

Abstract
This review aims to cover the topic of polycarbonate synthesis via ring-opening polymerization (ROP) of cyclic carbonates. We report a wide variety of ROP-initiating systems along with their detailed mechanisms. We focus on the challenges of preparing the polymers; the precise control of the properties of the materials, including molecular weight; the compositions of the copolymers and their structural characteristics. There is no one approach that works for all scales in cyclic carbonates ROP. A green process to produce polycarbonates is a luring challenge in terms of CO2 utilization and the targeted domains for application. The main resolution seems to be the use of controlled incorporation of functional/reactive groups into polymer chains that can tailor the physicochemical and biological properties of the polymer matrices, producing what appears to be an unlimited field of applications. Glycerol carbonate (GC) is prepared from renewable glycerol and considered as a CO2 fixation agent resulting in GC compound. This family of five-membered cyclic carbonates has attracted the attention of researchers as potential monomers for the synthesis of polycarbonates (PCs). This cyclic carbonate group presents a strong alternative to Bisphenol A (BPA), which is used mainly as a monomer for the production of polycarbonate and a precursor of epoxy resins. As of December 2016, BPA is listed as a substance of very high concern (SVHC) under the REACH regulation. In 2006, Mouloungui et al. reported the synthesis and oligomerization of GCs. The importance of GCs goes beyond their carbonate ring and their physical properties (high boiling point, high flash point, low volatility, high electrical conductivity) because they also contain a hydroxyl group. The latter offers the possibility of producing oligo and/or polycarbonate compounds that have hydroxyl groups that can potentially lead to different reaction mechanisms and the production of new classes of polycarbonates with a wide range of applications.
Keywords: six- and five-membered cyclic carbonates, glycerol, ring-opening polymerization mechanism, ROP catalysis, green chemistry
1. Introduction
Polycarbonates (PCs) are polymers with carbonate groups (-O-CO-O-) in their main chain. They have been classified as biodegradable [1] and biocompatible polymers [2], and they can be produced from renewable substances [3]. Thus, their use offers the possibility of developing sustainable technologies that are economically and ecologically attractive [4]. To date, polyesters have held the leading status in the field of polymers. However, PCs provide polydiester functionality, and their lack of acidic compounds that are related to biodegradation gives them a significant advantage over polyesters. In addition, the degradation rate of PCs is slower than that of polyesters. Due to all of these promising advantages, PCs have attracted great interest [5] concerning their potential application in many areas.
PCs have attracted the most attention over the past two decades for their significant potential in environmental and biomedical applications [6] due to their biocompatibility, biodegradability, bioresorbable activity [7], and the weak inflammatory properties of the degraded products [8,9]. There are several important biomedical uses of biodegradable polymers, some of which are listed below:
Packaging is potentially the largest use of biodegradable polymers [10]. Biodegradable polymeric materials and blends that have passed toxicological and cytocompatibility testing for in vivo use have obvious benefits as biodegradable materials for pharmaceutical products, drugs, dressings for wounds, and surgical fixation (sutures, clips, bone pins, and plates). Biodegradable implants can be used as alternatives to metal implants for the fixation of fractured bones and joints [11].
Controlled drug delivery may be the most important and versatile application of these polymers [12]. It has applications in the medical, veterinary, and agrochemical fields. Active ingredients, ranging from pesticides to contraceptives, can be delivered by sustained release followed by the ultimate biodegradation of the carrier medium.
Moreover, there are applications for PCs in the automobile industry as well as in lighting fixtures, textiles, microelectronic components, data storage, and construction-materials areas [13]. They also are used in the plastics industry because of their high impact resistance and thermal stability, due in part to the work of Nagahata et al. [14,15], by the solid-state, ring-opening polymerization of macrocyclic carbonates, which resulted in ultrahigh-molecular-weight bisphenol A-based polycarbonate (BPA-based polycarbonate) (MW > 2000 KDa). These polycarbonates are fabricated from BPA, and they have been in commercial use by Bayer and General Electric since 1958 [16]. However, because their biodegradation liberates the BPA, the World Health Organization (WHO) and Food and Drug Administration (FDA) [17] have expressed some concerns about the potential effects of BPA on the brain, human behavior, and the prostate gland in fetuses, infants, and young children. Therefore, FDA has declared that it supports the industry’s actions to stop producing BPA-containing baby bottles and infant feeding cups, baby formula packages, whereas the European Chemicals Agency (ECHA) has banned the use of BPA in thermal paper starting from January 2020.
Glycerol is one of the top 10 building blocks driven from biomass. Glycerol carbonate presents a multifunctional derivative of glycerol with a wide range of interesting properties, hence areas of applications. GC can be produced by integrating CO2 into glycerol, opening a new opportunity for carbon capture and utilization. Another point that makes glycerol carbonate more attractive is that some of its properties are placed between those of the cyclic alkylene carbonates (ethylene and propylene carbonate) and those of glycerol. Glycerol carbonate has a high boiling point (351 C), a low melting point (66.7 C), and a high dipole moment (l = 5.4 D) and a high dielectric constant value (e = 109.7) relative to many other organic compounds. Glycerol carbonate is also nontoxic and can be used as a polymer stabilizer and as a synthetic intermediate in organic reactions.
Additionally, at the polycarbonate-production level, polycondensation of cyclic carbonate has several challenging aspects, including poor control over the molecular parameters, limitation of molecular weights, and the use of toxic phosgene and pyridine [18]. The process also involves the phosgenation of hydroxyl compounds with KOH or pyridine as a catalyst or the transesterification of diols with lower dialkyl carbonates in the presence of a basic catalyst.
Considering the importance of greener routes for polycarbonates synthesis and the gravity of some related hazards, this review is focusing on the chemistry of the ring-opening polymerization of cyclic carbonates, presenting the advancements and the setbacks found in the literature. Overviewing the existing initiating systems with a deep comprehension of the associated mechanism shall incite the exploration of novel pathways in the field of polycarbonate industry.
2. Synthesis of Polycarbonates and ROP
Polycarbonates are divided into aliphatic polycarbonates (APCs) and aromatic polycarbonates (ArPCs), and the APCs are generally synthesized by either polycondensation or ring-opening polymerization. Polycondensation is performed by using activated carbonate derivatives with diphenols or diols [19] via a step-growth mechanism. Ring-opening polymerization (ROP) of cyclic carbonates proceeds in the presence of a catalyst and/or an initiator via a chain-growth mechanism that includes chain initiation and chain propagation; in the ideal case, there is no termination reaction. ROP offers the possibility of controlling the molecular weight, the distribution of molecular weights, the microstructure of the polymer, and the nature of its end groups. The importance of ROP has led us to explore all the ROPs associated with 5- and 6-membered cyclic carbonates for the preparation of polycarbonates.
The present review outlines the different synthesis pathways of polycarbonates, with the goal of investigating green synthesis routes for producing polycarbonates from bioresourced monomers. We focused on 5- and 6-membered cyclic carbonates as the “case study” monomers; the mechanisms of anionic, coordination-insertion, and cationic polymerization, and we presented the corresponding initiating systems.
2.1. Kinetic and Thermodynamic Aspects of Cyclic Carbonates Ring-Opening Polymerization
The ROP of cyclic carbonates depends on both kinetic and thermodynamic factors, and it also depends on the reaction mechanisms associated with different catalyst/initiator systems.
The ROP mechanism of cyclic carbonates includes two steps [20], i.e., (1) initiation by means of ionic- (anionic or cationic) and insertion-type initiators and (2) regulation of the chain-growth mechanism by the equilibrium between kinetically controlled products and thermodynamically controlled products (Figure 1).
Figure 1.

