.1, 156.2, 154.3, 143.7, 139.4, 138.3, 136.9, 133.1, 127.2 (2 × C), 126.9, 126.1, 123.1, 121.0, 119.8, 116.1, 99.4, 48.5, 45.3, 18.5. HRMS (TOF MS ES+): found M + 1: 480.0885, C21H18N5OSF3Cl requires MH+, 480.0873.
1-(1-((7-Chloroquinolin-4-yl)amino)propan-2-yl)-3-(6-fluorobenzo[d]thiazol-2-yl)urea (6v). Yellow brown powder; yield: 87.52%; m.p. 228–230 °C; IR (cm−1): 3356, 1547 (NH), 1676 (C=C), 1576 (C=O), 1377 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 9.57 (NH, brs, 1H), 8.58 (H2 and H-1′, m, 2H), 8.31 (H4′, s, 1H), 7.96 (H8, d, J 2.0 Hz, 1H), 7.73 (H5, d, J 8.8 Hz, 1H), 7.63 (H6, dd, J 8.8, 2.0 Hz, 1H), 7.36 (H2′, d, J 7.7 Hz, 1H), 7.07 (H3, d, J 7.1 Hz, 1H), 4.16 (H-10, m, 1H), 3.73 (H9a, m, 1H), 3.62 (H9b, m, 1H), 1.26 (H11, d, J 6.7 Hz, 3H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 207.2, 157.3, 152.9, 149.6, 141.5, 136.3, 135.5, 134.0, 131.3, 129.7, 126.3, 125.1, 124.3, 123.1, 122.5, 99.8, 32.3, 27.1, 18.2. HRMS (TOF MS ES+): found M + 1: 430.0910, C20H18N5OSFCl requires MH+, 430.0905.
1-(1-((7-Chloroquinolin-4-yl)amino)propan-2-yl)-3-(6-bromobenzo[d]thiazol-2-yl)urea (6w). Yellow powder; yield: 89.99%; m.p. 246–248 °C; IR (cm−1): 3245, 1525 (NH), 1672 (C=C), 1571 (C=O), 1335 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 8.45 (H2, d, J 8.7 Hz, 1H), 8.29 (H1′, d, J 9.0 Hz, 1H), 7.99 (H4′, s, 1H), 7.70 (H8, s, 1H), 7.61 (H5, d, J 7.9 Hz, 1H), 7.51 (H2′, d, J 9.0 Hz, 1H), 7.39 (H6, d, J 7.7 Hz, 1H), 6.89 (H3, d, J 8.5 Hz, 1H), 6.64 (NH, s, 1H), 4.14 (H-10, m, 1H), 3.48 (H9a, m, 1H and *H9b overlaps with water peak), 1.23 (H-11, d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 207.2, 157.3, 152.9, 149.6, 141.5, 136.3, 135.5, 134.0, 131.3, 129.7, 126.3, 125.1, 124.3, 123.1, 122.5, 99.8, 32.3, 27.1, 18.2. HRMS (TOF MS ES+): found M + 1: 490.0103, C20H18N5OSBrCl requires MH+, 490.0104.
1-(1-((7-Chloroquinolin-4-yl)amino)propan-2-yl)-3-(6-methylbenzo[d]thiazol-2-yl)urea (6x). Brown yellow powder; yield: 78.81%; m.p. 229–231 °C; IR (cm−1): 3359, 1542 (NH), 1672 (C=C), 1615 (C=O), 1346 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 9.67 (NH, t, J 5.1 Hz, 1H), 8.66 (H2, d, J 9.1 Hz, 1H), 8.57 (H4′, s, 1H), 8.04 (H1′, J 2.1 Hz, 1H), 7.74 (H5, d, J 8.3 Hz, 1H), 7.62 (H8, s, 1H), 7.53 (H2′ and NH, m, 2H), 7.16 (H6, d, J 8.3 Hz, 1H), 7.08 (H3, d, J 9.1 Hz, 1H), 4.14 (H10, m, 1H), 3.70 (H9a, m, 1H), 3.56 (H9b, m, 1H), 2.36 (H5′, s, 3H), 1.26 (H11, d, J 6.7 Hz, 3H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 159.3, 157.2, 156.4, 146.3, 144.6, 138.9, 138.6, 132.4, 131.6, 129.9, 129.6, 129.3, 127.7, 122.5, 119.4, 116.9, 99.3, 48.5, 45.1, 21.3, 18.6. HRMS (TOF MS ES+): found M + 1: 426.1154, C21H21N5OSCl requires MH+, 426.1155.
