

Abstract
Per- and polyfluoroalkyl substances (PFAS) are organic chemicals with wide industrial and consumer uses. They are found ubiquitously at low levels in the environment and are detectable in humans and wildlife. Perfluorobutane Sulfonate (PFBS) is a short-chained PFAS used to replace perfluorooctane sulfonate in commerce. In general, the rate of clearance for the short-chained PFAS is faster than that for the long-chained congeners. This study evaluated the pharmacokinetic properties of PFBS and its hepatic transcriptional responses in CD-1 mice. Males and females were given PFBS by oral gavage at 30 or 300 mg/kg; controls received 0.5% Tween-20 vehicle. Trunk blood was collected at 0.5, 1, 2, 4, 8, 16 and 24 h thereafter; liver and kidney were also harvested. Serum and tissue concentrations of PFBS were determined by HPLC-MS-MS. Expression of several hepatic nuclear receptor target genes was determined by qPCR. The half-life of PFBS was estimated as 5.8 h in the males and 4.5 h in the females. Tmax was reached within 1–2 h. Volume of distribution was similar between the two sexes (0.32–0.40 L/kg). The rate of PFBS clearance was linear with exposure doses. Within 24 h, serum PFBS declined to less than 5% of Cmax. PFBS was detected in liver or kidney, although tissue levels of the chemical were only a fraction of those in serum. At 24 h after administration of 300 mg/kg PFBS, elevated expression of several hepatic genes targeted for PPARα, PPARy, and PXR but not by AhR, LXR or CAR was observed, with responses indistinguishable between males and females. Little to no transcriptional response was seen with the 30 mg/kg dose. The short serum half-lives of PFBS (4–5 h) in mice were comparable to those reported in rats. Although detection of PFBS in liver was low compared to that in serum even at the 300 mg/kg dose, the tissue level was sufficient to activate several hepatic nuclear receptors, which may represent an acute response to the chemical at a high dose.
Keywords: perfluorobutane sulfonate, mouse, pharmacokinetics
Introduction
Perfluorobutane Sulfonate (PFBS) is a member of the perfluoroalkyl acids (PFAAs), which are a family of synthetic organic compounds widely used in a variety of consumer and industrial applications for over half a century. PFAAs possess a carbon backbone (typically varying from C4 to C14) that is fully fluorinated, and a functional group (usually carboxylic acid or sulfonic acid). Discovery and recent detection of polyfluoroalkyl ethers in the environment add to the legacy PFAAs for a new term of “per- and polyfluoroalkyl substances (PFAS)”. PFAS in general, and PFAAs in particular are exceptionally stable to metabolic and environmental degradation. They possess strong surfactant properties that make them ideal lubricants as well as water and oil repellents. Exposure to PFAS can occur through dietary intake, food packaging, drinking water, house dust and use of consumer products. The most common PFAAs in commerce were the 8-carbon products that can be metabolized and/or degraded to form perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA), until studies showed their extraordinary prevalence and persistence in the environment (Giesy et al., 2001), resulting in the voluntary termination of their production in the U.S. The adverse effects of PFAAs shown in animal studies appear to be related to their persistence in the body and their biological potency, which are in turn correlated with the carbon-chain length and functional group. Consequently, PFOS (C8) was replaced by the short-chain congener, PFBS (C4) in commerce due to the faster elimination rates of the latter chemical in rats and in humans. However, recent in vitro and in vivo laboratory findings suggest potential adverse effects of PFBS (Oldham et al., 2012; Feng et al., 2017; Qi et al., 2018; Chen et al., 2018a; Chen et al., 2018b; Sant et al., 2019), prompting an assessment for human health risks of this chemical by the U.S. EPA.
A key factor for PFAS adverse effects is the extent of their body burden (internal dosimetry), which is in part determined by their extensive binding to serum and tissue proteins, enterohepatic uptake and excretion (primarily urinary). However, species differences in the pharmacokinetic disposition of several PFAAs such as PFOA, perfluorononanoate (PFNA), and perfluorohexane sulfonate (PFHxS) have been shown previously, where a significant sex difference was noted in rats but not in humans, thereby hampering a direct extrapolation of data from the rat model to humans. PFBS also falls into this category. Studies by Chengelis et al. (2009) and Huang et al. (2019), and to a less extent by Olsen et al. (2009) indicated a clear sex difference in the rate of PFBS elimination in rats. In contrast, the mouse appears to lack such sex difference for several PFAAs examined (Lou et al., 2009; Tatum-Gibbs et al., 2011; Sundstrom et al., 2012). The present study therefore extends the investigation of PFBS to a mouse model. In addition, the ability of PFBS to activate hepatic nuclear receptors (a known action of PFAAs) was evaluated. The results were compared to previous findings with rats and other long-chain PFAAs.
Methods
Materials.
Perfluorobutane Sulfonate (C4F9SO3) Potassium Salt (97% pure, L7038-lot 2) was provided by the 3M Company (Maplewood, MN) and was prepared in 0.5% Tween-20. Stable 13C labeled isotope, sodium perfluoro-1-[2,3,4-13C3] butane sulfonate, was supplied as free acid in methanol (50 μg/ml) by Wellington Laboratories (Guelph, Ontario, Canada) and used as the internal standard for liquid chromatography/tandem mass spectrometric (LC-MS/MS) analysis of PFBS. All other chemicals and reagents used for this study were of analytical grade purchased from VWR (West Chester, PA).
Animal treatment.
All procedures involving the use of laboratory animals were conducted in accordance to the guidelines set forth by the U.S. EPA ORD/CPHEA Institutional Animal Care and Use Committee. Animal care procedures and facilities (AAALAC accredited) were consistent with the recommendations provided by the 2011 National Research Council’s “Guide for the Care and Use of Laboratory Animals”, the Animal Welfare Act, and the Public Health Service Policy on the Humane Care and Use of Laboratory Animals. CD-1 mice (8–10 weeks of age) were purchased from Charles River Laboratory (Raleigh, NC). Upon arrival, animals were housed individually in polycarbonate, solid bottom cages. Lab Diet 5001 pellet chow (PMI Nutrition International, Brentwood, MO) and tap water were provided ad libitum. Environmental controls for the animal facility were set to maintain a mean temperature of 20–24°C, relative humidity of 40–60%, and a 12-h light-dark cycle (lights off 7:00 p.m. – 7:00 a.m.). All animals were allowed one week to acclimate to their environment prior to the beginning of the study.
