

Abstract
Healthy mitochondria are essential for functional bioenergetics, calcium signaling, and balanced redox homeostasis. Dysfunctional mitochondria are a central aspect of aging and neurodegenerative diseases such as Alzheimer’s disease (AD). The formation and accumulation of amyloid beta (Aβ) and hyperphosphorylated tau (P-tau) play large roles in the cellular changes seen in AD, including mitochondrial dysfunction, synaptic damage, neuronal loss, and defective mitophagy. Mitophagy is the cellular process whereby damaged mitochondria are selectively removed, and it plays an important role in mitochondrial quality control. Dysfunctional mitochondria are associated with increased reactive oxygen species and increased levels of Aβ, P-tau and Drp1, which together trigger mitophagy and autophagy. Impaired mitophagy causes the progressive accumulation of defective organelles and damaged mitochondria, and it has been hypothesized that the restoration of mitophagy may offer therapeutic benefits to AD patients. This review highlights the challenges of pharmacologically inducing mitophagy through two different signaling cascades: 1) The PINK1/parkin-dependent pathway and 2) the PINK1/parkin-independent pathway, with an emphasis on abnormal mitochondrial interactions with Aβ and P-Tau, which alter mitophagy in an age-dependent manner. This article also summarizes recent studies on the effects of mitophagy enhancers, including urolithin A, NAD+, actinonin, and tomatidine, on mutant APP/Aβ and mutant Tau. Findings from our lab have revealed that mitophagy enhancers can suppress APP/Aβ-induced and mutant Tau-induced mitochondrial and synaptic dysfunctions in mouse and cell line models of AD. Finally, we discuss the mechanisms underlying the beneficial health effects of mitophagy enhancers like urolithin A, NAD+, resveratrol and spermidine in AD.
Keywords: Alzheimer’s disease, Aβ, Phosphorylated tau, Mitochondrial dysfunction, Mitophagy
1. Introduction
Alzheimer’s disease (AD) is the leading cause of dementia in the aging population, and it affects over 55 million people worldwide [1]. The World Alzheimer Report 2019 states that this number will escalate rapidly to 88 million by 2050 [2]. With increasing longevity, AD is becoming an increasingly urgent health concern for society. AD is a progressive neurodegenerative disease that leads to the irreversible loss of neurons and intellectual abilities and eventually causes death within a few years [3]. Currently there are no medical interventions capable of curing or delaying the progression of AD in patients.
The formation and accumulation of beta-amyloid (Aβ) and phosphorylated tau (P-tau) are the two major pathological changes in the brains of patients with AD [4–6]. Aβ is generated predominantly in forms containing 40 and 42 amino acids from amyloid precursor protein (APP) following sequential cleavages by β- and γ- secretases [7,8]. Indeed, Aβ is a neurotoxic peptide and its accumulation has been extensively reported in the development, progression and pathogenesis of AD. Hyperphosphorylation of tau, and its formation of neurofibrillary tangles, is another pathological hallmark and definitive diagnostic feature of AD. The neurons of AD patients undergo sequential functional modifications including inflammation, impaired energy metabolism, overwhelming oxidative stress, and synaptic dysfunction, leading to impaired axonal transport and neuronal death [9–12].
Many risk factors (especially diabetes, obesity, smoking and hypertension) are also associated with an increased risk of cognitive decline with age [13–18]. Like AD, these risk factors contribute to excessive oxidative stress, mitochondrial fragmentation and impaired autophagy and mitophagy.
The purpose of this article is to summarize the recent preclinical efforts to develop mitophagy enhancers as therapeutic drugs for AD. This article also discusses the mechanisms of mitophagy enhancers, particularly via the PINK1-Parkin dependent and independent pathways, in clearing dead and/or dying mitochondria from healthy and diseased neurons.
1.1. Mitochondria
Mitochondria are the powerhouses of the cell, which are responsible for ATP production, energy conversion, respiration and other important cellular events including proliferation, redox homeostasis, and apoptosis [19–22]. Mitochondrial function is maintained by balanced mitochondrial dynamics (consisting of mitochondrial fission and fusion) and biogenesis [23,24]. It is important to maintain mitochondrial dynamics and biogenesis for neuronal health. In many age-related neurocognitive disorders, including Alzheimer’s disease and Parkinson’s disease, the dysfunction of mitochondria is correlated with structural damage and defective dynamics, biogenesis, and mitophagy [25–27]. In AD, impaired mitochondrial dynamics, impaired energy metabolism, and overwhelming oxidative stress lead to the mitochondrial dysfunction observed in neurons [28,29].
Importantly, growing evidence indicates that accumulated Aβ and p-Tau within mitochondria can interact with mitochondrial outer membrane proteins like VDAC1, resulting in mitochondrial defects and playing a major role in AD pathogenesis [30]. Intramitochondrial Aβ also interacts with the electron transport chain (ETC), inducing reactive oxygen species (ROS) formation [31]. Accumulating evidence suggests that mitochondrial dysfunction associates with oxidative stress, aging, inactive lifestyle, and excessive caloric intake that can trigger amyloidogenic and ROS-mediated neuroinflammatory processes [32–37].
Mitophagy is a specialized form of autophagy that regulates the elimination of defective mitochondria. Programmed mitophagy plays a pivotal role in the natural turnover of mitochondria. It is regulated by protein machinery that engulfs damaged/aged mitochondria in an autophagosome, which is then fused with a lysosome to initiate degradation [38,39]. The age-dependent decline of mitophagy hampers the elimination of dysfunctional or damaged mitochondria and alters mitochondrial biogenesis [40–42]. Defective mitophagy is correlated with a series of pathophysiological events, such as Aβ- and P-tau-induced toxicities and mitochondrial defects. Aβ, P-tau and Drp1 interactions with PINK1 and Parkin cytosolic proteins, mitochondrial ROS, impaired biogenesis, and other factors are associated with unsuccessful mitophagy in age-related neurodegenerative diseases [43].
1.2. Mitochondrial abnormalities
Mitochondria are the centers of the signaling pathways which produce ATP, regulate calcium, and maintain redox homeostasis in healthy cells. Impaired mitochondrial dynamics results in excessive production of ROS, which in turn damages mitochondrial DNA (mtDNA) and ultimately causes defects in biogenesis, mitophagy, and apoptosis.
Mitochondria are dynamic organelles that fuse and divide to form constantly changing tubular networks in most eukaryotic cells. Furthermore, regulation of mitochondrial dynamics is crucial for the health of the cell [44–48]. At any point in the cell cycle, a mitochondrion may undergo fission to give rise to two separate mitochondria [49]. Simultaneously, mitochondria are undergoing fusion in which both the inner and outer membranes of the mitochondria break and rejoin to form a single intact mitochondrion. The mitochondrial fission and fusion processes maintain a dynamic reticular network that spreads throughout the cytosolic volume and responds to extrinsic and intrinsic pathological stress signals.
Mitochondria are dynamic organelles, and their shape is controlled by a balance of fusion and fission events, which are governed by the proteolysis and posttranslational modification of several proteins [50–52]. The known proteins that are involved in mitochondrial fission are Drp1 and Fis1, and mitochondrial fusion proteins include MFN1, MFN2, and OPA1 [53–56]. In a healthy state, Drp1 is important for mitochondrial division, distribution, and synaptic functions; In AD, abnormal interactions of Drp1 with Aβ and P-tau enhance the GTPase activity of Drp1, increasing mitochondrial fragmentation, impairing healthy mitochondrial dynamics, and ultimately leading to defective mitophagy and synaptic dysfunction in AD [57]. Aβ, P-tau, and Drp1 also interact with the mitochondrial outer membrane proteins like PINK1 and PARKIN, reducing mitophagy in AD neurons which prevents healthy turnover of old or damaged mitochondria [58,59]. Finally, AD neurons have diminished expression of PGC-1α, Nrf1, Nrf2, and TFAM, impairing the biogenesis of new mitochondria to replace those that have been damaged or removed [60].
2. Mitophagy
Mitophagy is a highly complex cellular process that regulates both mitochondrial quality and quantity. It is used to conveniently eliminate mitochondria that have been critically damaged [61–63].
Both morphological and biochemical markers have been described for mitophagy, and the use of a combination of approaches to measure mitophagy is strongly recommended. Despite the significant recent advances in our knowledge of selective mitophagy, more reliable and accurate quantitative assays to monitor mitophagy need to be developed. Interest in mitophagy has increased during recent years, mostly by virtue of the growing awareness that germline mutations in key mitophagy proteins, like PINK1 or Parkin, are frequently observed in patients with neurodegenerative diseases [64,65]. Mitophagy plays a pivotal role in the natural, regulated machinery of the cell cycle and disposes of the mitochondrial waste during mitosis. Mitophagy allows for the orderly degradation and recycling of damaged mitochondria and utilizes unique membrane trafficking processes [66,67]. Maintenance of mitochondrial quality and number is important for normal cellular metabolic homeostasis. Mitophagy promotes mitochondrial quality and prevents the accumulation of dysfunctional mitochondria that can lead to cellular degeneration.
Mitophagy begins with the formation of the phagophore which expands through lipid acquisition to become the autophagosome [68]. Following the selective engulfment of a defective mitochondria, the mitochondria is subsequently catabolized by fusion of the autophagosome to a lysosome to form an autolysosome where the enveloped contents are degraded [69–72]. How the autophagosome formation occurs is still not clear. However, studies have suggested that the plasma membrane, the Golgi complex, the endoplasmic reticulum (ER), and mitochondria are involved in the process [73]. Many studies have reported that autophagosomes that select and degrade defective mitochondria (sometimes called mitophagosomes) are formed by ER-mitochondria interactions in mammalian cells.
Nearly 30–45 autophagy-related genes (ATG genes) are involved in autophagosome formation in yeast. Autophagy involves the recruitment of regulatory protein complexes which include: (1) The ULK kinase complex: ULK1-ATG13-FIP200, (2) the phosphatidylinositol 3-kinase (Ptdlns3K) complex: Beclin1-ATG14-Vsp15-Vsp34-AMBRA1, (3) the ATG9-ATG2-ATG18-ATG3-ATG10 complex and (4) the ATG5-ATG12-ATG16 and ATG7/LC3 conjugation systems. Once assembled, the phagosome is ready to selectively engulf the defective or damaged mitochondria for degradation through mitophagy. Mitophagy is broadly classified into two types: a) PINK1/Parkin-dependent, and b) PINK1/Parkin-independent, as described in the following subsections.