1-(1-((7-Chloroquinolin-4-yl)amino)propan-2-yl)-3-(6-chlorobenzo[d]thiazol-2-yl)urea (6y). Brownish white powder; yield: 81.19%; m.p. 301–304 °C; IR (cm−1): 3254, 1532 (NH), 1672 (C=C), 1646 (C=O), 1343 (C-N); 1H NMR (400 MHz, DMSO-d6, ppm): δH 11.32 (NH, brs, 1H), 9.51 (NH, s, 1H), 8.69 (H2 and H1′, d, J 5.8 Hz, 2H), 8.39 (H4′, s, 1H), 7.96 (H8, s, 1H), 7.79 (H5 and NH, d, J 6.9 Hz, 2H), 7.70 (H2′, d, J 3.8 Hz, 1H), 7.32 (H6, d, J 7.1 Hz, 1H), 7.12 (H3, d, J 5.8 Hz, 1H), 4.12-4.06 (H-10, m, 1H), 3.43-3.01 (H9, m, 2H) 1.26 (H-11, d, J = 3.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6, ppm): δc 160.88, 154.77, 152.71, 149.18, 147.72, 147.69, 142.83 135.49, 133.61, 130.02, 127.09, 126.46, 125.53, 121.18, 118.30 117.21, 99.34, 48.28, 45.12, 18.70. HRMS (TOF MS ES+): found M + 1: 446.0699, C20H18N5OSCl2 requires MH+, 446.0609.
3.3. In Vitro Antitubercular Activity
The minimum inhibitory concentration (MIC) is determined using the standard broth microdilution method, according to which a 10 mL culture of Mycobacterium tuberculosis H37Rv pMSp12::GFP [54,55,56] is grown to an optical density (OD600) of 0.6–0.7. Cultures are diluted prior to the inoculation of assays as follows:
-
(i)
1:500 in middelbrook 7H9 supplemented with 0.03% casitone, 0.4% glucose, and 0.05% tyloxapol [57,58];
-
(ii)
1:500 in 7H9 supplemented with 10% Albumin Dextrose Catalase supplement (ADC), 0.05% Tween-80, 0.2% glucose [59,60].
The compounds to be tested are reconstituted to a concentration of 10 mM in DMSO. Two-fold serial dilutions of the test compound are prepared across a 96-well black microtiter plate, after which 50 μL of the diluted M. tuberculosis cultures is added to each well in the serial dilution in triplicate. The plate layout is a modification of the method previously described [61]. The assay controls used are a minimum growth control (rifampicin at 2 × MIC) and a maximum growth control (5% DMSO). The microtiter plates are sealed in a secondary container and incubated at 37 °C with 5% CO2 and humidification. Relative fluorescence (excitation 485 nM; emission 520 nM) is measured and the assay is set up in a black 96-well microtiter plate. Relative fluorescent readings are taken at day 7, using Spectramax i3x, and the lowest concentration of material displaying no visible growth is scored as the MIC90.
The microtiter plats are re-incubated for a further 7 days. Fluorescent readings are taken after 7 days again and reported as day 14 data.
Raw fluorescent data (relative fluorescent units) are acquired using a SpectraMax i3x Plate reader (serial No. 36370 3271), Molecular Devices Corporation 1311 Orleans Drive Sunnyvale, California 94089.