Non-fasted male and female mice weighing 31 g and 29 g, respectively (n = 6–12 per sex, per dose group, per time point, performed in 4 separate blocks) were given PFBS at 30 or 300 mg/kg body weight by oral gavage as a single dose. Controls received 0.5% Tween-20 vehicle. The relatively high doses were chosen based on the reported short half-life with rats, and to ensure that sufficient levels of chemicals were reached in all compartments for accurate determination of PFBS by the available analytical method. Mice were euthanized by CO2 asphyxiation and decapitated at 0.5, 1, 2, 4, 8, 16 or 24 h after PFBS administration. Trunk blood was collected and serum was prepared by centrifugation at 2000 × g for 30 minutes and stored at −80°C until analysis. Liver and kidneys were removed, weighed, flash-frozen immediately on dry-ice, and stored at −80°C until PFBS analysis. At the 24h time point, a section of the large lobe of liver (~100 mg) from each mouse was placed in 1 mL of RNAlater (ThermoFisher, AM7021) and stored frozen for gene expression analysis by qPCR.
Sample preparation for PFBS analysis.
Individual mouse liver and kidney samples (approximately 300 mg) were thawed, homogenized (polytron) in 4 volumes of picopure water, and stored at −80°C for PFBS analysis. For sample preparation, 25 μL of serum or tissue homogenate were aliquoted into polypropylene tubes, to which 100 μL of 0.1 M formic acid fortified with 45 ng/mL of internal standard (13C3-labeled PFBS) and 1 mL of cold (−20°C) acetonitrile (ACN) were added sequentially, with vigorous mixing (vortex) between each addition. Samples were then capped and centrifuged at 12,500 ×g for 3 min at room temperature. One-hundred μL of the supernatant was transferred to an HPLC auto-sampling vial, followed by the addition of 300 μL of 2 mM ammonium acetate to match the starting conditions of the LC/MS-MS analytical run. To ensure consistent accuracy and precision, each extraction set included quality control (QC) samples consisting of a background blank (acetonitrile in 2 mM ammonium acetate) that did not go through the extraction process, a method blank that went through extraction, a matrix blank (serum or tissue homogenate), and standards (spiked into appropriate matrices). Internal standard was added to all samples except the background blank.
Analytical procedures.
PFBS analysis in serum was performed using a modification of a method originally reported by Dewitt et al. (2008) and further refined by Reiner et al. (2009) for other PFAS. Analysis of samples was performed using an LC-MS/MS system consisting of a Waters Acuity UPLC (Milford, MA) interfaced with a triple quadrupole mass spectrometer. The mass spectrometer was operated in negative electrospray mode with mass transitions for PFBS (quantification 299→99, confirmation 299→80) and 13C3-PFBS (302→99) monitored using multiple-reaction-monitoring (MRM). Analyte-specific mass spectrometer parameters were optimized for both compounds. Samples were quantified with extracted matrix-matched standard curves and with isotope dilution prepared by spiking PFBS into the appropriate matrix, i.e., purchased mouse serum (Pel Freeze Biologicals, Rogers, AR) or control mouse liver and kidney homogenates previously prepared in our laboratory to serve as matrix-matched blanks. The range of the standard curves were 5 to 500,000 ng/ml (depending on the dose group and time point of samples being analyzed). QC samples were included in each analytical batch as specified above. QC check samples were analyzed concurrently with each analytical run. Overall QC recoveries were between 70% and 130% of the theoretical value for all samples employed throughout the study. When a sample exceeded the quality control parameters, it was re-extracted and reanalyzed to ensure proper quantification within the range of the standard curve. The limit of quantification (LOQ) for analysis was set to the lowest calibration point on the standard curve. Upon completion of analysis, data were deemed acceptable if they followed certain criteria that included: (a) background blanks and matrix blanks that had no significant PFBS peaks approaching the LOQ; (b) correlation coefficients > 0.98; and (c) fortified standard curve points and QC check samples that were within 70–130% of the theoretical values. For samples below LOQ (primarily background controls), we assigned a value equal to the LOQ divided by the square root of two (Hornung and Laurence, 1990). Analyst 1.4.2 software was used for quantification of all compounds (Applied Biosystems/MDS Sciex, Foster, CA). Analyte to internal peak area ratios were regressed against concentration to establish quadratic calibration curves using a 1/x weighting.
Gene expression analysis by qPCR.
Liver samples were homogenized in 1 mL of TRI reagent (Sigma/Aldrich, T9424) with 1 mm zirconium silicate beads in a Bullet Blender 24 (Next Advance, Inc.). Extraction was performed according to the manufacturer’s protocol. RNA pellets were resuspended in 100 mL of nuclease-free H2O, and RNA concentrations determined with Nanodrop 1000 (Nanodrop Technologies). RNA samples were treated with DNase I (Promega, M6101) and quantified using the Ribogreen Quantitation Kit (ThermoFisher, R11490). DNase I-treated RNA was reverse-transcribed with the ABI cDNA Archive Kit (ThermoFisher, 4322171) and 25 ng equivalent cDNA was amplified in a 12 μL volume using ABI TaqMan Gene Expression Assays and ABI Universal Master Mix (ThermoFisher, 4304437). Amplification was performed on an ABI model 7900HT sequence detection system using standard Taqman cycling parameters. Beta-2-microglobulin (B2m) was used as reference gene because it did not show significant differences between control and treated groups by single factor ANOVA. Assay identification for each target gene was as follows: Cyp1a1, Mm00487218_m1; Cyp2b10, Mm00456591_m1; Cyp7a1, Mm00484152_m1; Acox1, Mm00443579_m1; Ehhadh, Mm00470091_s1; Pdk4, Mm00443325_m1; Fabp4, Mm00445878_m1; Mogat1, Mm00503358_m1; Cyp3a11, Mm00731567_m1; B2m, Mm00437762_m1. All samples were run in technical duplicate. Data were analyzed by relative gene quantification using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Statistical Analysis
Pharmacokinetic Data. Pharmacokinetic model parameters were estimated for PFBS using a single uniform statistical analysis method developed in the R statistical language v3.5.2. The open source analysis software, a custom developed R package “invivoPKfit”, is freely available at https://github.com/USEPA/CompTox-ExpoCast-invivoPKfit. A one-compartment toxicokinetic model (O’Flaherty, 1981) was used to separately describe the toxicokinetics in the serum, kidney, and liver (see Supplemental Figure S1). Each tissue was characterized by a primary compartment in which the concentration is given by the amount (mg/kg body weight) of chemical in the compartment divided by an apparent volume of distrbution (Vdist, L/kg body weight). Elimination from each tissue was described by a single, first-order (proportional to concentration) elimination rate (kelim, 1/h). Exposure was modeled through an additional first-order absorption rate (kabs, 1/h) from a separate compartment holding the dose. Since the absorption phase is difficult to interpret for liver and kidney tissues, the parameters for this phase were not analyzed. The fraction absorbed (Fabs) was assumed to be one because PFAS are typically well absorbed and no intravenous data were available for its estimation.