ROP of 2,2-dimethyltrimethylene carbonate with a nucleophilic initiator [20].
In a spontaneous ROP, the Gibbs free energy must be negative (ΔGp < 0), i.e., the enthalpy of ring-opening polymerization, ΔHp, must be negative (Equation (1)), where ΔS is the polymerization entropy.
| (1) |
2.1.1. Kinetic Control
In the regime of kinetic control, the chain propagation reaction is predominant, and a high-molecular-weight polymer (Pn) is formed via Equation (2).
| (2) |
The polymerization conditions, such as solvent, temperature, concentration of the monomer, and especially, the nature of the active site must be adjusted to enhance the kinetically controlled regime of the reaction (where the polymer is formed) over the thermodynamically controlled regime (where a ring-chain equilibrium and the most probable chain-length distribution is established by means of transesterification reactions). For example, with Li+ and K+ as counter ions, the polymerization rates are much greater than the rates for Al3+ and Zn2+ in the active site [21].
Olsen et al. [22] have recently developed a switchable ROP of functional 6-member cyclic carbonate and ε-caprolactone monomers (i.e., AOMEC and εCL) catalyzed by TBD (1,5,7-triazabicyclo[4.4.0]-dec-5-ene). The synthesized nine-block copolymer was obtained in only 2.5 h. The switchable property was achieved by alteration of temperature during polymerization from +30 to −40 °C. A monomer-specific kinetic response arises for polymerization of εCL and retardation of the AOMEC polymerization.
2.1.2. Thermodynamic Control
In the regime of thermodynamic control, back-biting reactions (loss of CO2 from carbonate to give an ether linkage) occur to form a low-molecular-weight fraction of cyclic oligomers (Mx, Figure 1).
The monomer-polymer equilibrium is established in the kinetically controlled regime (Equation (3)):
| (3) |
This equilibrium is characterized by equilibrium constant K (Equation (4)):
| (4) |
K is approximately equal to the inverse of the equilibrium concentration of the monomer ([M]e). The relationship between the equilibrium concentration of the monomer and the free enthalpy of polymerization is presented by Equation (5):
| (5) |
For standard conditions and above a critical temperature, no polymer is obtained. The critical temperature is defined as the ceiling temperature (Tc = ΔH/ΔS) when both ΔH° and ΔS° are negative.
In parallel, another equilibrium is established in a thermodynamically controlled regime. Ring-chain equilibrium (Equation (6)) is attained in function of Kx, where Kx (Kx = [Pn−x][Mx]/[Pn] ≈ [Mx]e) is the equilibrium constant that determines the concentration of the cyclic oligomer with degree of polymerization x.
| (6) |
The equilibrium constant of an oligomer (Kx) (Kx = [Pn−x][Mx]/[Pn] ≈ [Mx]e) is approximately equal to the equilibrium concentration of the oligomer ([Mx]e), and Kx is proportional to the degree of polymerization to the −2.5 power [23,24].
In the thermodynamically controlled regime, further intermolecular transesterification reaction occurs, and the selectivity of the reaction and the microstructure of the polymer (in the case of copolymerization) depend on the ratios Kp/Kb and Kp/Kte.
Kp: rate constant of propagation
Kb: rate constant of back-biting
Kte: rate constant of transesterification
These rate constants [25] are determined by the monomer that is used and by the nature of the active sites. It is of significant importance to understand the kinetic and thermodynamic aspects of ROP, as well as its different mechanisms.
2.2. Mechanistic Aspects
2.2.1. Anionic Polymerization
The anionic ROP behavior of the cyclic carbonates depends on the size of their rings. Six- or seven-membered cycles tend to polymerize smoothly to yield the corresponding polycarbonates at lower temperatures (<100 °C) by anionic initiators. In contrast, the anionic ROP of the 5-membered ring is thermodynamically unfavorable and proceeds at higher temperatures (>150 °C, ethylene carbonate), causing elimination of carbon dioxide to give a copolymer that consists of both carbonate and ether linkages [26]. Anionic polymerization has been investigated in various solvents, such as tetrahydrofuran (THF), dimethylformamide (DMF), and toluene. In addition, bulk polymerization has been reported and the presence of a solvent in addition to the temperature at which the polymerization occurs enhances kinetic products over thermodynamic products. Anionic initiation of polymerization consists of a nucleophilic attack (Figure 2) of the initiator at the carbon atom of the carbonyl of the carbonate moiety, followed by an acyl-oxygen cleavage. The formation of active species, such as the alkoxide anion (alcoholate), was demonstrated by means of 1H and 13C NMR [25].
Figure 2.

Opening polymerization of 2,2-dimethyltrimethylene carbonate via anionic initiation [20].
The nucleophilicity of the initiator and the electron affinity of the monomer control the efficiency of the initiation step. In the chain-growth reaction, the nucleophilic attack is repeated until the conversion of the monomer reaches a certain percentage.
2.2.2. Coordination-Insertion Mechanism
In the insertion-coordination mechanism, different types of initiators can be used to start polymerization. However, detailed studies of the mechanism for cyclic esters have been conducted [27]; the findings in this research [28,29] will be extended to the field of polycarbonates with Kricheldorf et al. [30] who reported the ring-opening polymerization of trimethylene carbonate (TMC) with the same initiator used with cyclic esters, i.e., the tin octanoate (Sn-oct2) in chlorobenzene. They proposed the following mechanism for the initiation and propagation of the chain of the polymer (Figure 3)
Figure 3.

Coordination-insertion mechanism [30].
In the step (1), they proposed a coordination and insertion of the TMC into the Sn-oct2 bond, followed by step (2), decarboxylation of the mixed anhydride that was formed, to produce an octanoate ester end group, while the chain growth continues as shown in step (3) via the normal coordination-insertion mechanism involving the Sn-alkoxide group. This mechanism was only proved for reactions that were conducted at temperatures of 140, 160, and 180 °C. The presence of a small amount of water or 1,3 diol as impurities with Sn-oct2 leads to a one-step, insertion-coordination mechanism instead of two steps (Figure 4), because water or alcohol is needed as a coinitiator for a sufficiently rapid polymerization that incorporates an alkyl (R in case of alcohol ROH) or a hydrogen atom (H in case of water) into the chain and forms carbonate end group (in the case of alcohol) [30]. This mechanism occurs at reaction temperatures that range between 100 and 120 °C.
Figure 4.

Coordination-insertion mechanism in the presence of alcohol [30].
Höcker et al. reported that a special initiator with an aluminum alcoholate group (Figure 5) is that tetraphenylporphyrin aluminum. This initiator also is known as the ‘Inoue catalyst’, and it is active for various monomers and is used for the preparation of block copolymers with one or two polycarbonate blocks [21]. Initiation occurs by the nucleophilic addition of alkoxide-aluminum-tetraphenylporphyrin (RO-Al-TPP) to the carbonyl group of the carbonate. The functional characteristics of this catalyst are of great interest because it is responsible for the initiation step of the ROP, and it also prohibits the backbiting by reintegrating the CO2 that is liberated during the ring opening back into the polymeric chain.
Figure 5.

Structure of alkoxide-aluminum-tetraphenylporphyrin [21].
2.2.3. Cationic Polymerization
Under cationic conditions, polymerization is accompanied by partial elimination of carbon dioxide, producing polycarbonates with an ether unit [31]; this mechanism of partial loss also was observed during the ring-opening polymerization of cyclic sulfite, during which sulfur dioxide was eliminated [32]. A cationic ROP of thiocarbonate with no loss was reported by Kameshima [33], and this led to additional investigation of the role of the catalyst. It was found that decarboxylation depends on the nature of the counter-ion present in the initiator [34]. Ariga et al. [35] proved that halide counter ions ensure cationic polymerization that suppresses the elimination of carbon dioxide. It is also important to control the reaction kinetics such that the initiation step is not faster than the propagation step; otherwise, the concentration of the monomer (TMC in Scheme 1) would not be high enough to ensure a sufficient nucleophilic attack on the activated monomers (1 or 2 in Scheme 1) [36].
Scheme 1.

Cationic polymerization pathways of a 6-membered cyclic carbonate [35].
In Scheme 1, two possible pathways are explained for the cationic polymerization of trimethylene carbonate initiated with methyl triflate (A+Y− = MeOTf):
Use of excess methyl triflate leads to formation of (1) where the counter-ion is covalently bonded to the initiated monomer, a nucleophilic attack on the covalent counter-ion adduct results in (3), and this attack gives an intermediate oxycarbonyl cation (4).
Formation of (2) via alkylation of the exocyclic oxygen atom of the carbonate linkage, generating a trioxocarbenium ion, which undergoes a nucleophilic attack from another monomer to give adduct (4).
Adduct (4) liberates carbon dioxide through the propagation steps to produce a polycarbonate (5) with ether units in its main chain. When alkyl halide was used as the initiator, a polycarbonate (Mn = 3000–5000) with a halide end group and no ether units was obtained.
2.2.4. Challenge of Decarboxylation (Back-Biting)
The inconvenience of ionic polymerization is the decarboxylation (loss of CO2). Usually this is due to side reaction competing with propagation reaction or simply due to high reaction temperature. The advantage of the insertion-coordination mechanism is the absence of backbiting even at high temperatures (up to 180 °C) [36].
The decarboxylation from the carbonate group in the polymer chain can be both inter- and intramolecular, since it is always provoked by the oxide anion found at the end of the chain or that belongs to another polymer chain. Ariga et al. [35] studied the decarboxylation during chain propagation. They calculated the enthalpy associated with the formation of intermediate cations and proved that decarboxylation from the monomer is thermodynamically and kinetically favorable over that from the linear carbonate or polymer. On the other hand, Lee et al. studied the polymerization of ethylene carbonate, and they proposed a mechanism [37] (Figure 6) for the degradation of the polymer via decarboxylation.
Figure 6.

Decarboxylation mechanism (backbiting) of a polycarbonate [37].
The oxide anion takes a hydrogen atom, and then an electron-pair rearrangement occurs that gives a vinyl moiety (detected by 1H NMR) and a hydroxyl moiety. The other part of the polymeric unity loses a CO2 molecule to give an oxide anion at the end of the chain, which, in turn, can form a low-molecular-weight fraction of cyclic oligomers or ether-linkage units in the chain of the polymer. In summary the decarboxylation is occurring on several levels: starting with monomer initiation, continuing along chain propagation, and during chain degradation.
2.3. Cyclic Carbonate Monomers
One of the most interesting aspects of cyclic carbonates as monomers for ROP is their volume expansion aspect, since volume shrinkage during the polymerization process is a fundamental problem in the field of material science.
Takata and Endo [24,38] reported that cyclic carbonates polymerize with a considerable expansion in volume from 1.1 to 7.7% in contrast to common monomers that undergo volume shrinkage upon polymerization (methylmethacrylate or propylene oxide, for example). This was related to the overall decrease in density, since the decrease in density produced by the loss of intermolecular interaction caused by polymerization is more important than the increase in density due to the conversion of the monomer during polymerization (Figure 7).
Figure 7.

Decrease in intermolecular interaction due to polymerization [24].
The most common commercially available five- and six-membered cyclic carbonate monomers polymerized via ring-opening polymerization are listed below (Figure 8):
Figure 8.

Structures of five- and six-membered cyclic carbonates.
The TMC and DTC polymerizes faster than EC and PC due to less ring strain and thermodynamic conflicts. However, ROP of these monomers varies along with the applied catalyst-initiator system, leading to different mechanisms and products.
3. Catalyst-Initiator Systems in ROP of Six-Membered Cyclic Carbonates
The polymerization of six-membered cyclic carbonate (Scheme 2) was first reported in the 1930s by Carothers et al., and it was obtained by heating trimethylene carbonate with a small amount of K2CO3, which was used as the initiator [39]. The molecular weights of the polycarbonates did not exceed 4 kilodalton (KDa). Later, other initiation systems, including organometallic and organic components, were investigated.
Scheme 2.

