

Abstract
Poxviruses are a large and complex family of viruses with members such as monkeypox virus and variola virus. The possibility of an outbreak of monkeypox virus (or a related poxvirus) or the misuse of variola virus justifies the development of countermeasures. Furthermore, poxviruses can be a useful surrogate for developing technology involving antibody therapies. In our experiments, we explored the feasibility of utilizing unmodified mRNA that encodes three previously described monoclonal antibodies, c8A, c6C, and c7D11, as countermeasures to smallpox in a relatively large (>3 kg) laboratory animal (rabbits). We confirmed in vitro translation, secretion, and biological activity of mRNA constructs and identified target monoclonal antibody levels from a murine vaccinia virus model that provided a clinical benefit. Individually, we were able to detect c7D11, c8A, and c6C in the serum of rabbits within 1 day of an intramuscular jet injection of lipid nanoparticle (LNP)-formulated mRNA. Injection of a combination of three LNP-formulated mRNA constructs encoding the three different antibodies produced near equivalent serum levels compared with each individual construct administered alone. These data are among the first demonstrating the feasibility of launching multiple antibodies using mRNA constructs in a large, nonrodent species. Based on empirically derived target serum level and the observed decay rate, the antibody levels attained were unlikely to provide protection.
Keywords: MT: Delivery strategies, RNA, monoclonal antibodies, nucleic acid, neutralizing antibody, rabbits, poxvirus, lipid nanoparticle
Graphical abstract
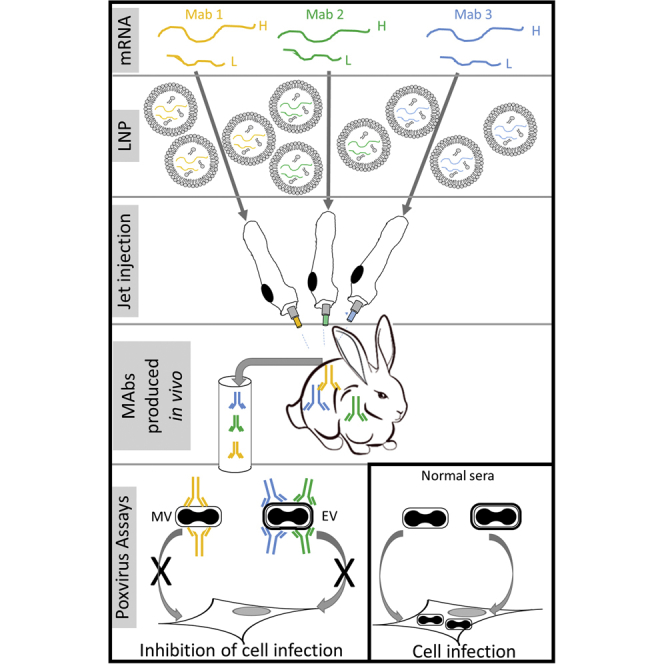
Hooper and colleagues demonstrate the feasibility of simultaneously delivering three monoclonal antibodies (mAbs) via intramuscular administration of mRNA to relatively large animals (rabbits). The unmodified mRNAs were LNP formulated and delivered via needle-free jet injection. All three anti-poxvirus mAbs were detected in sera within 1 day of mRNA injection and were biologically active.
Introduction
Poxviruses are known to cause disease in humans and animals. From the perspective of human disease, the most notable members are variola virus and monkeypox virus. Smallpox, the disease manifested by variola virus, was responsible for the death and maiming of millions until its eradication from nature in the latter part of the 20th century. It has been over 40 years since the general population received vaccination, and the protective benefit of the vaccine to smallpox and other orthopoxviruses wanes over time. Therefore, there are many at risk for zoonotic poxvirus infections, such as monkeypox virus, or reintroduction of variola virus, prompting the need for vaccines and therapeutics.