RFU Data are analysed using Softmax® Pro 6 software: (Version 6.5.1, serial No. 1278552768867612530), Molecular Devices Corporation 1311 Orleans Drive Sunnyvale, California 94089.
The onboard fluorescent intensity endpoint protocol is used in conjunction with the following wavelength filters: excitation: 485; emission: 520. The Softmax® Pro 6, 4-parameter curve fit protocol is used to generate a calculated MIC90. Raw RFU data are normalised to the minimum and maximum inhibition controls to generate a dose–response curve (percent inhibition), using the Levenberg–Marquardt damped least-squares method, from which the MIC90 is calculated. The lowest concentration of drug that inhibits 90% of growth of the bacterial population is considered to be the MIC90.
3.4. In Vitro MTT Cytotoxicity Studies
HepG2 liver cells (Highveld Biological, Johannesburg, South Africa) are characterised as non-tumorigenic cells with high proliferation rates. Originally derived from a Caucasian male, the cells have retained an epithelial-like morphology and key differentiated liver function, and are thus deemed a suitable model for cytotoxicity testing [62,63].
HepG2 were aseptically maintained at 37 °C (5% CO2) until confluent in complete culture medium (CCM) comprising Dulbecco’s Minimum Essential Medium, 10% foetal bovine serum, 1% L-glutamine, and 1% pen–strep–fungizone. The confluent HepG2 cells were rinsed with 0.1 M phosphate-buffered saline (PBS), then trypsinised and counted to facilitate their transfer to a 96-well microtiter plate (2 × 104 cells/150 µL CCM/well). After incubation overnight to ensure adherence to the plate wells, the CCM was discarded and the cells were treated with varying concentrations (0–200 µM) of the synthesized compounds for 24 h (37 °C, 5% CO2). A vehicle control (1% DMSO in CCM) was included. The treatment medium was removed and the wells were replenished with 20 µL MTT solution (5 mg/mL MTT in PBS) and 100 µL CCM. After 4 h (37 °C, 5% CO2), the MTT/CCM solution was replaced with 100 µL DMSO (1 h, 37 °C, 5% CO2) to solubilize the formazan crystals. The absorbance was read at 570 nm/690 nm using the SPECTROstar Nano (BMG Labtech, Ortenberg, Germany). Percentage cell viability was calculated using the absorbance values ( × 100). A nonlinear regression analysis (GraphPad Prism V5.0, GraphPad PRISM®, La Jolla, CA, USA) for cell viability vs. log treatment concentration was performed to extrapolate the 20% inhibitory concentration (IC20); at this concentration cells retained 80% cell viability. The cell viability at the respective 90% Mtb inhibitory concentration (MIC90 in the CAS assay) was also extrapolated from the curve.
3.5. In Silico ADME Profiling
QikProp (Schrödinger, LLC, New York, NY, USA) was utilized for predicting physically relevant descriptors for predicting potential drug-likeness, such as human oral absorption, QlogHERG, QlogBB, QlogKhsa, and the compliance to the Lipinski rule of five.
4. Conclusions
In this study, 25 new quinoline–urea–benzothiazole hybrids (6a–y) have been successfully synthesized via a three-step synthetic protocol in yields ranging from 50 to 90%. These compounds were then evaluated for their MIC90 anti-Mtb activity, in protein-rich (ADC) and -poor (CAS) media, with activities ranging from 0.97–62.5 μM. The anti-Mtb activities suggest that the type of the diamine used and its chain length, as well as the substituent on the benzothiazole moiety, influences activity. For example, (i) CF3-containing compounds (6b, 6i and 6u) on the benzothiazole moiety displayed the most consistent activities across the two media, with 6u being the most active of all the compounds in this study; and (ii) the propane-1,2-diamine (sub-series 6u–y) was the most preferred diamine, as this produced compounds with activities less than 15 µM across the board, even though 6u exhibited a potential for protein binding. The hybridization strategy was also found to be advantageous and the intermediate making up the hybrids appeared to function in a synergistic manner since on their own and in combination they are devoid of any activity. Lastly, based on the data, compound 6u seems to be a very good starting point in the quest for the next generation of hybrid compounds.