Trimethylene carbonate polymerization [40].
3.1. Organometallic Initiating Systems
Organometallic initiating systems are widely used in the technical production of polylactides, copolyesters of lactic acid, and the preparation of aliphatic polycarbonates. The high efficiency of these systems prompted several research groups to study the catalytic activity toward ring-opening polymerization of several monomers
3.1.1. Main Group Metals and Transition Metals
Kricheldorf et al. [30] were the first to report the ring-opening polymerization of TMC initiated with Sn-oct2. The use of this catalyst was also reported later by other researchers [41,42,43,44]. The temperature range of 100–120 °C seemed to have an important role for the initiation mechanism. At lower temperatures, a coinitiator of R-OH type was needed for sufficiently rapid polymerization. This coinitiator was incorporated into the chain and formed carbonate end groups. Both initiation and propagation followed the pattern of the normal coordination-insertion mechanism. At temperatures above 120 °C, the TMC can undergo direct insertion into the Sn-Oct bond, thus polyTMC chains that had one octanoate ester end group per chain were formed. Furthermore, the molecular weights (Mn) can be controlled via the monomer/initiator ratio, and high-molecular-weight polymers can be obtained with short time reactions at 160 °C.
Darensbourg et al. [40,45,46] reported the use of biocompatible metals (such as Mg, Ca, Fe, and Zn) with obvious advantages over the use of tin complexes [47]. Their work focused on Schiff base derivatives known as Salen complexes (Figure 9). Since M(II) salen complexes do not possess internal nucleophiles for the chain initiation step, the presence of an anion initiator (cocatalyst) derived from n-Bu4N+Cl− (tetrabutylammonium chloride) or PPN+N3− [µ-nitrido-bis(triphenylphosphine)] is necessary to form an effective catalyst system for the ring-opening polymerization of trimethylene carbonate (TMC).
Figure 9.
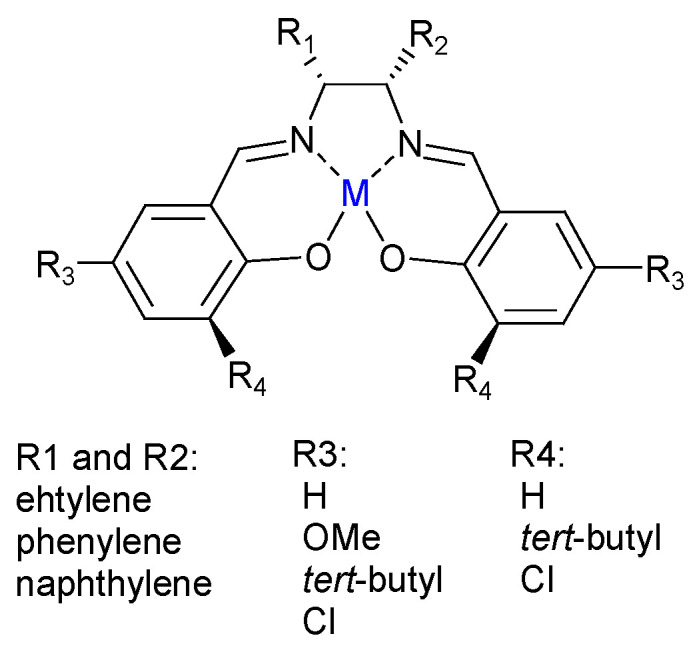
Structure of a metal-salen complex [40].
Their work demonstrated the catalytic activity of metals in the following order: Ca2+ > Mg2+ ≈ C2H5Al2+ > Zn2+, where the most effective catalytic system (with a TOF of 1286 h−1 at 86 °C) seemed to be Ca2+ complexed to a salen ligand with tert-butyl substituents in the 3,5-positions of the phenolate rings and an ethylene backbone for the diimine groups in addition to PPN+N3− as a cocatalyst. Poly(TMC) was obtained with molecular weights ranging from 30 to 63 KDa and a narrow distribution band of molecular weights (1.48–1.76) [40].
Dobrzinsky [48,49] reported TMC and DMC polymerization with the use of acetylacetonates of low-toxic metals, i.e., iron, zinc, and zirconium. Zinc (II) acetylacetonate, Zn (acac)2, was proven to be a very good initiator of homopolymerization. The reaction proceeded via an insertion-coordination mechanism, with which almost complete conversion of the monomer (TMC) was observed after five minutes, and the polymers had high molecular weights (98.2–195.1 KDa) with a narrow distribution band of molecular weights (1.6–1.7). The reaction was conducted at 110 °C for a period ranging between 0.2 and 1.5 h, with a monomer conversion of 98% [48]. In correlation to the undesired backbiting, the study of the homopolymerization of six-membered cyclic carbonates and the copolymerization with cyclic esters using Li, K, Mg, Al, Zn, and Sn based catalysts, led to the conclusion that systems with alkali metals, i.e., Mg and Sn, were found to produce cyclic oligomers during polymerization, i.e., the rate of backbiting was of the same order of magnitude as the rate of polymerization. On the other hand, Al- and Zn-based catalysts resulted in ROP without cyclic oligomers formation [50], so the backbiting reaction rate was slower than the chain-growth reaction. The unavoidable inconvenience for this group of catalyst-initiators is that intramolecular and intermolecular transesterification reactions take place along with the polymerization reaction.
3.1.2. Lanthanides
This group was studied thoroughly by Sheng et al. [51]. They worked with mixed-metal alkoxide clusters of lanthanide and sodium, such as [Ln2Na8(OCH2CH2NMe2)12(OH)2] and anionic lanthanide phenoxide complexes [52], such as [Ln(OAr)4][Na(DME)3]. These groups were found to be extremely active and presented a single-component initiator for the ring-opening polymerization of ε-caprolactone (ε-CL) and trimethylene carbonate. Polymerization proceeded through the cooperation of Ln and Na via a coordination-insertion mechanism to yield a high-molecular-weight polymer (23 × 104 Da for ε-CL and 6.71 × 104 Da for TMC) and a narrow distribution band of molecular weights (1.5–1.8 for poly(ε-caprolactone) and 1.6–2.2 for PTMC) taking into consideration that the reaction takes place at temperatures of 20 °C for ε-CL and 55 °C for TMC for short periods of three minutes for ε-CL and 30 min for TMC. Various research teams have studied and explored other lanthanide [53,54,55,56] complexes. The results shows a proven effectiveness for lanthanide complexes as promising initiators for the polymerization and copolymerization of TMC, DMC, and lactones [57,58,59]. The main issue facing the wide use of lanthanide complexes in ring-opening polymerization is the two required additional steps, i.e., (1) the preparation of these complexes since they are not commercially available; and (2) the purification process for the complexes [60,61].
3.1.3. Lewis Acids
Lewis acids based on metal groups 2, 3, 4, 12, and 13, such as magnesium, calcium, scandium, yttrium, cerium, samarium, ytterbium, zirconium, zinc, aluminum, and tin triflates or triflimidates, in addition to rare-earth metal complexes [62,63,64,65,66,67], are known to be effective catalysts in combination with a proton source, such as an alcohol or a carboxylic acid, for the ROP of cyclic esters, such as ε-caprolactone and lactide. In general, using several bicomponent systems based on metallo-organic and inorganic catalysts is efficient in ring-opening polymerization, yet at different temperatures and with different mechanism and/or productivities [68]. The use of organometallic initiating systems in ring-opening polymerization has the advantage of a controlled polymerization of cyclic carbonate [69]. However, it has the problem of removing the metallic residues from the final polymer, which is especially important when the target application is drug delivery.
3.2. Organic Initiating Systems
Organic initiating systems initiate polymerization under mild conditions, thereby offering the possibility of obtaining high-molecular-weight polycarbonate without toxic initiators, such as heavy metal ions. Thus, such systems permit the synthesis of biodegradable or biocompatible polymers without toxic impurities. These initiating systems were based on:
3.2.1. Brönsted Bases
The use of organic compounds such as amines [70] or guanidines [71] (Figure 10) in the copolymerization and immortal polymerization of cyclic carbonates was reported. Moreover, poly(ethylene glycol) [72,73] and natural amino acids also were used as catalysts for ROP of DTC and TMC [74]. The work of Nederberg [75] and Mindemark [76] represents a case study for the use of organic bases as catalysts for the ring-opening polymerization of TMC.
Figure 10.

Organic bases used as catalysts in ring-opening polymerization.
The polymers that were synthesized were isolated by precipitation; they had molecular weights up to 50 KDa and a polydispersity index (PDI) of 1.08. The presence or the absence of organocatalyst residues in the final product was not reported. Benzyl alcohol was used as the initiator in the reaction (Figure 11) to produce polymers with precise molecular weights and narrow distributions of molecular weights detected by gel-permeation chromatography (GPC). Moreover, the end groups were confirmed by 1H and 13C NMR.
Figure 11.