The US Food and Drug Administration has licensed four countermeasures for smallpox. ACAM2000 comprises a less virulent clone of Dryvax, the progenitor to ACAM2000, which was selected in an effort to reduce adverse events associated with the progenitor vaccine. Unfortunately, the ACAM2000 vaccine still has the same issues and contraindications as Dryvax. MVA (JYNNEOS), a licensed third-generation vaccine, is a highly attenuated nonreplicating vaccine.1 Although seemingly safer, there are concerns about the protective capability of these vaccines. Unlike Dryvax, ACAM2000 and MVA have not been battle tested against smallpox in humans. Two antivirals, TEMBEXA (brincidofovir) and TPOXX (tecovirimat or ST-246), have been approved for treatment of smallpox. These small-molecule inhibitors work via independent mechanisms, one interfering with poxvirus DNA synthesis and the other morphogenesis, respectively. Neither TEMBEXA nor TPOXX has been approved for prophylactic treatment to prevent smallpox.
Vaccinia immunoglobin (VIG) has been licensed for complications associated with the smallpox vaccine. VIG was shown to have efficacy against certain vaccine complications.2,3 Among other things, the potency of VIG has been questioned, and potential replacements have been postulated.4 For instance, polyclonal antibodies that were generated from single5,6 or multiple antigens,5 as well as monoclonal antibodies/cocktails,6, 7, 8 have been shown to be more effective in animal models.
Whether developing a new vaccine or other countermeasures, it is generally agreed that targeting both the intracellular and the extracellular form of the virus is necessary. Morphogenesis of poxviruses produces two “general” forms of the virus, referred to as extracellular enveloped virions (EV) and mature virions (MV). The EVs have an additional envelope in which the extracellular surface is antigenically distinct from that of the MV. It has been shown that vaccines lacking or unable to produce viral proteins (antigens) unique to EV, as well as antibodies to these antigens, provide less protection than those including both.5,6,9, 10, 11
More recent advances in nucleic acid technology and delivery have created the potential for utilizing gene transfer as a therapeutic tool against viral diseases.12, 13, 14, 15, 16, 17 In this study, we attempted to develop nucleic acid-based monoclonal antibodies capable of producing therapeutic levels of circulating anti-poxvirus antibody in a relatively large laboratory animal (>3 kg rabbits). We utilized mRNA sequences of mouse-human chimeric and chimpanzee-human chimeric monoclonal antibodies (mAbs) with known therapeutic capacity in animal models and directed against both the MV, c7D11 (anti-L1),18,19 and the EV, c8A (anti-B5) and c6C (anti-A33),20,21 forms of poxviruses. From these studies, we found that it was possible to produce three functional mAbs with different antigen specificities in vivo, either when the constructs were administered to one animal or when all three were administered concomitantly in the same animal. Based on empirically derived target serum level and the observed decay rate, the antibody levels attained were unlikely to provide protection.
Results
The goal of these experiments was to develop a treatment consisting of at least three mRNA constructs encoding three distinct humanized mAbs. Furthermore, the mRNA would have to be protective in a larger, nonrodent model as a barometer to test the technological success (e.g., increase efficiency in delivery/potency to overcome difficulties in scaling up to humans). Therefore, we constructed and characterized the function of mRNA constructs in vitro. Once successful, we utilized passive transfer of our most potent manufactured (“protein”) mAb in two mouse models of orthopoxviral disease, a fully lethal and a semi-lethal model, to determine circulating levels of antibody that correlated to statistically significant amelioration of disease. Mice were utilized for multiple reasons, but mainly because of the power (n) required to determine a minimal effect in two models and over multiple doses. Knowing that the answer from these studies equated to a low level of benefit for our single antibody, we were confident that the other two mAbs (c6C and c8A) would add to that benefit either cumulatively or synergistically (BioFactura, Inc., personal communication). Once our target level of c7D11 was established, we attempted to obtain those levels in rabbits using our key mRNA construct encoding c7D11, the other mRNA constructs encoding c6C and c8A, and finally all three simultaneously. Since we did not achieve our target goal with c7D11 using multiple methods, we never took the next step to determine whether that treatment afforded protection against rabbitpox virus in the rabbits. Below are the more specific results of our endeavors.
In vitro analysis of antibody-encoding mRNAs
To achieve biologically functional antibodies that target poxviruses from RNA constructs, we first generated mRNA encoding the heavy and light chains of each of the antibodies. To show that the designed constructs were functional, we conducted an in vitro expression study in BHK cells. Twenty-four hours after performing Lipofectamine-based transfections of each of the light- and heavy-chain mRNA mixtures at a weight ratio of 2:1 (heavy chain to light chain construct), we determined the titers in the cell supernatant by IgG-specific ELISA and western blot analysis. All three antibodies c7D11, c8A, and c6C (Figure 1), were detected at high levels in the supernatant of the cells within a range of 1,000–4,000 ng/mL.
