

Introduction
Cancer biomarkers tests, designated as tumor biomarker tests (TBTs), can inform oncology treatment decisions to improve therapeutic outcomes in patients with cancer.1 The term TBT has been broadened to refer to any circumstance related to cancer screening, prevention, diagnosis, prognosis, prediction of therapeutic effects, or monitoring. Many TBTs proposed for clinical use lack proper vetting. In this regard, the Evaluation of Genomic Applications in Practice and Prevention Working Group proposed that a clinical genetic assay must demonstrate analytical validity, clinical validity, and clinical utility to warrant testing in clinical practice.2 This set of semantics has been widely adopted, including by the Institution of Medicine (now the National Academy of Medicine).1
Analytical validity, that a test accurately measures a biomarker, and clinical validity, that a TBT divides one population into one or more groups that have differing biologic or clinical outcomes, are necessary but not sufficient for a TBT to be valuable in patient care. By contrast, clinical utility is defined as “evidence of improved measurable clinical outcomes, and [a test's] usefulness and added value to patient management decision-making compared with current management without [omics] testing.”1 Clinical utility implies that the TBT can be useful to inform treatment decisions in that clinical context.3,4
Recently, one of us (D.F.H.) published a commentary proposing that six interdependent factors need to be considered to determine the clinical utility of a TBT: analytical validity, end point, decision option (previously referred to as use context or intended use), magnitude of difference (effect size) in the end point of interest between patients for whom the biomarker test results differ (high v low, positive v negative, etc), risk tolerance, and stakeholders.5 This perspective focused primarily on tumor characteristics that are informative for cancer prognosis or prediction of treatment benefits.5 There are additional cancer TBTs that are not measured within the tumor, and are therefore not technically TBT, but can be assessed for clinical utility through similar considerations. For example, inherited genetic variants within the patient's germline genome in BRCA1 or BRCA2 have clinical utility to estimate a patient's risk of developing cancer6 or to predict their response to poly (ADP-ribose) polymerase inhibitors.7
Germline Indicators of Toxicity Risk: An Overview
Inherited germline genetic variants may also serve as biomarkers to predict a patient's likelihood of treatment-related toxicity.8 We will refer to these biomarkers as germline indicators of toxicity risk (GITR). These germline variants often take the form of single nucleotide polymorphisms (SNPs), but GITR can include tests that detect other forms of germline genetic variation such as insertion/deletions and copy number variations.
GITR can be divided into two mechanistic categories. If a SNP affects systemic or cellular drug concentrations, or pharmacokinetics, we designate a test for it as a GITR-pharmacokinetic (GITR-PK, Fig 1A).8 For example, failure to metabolically eliminate the drug because of a SNP in a gene encoding a drug-metabolizing enzyme will cause toxicity by increasing systemic exposure of the drug. Alternatively, if the SNP directly enhances the sensitivity of a patient's nonmalignant tissues to toxicity, via a pharmacodynamic (PD) effect, then we refer to the test as a GITR-PD (Fig 1C). A GITR-PD indicates whether a patient is more susceptible to the toxic effects of therapy without affecting drug concentrations.9
FIG 1.
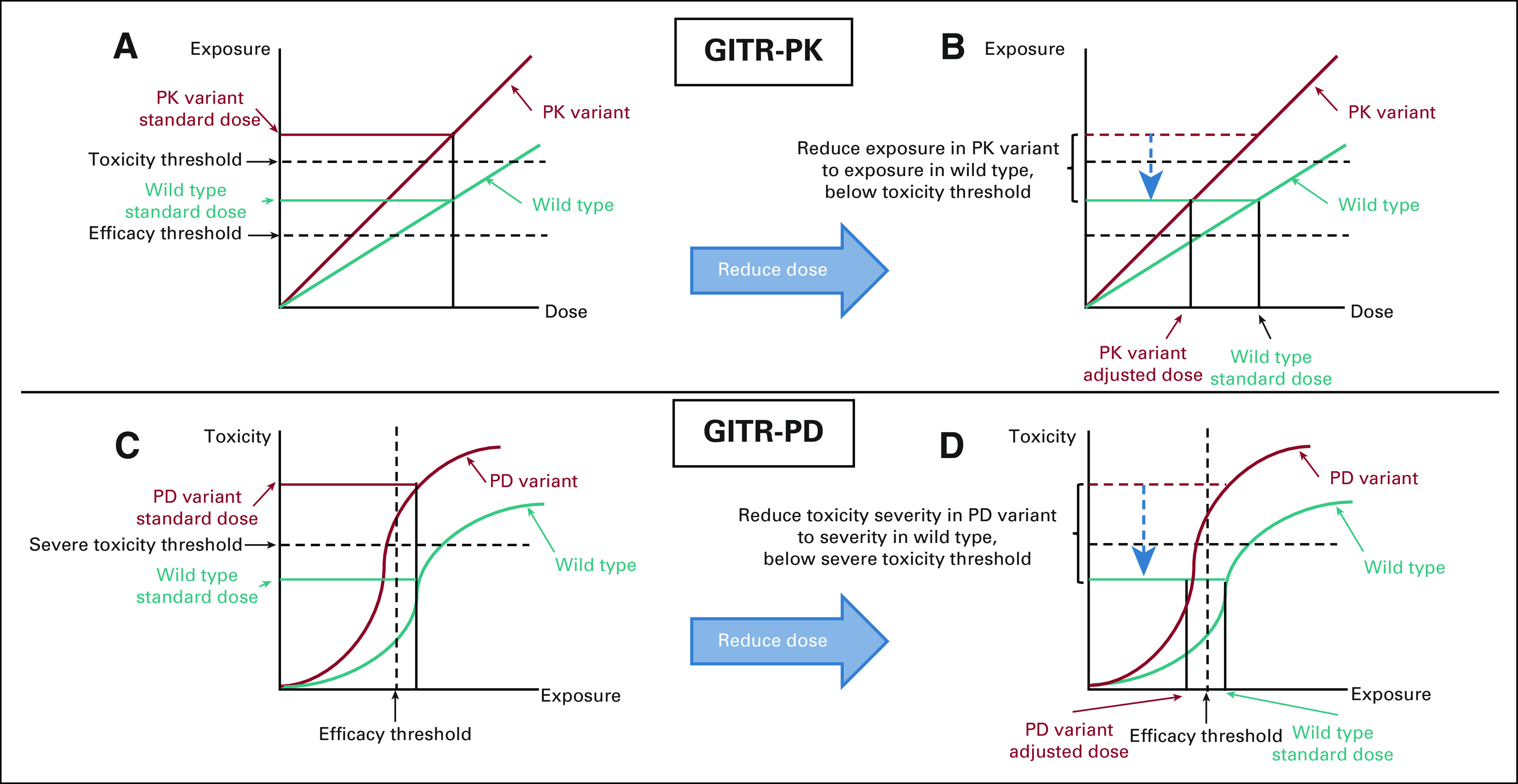
Dose reduction prevents toxicity via different mechanisms in GITR-PK and GITR-PD. (A) GITR-PK increase systemic drug exposure. (B) Decreasing dosing normalizes drug exposure, decreasing toxicity while maintaining efficacy. (C) GITR-PD increase the patient's toxicity sensitivity, meaning that they experience more severe toxicity at a given exposure. (D) Decreasing dosing normalizes their toxicity severity but reduces exposure and may reduce efficacy. See the text for details. GITR-PD, germline indicators of toxicity risk-pharmacodynamic; GITR-PK, germline indicators of toxicity risk-pharmacokinetic.