2.1. PINK1/Parkin-dependent mitophagy
In PTEN-induced putative kinase 1 (PINK1)/Parkin-dependent mitophagy, mitochondrial ubiquitin chains are recognized by autophagic ubiquitin receptors. Dysfunctional mitochondria tend to have depolarized membranes with reduced proton gradients across their inner mitochondrial membranes (IMM). This depolarization allows PINK1 kinase to accumulate at the outer mitochondrial membrane (OMM) and phosphorylate mitochondrial surface proteins including PINK1, Parkin, Mfn1 and Mfn2. Phosphorylation and ubiquitination by PINK1 of Ser65 of the E3 ubiquitin ligase Parkin initiates the ubiquitination of several substrates in the outer mitochondrial membrane [74]. Phosphorylation of ubiquitin chains leads to the recruitment of several autophagy adapter proteins, which interact, with the ubiquitin chains and LC3, such as the adapter proteins p62, OPTN, and NDP52, which drive mitophagy by binding to a phagophore. LC3 also functions as an adapter protein to recruit selective cargo to the autophagosome via interactions with lysosome cargo receptors. PINK1 and Parkin-mediated mitophagy is triggered by poor mitochondrial quality, which is in turn determined by the cumulative activities of other mitochondrial proteins such as Drp1, Fis1, Mfn1, Mfn2, and PGC1a. Dysfunctional mitochondria fail to maintain their membrane potentials. This activates PINK1, which subsequently leads to Parkin recruitment, polyubiquitination and adapter recruitment, phagophore LC3 binding, engulfment by the autophagosome, and lysosomal digestion [75,76] (Fig. 1).
Fig. 1. Mechanism of PINK1–Parkin-dependent mitophagy.
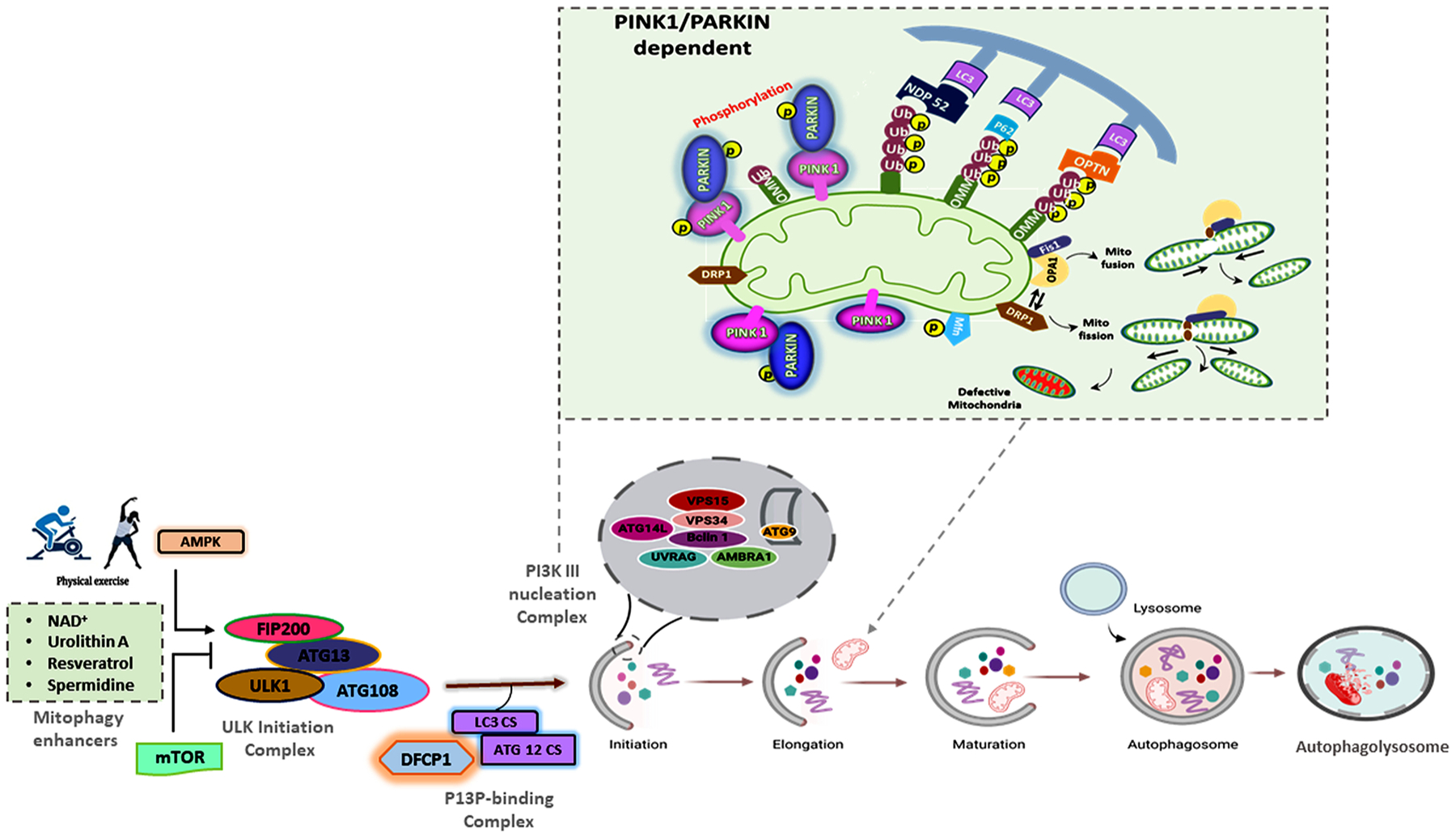
(A) Mitophagy induction is normally activated by AMPK and inhibited by mTOR. Here, the pharmacological induction of mitophagy by NAD+, urolithin A, resveratrol, or spermidine will activate the ULK1 complex (FIP200, ATG13, ULK1 and ATG108) to initiate the P13P-binding complex. The PI3KIII nucleation complex activates VPS34, VPS15 lipid kinase, beclin, UVRAG, and AMBRA1 which participate in these processes, including the stabilization of ULK1 and binding to the PI3KIII complex. PINK1 on the outer mitochondria membrane (OMM) is activated by mitochondrial depolarization and autophosphorylation. Activated PINK1 phosphorylates and recruits Parkin which ubiquitinates several OMM components. Ubiquitin chains attached to the OMM are subsequently phosphorylated by PINK1 serving as a signal for the autophagic machinery. The adapter proteins p62, OPTN, and NDP52 recognize phosphorylated poly-Ub chains on mitochondrial proteins and initiate autophagosome formation through binding with LC3B. Formation of a double-layered membrane phagophore formation within the cytosol requires the PI3KC3 complex. The complex then binds ATG proteins, resulting in the recruitment of lipids to form the phagophore; cytoplasm and organelles are engulfed during the elongation of the phagophore, forming an autophagosome. In the final step of the process, lysosomes fuse with the autophagosome, releasing lysosomal hydrolases into the interior, resulting in degradation of the vesicle contents, including mitochondria.
2.2. PINK1/Parkin-independent mitophagy
In response to hypoxic conditions PINK1/Parkin-independent mitophagy is initiated by the OMM proteins. Hypoxia leads to transcriptional upregulation of BNIP3 and Nix by the HIF 1/2 transcription factors, which are stabilized under hypoxic conditions. This leads to subsequent mitophagy events. Structurally, BNIP3 and Nix are single-pass transmembrane proteins targeted to the mitochondria, and the cytosolic tails contain LIR motifs. Phosphorylation at the Ser34 and 35 residues, and the Ser17 and 24 residues, flanking the LIR motifs in Nix and BNIP3 respectively, promote interactions with LC3 under stress conditions. LC3 can be recognized by mitochondrial receptor proteins including FUNDC1, BNIP, and NIX to promote the completion of the phagophore, thus targeting the mitochondrion for mitophagy [76]. NIX protein plays an important role in mitochondrial quality checks. NIX accumulation on the mitochondrial surface flags mitochondria for mitophagic degradation [74]. BNIP3 is also able to bind phagophore LC3 to initiate mitophagy through Opa1, in conjunction with Fis1 and Drp1 which initiate fission [75]. The FUNDC1 receptor also promotes mitochondrial clearance during hypoxic conditions; it interacts with the ER and recruits Drp1 during mitochondrial fragmentation. FUNDC1 is a good protein marker for stress-induced mitophagy [76] (Fig. 2).
Fig. 2. Mechanism of PINK1–Parkin-independent mitophagy.
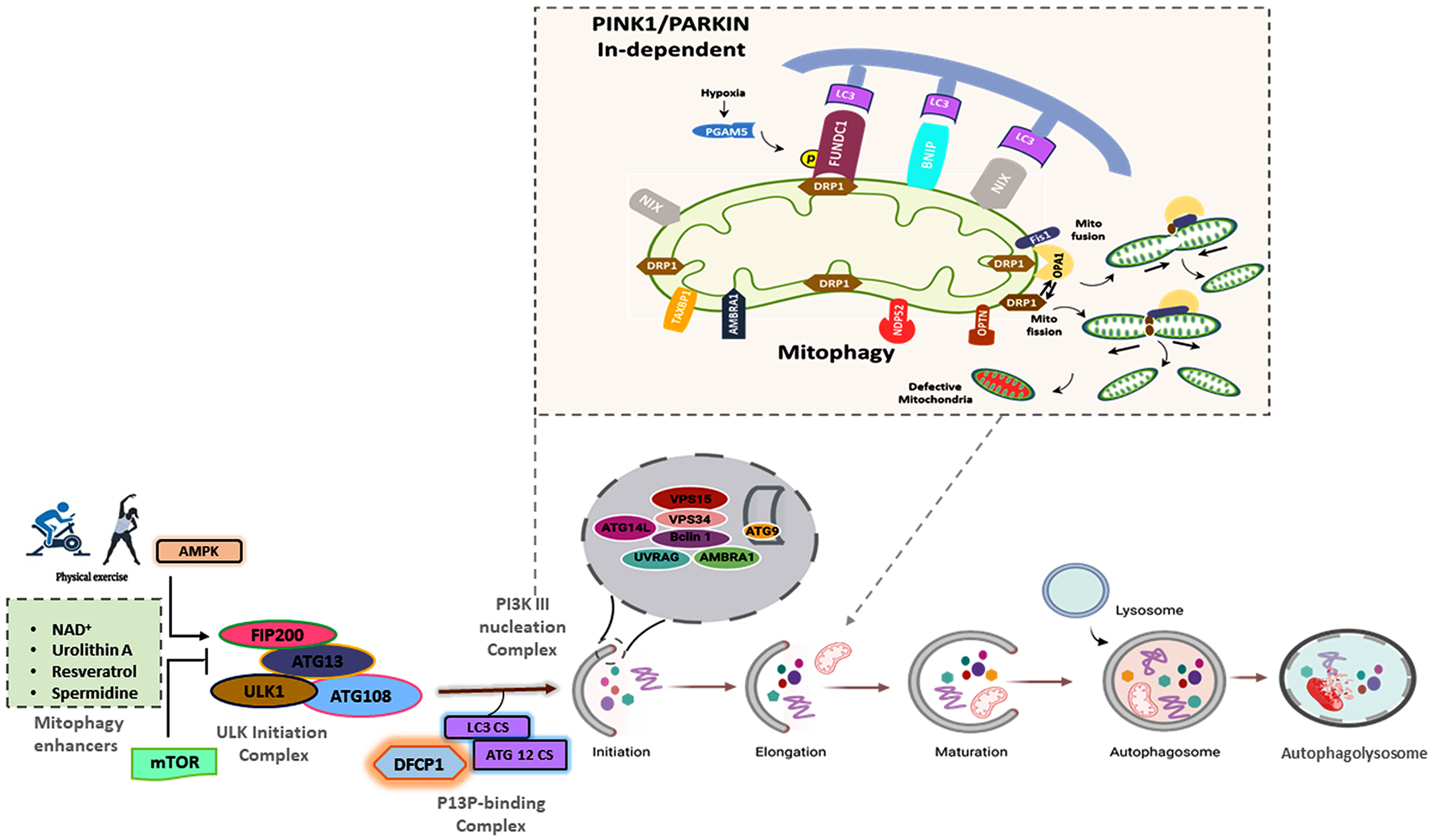
Specialized receptors, like NIX, BNIP3, and FUNDC1 are expressed on the OMM in response to stress stimuli. These receptors directly interact with LC3 to mediate mitochondrial elimination. In response to hypoxia, phosphorylation of NIX and BNIP3 enhances their associations with LC3. FUNDC1 also recruits LC3 in response to hypoxia, and FUNDC1 phosphorylation promotes fission of damaged mitochondria through the recruitment of DRP1 and Opa1 on the mitochondrial surface. Formation of a double-layered membrane phagophore within the cytosol requires the PI3KC3 complex. The complex then binds ATG resulting in LC3-II conjugation to phosphatidylethanolamine (PE) which drives autophagosomal membrane expansion. The membrane grows to enwrap a portion of the cytosol, forming an autophagosome. In the final step of the process, lysosomes fuse with the autophagosome, releasing lysosomal hydrolases into the interior, resulting in degradation of the vesicle contents, including mitochondria.