Figure 1.

In vitro expression of mRNA-encoded antibodies
(A) mRNA design to encode heavy- and light-chain molecules of c7D11, c8A, or c6C constructs contained a 5′ cap structure (CAP), 5′ untranslated region (UTR), open reading frame, 3′ UTR, and poly(A) (A(n)) sequence followed by a C30 stretch followed by a histone stem-loop sequence. Open reading frame: signal peptide (SP) and variable heavy (VH) regions (top) or variable light (VL) and constant light (CL) regions (bottom). (B) Quantification of mAb levels in supernatants of BHK cells by IgG-specific ELISA. Titers were determined in triplicate. (C) Western blot analysis showing expression of heavy and light chains in supernatants after mRNA transfection of BHK cells. Equal amounts of three replicates were pooled and loaded on denaturing SDS-PAGE. WFI, water for injection.
Functionally, the antibodies derived from cell culture were assessed utilizing a quantitative, immunogen-specific ELISA and a vaccinia virus plaque reduction neutralization test (PRNT). The supernatant containing c7D11 was found to be functional in both attaching to the L1 antigen (ELISA) and neutralizing the vaccinia virus (Figure 1 and Table 1). Not surprisingly, no activity was detected in the supernatant containing c8A or c6C in either assay, as these specific antibodies do not react with L1, nor do they neutralize the intracellular form of the virus. Quantitation of the c7D11 supernatant using three different standards (c7D11) on both the ELISA and the PRNT produced values that were in agreement at approximately 4 μg/mL (Table 1).
Table 1.
Results of the plaque reduction neutralization test and ELISA on mRNA-launched monoclonal antibodies in vitro
| Sample | Standard | PRNT (μg/mL) | Anti-L1 ELISA (μg/mL) | PRNT50 titer |
|---|---|---|---|---|
| c7D11 | 1 | 3.87 | 3.49 | 50 |
| 2 | 4.12 | 3.59 | – | |
| 3 | 3.21 | 4.62 | – | |
| c8A and c6C | 1 | negative results | BLOD/BLOQ | N/A |
| 2 | ||||
| 3 |
PRNT, plaque reduction neutraliation test 50% inhibitory concentration; PRNT50 titer, reciprocal of highest dilution resulting in ≥50% plaque reduction; BLOD/BLOQ, below the limit of detection/below the limit of quantitation; N/A, not applicable.
The biological activity of RNA-launched antibodies c8A and c6C in the medium from transfected cells was assessed by an EV neutralization assay. Briefly, vaccinia virus IHDJ was propagated and the supernatant (containing the extracellular form of the virus) was incubated with a dilution series of heat-inactivated cell culture supernatants, alongside manufactured mAbs c6C and c8A as controls. The assay was completed using medium with or without 5% human complement. In the presence of human complement, c8A and c6C neutralized 31.7% and 31.1% of the EV present in the assay. No activity was detectable in these samples in the absence of complement. The data suggest that there is biological activity, although the concentration was not sufficient to produce an IC50.
Because of the low qualitative EV neutralizing effect observed in the samples, we attempted to ascribe a quantitative value (concentration) based on the PRNT assay and compare with our more sensitive, quantitative ELISA. Using the manufactured mAbs as standard curves in the EV neutralization assay, we were able to extrapolate a concentration in the samples of approximately 2.5 μg/mL for c6C and 0.336 μg/mL for c8A. In comparison, data generated by the quantitative ELISA values were 2.42 and 1.54 μg/mL, respectively. These data help confirm our assessment that the samples contained the mAbs and most likely had low-level neutralization consistent with a low level of mAb in the samples.
In all, these data confirm that the products of our mRNA constructs functionally bind their respective antigens and have neutralizing activity, albeit less so for c6C and c8A.