Outside of oncology, GITR have clearly demonstrated clinical utility and are part of the standard of care (SOC). For example, genetic screening for HLA-B*5701 before the administration of abacavir is required to prevent fatal hypersensitivity reactions.10 Within oncology, some GITR have confirmed clinical validity (Table 1), but few have been demonstrated to have clinical utility. TPMT testing to prevent severe thiopurine toxicity is used within the Children's Oncology Group protocols and recommended within National Cancer Center Network guidelines.34 Similarly, DPYD testing to prevent severe fluoropyrimidine toxicity has been recommended by European Society of Medical Oncology guidelines35 and adopted throughout Europe. However, DPYD testing has not been recommended by the National Cancer Center Network or adopted within the United States.36
TABLE 1.
Germline Indicators of Antineoplastic Agent Toxicity Risk (GITR) Currently Considered to Have Clinical Validity

The US Food and Drug Administration (FDA) has used a patient safety rationale for recommending the GITR HLA-B*5701 for abacavir and the somatic TBT kRAS for cetuximab. kRAS mutations indicate a lack of cetuximab benefit, and thus, a patient can forego cetuximab to avoid toxicity without sacrificing efficacy. However, the FDA has not used this patient safety rationale to recommend testing for TPMT, DPYD, or other GITR in oncology. The lack of a framework for evaluating the clinical utility of GITR is an important contributor to their inconsistent approval and utilization. The intent of this article is to build upon the previously published framework for assessing clinical utility of TBT5 to discuss specific considerations relevant to the evaluation of clinical utility of GITR. These principles should facilitate discussions of study designs that might be used to establish clinical utility of GITR and eventually enhance the adoption of GITR in clinical practice to improve treatment outcomes in patients with cancer.
Consideration of Factors Important to Determine Clinical Utility of GITR
To assess the clinical utility of a TBT, one must consider the end point that will be affected by taking action indicated by the result.5 In the treatment of cancer, there are two primary clinical end points: improvement in overall survival (OS) and quality of life (QOL).37 OS and other efficacy indicators are of paramount concern when evaluating the clinical utility of a therapeutic agent or a somatic TBT. By contrast, QOL is affected by both efficacy and toxicity of treatment. Toxicities can be separated into those that are unpleasant but usually manageable and reversible (National Cancer Institute's [NCI's] Common Terminology Criteria for Adverse Events grade 1-2) and those that are potentially persistently life-altering, life-threatening, or life-taking (grades 3-5). Toxicity can also be assessed via patient-reported outcomes scales, which provide a more detailed description of the toxicity severity, frequency, and symptomology38 and reveal differences in patient's tolerability to toxicities.39
The primary reason to incorporate a GITR into clinical care is to reduce toxicity (ie, benefit); however, there may be undesirable secondary effects (ie, risks) of doing so. These undesirable effects include potential reductions of efficacy or enhancement of a different toxicity including cost (ie, financial toxicity). Both the benefits (of reduced toxicity) and risks (of reduced efficacy or enhanced other toxicity) must be considered when evaluating the clinical utility of a GITR.
The magnitude of these potential risks can be broadly predicted on the basis of the GITR category (GITR-PK v GITR-PD) and the GITR decision option (previously referred to as use context).5 There are at least three decision options: Opt-In, Opt-Out, or Opt-Alt, each of which is based on the effect of the TBT result on the existing SOC paradigm for the patient.5,40 If the SOC is to do nothing further, but the results could indicate that further testing or treatment should be pursued, then the biomarker test is used to Opt-In to the subsequent strategy. By contrast, an Opt-Out test will result in the clinician not using some SOC strategy. Finally, if there are several SOC options, then an Opt-Alt test will guide the clinician to choose one option.
We propose a modified version of these semantics specific to GITR. In general, a positive GITR result indicates that a patient has elevated toxicity risk. Thus, it is difficult to conceive of an Opt-In GITR. However, a clinician might use a GITR to Opt-Out of a therapy that is associated with unacceptable toxicity, particularly if the efficacy reduction is expected to be limited. A theoretical example of an Opt-Out GITR involves the risk of anthracycline-related cardiotoxicity. SNPs have been identified, which may be associated with higher risk of congestive heart failure caused by anthracyclines in patients with early-stage breast cancer.41 If these can be validated, the small benefit from adding anthracyclines to adjuvant chemotherapy in breast cancer42 would probably be outweighed by the risk of congestive heart failure in such patients, and they could be treated without the anthracycline component.
A GITR will most commonly be an Opt-Alt biomarker, allowing the clinician to select a safer alternative in a patient at high risk of toxicity. We propose that there are at least three Opt-Alt strategies available for a GITR: Opt-Alt-Drug, Opt-Alt-Dose/Schedule, and Opt-Alt-Support (Table 2). In an Opt-Alt-Drug circumstance, the GITR indicates that drug A1 has unacceptable toxicity, but other drugs, either within the same class (Drug A2) or from a different class (Drug B or C), are less likely to cause that toxicity. A substitution within drug class (A1 → A2) will likely,43 although not always,44 be similarly effective and have similar toxicities and costs. A substitution to an alternative drug class (A1 → B) will have greater concern for decreased efficacy, although there are clinical situations for which multiple similarly effective regimens are available,45 and for increasing risk of other toxicities.