2.3. Pharmacological modulation of mitophagy
Defective mitochondrial biogenesis and accumulation of dead mitochondria are the main phenomena observed in age-associated diseases such as AD. Newly developed pharmacological treatments and novel chemical modulators like urolithin A [77], tomatidine [78,79], and actinonin [80] might be used to promote the efficient removal of dead and/or damaged mitochondria and restore energy homeostasis. Pharmacologic agents and natural supplements that target mitophagy are potential drugs which may enhance the quality of mitochondria in aging individuals and ameliorate age-related diseases. To this end, several synthetic chemicals and natural compounds have been studied which help to regulate mitophagy and eliminate dysfunctional mitochondria.
2.4. Mitophagy enhancer NAD+
Nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADPH) are essential cofactors involved in several cellular redox reactions [81]. Nicotinamide adenine dinucleotide is a well-known coenzyme found in all living cells and is central to metabolism. NAD exists in two forms: the oxidized NAD+ form and the reduced NADH form [82]. NAD+ plays an important role in reductive reactions such as lipid biosynthesis and the synthesis and repair of DNA. The balance state of NAD is defined by the NAD+/NADH ratio, which is essential for the normal redox state of a cell. The decline of this ratio with age has been reported in multiple organs, including brain, liver, muscles, and adipose tissue [83].
NAD is involved in oxidation–reduction reactions critical for glycolysis, fatty acid oxidation, the TCA cycle, and, importantly, complex I of the mitochondrial respiratory chain which is a key regulator of autophagy. NAD+ protects against age-dependent metabolic impairment and promotes longevity through the NAD+ dependent enzymes like silent information regulators (sirtuins). Currently, seven sirtuins have been reported [84], each exhibiting distinct subcellular localization and performing specific functions.
NAD+-dependent transcriptional regulation of sirtuins is important to the study of both NAD+ sensors and of NAD+ regulatory mediators. The enzyme-substrate mechanisms of NAD+-consuming proteins, such as sirtuins, poly (ADP-ribosyl) polymerases (PARPs), and cyclic ADP-ribose synthases (CD38 and CD157) have been well studied. These include NAD+ consuming sirtuins that regulate a broad range of cellular functions via NAD+-dependent deacetylation, and enzymes that generate bioactive second messengers including cyclic AMP, cyclic GMP, inositol triphosphate, diacylglycerol (Fig. 3). Sirtuins are directly linked to NAD+ signaling, and have shown profound consequences for lifespan and the development of age-associated diseases including AD. NAD+-dependent sirtuin activation of autophagic processes occurs when rate-dependent increases in the NAD+/NADH ratio activate SIRT1, which then activates autophagy by directly deacetylating ATG proteins.
Fig. 3.
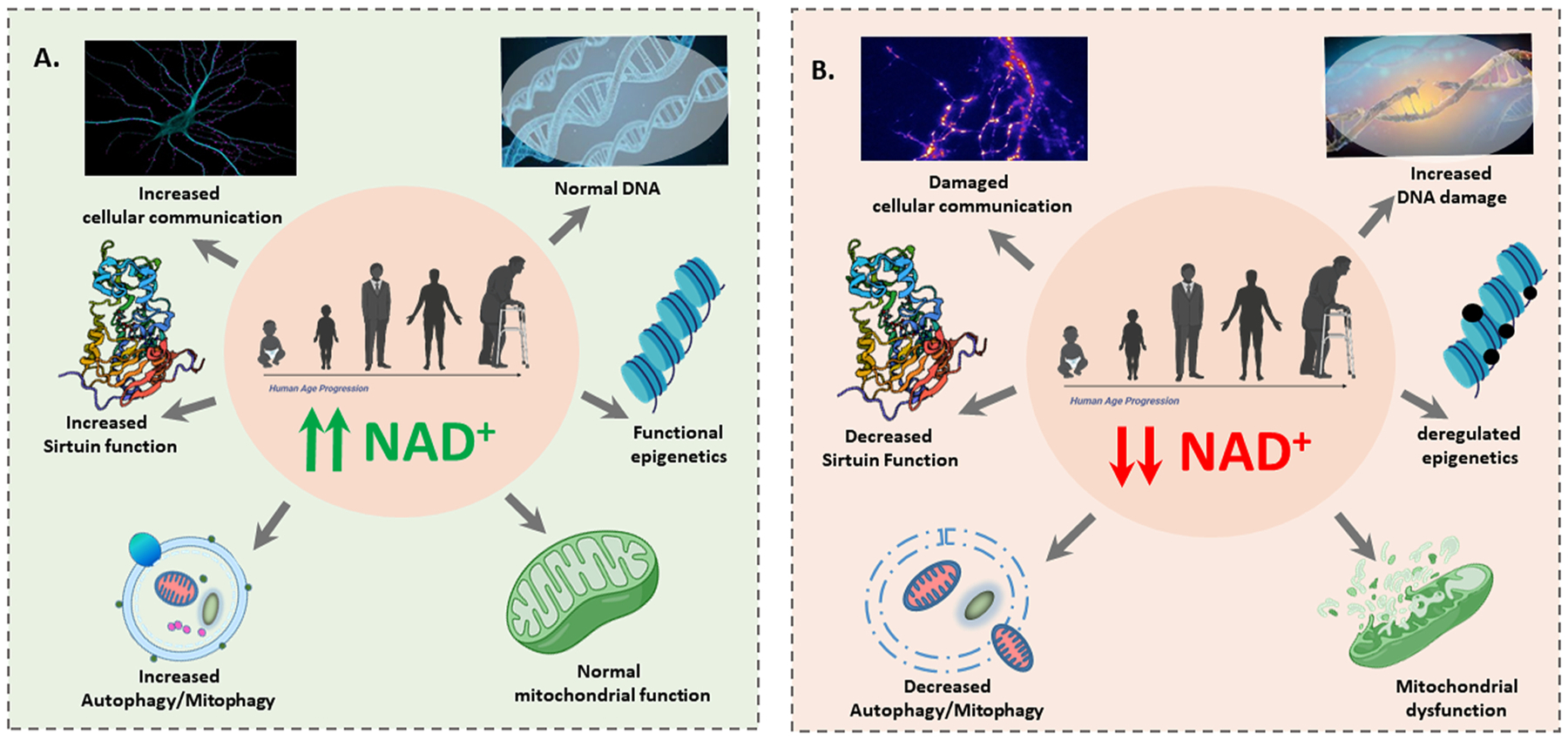
A. Normal NAD+ levels are important for health. A schematic representation of healthy NAD+ levels, which are required for several functions including repair of DNA, epigenetic regulation of gene expression, mitochondrial functions, effective mitophagy/autophagy, and Increased Sirtuin function. B. The decline of NAD+ is a core hallmark of Alzheimer’s disease and aging. A decline in NAD+ levels contributes to defects in DNA repair, deregulated epigenetics, mitochondrial dysfunction, deregulation of mitophagy/autophagy, and decreased Sirtuin function.
NADPH eliminates cytoplasmic reactive oxygen species, which can also activate autophagy [85]. These events, in turn, activate SIRT1-linked transhydrogenase activity resulting in the interconversion of NADPH and NAD+ into NADP+ and NADH [86]. In the diseased state, this activity is inhibited by reactive oxygen species which accumulate following the loss of the glutathione peroxidase-glutathione reductase system [87].
Increased production of cytoplasmic reactive oxygen species in diseased/aged cells is caused by various pathological protein interactions with Aβ or P-tau, including the interaction of Aβ/P-tau with Drp1 and the interaction of Aβ/P-tau with VDAC1. Increased Drp1 in cells leads to defective mitophagy [117]. These effects lead to suppressed glutamate dehydrogenase expression and suppressed autophagy by mTORC1-mediated signaling. Pharmacological modulation of intra-cellular NAD+ via supplements and/or treatments stimulates a rise in the NAD+-SIRT1 level which has been shown to enhance mitophagy in models of neurodegenerative diseases.
2.5. Urolithins
Urolithin is a microflora-derived metabolite produced by gut bacteria such as Gordonibacter urolithinfaciens and Gordonibacter pamelaeae, which is converted from ellagic acids (EA) and ellagitannins (ETs) into urolithin A [88]. Ellagitannins (ETs) and ellagic acid (EA) are complex polyphenols abundant in foods such as pomegranate, berries, and nuts (Fig. 4).
Fig. 4. Several foods contain the natural polyphenols ellagitannins (ETs) and ellagic acid (EA).