Benzyl alcohol initiator integrating with polymer structure.
In general, the catalytic activity of these base catalysts was exhibited towards the ROP of cyclic esters and carbonates. The guanidines, amidines, tertiary amines, N-heterocyclic carbenes, and thiourea tertiary amines proved similar activity in terms of monomer conversion percentage, but the highest DP, up to 420 KDa in the ring-opening polymerization of TMC, was only attained with the use of TBD [75]. This was due to the unique dual reception-donor functionality offered by the TBD catalyst; this dual function assures the activation of ROP without being directly inserted the mechanism of ROP through amide entity formation, as it is the case for the other amine-based catalysts [77].
The N-heterocyclic olefins (NHOs) are an emerging class of organopolymerization catalyst [78]. NHOs catalytic activity was investigated during the ROP of lactones and trimethylene carbonate (TMC). The reaction proceeds in the presence of Bn-OH at room temperature for a duration between 3.5 and 18 h; the conversion rates obtained did not exceed 76%, while the Mn of obtained polymers ranged between 5.8 kDa (for 3.5 h) and 7.2 kDa (for 18 h). Although the findings of these investigation places TBD as the first choice of catalyst, the findings related to the effect of polar moieties on the ring-opening polymerization could assist in developing tools for controlled polymerization.
3.2.2. Brönsted Acids
The organic initiating systems can also be based on strong Brönsted acids that allow electrophilic activation of the monomer. Triflic acid (TfOH), methanesulfonic acid (MSA), trifluoroacetic acid (TFA) [31,38,79], Diphenyl Phosphate (DPP) [80], and imidodiphosphoric acid (IDPA) [81] have been reported as catalysts for the ring-opening polymerization of TMC, where alcohol or water was used as the initiator [82].
Matsuo et al. [79] reported the polymerization of six- and seven-membered cyclic carbonates, with TFA as a catalyst to promote the nucleophilic attack of alcohols. Less sterically hindered alcohols showed higher reactivity towards ring-opening polymerization, which produced polycarbonates (Mn = 2500–6800 Da) with hydroxyl end groups. Endo et al. [31] reported that ester-substituted, six-membered cyclic carbonates polymerize faster than six-membered cyclic carbonates. They used triflic acid as an initiator at a reaction temperature of 20 °C for a long period (24 h). This produced a polycarbonate/ether polymer with a carbonate-to-ether linkage ratio of 7/3, molecular weights Mn = 1800–5000 Da, and a narrow distribution of molecular weights of Mw/Mn = 1.10–1.29. Furthermore, Delcroix et al. [82] reported the ring-opening polymerization of TMC catalyzed by triflic acid and methanesulfonic acid, using alcohol or water as initiators in stoichiometric amounts. The MSA was found to have similar polymerization kinetics to TfOH, although it is less acidic. Therefore, activity does not simply correlate with acidity. Moreover, MSA did not induce decarboxylation, and a polyTMC ether linkage-free polymer was produced with a molecular weight Mn = 2350–5800 Da and a narrow distribution of molecular weights of Mw/Mn = 1.08–1.17. The use of IDPA led to formation of star-shaped polycarbonates [81] after 34 h reaction at room temperature. Alternating the benzyl alcohol with other polyols did not have much effect on the molar mass of the polymer (Mn ≈ 5 kDa), while the narrowest distribution was obtained with BnOH (Mw/Mn =1.13).
3.3. Enzymes
It is noteworthy that enzymatic ring-opening polymerization has been studied extensively for cyclic esters with lipase as the enzymatic initiator [83,84,85,86]. It was reported that this approach is applicable for certain cyclic carbonates (Figure 12).
Figure 12.

Structure of cyclic carbonates applicable in enzymatic ring-opening polymerization.
An interesting result was obtained for the polymerization of DTC catalyzed with immobilized porcine pancreas lipase (IMPPL). When the polymerization proceeded at 120 °C for four days, poly(DTC) was obtained with a high molecular weight (15.7 KDa) and a narrow distribution band of molecular weights (PDI = 1.4) and the monomer conversion efficiency was 84.9% [87]. Better results were achieved using the second recycled IMPPL as catalyst, including higher molecular weight (26.2 KDa) and improved yield (97.6%) of polycarbonate.
4. Catalyst-Initiator Systems in ROP of Five-Membered Cyclic Carbonates
It is well known that five-membered alkylene carbonates undergo ROP with difficulty due to thermodynamic requirements [77]. Ethylene carbonate and propylene carbonate ROP have been studied extensively. Their polymerization occurs above 100 °C; it always results in poly (alkylene ether-carbonate) [88] polymer due to the loss of CO2 during the reaction. This decarboxylation affects the thermodynamic aspects of ROP by making the entropy positive, thus causing the polymerization to take place at high temperatures. Furthermore, the loss of CO2 during ROP depends on the mechanism imposed by the used initiating systems.
4.1. Main Group Metals and Transition Metals Initiating System
The reaction proceeds in the presence of metal alkoxide and metal acetylacetonates, as well as metal alkyls, as the catalyst. The optimized reaction temperature was found to be 170 °C for EC and 180 °C for PC. The homopolymerization for 5-membered cyclic carbonates was not possible since the polymerization is always accompanied by decarboxylation that leads to poly(alkylene ether-carbonate), and the loss of carbon dioxide during the polymerization exceeds 50 mol%; thus, the obtained copolymer content of carbonate units is less than 50 mol%.
The polymerization of five cyclic carbonates has been reported by several researchers. Among these is the significant work of Vogdanis et al. [88], on the ethylene carbonate monomer. They reported that the composition of poly(ether-carbonate) was strongly dependent on the catalyst that is chosen, and they used several catalysts, including dibutyltin dimethoxide, and butyllithium. They observed that CO2 retention decreased as the relative basicity of the catalyst increased.
The carbonate component of the product did not exceed 50 mol% in any experiment. Their explanation was that the polymerization occurred above the ceiling temperature (Tc) for the formation of pure poly(ethylene carbonate) and that propagation occurred via monomer insertion between the growing end of the polymer chain and a catalyst fragment (Scheme 3).
Scheme 3.

Propagation equilibrium during EC ring-opening polymerization [88].
Above Tc, equilibrium (1) must be shifted to the left since the sequence length of ethylene carbonate units cannot exceed one [89] Ethylene carbonate can be added when one carbon dioxide is lost. This equation shows the importance of the catalyst in CO2 retention (Equations (2) and (3), Scheme 4).
Scheme 4.

Propagation mechanism: Equation (2), loss of CO2; Equation (3), retention of CO2 [88].
A novel mechanistic vision was presented via NMR investigations during the reaction time in that the results indicated that there were two possible routes to react the monomer. In the first step, the growing polymer can attack the carbonyl carbon of the carbonate (Scheme 5).
Scheme 5.

Ring-opening polymerization via carbonyl carbon attack [37].
The other possible route would occur by attack at the alkylene carbon (Scheme 6). Kinetically, the carbonyl attack is favored over the alkylene attack. However, the carbonyl attack is reversible. Thermodynamically, the cyclic monomer is favored over the polymer, and the attack on the alkylene carbon is irreversible and accompanied with CO2 loss; therefore, the most probable mechanism includes both an alkylene carbon attack and a carbonyl carbon attack.
Scheme 6.