TABLE 2.
Opt-Alt Strategies and Anticipated Effect on Other Clinical Outcomes

In the Opt-Alt-Dose/Schedule circumstance, the GITR indicates that adjustment of the dose or schedule of Drug A1 would decrease toxicity risk. The risks for decreased efficacy and increased toxicity depend on the GITR category (GITR-PK or GITR-PD) and whether the dose or schedule is changed. There are several examples of GITR-PK that have sufficiently high levels of evidence to implement in clinical practice, including TPMT/NUDT15/thiopurines13 and DPYD/fluoropyrimidines.16 Both these GITR can guide dose reduction to normalize systemic drug concentrations in GITR-positive patients with those observed in GITR-negative patients receiving SOC doses (Fig 1B), resulting in an expected normalization in efficacy and toxicity. Such a personalized dosing strategy is conceptually similar to therapeutic drug monitoring, a technique commonly used outside of oncology in which systemic drug concentrations are monitored to guide personalized dosing to achieve target exposures that optimize treatment outcomes.46
By contrast, dose reductions dictated by a GITR-PD in a toxicity-sensitive patient will reduce tumor exposure as well, causing greater concern for decreased treatment efficacy (Fig 1D). However, it is possible that the GITR-PD could also render the tumor hypersensitive to the drug. If so, decreasing drug exposure would still result in high levels of antitumor activity,47 as has been proposed, but not confirmed, for the sensitivity to vincristine-induced neuropathy caused by CEP72 genotype.48
A third strategy within Opt-Alt-Dose/Schedule is to reduce the frequency of dosing or total number of doses administered. It would be expected that reducing dosing frequency would cause minimal efficacy reduction, whereas reducing administered doses could have a larger effect, but the actual effect may be drug-dependent and context-dependent.49
In an Opt-Alt-Support scenario, the high toxicity risk could be abrogated by addition of an ancillary supportive care strategy. For example, patients with germline deleterious SNPs in UGT1A1*28 who receive irinotecan are at high risk for severe neutropenia and diarrhea, which could be ameliorated with hematopoietic growth factor support and an antidiarrheal agent, respectively.18 Alternatively, one might simply enhance clinical monitoring for toxicity, such as that recommended by the FDA for patients with UGT1A1*28 receiving pazopanib.27 The addition of toxicity prophylaxis or monitoring should not affect antineoplastic dosing, exposure, or efficacy, but may increase treatment cost or burden.
The next consideration for determining clinical utility of a TBT is the magnitude of difference, or effect size, in testing, specifically, the absolute risk reduction in the end point(s) that result from switching SOC to biomarker-guided treatment.5 Again, we note that the primary objective for a somatic TBT is to estimate the expected efficacy, and the major consideration is whether the SOC or alternative treatment is more effective in one TBT-identified subgroup than another (eg, TBT-positive v TBT-negative). However, clinical utility of GITR is determined by the benefit from toxicity reduction and risk, primarily from the loss of treatment effect. Furthermore, estimation of the benefit of reducing toxicity must consider both the relative risk for and severity (ie, unpleasant but reversible v life-changing/life-threatening/life-taking) of the toxicity, as described above.
Figure 2 illustrates how these considerations may affect the clinical utility of using a GITR. The spectrum of possible clinical utilities ranges from very high (upper green region) to very low (lower red region). A GITR has the greatest clinical benefit if its use substantially reduces severe toxicity and minimally reduces efficacy (green area). By contrast, a GITR that indicates switching to an alternative strategy that substantially reduces an unpleasant but manageable toxicity, marginally reduces a severe toxicity, or substantially reduces efficacy would have a much smaller clinical benefit and would be less likely to have clinical utility (red area).
FIG 2.

Modeled clinical utility of GITR determined by benefits and risks. The clinical utility of GITR is determined by the net benefit, from toxicity avoidance, and net risk, primarily from the loss of treatment effect, from switching the standard of care to alternative treatment. See the text for details. Green, yellow, and red regions indicate high, moderate, and low clinical utility, respectively. Black indicators: examples of (A and C) high, (B or D) moderate, or (E) low clinical utility. GITR, germline indicators of toxicity risk.
The clinical context in which the GITR will be used is critical, as has been discussed for somatic TBT.5 For example, in patients with metastatic breast cancer harboring a DPYD SNP, switching from a fluoropyrimidine to an equally effective chemotherapeutic alternative (Opt-Alt-Drug), such as a taxane, avoids excessive toxicity without sacrificing efficacy (Fig 2, green area and black indicator A).50 Alternatively, switching adjuvant fluoropyrimidine to a less effective alternative (Opt-Alt-Drug) in a DPYD SNP carrier with early-stage colon cancer has greater risk of reduced efficacy and thus only moderate clinical benefit (Fig 2, yellow area and black indicator B). By contrast, clinical dosing guidelines16 recommend lowering the fluoropyrimidine dose in DPYD carriers (Opt-Alt-Dose/Schedule) and preserving the systemic exposure and efficacy of the drug while reducing toxicity risk (Fig 2, green area and black indicator C). In cases A and B, the same test (ie, DPYD) and strategy (Opt-Alt-Drug) have differing clinical utility because of the differing clinical contexts, whereas in cases A and C, the same test (ie, DPYD) leads to a different strategy (Opt-Alt-Drug v Opt-Alt-Dose/Schedule), but these have similarly high clinical utility.
A second example illustrates a theoretical GITR for cisplatin, which is palliative in metastatic non–small-cell lung cancer (NSCLC) but curative in metastatic testicular cancer. A SNP in MYH14 (rs1377817) has been reported to be associated with higher risk of cisplatin-induced neuropathy.51 If validated, this GITR-PD might be useful to avoid cisplatin in some patients with NSCLC, given the availability of other palliative agents for this disease (Fig 2, yellow area and black indicator D). On the other hand, men with testicular cancer would be unlikely to forego cisplatin to avoid neuropathy because of its 90% cure rate (Fig 2, black indicator E). Thus, the clinical utility of this SNP, if validated, would vary by clinical context because of differences in cisplatin effectiveness.