Overall, minimal cytotoxicity to HepG2 cells (cell viability above 75%) was observed in 11 compounds, indicating that they may be exploited for potential antimycobacterial effects, including activity under different growth conditions thought to mimic human infection, for example, nutrient starvation, hypoxia, and/or low pH. The cytotoxicity data also demonstrate the anti-proliferative activities of the synthesized compounds in HepG2 cells. Thus, further investigation into the possible anticancer effects of the synthesized compounds and their possible anti-Mtb mode of action are warranted in future. Generally, all the synthesized compounds showed favourable predicted in silico profiles against the majority of the selected descriptors. More importantly, these compounds were predicted to have a reduced potential to inhibit the cardiac potassium hERG channel (QlogHERG > −5).
Acknowledgments
We are grateful to Nombulelo Tracy Nyoni for re-running some of the NMR spectra and Stellenbosch University for conducting the HRMS. Furthermore, Clinton Veale (UKZN) and Marietjie Stander (Stellenbosch University) are also acknowledged for the pivotal role they played in acquiring the HRMS data.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph15050576/s1, Figures S1–S74: 1H, 13C NMR and HRMS for 6a–y.
Author Contributions
Conceptualization, M.T.; methodology, R.M., C.M., N.B.N., A.J. and R.K.; software, M.T.; validation, M.T., M.V.M., G.H.R., R.K. and D.F.W.; formal analysis, M.T. and M.V.M.; investigation, M.T., R.K. and D.F.W.; resources, M.T. and M.O.B.; writing—original draft preparation, R.M. and C.M.; writing—review and editing, M.T., M.V.M., M.O.B. and R.K.; supervision, M.T., M.V.M., M.O.B. and D.F.W.; project administration, M.T.; funding acquisition, M.T. and M.O.B. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Material.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
The South African National Research Foundation (NRF)’s Competitive Support for unrated research grants (CSUR: 116282 (MT) and 114369 (MOB)), UKZN, TUT, and CSIR are gratefully acknowledged for financial and other valuable support. In addition, the NRF is further acknowledged for the grant holder-linked bursaries to RM, CM, and NBN.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Osman M., Meehan S.-A., von Delft A., Preez K.D., Dunbar R., Marx F.M., Boulle A., Welte A., Naidoo P., Hesseling A.C. Early mortality in tuberculosis patients initially lost to follow up following diagnostic in provincial hospitals and primary health facilities in Western Cape, South Africa. PLoS ONE. 2021;16:e0252084. doi: 10.1371/journal.pone.0252084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Global Tuberculosis Report 2020. Geneva, World Health Organization, 2020, Licence: CC BY-NC-SA IGO. [(accessed on 14 February 2022)]. Available online: htpps://www.who.int/publication/i/item/9789240013131.