Model parameters were estimated by maximizing a likelihood function that assumed that the data were log-normally distributed around the concentrations predicted by the TK model (Cox and Hinkley, 1979). Confidence intervals on the estimated parameters were calculated using the Hessian of the likelihood function to estimate standard deviations of the log parameter (Bartlett, 1953a, b). Standard deviations (sd) around the optimized parameters are reported and remain on the log scale – a 95% confidence interval may be calculated as [eln x−1.97*sd, eln x+1.97*sd]. Because the log of the parameter was optimized, both arithmetic and geometric means and standard deviations could be calculated, but only geometric means and standard deviations are reported.
The R package optimx (Nash et al., 2011; Nash, 2014) version 2013.8.7 was used to perform a bounded optimization of the likelihood function using the method “L-BFGS-B” that implements the bounded (i.e., limits on lower and upper values) approach (Byrd et al., 1995). The log-transformed parameters were estimated. Rate constant of elimination (kelim) was estimated with an upper bound of 1000 1/h (i.e., very fast). Fractions were estimated with an upper limit of 1. Volumes of distribution (Vd) were estimated using a lower bound of 0.01 L. Statistical comparison of parameter estimates was performed using Welch’s modified two-sample t-test (R package BSDA), with significance defined as p < 0.05.
Transcriptomic Data. All gene expression data were analyzed with mixed effects linear models using the Mixed procedure in SAS/Stat software (2013). The Δ−CT values were analyzed, and fold change values were calculated from the Δ−CT estimates. For each gene, a two-way factorial ANOVA was performed to determine significant effects of dose (0, 30 or 300 mg/kg) or sex, and any interaction between dose and sex. A factor for block was included in the ANOVA to adjust for any systematic differences in block of animals or PCR run. In cases where PCR samples were evaluated more than once for the same gene, a random effect for sample was also included in the model. Data are expressed as means ± 1 standard error. Statistical significance is defined as p < 0.05.
Results
Perfluorobutane sulfonate, even at 300 mg/kg, did not alter body, liver or kidney weight within 24 h after treatment (Table 1). Background levels of PFBS were low. In control female mice (n = 9), serum PFBS was 5.9 ± 0.6 ng/mL, liver PFBS was 3.5 ± 0.1 ng/g, and kidney PFBS was 4.5 ± 0.8 ng/g. In control males, (n = 9), serum PFBS was 8.6 ± 2.9 ng/mL, liver PFBS was 3.5 ± 0.1 ng/g and kidney PFBS was 9.4 ± 3.2 ng/g. The chemical was absorbed readily, reaching maximal serum levels (Tmax) between 1–2 h after oral gavage (Fig. 1, Table 2), and distributed effectively to kidney and liver in both male and female mice in a dose-dependent manner. Comparing the Cmax estimates, the percent of renal distribution was similar in both sexes, regardless of dose (8–13% of serum levels). In contrast, the distribution to the liver (ranging from 27% to 59% of serum levels) was higher than that to the kidney, was dose-dependent (about 50–60% higher in the 300 mg/kg than in the 30 mg/kg dose) and appeared to be higher in the males than females by ~30–40%. PFBS levels in these three compartments then declined rapidly, regardless of sex or dose. Pharmacokinetic parameter estimates for the data, stratified on compartment, sex, and dose are described in Table 2. In general, the rates of elimination and half-life estimates of all three compartments did not differ significantly between doses; for instance, the serum half-life of PFBS at 30 mg/kg in female mice is 3.0 h and at 300 mg/kg is 3.9 h, and in males, 4.8 h and 5.4 h, respectively. Hence, these values can be combined across doses for a comparison between sex. The estimated half-life of PFBS in female mice (4.5 h) is slightly (26–34%) but statistically significantly shorter than that in males (5.8 h). Correspondingly, significantly higher estimates of AUC for serum, kidney and liver compartments were detected in males than in females (Table 2). Optimization of a single set of parameters for both doses and genders (solid line in the right-hand column of Figure 1) found that the data were reasonably well-described by a one-compartment model, with coefficients of determination (R2) of 0.76, 0.77, and 0.8 for the serum, liver, and kidney respectively (see Supplemental Figures S2–S4). However, due to large variability between individual mice, the average fold errors (AFEs) were 4.67, 3.42, and 3.95 for serum, liver, and kidney respectively.
Table 1.