Ring-opening polymerization via alkylene carbon attack [37].
4.2. Brönsted Bases Initiating Systems
A Brönsted base catalyst was also used to accelerate the polymerization reaction, but a significant loss of CO2 (carbonate units) was detected. Two parameters could be affected by this loss, i.e., (1) the polymerization reaction becomes more favorable from a thermodynamic point of view because the enthalpy of the reaction decreases as CO2 is lost; and (2) the molar fraction of carbonate units within the polymeric chain decreases (0.22–0.17).
The polymerization of ethylene carbonate with basic initiators has rarely been studied due to the basicity effect on CO2 during polymerization, but Lee et al. [37] studied the polymerization of ethylene carbonate (EC) initiated by KOH at various temperatures (150–200 °C) and ratios of carbonate to initiator (1000:1 to 20:1) and discovered that the reaction can be described as a two-step process. In this manner, the final polymer is the result of initiation and propagation as well as chain cleavage. In the first step, the molecular weight of the polymer increases with reaction time to a maximum in the range of 1000–9000 Da, depending on the carbonate-to-initiator ratio that is used. Generally, the polymer has a carbonate-to-ether linkage ratio of 2 (i.e., 30–32% carbonate), which remains fairly constant until approximately 90–100% of the monomer has been converted. In the second step, the molecular weight and carbonate content of the polymer decrease significantly with continuous heating.
Furthermore, ethylene carbonate and propylene carbonate undergo polymerization in the presence of bispenol A/KHCO3 as an initiator/catalyst system, as described by Kéki et al. [90] and by Soos et al. [91]. The EC formed polyether oligomers, while the polycarbonate-ether oligomers resulted from PC polymerization with limited yield. When KHCO3 was replaced by an organic base, as described by Wu et al. [92], polyether diol was formed as a result of PC polymerization. Conversely, when tert-butylphenol was used with KHCO3 in the co-oligomerization of EC, PC, and ε-caprolactone, the result was co-oligomers with t-butylphenol and OH head groups with two types of co-oligomers, i.e., cyclic and linear oligomers with low carbonate linkage compared to the co-oligomers without carbonate linkage [93].
4.3. Dual Initiating Systems-Lewis Base-Lewis Acid
Lewis acid catalysts were never reported as a meaningful catalyst for EC polymerization. While Lewis base catalysts are not known to play an important role in accelerating the polymerization reaction of ethylene carbonate (reaction period between 72 and 96 h), but they were important for the retention of CO2, i.e., carbonate units, where the detected molar fraction of ethylene carbonate present in the polymeric chain was up to 40% [88].
Recently, a newly emerging approach employing dual catalytic polymerization catalysis was reported [94]. The catalytic activity towards EC polymerization of a combination of cooperatively acting Lewis bases ((including N-heterocyclic olefins, phosphazenes and nitrogen bases)) and Lewis acids (such as LiCl, MgF2, BEt3), has been proved. The polymerizations were conducted under microwave irradiation at T = 160–200 °C with low catalyst load ranges between 0.4–0.005 mol%, resulting polymers of molar masses up to 10,000 g/mol.
4.4. Glycerol Carbonate RO-Polymerization/Oligomerization
The ring-opening polymerization of glycerol carbonate has not been studied extensively. The GC monomer has so far been used to produce hyperbranched polyglycerol [95]. The polymerization of glycerol carbonate proceeds via anionic approach with simultaneous decarboxylation. A partially deprotonated trimethylolpropane (TMP) was used as an initiator. The product of this process is a hyperbranched polyether with pendant hydroxyl groups. The reaction occurs at 170 °C, over 12 h, with 10% (wt/wt) of initiator. The obtained oligomer attained an Mn up to 1000 Da.
A different approach with a focus on CO2 retention was studied and patented in the work of Mouloungui et al. [96]. The oligomerization of glycerol carbonate was achieved via a one-pot in situ approach. The starting reagents were glycerol, urea [3:2 molar ratio], and monohydrated zinc sulfate (1.3% wt/wt) was used as catalyst. The reaction proceeds at 140 °C for 3 h, and a pressure of 4 kPa. This step is meant to produce glycerol carbonate while eliminating the ammonia formed. The absence of ammonia flow would indicate the end of the reaction [97]. The reactional mixture includes the produced glycerol carbonate, excess glycerol, and the catalyst. Then, oligomerization reaction proceeds in situ by increasing the temperature to 160 °C for 150 min, at atmospheric pressure. This results in polyhydroxylated oligomers (Mw < 1000 Da) rich in linear carbonate groups.
In view of all the reported work of 5-membered cyclic carbonate ROP, the challenge remains controlling decarboxylation reaction that occurs in parallel with polymerization reaction. Attaining the highest level of carbonate retention is beneficial in terms of intensifying the desired properties of linear carbonates. Thus, it is of great interest to find a catalyst/initiator system that assures a maximum retention of CO2 units within the polymeric chain, since all of the previous work has reported the production of a heteropolymer that contains both carbonate and ether units. A promising answer may be found in the work of Inoue et al. [98,99,100] who reported the synthesis of aliphatic polycarbonate starting from an epoxide, while bubbling CO2 at 1 atm in the presence of aluminum tetraphenylporphyrine acetate as a catalyst at room temperature. The Inoue catalyst demonstrated a bifunctional activity that should be capable of initiating the ROP of- membered cyclic carbonate and reintegrating the released CO2 units into the polymeric chain.
5. Conclusions
An inclusive review for the initiating systems of ring-opening polymerization of cyclic carbonates has been reported. It is apparent that there is no ‘perfect’ system that is preferred for all applications. Significant differences exist between the polymerization of 5-membered and 6-membered cyclic carbonates. The challenge of having decarboxylation reaction along with the ROP could be avoided via the use of Inoue’s kind of catalyst, which can guarantee minimum decarboxylation at mild-pressure conditions. All the reported synthetic routes and results represent a source of inspiration for us; many applications that target the transport of biologically active substances through biological membranes require biosourced and biodegradable materials. One important parameter that has a key role in enhancing the mobility through biological membranes is the molar volume of the carrier (or the transporter of the active substance); the mobility of the carrier increases as its size decreases, and with this kind of linear relationship between molar volume and mobility, we should direct our focus towards development of new, oligomeric structures rather than polymeric structures. Seeking oligomers through ring-opening oligomerization would overcome the inconvenience of long reaction periods for 5-membered cyclic carbonate, and the glycerol carbonate monomers represent an attractive candidate that is compatible from both synthesis and application perspectives.
Acknowledgments
The authors gratefully acknowledge the American University of the Middle East for supporting the writing of this review paper. Many thanks are addressed to Nimer Murshid for his valuable help with the editorial work.
Author Contributions
Conceptualization, methodology, investigation, writing—original draft preparation, project administration, Z.A.B.; validation, writing—review and editing, Z.A.B., H.D. and T.S.; supervision, Z.A.B.; funding acquisition, Z.A.B., H.D. and T.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research received no external funding.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Artham T., Doble M. Biodegradation of Aliphatic and Aromatic Polycarbonates. Macromol. Biosci. 2008;8:14–24. doi: 10.1002/mabi.200700106. [DOI] [PubMed] [Google Scholar]
- 2.Scharfenberg M., Hilf J., Frey H. Functional Polycarbonates from Carbon Dioxide and Tailored Epoxide Monomers: Degradable Materials and Their Application Potential. Adv. Funct. Mater. 2018;28:1704302. doi: 10.1002/adfm.201704302. [DOI] [Google Scholar]
