

Abstract
Lianhuaqingwen (LH), one traditional Chinese medicine (TCM), has been used to treat the coronavirus disease 2019 (COVID-19), but its ecotoxicity with potential human health security has not been well investigated. To overcome such adverse effects and improve its medication efficacy, an intelligent multi-method integrated dietary scheme, screening, and performance evaluation approach was developed. Thirteen LH compounds were selected, and the main protease (Mpro) was used as the potential drug target. Resulted information showed that the more compounds of LH added, the higher medication efficacy obtained using multi-method integrated screening system, expert consultation method, and molecular dynamics simulation. Pharmacodynamic mechanism analysis showed that low total energy and polar surface area of LH active compound (i.e., β-sitosterol) will contribute to the best therapeutic effect on COVID-19 using quantitative structure-activity relationships (QSAR) and sensitivity models. Additionally, when mild COVID-19 patients take LH with the optimum dietary scheme (i.e., β-lactoglobulin, α-lactalbumin, vitamin A, vitamin B, vitamin C, carotene, and vitamin E), the medication efficacy were significantly improved (23.58%). Pharmacokinetics and toxicokinetics results showed that LH had certain human health risks and ecotoxicity. This study revealed the multi-compound interaction mechanism of LH treatment on COVID-19, and provided theoretical guidance for improving therapeutic effect, evaluating TCM safety, and preventing human health risk.
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1007/s11356-022-21125-w.
Keywords: Lianhuaqingwen, COVID-19, Multi-order interaction mechanism, Dietary scheme, Risk assessment
Introduction
The novel coronavirus (SARS-CoV-2) causing coronavirus disease 2019 (COVID-19) was identified in Wuhan, China, in December 2019 (Brauer et al. 2020; Li et al. 2020a, b; El-Gammal et al. 2021; WHO 2020a). An emergent SARS-CoV-2 lineage which was more infectious was defined in the UK, in December 2020. A new variant of the virus, named Alpha, was then discovered in some countries (e.g., Spain, Sweden, and Canada) (WHO 2020b). Lianhuaqingwen (LH), as a representative Chinese patent medicine for public health events of the respiratory system, has played a therapeutic role equivalent to oseltamivir phosphate in the treatment of influenza A (Duan et al. 2011; Liu et al. 2020). Clinical research has confirmed that LH can significantly relieve and shorten the course of the disease by reducing its clinical symptoms like fever, cough, expectoration, shortness of breath, and other common symptoms of COVID-19 (National Health Commission 2020; Lv et al. 2020). However, the multi-compound and multi-target actions of traditional Chinese medicine (TCM) are still unclear (Wang et al. 2019). Therefore, the study on the multi-order interaction and mechanism of LH’s active compounds is of great significance with more comprehensive application in COVID-19 treatment.
The process of consuming TCM is divided into different dosage due to its various reaction mechanisms. The corresponding administration method was selected for the different dosages to improve its efficacy (Zhang et al. 2018). Studies have shown that good food and medicine compatibility is the key to enhance the effect of drugs. Conversely, drugs’ therapeutic effect will be reduced or weakened (Li and Li 2000). In addition, considering the difficulty of conducting experiments on the human body and collecting human samples, it may be an appropriate way to explore LH active compounds’ mechanism in the treatment of COVID-19 in vitro. Molecular dynamics (MD) simulation assisted by molecular docking is a theoretical method to study the interaction between receptor macromolecules and small ligand molecules (Scott et al. 1999). This method predicts small molecule drug targets, search small molecule–binding proteins, and predict the best binding mode between ligand and receptor (Mukherjee and Majumder 2009; Zhao et al. 2009). There are few studies to evaluate the change of ligand-receptor interaction mode by changing the external stimulation conditions. Therefore, this study used the above methods to explore the interaction ability changes between active compounds of LH and potential targets for the treatment of COVID-19 by changing external stimulation conditions (dietary structure) for further LH therapeutic improvement on COVID-19.
The exploration of TCM chemical composition could help clarify the compound interaction, pharmacological mechanism, potential compounds, compatibility law, and other follow-up work (Jiao et al. 2021). Studies have shown that the interaction of TCM multiple compounds could affect the absorption, distribution, metabolism, and other drug stages in humans. It is also the main reason for clinical drug treatment failure and an important factor for side effects (Lu and Di 2019; Patel et al. 2019). LH is composed of 13 TCM (Wu 2005), but the material basis of its efficacy is still unclear. The first step of the study on the material basis of its efficacy is to study its compounds' interaction (Jia et al. 2015). Most studies have explored the interaction between each LH compound and the potential target protein of COVID-19, or the interaction of the 13 active compounds with the target protein simultaneously, to determine the material basis and mechanism of LH against COVID-19 (Li et al. 2020a, b). There are few studies on the independent assortment of 13 LH compounds to determine its multi-order interaction, assess each order’s interaction mechanism on the treatment of COVID-19, and the most effective combination. Furthermore, the TCM compounds’ structure and properties are the important factors that affect the interaction between the compounds and the potential target proteins. Quantitative structure-activity relationships (QSAR) is a common method to build the database of physicochemical properties of chemical substances based on the molecular structure descriptors calculated by quantum chemical methods. The established QSAR model can predict the physicochemical properties of compounds and quantify molecular structure parameters’ influence on the above properties (Galimberti et al. 2020; TWG on Pesticide 2012). However, most QSAR models are only used to predict physicochemical properties. The research on the influencing factors, especially the pharmacodynamic mechanism, is relatively weak. Therefore, this study explored the multi-order interaction of the LH compounds and analyzed the factors affecting their efficacy based on the molecular structure to screen the most effective combination of the compounds for COVID-19 treatment.
Studies have shown that more than 200000 cases of TCM’s adverse reactions were reported in 2009–2013. TCM is regarded as a natural product that is safer than synthetic drugs, while its toxicity is ignored (Li 2016). With the development and broad application of the TCM industry in China, its wastewater discharge has increased, seriously damaging the environment (Tong 2016; Liu et al. 2018). Therefore, it is vital to analyze its absorption and metabolism in humans and predict its human and environmental risks while improving the therapeutic effect of lotus leaf blast on COVID-19. Pharmacokinetic properties reflect the laws of absorption (A), distribution (D), metabolism (M), and excretion (E) of drugs in vivo, an important part of preclinical and clinical research of drugs (Hadni and Elhallaoui 2020). The prediction of absorption (A), distribution (D), metabolism (M), and excretion (E) of drugs in-vivo and toxicity (T) (abbreviated as ADMET) provided theoretical guidance for the selection of the most promising compounds (Kuthyala et al. 2019; Zhang et al. 2021). The toxicity of compounds is closely related to their structures. According to their structures, the structure-toxicity database (TOPKAT) can quantitatively evaluate the compounds’ toxicity and then monitor their human and ecological risks (Pizzo et al. 2013; Li et al. 2021). In drug development, some studies are currently limited to the ADMET prediction of its compounds and then screening the drug compounds with high efficacy and safety (Prival 2001). Nevertheless, few reports are found on screening and monitoring drug absorption, metabolism, and human and environmental risks through ADMET and TOPKAT. Therefore, in this study, ADMET and TOPKAT were used to predict LH’s absorption, metabolism, and health risk in vivo and monitor its ecological risk. Thus, it guided the screening of environmentally friendly LH compounds with high efficacy and safety, with practical significance for detecting its risk control. This study aimed to explore the multi-order interaction and mechanism of LH active compounds in the treatment of COVID-19. And then, the dietary scheme to improve LH's effect on COVID-19 was determined, and LH’s side effects on humans were analyzed, which will provide the guidance in reducing and eliminating drug-induced diseases.
Materials and methods
Structure and data sources of LH active compounds and receptor proteins
In this study, 13 active compounds (i.e., quercetin, kaempferol, luteolin, β- sitosterol, indigo, wogonin, tryptamine, benzylidene acetone, 1-methyl-2-nonyl-4(1H)-quinolone, stigmasterol, naringenin, glycyrrhetinic acid, and deserpidine) of LH were selected, and the main protease of COVID-19 (Mpro) was used as the potential target. In this study, LH’s 13 main constituents (quercetin, kaempferol, luteolin, β- sitosterol, indigo, wogonin, tryptamine, benzylidene acetone, 1-methyl-2-nonyl-4(1H)-quinolone, stigmasterol, naringenin, glycyrrhetinic acid, and deserpidine) were selected as the research objects. Proteinase (Mpro) was used as the potential drug target of COVID-19 to explore LH multi-compound mechanisms in the treatment of COVID-19 (Wu 2005; Khaerunnisa et al. 2020). The molecular structures of LH 13 main compounds were obtained from the PubChem of the National Institutes of Health (NIH) (http://pubchem.ncbi.nlm.nih.gov). The protein structure of Mpro was obtained from the protein data bank.
Studies have shown that when the binding energy between ligand and receptor is less than 0, a ligand and receptor can bind spontaneously. The lower binding energy indicates the greater absolute value of binding energy (Qiu and Xiao 2009). This means that the more stable binding conformation will lead to a greater possibility of occurrence (i.e., better drug effect) (Zhou et al. 2020). Therefore, the binding energy between the molecule and target protein was selected as the LH index for the treatment of COVID-19.
