

Abstract
P-glycoprotein (P-gp) plays a critical role in drug oral bioavailability and modulation of this transporter can alter the safety and/or efficacy profile of substrate drugs. Individual oral molecular excipients that inhibit P-gp function have been considered as a mechanism for improving drug absorption, but a systematic evaluation of the interaction of excipients with P-gp is critical for informed selection of optimal formulations of proprietary and generic drug products. A library of 123 oral molecular excipients was screened for their ability to inhibit P-gp in two orthogonal cell-based assays. β-Cyclodextrin and Light green SF yellowish were identified as modest inhibitors of P-gp with IC50 values of 168 μM (95% CI, 118–251 μM) and 204 μM (95% CI, 5.9–1745 μM), respectively. The lack of effect of most of the tested excipients on P-gp transport provides a wide selection of excipients for inclusion in oral formulations with minimal risk of influencing the oral bioavailability of P-gp substrates.
Keywords: P-glycoprotein, oral excipients, Calcein-AM assay, digoxin flux, screening assays
Introduction
P-glycoprotein (P-gp, also known as Multidrug Resistance Protein 1 (MDR1) or ATP-binding cassette transporter B1 (ABCB1)) is an ABC transporter with established roles in drug disposition, efficacy and drug-drug interactions (DDIs) [1, 2]. Its ability to transport numerous structurally diverse compounds, including xenobiotics and endogenous substrates, and its ubiquitous expression in humans in tissues such as the intestine, kidney, liver, and brain underlie its critical role in pharmacology [3–5]. P-gp is well recognized for its ability to cause multidrug resistance in cancer through efflux of drugs out of the tumor [6]. In addition, P-gp plays an important role in intestinal absorption and hepatic and urinary excretion of drugs and serves as a biochemical barrier to the entry of xenobiotics into the central nervous system, the placenta and the testis [1, 7]. The contribution of P-gp to the pharmacokinetics and pharmacodynamics of many drug substrates has led to significant interest in understanding the regulation of P-gp function by drugs and other xenobiotics [2, 3].
Oral formulations are the most common, convenient and preferred route for drug administration and may involve drug release at the specific target site of absorption in a controlled manner [8, 9]. To improve the absorption of a drug molecule with suboptimal physiochemical properties (e.g., low solubility, limited permeability or high metabolism), molecular excipients are included in the oral dosage form [10, 11]. Excipients are substances other than the active pharmaceutical ingredient that are added to the formulation to increase stability, solubility, swellability, viscosity, biodegradability or buffering capacity. In addition, excipients can add nutrients, flavor or color, increase absorption, bioavailability or shelf life of the active ingredient, and add pH dependency or oxidation-reduction potential for its mechanistic action at a specific site [10, 12, 13]. Different excipients play distinct roles in oral dosage forms and significantly influence characteristics of the final product [14–16].
Oral molecular excipients are considered safe additives to drugs with the potential to interact with cellular proteins and alter their function. A recent study using computational predictions, cell-based assays and in vivo rodent studies identified excipients that interact with numerous target proteins and clinical safety targets [17]. One concern is that interactions between P-gp and excipients may lead to variability in absorption between drug formulations. Previous studies have largely focused on the identification of excipients that inhibit P-gp to increase drug bioavailability. The most widely studied excipients identified as P-gp inhibitors are polyethylene glycols, polysorbates, Cremophor EL and pluronics [18–27]. The inhibitory interaction of Vitamin E-PEG with P-gp has been exploited to increase the oral absorption of paclitaxel [28]. Similarly, the absorption of ganciclovir has been enhanced using excipients such as Cremophor EL-35, Pluronic block copolymer F68, PEG-400, Tween-80 and Labrasol, and the bioavailability of ranitidine was significantly increased with PEG-400 [29–31].
Still, many excipients have not been screened for interactions with P-gp. In the current study, 123 orally administered molecular excipients listed in the FDA Inactive Ingredient Database (https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm) [32] were screened using two orthogonal systems, HEK and MDCK cells overexpressing human P-gp. These findings expand our understanding of the interaction of oral molecular excipients with P-gp.
Materials and Methods
Materials
Calcein-AM (ENZ-52002) was purchased from Enzo Life Sciences. Poly D-lysine (P6407), cyclosporine A (C1832), elacridar (SML0486), digoxin (D6003) and hygromycin B (10843555001) were all purchased from Sigma Aldrich. Verapamil (06–541-G) was purchased from Fisher Scientific. Dulbecco’s Modified Eagle Medium with Glucose (DME-H21, CCFAA005), fetal bovine serum (FBS, CCFAP004), penicillin-streptomycin (CCFGK004), trypsin-EDTA (CCFGP001) and phosphate-buffered saline (PBS, CCFAL001) were all purchased from the UCSF Cell Culture Facility. Dulbecco’s PBS (DPBS) without calcium chloride and magnesium chloride was from Gibco (14190) and 3H-labeled digoxin (NET222250UC) was purchased from Perkin Elmer. Purchasing information for excipients was reported previously [33].
Cell lines and culture
Flp-In™-293 human embryonic kidney cells transfected with pcDNA5/FRT plasmid with (HEK293-MDR1) or without (HEK293-EV) the ABCB1 cDNA were described previously [34]. Cells were cultured in T-25 flasks in DMEM medium with 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml) and hygromycin B (75 μg/ml) at 37°C in a humidified incubator with 5% CO2. MDCK-hMDR1-cMDR1-KO cells have endogenous canine MDR1 knocked out and stably express human MDR1 and were kindly provided by Dr. Per Artursson (Uppsala University) along with the corresponding MDCK-cMDR1-KO cells [35, 36]. MDCK-hMDR1-cMDR1-KO cells were grown in similar conditions as the HEK cells except that hygromycin B was added at 400 μg/ml. Cells were passaged every 3–4 days after reaching 80–90% confluency by incubating with 1 ml of 0.25% Trypsin-EDTA at 37°C without shaking for 5–10 min, resuspending in DMEM medium and transferring to a 15 ml Falcon tube. The cell suspension was centrifuged at 100g for 5 min, and the resulting cell pellet was resuspended in 4–5 ml of supplemented DMEM medium as described above, then diluted at a 1:4 or 1:5 ratio in a final volume of 8 ml and placed into a T-25 flask. Cells were not used beyond passage 10.
Calcein accumulation assay
A calcein accumulation assay was performed as described previously [37]. In brief, HEK-MDR1 and HEK-EV cells were seeded onto 96-well plates coated with 50 μg/ml poly-D-lysine at a density of 8 × 105 cells/0.32 cm2, 24 hours prior to experiments. Cells were washed thrice with ice-cold PBS and incubated with calcein-AM (5 μM) in the presence and absence of the studied excipient or the known P-gp inhibitor verapamil (500 μM) at 37°C for 1 hr. The calcein accumulation assays were terminated by washing cells thrice with ice-cold PBS. The amount of accumulated calcein was quantified by measuring intracellular fluorescence at an excitation/emission of 485 nm/590 nm using a Genios Pro fluorescence plate reader (Tecan, Switzerland). The inhibitory effect of tested excipients on P-gp-mediated specific transport of calcein-AM was expressed as a normalized calcein accumulation ratio using the following equation:
where (P-gp + I) and (P-gp − I) represent the fluorescence of accumulated calcein in HEK293-MDR1 cells in the presence and absence of the excipient or known inhibitor, respectively and (EV + I) and (EV − I) represent the fluorescence of accumulated calcein in the HEK293-EV cells in the presence and absence of the excipient or known inhibitor, respectively.
Digoxin flux assay
For transwell assays, procedures were modified from published reports [36, 38]. Briefly, 2.1 × 104 MDCK-hMDR1-cMDR1-KO cells/well were seeded onto Falcon 24 multiwell inserts with a microporous polyethylene terephthalate membrane (351181, Corning Life Sciences) and complementary 24 well plate (353047, Corning Life Sciences). During differentiation, the volume of medium was 300 μl in the apical chamber and 1000 μl in the basal chamber. The cells were differentiated for 7–10 days and lucifer yellow permeability assays were carried out to check the integrity of the cell monolayer before the digoxin flux assays. Cells were washed twice with Hanks’ balanced salt solution (HBSS) at pH 7.4 with calcium chloride and magnesium chloride (Gibco 14025). Lucifer yellow (0.1 mg/ml) in HBSS was added to the apical chamber and the basal chamber was filled with fresh HBSS. The assembled plate was kept at 37°C for 1 hour and 15 μl aliquots were taken from the basal chamber for fluorescence measurements. The % permeability was calculated on a per-well basis as
where Fw is the fluorescence of a sample from an individual well, Fb is the fluorescence of HBSS and Fly is the fluorescence of the lucifer yellow solution. Wells with cell monolayer permeability of >3% were not used for the digoxin flux assay. After lucifer yellow measurements, cells were washed with HBSS before the addition of fresh medium to the apical and basal chambers. The cells recovered overnight and were used the next day for digoxin flux assays.