Clinical utility of GITR with moderate clinical benefit will depend upon the risk tolerance of the decision maker.5 Consider again the theoretical patient with NSCLC for whom MYH14 may predict cisplatin-induced neuropathy, but for whom an alternative agent would be somewhat less effective (black indicator D). If the patient prioritizes QOL over a small OS benefit, then the alternative agent might be preferred and GITR testing may have clinical utility. Another patient in a similar clinical context who prioritizes OS over QOL may prefer the SOC regardless of their GITR status, in which case, there is no clinical utility of testing. In addition to different prioritization of efficacy and QOL, different patients may have different tolerance for cost. Patients with comprehensive insurance may not consider cost when making treatment decisions, whereas those with limited resources and high co-pays, or no insurance at all, will almost certainly make cost an important component in their decisions to avoid financial toxicity.52
Moreover, these decisions may not be consistent between different stakeholder groups. In general, patients seek to maximize treatment outcomes, whereas payers tend to maximize financial interests, and regulatory bodies maximize societal utility.5 Although an insured patient may wish to undergo a GITR that would recommend adding a costly supportive treatment, such as hematopoietic growth factor support, their payer may not cover expensive ancillary treatments to prevent non–life-altering toxicities53 and would therefore not cover testing.
Levels of Evidence to Support Clinical Utility of GITR
Determination of clinical utility of a cancer biomarker test depends on the quality and strength of evidence that testing patients to inform treatment provides a clinical benefit.54 The National Academy of Medicine has adopted two pathways for demonstrating clinical utility of a cancer biomarker1: a prospective clinical trial in which the biomarker guides treatment55,56 or multiple well-conducted retrospective analyses of prospective clinical trials, so-called prospective-retrospective studies.1,57
Several trial designs have been proposed for prospective clinical trials in which the TBT is the primary objective of the trial (Fig 3 and Table 3).55-59 The biomarker-strategy design incorporates random assignment of patients to either use or not to use the TBT results for their care (Fig 3A).55,56 Although intuitively appealing, this trial design is inherently inefficient, especially if the frequency of GITR-positive patients is low, since most enrolled patients (eg, all patients in the arm in which the GITR is not used and the large subset of GITR-negative patients in the arm in which the GITR is used) will be treated with the identical SOC.60
FIG 3.

Prospective clinical trial designs to test clinical utility of GITR: (A) biomarker strategy design, (B) biomarker-enrichment design, (C) biomarker-stratified design, and (D) nonrandomized biomarker-guided design. See the text and Table 3 for description and limitations of each study design. Black box (R) indicates randomization. GITR, germline indicators of toxicity risk; SOC, standard-of-care.
TABLE 3.
Advantages and Disadvantages of Trial Designs to Test Clinical Utility of GITR

A biomarker-enrichment design tests all potential patients for the TBT, and only those who are positive (or negative, depending on the intended use) are enrolled and randomly assigned to SOC or alternative treatment (Fig 3B). This design of randomly assigning TBT-positive (eg, human epidermal growth factor receptor 2 [HER2]–positive breast cancer) patients to SOC (eg, chemotherapy) versus TBT-guided alternative (eg, adding trastuzumab to chemotherapy) treatment has been used to demonstrate clinical utility for the majority of somatic TBTs with companion diagnostics. The application of this design to GITR evaluation has major ethical concerns since patients who are GITR-positive and receive SOC treatment have already been demonstrated to have high risk of severe toxicity.61
A third prospective clinical trial design is the biomarker-stratified design, in which patients are tested for the TBT and both the TBT-positive and TBT-negative strata are randomly assigned to SOC or alternative treatment (Fig 3C). This design was used recently to test the clinical benefit of the poly (ADP-ribose) polymerase inhibitor veliparib in patients who carry BRCA variants and included a BRCA noncarrier group to assess the potential benefit in those patients as well.62 This design is also problematic for testing GITR. As in the enrichment design, randomly assigning a known GITR-positive patient to SOC treatment from which they have a high risk of severe toxicity raises serious ethical concerns. In addition, this design randomly assigns known GITR-negative patients to an alternative treatment that is expected to be worse than the SOC in those patients (eg, reduced dose in a patient with normal metabolic activity for a GITR-PK).
In the absence of equipoise concerning trade-offs between toxicity and potential efficacy reduction related to GITR results, none of the above randomized trial designs would be ethical. In that circumstance, the only option might be a less optimal, but potentially acceptable, nonrandomized prospective trial (Fig 3D). A prospective trial of GITR-based treatment could demonstrate that alternative treatment in GITR-positive patients is similarly toxic and effective compared with SOC treatment in GITR-negative patients. There are potential concerns about confounding factors, such as unequal distribution of prognostic characteristics, when directly comparing two different patient groups receiving two different treatments. Nevertheless, in a situation in which GITR-positive patients have unacceptable toxicity to SOC treatment, an alternative treatment with acceptable toxicity for these patients and similar efficacy to SOC treatment in GITR-negative patients should surpass a reasonable threshold of clinical utility, depending on the clinical context and stakeholder perspective. A possible derivative of this design would be to exclude GITR-negative patients and conduct a single-arm trial of alternative treatment in GITR-positive patients. The toxicity and efficacy of treatment could be compared with a prespecified, context-appropriate consensus threshold on the basis of historical controls and/or clinical judgment. This approach has been used recently to demonstrate adequate efficacy of adjuvant paclitaxel and trastuzumab in patients with very low anatomic risk (node-negative, relatively small) HER2-positive breast cancers.63 This example represents the same strategy of treatment de-escalation to avoid toxicity while maintaining apparent benefit, in the context of a TBT (HER2) that may also be acceptable for GITR.
Considering the resource intensiveness of prospective clinical trials, alternative study designs should be explored to evaluate the potential clinical utility of GITR. Investigators could leverage large databases of real-world evidence collected from sites that are conducting GITR testing and using results to guide treatment. Although this is lesser quality evidence,64 this information could be useful to glean evidence of the efficacy and toxicity of alternative treatment in GITR-positive patients. Relatedly, there may be an opportunity to use these real-world data to conduct synthetic clinical trials65 that mimic a prospective randomized trial to estimate the reduced toxicity and/or efficacy from alternative treatment compared with SOC. A similar strategy was pursued using real-world SEER data to evaluate clinical utility of the TBT Oncotype Dx for patients with hormone receptor–positive, HER2-negative breast cancer.66 Although such studies are subject to both anticipated and unanticipated biases, they are being used for TBT and might provide insights into the impact of GITR-guided changes to therapy.