Characterization of binding ability between the 13 main compounds of LH and potential drug targets in the treatment of COVID-19
Stratified sampling assisted by Taguchi experimental design and expert consultation
Due to TCM’s multi-compound and multi-target effect, this study used Taguchi experimental design and stratified sampling to screen LH’s effective compounds for treating COVID-19. Therefore, computer-stratified sampling assisted by the Taguchi experimental design and expert consultation method was used to investigate the 13 active compounds’ multi-order interaction. The MD simulation was used to simulate the binding ability between molecules with Mpro protein in various interactions. The best combination of active compounds was then screened out and selected for the treatment of COVID-19. Taguchi experimental design, a special orthogonal method, can analyze several variables with a small number of experiments (Castorena-Cortés et al. 2009). Taguchi experimental design, a special orthogonal method, can analyze several variables with a small number of experiments (Castorena-Cortés et al. 2009). This study conducted a design of 13 factors and two levels L32 (213) of the Taguchi orthogonal experiment. Thirteen molecules were selected as the variables, and molecules related to the incoming protein/molecules non-related to the incoming protein were set up as the experimental levels. The orthogonal experimental table was then generated using the selected variables and experimental levels. The binding energy of combinations formed through the Taguchi experimental design can be obtained using molecular docking and molecular dynamics simulation. Then, MD simulation (“Calculation of binding ability of 192 groups' multi-order interaction between LH active compounds and receptor proteins based on molecular docking and MD simulation”) was carried out to select LH’s effective compounds in the treatment of COVID-19. The active compounds and Mpro protein’s binding ability was used to assess the efficacy of different active compounds. However, the combination produced by Taguchi’s experimental design is limited.
Therefore, this study considered whether the 13 drug molecules of LH jointly could treat COVID-19 and selected as a variable to determine the multi-order interactions between the drug molecules. Among them, 8191 combinations of 13 drug molecules were obtained (: corresponding to select m from n molecules), which is too large. Firstly, the 13 drug molecules’ different order interactions were analyzed by the Taguchi experimental design of L32(213). It was found that the 32 designed sets of combinations lacked some high-order interaction information. Stratified random sampling to reduce the sample size was used to obtain more representative and authoritative results. For this purpose, error (E) was set to 0.05, the confidence interval to 95%, and the P-value (total sample ratio) to 15%. The sample size (n) was calculated according to formula (1), and the Delphi method was used to complete expert consultation (Banno et al. 2020) (Table S6).
| 1 |
Calculation of binding ability of 192 groups’ multi-order interaction between LH active compounds and receptor proteins based on molecular docking and MD simulation
In this study, 13 active molecules of LH and Mpro receptor (1P9U; PDB ID: 12746549) were loaded into Discovery Studio 4.0 software. The “LibDock module” was used to finish the docking. Details are included in Supporting Information.
MD simulation of the LH was studied by exercising the Dell PowerEdge R7425 server and Gromacs software. Detailed MD settings are available in Supplement Information. MD simulation was used to simulate the binding of LH compounds and Mpro protein. The Molecular Mechanics/Poison-Boltzmann Surface Area (MMPBSA) method was used to calculate the composite system’s binding energy to determine the optimal combination of LH’s potential compounds in treating COVID-19. The binding energy between the Mpro protein and the active compounds represents the strength of the interaction. Binding energy is used to determine which multi-order interaction has the best binding effect with Mpro protein. The greater the absolute value of binding energy, the stronger the interaction between Mpro protein and its active compounds. The formula for the combined binding energy by MMPBSA was obtained in the Supplement Materials.
Pharmacodynamic mechanism analysis of LH active compound in the treatment of COVID-19 based on 2D-QSAR model
Descriptor screening of the interaction between the active compound and drug targets based on cluster analysis
The QSAR model was established to analyze the mechanism of the best active compounds of LH on covid-19 and determine the dietary scheme, which promoted the efficacy of LH clearing and blast treatment for COVID-19. First, the geometric descriptors (dipole moment μ, quadrupole moment QXX, QYY, QZZ, QXY, QYZ, QXZ and molecular weight), electronic descriptors (total energy; the number of charges: the most positive Millikan charge (q+, E), the most negative Millikan charge (q−, E), the most positive Millikan hydrogen charge (qH+, E); molecular orbital energy: the lowest occupied orbital energy (ELUMO, eV), the highest occupied orbital energy (EHOMO, eV), and energy gap) and spectral descriptors (frequency) of 13 Lianhuaqingwen active compounds were obtained using Gaussian 09 based on density functional theory (DFT) (Qu et al. 2017; Qu et al. 2018). Then, the physicochemical descriptors (Henry’s constant, HL, and oil-water distribution coefficient, logP) and topology descriptors (polar surface area, TPSA) were calculated using ChemDraw 12.0 software (Du et al. 2019).
SPSS Statistics 24 software was used for variable clustering analysis of 13 molecular descriptors. Intergroup connection was selected for clustering method, Pearson’s correlation for measurement interval, and Z scoring method for standardization method. Descriptors with high correlation were classified into one class after cluster analysis, convenient for analysis and screening.
Establishment of the 2D-QSAR model affecting efficacy of LH active compounds combination in the treatment of COVID-19
SPSS Statistics 24 was used based on multiple linear regression. The docking binding energy of the main active compounds of LH to Mpro protein was used as the dependent variable. The molecular descriptor was the independent variable to establish a 2D-QSAR for the effect of active compounds in the treatment of COVID-19. The molecular properties that affect its therapeutic effect were analyzed based on the model information. In addition, based on the arm ratio method, the model’s application domain was calculated to evaluate the application scope of the model (Qin et al. 2020). The calculation formula was as follows:
| 2 |
| 3 |
where hi represents the lever of the ith compound, Zi is the descriptor matrix of the ith compound, Z is the matrix of the compound descriptor, ZT is the transpose matrix of Z, ZiT is the transpose matrix of Zi, h* is the warning lever, which is the upper limit of hi value, p is the number of model descriptors, and n is the number of molecules.
Determination of the dietary scheme to enhance the treatment effect
According to pharmacodynamics and expert recommendations, patients with mild conditions should consume food high in protein and vitamin C (VC) to prevent or interfere with COVID-19. Therefore, MD simulation (“Calculation of binding ability of 192 groups' multi-order interaction between LH active compounds and receptor proteins based on molecular docking and MD simulation”) was first used to simulate the effects of nutrients from the three typical types of foods (high protein: milk and eggs; vegetables and fruits rich in VC: carrot, spinach, broccoli, apple, kiwi, grape, orange, winter jujube; immune-enhancing drinks: tea, wolfberry, ginseng, honey) on the binding ability between the 13 main compounds of LH and Mpro protein. Furthermore, the main nutrients that enhance the binding ability between active compounds and Mpro proteins were screened. Among them, the control simulation experiment was set up. One was the control group without adding the control stimulus conditions. The other was the experimental group with the control stimulus conditions (above 14 foods). According to the binding energy, the main food components that significantly enhanced the active compounds' binding ability with Mpro receptors were screened. Taguchi experimental design (“Stratified sampling assisted by Taguchi experimental design and expert consultation”) was used to determine the nutritive composition collocation scheme that significantly enhanced the binding between the active compounds and Mpro receptors. In this experiment, the food components that were selected to enhance the binding ability significantly were used as variables. Whether to add each variable was used as the experimental level. The above MD simulation process was repeated to determine the optimal nutritional composition scheme that significantly enhanced the above complexes' binding ability. Furthermore, the best recommended dietary scheme was determined to improve LH therapy’s efficacy for COVID-19 significantly, to improve its therapeutic effect.
Pharmacodynamics, human health, and ecological risk assessment method of the best combination of LH active compounds in the treatment of COVID-19
Pharmacodynamic assessment based on ADMET
In this study, the pharmacokinetic ADMET module of Discovery Studio 4.0 was used to evaluate the efficacy of 13 active compounds of LH. ADMET module provides five pharmacodynamic evaluation models for chemical structure prediction of absorption, distribution, metabolism, and excretion (passive intestinal absorption, aqueous solubility at 25°C, blood-brain barrier permeability, cytochrome P450 2D6 inhibition, and plasma protein binding). Firstly, 13 active compounds (SDF format) of lotus were imported into the DS software. In the “small molecules” module, select “calculate molecular properties” and click ADMET descriptors (aqueous stability, blood-brain barrier pension, CYP2D6 binding, intelligent absorption, and plasma protein binding) and click Run to predict the properties of 13 active molecules.