Digoxin flux from the basal to apical side was measured as described in previous studies [38]. Cells were washed twice with HBSS before addition to the basal chamber (700 μl) of an HBSS solution of digoxin (6.35 nM 3H-labeled, final concentration 2.5 μM) in the presence or absence of the known P-gp inhibitor cyclosporine A (10 μM) or the indicated concentration of studied excipient. The same concentration of known inhibitor or excipient and 0.1 mg/ml lucifer yellow were added to the apical chamber in HBSS (225 μl). Plates were incubated at 37°C with shaking at 300 rpm and 25 μl aliquots were removed from the apical compartment at 1, 1.5 and 2 hours and replaced with the same volume of apical solution. Samples (25 μl) were also taken from the basal chambers before the start and at the end of each experiment to calculate the dpm/pmole ratio and for mass balance calculations. Scintillation fluid (2.5 ml, 6013141, Perkin Elmer) was added to all apical and basal samples and radioactivity was measured by liquid scintillation counting (Beckman LS6500). Additionally, 15 μl samples were collected from the basal chamber at the end of the assay for lucifer yellow fluorescence measurements. For data analysis, digoxin dpm counts from the apical samples were converted to pmoles using the measured radioactivity in the substrate solution. The rate of basal to apical transport was calculated by linear regression using the timed samples and the effect of each excipient was expressed as a fold change in transport rate compared to digoxin alone. Fold change in rate was calculated as
where R(B-A) + I and R(B-A) − I are the rates of digoxin transport from the basal to apical side in the presence and absence of known inhibitor or excipient, respectively. The permeability of the cell monolayer at the end of the digoxin flux assay was calculated using lucifer yellow as described above.
Excipient screening
An initial pool of 123 excipients for screening was a subset of 136 FDA-approved excipients that have been described previously [33]. Excipients which are no longer used, commercially unavailable, poorly soluble or formulated for inhalation were not included in the screening. The excipients were screened at a concentration of 200 μM except those with limited solubility (10 μM for Yellow 62 and 50 μM for docusate sodium salt, sodium lauryl sulfate, cetyl pyridinium chloride, D&C Red No. 6, Oil Orange SS, propylparaben, glyceryl caprylate and Yellow AB) and sugars (1 mM for sucrose, dextrose, D-tagatose, mannose, galactose, maltose, fructose and sucralose). The screening of excipients included three technical replicates for the calcein accumulation assays and two technical replicates for the digoxin flux assays.
Dose-response analysis
Potential P-gp inhibitors identified in each respective screen were further evaluated over a range of excipient concentrations using the same assays. IC50 estimates were obtained by fitting the dose-response data using the log (inhibitor) vs. response – variable slope (four parameters) relationship in GraphPad Prism 9.
Results
Screening overview
A total of 123 molecular excipients were screened for their ability to inhibit P-gp transport function in two different assays, a calcein-AM fluorescence-based accumulation assay and a digoxin flux assay. The screened excipients represent diverse functional classes and include flavoring agents (25%), dyes (20%), antimicrobial agents (12%), buffering agents (8%), nutrient supplements (6%), solubilizing agents (4%), surfactants (4%), antioxidants (3%), diluents (3%), coating agents (2%) and plasticizing agents (2%). The overview of the screening process is outlined in Figure 1. Potential P-gp inhibitors were subsequently examined in dose-response studies to estimate IC50 values. Finally, the potency of excipients confirmed as inhibitors from either assay was compared to estimated intestinal concentrations of the tested excipients to evaluate the potential for inhibition of human P-gp in vivo.
Figure 1.
Design of screening for interaction of oral molecular excipients with P-gp.
Inhibition of calcein accumulation by excipients
Calcein accumulation was ~3-fold higher in the HEK-EV cells compared to the HEK-MDR1 cells and was robustly inhibited in the HEK-MDR1 cells by the known P-gp inhibitor verapamil (Figure 2A). There was minimal effect of verapamil on calcein accumulation in the HEK-EV cells (Figure 2A) and the normalized calcein accumulation ratio was 2.2 for verapamil (Figure 2B). Verapamil was included as a positive control in all screening assays. The distribution of normalized calcein accumulation ratios is shown in Figure 2C and the accumulation ratios are tabulated for all tested excipients in Table I. A threshold of 40% increase in normalized accumulation ratios was used to identify the excipients with the most potent effects on P-gp activity and identified 10 potential inhibitors. Individual inspection of the +excipient/-excipient calcein ratios in each cell line for these potential inhibitors differentiated between specific effects on P-gp activity from non-specific effects, possibly due to cytotoxicity. Only two excipients (D&C Red No. 6 and D&C Brown No. 1) inhibited calcein flux at least 40% in the MDR1-overexpressing cells and had no effect in the EV cells (Figure 2D). Seven excipients were eliminated as potential inhibitors due to a non-specific effect resulting in a decrease in calcein ratios (+excipient/-excipient) in both the control and MDR1-overexpressing cells (lower left quadrant in Figure 2D) that translated into increased normalized calcein accumulation ratios (benzalkonium chloride, docusate sodium, D&C Red No. 33, D&C Red No. 27, D&C Red No. 28, D&C Orange No. 4 and D&C Red No. 3). The remaining excipient with a normalized accumulation ratio >1.4 (NaHCO3) was also eliminated since it demonstrated <40% increase in calcein accumulation in the MDR1-overexpressing cells. Dose-response studies with the two putative inhibitors failed to validate either of these dyes as an inhibitor of P-gp at concentrations <300 μM (Supplemental Figure 1), which is more than 100-fold higher than the maximum predicted intestinal concentration based on the maximum potency per unit dose listed in the FDA Inactive Ingredient Database [32].
Figure 2.
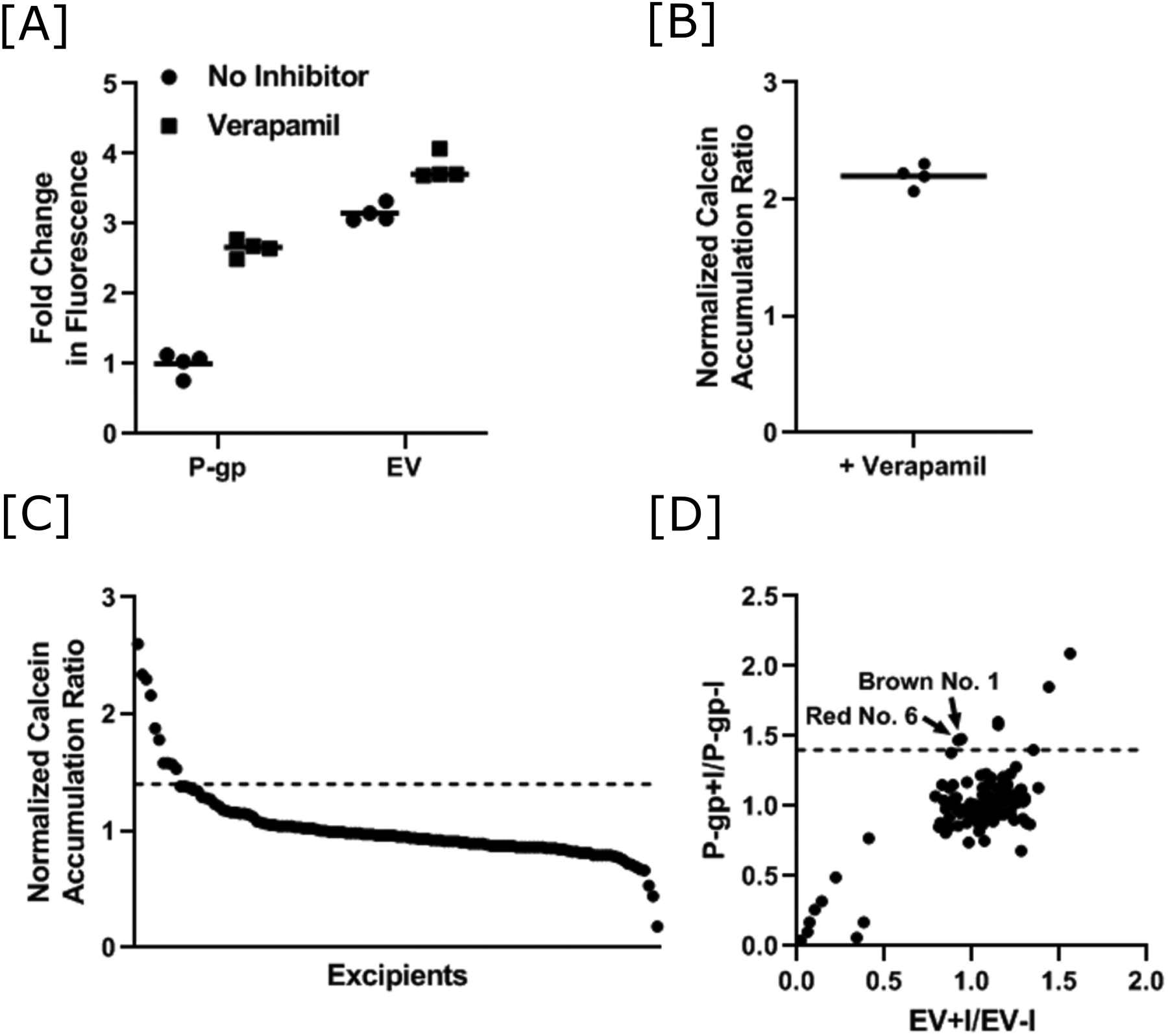
Identification of oral molecular excipients as P-gp inhibitors using a calcein-AM assay. [A] Difference in calcein fluorescence of HEK-empty vector (EV) and HEK-MDR1 (P-gp) cells after incubation with 5 μM calcein-AM in the absence and presence of the known P-gp inhibitor verapamil (62.5 μM). The mean of four replicates for the HEK-MDR1 cells was normalized to 1. Values are shown for each replicate and the lines represent the mean value. [B] The data in panel A were transformed into normalized calcein accumulation ratios by taking the ratio of fluorescence in the presence and absence of verapamil in the HEK-MDR1 cells to the ratio of fluorescence in the presence and absence of verapamil in the HEK-EV cells. Values are shown for each replicate and the line represents the mean value. [C] Normalized calcein accumulation ratios for 123 oral excipients are shown in decreasing order. The dashed line at 1.4 represents the cut-off used to assign potential inhibitors. [D] A scatter plot of the P-gp ratio with the EV ratio for the 123 molecular excipients that were screened is shown. The dashed line at 1.4 represents the cut off used to assign inhibition of P-gp. Excipients that inhibit P-gp at least 40% in the HEK-MDR1 cells and have no effect in the HEK-EV cells are labeled.