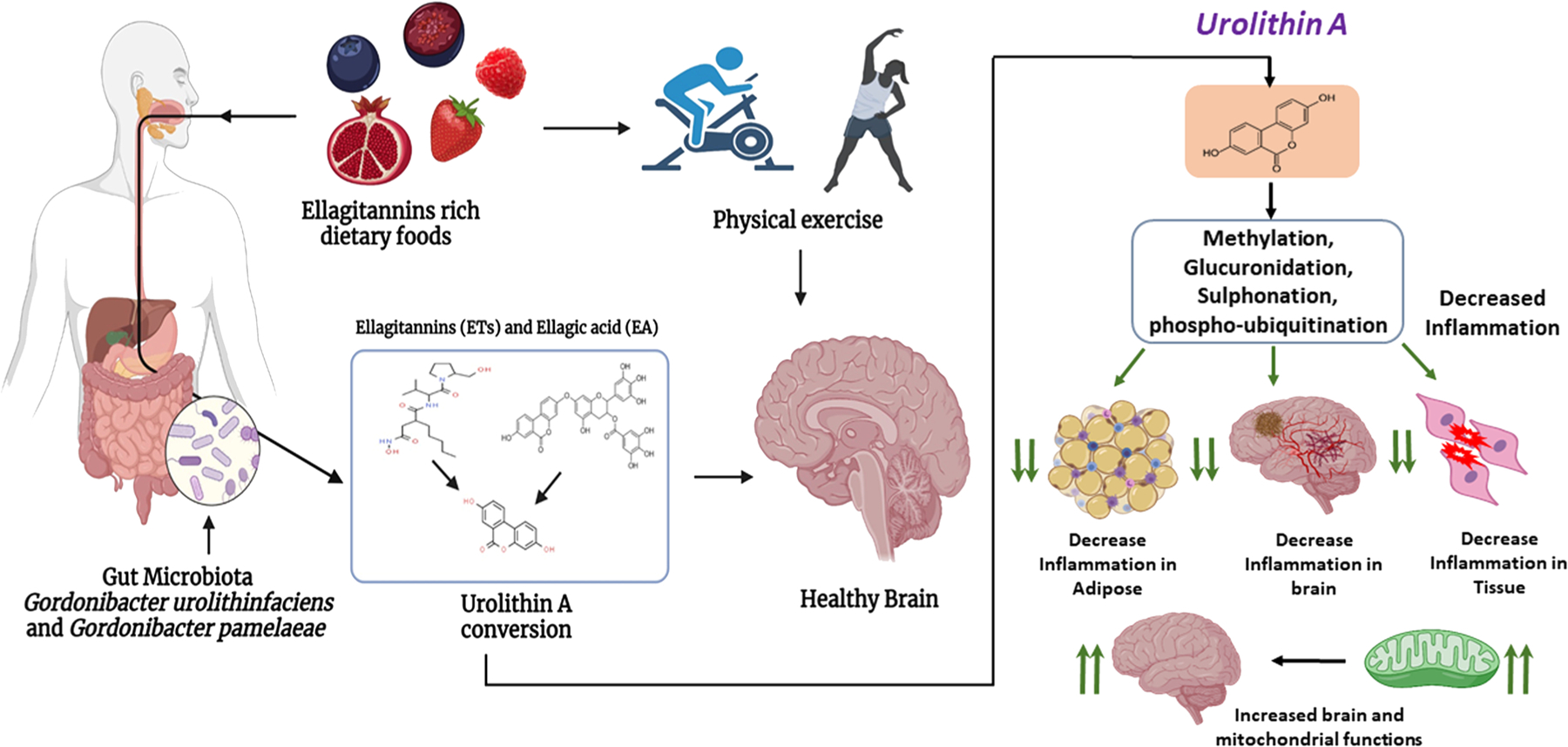
Ellagic-acid containing foods undergo gut microbial conversion to yield various forms of Urolithins. This conversion occurs only in the presence of microbes Gordonibacter urolithinfaciens and Gordonibacter pamelaeae. Urolithin supplementation improves mitochondrial and cellular health in age-related conditions, including AD. Urolithin A (UA) undergoes methylation, glucuronidation, and sulfation to yield conjugated forms of UA which decrease adipose, neuronal, and tissue inflammation. Decreased inflammation promotes healthy aging by improving mitochondrial function in the brain.
Urolithins belong to the class of organic compounds known as benzo-coumarins containing a 1-benzopyran moiety with a ketone group at the C2 carbon atom [89]. Hydrolysis of ellagitannins in the gut releases ellagic acid, which is further processed to urolithins by the loss of a lactone ring. Urolithins are absorbed into the intestine where they enter circulation. These circulatory urolithins are subjected to additional chemical transformations, including glucuronidation, methylation, and sulfation [90]. Urolithin formation depends on age, health status, dietary intake, and, importantly, the gut microbiome.
Glucuronidation occurs on Urolithin hydroxyl and amino groups in the presence of a glucuronyl transferase enzyme [91]. Glucuronidation helps in detoxifying foreign substances in humans and many animals. Methylation is involved in many biological functions including the regulation of gene expression, protein functions, and RNA processing. Methylated Urolithin A (UA) shows significant biological effects, including cell apoptosis, mitochondrial depolarization and down-regulation of Bcl-2/Bax [92]. Studies have shown that treatments of UA slow aging, reduce age-related diseases, and improve quality of mitochondria and overall health status [93]. UA prevents inflammation and promotes mitochondrial health, biogenesis and dynamics in different cell types and in different species including worms, mice, and humans [94–96].
Sulfated UA can in turn sulfate other cellular targets, improving multiple biological functions, such as the posttranslational modification of mitochondrial proteins, apoptosis, and mitophagy. The sulfate is transferred from UA to the tyrosine residues of target proteins by tyrosylprotein sulfotransferase enzymes of the Golgi apparatus. UA sulfation plays a role in protein-protein interactions, regulating G-protein-coupled receptors, coagulation factors, serine protease inhibitors, extracellular matrix proteins, and hormones. PINK1 and phospho-ubiquitin accumulation was observed in C2C12 mouse muscle myoblasts after treatment with UA [96], and increased levels of ubiquitinated and phospho-ubiquitinated mitochondrial proteins were observed following administration of UA to wild-type rodents, supporting the improvement of mitophagy by UA treatment [97].
Dietary consumption of EA-rich food has been demonstrated to suppress inflammatory cytokine release in the brains of AD mice [98]. Studies in rats reported that UA attenuates inflammatory IL-1β signaling by reducing the production of inflammatory mediators via the ERK, JNK, p38, and NF-kB pathways. Another study reported that UA induces mitophagy in vivo following oral consumption [98], and improvements in exercise capacity were observed in different models of age-related muscle atrophy [99]. UA supplementation studies in humans showed clear evidence that the functions of electron transport chain complexes I, II, and IV were increased, indicating improved mitochondrial health [100,101]. This finding was supported by evidence that dietary UA increased the mRNA and protein expression of autophagy/mitophagy and mitochondrial biogenesis genes [102]. Additional research on the beneficial effects of UA on mitochondrial function is emerging. UA supplementation or pharmacological treatments that target mitophagy pathways to improve mitochondrial health are proving to have beneficial effects in aging and age-related diseases, including AD and Parkinson’s disease (Fig. 4).
2.6. Resveratrol
Resveratrol is a naturally occurring phenol anti-aging agent, present in the skins of grapes, blueberries, raspberries, and mulberries. It exhibits numerous pharmacological properties including antitumor, antioxidant, antiviral, and neuroprotective effects in AD. Resveratrol reduces proinflammatory NF-kB signaling, restores normal expression of CREB protein in cells, and activates the Sirt1 pathway [103]. Activation of Sirt1 by resveratrol affects autophagy and mitophagy in both healthy and diseased states (Fig. 5A). Resveratrol also inhibits mitogen-activated protein kinases, and modulates the calcium-activated potassium channels, which are key for mitochondrial dynamics and biogenesis in aging and AD [104].
Fig. 5. Protective effects of resveratrol.
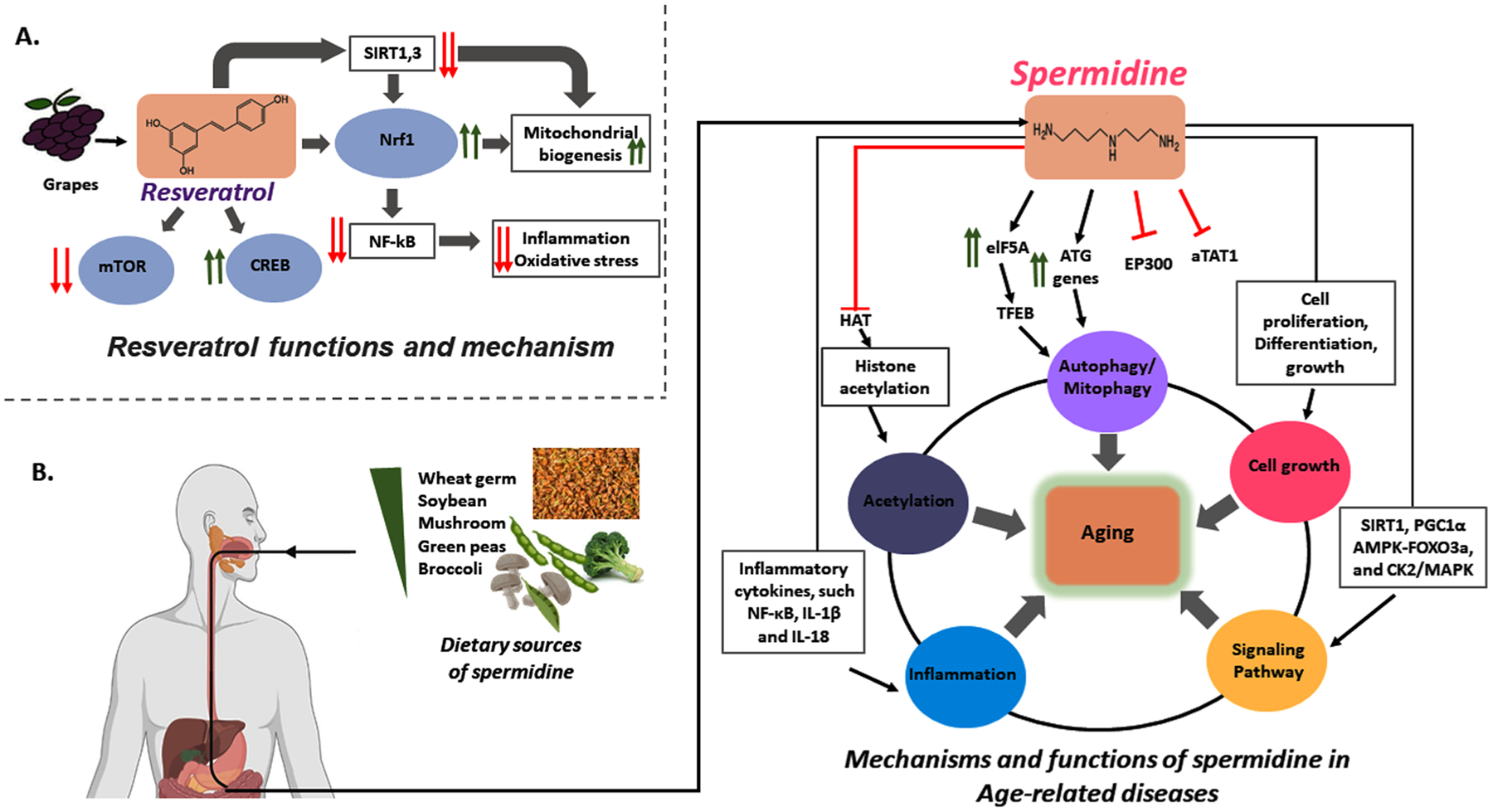
(A) Scheme of resveratrol’s actions to decrease inflammation and oxidative stress and to increase mitochondrial biogenesis by increasing SIRT1 and SIRT3 and by enhancing Nrf1 activation which inhibits Nf-kB signaling. Resveratrol counteracts mTOR signaling by activation of CREB, leading to improved mitochondrial biogenesis and healthy aging. (B) Several foods contain spermidine, which may improve mitochondrial function in AD and other age-related diseases. Spermidine induces autophagy by modulating the expressions of the ATG genes and the transcription factor elF5A. elF5A promotes the expression of the transcription factor TFEB which activates the expression of autophagy genes. Spermidine inhibits EP300, which directly promotes the acetylation of ATG proteins and indirectly stimulates deacetylation of tubulin due to inhibition of aTAT1. Spermidine exerts potent anti-inflammatory effects by suppressing multiple inflammatory cytokines, such as ROS, NF-κB, IL-1β and IL-18. Spermidine also suppresses histone acetylation. On the other hand, it can normalize signaling pathways affected by aging, such as SIRT1/PGC-1α, insulin/IGF, AMPK-FOXO3a, and CK2/MAPK.