Body weight, absolute and relative liver and kidney weight at 24 h after PFBS administration.
| Sex | PFBS (mg/kg) | N | Body Wt (g) | Liver Wt (g) | Rel Liver Wt (%) | Kidney Wt (g) | Rel Kidney Wt (%) |
|---|---|---|---|---|---|---|---|
| Female | 0 | 9 | 26.4 ± 0.7 | 1.20 ± 0.08 | 4.55 ± 0.21 | 0.33 ± 0.01 | 7.35 ± 0.32 |
| 30 | 3 | 27.1 ± 0.7 | 1.33 ± 0.04 | 4.92 ± 0.01 | 0.35 ± 0.01 | 7.03 ± 0.28 | |
| 300 | 12 | 25.6 ± 0.7 | 1.23 ± 0.06 | 4.80 ± 0.16 | 0.32 ± 0.01 | 6.76 ± 0.24 | |
| Male | 0 | 9 | 27.2 ± 0.9 | 1.38 ± 0.05 | 5.09 ± 0.11 | 0.46 ± 0.02 | 9.01 ± 0.45 |
| 30 | 3 | 32.0 ± 1.3 | 1.63 ± 0.07 | 5.09 ± 0.22 | 0.47 ± 0.02 | 9.26 ± 0.24 | |
| 300 | 12 | 27.9 ± 1.2 | 1.66 ± 0.11 | 5.91 ± 0.18 | 0.44 ± 0.02 | 7.41 ± 0.39 |
Data represent means ± S.E. for number of mice evaluated (N).
Fig. 1.
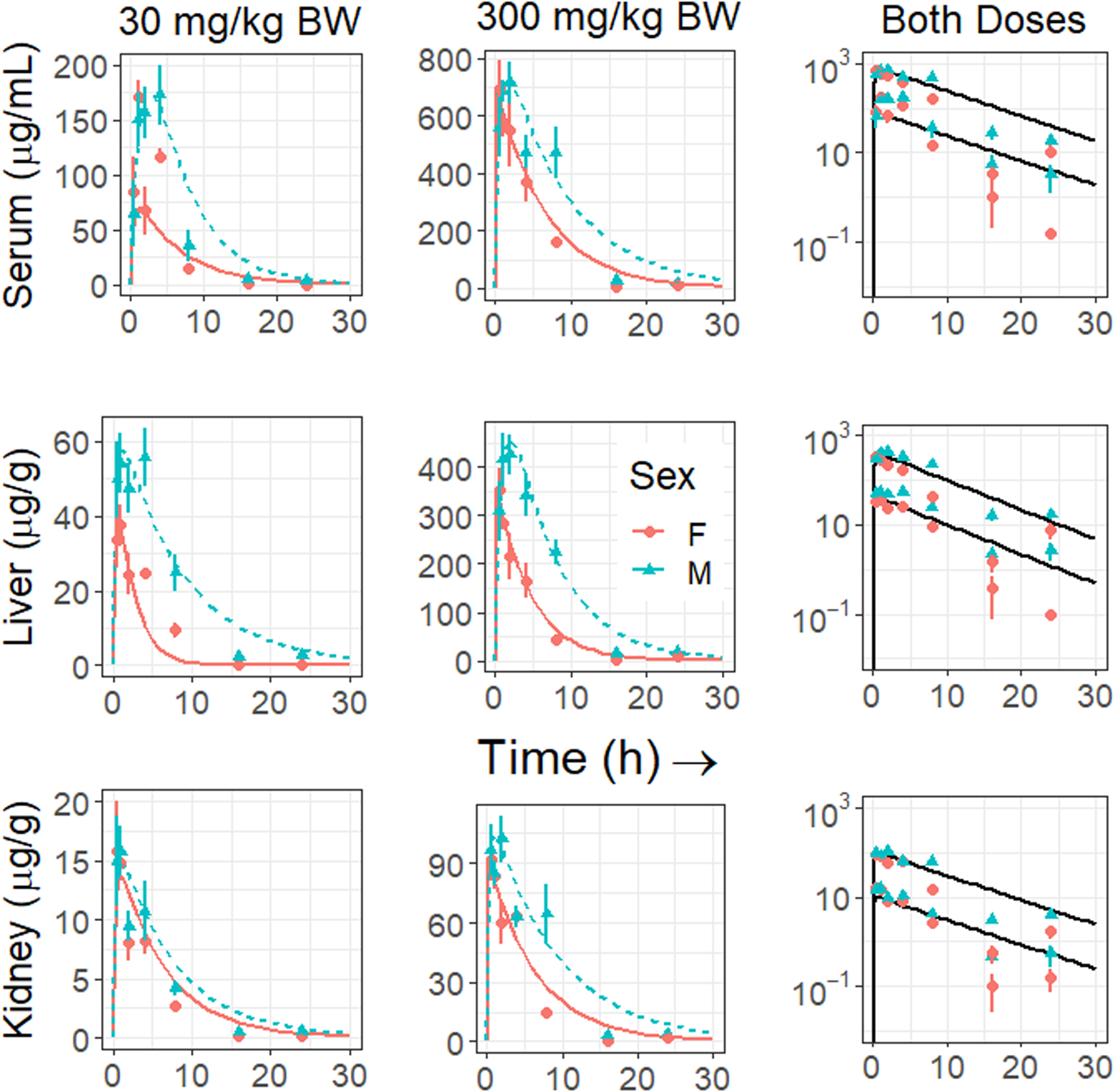
Serum, kidney and liver concentrations of PFBS during 24 h after a single oral gavage of the chemical at 30 (left panels) or 300 mg/kg (middle panels) in male and female mice. The right panels illustrate the combined doses. Data are expressed as means and standard errors of 6–12 animals. Males are illustrated by blue triangles and females by pink circles; curves are fitted by the computational model. Predictions for the optimized sets of parameters are shown for separate optimizations for females (solid line) and males (dashed line) but the same parameters for both doses in the first two-columns. In the third column a single set of parameters optimized for both sexes and doses is used to make the predictions (solid line); these predictions are further evaluated in Supplemental Figures 2–4
Table 2.