In vivo evaluation of the mRNAs encoding the three different smallpox-specific antibodies
Before testing our three constructs in vivo, we needed to determine a target dose of circulating antibody that would provide benefit. To answer this question, we evaluated prophylactic treatments of our most potent mAb, c7D11, in a murine model of poxviral disease. More specifically, we tested multiple subcutaneous doses of c7D11 administered 24 h prior to intranasal vaccinia virus strain IHDJ (Figure S1). Based on statistical amelioration of disease (mortality and/or weight loss) in lethal and semi-lethal models (Tables S1–S4), a prophylactic dose of mAb was chosen to further evaluate systemic levels of c7D11 (Figure S2). We found that a dose of 10 μg per mouse provided benefit, and this correlated with a maximum systemic concentration of 4.1 μg/mL. More definitive protection was shown when 50 μg was administered per mouse, and animals had a mean systemic level of 27.3 μg/mL. Therefore, we hoped to achieve >4.1 μg/mL of circulating antibodies.
Based on the in vitro and mouse efficacy studies, we proceeded to test mRNA constructs in animals. Rabbits were chosen as a model for multiple reasons. For instance, jet injection for intramuscular administration requires an animal with adequate muscle size, and the number of animals required precluded nonhuman primates. Because of the larger size, producing adequate levels of effector molecule systemically in rabbits is much more challenging, helping us to evaluate the feasibility of the technology as a future human treatment.
Because c7D11 was our most potent mAb and a systemic target dose was empirically derived from disease models, we initially focused on the c7D11 mRNA construct in an effort to maximize systemic effector antibody concentrations to theoretically beneficial levels. To do so, mRNA mixtures of heavy and light chain-coding RNAs were formulated into lipid nanoparticles for delivery via intramuscular administration in a rabbit host. More specifically, four rabbits received 1 mg each via a single jet injection into one hindleg (average per group of 0.28 mg/kg); 2 mg per rabbit via administration of two 1 mg jet injections, one per hindleg (average per group of 0.67 mg/kg); or 4 mg per rabbit via four jet injections, two per hindleg (average per group of 0.9 mg/kg) of the c7D11 construct. We found that levels of circulating antibody detected by immunogen-L1-specific ELISA were proportional to the number of injections (i.e., the amount of RNA injected), with peak mean concentrations for one (group 2), two (group 3), and four injections (group 4) of 412, 1,553, and 3,495 ng/mL, respectively (Figure 2A). The animal with the highest ELISA titer, 9,165 ng/mL, was animal 29 from group 4. The highest group ELISA concentrations (mean) were observed either on day 1 (two injections) or on day 3 (remainder of groups). A 50% clearance of the maximum ELISA titers appeared to occur between days 5 and 7. Complete clearance of the antibody seemed animal specific, as opposed to group specific, and ranged from day 7 (one animal from each of groups 3 and 4) to beyond the last time point evaluated.
Figure 2.

Intramuscular administration of mRNA constructs in rabbits
(A) Design outline: mRNA encoding c7D11, c8A, or c6C was injected once (groups 1, 2, 5, 6, and 7), twice (group 3), or four times (group 4) by either needle (group 1) or jet injection (groups 2, 3, 4, and 7). (B and E) Temporally collected sera from rabbits were diluted and analyzed by a quantitative, immunogen-specific ELISA using poxvirus antigen, L1 (B) or c6C (E) or c8A (E). (C and F) Serial dilutions of sera from select rabbits and time points were tested for the ability to neutralize the intracellular (MV) form (C) or the extracellular (EV) form (F) of VacV-IHDJ using plaque reduction neutralization test/assay (PRNT). Purified monoclonal antibodies (mAbs) c7D11 (at 10 μg/mL), c6C (at 1 μg/mL), and c8A (at 1 μg/mL) were utilized as controls. (D) L1-immunogen-specific ELISA mean titers corresponding to samples utilized for neutralization testing in (C). (G) Mean peak concentrations as determined by ELISA were normalized by calculating the total theoretical antibody per rabbit (average peak concentration per group multiplied by the average weight of rabbits multiplied by the average blood volume per kilogram [56 mL/kg]) and dividing by the amount of construct (micrograms) administered. Mean and standard error of the mean are given for (B) and (E). Mean and standard deviation are given for (C) and (F).