2.7. Spermidine
Polyamines are essential for cell proliferation and growth, and important for cellular metabolism [105]. Spermidine is a polyamine known to regulate various cellular processes, including DNA stability, cellular growth, differentiation, and apoptosis in aging and age-related diseases such as AD. Wheat germ, soybeans, mushrooms, rice bran, green peas and broccoli are rich sources of spermidine and arginine, which is a precursor of polyamines that increases the intestinal production of polyamines [106,107]. Studies in both animals [108] and humans [109] have reported that tissue concentrations of spermidine decline with age. Spermidine influences plasma membrane potentials via its effects on the Na+/K+-ATPase transporter and on Ca2+ influx by the glutamatergic N-methyl-D-aspartate receptor (NMDA receptor). Researchers have also found that spermidine helped to maintain the mitochondrial membrane potential and increase oxidative phosphorylation in isolated neuronal rat mitochondria [110]. Spermidine inhibits EP300, an acetyltransferase that inhibits the elongation of autophagosome membranes by acetylation of ATG proteins. This results in reduced ATG acetylation which induces autophagy, drawing further attention to the possible beneficial use of spermidine in AD and other age-related diseases [111].
Finally, EP300 has histone acetyltransferase activity, and thus has an epigenetic role as a transcriptional co-activator which is inhibited by spermidine. This finding, along with the finding that spermidine regulates the FOXO transcription factor, shows that spermidine may also promote mitophagy via epigenetic and transcriptional regulation in healthy and aged cells [112,113] (Fig. 5B).
2.8. Mitophagy enhancers with therapeutic applications
Recently, our lab investigated the protective effects of mitophagy enhancers against mutant APP and Aβ-induced mitochondrial and synaptic toxicities in cell models of AD [114]. We optimized doses of the mitophagy enhancers UA, actinonin, tomatidine, and nicotinamide riboside in immortalized mouse primary hippocampal (HT22) neurons. We transfected HT22 cells with a mutant APP overexpression plasmid, treated with the mitophagy enhancers, and assessed the mRNA and protein expression levels of mitochondrial dynamics, mitochondrial biogenesis, mitophagy and synaptic genes. We additionally assessed cell survival by MTT assay and mitochondrial respiration using a Seahorse Bioanalyzer in treated and control mAPP-HT22 cells. Finally, we assessed mitochondrial length and number in treated and control mAPP-HT22 cells using transmission electron microscopy [114].
In a second study, we evaluated the protective effects of mitophagy enhancers against P-tau-induced mitochondrial and synaptic toxicities in AD [115]. We transfected HT22 with a mutant tau expression plasmid and treated the cells with nicotinamide riboside, tomatidine, actinonin, and UA [115]. We assessed the mRNA and protein expression levels of mitochondrial dynamics, mitochondrial biogenesis, mitophagy and synaptic genes [115]. We also assessed cell survival, mitochondrial respiration, and mitochondrial morphology in treated and control mTau-HT22.
The results of the two studies were very similar. Relative to the control cells, the mAPP-HT22 and mTau-HT22 cells had increased expression of mitochondrial fission genes, decreased expression of mitochondrial fusion, synaptic and mitophagy genes, reduced cell viability, defective mitochondrial respiration, and excessively fragmented and smaller mitochondria. However, these events were reversed when the mAPP-HT22 and mTau-HT22 cells were treated with combinations of mitophagy enhancers. Cell survival was significantly increased, mRNA and protein levels of mitochondrial fusion, synaptic and mitophagy genes were increased, and mitochondrial fragmentation was reduced, resulting in mitochondria that were larger and fewer in number [114]. Of the treatments tested, UA showed the strongest protective effects against mutant APP- and Tau-induced mitochondrial and synaptic toxicities.
We then tested UA in combination with epigallocatechin gallate (EGCG), an abundant polyphenol in green tea which has been studied for its beneficial anti-neoplastic, anti-microbial and neuroprotective effects [116]. We found that the addition of EGCG to UA resulted in enhanced anti-AD effects relative to either single agent in the mTau-HT22 model [115]. These observations, together with findings from other research groups, indicate that mitophagy enhancers are promising as therapeutics for AD, and further studies are warranted.
Impaired mitophagy contributes to many pathological states, including synaptic dysfunction and cognitive deficits. Abnormal accumulation of Aβ and P-tau interacts with Drp1, causing excessive mitochondrial fragmentation and depletion of PINK1 and Parkin, leading to defective mitophagy in AD [117,118]. Pharmacological drugs and natural supplements like NAD+, urolithin, and resveratrol, along with daily exercise, can restore the functional aspects of mitophagy and help to eliminate diseased/aged mitochondria from neurons.
Evidence increasingly suggests that autophagy enhancing drugs like rapamycin, CCI-779, Glc, Glc-6-P, Torin1, perhexiline, niclosamide, and rottlerin attenuate mTORC1 signaling and enhance mitochondrial biogenesis in both diseased and healthy states [119]. Natural compounds like NAD+ [120], urolithins, and resveratrol have been shown to alter mitophagy via FOXO3a signaling, which potentiates parkin-PINK1-dependent mitophagy in diseased and stressed cells [121,122].
Resveratrol mitigates the effects of defective mitophagy by activating mitochondrial biogenesis via PGC-1α-SIRT1-AMPK signaling and restoring mitochondrial oxidative phosphorylation [123]. In addition, resveratrol inhibits the NF-kB pathway, reducing proinflammatory signaling. Another mitophagy enhancer, spermidine, was reported to attenuate mitochondrial depolarization by regulating the Ca2+ levels in mitochondria and, in turn, promote healthy mitochondrial biogenesis along with increased levels of PGC-1α, SIRT1, and TFAM [124,125]. Identification of compounds, validation of mitophagy enhancing effects, and characterization of the signaling and transcriptional pathways which mediate those effects will result in promising mitophagy enhancers that can be clinically evaluated as therapeutics to improve outcomes in aging and age-related diseases [126,127] (Fig. 6).
Fig. 6. Pharmacological modulation of healthy aging with urolithin A, NAD+, resveratrol and spermidine.
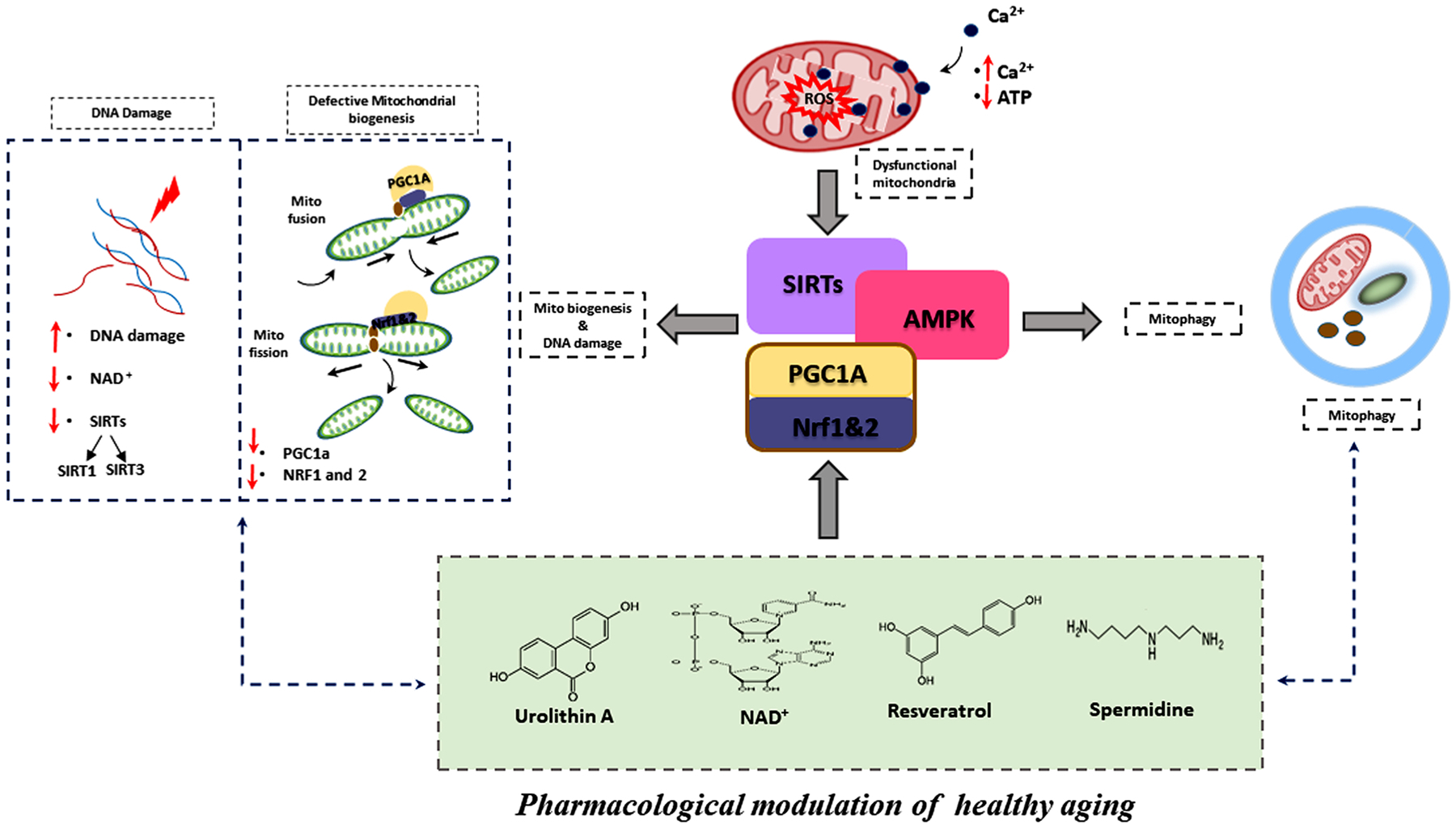
Mitochondrial dysfunction is characterized by elevated cytoplasmic calcium levels, ATP depletion, mitochondrial membrane potential failure, and increased reactive oxygen species production. Mitochondrial dysfunction is also associated with DNA damage and defective mitochondrial biogenesis. Mitophagy enhancers like Urolithin A, NAD+, Resveratrol and Spermidine, and physical exercise, promote effective mitophagy, mitochondrial dynamics, and mitochondrial biogenesis through the Sirtuin pathway. Pharmacological induction of PGC-1a through the AMPK-SIRT1 pathway stimulates mitochondrial biogenesis and improves mitochondrial health. PGC-1a and NRF2 activity affects mitochondrial detoxification and mitophagy induction. Pharmacological modulation of mitophagy and mitochondrial biogenesis may play an important role in mitochondrial homeostasis during stress, promoting healthy aging.