- 3.Amass W., Amass A., Tighe B. A Review of Biodegradable Polymers: Uses, Current Developments in the Synthesis and Characterization of Biodegradable Polyesters, Blends of Biodegradable Polymers and Recent Advances in Biodegradation Studies. Polym. Int. 1998;47:89–144. doi: 10.1002/(SICI)1097-0126(1998100)47:2<89::AID-PI86>3.0.CO;2-F. [DOI] [Google Scholar]
- 4.Dubois P., Coulembier O., Raquez J.-M. Handbook of Ring-Opening Polymerization. John Wiley & Sons; Hoboken, NJ, USA: 2009. [Google Scholar]
- 5.Taraghi I., Paszkiewicz S., Grebowicz J., Fereidoon A., Roslaniec Z. Nanocomposites of Polymeric Biomaterials Containing Carbonate Groups: An Overview. Macromol. Mater. Eng. 2017;302:1700042. doi: 10.1002/mame.201700042. [DOI] [Google Scholar]
- 6.Fukushima K. Poly(Trimethylene Carbonate)-Based Polymers Engineered for Biodegradable Functional Biomaterials. Biomater. Sci. 2016;4:9–24. doi: 10.1039/C5BM00123D. [DOI] [PubMed] [Google Scholar]
- 7.Han Y., Jin X., Yang J., Fan Z., Lu Z., Zhang Y., Li S. Totally Bioresorbable Composites Prepared from Poly(l-Lactide)-Co-(Trimethylene Carbonate) Copolymers and Poly(l-Lactide)-Co-(Glycolide) Fibers as Cardiovascular Stent Material. Polym. Eng. Sci. 2012;52:741–750. doi: 10.1002/pen.22137. [DOI] [Google Scholar]
- 8.Mohajeri S., Amsden B.G. In Vivo Degradation Mechanism and Biocompatibility of a Biodegradable Aliphatic Polycarbonate: Poly(Trimethylene Carbonate- Co -5-Hydroxy Trimethylene Carbonate) ACS Appl. Bio Mater. 2021;4:3686–3696. doi: 10.1021/acsabm.1c00160. [DOI] [PubMed] [Google Scholar]
- 9.Brannigan R.P., Dove A.P. Synthesis, Properties and Biomedical Applications of Hydrolytically Degradable Materials Based on Aliphatic Polyesters and Polycarbonates. Biomater. Sci. 2017;5:9–21. doi: 10.1039/C6BM00584E. [DOI] [PubMed] [Google Scholar]
- 10.Xu Y., Zhou F., Zhou D., Mo J., Hu H., Lin L., Wu J., Yu M., Zhang M., Chen H. Degradation Behaviors of Biodegradable Aliphatic Polyesters and Polycarbonates. J. Biobased Mater. Bioenergy. 2020;14:155–168. doi: 10.1166/jbmb.2020.1958. [DOI] [Google Scholar]
- 11.Ploypetchara N., Suppakul P., Atong D., Pechyen C. Blend of Polypropylene/Poly (Lactic Acid) for Medical Packaging Application: Physicochemical, Thermal, Mechanical, and Barrier Properties. Energy Procedia. 2014;56:201–210. doi: 10.1016/j.egypro.2014.07.150. [DOI] [Google Scholar]
- 12.Chen W., Meng F., Cheng R., Deng C., Feijen J., Zhong Z. Advanced Drug and Gene Delivery Systems Based on Functional Biodegradable Polycarbonates and Copolymers. J. Controlled Release. 2014;190:398–414. doi: 10.1016/j.jconrel.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Abts G., Eckel T., Wehrmann R. Polycarbonates. In: Wiley-VCH Verlag GmbH & Co. KGaA, editor. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2014. pp. 1–18. [Google Scholar]
- 14.Sugiyama J., Nagahata R., Goyal M., Asai M., Ueda M., Takeuchi K. Polymerization of Uniform Macrocyclic Carbonate Initiated by Neutral or Weak Basic Salt. Vol. 218. American Chemical Society; Washington, DC, USA: 1999. pp. U456–U457. [Google Scholar]
- 15.Nagahata R., Sugiyama J., Goyal M., Asai M., Ueda M., Takeuchi K. Synthesis of Ultra High-molecular-weight Polycarbonate. Polym. Adv. Technol. 2000;11:727–732. doi: 10.1002/1099-1581(200008/12)11:8/12<727::AID-PAT50>3.0.CO;2-J. [DOI] [Google Scholar]
- 16.Serini V. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH; Weinheim, Germany: 2000. Polycarbonates. [Google Scholar]
- 17.Shelnutt S., Kind J., Allaben W. Bisphenol A: Update on Newly Developed Data and How They Address NTP’s 2008 Finding of “Some Concern”. Food Chem. Toxicol. 2013;57:284–295. doi: 10.1016/j.fct.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 18.Suriano F., Coulembier O., Hedrick J.L., Dubois P. Functionalized Cyclic Carbonates: From Synthesis and Metal-Free Catalyzed Ring-Opening Polymerization to Applications. Polym. Chem. 2011;2:528–533. doi: 10.1039/C0PY00211A. [DOI] [Google Scholar]
- 19.Yokoe M., Aoi K., Okada M. Biodegradable Polymers Based on Renewable Resources. VII. Novel Random and Alternating Copolycarbonates from 1, 4: 3, 6-dianhydrohexitols and Aliphatic Diols. J. Polym. Sci. Part Polym. Chem. 2003;41:2312–2321. doi: 10.1002/pola.10772. [DOI] [Google Scholar]
- 20.Keul H. Polycarbonates. In: Dubois P., Coulembier O., Raquez J.-M., editors. Handbook of Ring-Opening Polymerization. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2009. pp. 307–327. [Google Scholar]
- 21.Hovestadt W., Keul H., Höcker H. Tetraphenylporphyrin-Aluminium Compounds as Initiators for the Ring-Opening Polymerization of 2, 2-Dimethyltrimethylene Carbonate: Synthesis of Homopolymers and Copolymers with ε-Caprolactone, Ethylene Oxide and Propylene Oxide. Polymer. 1992;33:1941–1948. doi: 10.1016/0032-3861(92)90497-K. [DOI] [Google Scholar]
- 22.Olsén P., Odelius K., Keul H., Albertsson A.-C. Macromolecular Design via an Organocatalytic, Monomer-Specific and Temperature-Dependent “On/Off Switch”. High Precision Synthesis of Polyester/Polycarbonate Multiblock Copolymers. Macromolecules. 2015;48:1703–1710. doi: 10.1021/acs.macromol.5b00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clements J.H. Reactive Applications of Cyclic Alkylene Carbonates. Ind. Eng. Chem. Res. 2003;42:663–674. doi: 10.1021/ie020678i. [DOI] [Google Scholar]
- 24.Takata T., Sanda F., Ariga T., Nemoto H., Endo T. Cyclic Carbonates, Novel Expandable Monomers on Polymerization. Macromol. Rapid Commun. 1997;18:461–469. doi: 10.1002/marc.1997.030180602. [DOI] [Google Scholar]
- 25.Keul H., Höcker H. Expected and Unexpected Reactions in Ring-opening (Co) Polymerization. Macromol. Rapid Commun. 2000;21:869–883. doi: 10.1002/1521-3927(20000801)21:13<869::AID-MARC869>3.0.CO;2-U. [DOI] [Google Scholar]
- 26.Haba O., Tomizuka H., Endo T. Anionic Ring-Opening Polymerization of Methyl 4, 6-O-Benzylidene-2, 3-O-Carbonyl-α-d-Glucopyranoside: A First Example of Anionic Ring-Opening Polymerization of Five-Membered Cyclic Carbonate without Elimination of CO2. Macromolecules. 2005;38:3562–3563. doi: 10.1021/ma0476745. [DOI] [Google Scholar]
- 27.Duda A., Kowalski A., Libiszowski J., Penczek S. Thermodynamic and Kinetic Polymerizability of Cyclic Esters. Macromol. Symp. 2005;224:71–84. doi: 10.1002/masy.200550607. [DOI] [Google Scholar]
- 28.Kowalski A., Duda A., Penczek S. Kinetics and Mechanism of Cyclic Esters Polymerization Initiated with Tin(II) Octoate. 3. Polymerization of l, l -Dilactide. Macromolecules. 2000;33:7359–7370. doi: 10.1021/ma000125o. [DOI] [Google Scholar]
- 29.Duda A., Penczek S. On the Difference of Reactivities of Various Aggregated Forms of Aluminium Triisopropoxide in Initiating Ring-Opening Polymerizations. Macromol. Rapid Commun. 1995;16:67–76. doi: 10.1002/marc.1995.030160112. [DOI] [Google Scholar]
- 30.Kricheldorf H.R., Stricker A. Polymers of Carbonic Acid, 28. SnOct2-Initiated Polymerizations of Trimethylene Carbonate (TMC, 1,3-Dioxanone-2) Macromol. Chem. Phys. 2000;201:2557–2565. doi: 10.1002/1521-3935(20001101)201:17<2557::AID-MACP2557>3.0.CO;2-F. [DOI] [Google Scholar]
- 31.Nemoto N., Sanda F., Endo T. Cationic Ring-Opening Polymerization of Six-Membered Cyclic Carbonates with Ester Groups. J. Polym. Sci. Part Polym. Chem. 2001;39:1305–1317. doi: 10.1002/pola.1108. [DOI] [Google Scholar]
- 32.Azuma N., Takata T., Sanda F., Endo T. Cationic Ring-Opening Polymerization Behavior of Six-Membered Cyclic Sulfite. Macromolecules. 1995;28:7331–7334. doi: 10.1021/ma00126a007. [DOI] [Google Scholar]
- 33.Kameshima H., Nemoto N., Sanda F., Endo T. Cationic Ring-Opening Polymerization of Five-Membered Cyclic Thiocarbonate Bearing an Adamantane Moiety via Selective Ring-Opening Direction. Macromolecules. 2002;35:5769–5773. doi: 10.1021/ma012094i. [DOI] [Google Scholar]
- 34.Rokicki G. Aliphatic Cyclic Carbonates and Spiroorthocarbonates as Monomers. Prog. Polym. Sci. 2000;25:259–342. doi: 10.1016/S0079-6700(00)00006-X. [DOI] [Google Scholar]
- 35.Ariga T., Takata T., Endo T. Cationic Ring-Opening Polymerization of Cyclic Carbonates with Alkyl Halides to Yield Polycarbonate without the Ether Unit by Suppression of Elimination of Carbon Dioxide. Macromolecules. 1997;30:737–744. doi: 10.1021/ma9607325. [DOI] [Google Scholar]
- 36.Kricheldorf H.R., Weegen-Schulz B., Jenssen J. Cationic Polymerization of Aliphatic Cyclocarbonates. Macromol. Symp. 1998;132:421–430. doi: 10.1002/masy.19981320139. [DOI] [Google Scholar]
- 37.Lee J.-C., Litt M.H. Ring-Opening Polymerization of Ethylene Carbonate and Depolymerization of Poly(Ethylene Oxide-Co-Ethylene Carbonate) Macromolecules. 2000;33:1618–1627. doi: 10.1021/ma9914321. [DOI] [Google Scholar]