Table I.
Summary of results from calcein-AM and digoxin screening assays
| Excipient | 1Functional Category | Calcein-AM Assay | Digoxin Assay | |||||
|---|---|---|---|---|---|---|---|---|
| Normalized Accumulation Ratio2 | EV Ratio3 | MDR1 Ratio4 | Classification5 | Fold Change in Flux6 | Lucifer Yellow Permeability (%)7 | Classification8 | ||
| D&C Red No. 28 | Dye | 2.60 | 0.10 | 0.26 | False positive | 0.45 | 186 | Fluorescent |
| Benzalkonium chloride | Antimicrobial | 2.34 | 0.07 | 0.17 | False positive | 3.87 | 7.79 | Permeabilizer |
| D&C Red No. 27 | Dye | 2.30 | 0.14 | 0.32 | False positive | 0.75 | 211 | Fluorescent |
| Docusate sodium | Surfactant | 2.16 | 0.22 | 0.49 | False positive | 2.47 | 7.6 | Permeabilizer |
| D&C Red No. 3 | Dye | 1.88 | 0.41 | 0.77 | False positive | 1.02 | 65.6 | Fluorescent |
| D&C Red No. 33 | Dye | 1.78 | 0.02 | 0.04 | False positive | 1.18 | 0.11 | Inert |
| D&C Red No. 6 | Dye | 1.58 | 0.92 | 1.47 | Inhibitor | 1.20 | 0.14 | Inert |
| D&C Brown No. 1 | Dye | 1.58 | 0.94 | 1.48 | Inhibitor | 0.68 | 0.15 | Inert |
| NaHCO3 | Buffering agent | 1.57 | 0.88 | 1.38 | Inert | 1.55 | 0.12 | Non-specific |
| D&C Orange No. 4 | Dye | 1.53 | 0.06 | 0.10 | False positive | 0.96 | 0.15 | Inert |
| Naphthol blue black | Dye | 1.38 | 1.15 | 1.60 | Inert | 1.08 | 0.01 | Inert |
| FD&C Red No. 4 | Dye | 1.38 | 0.83 | 1.15 | Inert | 1.28 | 0.12 | Inert |
| Butylparaben | Antimicrobial | 1.37 | 1.15 | 1.58 | Inert | 1.08 | 0.38 | Inert |
| Epilactose | Other | 1.35 | 0.79 | 1.07 | Inert | 0.80 | 0.08 | Inert |
| Acid blue 9 | Dye | 1.34 | 1.56 | 2.09 | Inert | 0.67 | 0.22 | Inert |
| Sodium 1,2-ethanedisulfonate | Salt forming | 1.29 | 0.87 | 1.13 | Inert | 1.11 | 0.11 | Inert |
| Light green CF yellowish | Dye | 1.28 | 1.44 | 1.85 | Inert | 0.55 | 0.31 | Inhibitor |
| Dextrose | Flavoring agent | 1.27 | 0.89 | 1.15 | Inert | 0.99 | 0.11 | Inert |
| FD&C Yellow No. 5 | Dye | 1.23 | 0.84 | 1.04 | Inert | 1.03 | 0.57 | Inert |
| Rhodamine B | Dye | 1.21 | 0.97 | 1.17 | Inert | 0.38 | 11.3 | Permeabilizer |
| Fumaric acid | Flavoring agent | 1.18 | 0.90 | 1.06 | Inert | 0.69 | 0.11 | Inert |
| Sodium salicylate | Other | 1.17 | 0.87 | 1.02 | Inert | 1.15 | 1.25 | Inert |
| Dibutyl sebacate | Plasticizer | 1.16 | 1.05 | 1.22 | Inert | 0.96 | 0.11 | Inert |
| Ethanol | Antimicrobial | 1.16 | 0.91 | 1.06 | Inert | 0.86 | 0.06 | Inert |
| Indigo carmine | Dye | 1.15 | 0.85 | 0.98 | Inert | 1.06 | 0.08 | Inert |
| Erythritol | Flavoring agent | 1.15 | 0.90 | 1.03 | Inert | 0.93 | 0.05 | Inert |
| Ethyl vanillin | Flavoring agent | 1.14 | 1.08 | 1.23 | Inert | 1.07 | 0.19 | Inert |
| Ethyl maltol | Flavoring agent | 1.12 | 0.90 | 1.00 | Inert | 0.96 | 0.09 | Inert |
| Tributyl-2-acetyl citrate | Plasticizer | 1.08 | 0.91 | 0.98 | Inert | 1.10 | 0.13 | Inert |
| Sucrose | Coating agent | 1.07 | 0.82 | 0.88 | Inert | 1.03 | 0.20 | Inert |
| Sodium bitartrate | Flavoring agent | 1.06 | 1.06 | 1.13 | Inert | 1.28 | 0.29 | Inert |
| Oil orange SS | Dye | 1.05 | 0.87 | 0.92 | Inert | 1.89 | 0.38 | Non-specific |
| L-ascorbic acid | Antioxidant | 1.05 | 0.81 | 0.85 | Inert | 1.08 | 0.29 | Inert |
| Butyl alcohol | Solubilizer | 1.04 | 1.09 | 1.15 | Inert | 1.19 | 0.13 | Inert |
| FD&C Red No. 40 | Dye | 1.04 | 1.09 | 1.15 | Inert | 1.25 | 0.03 | Inert |
| Ammonium glycyrrhizate | Flavoring agent | 1.04 | 1.11 | 1.2 | Inert | 1.54 | 0.11 | Non-specific |
| Na3PO4 | Buffering agent | 1.04 | 1.35 | 1.40 | Inert | 1.48 | 0.09 | Non-specific |
| Succinic acid | Flavoring agent | 1.03 | 0.95 | 0.98 | Inert | 0.86 | 0.13 | Inert |
| Ponceau 3R | Dye | 1.03 | 0.99 | 1.02 | Inert | 0.97 | 0.05 | Inert |
| 2-Ethoxyethanol | Solvent | 1.02 | 0.93 | 0.95 | Inert | 1.37 | 0.08 | Inert |
| Pigment yellow 62 | Dye | 1.02 | 1.18 | 1.21 | Inert | 0.99 | 0.05 | Inert |
| Carmine | Dye | 1.02 | 1.25 | 1.28 | Inert | 1.26 | 0.16 | Inert |
| Ponceau xylidine | Dye | 1.01 | 1.06 | 1.08 | Inert | 1.05 | 0.09 | Inert |
| Propyl gallate | Antimicrobial | 1.00 | 1.22 | 1.23 | Inert | 1.54 | 0.31 | Non-specific |
| Cetyl alcohol | Coating agent | 1.00 | 1.18 | 1.18 | Inert | 0.82 | 0.09 | Inert |
| Sodium benzoate | Antimicrobial | 0.99 | 0.84 | 0.84 | Inert | 0.99 | 0.18 | Inert |
| Triethyl citrate | Plasticizer | 0.99 | 0.86 | 0.86 | Inert | 1.04 | 0.33 | Inert |
| EDTA | Chelator | 0.99 | 1.01 | 1.01 | Inert | 0.93 | 0.09 | Inert |
| L-arginine | Nutrient suppl. | 0.99 | 1.14 | 1.13 | Inert | 1.33 | 0.07 | Inert |
| D-tagatose | Flavoring agent | 0.98 | 1.03 | 1.01 | Inert | 1.08 | 0.06 | Inert |
| Acesulfame potassium | Flavoring agent | 0.98 | 0.86 | 0.85 | Inert | 1.04 | 2.08 | Inert |
| Neotame | Flavoring agent | 0.98 | 1.08 | 1.07 | Inert | 0.97 | 0.11 | Inert |
| Vanillin | Flavoring agent | 0.98 | 0.97 | 0.95 | Inert | 1.33 | 0.04 | Inert |
| Propylparaben | Antimicrobial | 0.97 | 1.19 | 1.15 | Inert | 1.43 | 0.20 | Inert |
| Xylitol | Flavoring agent | 0.97 | 1.10 | 1.07 | Inert | 1.37 | 0.06 | Inert |
| Ext D&C yellow No. 7 | Dye | 0.97 | 1.20 | 1.16 | Inert | 1.23 | 0.14 | Inert |
| Potassium sorbate | Antimicrobial | 0.96 | 1.19 | 1.15 | Inert | 1.45 | 0.10 | Non-specific |
| Sorbitol | Diluent | 0.96 | 0.87 | 0.85 | Inert | 0.97 | 0.13 | Inert |
| Glycerin | Antimicrobial | 0.96 | 0.85 | 0.82 | Inert | 0.88 | 0.28 | Inert |
| Sodium gluconate | Nutrient suppl. | 0.96 | 1.10 | 1.06 | Inert | 1.10 | 0.18 | Inert |
| Propylene glycol | Antimicrobial | 0.96 | 0.85 | 0.81 | Inert | 1.32 | 0.27 | Inert |