3. Conclusions and future directions
Mitochondrial abnormalities and synaptic damage are early events in the disease processes of AD. In AD, the accumulation of Aβ and P-tau plays a key role in mitochondrial dysfunction, synaptic damage, and neuronal loss. Dysfunctional mitochondria produce higher levels of reactive oxygen species, and the interactions of Aβ and P-tau with the mitochondrial proteins Drp1, VDAC, CypD, ABAD, PINK1, and Parkin enhance mitochondrial fragmentation and reduce mitophagy, leading to defective mitochondria in AD. Recent studies also suggest that mitophagy enhancers such as urolithin A, actinonin, tomatidine, and nicotinamide riboside are potential drug candidates that increase PINK1 and Parkin levels and increase the clearance of dead mitochondria [126,127].
Using immortalized mouse primary hippocampal (HT22) neurons transfected with mutant APP and mutant Tau expression plasmids, our lab recently investigated the protective effects of mitophagy enhancers against mutant Aβ- and mutant Tau-induced mitochondrial and synaptic toxicities in AD [114,115]. Mutant APP and mutant Tau-expressing HT22 cells showed increased expression of mitochondrial fission genes, decreased expression of mitochondrial fusion, synaptic and mitophagy genes, reduced cell survival, defective mitochondrial respiration, and excessively fragmented mitochondria of reduced lengths [114,115]. However, these events were reversed following treatment with mitophagy enhancers. In our work, UA showed the strongest protective effects against mutant APP and P-tau-induced mitochondrial and synaptic toxicities.
These and other observations [128,129] strongly suggest mitophagy enhancers are promising drugs to treat patients with AD and other tauopathies. However, these drugs need to be tested carefully using mouse models, particularly recently developed humanized Abeta knock-in (hAbKI) mice that express human amyloid beta peptide and exhibit late-onset AD features [130,131]. Currently, we are several mitophagy enhancers in late-onset hAbKI mice. Prior findings in other models support the safety and efficacy of this strategy; we expect that clinical trials will soon be warranted.
Funding
The research presented in this article was supported by NIH Grants AG042178, AG047812, NS105473, AG060767, AG069333 and AG066347 (to PHR).
Abbreviations:
- AD
Alzheimer’s disease
- ROS
Reactive Oxygen Species
- mtDNA
mitochondrial DNA
- Aβ
Amyloid beta
- Drp1
Dynamin-1-like protein
- ER
Endoplasmic reticulum
- TEM
Transmission electron microscopy
- Fis1
fission 1 protein
- Miro
Mitochondrial Rho GTPase
- Opa1
optic atrophy 1
- Mfn1
Mitofusin-1
- Mfn2
Mitofusin-2
- Mid49
Mitochondrial dynamics protein MID49
- Mid51
Mitochondrial dynamics protein MID51
- Mff
Mitochondrial fission factor
- NMDA
glutamatergic N-methyl-d-aspartate receptor
- PGC-1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- NRF1
nuclear respiratory factor 1
- NRF2
nuclear respiratory factor 2
- TFAM
transcription factor A, mitochondrial
- mAPP
Mutant amyloid beta precursor protein
- PINK1
PTEN-induced kinase 1
- BNIP3
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- OPTN
Optineurin
- FUNDC1
FUN14 domain containing 1
- TBK1
TANK-binding kinase 1
- SIAH1
E3 ubiquitin-protein ligase SIAH1
- GSK3β
Glycogen synthase kinase 3 beta
- CDK
Cyclin-dependent kinases
- PKA
Protein kinase A
- EF
helix-loop-helix structural domain
- ERMCS
ER mitochondria encounter structures
- LRRK2
leucine-rich repeat kinase 2
- PARL
Proteases like presenilin-associated
- OMA1
Metalloendopeptidase 1
- AGEs
Advanced glycation end products
- OMM
Outer membrane of mitochondria
- ETC
Electron transport chain
- SIRT1
Sirtuin 1
- VDAC
Voltage-dependent anion channels
- HT22
Mouse Hippocampal Neuronal Cell Line
- mAPP
Mutant Amyloid Precursor Protein
- P-tau
Phosphorylated Tau
Footnotes
CRediT authorship contribution statement
J.A.P. and P.H.R. contributed to the Conceptualization and formatting of the article. J.A.P., A.H., S. K. and P.H.R are responsible for Writing – original draft, and finalization of the manuscript. P.H.R. is responsible for Funding acquisition.
Compliance to ethical standards
Presented research is in compliance with ethical standards.
Consent to publication
All authors agreed to publish the contents.
Declaration of Competing Interest
Authors would like to inform you that they do not have conflict of interest with contents of our manuscript.
Data availability
No data was used for the research described in the article.
References
- [1].2021 Alzheimer’s disease facts and figures, Alzheimers Dement. 17 (2021) 327–406. [DOI] [PubMed] [Google Scholar]
- [2].A.s. Association, Alzheimer’s disease facts and figures, Alzheimer’s Dement. 15 (2019) 321–387. [Google Scholar]
- [3].Lee HG, Zhu X, Nunomura A, Perry G, Smith MA, Amyloid beta: the alternate hypothesis, Curr. Alzheimer Res 3 (2006) 75–80. [DOI] [PubMed] [Google Scholar]
- [4].Murphy MP, LeVine H 3rd, Alzheimer’s disease and the amyloid-beta peptide, J. Alzheimers Dis 19 (2010) 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bloom GS, Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis, JAMA Neurol. 71 (2014) 505–508. [DOI] [PubMed] [Google Scholar]
- [6].Furcila D, DeFelipe J, Alonso-Nanclares L, A study of amyloid-β and phosphotau in plaques and neurons in the hippocampus of Alzheimer’s disease patients, J. Alzheimers Dis 64 (2018) 417–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chow VW, Mattson MP, Wong PC, Gleichmann M, An overview of APP processing enzymes and products, Neuromol. Med 12 (2010) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tabaton M, Zhu X, Perry G, Smith MA, Giliberto L, Signaling effect of amyloid-beta(42) on the processing of AbetaPP, Exp. Neurol 221 (2010) 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fang C, Bourdette D, Banker G, Oxidative stress inhibits axonal transport: implications for neurodegenerative diseases, Mol. Neurodegener 7 (29) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou B, Cai Q, Xie Y, Sheng ZH, Snapin recruits dynein to BDNF-TrkB signaling endosomes for retrograde axonal transport and is essential for dendrite growth of cortical neurons, Cell Rep. 2 (2012) 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tammineni P, Jeong YY, Feng T, Aikal D, Cai Q, Impaired axonal retrograde trafficking of the retromer complex augments lysosomal deficits in Alzheimer’s disease neurons, Hum. Mol. Genet 26 (2017) 4352–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Reddy PH, Shirendeb UP, Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease, Biochim. Biophys. Acta 2012 (1822) 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Leritz EC, McGlinchey RE, Kellison I, Rudolph JL, Milberg WP, Cardiovascular disease risk factors and cognition in the elderly, Curr. Cardiovasc. Risk Rep 5 (2011) 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cournot M, Marquíe JC, Ansiau D, Martinaud C, Fonds H, Ferrières J, Ruidavets JB, Relation between body mass index and cognitive function in healthy middle-aged men and women, Neurology 67 (2006) 1208–1214. [DOI] [PubMed] [Google Scholar]
- [15].Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB, Obesity, diabetes and cognitive deficit: the Framingham heart study, Neurobiol. Aging 26 (2005) 1. [DOI] [PubMed] [Google Scholar]
- [16].Kilander L, Nyman H, Boberg M, Lithell H, Cognitive function, vascular risk factors and education. A cross-sectional study based on a cohort of 70-year-old men, J. Intern. Med 242 (1997) 313–321. [DOI] [PubMed] [Google Scholar]
- [17].Wolf PA, Beiser A, Elias MF, Au R, Vasan RS, Seshadri S, Relation of obesity to cognitive function: importance of central obesity and synergistic influence of concomitant hypertension. The Framingham Heart Study, Curr. Alzheimer Res 4 (2007) 111–116. [DOI] [PubMed] [Google Scholar]
- [18].Sturman MT, de Leon CF, Bienias JL, Morris MC, Wilson RS, Evans DA, Body mass index and cognitive decline in a biracial community population, Neurology 70 (2008) 360–367. [DOI] [PubMed] [Google Scholar]
- [19].McBride HM, Neuspiel M, Wasiak S, Mitochondria: more than just a powerhouse, Curr. Biol 16 (14) (2006) R551–R560. [DOI] [PubMed] [Google Scholar]
- [20].Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC, Mitochondrial regulation of cell cycle and proliferation, Antioxid. Redox Signal 16 (10) (2012) 1150–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Anderson AJ, Jackson TD, Stroud DA, Stojanovski D, Mitochondria-hubs for regulating cellular biochemistry: emerging concepts and networks, Open Biol. 9 (8) (2019), 190126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Reddy PH, Beal MF, Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res. Brain Res. Rev 49 (2005) 618–632. [DOI] [PubMed] [Google Scholar]
- [23].Herst PM, Rowe MR, Carson GM, Berridge MV, Functional mitochondria in health and disease, Front. Endocrinol 8 (2017) 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH, Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease, Hum. Mol. Genet 20 (2011) 4515–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang Y, Liu HH, Cao YT, Zhang LL, Huang F, Yi C, The role of mitochondrial dynamics and mitophagy in carcinogenesis, metastasis and therapy, Front. Cell Dev. Biol 8 (2020) 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Reddy PH, Oliver DM, Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease, Cells 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rai N, Singh C, Singh A, Singh MP, Singh BK, Mitochondrial dysfunction: a potential therapeutic target to treat Alzheimer’s disease, Mol. Neurobiol 57 (2020) 3075–3088. [DOI] [PubMed] [Google Scholar]
- [28].Cenini G, Lloret A, Cascella R, Oxidative stress in neurodegenerative diseases: from a mitochondrial point of view, Oxid. Med. Cell. Longev (2019), 2105607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Grimm A, Eckert A, Brain aging and neurodegeneration: from a mitochondrial point of view, J. Neurochem 43 (2017) 418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Camara AKS, Zhou Y, Wen PC, Tajkhorshid EW, Kwok M, Mitochondrial VDAC1: a key gatekeeper as potential therapeutic target, Front. Physiol 8 (2017) 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pagani L, Eckert A, Amyloid-beta interaction with mitochondria, Int. J. Alzheimers Dis (2011), 925050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF, Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms, Trends Neurosci. 40 (2017) 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chakravorty A, Jetto CT, Manjithaya R, Dysfunctional mitochondria and mitophagy as drivers of Alzheimer’s disease pathogenesis, Front. Aging Neurosci 11 (2019) 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Uddin MS, Mamun AA, Labu ZK, Hidalgo-Lanussa O, Barreto GE, Ashraf GM, Autophagic dysfunction in Alzheimer’s disease: cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis, J. Cell. Physiol 234 (2019) 8094–8112. [DOI] [PubMed] [Google Scholar]