- 38.Endo T., Sanda F. Novel Ring-Opening Polymerization and Its Application to Polymeric Materials. Macromol. Symp. 2000;159:1–8. doi: 10.1002/1521-3900(200010)159:1<1::AID-MASY1>3.0.CO;2-Z. [DOI] [Google Scholar]
- 39.Carothers W.H., Natta F.J.V. Studies on Polymerization and Ring Formation. III Glycol Esters of Carbonic Acid. J. Am. Chem. Soc. 1930;52:314–326. doi: 10.1021/ja01364a045. [DOI] [Google Scholar]
- 40.Darensbourg D.J., Choi W., Ganguly P., Richers C.P. Biometal Derivatives as Catalysts for the Ring-Opening Polymerization of Trimethylene Carbonate. Optimization of the Ca(II) Salen Catalyst System. Macromolecules. 2006;39:4374–4379. doi: 10.1021/ma0603433. [DOI] [Google Scholar]
- 41.Wang X.-L., Zhuo R.-X., Huang S.-W., Liu L.-J., He F. Synthesis, Characterization and In Vitro Cytotoxicity of Poly[(5-Benzyloxy-Trimethylene Carbonate)-Co-(Trimethylene Carbonate)] Macromol. Chem. Phys. 2002;203:985–990. doi: 10.1002/1521-3935(20020401)203:7<985::AID-MACP985>3.0.CO;2-0. [DOI] [Google Scholar]
- 42.Mei H., Zhong Z., Long F., Zhuo R. Synthesis and Characterization of Novel Glycerol-Derived Polycarbonates with Pendant Hydroxyl Groups. Macromol. Rapid Commun. 2006;27:1894–1899. doi: 10.1002/marc.200600484. [DOI] [Google Scholar]
- 43.Stridsberg K.M., Ryner M., Albertsson A.-C. Degradable Aliphatic Polyesters. Springer; Berlin/Heidelberg, Germany: Controlled Ring-Opening Polymerization: Polymers with Designed Macromolecular Architecture; pp. 41–65. [Google Scholar]
- 44.Chen W., Yang H., Wang R., Cheng R., Meng F., Wei W., Zhong Z. Versatile Synthesis of Functional Biodegradable Polymers by Combining Ring-Opening Polymerization and Postpolymerization Modification via Michael-Type Addition Reaction. Macromolecules. 2010;43:201–207. doi: 10.1021/ma901897y. [DOI] [Google Scholar]
- 45.Darensbourg D.J., Ganguly P., Billodeaux D. Ring-Opening Polymerization of Trimethylene Carbonate Using Aluminum(III) and Tin(IV) Salen Chloride Catalysts. Macromolecules. 2005;38:5406–5410. doi: 10.1021/ma050666j. [DOI] [Google Scholar]
- 46.Darensbourg D.J., Ganguly P., Choi W. Metal Salen Derivatives as Catalysts for the Alternating Copolymerization of Oxetanes and Carbon Dioxide to Afford Polycarbonates. Inorg. Chem. 2006;45:3831–3833. doi: 10.1021/ic052109j. [DOI] [PubMed] [Google Scholar]
- 47.Helou M., Miserque O., Brusson J.-M., Carpentier J.-F., Guillaume S.M. Highly Effective and Green Catalytic Approach Toward α, ω-Dihydroxy-Telechelic Poly(Trimethylenecarbonate) Macromol. Rapid Commun. 2009;30:2128–2135. doi: 10.1002/marc.200900498. [DOI] [PubMed] [Google Scholar]
- 48.Dobrzynski P., Pastusiak M., Bero M. Less Toxic Acetylacetonates as Initiators of Trimethylene Carbonate and 2,2-Dimethyltrimethylene Carbonate Ring Opening Polymerization. J. Polym. Sci. Part Polym. Chem. 2005;43:1913–1922. doi: 10.1002/pola.20670. [DOI] [Google Scholar]
- 49.Dobrzynski P., Kasperczyk J. Synthesis of Biodegradable Copolymers with Low-Toxicity Zirconium Compounds. V. Multiblock and Random Copolymers OfL-Lactide with Trimethylene Carbonate Obtained in Copolymerizations Initiated with Zirconium(IV) Acetylacetonate. J. Polym. Sci. Part Polym. Chem. 2006;44:3184–3201. doi: 10.1002/pola.21428. [DOI] [Google Scholar]
- 50.Brignou P., Guillaume S.M., Roisnel T., Bourissou D., Carpentier J.-F. Discrete Cationic Zinc and Magnesium Complexes for Dual Organic/Organometallic-Catalyzed Ring-Opening Polymerization of Trimethylene Carbonate. Chem.-Eur. J. 2012;18:9360–9370. doi: 10.1002/chem.201200336. [DOI] [PubMed] [Google Scholar]
- 51.Sheng H., Xu F., Yao Y., Zhang Y., Shen Q. Novel Mixed-Metal Alkoxide Clusters of Lanthanide and Sodium: Synthesis and Extremely Active Catalysts for the Polymerization of ε-Caprolactone and Trimethylene Carbonate. Inorg. Chem. 2007;46:7722–7724. doi: 10.1021/ic701238w. [DOI] [PubMed] [Google Scholar]
- 52.Sheng H., Zhou L., Zhang Y., Yao Y., Shen Q. Anionic Lanthanide Phenoxide Complexes as Novel Single-Component Initiators for the Polymerization of ε-Caprolactone and Trimethylene Carbonate. J. Polym. Sci. Part Polym. Chem. 2007;45:1210–1218. doi: 10.1002/pola.21888. [DOI] [Google Scholar]
- 53.Yasuda H., Aludin M.-S., Kitamura N., Tanabe M., Sirahama H. Syntheses and Physical Properties of Novel Optically Active Poly(Ester−carbonate)s by Copolymerization of Substituted Trimethylene Carbonate with ε-Caprolactone and Their Biodegradation Behavior. Macromolecules. 1999;32:6047–6057. doi: 10.1021/ma981904w. [DOI] [Google Scholar]
- 54.Zhou L., Sun H., Chen J., Yao Y., Shen Q. Homoleptic Lanthanide Guanidinate Complexes: The Effective Initiators for the Polymerization of Trimethylene Carbonate and Its Copolymerization with?—Caprolactone. J. Polym. Sci. Part Polym. Chem. 2005;43:1778–1786. doi: 10.1002/pola.20644. [DOI] [Google Scholar]
- 55.Nakayama Y., Yasuda H., Yamamoto K., Tsutsumi C., Jerome R., Lecomte P. Comparison of Sm Complexes with Sn Compounds for Syntheses of Copolymers Composed of Lactide and Cyclic Carbonates and Their Biodegradabilities. React. Funct. Polym. 2005;63:95–105. doi: 10.1016/j.reactfunctpolym.2005.02.012. [DOI] [Google Scholar]
- 56.Li C., Wang Y., Zhou L., Sun H., Shen Q. Homoleptic Lanthanide Amidinate Complexes: A Single-Component Initiator for Ring-Opening Polymerization of Trimethylene Carbonate and Copolymerization with ε-Caprolactone. J. Appl. Polym. Sci. 2006;102:22–28. doi: 10.1002/app.24109. [DOI] [Google Scholar]
- 57.Zhao B., Lu C.R., Shen Q. Ring-Opening Polymerization of Trimethylenecarbonate and Its Copolymerization with ε-Caprolactone by Lanthanide (II) Aryloxide Complexes. J. Appl. Polym. Sci. 2007;106:1383–1389. doi: 10.1002/app.26761. [DOI] [Google Scholar]
- 58.Palard I., Schappacher M., Belloncle B., Soum A., Guillaume S.M. Unprecedented Polymerization of Trimethylene Carbonate Initiated by a Samarium Borohydride Complex: Mechanistic Insights and Copolymerization with ɛ-Caprolactone. Chem.-Eur. J. 2007;13:1511–1521. doi: 10.1002/chem.200600843. [DOI] [PubMed] [Google Scholar]
- 59.Liu B., Cui D. Polymerization of 2,2′-Dimethyltrimethylene Carbonate by Lutetium Complexes Bearing Amino-Phosphine Ligands. J. Appl. Polym. Sci. 2009;112:3110–3118. doi: 10.1002/app.29534. [DOI] [Google Scholar]
- 60.Zhang L., Wang Y., Shen L., Zhang T. Ring-Opening Copolymerization of 2,2-Dimethyltrimethylene Carbonate and ε-Caprolactone Using Rare Earth Aryloxides Substituted by Various Alkyl Groups. Chin. J. Chem. 2010;28:1019–1026. doi: 10.1002/cjoc.201090160. [DOI] [Google Scholar]
- 61.Xu N., Liu X., Liu Y., Zhu W., Chen F., Shen Z. Synthesis and Properties of Random Copolymers of 2,2-Dimethyltrimethylene Carbonate and Ethylene Carbonate Catalyzed by Lanthanide Tris(2,6-Di- Tert -Butyl-4-Methylphenolate) J. Appl. Polym. Sci. 2010;115:46–51. doi: 10.1002/app.30514. [DOI] [Google Scholar]
- 62.Nomura N., Taira A., Tomioka T., Okada M. A Catalytic Approach for Cationic Living Polymerization: Sc(OTf) 3 -Catalyzed Ring-Opening Polymerization of Lactones. Macromolecules. 2000;33:1497–1499. doi: 10.1021/ma991580r. [DOI] [Google Scholar]
- 63.Kunioka M., Wang Y., Onozawa S. Poly(Lactic Acid) Polymerized by Aluminum Triflate. Macromol. Symp. 2005;224:167–180. doi: 10.1002/masy.200550615. [DOI] [Google Scholar]
- 64.Gorczynski J.L., Chen J., Fraser C.L. Iron Tris(Dibenzoylmethane)-Centered Polylactide Stars: Multiple Roles for the Metal Complex in Lactide Ring-Opening Polymerization. J. Am. Chem. Soc. 2005;127:14956–14957. doi: 10.1021/ja0530927. [DOI] [PubMed] [Google Scholar]
- 65.Nomura N., Taira A., Nakase A., Tomioka T., Okada M. Ring-Opening Polymerization of Lactones by Rare-Earth Metal Triflates and by Their Reusable System in Ionic Liquids. Tetrahedron. 2007;63:8478–8484. doi: 10.1016/j.tet.2007.05.073. [DOI] [Google Scholar]
- 66.Li H., Yao Y., Yao C., Sheng H., Shen Q. Homo- and Copolymerization of 2,2-Dimethyltrimethylene Carbonate Promoted by Samarium Thiolate Derivatives: Novel and Versatile Initiators. J. Polym. Sci. Part Polym. Chem. 2005;43:1312–1316. doi: 10.1002/pola.20620. [DOI] [Google Scholar]
- 67.Yu C., Zhang L., Shen Z. Random Copolymerization of 2,2-Dimethyltri Methylene Carbonate Andɛ-Caprolactone Using a Rare-Earth Tris(4-Tert-Butylphenolate)s as a Single-Component Initiator. Polym. Int. 2004;53:1485–1490. doi: 10.1002/pi.1568. [DOI] [Google Scholar]
- 68.Guillaume S.M., Carpentier J.-F. Recent Advances in Metallo/Organo-Catalyzed Immortal Ring-Opening Polymerization of Cyclic Carbonates. Catal. Sci. Technol. 2012;2:898. doi: 10.1039/c2cy00507g. [DOI] [Google Scholar]