- 3.Eshetie S., Gizachew M., Dagnew M., Kumera G., Woldie H., Ambaw F., Tesema B., Moges F. Multidrug resistant tuberculosis in Ethiopian settings and its association with previous history of antituberculosis treatment: A systematic review and meta-analysis. BMB Infect. Dis. 2017;17:219. doi: 10.1186/s12879-017-2323-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matteeli A., Roggi A., Carvhalo A.C.C. Extensive drug-resitant tuberculosis: Epidemiology and management. Clin. Epidemiol. 2014;6:111–118. doi: 10.2147/CLEP.S35839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conradie F., Diacon A.H., Ngubane N., Howell P., Everitt D., Crook A.M., Mendel C.M., Egizi E., Moreira J., Timm J., et al. Treatment of highly drug-resitsat pulmonary tiberculosis. N. Engl. J. Med. 2020;382:893–902. doi: 10.1056/NEJMoa1901814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batt S.M., Minnikin D.E., Besra G.S. The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host’s immune system. Biochem. J. 2020;477:1983–2006. doi: 10.1042/BCJ20200194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batt S.M., Burke C.E., Moorey A.R., Besra G.S. Antibiotic and resistance: The two sided coin of the mycobacterial cell wall. Cell Surf. 2020;6:100044. doi: 10.1016/j.tcsw.2020.100044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paomino J.C., Martin A. Drug resistance mechanism in mycobacterium tuberculosis. Antibiotics. 2014;3:317–340. doi: 10.3390/antibiotics3030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith T., Wolff K.A., Nguyen L. Molecular biology of rug resistance in mycobacterium tuberculosis. Curr. Top. Microbiol. Immunol. 2013;374:53–80. doi: 10.1007/82_2012_279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morlock G.P., Plikaytis B.B., Crawford J.T. Charaxterization of spontaneous in vitro selected rifampin-resistant mutantas of mycobacterium tuberculosis strain H37Rv. Antimicrob. Agents Chemother. 2000;44:3298–3302. doi: 10.1128/AAC.44.12.3298-3301.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaw M.T., Emran N.A., Lin Z. Mutations inside rifmpcicin-resistance determinig region of rpoB gene associated with rifampicin-resistance in mycobactrium tuberculosis. J. Infect. Public Health. 2018;11:605–610. doi: 10.1016/j.jiph.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Soni I., de Groote M.A., Dasgupta A., Chopra S. Challenges facing the drug discovery pipeline for non-tuberculous mycobacteria. J. Med. Microbiol. 2016;65:1–8. doi: 10.1099/jmm.0.000198. [DOI] [PubMed] [Google Scholar]
- 13.Libardo M.D.J., Boshoff H.I., Barry C.E., III The present state of the tubeculosis drug development pipeline. Curr. Opin. Pharmacol. 2018;42:81–94. doi: 10.1016/j.coph.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buhuguna A., Rawat D.S. An overview of new antitubercular drugs, drug candidayes, and their targets. Med. Res. Rev. 2020;40:263–292. doi: 10.1002/med.21602. [DOI] [PubMed] [Google Scholar]
- 15.Tahlan K., Wilson R., Kastrinsky D.B., Arora K., Nair V., Fischer E., Barnes S.W., Walker J.R., Alland D., Barry C.E., II, et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycloic acid donation to the cell wall core of mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012;56:1798–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shetye G.S., Franzblau S.G., Cho S. New tuberculosis drugs targets, their inhibitors, and potentail therapeutic impact. Transl. Res. 2020;220:68–97. doi: 10.1016/j.trsl.2020.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Li W., Upadhyay A., Fontes F.L., North E.J., Wang Y., Crans D.C., Grzegorzerics A.E., Jones V., Franzblau S.G., Lee R.E., et al. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharamrcophoes in mycobactrium tuberculosis. Antimicrob. Agents Chemother. 