Human health and ecological risk assessment based on ADMET and TOPKAT
Pharmacokinetics and toxicokinetics were used to predict the human health and ecological risk of the LH optimal combination in COVID-19 treatment, which will assess medication safety, as well as prevent, reduce, and eliminate drug-induced diseases. The hepatotoxicity of 13 active compounds of lotus leaf blast was predicted using the ADMET module of Discovery Studio 4.0 software and setting 2.3.1 ADMET descriptors as hepatotoxicity. In addition, 13 kinds of active compounds (SDF format) of LH blast clearing active compounds were imported into DS software using the TOPKAT module. In the “small molecules” module, select “calculate molecular properties,” and click “toxicity prediction” to set the parameters, and then click Run. TOPKAT included 14 toxicity-related endpoints. In this study, rodent carcinogenicity, developmental toxicity potential (DTP), mutagenicity (Ames test), skin sensitization (GPMT), skin irritancy, and ocular irritation were mainly selected. Fathead minnow LC50 and Daphnia Magna EC50 were selected as the toxicity endpoint parameters for ecological risk assessment. The smaller the LC50 and EC50 are, the greater the toxicity will be (Chen et al. 2019).
Results and discussion
Calculation of the binding ability between 13 main compounds of LH and potential drug targets
Determination of multi-order interaction combination
While paying attention to the changes of the efficacy of traditional Chinese medicine (LH), the interaction mechanisms of 13 components of LH should be deeply revealed as well. Besides, the relationship between the interaction of 13 components with protein receptor and its therapeutic effect on COVID-19 should also be explained. Thus, a clear definition of the interaction between the LH’s components is the premise of exploring the interaction between components and receptor protein. It is also an effective measure to help put forward the enhancement of the LH capsule. Previous studies have shown that ligand and receptor can spontaneously bind when ligand and receptor binding energy is less than 0. When the binding energy is lower (the absolute value of binding energy is greater), the binding conformation is more stable with a greater possibility of occurrence (i.e., better drug effect) (Zhou et al. 2020). The 13 drug molecules were combined as a variable to explore the multi-order interaction of 13 main compounds of LH in the treatment of COVID-19, but 8191 combinations were observed. The amount of data was too large; therefore, the interaction of component molecules in different orders was analyzed by Taguchi’s experimental design of L32 (213). According to the Taguchi orthogonal experimental table of L32 (213), molecular docking was used to complete the docking of 13 compounds with Mpro protein. The binding energy of 32 combinations of molecular protein complex systems was calculated by the MD method (Fig. 1) to screen the effective compound combination of LH in the treatment of COVID-19.
Fig. 1.

Binding energy (kJ/mol) of 13 active compounds of LH activity to Mpro protein based on Taguchi experimental design
In the Taguchi experimental design, the signal-to-noise ratio (SNR) parameter with small calculation error was selected as the evaluation standard of binding energy (Fig. 1). The trend of SNR size was consistent with that of binding energy in 32 experimental design (Table S1). Therefore, according to Fig. 1, it was found that the binding energy of the complexes of the 13 compounds of LH and Mpro protein was the largest (−1381.123 kJ/mol), followed by 7/8 active compounds (7th order/8th order interaction) with Mpro protein. In addition, Fig. 1 showed that the more active compounds docking with Mpro protein, the greater the binding energy of the molecular protein complex; that is, the effect of LH in the treatment on COVID-19 is better.
It was found that the 32 combinations of Taguchi experimental design lacked some high-order interaction information (Fig. 1). A stratified random sampling method was proposed to reduce the sample size and obtain more representative and authoritative results. According to formula (1), the error E was set as 0.05, the confidence interval was 95%, the P-value (proportion of total samples) was 15%, and N is the combination of whether 13 kinds of drug molecules were added or not. There were 8191 combinations in total. The sample size (n) representing the whole population was calculated, and N was solved as 192 groups. The principle of weight increasing with the number of drug molecules in each layer combination was adopted when stratified random sampling combined with the multi-compound and multi-target effect of TCM. Table 1 shows the weight samples of stratified random sampling with a total of 192 groups verified by field expert consultation method. In addition, according to the weight setting in Table 1, a stratified random sampling program for 8191 groups was compiled to ensure the same probability of each sampling unit being selected. Finally, 192 groups of 13 multi-order interactive combinations of active compounds of lotus blast cleaning were determined. The molecular docking method was used to complete the docking of the active molecules and Mpro protein in 192 combinations.
Table 1.
Example of weight setting of random stratified sampling
| Combination | Combination number | Weight | Sampling number |
|---|---|---|---|
| C(13,1) | 13 | 4.00% | 1 |
| C(13,2) | 78 | 4.00% | 3 |
| C(13,3) | 286 | 0.55% | 2 |
| C(13,4) | 715 | 0.55% | 4 |
| C(13,5) | 1287 | 0.55% | 7 |
| C(13,6) | 1716 | 0.60% | 10 |
| C(13,7) | 1716 | 2.45% | 42 |
| C(13,8) | 1287 | 3.00% | 39 |
| C(13,9) | 715 | 3.50% | 25 |
| C(13,10) | 286 | 8.00% | 23 |
| C(13,11) | 78 | 30.00% | 23 |
| C(13,12) | 13 | 90.00% | 12 |
| C(13,13) | 1 | 100.00% | 1 |
| Total | 8191 | 192 |
Analysis and verification of the binding ability of 192 groups between main compounds and receptor proteins
MD simulation was used to simulate the multi-order interaction of 192 groups, LH main compounds to treat COVID-19. The binding energy between active molecules and drug target protein (Mpro) was used as the evaluation index (Table S2). As shown in Fig. 2, the binding energies of the first- to thirteenth-order interactions with Mpro protein are as follows: −75.383 to −308.928 kJ/mol, −45.181 to −214.231 kJ/mol, −71.095 to −382.138 kJ/mol, −120.532 to −524.831 kJ/mol, −279.512 to −687.735 kJ/mol, −331.67 to −968.17 kJ/mol, −467.564 to −995.648 kJ/mol, −567.513 to −975.612 kJ/mol, −594.558 to −1162.41 kJ/mol, −824.815 to −1249.417 kJ/mol, −927.954 to −1326.583 kJ/mol, −1042.275 to −1358.765 kJ/mol, and −1399.826 kJ/mol. Results showed that with the increase in active molecules, the greater the absolute value of protein molecular binding energy, and the better the efficacy of LH in the treatment of COVID-19. The 13 active compounds showed the best effect. The results further verified LH's effectiveness in the treatment of COVID-19 by targeting the virus through multiple compounds, targets, and pathways. In addition, the results obtained by calculating the binding energies between 13 active components and Mpro protein (first-order interaction) showed that the therapeutic effects of 13 active components on COVID-19 from strong to weak were as follows: β-sitosterol > benzylidene acetone > tryptamine > stigmasterol > kaempferol > naringenin > quercetin > 1-methyl-2-nonyl-4 (1H) - quinolone > luteolin > indigo > hanhuangcensu > snake Root Pingding > glycyrrhetinic acid. According to 192 kinds of multi-order interaction combinations (Fig. 2 and Table S2), it was found that the effect of several active components of LH was not the sum of the effects of each single component. Antagonistic or synergistic effects existed among different active components, and antagonism accounted for 72% of the 192 multi-order interactions. The effect values of antagonism and synergism were 68.06% and 31.94%, respectively. Therefore, it can be found that although the 13 active components have the strongest effect on the treatment of COVID-19, the main molecular antagonism among the 13 components indicates that the active components of LH play a common role and the antagonistic effects will occur among the components in varying degrees. This result is consistent with that TCM is not only the simple addition of each single herb, but also produces new effective substances and new functions after interaction in terms of curative effect (Chen et al. 2015; Lei et al. 2014). What’s more, Fig. 2 showed that in addition to the 13 active components, the molecules of active components 1, 2, 3, 5, 6, 7, 8, 9, 10, 11, 12, 13 in the 12th-order interaction, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 13 in the 11th-order interaction, and 1, 2, 3, 6, 7, 8, 9, 10, 11, 13 in the 10th-order interaction also have the significant therapeutic effect of COVID-19. Their binding energies were −1358.765 kJ/mol, −1326.583 kJ/mol, and −1249.417 kJ/mol, respectively, which were −2.93%, −5.23%, and −10.74% lower than that of 13 active components. Therefore, this study will use the mechanism analysis to further explore the factors affecting the effectiveness of each component molecular therapy for COVID-19, so as to improve the efficacy of LH in the treatment of COVID-19.
Fig. 2.