- 69.Möller M., Kånge R., Hedrick J.L. Sn (OTf) 2 and Sc (OTf) 3: Efficient and Versatile Catalysts for the Controlled Polymerization of Lactones. J. Polym. Sci. Part Polym. Chem. 2000;38:2067–2074. doi: 10.1002/(SICI)1099-0518(20000601)38:11<2067::AID-POLA150>3.0.CO;2-1. [DOI] [Google Scholar]
- 70.Cervellera R., Ramis X., Salla J.M., Mantecón A., Serra A. N,N-Dimethylaminopyridine as Initiator in the Copolymerization of Diglycidylether of Bisphenol A with Six-Membered Cyclic Carbonates. J. Polym. Sci. Part Polym. Chem. 2006;44:2873–2882. doi: 10.1002/pola.21393. [DOI] [Google Scholar]
- 71.Helou M., Miserque O., Brusson J.-M., Carpentier J.-F., Guillaume S.M. Organocatalysts for the Controlled “Immortal” Ring-Opening Polymerization of Six-Membered-Ring Cyclic Carbonates: A Metal-Free, Green Process. Chem.-Eur. J. 2010;16:13805–13813. doi: 10.1002/chem.201001111. [DOI] [PubMed] [Google Scholar]
- 72.Morinaga H., Ochiai B., Endo T. Synthesis and Properties of Star-Shaped Polymers by the Ring-Opening Polymerization of Cyclic Carbonate Initiated with a Trifunctional, Poly(Ethylene Glycol)-Based Surfactant. J. Polym. Sci. Part Polym. Chem. 2006;44:6633–6639. doi: 10.1002/pola.21767. [DOI] [Google Scholar]
- 73.Morinaga H., Ochiai B., Mori H., Endo T. Synthesis and Characterization of Block Copolymers by Metal- and Solvent-Free Ring-Opening Polymerization of Cyclic Carbonates Initiated from PEG-Based Surfactants. J. Polym. Sci. Part Polym. Chem. 2006;44:1985–1996. doi: 10.1002/pola.21306. [DOI] [Google Scholar]
- 74.Liu J., Zhang C., Liu L. Ring Opening Polymerization of Aliphatic Cyclic Carbonates in the Presence of Natural Amino Acids. J. Appl. Polym. Sci. 2008;107:3275–3279. doi: 10.1002/app.27440. [DOI] [Google Scholar]
- 75.Nederberg F., Lohmeijer B.G.G., Leibfarth F., Pratt R.C., Choi J., Dove A.P., Waymouth R.M., Hedrick J.L. Organocatalytic Ring Opening Polymerization of Trimethylene Carbonate. Biomacromolecules. 2007;8:153–160. doi: 10.1021/bm060795n. [DOI] [PubMed] [Google Scholar]
- 76.Mindemark J., Hilborn J., Bowden T. End-Group-Catalyzed Ring-Opening Polymerization of Trimethylene Carbonate. Macromolecules. 2007;40:3515–3517. doi: 10.1021/ma0629081. [DOI] [Google Scholar]
- 77.Nifant’ev I., Shlyakhtin A., Bagrov V., Lozhkin B., Zakirova G., Ivchenko P., Legon’kova O. Theoretical and Experimental Studies of 1,5,7-Triazabicyclo[4.4.0]Dec-5-Ene-Catalyzed Ring Opening/Ring Closure Reaction Mechanism for 5-, 6- and 7-Membered Cyclic Esters and Carbonates. React. Kinet. Mech. Catal. 2016;117:447–476. doi: 10.1007/s11144-015-0952-y. [DOI] [Google Scholar]
- 78.Naumann S., Thomas A.W., Dove A.P. Highly Polarized Alkenes as Organocatalysts for the Polymerization of Lactones and Trimethylene Carbonate. ACS Macro Lett. 2016;5:134–138. doi: 10.1021/acsmacrolett.5b00873. [DOI] [PubMed] [Google Scholar]
- 79.Matsuo J., Nakano S., Sanda F., Endo T. Ring-Opening Polymerization of Cyclic Carbonates by Alcohol-Acid Catalyst. J. Polym. Sci. Part Polym. Chem. 1998;36:2463–2471. doi: 10.1002/(SICI)1099-0518(199810)36:14<2463::AID-POLA4>3.0.CO;2-U. [DOI] [Google Scholar]
- 80.Makiguchi K., Ogasawara Y., Kikuchi S., Satoh T., Kakuchi T. Diphenyl Phosphate as an Efficient Acidic Organocatalyst for Controlled/Living Ring-Opening Polymerization of Trimethylene Carbonates Leading to Block, End-Functionalized, and Macrocyclic Polycarbonates. Macromolecules. 2013;46:1772–1782. doi: 10.1021/ma4000495. [DOI] [Google Scholar]
- 81.He X., Ji Y., Jin Y., Kan S., Xia H., Chen J., Liang B., Wu H., Guo K., Li Z. Bifunctional Imidodiphosphoric Acid-Catalyzed Controlled/Living Ring-Opening Polymerization of Trimethylene Carbonate Resulting Block, α,ω-Dihydroxy Telechelic, and Star-Shaped Polycarbonates. J. Polym. Sci. Part Polym. Chem. 2014;52:1009–1019. doi: 10.1002/pola.27082. [DOI] [Google Scholar]
- 82.Delcroix D., Martín-Vaca B., Bourissou D., Navarro C. Ring-Opening Polymerization of Trimethylene Carbonate Catalyzed by Methanesulfonic Acid: Activated Monomer versus Active Chain End Mechanisms. Macromolecules. 2010;43:8828–8835. doi: 10.1021/ma101461y. [DOI] [Google Scholar]
- 83.Kobayashi S., Makino A. Enzymatic Polymer Synthesis: An Opportunity for Green Polymer Chemistry. Chem. Rev. 2009;109:5288–5353. doi: 10.1021/cr900165z. [DOI] [PubMed] [Google Scholar]
- 84.Yamamoto Y., Kaihara S., Toshima K., Matsumura S. High-Molecular-Weight Polycarbonates Synthesized by Enzymatic ROP of a Cyclic Carbonate as a Green Process. Macromol. Biosci. 2009;9:968–978. doi: 10.1002/mabi.200900039. [DOI] [PubMed] [Google Scholar]
- 85.Tasaki H., Toshima K., Matsumura S. Enzymatic Synthesis and Polymerization of Cyclic Trimethylene Carbonate Monomer with/without Methyl Substituent. Macromol. Biosci. 2003;3:436–441. doi: 10.1002/mabi.200350013. [DOI] [Google Scholar]
- 86.Namekawa S., Uyama H., Kobayashi S., Kricheldorf H.R. Lipase-Catalyzed Ring-Opening Polymerization and Copolymerization of Cyclic Dicarbonates. Macromol. Chem. Phys. 2000;201:261–264. doi: 10.1002/(SICI)1521-3935(20000201)201:2<261::AID-MACP261>3.0.CO;2-Q. [DOI] [Google Scholar]
- 87.Yu X.-H., Zhuo R.-X., Feng J., Liao J. Enzymatic Ring-Opening Polymerization of 2,2-Dimethyltrimethylene Carbonate Catalyzed by PPL Immobilized on Silica Nanoparticles. Eur. Polym. J. 2004;40:2445–2450. doi: 10.1016/j.eurpolymj.2004.06.026. [DOI] [Google Scholar]
- 88.Vogdanis L., Martens B., Uchtmann H., Hensel F., Heitz W. Synthetic and Thermodynamic Investigations in the Polymerization of Ethylene Carbonate. Makromol. Chem. 1990;191:465–472. doi: 10.1002/macp.1990.021910301. [DOI] [Google Scholar]
- 89.Vogdanis L., Heitz W. Carbon Dioxide as a Monomer, 3. The Polymerization of Ethylene Carbonate. Makromol. Chem. Rapid Commun. 1986;7:543–547. doi: 10.1002/marc.1986.030070901. [DOI] [Google Scholar]
- 90.Kéki S., Török J., Deák G., Zsuga M. Ring-Opening Oligomerization of Propylene Carbonate Initiated by the Bisphenol A/KHCO 3 System: A Matrix-Assisted Laser Desorption/Ionization Mass Spectrometric Study of the Oligomers Formed. Macromolecules. 2001;34:6850–6857. doi: 10.1021/ma0100302. [DOI] [Google Scholar]
- 91.Soós L., Deák G., Kéki S., Zsuga M. Anionic Bulk Oligomerization of Ethylene and Propylene Carbonate Initiated by Bisphenol-A/Base Systems. J. Polym. Sci. Part Polym. Chem. 1999;37:545–550. doi: 10.1002/(SICI)1099-0518(19990301)37:5<545::AID-POLA4>3.0.CO;2-T. [DOI] [Google Scholar]
- 92.Wu M., Guo J., Jing H. Organic Base Catalyzed Oligomerization of Propylene Carbonate and Bisphenol A: Unexpected Polyether Diol Formation. Catal. Commun. 2008;9:120–125. doi: 10.1016/j.catcom.2007.05.018. [DOI] [Google Scholar]
- 93.Kéki S., Török J., Deák G., Zsuga M. Mechanism of Ring-Opening and Elimination Cooligomerization of Cyclic Carbonates and ε-Caprolactone: Formation of Cyclic Cooligomers. Eur. Polym. J. 2005;41:1478–1483. doi: 10.1016/j.eurpolymj.2005.02.010. [DOI] [Google Scholar]
- 94.Von Seggern N., Schindler T., Naumann S. Dual Catalytic Ring-Opening Polymerization of Ethylene Carbonate for the Preparation of Degradable PEG. Biomacromolecules. 2020;21:2661–2669. doi: 10.1021/acs.biomac.0c00360. [DOI] [PubMed] [Google Scholar]
- 95.Rokicki G., Rakoczy P., Parzuchowski P., Sobiecki M. Hyperbranched Aliphatic Polyethers Obtained from Environmentally Benign Monomer: Glycerol Carbonate. Green Chem. 2005;7:529–539. doi: 10.1039/b501597a. [DOI] [Google Scholar]
- 96.Mouloungui Z., Marechal P., Troung Dinh N. Glycerol Polycarbonate, Organic Compositions Containing Same and Method for Obtaining Said Compositions. US7928182B2. U.S. Patent. 2009 February 26;:38.
- 97.Claude S., Mouloungui Z., Yoo J.-W., Gaset A. Method for Preparing Glycerol Carbonate. US6025504A. U.S. Patent. 2000 February 15;
- 98.Hino T., Inoue N., Endo T. Detailed Study of the Ring-Opening Metathesis Polymerization of Norbornene Bearing a Five- or Six-Membered Ring Cyclic Carbonate along with Volume Expansion. J. Polym. Sci. Part Polym. Chem. 2006;44:395–405. doi: 10.1002/pola.21073. [DOI] [Google Scholar]
- 99.Sugimoto H., Ohshima H., Inoue S. Alternating Copolymerization of Carbon Dioxide and Epoxide by Manganese Porphyrin: The First Example of Polycarbonate Synthesis from 1-Atm Carbon Dioxide. J. Polym. Sci. Part Polym. Chem. 2003;41:3549–3555. doi: 10.1002/pola.10835. [DOI] [Google Scholar]
- 100.Ochiai B., Inoue S., Endo T. One-Pot Non-Isocyanate Synthesis of Polyurethanes from Bisepoxide, Carbon Dioxide, and Diamine. J. Polym. Sci. Part Polym. Chem. 2005;43:6613–6618. doi: 10.1002/pola.21103. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.