2014;58:6413–6423. doi: 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bukhdruker S., Varaksa T., Grabovec I., Martin E., Shabunya P., Kadukova M., Grudinin S., Kavaleuski A., Gusach A., Gilep A., et al. Hydroxylation of antitubecular candidate, SQ109, by mycobacterial cytochrome P450. Int. J. Mol. Sci. 2020;21:7683. doi: 10.3390/ijms21207683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petkova Z., Valcheva V., Momekov G., Petrov P., Dimitrov V., Doytchinova I., Stavrakov G., Stoyanova M. Antimycobacterial activity of chiral aminoalcohols with camphane scaffold. Eur. J. Med. Chem. 2014;81:150–157. doi: 10.1016/j.ejmech.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Arnott J.A., Planey S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Disc. 2012;7:909–921. doi: 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- 21.Lobo S. Is there enough focus on lipophilicity in drug discovery. Expert. Opin. Drug Disc. 2020;15:261–263. doi: 10.1080/17460441.2020.1691995. [DOI] [PubMed] [Google Scholar]
- 22.Machado D., Azzali E., Couto I., Costantino G., Pieroni M., Viveiros M. Adjuvant therapies against tuberculosis: Discovery of a 2-aminothiazole targeting mycobacterium tuberculosis energetics. Future Microbiol. 2018;13:1383–1402. doi: 10.2217/fmb-2018-0110. [DOI] [PubMed] [Google Scholar]
- 23.Angula K.T., Legoabe L.J., Beteck R.M. Chemical classes presenting novel antituberculosis agents in different phases of drug development: A 2010–2020 review. Pharmaceuticals. 2021;14:461. doi: 10.3390/ph14050461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Field S.K. Bedaquiline for treatment of multidrug -resistant tuberculosis: Great promise or disappointment? Ther. Adv. Chronic. Dis. 2015;6:170–184. doi: 10.1177/2040622315582325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koul A., Dendouga N., Vergauwen K., Molenberghs B., Vranckx L., Willebrordz R., Ristic Z., Lill H., Dorange I., Guillemont J., et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007;3:323–324. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 26.Hoagland D., Liu J., Lee R.B., Lee R.E. New agents for the treatment of drug-resistant mycobacterium tuberculosis. Adv. Drg. Deliv. Res. 2016;102:55–72. doi: 10.1016/j.addr.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blaser A., Sutherland H.S., Tong A.S., Choi P.J., Conole D., Franzblau S.G., Cooper C.B., Upton A.M., Lotlikar M., Denny W.A., et al. Structure-activity relationships for unit C pyridyl analogues of the tuberculosis drug bedaquiline. Bioorg. Med. Chem. 2019;37:1283–1291. doi: 10.1016/j.bmc.2019.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karmakar M., Rodrigues C.H., Holt K.E., Dunstan S.J., Denholm J., Ascher D.B. Empirical ways to identify novel bedaquiline resistance mutations in AtpE. PLoS ONE. 2019;14:e0217169. doi: 10.1371/journal.pone.0217169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tukulula M., Sharma R.-K., Meurillon M., Mahajan A., Naran K., Warner D., Huang J., Mekonnen B., Chibale K. Synthesis and antiplasmodial and antimycobacterial evaluation of new nitroimidazole and nitroimidazooxazine derivatives. ACS Med. Chem. Lett. 2013;4:128–131. doi: 10.1021/ml300362a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tukulula M., Little S., Gut J., Rosenthal P.J., Wan B., Franzblau S.G., Chibale K. The design, synthesis, in silico ADME profiling, antiplasmodial and antimycobacterial evaluation of new arylaminoquinoline derivatives. Eur. J. Med. Chem. 2012;57:259–267. doi: 10.1016/j.ejmech.2012.08.047. [DOI] [PubMed] [Google Scholar]
- 31.Luo B., Li D., Zhang A.L., Gao J.-M. Synthesis, antifungal activities and molecular docking studies of benzoxazole and benzothiazole derivatives. Molecules. 2018;23:2457. doi: 10.3390/molecules23102457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heroun M., Tratrat C., Kositsi K., Tsolaki E., Petrou A., Aldhubiab B., Attimarad M., Harsha S., Geronikaki A., Venugopala K.N., et al. New benzothiazole-based thiazolidinones as potent antimicrobial agaents. Design, synthesis and biological evaluation. Curr. Top. Med. Chem. 2018;18:75–87. doi: 10.2174/1568026618666180206101814. [DOI] [PubMed] [Google Scholar]