Prospective-retrospective studies of previously conducted prospective treatment trials with available specimens for retrospective biomarker testing are another appealing approach for biomarker validation.57,67 This approach has been pursued successfully for somatic TBTs, including the previously discussed example of activating kRAS mutations to Opt-Out of cetuximab treatment.68 Importantly, these prospective-retrospective studies were possible because specimens were collected, processed, and archived for future correlative studies and because both SOC (ie, no cetuximab) and biomarker-informed alternative treatment (ie, cetuximab) were represented within the clinical trial arms. Few large prospective clinical trials collect blood or sputum for germline genetic analyses or detailed drug toxicity data, both of which are needed. In addition, few prospective clinical trials compare SOC with a relevant alternative treatment, particularly for Opt-Alt-Dose/Schedule or Opt-Alt-Support strategies.
Nonetheless, given the increasing interest in applying precision medicine in oncology and remarkable advances in genetic sequencing technology, we support efforts for future trials to collect these two critical elements: samples for germline genetic analysis and sufficient, well-justified drug toxicity data. Of interest, we and others have demonstrated acceptable accuracy of germline SNP analysis conducted within banked tumor samples69 although this strategy has been questioned70 and should be pursued cautiously. It would also be useful to establish a system similar to ClinicalTrials.gov to register prospective-retrospective biomarker studies with prespecified definitions of the GITR, primary end point, and formal analysis plan, as has been suggested for somatic TBTs.71 The Core Correlative Science Committee Review within the NCI Navigator system, which requires a prespecified analysis plan to access stored specimens from NCI-funded oncology clinical trials, could be adapted to serve this registration function.72
Special Considerations in the Setting of Existing GITR Information
Frameworks for determining clinical utility of medical tests, and therefore value and reimbursement strategies, typically assume that the tests would need to be ordered, as is routinely done for somatic TBT in cancer.5,40 However, unlike somatic TBTs, which may change as the tumor evolves, the germline genome is stable. Thus, germline information collected within the context of other genetic testing, such as matched germline testing during somatic analysis or hereditary cancer predisposition testing,73,74 can be used indefinitely for a patient. Since repurposing of existing GITR information has no testing cost, societal or payer concerns for resource allocation are irrelevant and only the expected benefits and risks of altering treatment need to be considered.75
Conclusion and Future Directions to Confirm Clinical Utility of GITR
This article proposes a system for consideration of the clinical utility of GITR in patients with cancer. One of the biggest challenges is estimation of the reduction of toxicity and, perhaps more importantly, efficacy caused by changing SOC to alternative treatment on the basis of the GITR. There are considerable logistical and ethical challenges in using widely accepted study designs for generating evidence of clinical utility. Further discussion among stakeholders is needed to determine what trial designs are necessary and practical to demonstrate clinical utility for GITR, potentially including prospective nonrandomized trials or synthetic trials using real-world data. Consideration of these issues would assist clinical guidelines committees and regulatory agencies with determining whether GITR have demonstrated clinical utility and should be recommended. This would provide clinicians with clear guidance for using GITR and activate implementation scientists to determine how best to integrate GITR within clinicalpractice.76 This road map for moving strong associations into clinical practice will assist oncologists and their patients with making evidence-based treatment decisions to improve clinical outcomes in patients with cancer.
Daniel L. Hertz
Research Funding: Disarm Therapeutics
Other Relationship: Advocates for Universal DPD/DPYD Testing (AUDT)
Uncompensated Relationships: PEPID, Saladax Biomedical
Lisa M. McShane
This author is a member of the Journal of Clinical Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Daniel F. Hayes
Stock and Other Ownership Interests: InBiomotion
Honoraria: Tempus
Consulting or Advisory Role: Cepheid, Freenome, Epic Sciences, Cellworks, BioVica, Oncocyte, Turnstone Bio, Predictus Biosciences, Guardant Health, L-Nutra, MacroGenics, Tempus
Research Funding: AstraZeneca (Inst), Pfizer (Inst), Merrimack (Inst), Menarini Silicon Biosystems (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from licensed technology, Diagnosis and Treatment of Breast Cancer. Patent No. US 8,790,878 B2. Date of Patent: July 29, 2014. Applicant Proprietor: University of Michigan. Dr Daniel F. Hayes is designated as an inventor/coinventor, Circulating Tumor Cell Capturing Techniques and Devices. Patent No.: US 8,951,484 B2. Date of Patent: February 10, 2015. Applicant Proprietor: University of Michigan. Dr Daniel F. Hayes is designated as an inventor/coinventor, Title: A method for predicting progression-free and overall survival at each follow-up timepoint during therapy of metastatic breast cancer patients using circulating tumor cells. Patent No. 05725638.0-1223-US2005008602
Travel, Accommodations, Expenses: Menarini Silicon Biosystems
Other Relationship: Menarini, UpToDate
Uncompensated Relationships: UpToDate
No other potential conflicts of interest were reported.
SUPPORT
Supported by the Fashion Footwear Charitable Foundation of New York/QVC Presents Shoes on Sale (D.F.H.).
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Defining Clinical Utility of Germline Indicators of Toxicity Risk: A Perspective
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Daniel L. Hertz
Research Funding: Disarm Therapeutics
Other Relationship: Advocates for Universal DPD/DPYD Testing (AUDT)
Uncompensated Relationships: PEPID, Saladax Biomedical
Lisa M. McShane
This author is a member of the Journal of Clinical Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Daniel F. Hayes
Stock and Other Ownership Interests: InBiomotion
Honoraria: Tempus
Consulting or Advisory Role: Cepheid, Freenome, Epic Sciences, Cellworks, BioVica, Oncocyte, Turnstone Bio, Predictus Biosciences, Guardant Health, L-Nutra, MacroGenics, Tempus
Research Funding: AstraZeneca (Inst), Pfizer (Inst), Merrimack (Inst), Menarini Silicon Biosystems (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from licensed technology, Diagnosis and Treatment of Breast Cancer. Patent No. US 8,790,878 B2. Date of Patent: July 29, 2014. Applicant Proprietor: University of Michigan. Dr Daniel F. Hayes is designated as an inventor/coinventor, Circulating Tumor Cell Capturing Techniques and Devices. Patent No.: US 8,951,484 B2. Date of Patent: February 10, 2015. Applicant Proprietor: University of Michigan. Dr Daniel F. Hayes is designated as an inventor/coinventor, Title: A method for predicting progression-free and overall survival at each follow-up timepoint during therapy of metastatic breast cancer patients using circulating tumor cells. Patent No. 05725638.0-1223-US2005008602
Travel, Accommodations, Expenses: Menarini Silicon Biosystems
Other Relationship: Menarini, UpToDate
Uncompensated Relationships: UpToDate
No other potential conflicts of interest were reported.