Summary of estimated pharmacokinetic parameters.
| Tissue | Dose (mg/kg) | Sex | Kelim (1/h) | Half-Life (h) | Clearance-tot (L/Kg/h) | Vd (L/Kg) | Tmax (h) | Cmax (μg/mL) or (μg/g) | AUC (μg/mL*h) |
|---|---|---|---|---|---|---|---|---|---|
| Serum | 30 | M | 0.186 (0.105, 0.330) | 3.7 (2.1, 6.6) | 0.019 (0.009, 0.042) | 0.129 | 2.7 | 175 | 1515 |
| 300 | M | 0.115 (0.095, 0.139) | 6.0 (5.0, 7.3) | 0.039 (0.030, 0.050) | 0.291 | 1.7 | 731 | 7178 | |
| 30/300 | M | 0.120 (0.102, 0.141) | 5.8 (4.9, 6.8) | 0.038 (0.031, 0.048) | 0.275 | -- | -- | -- | |
| 30 | F | 0.158 (0.081, 0.309) | 4.4 (2.2, 8.5) | 0.056 (0.022, 0.144) | 0.145 | 1.3 | 68.8 | 520 | |
| 300 | F | 0.152 (0.123, 0.188) | 4.6 (3.7, 5.6) | 0.064 (0.011, 0.385) | 0.308 | < 2* | 700 | 4587 | |
| 30/300 | F | 0.156 (0.129, 0.188) | 4.5 (3.7, 5.4) | 0.063 (0.010, 0.406) | 0.278 | -- | -- | -- | |
| Kidney | 30 | M | 0.135 (0.074, 0.247) | 5.1 (2.8, 9.4) | -- | -- | 1.2 | 14.6 | 122 |
| 300 | M | 0.111 (0.090, 0.137) | 6.2 (5.1, 7.7) | -- | -- | 1.1 | 104 | 982 | |
| 30/300 | M | 0.119 (0.100, 0.142) | 5.8 (4.9, 7.0) | -- | -- | -- | -- | -- | |
| 30 | F | 0.162 (0.082, 0.318) | 4.3 (2.2, 8.4) | -- | -- | 1.0 | 13.6 | 97.4 | |
| 300 | F | 0.154 (0.119, 0.199) | 4.5 (3.5, 5.8) | -- | -- | < 2* | 92.0 | 597 | |
| 30/300 | F | 0.157 (0.126, 0.197) | 4.4 (3.5, 5.5) | -- | -- | -- | -- | -- | |
| Liver | 30 | M | 0.119 (0.040, 0.351) | 5.8 (2.0, 17.2) | -- | -- | 1.3 | 57.2 | 527 |
| 300 | M | 0.155 (0.111, 0.215) | 4.5 (3.2, 6.2) | -- | -- | 2.0 | 456 | 3917 | |
| 30/300 | M | 0.159 (0.118, 0.213) | 4.4 (3.3, 5.9) | -- | -- | -- | -- | -- | |
| 30 | F | 0.449 (0.016, 12.3) | 1.5 (0.1, 42.1) | -- | -- | 1.1 | 37.9 | 120 | |
| 300 | F | 0.219 (0.139, 0.345) | 3.2 (2.0, 5.0) | -- | -- | 0.9 | 360 | 1687 | |
| 30/300 | F | 0.229 (0.147, 0.357) | 3.0 (1.9, 4.7) | -- | -- | -- | -- | -- |
Data are expressed as means and 95% confidence intervals. Statistical analysis did not indicate a significant difference between doses (30 and 300 mg/kg), therefore, estimates of combined doses (30/300 mg/kg) are also presented. Welch’s modified two-sample t-test indicated a significant difference (p < 0.05) between sex in estimated half-life and AUC. Asterisks denote values with imprecise estimation due to a noisy absorption phase.
Expression of target genes for several hepatic nuclear receptors was examined at 24 h after PFBS treatment. Expression of these genes was not altered by the 30 mg/kg dose (data not shown); responses to the 300 mg/kg dose are shown in Table 3. Genes involved in the peroxisome proliferator-activated receptor (PPAR) pathways, including both alpha and gamma types, were significantly (and some robustly) upregulated, compared to controls. In addition, genes targeted by pregnane X receptor (PXR) were similarly upregulated. PFBS did not activate the target genes involved in the aryl hydrocarbon receptor (AHR), the constitutive androstane receptor (CAR) or the liver X receptor (LXR) pathways.
Table 3.
Expression of candidate genes for hepatic nuclear receptors 24 h after exposure to 300 mg/kg PFBS.
| Target Gene | Putative Receptor Pathway | N | Fold Change |
|---|---|---|---|
| Cyp1a1 | Aryl Hydrocarbon Receptor | 24 | 1.37 (1.12, 1.87) |
| Cyp2b10 | Constitutive Androstane Receptor | 12 | 2.71 (1.63, 4.51) |
| Cyp7a1 | Liver X Receptor | 12 | 2.06 (1.17, 3.64) |
| Acox1 | Peroxisome Proliferator Activated Receptor- α | 18 | 3.48 (2.88, 4.20)* |
| Ehhadh | Peroxisome Proliferator Activated Receptor- α | 24 | 8.43 (6.24, 11.4)* |
| Pdk4 | Peroxisome Proliferator Activated Receptor- α | 6 | 3.28 (2.43, 4.42)* |
| Fabp4 | Peroxisome Proliferator Activated Receptor- γ and α | 18 | 1.49 (1.23, 1.82)* |
| Mogat1 | Peroxisome Proliferator Activated Receptor- γ | 24 | 7.55 (5.91, 9.65)* |
| Cyp3a11 | Pregnane X Receptor | 18 | 3.23 (2.22, 4.72)* |
Two-way ANOVA did not indicate a sex and response interaction. Therefore, responses from male and female mice were combined for analysis. Data are expressed in mean fold changes from controls (set at 1) ± 1 standard error of various numbers of mice (N). Asterisks denote statistically significant difference (p < 0.05).