- 33.Singh M., Singh S.K. Benzothiazoles: How relevant in cancer drug design strategy? Anticancer. Agents Med. Chem. 2014;14:127–146. doi: 10.2174/18715206113139990312. [DOI] [PubMed] [Google Scholar]
- 34.Eshkil F., Eshghi H., Saljooghi A.S., Bakavoli M., Rahimizadeh M. Benzothiazolesole thiourea derivatives as anticancer agents: Design, synthesis and biological screening. Russ. J. Bioorg. Chem. 2017;43:576–582. doi: 10.1134/S1068162017050065. [DOI] [Google Scholar]
- 35.Venugopala K.N., Khedr M.A., Pillau M., Nayak S.K., Chandrashekharappa S., Aldhubiab B.E., Harsha S., Attimard M., Odhav B. Benzothiazole analogs as potential anti-TB agents: Computation input and molecular dynamics. J. Biomol. Struct. Dyn. 2019;37:1830–1842. doi: 10.1080/07391102.2018.1470035. [DOI] [PubMed] [Google Scholar]
- 36.Bhat M., Shiddappa B.L. Synthesis, in vitro and in silico studies of benzothiazole azo-ester derivatives as anti-TB agents. Anti-Infect. Agents. 2020;18:15–23. doi: 10.2174/2211352517666190126160534. [DOI] [Google Scholar]
- 37.Netalkar P.P., Netalkar S.P., Budagumpi S., Revankar V.K. Synthesis, crystal structures and characterization of late first row transition metal complexes derived from benzothiazole core: Antitubercular activity and special emphasis on DNA binding and cleavage property. Eur. J. Med. Chem. 2014;22:47–56. doi: 10.1016/j.ejmech.2014.03.083. [DOI] [PubMed] [Google Scholar]
- 38.Cho Y., Loerger T.R., Sacchettini J.C. Discovery of novel nitrobenzothiazole inhibitors for mycobacterium tuberculosis ATP Phorsphoribosyly transferase (HisG) through virtual screening. J. Med. Chem. 2008;51:5984–5992. doi: 10.1021/jm800328v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pellet A. Nitrobenzothiazole Derivatives, Preparation Thereof and Therapeutic Applications Thereof. No. 8993561B2. U.S. Patent. 2015 March 31;
- 40.Sharma P.C., Sinhmar A., Sharma A., Rajak H., Pathak D.P. Medicinal significance of benzothiazole scaffold: An insight view. J. Enzyme Inhib. Med. Chem. 2013;28:240–266. doi: 10.3109/14756366.2012.720572. [DOI] [PubMed] [Google Scholar]
- 41.Chikhale R., Menghani S., Babu R., Bansode R., Bhargavi G., Karodia N., Rajasekharan M.V., Paradkar A., Khedekar P. Development of selectibe DprE1 inhibitors: Design, synthesi, crystal structure and antitubercular activity of benzothiazoylpyrimidine-5-carbaxamides. Eur. J. Med. Chem. 2015;96:30–46. doi: 10.1016/j.ejmech.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 42.Venugopala K.N., Chandrashekharappa S., Pillay M., Bhandary S., Kandeel M., Mahommodally F.M., Morsy M.A., Chopra D., Aldubiab B.E., Attimarad M., et al. Synthesis and structural elucidation of novel benzothiazole derivatives as antitubercular agents: In silico screening for possible target identification. Med. Chem. 2019;15:311–326. doi: 10.2174/1573406414666180703121815. [DOI] [PubMed] [Google Scholar]
- 43.Landge S., Mullick A.B., Nagalapur K., Neres J., Subbulakshmi V., Murugan K., Ghosh A., Sadler C., Fellows M.D., Humnabadkar V., et al. Discover of bezothiazole antimycobacterial agents: Synthesis, structure-activity relationships and binding studies with mycobacterial tuberculosis decarprenylphosphoryl-β-D-ribose 2′-oxidase. Bioorg. Med. Chem. 2015;15:7694–7710. doi: 10.1016/j.bmc.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 44.Hamdy A.-M., Morsy M.A. Novel benzothiazolyl urea and thiourea derivatives with potential cytotoxicity and antimicrobial activities. J. Enzyme Inhib. Med. Chem. 2007;22:57–64. doi: 10.1080/14756360600991082. [DOI] [PubMed] [Google Scholar]
- 45.Ndjoubi K.O., Sharma E., Badmus J.A., Jacobs A., Jordaan A., Marnewick J., Warner D.F., Hussein A.A. Antimycobacterial, cytotoxic, and antioxidant activities of abietane diterpenoids isolated from Plectranthus madagascariensis. Plant. 2021;10:175. doi: 10.3390/plants10010175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perkin Elmer Chemdraw Professional V18.0.0.231. [(accessed on 2 March 2022)]. Available online: https://perkinelmerinformatics.com/products/research/chemdraw/