REFERENCES
- 1.Institute of Medicine . Evolution of Translational Omics: Lessons Learned and the Path Forward. Washington, DC: The National Academies Press; 2012. [PubMed] [Google Scholar]
- 2. Teutsch SM, Bradley LA, Palomaki GE, et al. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: Methods of the EGAPP working group. Genet Med. 2009;11:3–14. doi: 10.1097/GIM.0b013e318184137c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pritchard D, Goodman C, Nadauld LD. Clinical utility of genomic testing in cancer care. JCO Precis Oncol. doi: 10.1200/PO.21.00349. 10.1200/PO.21.00349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hayeems RZ, Dimmock D, Bick D, et al. Clinical utility of genomic sequencing: A measurement toolkit. NPJ Genom Med. 2020;5:56. doi: 10.1038/s41525-020-00164-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hayes DF. Defining clinical utility of tumor biomarker tests: A clinician's viewpoint. J Clin Oncol. 2021;39:238–248. doi: 10.1200/JCO.20.01572. [DOI] [PubMed] [Google Scholar]
- 6. Robson M. Breast cancer surveillance in women with hereditary risk due to BRCA1 or BRCA2 mutations. Clin Breast Cancer. 2004;5:260–268. doi: 10.3816/cbc.2004.n.029. discussion 269-271. [DOI] [PubMed] [Google Scholar]
- 7. Tutt ANJ, Garber JE, Kaufman B, et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N Engl J Med. 2021;384:2394–2405. doi: 10.1056/NEJMoa2105215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hertz DL, McLeod HL. Use of pharmacogenetics for predicting cancer prognosis and treatment exposure, response and toxicity. J Hum Genet. 2013;58:346–352. doi: 10.1038/jhg.2013.42. [DOI] [PubMed] [Google Scholar]
- 9. Hertz DL, McLeod HL. Using pharmacogene polymorphism panels to detect germline pharmacodynamic markers in oncology. Clin Cancer Res. 2014;20:2530–2540. doi: 10.1158/1078-0432.CCR-13-2780. [DOI] [PubMed] [Google Scholar]
- 10. Martin MA, Kroetz DL. Abacavir pharmacogenetics–from initial reports to standard of care. Pharmacotherapy. 2013;33:765–775. doi: 10.1002/phar.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 12. Yang JJ, Landier W, Yang W, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol. 2015;33:1235–1242. doi: 10.1200/JCO.2014.59.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Relling MV, Schwab M, Whirl-Carrillo M, et al. Clinical pharmacogenetics implementation consortium guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin Pharmacol Ther. 2019;105:1095–1105. doi: 10.1002/cpt.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Relling MV, Pui C-H, Cheng C, et al. Thiopurine methyltransferase in acute lymphoblastic leukemia. Blood. 2006;107:843–844. doi: 10.1182/blood-2005-08-3379. [DOI] [PubMed] [Google Scholar]
- 15. Meulendijks D, Henricks LM, Sonke GS, et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16:1639–1650. doi: 10.1016/S1470-2045(15)00286-7. [DOI] [PubMed] [Google Scholar]
- 16. Amstutz U, Henricks LM, Offer SM, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103:210–216. doi: 10.1002/cpt.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deenen MJ, Meulendijks D, Cats A, et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: A safety and cost analysis. J Clin Oncol. 2016;34:227–234. doi: 10.1200/JCO.2015.63.1325. [DOI] [PubMed] [Google Scholar]
- 18. Innocenti F, Undevia SD, Iyer L, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 19. Swen JJ, Nijenhuis M, de Boer A, et al. Pharmacogenetics: From bench to byte–an update of guidelines. Clin Pharmacol Ther. 2011;89:662–673. doi: 10.1038/clpt.2011.34. [DOI] [PubMed] [Google Scholar]
- 20. Innocenti F, Schilsky RL, Ramírez J, et al. Dose-finding and pharmacokinetic study to optimize the dosing of irinotecan according to the UGT1A1 genotype of patients with cancer. J Clin Oncol. 2014;32:2328–2334. doi: 10.1200/JCO.2014.55.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujii H, Yamada Y, Watanabe D, et al. Dose adjustment of irinotecan based on UGT1A1 polymorphisms in patients with colorectal cancer. Cancer Chemother Pharmacol. 2019;83:123–129. doi: 10.1007/s00280-018-3711-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Catenacci DVT, Chase L, Lomnicki S, et al. Evaluation of the association of perioperative UGT1A1 genotype-dosed gFOLFIRINOX with margin-negative resection rates and pathologic response grades among patients with locally advanced gastroesophageal adenocarcinoma: A phase 2 clinical trial. JAMA Netw Open. 2020;3:e1921290. doi: 10.1001/jamanetworkopen.2019.21290. [DOI] [PubMed] [Google Scholar]
- 23. Goey AK, Sissung TM, Peer CJ, et al. Effects of UGT1A1 genotype on the pharmacokinetics, pharmacodynamics, and toxicities of belinostat administered by 48-hour continuous infusion in patients with cancer. J Clin Pharmacol. 2016;56:461–473. doi: 10.1002/jcph.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peer CJ, Goey AK, Sissung TM, et al. UGT1A1 genotype-dependent dose adjustment of belinostat in patients with advanced cancers using population pharmacokinetic modeling and simulation. J Clin Pharmacol. 2016;56:450–460. doi: 10.1002/jcph.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Motzer RJ, Johnson T, Choueiri TK, et al. Hyperbilirubinemia in pazopanib- or sunitinib-treated patients in COMPARZ is associated with UGT1A1 polymorphisms. Ann Oncol. 2013;24:2927–2928. doi: 10.1093/annonc/mdt394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu CF, Reck BH, Xue Z, et al. Pazopanib-induced hyperbilirubinemia is associated with Gilbert's syndrome UGT1A1 polymorphism. Br J Cancer. 2010;102:1371–1377. doi: 10.1038/sj.bjc.6605653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Votrient (Pazopanib) [package insert] Basel, Switzerland: Novartis; 2009. [Google Scholar]