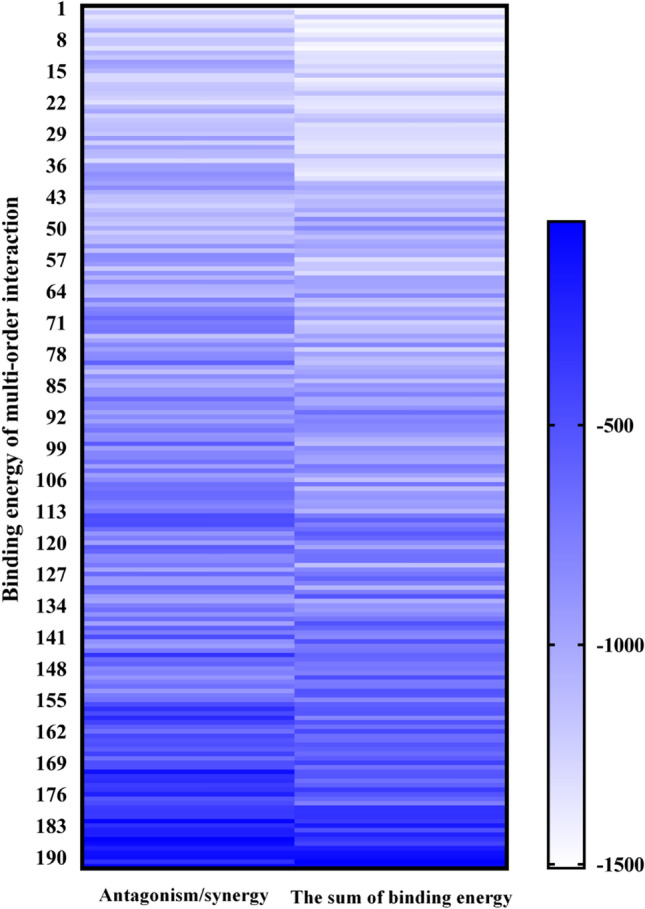
Distribution of binding energy between LH active components and Mpro protein in 192 group multi-order interactions
Studies have shown that the mutated new coronavirus enhances its binding ability to human ACE2 protein (Rambaut et al. 2020; Starr et al. 2020). What’s more, in order to further verify the reliability of above results, another potential drug target proteins (ACE2) (Alexandre et al. 2020) was selected as the receptor to carry out the molecular docking with 13 active components. In this process, we used the fractional factorial experiment design method with resolution V, 13 active components were selected as the variables, whether the above molecules were connected to the protein was taken as the experimental level. According to the generated orthogonal experimental table, the docking of 13 active components with ACE2 protein molecule was finished. And then the MD simulation was used to screen the LH active components of the treatment on COVID-19. According to the binding ability of different active components and ACE2 protein, we can judge the efficacy of different active components in the treatment of COVID-19, and then the main active components will be determined. The results showed that (Fig. 3), the binding energy of 13 active components acting on ACE2 protein was the highest, that is to say, the therapeutic effect on COVID-19 was the best. Moreover, the therapeutic effect of LH on ACE2 was greater than that on Mpro (the absolute value of binding energy increased 29.22%). The results further verified the reliability of the comprehensive screening system of Taguchi experimental design, MD simulation and computer-aided stratified sampling method intelligent integration for screening the most effective components of LH for COVID-19, and also verified that LH used multi-component, multi-target, and multi-channel action on COVID-19. Results showed that 13 active components had the best therapeutic effect on COVID-19 for the two potential target proteins (Mpro and ACE2), and it also can be speculated that LH had a certain effect on the prevention or treatment of COVID-19 caused by mutant virus. In addition, the comprehensive screening system established by multiple methods is helpful to screen the components with the best therapeutic effect of other TCM, and provides a new idea for improving the therapeutic effect of TCM.
Fig. 3.

Distribution map of binding energy between active components and ACE2 protein in 128 multi-order interactions (fractional factorial results)
Analysis of the mechanism of the active compounds of LH in the treatment of COVID-19
In order to explore the reasons for the most significant effect of 13 LH active components on COVID-19 in section “Calculation of the binding ability between 13 main compounds of LH and potential drug targets,” this study established a QSAR model of LH's active compounds mechanism in the treatment of COVID-19 using variable clustering and sensitivity analysis. The mechanism of the active compound of LH in the treatment of COVID-19 based on the molecular structure level was further analyzed.
Screening of parameters affecting LH molecules efficacy based on variable cluster analysis
The binding energy of the main active compounds of LH and Mpro protein and each descriptor variable’s data is shown in the Table S2. The result of cluster analysis of the descriptor variables is shown in Fig. 4. The correlation between the dependent and independent variable (molecular descriptor) of the 2D-QSAR model should not be too high. This study used a tree based on cluster analysis to screen the descriptors classified into one category to reduce the independent variable number and correlation between the dependent and the independent variable, thus increasing the model accuracy. In Fig. 4, a vertical red line was drawn, and three horizontal lines on the left side of that line connect three types of descriptors. In the descriptor screening process, the descriptor variables that were not significantly related to the dependent variable were replaced by descriptors classified as the same type. The descriptor combination of the model was determined, and the qualified 2D-QSAR model was established. The descriptors, TPSA, logP, and TE, were screened as independent variables to build the 2D-QSAR model.
Fig. 4.

Cluster analysis tree of main active compounds of LH molecular parameters
Analysis of the pharmacodynamic mechanism of the active compounds of LH based on QSAR model and sensitivity evaluation
In this study, the binding energy of the 13 main active compounds of LH, docking with the Mpro protein, was used as the dependent variable and the molecular descriptor as the independent variable. A 2D-QSAR mechanism model was constructed to explore the effects of the 13 active compounds of LH in the treatment of COVID-19. The model formula is as follows:
| 4 |
The coefficient R was 0.808, higher than the critical value of 0.685 (confidence level p = 0.01), indicating that the established regression equation is significant. The model was found significant (sig. = 0.019) (<0.05) (Yang and Liu 2019). It is necessary to evaluate the application domain of the QSAR model to evaluate the rationality of the established model for the molecules. William’s diagram of the model was drawn by calculating each molecular parameter’s (residual error and leverage value) (Fig. 5). In the Williams diagram, the molecules with residual error within ±3 (-3 < δ < 3) and the leverage value (hi) less than the warning value (h*) were in the application domain of the model and can reliably predict and analyze them (Li et al. 2022). As shown in Fig. 5, all molecules were in the model application domain, indicating that the established model had good applicability and can be used to predict this type of compound’s binding energy.
Fig. 5.

Williams diagram of the QSAR model
The QSAR model sensitivity was calculated to evaluate the molecular parameters’ relative contribution and further analyze the influence of molecular structure parameters (descriptors) and binding energy in the QSAR model. It revealed the interaction mechanism of multi-compound molecules in the treatment of COVID-19 from the perspective of molecular structure and provided guidance for improving its effectiveness in treating COVID-19. The model sensitivity calculation formula is as follows:
| 5 |
In formula (5), SCi represents the sensitivity coefficient of the input descriptor parameter i, ΔXi/Xi, and ΔYi/Yi, respectively, represent the change rate of the input parameter and the corresponding prediction result (Yang and Liu 2019). The sensitivity coefficients of TPSA, TE, and logP were −1.71, −3.05, and -1.31, respectively. The order of the descriptor influence was total energy (TE) > the polar surface area (TPSA) > oil-water partition coefficient (logP). The descriptor that had the most significant impact on the model was the TE of the molecule, with a negative effect on the drug effect, which means the smaller the total energy of the molecule, the greater the impact on improving the effectiveness of the active compound. In addition, the polar surface area (TPSA) and the oil-water partition coefficient (logP) also impact the active compound’s efficacy. As these two properties increase, the active compound’s efficacy decreases. According to the sensitivity analysis of the 2D-QSAR model, the low total energy of a molecule reflects in its higher efficacy. Table S2 showed that the 13 active components of LH generally have the lowest binding energy values. For example, β-sitosterol with the lowest binding energy (−1953.52 kJ/mol) and the smallest TPSA has the highest binding energy (−308.93 kJ/mol). In addition, Table S3 showed that the hydrophobicity (logP) of molecules has little effect on the efficacy of each component. In summary, molecules with lower total molecular energy and higher polar surface area have higher efficacy in treating COVID-19 and the molecules studied have corresponding characteristics. These combinations could theoretically make LH with a high therapeutic effect, consistent with the results obtained in chapter “Calculation of the binding ability between 13 main compounds of LH and potential drug targets.”
The above mechanism showed that TPSA, TE, and logP were the main factors affecting the molecular therapeutic effect of each component of LH. The existence of oxygen and nitrogen atoms in the molecule is the main reason affecting the size of TPSA. In addition, eating more high protein foods, vegetables, and fruits rich in vitamin C can prevent or improve the control effect of COVID-19., according to experts and hospital recommendation. Therefore, it can be further speculated that the treatment effect of LH on COVID-19 can be improved by taking high protein and vitamin nutrients rich in nitrogen or oxygen atoms at the same time.
Analysis of complementary nutrition scheme based on molecular dynamics simulation to improve the efficacy of LH
Taking Mpro receptor as an example, this study designed and screened an optimum complementary nutrition scheme that is conducive to improving the effectiveness of LH in the treatment of COVID-19. And then, the best complementary nutrition scheme was applied to ACE2 protein to verify its applicability and feasibility. The lower the binding energy, the more stable the conformation of ligand binding to the receptor (Mishra et al. 2021). The binding energy ≤ -5.0 kcal/mol indicates that the two can bind, while ≤ −7.0 kcal/mol indicates a good binding ability. The stronger the binding ability of the active compounds of LH to Mpro protein, the better its efficacy in treating COVID-19. At present, according to the recommendations of experts and hospitals for the treatment to prevent or treat novel coronavirus pneumonia, it is necessary to eat more high-protein foods and fruits and vegetables rich in vitamin C (VC) in the diet. In this study, a MD simulation was used to simulate the effects of nutrients in three types of foods (high protein, fruits, and vegetables rich in VC, enhance immunity drinks) on the binding ability of 13 main compounds of LH and Mpro protein and explore their interaction mechanism. Furthermore, the optimal combination of food and nutrition conducive to promote the treatment of COVID-19 by LH can also provide a reference for the COVID-19 treatment by TCM (National Health Commission 2020).