To assess any potential advantage of jet injection, four animals (group 1) also received one needle injection of 1 mg (0.25 mg/kg) (Figure 2A). A peak mean titer of 230 ng/mL occurred on day 3 post injection, and levels tended to trend similar to group 2, with the exception that the magnitude of group 2 was slightly higher (peak of 412 ng/mL), but a single animal in group 1 seemed to have anti-L1 antibody that persisted in the serum for a longer time.
To confirm “immediate” biological activity, we tested the neutralizing capacity of heat-inactivated serum samples acquired from our earliest time point, day 1. To show activity, given the lack of sensitivity of the c7D11 neutralization relative to antigen-specific L1-ELISA (for c7D11), serum samples that had the highest (ELISA-derived) concentration of mAb on day 1 (i.e., group 4 animals 5, 27, 28, and 29) (Figure 2B) were chosen. We also included the day 3 sample of animal 29 for two reasons: it had the overall highest ELISA titers of any animal, and we also wanted to confirm the ELISA data that showed an increase of more than 5 μg/mL from day 1 to day 3. Similar to our in vitro samples (Table 1), all samples neutralized the intracellular mature form of vaccinia virus strain IHDJ (VacV-IHDJ) with a magnitude corresponding to the ELISA titers (Figures 2B and 2C). In agreement with our ELISA, differences in neutralization were noted between day 1 and day 3 samples from rabbit 29 and were 1.6-fold (ELISA) versus 1.8-fold (PRNT50). More specifically, the interpolated values for days 1 and 3 from our neutralization assays were 200 and 358 ng, respectively. These interpolated PRNT50 data from rabbit 29 (day 3) were very similar to the 10 μg/mL c7D11 mAb control (interpolated PRNT50 titer of 292) used in the neutralization assay and were consistent with our predicted concentration based on ELISA.
To test constructs encoding mAbs c8A and c6C, four rabbits were given a single fixed dose of 1 mg jet injection of construct c8A at 0.25 mg/kg (group 5) or c6C at 0.28 mg/kg (group 6). Similar to our experiment with the c7D11 mRNA (Figure 2A), sera from multiple days were analyzed by either B5- or A33-immunogen-specific ELISA (Figure 2D), respectively, and select samples were chosen to evaluate neutralization of the EV form of vaccinia virus (Figure 2E). Levels of circulating c8A (383 ng/mL) and c6C (622 ng/mL) peaked on days 1 and 3, respectively (Figure 2D). There was no detection of either c6C or c8A antibody after day 7. A summary is provided for comparing the different groups, which utilizes the average of the highest ELISA titers (one titer per animal) obtained for each group and average weights, normalized to the total circulating (sera) concentration of antibody to the amount of mRNA administered (Figure 2F).
We next performed neutralization assays utilizing EVs from vaccinia virus strain IHDJ. Again, samples containing larger amounts of c6C or c8A, as determined by antigen-specific ELISA, were chosen. This is even more important for the EV neutralization assay as it is less responsive than both the MV neutralizing assay and the ELISA. Samples with the highest ELISA concentrations from each of the two groups (rabbit 20 day 3, 693 ng/mL, and rabbit 20 day 1, 844 ng/mL) and the mAb controls. Rabbit 20 (c8A) on day 3 had approximately 50% neutralization of EV, but rabbit 7 (c6C) had negligible activity (approximately 13%). Similarly, 1 μg/mL c8A mAb neutralized 60% of the EV, while 1 μg/mL c6C mAb neutralized only 42% (Figure 1E).
Together, these data show that after intramuscular injection of mRNA, we can obtain biologically active human mAbs. All three mRNA construct products (mRNAs c7D11, c6C, and c8A) tested were quantifiable in the sera and had neutralizing activity relative to the magnitude of the ELISA quantitation and mAb standards. Moreover, the use of jet injection for the mRNA constructs slightly increased peak serum concentrations of c7D11 (about 2-fold) compared with needle injection of the same material.