- 28.TRODELVY (Sacituzumab Govitecan-Hziy) [package insert] Foster City, CA: Gilead; 2020. [Google Scholar]
- 29. Xu CF, Johnson T, Wang X, et al. HLA-B 57:01 confers susceptibility to pazopanib-associated liver injury in patients with cancer. Clin Cancer Res. 2016;22:1371–1377. doi: 10.1158/1078-0432.CCR-15-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schaid DJ, Spraggs CF, McDonnell SK, et al. Prospective validation of HLA-DRB1*07:01 allele carriage as a predictive risk factor for lapatinib-induced liver injury. J Clin Oncol. 2014;32:2296–2303. doi: 10.1200/JCO.2013.52.9867. [DOI] [PubMed] [Google Scholar]
- 31.Tykerb (Lapatinib) [package insert] Basel, Switzerland: Novartis; 2007. [Google Scholar]
- 32. Diouf B, Crews KR, Lew G, et al. Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA. 2015;313:815–823. doi: 10.1001/jama.2015.0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wright GEB, Amstutz U, Drogemoller BI, et al. Pharmacogenomics of vincristine-induced peripheral neuropathy implicates pharmacokinetic and inherited neuropathy genes. Clin Pharmacol Ther. 2019;105:402–410. doi: 10.1002/cpt.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burnett HF, Tanoshima R, Chandranipapongse W, et al. Testing for thiopurine methyltransferase status for safe and effective thiopurine administration: A systematic review of clinical guidance documents. Pharmacogenomics J. 2014;14:493–502. doi: 10.1038/tpj.2014.47. [DOI] [PubMed] [Google Scholar]
- 35. Argilés G, Tabernero J, Labianca R, et al. Localised colon cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31:1291–1305. doi: 10.1016/j.annonc.2020.06.022. [DOI] [PubMed] [Google Scholar]
- 36.Referenced From the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Colon Cancer V.2.2019. National Comprehensive Cancer Network; 2019. www.nccn.org [Google Scholar]
- 37. Hudis CA, Barlow WE, Costantino JP, et al. Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: The STEEP system. J Clin Oncol. 2007;25:2127–2132. doi: 10.1200/JCO.2006.10.3523. [DOI] [PubMed] [Google Scholar]
- 38. Basch E, Reeve BB, Mitchell SA, et al. Development of the National Cancer Institute's patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE) J Natl Cancer Inst. 2014;106:dju244. doi: 10.1093/jnci/dju244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Coens C, Pe M, Dueck AC, et al. International standards for the analysis of quality-of-life and patient-reported outcome endpoints in cancer randomised controlled trials: Recommendations of the SISAQOL consortium. Lancet Oncol. 2020;21:e83–e96. doi: 10.1016/S1470-2045(19)30790-9. [DOI] [PubMed] [Google Scholar]
- 40. Dinan MA, Lyman GH, Schilsky RL, et al. Proposal for value-based, tiered reimbursement for tumor biomarker tests to promote innovation and evidence generation. JCO Precis Oncol. doi: 10.1200/PO.19.00210. 10.1200/PO.19.00210 [DOI] [PubMed] [Google Scholar]
- 41. Schneider BP, Shen F, Jiang G, et al. Impact of genetic ancestry on outcomes in ECOG-ACRIN-e5103. JCO Precis Oncol. doi: 10.1200/PO.17.00059. 10.1200/PO.17.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peto R, Davies C, Godwin J, et al. Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet. 2012;379:432–444. doi: 10.1016/S0140-6736(11)61625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Budd GT, Barlow WE, Moore HC, et al. SWOG S0221: A phase III trial comparing chemotherapy schedules in high-risk early-stage breast cancer. J Clin Oncol. 2015;33:58–64. doi: 10.1200/JCO.2014.56.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lokich J, Anderson N. Carboplatin versus cisplatin in solid tumors: An analysis of the literature. Ann Oncol. 1998;9:13–21. doi: 10.1023/a:1008215213739. [DOI] [PubMed] [Google Scholar]
- 45. McAndrew NP, Finn RS. Management of ER positive metastatic breast cancer. Semin Oncol. 2020;29:30082–30088. doi: 10.1053/j.seminoncol.2020.07.005. [DOI] [PubMed] [Google Scholar]
- 46. Beumer JH, Chu E, Allegra C, et al. Therapeutic drug monitoring in oncology: International association of therapeutic drug monitoring and clinical toxicology recommendations for 5-fluorouracil therapy. Clin Pharmacol Ther. 2019;105:598–613. doi: 10.1002/cpt.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hertz DL, Ramsey LB, Gopalakrishnan M, et al. Analysis approaches to identify pharmacogenetic associations with pharmacodynamics. Clin Pharmacol Ther. 2021;110:589–594. doi: 10.1002/cpt.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Diouf B, Evans WE. Pharmacogenomics of vincristine-induced peripheral neuropathy: Progress continues. Clin Pharmacol Ther. 2019;105:315–317. doi: 10.1002/cpt.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hertz DL, Childs DS, Park SB, et al. Patient-centric decision framework for treatment alterations in patients with Chemotherapy-induced Peripheral Neuropathy (CIPN) Cancer Treat Rev. 2021;99:102241. doi: 10.1016/j.ctrv.2021.102241. [DOI] [PubMed] [Google Scholar]
- 50. Stavraka C, Pouptsis A, Okonta L, et al. Clinical implementation of pre-treatment DPYD genotyping in capecitabine-treated metastatic breast cancer patients. Breast Cancer Res Treat. 2019;175:511–517. doi: 10.1007/s10549-019-05144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Trendowski MR, El-Charif O, Ratain MJ, et al. Clinical and genome-wide analysis of serum platinum levels after cisplatin-based chemotherapy. Clin Cancer Res. 2019;25:5913–5924. doi: 10.1158/1078-0432.CCR-19-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cherny NI, de Vries EGE, Dafni U, et al. Comparative assessment of clinical benefit using the ESMO-magnitude of clinical benefit scale version 1.1 and the ASCO value framework net health benefit score. J Clin Oncol. 2019;37:336–349. doi: 10.1200/JCO.18.00729. [DOI] [PubMed] [Google Scholar]