| Sodium acetate | Antimicrobial | 0.95 | 1.20 | 1.15 | Inert | 1.16 | 0.11 | Inert |
| Butylated hydroxytoluene | Antioxidant | 0.95 | 0.99 | 0.95 | Inert | 1.45 | 2.57 | Non-specific |
| Calcium pyrophosphate | Nutrient suppl. | 0.95 | 1.07 | 1.02 | Inert | 1.05 | 0.05 | Inert |
| Transanethol | Flavoring agent | 0.94 | 1.20 | 1.14 | Inert | 1.28 | 0.31 | Inert |
| Maltitol | Flavoring agent | 0.94 | 1.05 | 0.98 | Inert | 1.01 | 0.09 | Inert |
| Lactose monohydrate | Diluent | 0.93 | 0.99 | 0.93 | Inert | 1.25 | 0.09 | Inert |
| Isopropanol | Antimicrobial | 0.93 | 1.04 | 0.97 | Inert | 1.01 | 0.10 | Inert |
| Sodium lauryl sulfate | Surfactant | 0.93 | 0.92 | 0.86 | Inert | 2.23 | 7.98 | Permeabilizer |
| Glycine | Buffering agent | 0.92 | 1.06 | 0.97 | Inert | 1.36 | 0.11 | Inert |
| L-Malic acid | Flavoring agent | 0.92 | 1.17 | 1.08 | Inert | 1.21 | 0.12 | Inert |
| Acetophenone | Solubilizer | 0.92 | 1.14 | 1.05 | Inert | 1.20 | 0.04 | Inert |
| Mannose | Flavoring agent | 0.91 | 1.14 | 1.04 | Inert | 1.00 | 0.06 | Inert |
| Methanolamine | Buffering agent | 0.91 | 1.13 | 1.03 | Inert | 1.11 | 0.11 | Inert |
| Sodium thiosulfate | Antioxidant | 0.91 | 0.97 | 0.88 | Inert | 0.99 | 0.11 | Inert |
| Na2HPO4 | Buffering agent | 0.91 | 1.04 | 0.94 | Inert | 1.34 | 0.06 | Inert |
| Glyceryl caprylate | Surfactant | 0.90 | 1.08 | 0.97 | Inert | 1.05 | 0.13 | Inert |
| Methylparaben sodium | Antimicrobial | 0.90 | 1.10 | 0.99 | Inert | 1.59 | 0.07 | Non-specific |
| Sodium citrate | Buffering agent | 0.89 | 1.11 | 1.00 | Inert | 1.13 | 0.18 | Inert |
| Mannitol | Diluent | 0.89 | 1.22 | 1.08 | Inert | 1.24 | 0.13 | Inert |
| Methanol | Solubilizer | 0.89 | 1.02 | 0.92 | Inert | 1.05 | 0.10 | Inert |
| L-Histidine | Nutrient suppl. | 0.89 | 1.09 | 0.98 | Inert | 1.18 | 0.22 | Inert |
| Piperazine | Flavoring agent | 0.88 | 1.05 | 0.94 | Inert | 1.58 | 0.07 | Non-specific |
| Tromethamine | Buffering agent | 0.87 | 1.28 | 1.12 | Inert | 1.29 | 0.07 | Inert |
| Cinnamaldehyde | Flavoring agent | 0.87 | 1.10 | 0.96 | Inert | 3.14 | 103 | Fluorescent |
| DL-α-tocopherol acetate | Antioxidant | 0.87 | 1.19 | 1.05 | Inert | 1.50 | 0.09 | Non-specific |
| Meglumine | Buffering agent | 0.87 | 1.24 | 1.07 | Inert | 1.02 | 0.11 | Inert |
| Maleic acid | Buffering agent | 0.87 | 1.06 | 0.92 | Inert | 1.13 | 0.09 | Inert |
| Ethylparaben | Antimicrobial | 0.87 | 1.27 | 1.11 | Inert | 1.10 | 0.14 | Inert |
| Galactose | Flavoring agent | 0.86 | 1.09 | 0.94 | Inert | 1.34 | 0.06 | Inert |
| Malic acid | Flavoring agent | 0.86 | 1.12 | 0.97 | Inert | 1.14 | 0.08 | Inert |
| Tartaric acid | Flavoring agent | 0.86 | 1.14 | 0.98 | Inert | 1.47 | 0.08 | Non-specific |
| Glutamic acid | Nutrient suppl. | 0.86 | 1.18 | 1.02 | Inert | 0.72 | 0.12 | Inert |
| Lactitol | Diluent | 0.86 | 1.03 | 0.89 | Inert | 1.17 | 0.09 | Inert |
| Isomalt | Coating agent | 0.86 | 1.06 | 0.91 | Inert | 1.52 | 0.08 | Non-specific |
| Amarnath | Dye | 0.86 | 1.22 | 1.05 | Inert | 1.12 | 0.02 | Inert |
| NaH2PO4 | Buffering agent | 0.85 | 1.04 | 0.89 | Inert | 1.31 | 0.06 | Inert |
| Maltose | Flavoring agent | 0.85 | 1.11 | 0.95 | Inert | 1.61 | 0.10 | Non-specific |
| Urea | Surfactant | 0.85 | 1.28 | 1.08 | Inert | 1.21 | 0.07 | Inert |
| Neohesperidin dihyhrochalone | Flavoring agent | 0.84 | 1.13 | 0.96 | Inert | 1.61 | 0.18 | Non-specific |
| Sodium sulfite | Antimicrobial | 0.84 | 1.04 | 0.87 | Inert | 0.95 | 0.09 | Inert |
| Myristyl alcohol | Emollient | 0.83 | 1.24 | 1.03 | Inert | 0.90 | 0.08 | Inert |
| FD&C yellow No. 6 | Dye | 0.82 | 1.25 | 1.02 | Inert | 1.03 | 0.19 | Inert |
| Fructose | Flavoring agent | 0.82 | 1.06 | 0.87 | Inert | 1.36 | 0.14 | Inert |
| Methacrylic acid | Other | 0.81 | 1.38 | 1.13 | Inert | 1.10 | 0.08 | Inert |
| Acetone | Solubilizer | 0.81 | 1.14 | 0.93 | Inert | 1.08 | 0.06 | Inert |
| DL-Aspartic acid | Nutrient suppl. | 0.81 | 1.30 | 1.06 | Inert | 1.14 | 0.07 | Inert |
| Sodium 4-aminobenzoate | Other | 0.79 | 1.12 | 0.89 | Inert | 1.03 | 0.13 | Inert |
| Sucrose palmitate | Surfactant | 0.79 | 1.30 | 1.04 | Inert | 1.48 | 2.60 | Non-specific |
| Aspartame | Flavoring agent | 0.79 | 1.29 | 1.03 | Inert | 1.18 | 0.10 | Inert |
| Chloromethane | Other | 0.79 | 1.22 | 0.97 | Inert | 1.38 | 0.04 | Inert |
| β-Cyclodextrin | Solubilizer | 0.79 | 1.18 | 0.94 | Inert | 0.25 | 0.14 | Inhibitor |
| Maltol | Flavoring agent | 0.78 | 1.04 | 0.82 | Inert | 0.96 | 0.09 | Inert |
| MgSO4 | Nutrient suppl. | 0.77 | 1.22 | 0.94 | Inert | 0.84 | 0.04 | Inert |
| Sucralose | Flavoring agent | 0.75 | 0.98 | 0.74 | Inert | 1.11 | 0.16 | Inert |
| Sodium carbonate | Buffering agent | 0.72 | 1.24 | 0.90 | Inert | 1.39 | 0.19 | Inert |
| Ethyl acetate | Flavoring agent | 0.71 | 1.29 | 0.91 | Inert | 1.07 | 0.06 | Inert |
| Leucine | Flavoring agent | 0.69 | 1.07 | 0.75 | Inert | 1.40 | 0.06 | Inert |
| DL-Lactic acid | Flavoring agent | 0.67 | 1.31 | 0.88 | Inert | 1.15 | 0.07 | Inert |
| Cysteine hydrochloride | Nutrient suppl. | 0.66 | 1.33 | 0.87 | Inert | 1.05 | 0.06 | Inert |
| Sucrose monolaurate | Surfactant | 0.53 | 1.28 | 0.68 | Non-specific | 1.54 | 2.24 | Non-specific |
| Yellow AB | Dye | 0.44 | 0.38 | 0.17 | Non-specific | 1.81 | 0.14 | Non-specific |
| Cetylpyridinium chloride | Antimicrobial | 0.18 | 0.34 | 0.06 | Non-specific | 3.35 | 6.22 | Permeabilizer |
The most common use is noted for most excipients. For those marked ‘Other’ no clear indication was available.