Injection of all three constructs yields functionally similar circulating levels of each mAb relative to administration of a single construct in rabbits
Given the individual success of our constructs when administered by jet injection, we next asked whether we could simultaneously administer all three to one host without affecting the yield/quality of each individual construct (e.g., interference). As we have three constructs and are limited to four injections per rabbit, we decided to give one injection each of the c6C and c8A mRNA constructs and two injections of the c7D11 construct for multi-dosing the rabbits. Again, this design was an effort to maximize the levels of c7D11, our most potent mAb. For comparison, three groups of animals would receive an analogous regimen of only one of the constructs. More specifically, four groups of rabbits (n = 3/group) were injected twice with the c7D11 in the right hind quadriceps (groups 1 and 2), once with the c8A construct in the left hind quadriceps (groups 1 and 3), and/or once with the c6C construct in the left hind quadriceps (groups 1 and 4) (Figure 3A). Rabbits in group 1 received a total of 2 mg of mRNA (1 mg c7D11 construct, 0.5 mg c6C construct, and 0.5 mg c8A construct), whereas group 2 received 1 mg (c7D11 construct), and groups 3 and 4 received 0.5 mg (c6C or c8A construct, respectively) of mRNA. Temporal blood draws were performed and the resulting sera were utilized to evaluate the concentrations of individual mAbs c7D11, c8A, and c6C using immunogen-specific ELISAs (Figures 3B–3E). Although group 1 (all) had more variability, comparison of anti-L1 (c7D11) antibodies between groups 1 and 2 showed that mean peak concentrations were similar, 1,095 versus 965 ng/mL, respectively, and these peak concentrations occurred on the same sample day, day 3 (Figure 3B). A similar comparison of groups 1 (all) and 4 (c6C) for anti-A33 antibodies again showed similar peak concentrations (133 versus 152 ng/mL) on day 3, but, unlike anti-L1 antibodies, one animal from group 1 (all), animal 3, did not completely clear the antibody prior to the experiment’s conclusion (Figure 3C). The concentrations of anti-B5 antibodies (c8A) were 1.7 times lower (279 versus 464 ng/mL) in group 1 (all) versus group 3 (c8A) (Figure 3D). Again, mean peak concentrations were noted on day 3 in both groups and, once again, animal 3 from group 1 (all) did not clear anti-B5 antibodies before termination of the experiment. Statistical comparison by a two-way ANOVA and Sidak’s multiple comparison test between group 1 mAb concentrations and analogous groups showed no statistical difference, with the exception of group 1 (all) and group 3 (c8A) on days 3 and 5 (p = 0.0282 and p = 0.0312, respectively) (Table S6). Normalizing the delivered dose based on the average group weights of the rabbits revealed little difference between group 1 and similarly treated groups (c7D11, group 1, 0.228 mg/kg, versus group 2, 0.228 mg/kg; c8A, group 1, 0.114 mg/kg, versus group 3, 0.117 mg/kg; and group 1, 0.114 mg/kg versus 0.115 mg/kg), suggesting non-weight-related variations in the animals (Figures 2D and 3D). Examining the levels of each construct within group 1 (all) showed that anti-L1 antibodies were 4- to 8-fold greater than anti-B5 or anti-A33 antibodies on day 3 (Figure 3E).
Figure 3.

Co-injection of rabbits with all three mRNA poxvirus mAb constructs produces individually functional circulating c8A, c6C, and c7D11 similar in magnitude to control rabbits receiving only a single construct
(A) Outline of the experimental design: rabbits were jet injected with two injections of mRNA encoding c7D11 (groups 1 and 2) and/or a single injection of c6C (groups 1 and 4) and/or c8A (groups 1 and 3). (B–D) Immunogen-specific ELISAs were performed on temporally acquired serum samples. Immunogen and mAb targets were as follows: (B) L1, c7D11; (C) A33, c6C; and (D) B5, c8A. (E) A comparison of ELISA concentrations over time for each construct within rabbits that received all three constructs simultaneously (group 1). Mean and standard error are given for (B–E). Arrows denote the day of injection. When multiple injections were given, all injections were performed within 2 min of the first injection.