From the results of “Calculation of the binding ability between 13 main compounds of LH and potential drug targets,” it was found that when the 13 active compounds of LH act simultaneously, their binding ability to Mpro protein was the best, and the effect of treating COVID-19 was the most significant. Therefore, in this study, the MD method was used to simulate the changes of binding energy between Mpro protein and 13 kinds of LH active compounds in the process of adding the three typical foods (high protein: milk and eggs; vegetables and fruits rich in VC: carrot, spinach, broccoli, apple, kiwi, grape, orange, winter jujube; immune-enhancing drinks: tea, wolfberry, ginseng, honey), and determine the reason for the enhanced binding of active molecules to Mpro protein. According to the nutrition label, the main compounds of milk (whey protein) are β-lactoglobulin (50–55%) and α-lactoalbumin (20–25%); the main compounds of eggs are ovalbumin and ovotransferrin; the main compounds of apple are vitamin A, vitamin B, vitamin C, and carotene; the main compounds of kiwifruit are vitamin C and E; the main compounds of grape are glucose, fructose, sucrose, xylose, tartaric acid, malic acid, carotene, vitamin B, vitamin C, and vitamin P; the main compounds of oranges are vitamins A, B1, B2, C, D, and E, folic acid, biotin, carotene, and niacin; the main compounds of winter jujube are folic acid, biotin, vitamin B1 and B2, vitamin C, vitamin E, and niacin; the main compounds of carrot are sucrose, carotene, vitamin A, vitamin B1, vitamin B2, niacin, vitamin C, vitamin E, vitamin K, and folic acid; the main compounds of spinach are carotene, vitamin C, folic acid, thiamine, riboflavin, rutin, niacin, and oxalic acid; the main compounds of broccoli are vitamin C and carotene; the main compounds of tea are catechin, caffeine, vitamin C, vitamin E, pyrroloquinoline quinone, and folic acid; the main compounds of Chinese wolfberry are vitamin A, carotene, vitamin C, E, and B2; the main compounds of ginseng are ginsenoside (ginsenoside rg3; ginsenoside rh3; ginsenoside rb1; ginsenoside rg1); and the main compound of honey is vitamin B2, vitamin C, and niacin. The results showed that the binding energy (−1512.614 kJ/mol, −1476.682 kJ/mol, −1508.221 kJ/mol, −1424.144 kJ/mol, −1358.759 kJ/mol, −1338.677 kJ/mol, −1367.412 kJ/mol, −1471.626 kJ/mol, −1405.542 kJ/mol, −1302.986 kJ/mol, −1428.582 kJ/mol, −1344.649 kJ/mol, −1287.032 kJ/mol, and −1284.172 kJ/mol) of compounds with the main compounds of food added in the above order varied from −0.41 to 8.26% compared to without the main compounds of food (−1399.826 kJ/mol). The high-protein products add β-lactoglobulin and α-lactalbumin or ovalbumin and ovotransferrin, respectively; the vitamins add vitamin A, B, C, and carotene or vitamin C and E, respectively; and the binding energy of the complex reduced (1.74% to 8.06%) with the addition of catechin, caffeine, vitamin C, vitamin E, pyrrolidine, quinoline, quinone, and folic acid. Therefore, different complementary food collocation schemes generated by the Taguchi orthogonal experimental design table was used to repeat the molecular dynamics simulation binding energy calculation of Mpro protein and 13 active molecular systems (Fig. 6). Thus, confirming it was beneficial to promote the treatment of COVID-19 and the best recommended nutritional mix.
Fig. 6.
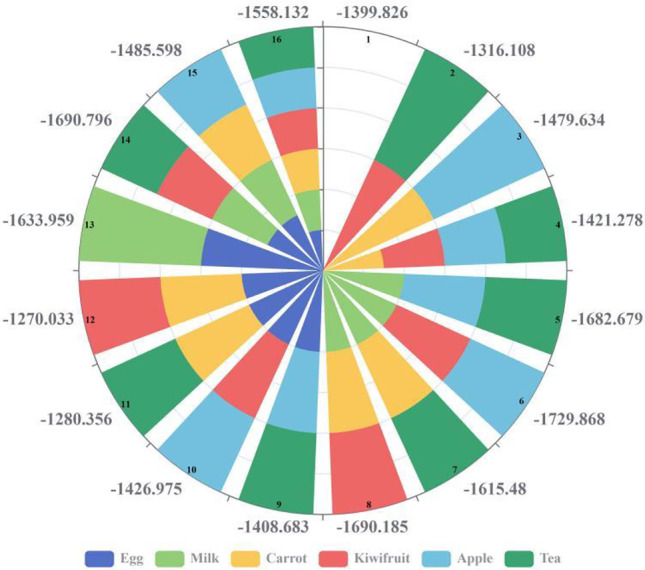
Calculation of binding energy (kJ/mol) of the docking system of Mpro protein and 13 kinds of active compound molecules of LH under different complementary food matching schemes
In this paper, the signal-to-noise ratio (SNR) with a small calculation error of experimental data was selected as the binding energy evaluation standard (Fig. 6 and Table S4). In a 6-factor 2-level orthogonal experimental design, 16 experimental groups were generated for a supplementary feeding scheme that significantly enhanced the binding ability of 13 active molecules with Mpro protein. The SNR of the experimental group was consistent with the absolute value of binding energy. It was observed that except for combinations 2, 11, and 12, the binding energy of the rest of the complementary food programs reduced compared with combination 1 (without adding any of the above food compounds), which promoted the binding of 13 active compound molecules to Mpro protein, the binding capacity, and the change rate of binding energy compared with combination 1 (−0.63 to −23.58%) (Fig. 4). In combination 6, when the main compounds of milk, apple, and kiwi (β-lactoglobulin, α-lactoalbumin, vitamin A, B2, C, carotene, and vitamin E) were added simultaneously, the binding ability of 13 active compounds to Mpro protein significantly enhanced (23.58%); thus, improving the efficacy of LH in the treatment of COVID-19. It was found that proteins, vitamins (especially VC), and carotene significantly promote the binding ability of 13 active compounds to Mpro protein. It can be concluded that high-protein and vitamins could be the best dietary supplement combination to promote LH’s therapeutic effect on COVID-19. Therefore, high-protein and vitamin-based foods can enhance human immunity to prevent COVID-19 and serve as a supplementary dietary and nutritional regimen to promote the treatment of COVID-19 by TCM (LH). In addition, the use of eggs, carrots, and tea should be circumvented at the same time.
In addition, in order to further improve the effect of LH on ACE2 protein in the treatment of COVID-19 and COVID-19 caused by mutant virus, the above optimal complementary nutritional combination was added into 13 active components and ACE2 protein scenarios. It was found that the binding energy (−2069.775kJ/mol) increased by 15.97% compared with the original (−1784.726kJ/mol), which confirmed the effectiveness of the supplementary nutrition scheme, and provided guidance for improving the efficacy of LH in the treatment of COVID-19 caused by mutant virus.
Efficacy and risk assessment of the best active compound of LH in the treatment of COVID-19 based on ADMET and TOPKAT models
Drug safety is a key issue related to people’s livelihood, and the ecological environment is the basis of sustainable development. Therefore, while paying attention to the efficacy of LH, it is also important to predict the human health risk and ecological environment risk after taking and discharging it.
Pharmacodynamic evaluation of the best active compound of LH based on ADMET model
In innovative drug development, pharmacokinetics, pharmacodynamics, and toxicology research are all important. At present, it has become an essential part of drug development and clinical research (Grossman 2009; Waterbeemd and Gifford 2003). In this study, the ADMET module was used to study the aqueous solubility, cytochrome P450 2D6 inhibition (CYP2D6), human intestinal absorption (HIA), and absorptivity (Alogp98) of 13 main active compounds of LH, to evaluate the absorption, distribution, metabolism, and excretion of the active compounds. Specifically, the solubility model uses a linear regression method to predict the solubility of each compound in the water at 25°C. The inhibition model of CYP2D6 is based on the two-dimensional chemical structure of compounds that predicts its inhibitory effect. As CYP2D6 is involved in the metabolism of various substrates in the liver, its inhibition experiment is considered an essential part of drug development (Xia et al. 2004; Burton et al. 2009).