Normalized accumulation ratio = where (P-gp + I) and (P-gp − I) represent the fluorescence of accumulated calcein in HEK-MDR1 cells in the presence and absence of the test compound, respectively and (EV + I) and (EV − I) represent the fluorescence of accumulated calcein in the HEK-EV cells in the presence and absence of the test compound, respectively.
EV ratio = where (EV + I) and (EV − I) represent the fluorescence of accumulated calcein in the HEK-EV cells in the presence and absence of the test compound, respectively.
MDR1 ratio = where (P-gp + I) and (P-gp − I) represent the fluorescence of accumulated calcein in the HEK-MDR1 cells in the presence and absence of the test compound, respectively.
Excipients were classified as inhibitors if the normalized accumulation ratio was ≥1.40 and the MDR1 ratio was ≥1.40. Excipients with a normalized accumulation ratio ≥1.40 with decreased ratios in both the EV and MDR1 cells were classified as false positives. Excipients with a normalized accumulation ratio ≤0.60 were classified as non-specific activators. All other excipients were considered inert.
Fold change in flux is the ratio of digoxin flux in the basal to apical direction in the presence and absence of the indicated excipient.
Lucifer yellow permeability is the percentage of lucifer yellow that moved from the apical to the basal chamber during the 2 hr flux assay.
Excipients were classified as inhibitors if the fold change in digoxin flux was ≤0.60 and lucifer yellow permeability was <3%. Excipients that interfered with the lucifer yellow measurement (permeability >50%) were classified as fluorescent and those that increased lucifer yellow permeability >3% and ≤50% were classified as permeabilizers. Excipients that increased digoxin flux ≥40% were classified as non-specific activators. All other excipients were considered inert.
Inhibition of digoxin flux by excipients
Basal to apical flux of digoxin was ~10-fold higher in the MDCK cells expressing human MDR1 compared to the control cells that do not expresscanine or human MDR1. Cyclosporine, a known inhibitor of P-gp, reduced digoxin flux in the human MDR1-overexpressing cells to similar levels as in the control cells, indicating specific transport of digoxin by P-gp (Figure 3A). Excipients were screened to identify potential inhibitors of digoxin flux, using a decrease in flux of at least 40% as the cutoff (Figure 3B and Table I). Four excipients were identified as potential inhibitors, two of which were not considered further because of their interference with fluorescence measurements of lucifer yellow (D&C Red No. 28) or increased lucifer yellow permeability (rhodamine B) (Figure 3C). The remaining two potential inhibitors were light green SF yellowish (45% decrease) and β-cyclodextrin (β-CD, 75% decrease). Dose-response analyses confirmed both of these excipients as modest inhibitors of digoxin flux, with IC50 estimates of 168 μM (95% CI, 118–251 μM, Figure 3D) and 204 μM (95% CI, 5.9–1745 μM, Figure 3E) for β-CD and light green SF yellowish, respectively (Figures 3D and 3E). In addition to D&C Red No. 28 and rhodamine B, seven more excipients could not be evaluated as P-gp inhibitors in this assay due to their effects on cell monolayer permeability (benzalkonium chloride, docusate sodium salt, sodium lauryl sulfate, cinnamaldehyde and cetyl pyridinium chloride) or interference with the lucifer yellow assay (D&C Red No. 27, D&C Red No. 3) (Figure 3C and Table I).
Figure 3.
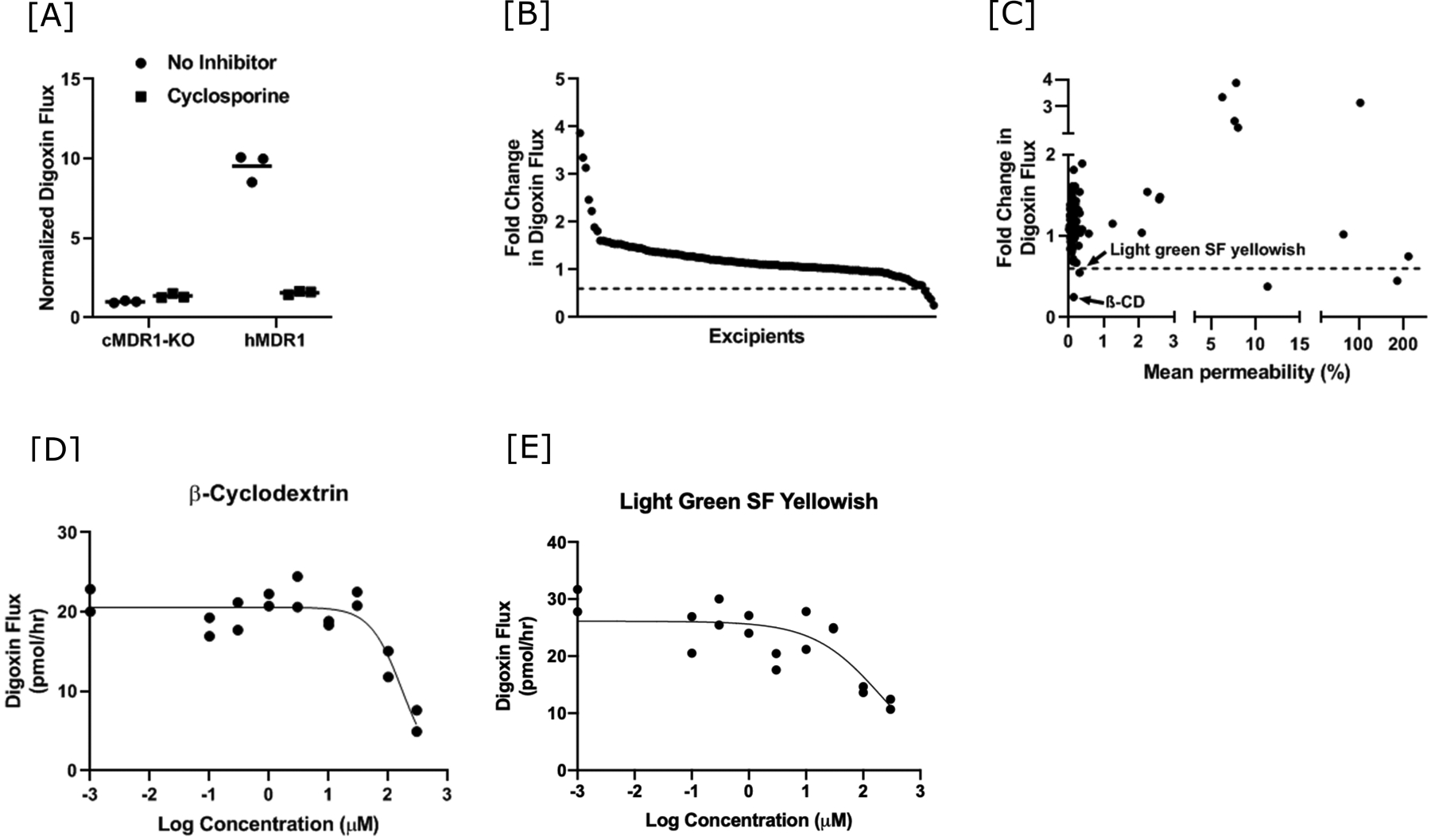
Identification of oral molecular excipients as P-gp inhibitors using a digoxin flux assay. [A] The rate of basal to apical digoxin flux in canine MDR1 knockout (cMDR1-KO) and canine MDR1 knockout overexpressing human MDR1 (hMDR1) MDCK cells is shown in the absence and presence of the known P-gp inhibitor cyclosporine (10 μM). Digoxin flux rates are normalized to the average in the absence of inhibitor in the cMDR1-KO cells. Values are shown for each replicate and the lines represent the mean value. [B] Fold change in digoxin flux rate for 123 oral molecular excipients in decreasing order. The dashed line at 0.6 represents the cut-off used to identify putative inhibitors. [C] Scatter plot of fold change in digoxin flux rate with respect to mean lucifer yellow permeability for the 123 oral excipients that were screened. Excipients that inhibit digoxin flux at least 40% and with lucifer yellow permeability <3% are labeled. Disruptions in both axes are used for better visualization of the data. [D and E] Representative excipient dose-response curves for the two putative P-gp inhibitors identified in the screening. Individual replicates from a single experiment are plotted. The curve was fit using the log(inhibitor) vs. response – variable slope (four parameters) relationship in GraphPad Prism 9. IC50 estimates were 168 μM (95% CI = 118–251 μM) for β-cyclodextrin and 204 μM (95% CI = 5.9–1745 μM) for light green SF yellowish.