- 47.van Loveren C. Antimicrobial activity of flouride and its in vivo importance: Identification of research question. Caries Res. 2001;35:65–79. doi: 10.1159/000049114. [DOI] [PubMed] [Google Scholar]
- 48.Mossman J. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 49.Standard I. Biological Evaluation of Medical Devices. Tests for In Vitro Cytotoxicity. ISO/TC; Geneva, Switzerland: 2009. p. 194. [Google Scholar]
- 50.Schrödinger Release 2021-2: QikProp. Schrödinger, LLC; New York, NY, USA: 2021. [(accessed on 21 September 2021)]. Available online: https://www.schrodinger.com/citations. [Google Scholar]
- 51.Prentis R.A., Lis Y., Walker S.R. Pharmaceutical innovations by the seven UK-owned pharmaceutical companies (1964–1985) Br. J. Clin. Pharmacol. 1988;25:387–396. doi: 10.1111/j.1365-2125.1988.tb03318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bocci G., Carosati E., Vayer P., Arrault A., Lozano S., Cruciani G. ADME properties: A new tool for medicinal chemistry to explore ADME properties. Sci. Rep. 2017;7:6359. doi: 10.1038/s41598-017-06692-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacology. Toxicol. Methods. 2000;44:235–249. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 54.Abrahams G.L., Kumar A., Savvi S., Hung A.W., Wen S., Abell C., Barry C.E., 3rd, Sherman D.R., Boshoff H.I., Mizrahi V.V. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 2012;19:844–854. doi: 10.1016/j.chembiol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Collins L.A., Torrero M.N., Franzblau S.G. Green fluorescent protein reporter microplate assay for high-throughput screening of compounds against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1998;42:344–347. doi: 10.1128/AAC.42.2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collins L., Franzblau S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997;41:1004–1009. doi: 10.1128/AAC.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Affolabi D., Odoun M., Sanoussi N.D., Martin A., Palomino J.C., Kestens L., Anagonou S., Portaels F. Rapid and inexpensive detection of multidrug-resistant Mycobacterium tuberculosis with the nitrate reductase assay using liquid medium and direct application to sputum samples. J. Clin. Micriobiol. 2008;46:3243–3245. doi: 10.1128/JCM.00083-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bi J., Wang Y., Yu H., Qian X., Wang H., Liu J., Zhang X. Modulation of central carbon metabolism by acetylation of isocitrate lyase in Mycobacterium tuberculosis. Sci. Rep. 2017;7:44826. doi: 10.1038/srep44826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Vos J.J., Rutter K., Schroeder B.G., Su H., Zhu Y., Barry C.E., 3rd The salicylate-derived mycobactin siderophores of mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.SFranzblau G., DeGroote M.A., Cho S.H., Andries K., Nuermberger E., Orme I.M., Mdluli K., Angulo-Barturen I., Dick T., Dartois V., et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis. 2012;92:453–488. doi: 10.1016/j.tube.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 61.Ollinger J., Bailey M.A., Moraski G.C., Casey A., Florio S., Alling T., Miller M.J., Parish T. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS ONE. 2013;4:e60531. doi: 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Donato M.T., Tolosa L., Gómez-Lechón M.J. Culture and functional characterization of human hepatoma HepG2 cells. Method Mol. Biol. 2015;1250:77–93. doi: 10.1007/978-1-4939-2074-7_5. [DOI] [PubMed] [Google Scholar]
- 63.Kehinde I., Khan R., Nlooto M., Gordon M. Modulatory influences of antiviral bioactive compounds on cell viability, m RNA and protein expression of cytochrome P450 3A4 and P-glycoprotein in HepG2 and HEK293 cells. Biorg. Chem. 2021;107:104573. doi: 10.1016/j.bioorg.2020.104573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is contained within the article and Supplementary Material.