- [35].Giorgi C, Bouhamida E, Danese A, Previati M, Pinton P, Patergnani S, Relevance of autophagy and mitophagy dynamics and markers in neurodegenerative diseases, Biomedicines 9 (2021) 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aman Y, Ryan B, Torsetnes SB, Knapskog AB, Watne LO, McEwan WA, Fang EF, Enhancing mitophagy as a therapeutic approach for neurodegenerative diseases, Int. Rev. Neurobiol 155 (2020) 169–202. [DOI] [PubMed] [Google Scholar]
- [37].Ma K, Chen G, Li W, Keep O, Zhu Y, Chen Q, Mitophagy, mitochondrial homeostasis, and cell fate, Front. Cell Dev. Biol 8 (2020) 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xian H, Liou YC, Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy, Cell Death Differ. 28 (2021) 827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Moreira OC, Estébanez B, Martínez-Florez S, de Paz JA, Cuevas MJ, González-Gallego J, Mitochondrial function and mitophagy in the elderly: effects of exercise, Oxid. Med. Cell. Longev (2017), 2012798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cao X, Wang H, Wang Z, Wang Q, Zhang S, Deng Y, Fang Y, In vivo imaging reveals mitophagy independence in the maintenance of axonal mitochondria during normal aging, Aging Cell 16 (2017) 1180–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hattori N, Saiki S, Imai Y, Regulation by mitophagy, Int. J. Biochem. Cell Biol 53 (2014) 147–150. [DOI] [PubMed] [Google Scholar]
- [42].Chen J, He H, Ye J, Feng Q, Wang F, Gu WWY, Han R, Xie C, Defective autophagy and mitophagy in Alzheimer’s disease: mechanisms and translational implications, Mol. Neurobiol 58 (2021) 5289–5302. [DOI] [PubMed] [Google Scholar]
- [43].Adiele RC, Adiele CA, Mitochondrial regulatory pathways in the pathogenesis of Alzheimer’s disease, J. Alzheimers Dis 53 (2016) 1257–1270. [DOI] [PubMed] [Google Scholar]
- [44].Mishra P, Chan DC, Mitochondrial dynamics and inheritance during cell division, development and disease, Nat. Rev. Mol. Cell Biol 15 (2014) 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Romanello V, Sandri M, Mitochondrial quality control and muscle mass maintenance, Front. Physiol 6 (422) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS, Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner, Biochim. Biophys. Acta 2012 (1823) 2297–2310. [DOI] [PubMed] [Google Scholar]
- [47].Huang DD, Fan SD, Chen XY, Yan XL, Zhang XZ, Ma BW, Yu DY, Xiao WY, Zhuang CL, Yu Z, Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner, Exp. Gerontol 119 (2019) 61–73. [DOI] [PubMed] [Google Scholar]
- [48].Suen DF, Norris KL, Youle RJ, Mitochondrial dynamics and apoptosis, Genes Dev. 22 (2008) 1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zorzano A, Claret M, Implications of mitochondrial dynamics on neurodegeneration and on hypothalamic dysfunction, Front. Aging Neurosci 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P, Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases, Brain Res. Rev 67 (2011) 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cheng Y, Bai F, The association of tau with mitochondrial dysfunction in Alzheimer’s disease, Front. Neurosci 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kandimalla R, Manczak M, Fry D, Suneetha Y, Sesaki H, Reddy PH, Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease, Hum. Mol. Genet 25 (2016) 4881–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tran M, Reddy PH, Defective autophagy and mitophagy in aging and Alzheimer’s disease, Front. Neurosci 14 (612757) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Oliver DMA, Reddy PH, Molecular basis of Alzheimer’s disease: focus on mitochondria, J. Alzheimers Dis 72 (2019) S95–S116. [DOI] [PubMed] [Google Scholar]
- [55].Rius-Pérez S, Torres-Cuevas I, Millán I, Ortega ÁL, Pérez S, PGC-1α, inflammation, and oxidative stress: an integrative view in metabolism, Oxid. Med. Cell. Longev (2020), 1452696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bragoszewski P, Turek M, Chacinska A, Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system, Open Biol. 7 (2017), 170007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ross JM, Olson L, Coppotelli G, Mitochondrial and ubiquitin proteasome system dysfunction in ageing and disease: two sides of the same coin? Int. J. Mol. Sci 16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wu Y, Chen M, Jiang J, Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling, Mitochondrion 49 (2019) 35–45. [DOI] [PubMed] [Google Scholar]
- [59].Cai Q, Jeong YY, Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases, Cells 9 (1) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liu L, Liao X, Wu H, Li Y, Zhu Y, Chen Q, Mitophagy and its contribution to metabolic and aging-associated disorders, Antioxid. Redox Signal 32 (2020) 906–927. [DOI] [PubMed] [Google Scholar]
- [61].Fritsch LE, Moore ME, Sarraf SA, Pickrell AM, Ubiquitin and receptor-dependent mitophagy pathways and their implication in neurodegeneration, J. Mol. Biol 432 (2020) 2510–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Harper JW, Ordureau A, Heo JM, Building and decoding ubiquitin chains for mitophagy, Nat. Rev. Mol. Cell Biol 19 (2018) 93–108. [DOI] [PubMed] [Google Scholar]
- [63].Roca-Agujetas V, de Dios C, Lestón L, Marí M, Morales A, Colell A, Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress, Oxid. Med. Cell. Longev 2019 (2019), 3809308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Glick D, Barth S, Macleod KF, Autophagy: cellular and molecular mechanisms, J. Pathol 221 (2010) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tan CC, Yu JT, Tan MS, Jiang T, Zhu XC, Tan L, Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy, Neurobiol. Aging 35 (2014) 941–957. [DOI] [PubMed] [Google Scholar]
- [66].Levine B, Kroemer G, Autophagy in the pathogenesis of disease, Cell 132 (2008) 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rajawat YS, Hilioti Z, Bossis I, Aging: central role for autophagy and the lysosomal degradative system, Ageing Res. Rev 8 (2009) 199–213. [DOI] [PubMed] [Google Scholar]
- [68].Stamatakou E, Wróbel L, Hill SM, Puri C, Son SM, Fujimaki M, Zhu Y, Siddiqi F, Fernandez-Estevez M, Manni MM, Park SJ, Villeneuve J, Rubinsztein DC, Mendelian neurodegenerative disease genes involved in autophagy, Cell Discov. 6 (2020) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yamano K, Matsuda N, Tanaka K, The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation, EMBO Rep. 17 (2016) 300–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Saito T, Sadoshima J, Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart, Circ. Res 116 (2015) 1477–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Johansen T, Lamark T, Selective autophagy mediated by autophagic adapter proteins, Autophagy 7 (2011) 279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Svenning S, Johansen T, Selective autophagy, Essays Biochem. 55 (2013) 79–92. [DOI] [PubMed] [Google Scholar]
- [73].Onishi M, Okamoto K, Mitochondrial clearance: mechanisms and roles in cellular fitness, FEBS Lett. 595 (2021) 1239–1263. [DOI] [PubMed] [Google Scholar]
- [74].Park JS, Koentjoro B, Sue CM, Commentary: nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease, Front. Mol. Neurosci 10 (2017) 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].van der Bliek AM, Shen Q, Kawajiri S, Mechanisms of mitochondrial fission and fusion, Cold Spring Harb. Perspect. Biol 5 (2013), a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, Han Z, Chen L, Gao R, Liu L, Chen Q, Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy, Autophagy 12 (2016) 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Esselun C, Theyssen E, Eckert GP, Effects of urolithin A on mitochondrial parameters in a cellular model of early Alzheimer disease, Int. J. Mol. Sci 22 (15) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Dyle MC, Ebert SM, Cook DP, Kunkel SD, Fox DK, Bongers KS, Bullard SA, Dierdorff JM, Adams CM, Systems-based discovery of tomatidine as a natural small molecule inhibitor of skeletal muscle atrophy, J. Biol. Chem 289 (2014), 14913–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Fang EF, Waltz TB, Kassahun H, Lu Q, Kerr JS, Morevati M, Fivenson EM, Wollman BN, Marosi K, Wilson MA, Iser WB, Eckley DM, Zhang Y, Lehrmann E, Goldberg IG, Scheibye-Knudsen M, Mattson MP, Nilsen H, Bohr VA, Becker KG, Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway, Sci. Rep 7 (2017) 46208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Richter U, Lahtinen T, Marttinen P, Suomi F, Battersby BJ, Quality control of mitochondrial protein synthesis is required for membrane integrity and cell fitness, J. Cell Biol 211 (2015) 373–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Xiao W, Wang RS, Handy DE, Loscalzo J, NAD(H) and NADP(H) redox couples and cellular energy metabolism, Antioxid. Redox Signal 28 (3) (2018) 251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yang Y, Sauve AA, NAD(+) metabolism: bioenergetics, signaling and manipulation for therapy, Biochim. Biophys. Acta 1864 (2016) 1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, Li C, Shen G, Zou B, NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential, Signal Transduct. Target Ther 5 (1) (2020) 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Grabowska W, Sikora E, Bielak-Zmijewska A, Sirtuins, a promising target in slowing down the ageing process, Biogerontology 18 (2017) 447–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Di Meo S, Reed TT, Venditti P, Victor VM, Role of ROS and RNS sources in physiological and pathological conditions, Oxid. Med. Cell. Longev (2016), 1245049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Houtkooper RH, Cantó C, Wanders RJ, Auwerx J, The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways, Endocr. Rev 31 (2010) 194–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez-Pérez P, Cadenas S, Lamas S, Antioxidant responses and cellular adjustments to oxidative stress, Redox Biol. 6 (2015) 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Piwowarski JP, Kiss AK, Granica S, Moeslinger T, Urolithins, gut microbiota-derived metabolites of ellagitannins, inhibit LPS-induced inflammation in RAW 264.7 murine macrophages, Mol. Nutr. Food Res 59 (2015) 2168–2177. [DOI] [PubMed] [Google Scholar]