The plasma protein binding model is used to predict whether compounds are highly bound to carrier proteins in the blood. Because the drug and plasma protein binding was temporarily blocked, which cannot be metabolized, the plasma protein binding of drug molecules directly affects drugs’ efficiency. On the other hand, compounds enter the blood circulation and bind to plasma proteins. In contrast, the non-binding, free compounds penetrate the biofilm and distribute to various tissues, and react with the corresponding target molecules to exert efficacy. Therefore, the binding of drugs with plasma proteins in vivo directly affects drugs’ efficacy and toxicity (Obach 2018). The absorption model is used to predict the absorption of the human intestinal tract after oral administration. Intestinal absorption is defined as the percentage of absorption, expressed by Alogp98. The prediction results of the above four model indexes are shown in Fig. 7.
Fig. 7.

The prediction results of 13 main active compounds of LH
The pharmacodynamic prediction results showed that (Fig. 7 and Table S5) the Alogp98 values of quercetin and other 12 main active compounds of LH were all in the range of [−2.0, 7.0], and that of wogonin was 8.084. Therefore, we believe that the absorption of quercetin and other 12 active compounds into the human intestinal tract by oral delivery was good. However, wogonin’s absorption effect was not ideal and not easily absorbed by the human gastrointestinal tract. In chemical drugs, the water solubility of molecules is one of the critical factors of patent medicines. The distribution of drugs, the transport of drug molecules in the human body, and the transmembrane all require water solubility. The water solubility prediction results at 25°C showed that the water solubility values of kaempferol and the other six main active compounds were [−4.0, −2.0], indicating a good water solubility. The predicted water solubility values of quercetin and the other four active compounds were [−6.0, −4.0], indicating a relatively low water solubility. The predicted water solubility values of wogonin, stigmasterol, and glycyrrhetinic acid were −8.256, −7.144, and −7.963, respectively, indicating extremely low water. In addition, after entering the target organism’s blood circulation system, the drug binds with plasma protein, while the non-binding, free compounds penetrate the biofilm and are distributed to various tissues due to the reaction of the corresponding target molecules and then exert the efficacy. Therefore, the plasma protein binding rates of the 13 active compounds were predicted. The predicted results showed that the binding values of 11 main active compounds, such as quercetin, with plasma protein, were less than 4, indicating that they were all bound to plasma protein. Besides, the drugs were free compounds after entering the human body with good activity. The binding values of wogonin and glycyrrhetinic acid with plasma protein were 8.594 and 8.010, respectively, indicating they combine with plasma protein after entering the blood system and lose their medication efficacy. The results of CYP2D6 inhibition prediction showed that quercetin and the other four main active compounds had inhibitory effects, while the remaining nine were all less than 0.5, indicating that quercetin had no inhibitory effect.
In conclusion, the pharmacodynamic evaluation results showed that among the 13 main active compounds of LH, tryptamine, and naringenin had the best pharmacokinetics that met the standards of drug research and development.
Human health risk assessment of the best active compound of LH based on ADMET and TOPKAT models
In this paper, ADMET and TOPKAT models were used to evaluate the potential human health risks of 13 main active compounds of LH in addition to assessing their efficacy. Each compound’s drug safety was determined by assessing the 13 main active compounds’ toxic effects on the human body. The human health risk assessment indicators mainly include hepatotoxicity, carcinogenicity, and teratogenicity (developmental toxicity potential, DTP), mutagenicity, skin sensitization, skin irritancy, and ocular irritancy (Cheng and Dixon 2003). The kinetic prediction of the above parameters is shown in Fig. 8.
Fig. 8.

Human health risk assessment of LH active components
The human health risk assessment results showed that the predicted hepatotoxicity values of 8 active compounds such as quercetin were less than 0.5, indicating no hepatotoxic effect. Thus, they show no adverse effects on human liver organs. However, the predicted hepatotoxicity values of the remaining five active compounds were greater than 0.5, indicating a potential impact on human liver organs. In terms of carcinogenicity, the predicted values of β-sitosterol, indigo, stigmasterol, and naringenin were 0.014, 0.000, 0.012, and 0.003, respectively. Their values were less than 0.3, indicating that their carcinogenic effects were relatively low after entering the human body. The predictive value of benzylidene acetone was 0.453, and its carcinogenic probability was uncertain. The remaining eight active compounds’ predicted values were all greater than 0.7, which showed that the probability of carcinogenic effect on the human body is very high. Only indigo and benzylidene acetone predicted values less than 0.3, indicating a low probability of teratogenicity. The remaining 11 active compounds showed a high probability of teratogenicity in the human body. The predicted values of the 7 active compounds, such as β-sitosterol were close to or equal to 0, indicating that their mutagenicity is negligible. The predicted values of six active compounds were greater than 0.7, suggesting that they might be mutagenic to the human body. In terms of skin sensitization, the predictive values of benzylidene acetone and 1-methyl-2-nonyl-4(1H)-quinolone were 0.006 and 0.000, respectively, indicating they were not sensitized to human skin or their possible sensitization is negligible. The predictive values of indigo, tryptamine, and deserpidine were 0.339, 0.437, and 0.489, respectively; however, their possible skin sensitization could not be determined. In addition, the predicted values of the remaining eight active compounds were all greater than 0.7, indicating that the possibility of sensitization to skin was very high. In terms of skin irritation, quercetin’s predictive values and the other seven active compounds were less than 0.3, indicating the possible irritation of quercetin to human skin could be ignored entirely. The remaining six active compounds’ predictive values were greater than 0.7, indicating that the probability of skin irritation was extremely high. For eye irritation, except for 1-methyl-2-nonyl-4, (1H)-quinolone, and deserpidine, the predicted values were close to 0, indicating that they had little irritation to the eyes.
To sum up, the 13 main active compounds of LH were found to have certain risks to the human body through the human health risk assessment results, such as liver damage, skin irritation, and so on. Therefore, we should also monitor the harmful effects of drugs on humans in the rational use of drugs.
Ecological risk assessment of the best active component of Lianhuaqingwen in the treatment of COVID-19 based on TOPKAT model
On the basis of pharmacodynamics and human health risk assessment, TOPKAT model was used to evaluate the ecological risks of 13 main active components of Lianhuaqingwen, including fatead minnow LC50, daphnia magna EC50, and aerobic biodegradability. The kinetic prediction results of the above parameters are shown in Table 2.
Table 2.
The ecological risks prediction results of 13 main active components in Lianhuaqingwen
| Compound | Fathead Minnow LC50(mg/L) | Daphnia magna EC50(mg/L) | Aerobic biodegradability |
|---|---|---|---|
| Quercetin | 0.185 | 2.000 | 0.000 |
| Kaempferol | 0.325 | 1.700 | 0.004 |
| Lteolin | 0.318 | 1.800 | 0.000 |
| Beta-sitosterol | 0.0014 | 1000000.000 | 0.000 |
| Indigo | 3.400 | 0.019 | 0.000 |
| Wogonin | 0.242 | 0.201 | 0.000 |
| Tryptanthrin | 3.700 | 210.900 | 0.000 |
| Benzalacetone | 35.100 | 11.500 | 0.840 |
| Quinolone | 0.062 | 566.200 | 0.000 |
| Stigmasterol | 0.004 | 686700.000 | 0.000 |
| Naringenin | 0.390 | 13.100 | 0.000 |
| Glycyrrhetinic-acid | 0.000 | 1000000.000 | 0.000 |
| Deserpidine | 0.000 | 0.207 | 1.000 |
In this paper, the ecological effects of 13 main active components in Lianhuaqingwen were evaluated according to the guidelines for hazard assessment of new chemical substances. The results of ecotoxicological evaluation showed that the 50% lethal concentration of 10 active components such as quercetin was less than 1. The results showed that the 10 active components were extremely harmful to the ecological environment, and the Fathead Minnow LC50 value of indigo was 3.400 mg/L, which indicated that indigo had high risk to the ecological environment. The Fathead Minnow LC50 value of benzylidene acetone was 35.100 mg/L, indicating that indigo was moderately harmful to the ecological environment. In addition, the EC50 values of indigo, wogonin, and serpentine to Daphnia magna were 0.019, 0.201, and 0.207 (less than 1), respectively, indicating that the three active components were highly harmful to the ecological environment. The EC50 values of quercetin, kaempferol, and luteolin to D. magna were 2.000, 1.700, and 1.800 (more than 1 and less than 10), respectively, which indicated that the three active components were highly harmful to the ecological environment. The EC50 values of benzylidene acetone and naringenin to D. magna were 11.500 and 13.100 (more than 10 and less than 100), respectively, which indicated that they were moderately harmful to the ecological environment. The EC50 values of the remaining five active components to D. magna were all greater than 100, indicating that they had low harm to the ecological environment. Finally, in terms of aerobic biodegradability, except for benzylidene acetone and serpentine, which were predicted to be 0.840 and 1.000, respectively, with high biodegradability, the remaining 11 active components did not have biodegradability (HJ/T 154-2004 2004-06-01).
Comprehensive analysis of the evaluation results of 13 main active components on ecotoxicology showed that the 13 main components of Lianhuaqingwen had strong toxicity to black headed fish and D. magna, and their random discharge was very likely to cause a large number of fish and fleas’ death in aquatic ecosystem, and seriously damage and affect the balance and stability of the ecosystem. In addition, the biodegradability of 13 main active components was not ideal, and most of them did not have the ability of biodegradation. Therefore, we suggest that the medical wastewater must be strictly treated by the sewage treatment plant before discharge, in order to reduce its possible adverse effects on the ecosystem and human health.