Discussion
A major consideration of the adverse effects of some PFAS is their slow elimination rate through renal clearance (because of renal resorption from the kidney tubules) that leads to extraordinary persistence in the body. Notably, the long-chain PFAAs such as PFOA and PFOS, are known to have serum half-life estimates ranging in weeks for rodents and in years for humans (Lau, 2015). In contrast, the short-chain PFAAs such as perfluorobutyrate (PFBA) and perfluorohexanoic acid (PFHxA) have much faster clearance rates, with serum half-lives estimated in hours for rodents and in days to weeks in humans. The pharmacokinetic profile of PFBS resembles the latter group of PFAAs. Chengelis et al. (2009) and Olsen et al. (2009) evaluated the serum elimination half-life of PFBS in male and female Sprague-Dawley rats after a single intravenous injection. The t½ estimates were substantially different between these two studies; Chengelis and coworkers reported a half-life of 2.1 h for male rats and 0.64 h for females that suggested a sex difference, while Olsen and colleagues reported estimates of 4.5 h and 4.0 h, respectively for males and females. Interestingly, the latter study also evaluated the pharmacokinetic parameters after a single oral treatment and suggested a significantly shorter terminal half-life of 4.7 h for males than that of 7.4 h for females. In contrast, a more recent study by Huang et al. (2019) with the same strain of rats reported yet a different set of half-life estimates after oral administration, with males ranging from 2.7 to 4.4 h (depending on dose) compared to females ranging from 1.1 to 1.5 h, which would support a trend toward the sex difference observed by Chengelis et al. (2009). Indeed, the reported sex-dependent rates of elimination among perfluoroalkyl sulfonates are quite inconsistent in the rat, with significantly shorter t½ estimated in females (1.8 days) than in males (29.1 days) for PFHxS (an elimination rate ~16 times faster in females) (Sundstrom et al., 2012), but much smaller difference between females (62–71 days) and males (38–41 days) for PFOS (difference in opposite direction, with an elimination rate being ~1.7 times faster in males) (Chang et al., 2012). By comparison, the sex difference in serum elimination of PFAAs for the mouse are much smaller (many are statistically indistinguishable) and are much more consistent among chemicals of various functional groups and chain-lengths, characteristics that more closely resemble those of humans (Lau, 2015).
The pharmacokinetic profile of PFBS for rats, monkeys and humans has been described previously (Chengelis et al., 2009; Olsen et al., 2009; Huang et al., 2019); this study focused on the mouse model. Linear pharmacokinetics of PFBS was seen in mice at administered doses up to 300 mg/kg, as no significant differences were seen in all estimated parameters between the 30 and 300 mg/kg doses. PFBS was rapidly absorbed in mice after oral exposure, with Tmax estimated between 1 to 2 h, which is in line with the values described by Huang et al. (2019) (2.8–4.4 h for males and 1.1–1.5 h for females). Estimates of Cmax and AUC are consistent among serum, kidney and liver compartments, and are approximately proportional to the different administered doses. Because there was no significant difference in half-life estimates between doses, values were combined for both groups. Serum half-life was 5.8 h for male mice and 4.5 h for females; although such sex difference is small (females about 28% shorter than males), it is nonetheless statistically significant. The half-life estimates of PFBS in kidney and liver mirrored those in serum. By comparison, the sex difference in PFBS elimination in mice (about 28% faster in females) observed in this study appears to be much smaller than those reported in rats (about 250–330% faster in females) (Chengalis et al., 2009; Huang et al., 2019). This trend of species difference is consistent with that for other PFAAs evaluated thus far (Sundstrom et al., 2012 for PFHxS; Tatum-Gibbs et al., 2011 for PFNA; Lou et al., 2009 for PFOA; and Russell et al., 2013 for PFHxA). The biological basis underpinning the difference in disposition of PFAS between rats and mice is not well understood, but in this regard, mice appear to resemble more closely the characteristics of humans and may provide a more suitable model than rats for species extrapolation.
For the long-chain PFAAs such as PFNA and PFOA in the mouse model, Cmax estimate and bioaccumulation of the chemicals tend to be significantly higher (by 2–3 folds) in the liver than in serum (Tatum-Gibbs, 2011; Lou et al., 2009). The converse is noted with the short-chain PFAA such as PFBA (Chang et al., 2008; Das et al., 2008) where the liver concentrations are only a fraction (~20–30%) of the serum levels. However, this finding differs from those reported by Bogdanska et al. (2014) where the mouse hepatic levels of PFBS were described to be 1.4-fold higher than the level in blood, although this ratio is substantially lower than those reported by Olsen et al. (2009) with rats (9.6-fold). On the other hand, the kidney:serum ratios of PFBS being less than one (0.1–0.2) in the present study are in general agreement with that reported in the Bogdanska study (0.7). The higher estimates of tissue:serum ratios from the Bogdanska 5-day dietary study compared to those from the current single gavage study may reflect a different manner of oral exposure.
The mechanisms of action for PFAS in general and PFBS specifically are not well understood, but PFAS evaluated thus far have been shown to activate hepatic nuclear receptors involved in fatty acid and energy metabolism, particularly the peroxisome proliferator-activated receptor-alpha (PPARα) in several species (Lau, 2015). However, in vitro studies indicated that the potency of PPARα activation varies among PFAAs, with the perfluoroalkyl sulfonates generally weaker than the perfluorocarboxylates, and the short-chain PFAAs less potent than the long-chain congeners (Wolf et al., 2012; Rosen et al., 2013). The potency of PFBS ranks below that of PFHxS, PFOS, and even PFBA (Wolf et al., 2012), and did not significantly activate human PPARα in an in vitro assay (Behr et al., 2020). In addition to the PPARs, other hepatic nuclear receptors pregnane X receptor (PXR) and constitutive androstane receptor (CAR) have been shown to be activated by PFAAs (Vanden Heuvel et al., 2006; Cheng and Klaassen, 2008; Martin et al., 2007; Ren et al., 2009; Bjork et al., 2011; Li et al., 2019), while activation of liver X receptor (LXR) appeared to be less effective (Vanden Heuvel et al., 2006; Bjork et al., 2011) and that of aryl hydrocarbon receptor (AhR) was absent (Cheng and Klaassen, 2008; Brewster and Birnbaum 1989). The question remains, however, whether PFBS would share this response profile of hepatic nuclear receptor activation. In the present study, exposure to 30 mg/kg PFBS failed to activate any of the nuclear receptor target genes in the liver; however, PPARα, PPARγ and PXR target genes were significantly upregulated at 300 mg/kg. These limited findings are largely consistent with previous studies. It must be noted that the effective PFBS dose of 300 mg/kg that produced a serum Cmax estimate of 360–456 mg/L is extraordinarily high relative to human exposure, which is below the detection limit of 0.1 μg/L in the general U.S. population (CDC, 2019). However, results from this study may implicate a common mechanism of action involving hepatic nuclear receptors for all PFAS, including those with short half-lives, provided that significant tissue levels are reached.