Discussion
Intestinal P-gp plays a critical role in limiting the bioavailability of numerous drugs and its inhibition or induction can result in clinically significant drug-drug interactions [2–4]. While drug-drug interactions involving P-gp are extensively characterized both during drug development and post marketing, P-gp interactions with excipients have largely been limited to a few functional classes [18, 21, 39–41]. Molecular excipients are commonly used to optimize oral drug formulations and are generally considered to be inert. This tenet of excipient inertness was tested in the current study with a panel of 123 oral molecular excipients evaluated for their ability to modulate P-gp function in vitro using two orthogonal methods, an accumulation assay and a flux assay that indirectly and directly measured P-gp transport, respectively. The tested oral excipients are used as flavoring agents, solubilizing agents, antimicrobials, buffering agents, dyes and as nutrient supplements; most were inert with respect to P-gp transport activity. Light green SF yellowish, a dye, and β-CD, a solubilizing agent, were confirmed inhibitors with modest potency that is not predicted to influence P-gp function in the human intestine. These findings provide useful information towards developing an excipient selection strategy for pharmacologically active ingredients in oral dosage forms.
Excipients can be added at high amounts relative to the active pharmaceutical ingredient and in some cases are predicted to reach high intestinal concentrations within the limited volume (~250 ml) of the small intestine [32]. The use of high screening concentrations in both assays was based on an interest in only the most potent inhibitors with likely clinical relevance and the expectation of relatively high gut concentrations of the screened excipients. Despite these high concentrations, very few excipients had any effect on P-gp-mediated transport of either calcein-AM or the cardiac glycoside digoxin. Only two excipients with a normalized calcein accumulation ratio >1.4, D&C Red No. 6 and D&C Brown No. 1, increased intracellular levels of calcein fluorescence 40% or greater in HEK cells overexpressing human MDR1, without an effect on the control cells. However, dose-response analyses did not confirm significant inhibition of P-gp-mediated transport using the calcein accumulation assay and neither of these dyes reached the inhibition threshold for basal to apical flux of digoxin. It is worth noting that D&C Brown No. 1 inhibited basal to apical flux of digoxin by 32% and may warrant further evaluation as a potential P-gp inhibitor. The two excipients identified as possible P-gp inhibitors in the digoxin flux assay, light green SF yellowish and β-CD, were weak inhibitors with IC50 values of 204 μM and 168 μM, respectively. Light green SF yellowish increased calcein accumulation 85% and 44% in the HEK-MDR1 and HEK-EV cells, respectively, resulting in a normalized accumulation ratio (1.28) slightly below the cutoff for inhibitor classification. Considering that the increase in calcein fluorescence with addition of light green SF yellowish in the MDR1-overexpressing cells was two-fold higher than in control cells, the results from this screen also support its classification as a P-gp inhibitor. In contrast, β-CD had no effect on calcein-AM efflux.
Most of the excipients screened in the present study have not been previously evaluated for their potential to inhibit P-gp. One exception is the solubilizing agent β-CD, a cyclic oligosaccharide consisting of (α−1,4)-linked α-D-glucopyranose units forming cages with hydrophobic cavities and hydrophilic outer surfaces [42]. β-CD has been reported to enhance the intestinal absorption of berberine hydrochloride, a P-gp substrate, not only by increasing its dissolution rate, but also by inhibiting substrate-stimulated P-gp ATPase activity [43]. Related cyclodextrin derivatives have been extensively studied as P-gp inhibitors. Dimethyl- and methyl-β-CD have been characterized as P-gp inhibitors in cellular efflux assays and in situ intestinal absorption studies [40, 41, 44–49]. One mechanism for this inhibition is release of P-gp from the cell membrane [44, 45, 47, 49]. Dimethyl-β-CD was also shown to improve the bioavailability of tacrolimus in rats [48], supporting the potential for cyclodextrins to inhibit P-gp and enhance bioavailability in humans. In contrast, hydroxypropyl-β-CD, an excipient in the oral solution of itraconazole commonly used in DDI studies, was recently demonstrated to reduce the permeability of fenebrutinib across an MDCK monolayer and to limit fenebrutinib absorption in dogs [50]. These findings suggest complicated interactions between drugs and cyclodextrins in humans, including the potential for unintended DDIs during studies undertaken to inform drug labeling.
A recent screen of the same excipient library against breast cancer resistance protein (BCRP) indicated little overlap between inhibition of BCRP and P-gp. Only light green SF yellowish was identified in both studies, with a much higher potency for inhibition of BCRP in membrane vesicles (IC50 = 1.0 μM) compared to inhibition of P-gp-mediated digoxin flux in the current study [51]. For drugs that are substrates of both P-gp and BCRP, the combined inhibition of these transporters by light green SF yellowish could enhance bioavailability through independent effects on the transporters. These findings may warrant caution for oral drug formulations containing light green SF yellowish.
Although the MDCK transwell flux assay is considered a reasonable model for P-gp-mediated efflux, the lack of host and microbial metabolizing enzymes, the inability to mimic the transit of drugs through the intestine, and artificially high levels of P-gp expression limit the direct extrapolation of these findings to humans. The maximum amount per unit dose of β-CD and light green SF yellowish in marketed drug formulations is 133 mg and 40 mg, respectively [32]. Considering an intestinal volume of 250 ml, this translates into a predicted maximum intestinal concentration (Imax) of 470 μM for β-CD and 214 μM for light green SF yellowish. The FDA regulatory guidance In Vitro Drug Interaction Studies - Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions classifies transporter inhibitors of potential clinical significance if the [Imax] to [IC50] ratio is >10 [52]. [Imax]/[IC50] values of 1.1 and 2.8 for light green SF yellowish and β-CD, respectively, suggest that neither of these in vitro P-gp inhibitors is likely to show clinically significant inhibition of P-gp in humans (Supplemental Table I). However, the incorporation of increased amounts of β-CD into new formulations should be carefully evaluated for potential P-gp interactions.
A normalized calcein accumulation ratio was utilized to identify specific effects on P-gp transport, as applied in earlier studies of P-gp inhibition to reverse drug sensitivity [53]. However, this ratio led to numerous false positives and negatives. It was expected that there would be no effect on calcein-AM efflux in the HEK-EV cells, which held true for both D&C Red No. 6 and D&C Brown No. 1. However, closer inspection of the other potential inhibitors based on the normalized calcein accumulation ratio identified a number of excipients that reduced fluorescence to a larger extent in HEK-EV cells than in HEK-MDR1 cells, such that the normalized ratio was >1.4, the cutoff for inhibition. These false positives could be due to toxicity of the compounds, which was consistent with permeability measurements of lucifer yellow across the MDCK cell monolayer for benzalkonium chloride and docusate sodium. Additionaly, since calcein-AM is also a substrate for other ABC transporters endogenously expressed in HEK293 cells [54–56], activating effects of these excipients on one or more of these transporters could lead to increased calcein-AM efflux and a corresponding decrease in fluorescence. Interestingly, a number of the excipients identified as false positives in the calcein accumulation assay (D&C Red No. 3, D&C Red No. 27, D&C Red No. 28, D&C Orange No. 4, D&C Red No. 33 and docusate sodium) led to confounding results in a similar screen against BCRP [51]. These excipients inhibited BCRP overexpressed in Sf9 membrane vesicles, but not in HEK293 cells overexpressing BCRP. While these BCRP results may be explained at least in part by differences in availability of the excipient in the Sf9 inside-out vesicles compared to HEK293 cells, this cannot explain the current results with P-gp since both assays used in this study required the excipients to be membrane permeable for transporter inhibition. In addition to false positives, the normalized accumulation ratio also leads to potential false negatives. For example, acid blue 9, naphthol blue black, and butylparaben did not reach the inhibitor criteria based on the normalized accumulation ratio, although each increased calcein fluorescence in the MDR1-overexpressing cells by at least 60%. Although these excipients had no effect on digoxin flux, further investigation is warranted for potential interactions with P-gp.