Conclusion
In this study, the multi-order interaction of LH multi-compound in the treatment of COVID-19 was discussed using Taguchi experimental design, expert consultation method of computer stratified sampling, and MD simulation method. Intelligent integration of the comprehensive screening system for TCM compounds was used to identify the most effective active compound molecules for the treatment of COVID-19. The best complementary food nutrition scheme was determined by the MD simulation method. The QSAR method and sensitivity model was used to reveal the mechanism of influencing each active molecule’s efficacy in the treatment of COVID-19. In addition, pharmacokinetics and toxicokinetics were used to predict the human health and ecological risk of the optimal combination of LH for COVID-19, assess its safety, prevent and eliminate drug-induced diseases, and also provide guidance for the regulation of ecological risk after drug use. In summary, tryptamine and naringenin had the best pharmacokinetic properties. Thus, they met the standards of drug research and development.
Supplementary information
(DOCX 21718 kb)
Author contribution
Wenwen Gu, Yuanyuan Zhao, and Luze Yang are the main contributor to this paper, and they are mainly responsible for the ideas’ design, data calculation and writing of this paper; Meijin Du is mainly responsible for assisting in drawing; Zhixing Ren and Qing Li are mainly responsible for sorting out data; Xixi Li is the main contributor to this paper and mainly responsible for supervising throughout the research and finalizing the manuscript.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Wenwen Gu, Yuanyuan Zhao, Luze Yang, Meijin Du, Qing Li, Zhixing Ren and Xixi Li have contributed equally to the study and they receive equal credit.
Contributor Information
Wenwen Gu, Email: gww0813@outlook.com.
Yuanyuan Zhao, Email: zyy0210@ncepu.edu.cn.
Luze Yang, Email: yanglz19@mails.jlu.edu.cn.
Meijin Du, Email: mjdu0401@outlook.com.
Qing Li, Email: lq0226@outlook.com.
Zhixing Ren, Email: RenzhixingRyy@outlook.com.
Xixi Li, Email: xixi.li@mun.ca.
References
- Alexandre J, Cracowski JL, Richard V, Bouhanick B. Renin-angiotensin-aldosterone system and COVID-19 infection. Ann Endocrinol-paris. 2020;81:63–67. doi: 10.1016/j.ando.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banno M, Tsujimoto Y, Kataoka Y. The majority of reporting guidelines are not developed with the Delphi method: a systematic review of reporting guidelines. J Clin Epidemiol. 2020;124:50–57. doi: 10.1016/j.jclinepi.2020.04.010. [DOI] [PubMed] [Google Scholar]
- Brauer M, Zhao JT, Bennitt FB, Stanaway JD. Global access to handwashing: implications for COVID-19 control in low-income countries. Environ Health Presp. 2020;5:057005. doi: 10.1289/EHP7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton I, Iijaali I, Petite F, Michel A, Vercauteren D. Virtual screening for cytochromes P450: successes of machine learning filters. Comb Chem High T Scr. 2009;12:369–382. doi: 10.2174/138620709788167935. [DOI] [PubMed] [Google Scholar]
- Castorena-Cortés T, Roldán-Carrillo I, Zapata-PeAsco J, Reyes-Avila L, Quej-Aké JM-C, Olguín-Lora P. Microcosm assays and Taguchi experimental design for treatment of oil sludge containing high concentration of hydrocarbons. Bioresource Technol. 2009;100:5671–5677. doi: 10.1016/j.biortech.2009.06.050. [DOI] [PubMed] [Google Scholar]
- Chen YY, Li Q, Pan CS, Yan L, Fan JY, He K, Sun K, Liu YY, Chen QF, Bai Y, Wang CS. QiShenYiQi Pills, a compound in Chinesd medicine, protects against pressure overload-induced cardiachypertrophy through a multi-component and multi-target mode. Sci Rep. 2015;5:11802. doi: 10.1038/srep11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, Wang ZY, Fu ZQ (2019) Environmental computational chemistry and toxicology, Science Press (Beijing). (In Chinese)
- Cheng A, Dixon SL. In silico models for the prediction of dose-dependent human hepatotoxicity. J Comp Aid Mol Des. 2003;17:811–823. doi: 10.1023/B:JCAM.0000021834.50768.c6. [DOI] [PubMed] [Google Scholar]
- Du MJ, Zhang D, Hou YL, Zhao XH, Li Y. Combined 2D-QSAR, principal component analysis and sensitivity analysis studies on fluoroquinolones’ genotoxicity. Int J Environ Res Public Health. 2019;16:1–14. doi: 10.3390/ijerph16214156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan ZP, Jia ZH, Zhang J, Liu S, Chen Y, Liang LC, Zhang CQ, Zhang Z, Yan S, Zhang SQ, Wang YY, Wu YL. Natural herbal medicine Lianhuaqingwen capsule anti-influenza A (H1N1) trial: a randomized, double blind, positive controlled clinical trial. Chin Med J. 2011;124:2925–2933. [PubMed] [Google Scholar]
- El-Gammal OA, El-Bindary AA, Mohamed FS, Rezk GN, El-Bindary MA. Synthesis, characterization, design, molecular docking, anti COVID-19 activity, DFT calculations of novel Schiff base with some transition metal complexes. J Mol Liq. 2021;346:117850. doi: 10.1016/j.molliq.2021.117850. [DOI] [Google Scholar]
- Galimberti F, Moretto A, Papa E (2020) Application of chemometric methods and QSAR models to support pesticide risk assessment starting from ecotoxicological datasets. Water Res 174:115583 [DOI] [PubMed]
- Grossman I. ADME pharmacogenetics: current practices and future outlook. Expert Opin Drug Metab Toxicol. 2009;5:449–462. doi: 10.1517/17425250902902322. [DOI] [PubMed] [Google Scholar]
- Hadni H, Elhallaoui M. 2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents. New J Chem. 2020;44:6553–6565. doi: 10.1039/C9NJ05767F. [DOI] [Google Scholar]
- Hanine H, Menana E. 3D-QSAR, docking and ADMET properties of aurone analogues as antimalarial agents. Heliyon. 2020;6:e03580. doi: 10.1016/j.heliyon.2020.e03580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia WN, Wang CH, Wang YF, Pan GX, Jiang MM, Zheng L, Yan Z. Qualitative and quantitative analysis of the major constituents in Chinese medical preparation Lianhua-Qingwen capsule by UPLC-DAD-QTOFMS. Sci World J. 2015;2015:1–19. doi: 10.1155/2015/731765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao XY, Jin X, Ma YY, Yang Y, Li JJ, Liang LY, Liu R, Li Z (2021) A comprehensive application: molecular docking and network pharmacology for the prediction of bioactive constituents and elucidation of mechanisms of action in component-based Chinese medicine. Comput Biol Chem 90:107402 [DOI] [PubMed]
- Khaerunnisa S, Kurniawan H, Awaluddin R, Suhartati S, Soetjipto S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Prepr. 2020;10:1–14. [Google Scholar]
- Kuthyala S, Hanumanthappa M, Kumar SM, Sheik S, Karikannar NG, Prabhu A. Crystal, Hirshfeld, ADMET, drug-like and anticancer study of some newly synthesized imidazopyridine containing pyrazoline derivatives. J Mol Struct. 2019;1197:65–72. doi: 10.1016/j.molstruc.2019.07.031. [DOI] [Google Scholar]
- Lei M, Jiang Z, Liu H, Cui Y, Ye X, Ji B, Guo X. Comparative pharmacokinetics study of a kaurane diterpenoid after oral administration of monomer and Siegesbeckiae pubescens Makino extract to rats. Biomed Chromatogr. 2014;28:673–679. doi: 10.1002/bmc.3088. [DOI] [PubMed] [Google Scholar]
- Li SX (2016) Data analysis of adverse drug reactions of traditional Chinese medicine in pharmaceutical manufacturers in Beijing (2009-2013). Beijing University of Chinese Medicine (In Chinese)
- Li XZ, Li Q. Discussion on diet regulation and efficacy in traditional Chinese medicine nursing. Yunnan J Tradit Chin Med Mater Med. 2000;21:45. [Google Scholar]
- Li DS, Sangion A, Li L. Evaluating consumer exposure to disinfecting chemicals against coronavirus disease 2019 (COVID-19) and associated health risks. Environ Int. 2020;145:106108. doi: 10.1016/j.envint.2020.106108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RF, Hou YL, Huang JH, Pan WQ, Ma QH, Shi YX, Li CF, Zhao J, Jia ZH, Jiang HM, Zheng K, Huang SX, Dai J, Li XB, Hou XT, Wang L, Zhong NS, Yang ZF. Lanhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2) Pharmacol Res. 2020;156:10476. doi: 10.1016/j.phrs.2020.104761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XX, Gu WW, Chen B, Zhu ZW, Zhang BY. Functional modification of HHCB: strategy for obtaining environmentally friendly derivatives. J Hazard Mater. 2021;416:126116. doi: 10.1016/j.jhazmat.2021.126116. [DOI] [PubMed] [Google Scholar]
- Li XX, He W, Zhao YY, Chen B, Zhu ZW, Kang Q, Zhang BY (2022) Dermal exposure to synthetic musks: Human health risk assessment, mechanism, and control strategy. Ecotoxicol Environ Saf 236:113463 [DOI] [PubMed]
- Liu LH, Yu Z, Yun ZJ, He B, Zhang QH, Hu LG, Jiang GB. Speciation and bioaccessibility of arsenic in traditional Chinese medicines and assessment of its potential health risk. Sci Total Environ. 2018;619:1088–1097. doi: 10.1016/j.scitotenv.2017.11.113. [DOI] [PubMed] [Google Scholar]
- Liu M, Ya G, Yuan Y, Yang K, Shi SZ, Tian JH, Zhang JH. Efficacy and safety of herbal medicine (Lianhuaqingwen) for treating COVID-19: a systematic review and meta-analysis. Integr Med Res. 2020;10:100644. doi: 10.1016/j.imr.2020.100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Di L. In vitro and in vivo methods to assess pharmacokinetic drug-drug interactions in drug discovery and development. Biopharm Drug Dispos. 2019;28:1–29. doi: 10.1002/bdd.2212. [DOI] [PubMed] [Google Scholar]
- Lv RB, Wang WJ, Li X (2020) Novel coronavirus pneumonia suspected cases treated with Lianhua Qingwen decoction: a clinical observation of 63 cases. J Tradit Chin Med 1–5 (In Chinese)
- Mishra D, Maurya RR, Kumar K, Munjal NS, Bahadur V, Sharma S, Singh P, Bahadur I. Structurally modified compounds of hydroxychloroquine, remdesivir and tetrahydrocannabinol against main protease of SARS-CoV-2, a possible hope for COVID-19: Docking and molecular dynamics simulation studies. J Mol Liq. 2021;335:116185. doi: 10.1016/j.molliq.2021.116185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Graphics and Modelling: 197-214. The guidelines for the hazard evaluation of new chemical substances (HJ/T 154-2004 2004-06-01). http://www.mee.gov.cn/image20010518/4342.pdf (accessed 13 July 2021).