Taken together, our results show that PFBS is more effectively eliminated than the long-chain PFAAs such as PFOS and PFOA in mice, in line with previous findings with rats. However, unlike the rat model, the sex differences detected with the pharmacokinetic parameters in mice is relatively small, suggesting that the mouse model may be more appropriate for extrapolation to humans.
Supplementary Material
Acknowledgments
The information in this document has been funded by the U.S. Environmental Protection Agency. It has been subjected to review by the Center for Public Health and Environmental Assessment (CPHEA) and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
References
- Bartlett MS 1953a. Approximate confidence intervals. Biometrika 40, 12–19. [Google Scholar]
- Bartlett MS 1953b. Approximate confidence intervals.II. More than one unknown parameter. Biometrika 40, 306–317. [Google Scholar]
- Behr AC, Plinsch C, Braeuning A and Buhrke T 2020. Activation of human nuclear receptors by perfluoroalkylated substances (PFAS). Toxicol In Vitro 62:104700. [DOI] [PubMed] [Google Scholar]
- Bjork JA, Butenhoff JL and Wallace KB 2011. Multiplicity of nuclear receptor activation by PFOA and PFOS in primary human and rodent hepatocytes. Toxicology 288:8–17. [DOI] [PubMed] [Google Scholar]
- Bogdanska J, Sundstrom M, Bergstrom U, Borg D, Abedi-Valugerdi M, Bergman A, DePierre J and Nobel S 2014. Tissue distribution of 35S-labelled perfluorobutanesulfonic acid in adult mice following dietary exposure for 1–5 days. Chemosphere 98, 28–36. [DOI] [PubMed] [Google Scholar]
- Brewster DW and Birnbaum LS 1989. The biochemical toxicity of perfluorodecanoic acid in the mouse is different from that of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol 99:544–554. [DOI] [PubMed] [Google Scholar]
- Byrd RH, Lu P, Nocedal J, and Zhu C 1995. A limited memory algorithm for bound constrained optimization. SIAM Journal on Scientific Computing 16, 1190–1208. [Google Scholar]
- CDC, 2019. https://www.cdc.gov/nchs/nhanes/
- Chang S, Das KP, Ehresman DJ, Ellefson ME, Gorman GS, Hart JA, Noker PE, Tan Y-M, Lieder PH, Lau C, Olsen GW and Butenhoff JL 2008. Comparative pharmacokinetics of perfluorobutyrate (PFBA) in rats, mice, and monkeys and humans and relevance to human exposure via drinking water. Toxicol. Sci 104, 40–53. [DOI] [PubMed] [Google Scholar]
- Chang SC, Noker PE, Gorman GS, Gibson SJ, Hart JA, Ehresman DJ, and Butenhoff JL 2012. Comparative pharmacokinetics of perfluorooctanesulfonate (PFOS) in rats, mice, and monkeys. Reprod. Toxicol 33, 428–440. [DOI] [PubMed] [Google Scholar]
- Chen F, Wei C, Chen Q, Zhang J, Wang L, Zhou Z Chen M and Liang Y 2018a. Internal concentration of perfluorobutane sulfonate (PFBS) comparable to those of perfluorooctane sulfonate (PFOS) induce reproductive toxicity in Caenorhabditis elegans. Ecotoxicol. Environ. Safety 158, 223–229. [DOI] [PubMed] [Google Scholar]
- Chen L, Hu C, Tsui MMP, Wan T Peterson DR, Shi Q, Lam PKS Au DWT, Lam JCW and Zhou B 2018b. Multigenerational disruption of the thyroid endocrine system in marine medaka after a life-cycle exposure to perfluorobutanesulfonate. Environ. Sci. Technol 52, 4432–4439. [DOI] [PubMed] [Google Scholar]
- Cheng X and Klaassen CD 2008. Perfluorocarboxylic acids induce cytochrome P450 enzymes in mouse liver through activation of PPAR-alpha and CAR transcription factors. Toxicol. Sci 106:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chengelis CP, Kirkpatrick JB, Myers NR, Shinohara M, Stetson PL and Sved DW 2009. Comparison of the toxicokinetic behavior of perfluorohexanoic acid (PFHxA) and nonafluorobutane-1-sulfonic acid (PFBS) in cynomolgus monkeys and rats. Reprod. Toxicol 27, 400–406. [DOI] [PubMed] [Google Scholar]
- Cox DR and Hinkley DV. 1979. Theoretical statistics. Chapman and Hall/CRC. [Google Scholar]
- Das KP, Grey BE, Zehr RD, Wood C, Butenhoff JL, Chang S-C, Ehresman DJ, Tan Y-M and Lau C 2008. Effects of perfluorobutyric acid exposure during pregnancy in the mouse. Toxicol. Sci 105, 173–181. [DOI] [PubMed] [Google Scholar]
- Dewitt JC, Copeland CB, Strynar MJ and Luebke RW 2008. Perfluorooctanoic acid-induced immunomodulation in adult C57BL/6J or C57BL/6N female mice. Environ. Health Perspect 116, 644–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Cao X, Zhao S, Wang X, Hua X, Chen L and Chen L 2017. Exposure of pregnant mice to perfluorobutanesulfonate causes hypothyroxinemia and developmental abnormalities in female offspring. Toxicol. Sci 155, 409–419. [DOI] [PubMed] [Google Scholar]
- Gannon SA, Fasano WJ, Mawn MP, Nabb DL, Buck RC, Buxton W, Jepson MP and Frame SR 2016. Absorption, distribution, metabolism, excretion, and kinetics of 2,3,3,3-tetrafluoro-2-(heptafluoropropoxy)propanoic ammonium salt following a single dose in rat, mouse, and cynomolgus monkey. Toxicology 340, 1–9. [DOI] [PubMed] [Google Scholar]