Differences in results with the two screening assays have several plausible explanations. First, two substrates were used which may have different mechanisms of transport via P-gp. While both calcein-AM and digoxin are highly hydrophobic, structural differences may result in interactions with distinct residues in P-gp during transport. Similarly, identified excipient inhibitors may interact with different residues in P-gp, making it reasonable that the detection of P-gp inhibition is substrate dependent. We have previously shown that P-gp genetic variants have different sensitivities to cyclosporine inhibition and that P-gp inhibition is substrate-dependent [34]. A second difference in the assays was the use of fluorescence versus radioactivity. Several of the dyes that were tested (D&C Red No. 28, D&C Red No. 27 and D&C Red No. 3) interfered withfluorescence measurements for calcein accumulation as well as permeability measurements using lucifer yellow. Differences in sensitivity of transport measurement between the two substrates are also possible, although the common use of both calcein-AM and digoxin for P-gp assays makes this less likely [57]. A final difference in the assays is the cell types, a human kidney epithelial cell for the calcein-AM assay and a canine kidney epithelial cell that is polarized on a semipermeable membrane for the digoxin assay. Differences in the permeability of the tested excipients between the two cell lines cannot be ruled out.
Despite these limitations, consistent results between the current findings and published studies support the validity of this screen. A recent evaluation of the effect of common food additives on P-gp function demonstrated minimal effects, including many that showed no inhibition in the current calcein-AM and digoxin screens (acesulfame K, aspartame, neohesperidin DC, neotame, sucralose, DL-malic acid, fumaric acid, methylparaben and ethylparaben) [58]. Additional excipients previously tested against P-gp with similar negative results as the current study include lactose monohydrate, sorbitol, sucrose palmitate, sucrose monolaurate and sodium lauryl sulfate [18, 21, 40, 41, 59–61]. However, excipients such as polyethylene glycols, polysorbates, Cremophor EL and pluronics have been shown to improve intestinal absorption of P-gp substrates using in situ and animal models by inhibiting P-gp activity [18–27, 61]. The current data expands the list of molecular excipients that have been characterized with respect to P-gp interactions, allowing for more informed decisions with regard to excipient selection for drug formulation.
Conclusion
In conclusion, a large number of oral molecular excipients were shown to not interact with human P-gp, a critical membrane transporter in the human intestine. These data suggest that diverse molecular excipients can be used in orally administered drug products with limited risk of influencing the bioavailability of P-gp substrates. While β-CD and light green SF yellowish modestly inhibited human P-gp in vitro and, thus, are not likely to have significant effects in humans, careful evaluation for their inclusion in new formulations may be warranted.
Supplementary Material
Acknowledgements
We thank Drs. Per Artutsson and Maria Kalgren for the MDCK-hMDR1-cMDR1-knock out stable cell lines. We also thank Drs. Xiaomin Liang (Gilead Sciences) and Eugene Chen (Genentech) for advice on the digoxin flux assays. Drs. Chenling Xiong, Katherina Chua, Josefina Priotti and Nura El-Haj provided insightful discussions of the data.
Funding Statement
This research was made possible by Grant U01FD004979/U01FD005978 from the US Food and Drug Administration (FDA), which supports the University of California, San Francisco - Stanford Center of Excellence in Regulatory Sciences and Innovation (UCSF-Stanford CERSI). Funding for the research described in the article was provided by the Office of Generic Drugs through the UCSF-Stanford CERSI. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the HHS or FDA. Ruchika Bajaj was partially supported by American Heart Association postdoctoral fellowship Award No. 19POST34370101.
Footnotes
Conflict of Interest Statement
The authors have no relevant financial or non-financial interests to disclose.
References
- 1.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lund M, Petersen TS, Dalhoff KP. Clinical implications of P-glycoprotein modulation in drug-drug interactions. Drugs. 2017;77:859–83. [DOI] [PubMed] [Google Scholar]
- 3.Kim RB. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34:47–54. [DOI] [PubMed] [Google Scholar]
- 4.Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet. 2003;42:59–98. [DOI] [PubMed] [Google Scholar]
- 5.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A. 1987;84:7735–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. [DOI] [PubMed] [Google Scholar]
- 7.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. [DOI] [PubMed] [Google Scholar]
- 8.Kaur G, Arora M, Ravi Kumar MNV. Oral drug delivery technologies-A decade of developments. J Pharmacol Exp Ther. 2019;370:529–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stewart KD, Johnston JA, Matza LS, Curtis SE, Havel HA, Sweetana SA, et al. Preference for pharmaceutical formulation and treatment process attributes. Patient Prefer Adherence. 2016;10:1385–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamman J, Steenekamp J. Excipients with specialized functions for effective drug delivery. Expert Opin Drug Deliv. 2012;9:219–30. [DOI] [PubMed] [Google Scholar]
- 11.Panakanti R, Narang AS. Impact of excipient interactions on drug bioavailability from solid dosage forms. Pharm Res. 2012;29:2639–59. [DOI] [PubMed] [Google Scholar]
- 12.Darji MA, Lalge RM, Marathe SP, Mulay TD, Fatima T, Alshammari A, et al. Excipient stability in oral solid dosage forms: A review. AAPS PharmSciTech. 2018;19:12–26. [DOI] [PubMed] [Google Scholar]
- 13.Palcso B, Zelko R. Different types, applications and limits of enabling excipients of pharmaceutical dosage forms. Drug Discov Today Technol. 2018;27:21–39. [DOI] [PubMed] [Google Scholar]
- 14.Dave VS, Saoji SD, Raut NA, Haware RV. Excipient variability and its impact on dosage form functionality. J Pharm Sci. 2015;104:906–15. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Arieta A Interactions between active pharmaceutical ingredients and excipients affecting bioavailability: impact on bioequivalence. Eur J Pharm Sci. 2014;65:89–97. [DOI] [PubMed] [Google Scholar]
- 16.Zarmpi P, Flanagan T, Meehan E, Mann J, Fotaki N. Biopharmaceutical aspects and implications of excipient variability in drug product performance. Eur J Pharm Biopharm. 2017;111:1–15. [DOI] [PubMed] [Google Scholar]
- 17.Pottel J, Armstrong D, Zou L, Fekete A, Huang XP, Torosyan H, et al. The activities of drug inactive ingredients on biological targets. Science. 2020;369:403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornaire G, Woodley J, Hermann P, Cloarec A, Arellano C, Houin G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int J Pharm. 2004;278:119–31. [DOI] [PubMed] [Google Scholar]
- 19.Cornaire G, Woodley JF, Saivin S, Legendre JY, Decourt S, Cloarec A, et al. Effect of polyoxyl 35 castor oil and polysorbate 80 on the intestinal absorption of digoxin in vitro. Arzneimittel-Forschung-Drug Research. 2000;50:576–9. [DOI] [PubMed] [Google Scholar]
- 20.Guan Y, Huang J, Zuo L, Xu J, Si L, Qiu J, et al. Effect of pluronic P123 and F127 block copolymer on P-glycoprotein transport and CYP3A metabolism. Arch Pharm Res. 2011;34:1719–28. [DOI] [PubMed] [Google Scholar]
- 21.Gurjar R, Chan CYS, Curley P, Sharp J, Chiong J, Rannard S, et al. Inhibitory effects of commonly used excipients on P-glycoprotein in vitro. Molecular Pharmaceutics. 2018;15:4835–42. [DOI] [PubMed] [Google Scholar]
- 22.Hugger ED, Audus KL, Borchardt RT. Effects of poly(ethylene glycol) on efflux transporter activity in Caco-2 cell monolayers. J Pharm Sci. 2002;91:1980–90. [DOI] [PubMed] [Google Scholar]
- 23.Hugger ED, Novak BL, Burton PS, Audus KL, Borchardt RT. A comparison of commonly used polyethoxylated pharmaceutical excipients on their ability to inhibit P-glycoprotein activity in vitro. Journal of Pharmaceutical Sciences. 2002;91:1991–2002. [DOI] [PubMed] [Google Scholar]
- 24.Katneni K, Charman SA, Porter CJ. Impact of cremophor-EL and polysorbate-80 on digoxin permeability across rat jejunum: delineation of thermodynamic and transporter related events using the reciprocal permeability approach. J Pharm Sci. 2007;96:280–93. [DOI] [PubMed] [Google Scholar]