- 53. Chow R, Chiu L, Herrstedt J, et al. Cost-effectiveness analysis of olanzapine-containing antiemetic therapy for the prophylaxis of chemotherapy-induced nausea and vomiting (CINV) in highly emetogenic chemotherapy (HEC) patients. Support Care Cancer. 2021;29:4269–4275. doi: 10.1007/s00520-020-05977-x. [DOI] [PubMed] [Google Scholar]
- 54. Luzum JA, Petry N, Taylor AK, et al. Moving pharmacogenetics into practice: It's all about the evidence! Clin Pharmacol Ther. 2021;110:649–661. doi: 10.1002/cpt.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sargent DJ, Conley BA, Allegra C, et al. Clinical trial designs for predictive marker validation in cancer treatment trials. J Clin Oncol. 2005;23:2020–2027. doi: 10.1200/JCO.2005.01.112. [DOI] [PubMed] [Google Scholar]
- 56. Freidlin B, McShane LM, Korn EL. Randomized clinical trials with biomarkers: Design issues. J Natl Cancer Inst. 2010;102:152–160. doi: 10.1093/jnci/djp477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Simon RM, Paik S, Hayes DF. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst. 2009;101:1446–1452. doi: 10.1093/jnci/djp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mandrekar SJ, Sargent DJ. Clinical trial designs for predictive biomarker validation: Theoretical considerations and practical challenges. J Clin Oncol. 2009;27:4027–4034. doi: 10.1200/JCO.2009.22.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mandrekar SJ, Grothey A, Goetz MP, et al. Clinical trial designs for prospective validation of biomarkers. Am J Pharmacogenomics. 2005;5:317–325. doi: 10.2165/00129785-200505050-00004. [DOI] [PubMed] [Google Scholar]
- 60. Shih WJ, Lin Y. Relative efficiency of precision medicine designs for clinical trials with predictive biomarkers. Stat Med. 2018;37:687–709. doi: 10.1002/sim.7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boisdron-Celle M, Capitain O, Metges J-P, et al. Prevention of 5-FU-induced health-threatening toxicity by pretherapeutic DPD deficiency screening: Medical and economic assessment of a multiparametric approach. J Clin Oncol. 2013;31 abstr 3601. [Google Scholar]
- 62. Sharma P, Rodler E, Barlow WE, et al. Results of a phase II randomized trial of cisplatin +/- veliparib in metastatic triple-negative breast cancer (TNBC) and/or germline BRCA-associated breast cancer (SWOG S1416) J Clin Oncol. 2020;38 abstr 1001. [Google Scholar]
- 63. Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med. 2015;372:134–141. doi: 10.1056/NEJMoa1406281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Duarte JD, Dalton R, Elchynski AL, et al. Multisite investigation of strategies for the clinical implementation of pre-emptive pharmacogenetic testing. Genet Med. 2021;23:2335–2341. doi: 10.1038/s41436-021-01269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zauderer MG, Grigorenko A, May P, et al. Creating a synthetic clinical trial: Comparative effectiveness analyses using an electronic medical record. JCO Clin Cancer Inform. 2019;3:1–10. doi: 10.1200/CCI.19.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Petkov VI, Miller DP, Howlader N, et al. Breast-cancer-specific mortality in patients treated based on the 21-gene assay: A SEER population-based study. NPJ Breast Cancer. 2016;2:16017. doi: 10.1038/npjbcancer.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pepe MS, Feng Z, Janes H, et al. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: Standards for study design. J Natl Cancer Inst. 2008;100:1432–1438. doi: 10.1093/jnci/djn326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sepulveda AR, Hamilton SR, Allegra CJ, et al. Molecular biomarkers for the evaluation of colorectal cancer: Guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J Clin Oncol. 2017;35:1453–1486. doi: 10.1200/JCO.2016.71.9807. [DOI] [PubMed] [Google Scholar]
- 69. Hertz DL, Kidwell KM, Thibert JN, et al. Genotyping concordance in DNA extracted from formalin-fixed paraffin embedded (FFPE) breast tumor and whole blood for pharmacogenetic analyses. Mol Oncol. 2015;9:1868–1876. doi: 10.1016/j.molonc.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ratain MJ, Nakamura Y, Cox NJ. CYP2D6 genotype and tamoxifen activity: Understanding interstudy variability in methodological quality. Clin Pharmacol Ther. 2013;94:185–187. doi: 10.1038/clpt.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Andre F, McShane LM, Michiels S, et al. Biomarker studies: A call for a comprehensive biomarker study registry. Nat Rev Clin Oncol. 2011;8:171–176. doi: 10.1038/nrclinonc.2011.4. [DOI] [PubMed] [Google Scholar]
- 72. Schilsky RL. Publicly funded clinical trials and the future of cancer care. Oncologist. 2013;18:232–238. doi: 10.1634/theoncologist.2012-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hertz DL, Glatz A, Pasternak AL, et al. Integration of germline pharmacogenetics into a tumor sequencing program. JCO Precis Oncol. doi: 10.1200/PO.18.00011. 10.1200/PO.18.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hertz DL, Sahai V. Including DPYD on cancer genetic panels to prevent fatal fluoropyrimidine toxicity. J Natl Compr Canc Netw. 2020;18:372–374. doi: 10.6004/jnccn.2019.7527. [DOI] [PubMed] [Google Scholar]
- 75. Altman RB. Pharmacogenomics: “Noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther. 2011;89:348–350. doi: 10.1038/clpt.2010.310. [DOI] [PubMed] [Google Scholar]
- 76. Weitzel KW, Alexander M, Bernhardt BA, et al. The IGNITE network: A model for genomic medicine implementation and research. BMC Med Genomics. 2016;9:1. doi: 10.1186/s12920-015-0162-5. [DOI] [PMC free article] [PubMed] [Google Scholar]