- Giesy JP, Kannan K and Jones PD 2001. Global biomonitoring of perfluorinated organics. Scientific World Journal, 1:627–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung RWR and Laurence D 1990. Estimation of average concentration in the presence of nondetectable values. Appl. Occup. Environ. Hyg 5: 46–51. [Google Scholar]
- Huang MC, Dzierlenga AL, Robinson VG, Waidyanatha S, DeVito MJ, Eifrid MA, Granville CA, Gibbs ST, Blystone CR 2019. Toxicokinetics of perfluorobutane sulfonate (PFBS), perfluorohexane-1-sulphonic acid (PFHxS), and perfluorooctane sulfonic acid (PFOS) in male and female Hsd:Sprague Dawley SD rats after intravenous and gavage administration. Toxicol. Rep 6, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C 2015. Perfluorinated compounds: An overview. Toxicological Effects of Perfluoroalkyl and Polyfluoroalkyl Substances, ed. DeWitt J, Humana Press, pp. 1–21. [Google Scholar]
- Li X, Wang Z and Klaunig JE 2019. The effects of perfluorooctanoate on high fat diet induced non-alcoholic fatty liver disease in mice. Toxicology 416:1–14. [DOI] [PubMed] [Google Scholar]
- Livak KJ and Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Lou I, Wambaugh J, Lau C, Hanson RG, Lindstrom AB, Strynar MJ, Zehr RD, Setzer RW and Barton HA 2009. Modeling single and repeated dose pharmacokinetics of PFOA in mice. Toxicol. Sci, 107, 331–341. [DOI] [PubMed] [Google Scholar]
- Martin MT, Breman R, Hu W, Ayanoglu E, Lau C, Ren H Wood CR, Corton JC, Kavlock RJ and Dix DJ 2007. Toxicogenomic study of triazole fungicides and perfluoroalkyl acids in rat livers accurately categorizes chemicals and identifies mechanisms of toxicity. Toxicol. Sci 97, 595–613. [DOI] [PubMed] [Google Scholar]
- Nash JC 2014. On best practice optimization methods in R. Journal of Statistical Software 60, 1–14. [Google Scholar]
- Nash JC, and Varadhan R 2011. Unifying optimization algorithms to aid software system users: optimx for R. Journal of Statistical Software 43, 1–14. [Google Scholar]
- O’Flaherty EJ 1981. Toxicants and Drugs: Kinetics and Dynamics. John Wiley & Sons. [Google Scholar]
- Oldham ED, Xie W, Farnoud AM, Fiegel J and Lehmler HJ 2012. Disruption of phosphatidylcholine monolayers and bilayers by perfluorobutane sulfonate. J. Phys. Chem. B 166, 9999–10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen GW, Chang SC, Noker PE, Gorman GS, Ehresman DJ, Lieder PH and Butenhoff JL 2009. A comparison of the pharmacokinetics of perfluorobutanesulfonate (PFBS) in rats, monkeys, and humans. Toxicology 256, 65–74. [DOI] [PubMed] [Google Scholar]
- Qi W, Clark JM, Timme-Laragy AR and Park Y 2018. Perfluorobutanesulfonic acid (PFBS) potentiates adipogenesis of 3T3-L1 adipocytes. Food and Chem. Toxicol 120, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner JL, Nakayama SF, Delinsky AD, Stanko JP, Fenton SE, Lindstrom AB and Strynar MJ 2009. Analysis of PFOA in dosed CD1 mice. Part 1. Methods development for the analysis of tissues and fluids from pregnant and lactating mice and their pups. Reprod. Toxicol 27, 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Vallanat BV, Nelson DM, Yeung LWY, Guruge KS, Lam PKS, Lehman-McKeeman LD and Corton JC 2009. Evidence for the involvement of xenobiotic-responsive nuclear receptors in transcriptional effects upon perfluoroalkyl acid exposure in diverse species. Reprod. Toxicol 27, 266–277. [DOI] [PubMed] [Google Scholar]
- Rosen MB, Das KP, Wood C, Wolf CJ, Abbott BD and Lau C 2013. Evaluation of perfluoroalkyl acid activity using primary mouse and human hepatocytes. Toxicology 308:129–137. [DOI] [PubMed] [Google Scholar]
- Russell MH, Nilsson H and Buck RC 2013. Elimination kinetics of perfluorohexanoic acid in humans and comparison with mouse, rat and monkey. Chemosphere 93, 2419–2425. [DOI] [PubMed] [Google Scholar]
- Sant KE,m Venezia OL, Sinno PP and Timme-Laragy AR 2019. Perfluorobutanesulfonic acid disrupts pancreatic organogenesis and regulation of lipid metabolism in the zebrafish, Danio rerio. Toxicol. Sci 167, 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAS/STAT® software v.13.1. 2013. SAS Institute Inc., Cary, NC, USA. [Google Scholar]
- Sundstrom M, Chang SC, Noker PE, Gorman GS, Hart JA, Ehresman DJ, Bergman A, and Butenhoff JL 2012. Comparative pharmacokinetics of perfluorohexanesulfonate (PFHxS) in rats, mice, and monkeys. Reprod. Toxicol 33, 441–451. [DOI] [PubMed] [Google Scholar]
- Tatum-Gibbs K, Wambaugh J Das KP, Zehr RD, Strynar MJ, Lindstrom AB, Delinsky A and Lau C 2011. Comparative pharmacokinetics of perfluorononanoic acid in rats and mice. Toxicology 281, 48–55. [DOI] [PubMed] [Google Scholar]
- Vanden Heuvel JP, Thompson JT, Frame SR and Gillies PJ 2006. Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptor-alpha, -beta, and -gamma, liver X receptor-beta, and retinoid X receptor-alpha. Toxicol. Sci 92:476–489. [DOI] [PubMed] [Google Scholar]
- Wolf CJ, Schmid JE, Lau C and Abbott BD 2012. Activation of mouse and human peroxisome proliferator-activated receptor-alpha (PPARα) by perfluoroalkyl acids (PFAAs): Further investigation of C4 – C12 compounds. Reprod. Toxicol 33, 546–551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.