- 25.Lo YL, Huang JD. Effects of sodium deoxycholate and sodium caprate on the transport of epirubicin in human intestinal epithelial Caco-2 cell layers and everted gut sacs of rats. Biochemical Pharmacology. 2000;59:665–72. [DOI] [PubMed] [Google Scholar]
- 26.Ma L, Wei Y, Zhou Y, Ma X, Wu X. Effects of Pluronic F68 and Labrasol on the intestinal absorption and pharmacokinetics of rifampicin in rats. Arch Pharm Res. 2011;34:1939–43. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H, Yao M, Morrison RA, Chong S. Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch Pharm Res. 2003;26:768–72. [DOI] [PubMed] [Google Scholar]
- 28.Varma MV, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci. 2005;25:445–53. [DOI] [PubMed] [Google Scholar]
- 29.Ashiru DA, Patel R, Basit AW. Polyethylene glycol 400 enhances the bioavailability of a BCS class III drug (ranitidine) in male subjects but not females. Pharm Res. 2008;25:2327–33. [DOI] [PubMed] [Google Scholar]
- 30.Li M, Si L, Pan H, Rabba AK, Yan F, Qiu J, et al. Excipients enhance intestinal absorption of ganciclovir by P-gp inhibition: assessed in vitro by everted gut sac and in situ by improved intestinal perfusion. Int J Pharm. 2011;403:37–45. [DOI] [PubMed] [Google Scholar]
- 31.Shen Y, Lu Y, Jv M, Hu J, Li Q, Tu J. Enhancing effect of Labrasol on the intestinal absorption of ganciclovir in rats. Drug Dev Ind Pharm. 2011;37:1415–21. [DOI] [PubMed] [Google Scholar]
- 32.Administration USFD. Inactive Ingredient Search for Approved Drug Products. U.S. Food & Drug Administration; 2020. [updated 01/21/2021. Available from: https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm. [Google Scholar]
- 33.Zou L, Spanogiannopoulos P, Pieper LM, Chien HC, Cai W, Khuri N, et al. Bacterial metabolism rescues the inhibition of intestinal drug absorption by food and drug additives. Proc Natl Acad Sci U S A. 2020;117:16009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gow JM, Hodges LM, Chinn LW, Kroetz DL. Substrate-dependent effects of human ABCB1 coding polymorphisms. J Pharmacol Exp Ther. 2008;325:435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karlgren M, Simoff I, Backlund M, Wegler C, Keiser M, Handin N, et al. A CRISPR-Cas9 generated MDCK cell line expressing human MDR1 without endogenous canine MDR1 (cABCB1): An improved tool for drug efflux studies. J Pharm Sci. 2017;106:2909–13. [DOI] [PubMed] [Google Scholar]
- 36.Simoff I, Karlgren M, Backlund M, Lindstrom AC, Gaugaz FZ, Matsson P, et al. Complete knockout of endogenous Mdr1 (Abcb1) in MDCK cells by CRISPR-Cas9. J Pharm Sci. 2016;105:1017–21. [DOI] [PubMed] [Google Scholar]
- 37.Levy ES, Samy KE, Lamson NG, Whitehead KA, Kroetz DL, Desai TA. Reversible inhibition of efflux transporters by hydrogel microdevices. Eur J Pharm Biopharm. 2019;145:76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hubatsch I, Ragnarsson EG, Artursson P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc. 2007;2:2111–9. [DOI] [PubMed] [Google Scholar]
- 39.Gerber W, Hamman JH, Steyn JD. Excipient-drug pharmacokinetic interactions: Effect of disintegrants on efflux across excised pig intestinal tissues. J Food Drug Anal. 2018;26:S115–S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takizawa Y, Goto N, Furuya T, Hayashi M. Influene of pharmaceutical excipients on the membrane transport of a P-glycoprotein substrate in the rat small intestine. Eur J Drug Metab Pharmacokinet. 2020;45:645–52. [DOI] [PubMed] [Google Scholar]
- 41.Takizawa Y, Kishimoto H, Nakagawa M, Sakamoto N, Tobe Y, Furuya T, et al. Effects of pharmaceutical excipients on membrane permeability in rat small intestine. Int J Pharm. 2013;453:363–70. [DOI] [PubMed] [Google Scholar]
- 42.Conceicao J, Adeoye O, Cabral-Marques HM, Lobo JMS. Cyclodextrins as excipients in tablet formulations. Drug Discov Today. 2018;23:1274–84. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Cui YL, Gao LN, Jiang HL. Effects of beta-cyclodextrin on the intestinal absorption of berberine hydrochloride, a P-glycoprotein substrate. Int J Biol Macromol. 2013;59:363–71. [DOI] [PubMed] [Google Scholar]
- 44.Arima H, Yunomae K, Hirayama F, Uekama K. Contribution of P-glycoprotein to the enhancing effects of dimethyl-beta-cyclodextrin on oral bioavailability of tacrolimus. J Pharmacol Exp Ther. 2001;297:547–55. [PubMed] [Google Scholar]
- 45.Arima H, Yunomae K, Morikawa T, Hirayama F, Uekama K. Contribution of cholesterol and phospholipids to inhibitory effect of dimethyl-beta-cyclodextrin on efflux function of P-glycoprotein and multidrug resistance-associated protein 2 in vinblastine-resistant Caco-2 cell monolayers. Pharm Res. 2004;21:625–34. [DOI] [PubMed] [Google Scholar]
- 46.Cai C, Zhu H, Chen J. Overexpression of caveolin-1 increases plasma membrane fluidity and reduces P-glycoprotein function in Hs578T/Dox. Biochem Biophys Res Commun. 2004;320:868–74. [DOI] [PubMed] [Google Scholar]
- 47.Kamau SW, Kramer SD, Gunthert M, Wunderli-Allenspach H. Effect of the modulation of the membrane lipid composition on the localization and function of P-glycoprotein in MDR1-MDCK cells. In Vitro Cell Dev Biol Anim. 2005;41:207–16. [DOI] [PubMed] [Google Scholar]
- 48.Pathak SM, Musmade P, Dengle S, Karthik A, Bhat K, Udupa N. Enhanced oral absorption of saquinavir with methyl-beta-cyclodextrin-Preparation and in vitro and in vivo evaluation. Eur J Pharm Sci. 2010;41:440–51. [DOI] [PubMed] [Google Scholar]
- 49.Yunomae K, Arima H, Hirayama F, Uekama K. Involvement of cholesterol in the inhibitory effect of dimethyl-beta-cyclodextrin on P-glycoprotein and MRP2 function in Caco-2 cells. FEBS Lett. 2003;536:225–31. [DOI] [PubMed] [Google Scholar]
- 50.Durk MR, Jones NS, Liu J, Nagapudi K, Mao C, Plise EG, et al. Understanding the effect of hydroxypropyl-beta-cyclodextrin on fenebrutinib absorption in an itraconazole-fenebrutinib drug-drug interaction study. Clin Pharmacol Ther. 2020;108:1224–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou L, Pottel J, Khuri N, Ngo HX, Ni Z, Tsakalozou E, et al. Interactions of oral molecular excipients with breast cancer resistance protein, BCRP. Mol Pharm. 2020;17:748–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Administration USFD. In Vitro Drug Interaction Studies - Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. U.S. Food & Drug Administration; 2020. [updated 05/07/2020. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. [Google Scholar]
- 53.Tiberghien F, Loor F. Ranking of P-glycoprotein substrates and inhibitors by a calcein-AM fluorometry screening assay. Anticancer Drugs. 1996;7:568–78. [DOI] [PubMed] [Google Scholar]
- 54.Caetano-Pinto P, Janssen MJ, Gijzen L, Verscheijden L, Wilmer MJ, Masereeuw R. Fluorescence-based transport assays revisited in a human renal proximal tubule cell line. Mol Pharm. 2016;13:933–44. [DOI] [PubMed] [Google Scholar]
- 55.Olson DP, Taylor BJ, Ivy SP. Detection of MRP functional activity: Calcein AM but not BCECF AM as a Multidrug Resistance-related Protein (MRP1) substrate. Cytometry. 2001;46:105–13. [DOI] [PubMed] [Google Scholar]
- 56.Reznicek J, Ceckova M, Ptackova Z, Martinec O, Tupova L, Cerveny L, et al. MDR1 and BCRP transporter-mediated drug-drug interaction between rilpivirine and abacavir and effect on intestinal absorption. Antimicrob Agents Chemother. 2017;61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwab D, Fischer H, Tabatabaei A, Poli S, Huwyler J. Comparison of in vitro P-glycoprotein screening assays: recommendations for their use in drug discovery. J Med Chem. 2003;46:1716–25. [DOI] [PubMed] [Google Scholar]
- 58.Sjostedt N, Deng F, Rauvala O, Tepponen T, Kidron H. Interaction of food additives with intestinal efflux transporters. Mol Pharm. 2017;14:3824–33. [DOI] [PubMed] [Google Scholar]
- 59.Kiss L, Hellinger E, Pilbat AM, Kittel A, Torok Z, Furedi A, et al. Sucrose esters increase drug penetration, but do not inhibit P-glycoprotein in caco-2 intestinal epithelial cells. J Pharm Sci. 2014;103:3107–19. [DOI] [PubMed] [Google Scholar]
- 60.Lo YL. Relationships between the hydrophilic-lipophilic balance values of pharmaceutical excipients and their multidrug resistance modulating effect in Caco-2 cells and rat intestines. J Control Release. 2003;90:37–48. [DOI] [PubMed] [Google Scholar]
- 61.Ruiz-Picazo A, Gonzalez-Alvarez M, Gonzalez-Alvarez I, Bermejo M. Effect of common excipients on intestinal drug absorption in Wistar rats. Mol Pharm. 2020;17:2310–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.