- [89].Espín JC, Larrosa M, García-Conesa MT, Tomás-Barberán F, Biological significance of urolithins, the gut microbial ellagic acid-derived metabolites: the evidence so far, Evid. Based Complement. Altern. Med (2013), 270418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kujawska M, Jodynis-Liebert J, Potential of the ellagic acid-derived gut microbiota metabolite – urolithin A in gastrointestinal protection, World J. Gastroenterol 26 (2020) 3170–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Tomás-Barberán FA, González-Sarrías A, García-Villalba R, Núñez-Sánchez MA, Selma MV, García-Conesa MT, Espín JC, Urolithins, the rescue of “old” metabolites to understand a “new” concept: metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status, Mol. Nutr. Food Res 61 (2017). [DOI] [PubMed] [Google Scholar]
- [92].Al-Harbi SA, Abdulrahman AO, Zamzami MA, Khan MI, Urolithins: the gut based polyphenol metabolites of Ellagitannins in cancer prevention, a review, Front. Nutr 8 (2021), 647582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].D’Amico D, Andreux PA, Valdés P, Singh A, Rinsch C, Auwerx J, Impact of the natural compound urolithin A on health, disease, and aging, Trends Mol. Med 27 (2021) 687–699. [DOI] [PubMed] [Google Scholar]
- [94].Esselun C, Theyssen E, Eckert GP, Effects of urolithin A on mitochondrial parameters in a cellular model of early Alzheimer disease, Int. J. Mol. Sci 22 (2021) 8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Toney AM, Fan R, Xian Y, Chaidez V, Ramer-Tait AE, Chung S, Urolithin A, a gut metabolite, improves insulin sensitivity through augmentation of mitochondrial function and biogenesis, Obesity 27 (2019) 612–620. [DOI] [PubMed] [Google Scholar]
- [96].Xia B, Shi XC, Xie BC, Zhu MQ, Chen Y, Chu XY, Cai GH, Liu M, Yang SZ, Mitchell GA, Pang WJ, Wu JW, Urolithin A exerts antiobesity effects through enhancing adipose tissue thermogenesis in mice, PLoS Biol. 18 (2020), e3000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lin J, Zhuge J, Zheng X, Wu Y, Zhang Z, Xu T, Meftah Z, Xu H, Wu Y, Tian N, Gao W, Zhou Y, Zhang X, Wang X, Urolithin A-induced mitophagy suppresses apoptosis and attenuates intervertebral disc degeneration via the AMPK signaling pathway, Free Radic. Biol. Med 150 (2020) 109–119. [DOI] [PubMed] [Google Scholar]
- [98].Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA, Williams EG, Jha P, Lo Sasso G, Huzard D, Aebischer P, Sandi C, Rinsch C, Auwerx J, Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents, Nat. Med 22 (2016) 879–888. [DOI] [PubMed] [Google Scholar]
- [99].Grevendonk L, Connell NJ, McCrum C, Fealy CE, Bilet L, Bruls YMH, Mevenkamp J, Schrauwen-Hinderling VB, Jörgensen JA, Moonen-Kornips E, Schaart G, Havekes B, de Vogel-van den Bosch J, Bragt MCE, Meijer K, Schrauwen P, Hoeks J, Impact of aging and exercise on skeletal muscle mitochondrial capacity, energy metabolism, and physical function, Nat. Commun 12 (2021) 4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Palikaras K, Daskalaki I, Markaki M, Tavernarakis N, Mitophagy and age-related pathologies: development of new therapeutics by targeting mitochondrial turnover, Pharm. Ther 178 (2017) 157–174. [DOI] [PubMed] [Google Scholar]
- [101].Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, Auwerx J, Singh A, Rinsch C, The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans, Nat. Metab 1 (2019) 595–603. [DOI] [PubMed] [Google Scholar]
- [102].Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, et al. , Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease, Nat. Neurosci 22 (2019) 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Tian Y, Ma J, Wang W, Zhang L, Xu J, Wang K, Li D, Resveratrol supplement inhibited the NF-κB inflammation pathway through activating AMPKα-SIRT1 pathway in mice with fatty liver, Mol. Cell. Biochem 422 (1–2) (2016) 75–84. [DOI] [PubMed] [Google Scholar]
- [104].Lin KL, Lin KJ, Wang PW, Chuang JH, Lin HY, Chen SD, Chuang YCST, Huang MM, Tiao JB, Chen PH, Huang CW, Liou TK, Lin, Resveratrol provides neuroprotective effects through modulation of mitochondrial dynamics and ERK1/2 regulated autophagy, Free Radic. Res 52 (2018) 1371–1386. [DOI] [PubMed] [Google Scholar]
- [105].Thomas T, Thomas TJ, Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications, Cell. Mol. Life Sci 58 (2001) 244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Tofalo R, Cocchi S, Suzzi G, Polyamines and gut microbiota, Front. Nutr 6 (2019) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Bekebrede AF, Keijer J, Gerrits WJJ, Boer VCJ, The molecular and physiological effects of protein-derived polyamines in the intestine, Nutrients 12 (2020) 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Das R, Kanungo MS, Activity and modulation of ornithine decarboxylase and concentrations of polyamines in various tissues of rats as a function of age, Exp. Gerontol 17 (1982) 95–103. [DOI] [PubMed] [Google Scholar]
- [109].Kiechl S, Pechlaner R, Willeit P, Notdurfter M, Paulweber B, Willeit K, Werner P, Ruckenstuhl C, Iglseder B, Weger S, Mairhofer B, Gartner M, Kedenko L, Chmelikova M, Stekovic S, Stuppner H, Oberhollenzer F, Kroemer G, Mayr M, Eisenberg T, Tilg H, Madeo F, Willeit J, Higher spermidine intake is linked to lower mortality: a prospective population-based study, Am. J. Clin. Nutr 108 (2018) 371–380. [DOI] [PubMed] [Google Scholar]
- [110].Zhang R, Ma XN, Liu K, Zhang L, Yao M, Exogenous spermine preserves mitochondrial bioenergetics via regulating Src kinase signaling in the spinal cord, Mol. Med. Rep 16 (2017). [DOI] [PubMed] [Google Scholar]
- [111].Hirose T, Saiki R, Yoshizawa Y, Imamura M, Higashi K, Ishii I, Toida T, Williams K Kashiwagi K Igarashi K Spermidine and Ca(2+), but not Na(+), can permeate NMDA receptors consisting of GluN1 and GluN2A or GluN2B in the presence of Mg(2+), Biochem. Biophys. Res. Commun 463 (2015) 1190–1195. [DOI] [PubMed] [Google Scholar]
- [112].Li G, Ding H, Yu X, Meng Y, Li J, Guo Q, Zhou H, Shen N, Spermidine suppresses inflammatory DC function by activating the FOXO3 pathway and counteracts autoimmunity, iScience 23 (2020), 100807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Madeo F, Eisenberg T, Pietrocola F, Kroemer G, Spermidine in health and disease, Science 359 (2018) eaan2788. [DOI] [PubMed] [Google Scholar]
- [114].Kshirsagar S, Sawant N, Morton H, Reddy AP, Reddy PH, Protective effects of mitophagy enhancers against amyloid beta-induced mitochondrial and synaptic toxicities in Alzheimer disease, Hum. Mol. Genet (2021) ddab262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Kshirsagar S, Sawant N, Morton H, Reddy AP, Reddy PH, Mitophagy enhancers against phosphorylated Tau-induced mitochondrial and synaptic toxicities in Alzheimer disease, Pharm. Res 105973 (2021), 105973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Wan ML, Ling KH, Wang MF, El-Nezami H, Green tea polyphenol epigallocatechin-3-gallate improves epithelial barrier function by inducing the production of antimicrobial peptide pBD-1 and pBD-2 in monolayers of porcine intestinal epithelial IPEC-J2 cells, Mol. Nutr. Food Res 60 (2016) 1048–1058. [DOI] [PubMed] [Google Scholar]
- [117].Kandimalla R, Manczak M, Pradeepkiran JA, Morton H, Reddy PH, A partial reduction of Drp1 improves cognitive behavior and enhances mitophagy, autophagy and dendritic spines in a transgenic tau mouse model of Alzheimer disease, Hum. Mol. Genet (2021) ddab360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Quinn PMJ, Moreira PI, Ambrósio AF, Alves CH, PINK1/PARKIN signalling in neurodegeneration and neuroinflammation, Acta Neuropathol. Commun 8 (2020) 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J, Jones RG, AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species, Cell Rep. 21 (2017) 1–9. [DOI] [PubMed] [Google Scholar]
- [120].Cantó C, Menzies KJ, Auwerx J, NAD (+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus, Cell Metab. 22 (2015) 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Mei Y, Zhang Y, Yamamoto K, Xie WT, Mak W, You H, FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation, Proc. Natl. Acad. Sci. USA 106 (2009) 5153–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Zhou B, Wang J, Zheng G, Qiu Z, Methylated urolithin A, the modified ellagitannin-derived metabolite, suppresses cell viability of DU145 human prostate cancer cells via targeting miR-21, Food Chem. Toxicol 97 (2016) 375–384. [DOI] [PubMed] [Google Scholar]
- [123].Valenti D, de Bari L, de Rasmo D, Signorile A, Henrion-Caude A, Contestabile A, Vacca RA, The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model, Biochim. Biophys. Acta 1862 (2016) 1093–1104. [DOI] [PubMed] [Google Scholar]
- [124].Zhou J, Yang Z, Shen R, Zhong W, Zheng H, Chen Z, Tang J, Zhu J, Resveratrol improves mitochondrial biogenesis function and activates PGC-1α pathway in a preclinical model of early brain injury following subarachnoid hemorrhage, Front. Mol. Biosci 8 (2021), 620683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Chuang YC, Chen SD, Hsu CY, Chen SF, Chen NC, Jou SB, Resveratrol promotes mitochondrial biogenesis and protects against seizure-induced neuronal cell damage in the hippocampus following status epilepticus by activation of the PGC-1α signaling pathway, Int. J. Mol. Sci 20 (4) (2019) 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Jayatunga DPW, Hone E, Khaira H, Lunelli T, Singh H, Guillemin GJ, Fernando B, Garg ML, Verdile G, Martins RN, Therapeutic potential of mitophagy-inducing microflora metabolite, urolithin A for Alzheimer’s disease, Nutrients 13 (2021) 3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Katayama H, Hama H, Nagasawa K, Kurokawa H, Sugiyama M, Ando R, Funata M, Yoshida N, Homma M, Nishimura T, Takahashi M, Ishida Y, Hioki H, Tsujihata Y, Miyawaki A, Visualizing and modulating mitophagy for therapeutic studies of neurodegeneration, Cell 181 (2020) 1176–1187 (.e16). [DOI] [PubMed] [Google Scholar]
- [128].Esselun C, Theyssen E, Eckert GP, Effects of urolithin A on mitochondrial parameters in a cellular model of early Alzheimer disease, Int. J. Mol. Sci 22 (2021) 8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Fang EF, Tao J, Targeting on the NAD+-mitophagy axis to treat cardiovascular disease, Aging Med. 3 (2020) 151–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Baglietto-Vargas D, Forner S, Cai L, Martini AC, Trujillo-Estrada L, Swarup V, Nguyen MMT, Do Huynh K, Javonillo DI, Tran KM, Phan J, Jiang S, Kramár EA, Nuñez-Diaz C, Balderrama-Gutierrez G, Garcia F, Childs J, Rodriguez-Ortiz CJ, Garcia-Leon JA, Kitazawa M, Shahnawaz M, Matheos DP, Ma X, Da Cunha C, Walls KC, Ager RR, Soto C, Gutierrez A, Moreno-Gonzalez I, Mortazavi A, Tenner AJ, MacGregor GR, Wood M, Green KN, LaFerla FM, Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer’s disease-like pathology, Nat. Commun 12 (2021) 2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Kshirsagar S, Alvir RV, Hindle A, Kumar S, Vijayan M, Pradeepkiran JA, Reddy AP, Ramasubramanian B, Reddy PH, Early cellular, molecular, morphological and behavioral changes in the humanized amyloid-beta-knock-in mouse model of late-onset Alzheimer’s disease, Cells 11 (2022) 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.