- Mukherjee S, Majumder D. Computational molecular docking assessment of hormone receptor adjuvant drugs: breast cancer as an example. Pathophysiology. 2009;16:19–29. doi: 10.1016/j.pathophys.2008.12.001. [DOI] [PubMed] [Google Scholar]
- National Health Commissiom, Nutritional advice on novel coronavirus infection in prevention and treatment of pneumonia. 2020
- National Health Commission, State Administration of traditional Chinese medicine. Diagnosis and treatment of novel coronavirus infection pneumonia (trial version 7). 2020-03-04
- Obach RS. Aldehyde oxidase in drug design and drug discovery. Drug Metab Pharmacok. 2018;33:S11. doi: 10.1016/j.dmpk.2017.11.053. [DOI] [Google Scholar]
- Patel O, Muller C, Joubert E, Rosenkranz B, Taylor MJ, Louw J, Awortwe C. Pharmacokinetic interaction of green rooibos extract with atorvastatin and metformin in rats. Front Pharmacol. 2019;10:1243. doi: 10.3389/fphar.2019.01243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo F, Lombardo A, Manganaro A, Benfenati E. In silico models for predicting ready biodegradability under REACH: a comparative study. Sci Total Environ. 2013;463:161–168. doi: 10.1016/j.scitotenv.2013.05.060. [DOI] [PubMed] [Google Scholar]
- Prival MJ. Evaluation of the TOPKAT system for predicting the carcinogenicity of chemicals. Environ Mol Mutagen. 2001;37:55–69. doi: 10.1002/1098-2280(2001)37:1<55::AID-EM1006>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Qin H, Chen JW, Wang Y, Wang B, Li XH, Li F, Wang YN. Development and assessment of quantitative structure activity relationship models for bioconcentration factors of organic pollutants, Chinese. Sci Bull. 2020;54:628–634. doi: 10.1007/s11434-009-0053-2. [DOI] [Google Scholar]
- Qiu L, Xiao H. Molecular dynamics study of binding energies, mechanical properties, and detonation performances of bicyclo-HMX-based PBXs. J Hazard Mater. 2009;164:329–336. doi: 10.1016/j.jhazmat.2008.08.030. [DOI] [PubMed] [Google Scholar]
- Qu RJ, Li CG, Pan XX, Zeng XL, Liu JQ, Huang QG, Wang ZY. Solid surface-mediated photochemical transformation of decabromodiphenyl ether (BDE-209) in aqueous solution. Water Res. 2017;125:114–122. doi: 10.1016/j.watres.2017.08.033. [DOI] [PubMed] [Google Scholar]
- Qu RJ, Li CG, Liu JQ, Xiao RY, Pan XX, Zeng XL, Wang ZY, Wu JC. Hydroxyl radical based photocatalytic degradation of halogenated organic contaminants and paraffin on silica gel. Environ Sci Technol. 2018;13:7220–7229. doi: 10.1021/acs.est.8b00499. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Loman N, Pybus O, Barclay W, Barrett J, Carabelli A, Connor T, Peacock T, Robertson DL, Volz E, on behalf of COVID-19 Genomics Consortium UK (CoG-UK) (2020) preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563. Accessed 13 July 2021
- Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KH, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC, King NP. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–1310. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J. Progress on pollution characteristics and treatment of traditional Chinese Medicine Wastewater. Engineering Technology. 2016;7:00320–00320. [Google Scholar]
- Scott WRP, Hünenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J, Torda AE, Huber T, Krüger P, Van Gunsteren WFJ. The GROMOS biomolecular simulation program package. Phys Chem A. 1999;103:3596–3670. doi: 10.1021/jp984217f. [DOI] [Google Scholar]
- Van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003;2:192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- Wang YL, Cui T, Li YZ, Liao ML, Zhang HB, Hou WB, Zhang TJ, Liu L, Huang H, Liu CX. Prediction of quality markers of traditional Chinese medicines based on network pharmacology. Chin Herb Med. 2019;11:349–356. doi: 10.1016/j.chmed.2019.08.003. [DOI] [Google Scholar]
- WHO (2020a) Pneumonia of unknown cause in China. https://www.who.int/csr/don/05-january-2020-pneumonia-of-unkown-cause-china/en/. Accessed 28 May 2022
- WHO (2020b) SARS-CoV-2 Variant - United Kingdom of Great Britain and Northern Ireland. https://www.who.int/emergencies/disease-outbreak-news/item/2020-DON304. Accessed 28 May 2022
- Wu YL. Interpretation of Lianhua-Qinwen capsule. Guide of China Medicine. 2005;3:120–121. [Google Scholar]
- Xia X, Maliski EG, Gallant P, Rogers D. Classification of kinase inhibitors using a Bayesian model. J Med Chem. 2004;47:4463–4470. doi: 10.1021/jm0303195. [DOI] [PubMed] [Google Scholar]
- Yang LZ, Liu M. 3D-QSAR model of polybrominated biphenyls tri-effect modified by standard deviation standardization method and its application in environmental friendly molecular modification. Chem J Chin Univ. 2019;40:2471–2479. [Google Scholar]
- Yao KT, Liu MY, Li X, Huang JH, Cai HB (2020) Retrospective clinical analysis on treatment of novel coronavirus-infected pneumonia with traditional Chinese medicine Lianhua Qingwen. Chin J Exp Tradit Med Formulae 11:1–7 (In Chinese)
- Zhang CF, Shen S, Song LQ, Ren J, Sun YB, Bi D (2018) Chemical components from Lianhua Qingwen capsules (II). Chin Tradit Herb Drug 49:3222–3225. (In Chinese)
- Zhang DC, Ye T, Yu T, Xing HD, Liu S, Zhang HY, Ding SZ, Cai PL, Sun DD, Zhang T, Hong YH, Dai HK, Tu WZ, Chen JN, Wu AB, Hu QN. A data-driven integrative platform for computational prediction of toxin biotransformation with a case study. J Hazard Mater. 2021;408:124810. doi: 10.1016/j.jhazmat.2020.124810. [DOI] [PubMed] [Google Scholar]
- Zhao XM, Xia LQ, Ding XZ, Yu ZQ, Lv Y, Tao WN. Homology Modeling of Cyt2Ca1 of Bacillus thuringiensis and its molecular docking with inositol monophosphate. Chin J Chem. 2009;27:2085. doi: 10.1002/cjoc.200990350. [DOI] [Google Scholar]
- Zhou WJ, Zhang M, Yan YC, Wang SS, Peng T, Li YY, Chai Z. Molecular mechanism of Huoxiang (Pogostemonis herba) in treatment of Corona virus disease 2019 (COVID-19) based on network pharmacology and molecular docking. Journal of Practical Traditional Chinese Internal Medicine. 2020;9:1–4. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 21718 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files].