Neutralization assays were performed to provide evidence for functional activity of the ELISA-detected antibodies. For reasons previously discussed, samples (animal/day) with the highest ELISA titers were chosen for each type of antibody. For group 1, animal 3 produced the highest ELISA titers for all three antibodies on the same day. Anti-MV and anti-EV assays were conducted specific for each antibody target, with the exception of animal 5, group 2, which was included to control for an animal that had anti-EV antibodies but also high anti-MV (c7D11) antibodies. Anti-MV assays utilizing VacV-IHDJ showed that serum samples from groups 1 (rabbit 3 day 3) and 2 (rabbit 5 day 3) had functional activity neutralizing 89% and 81% of VacV-IHDJ, respectively (Figure 4A). Utilizing VacV-IHDJ EV as the target of neutralization, minimal (if any) neutralization, of approximately 32%, 22%, and 30%, was found for rabbits 3 (group 1), 7 (group 3), and 12 (group 4), respectively (Figures 4B and 4D). It has been shown that VacV-IHDJ EV and VacV-WR EV have different susceptibilities to neutralization by c6C.22 Utilizing VacV-WR EV, neutralization increased for samples from rabbits 3 (56%) and 7 (41%), but only slightly increased for rabbit 12 (36%) (Figures 4C and 4D). In a similar way, c6C mAb control antibody poorly neutralized EV from VacV-IHDJ (54% peak) relative to VacV-WR (94% peak), whereas c8A had no change (97%) (Figure 4D). From these data, we show that intramuscular jet injections of multiple constructs per rabbit produce detectable and functional antibodies with little evidence of interference.
Figure 4.

Comparison of neutralizing activity of serum from rabbits jet injected with one or all three mRNA constructs
Rabbit serum samples with high ELISA titers were selected from each group and evaluated for neutralizing activity to (A) the mature virion and/or (B–D) the extracellular virion by PRNT using vaccinia virus strain IHDJ or strains IHDJ and WR, respectively. A summary of the maximum neutralization of the extracellular virion is given in (D). MV, mature virion; EV, extracellular virion. The mean and standard deviation are given for (A–C).
Discussion
In these studies, we were able to show the efficacy of the human-mouse chimeric mAb, c7D11, in a vaccinia challenge model (Figure S1 and Tables S1–S3) and adapted mRNA technology to produce the antibody both in vitro and in vivo. We applied the technology to produce two other poxvirus-specific mAbs, c6C and c8A, in rabbits. Finally, we successfully used an intramuscular jet injection device to administer three mRNA constructs encoding three mAbs to concomitantly produce all three functional antibodies in a single nonrodent animal.
Before launching mAbs in vivo from mRNA we needed to first determine the systemic level of antibodies that correlated with protection in a disease model. To increase our likelihood of success, we determined the lowest dose of mAb required for benefit against disease and utilized our most potent neutralizing antibody, mAb c7D11. The empirically derived systemic levels would become our target to achieve through mRNA injections. We found that prophylactic treatment with as little as 10 μg of c7D11 antibody (protein) per mouse, approximately 0.5 mg/kg, provided a statistical health benefit against vaccinia virus-induced disease parameters, mainly death and/or weight, depending on the model (Figures S1A and S1D, Tables S1 and S3). Although we did not do a direct comparison, it was previously reported that treatment with 100 μg of the native mouse antibody, 7D11, in the lethal vaccinia virus IHDJ model provides 100% protection from death.5 In our study, 54% survived using c7D11 at the same dose. Most likely, there is some decrease in efficacy due to replacing the mouse constant regions with human. We also found that neutralization of vaccinia virus by the chimeric antibody is approximately four to eight times lower than the reported 7D11 (concentration of mAb required for a 50% plaque reduction of 40–77 ng/mL versus 171-587 ng/mL, respectively).5,18
In surrogate smallpox disease models, we were able to show that there was a significant benefit based on body weight and survival of passively transferred mAb c7D11 at a dose of 0.5 mg/kg (10 μg per mouse) administered subcutaneously (Figures S1B and S1E, Tables S2 and S4). In our severe model of poxviral disease, statistical differences in weight between the treated and the vehicle control groups were noted as early as day 2 for 200 and 100 μg-dosed groups. These animals fared better than the 50 and 10 μg groups, where differences were not noted until day 3. Statistical differences were observed in all treated groups on day 3 when exposed to a semi-lethal dose of vaccinia virus relative to the vehicle control group. For both the uniformly lethal and the semi-lethal models, groups that had earlier and more sustained differences in weight from the control group had fewer deaths. These data allowed us to select a mAb dose of 0.5 mg/kg (protein) as our target dose and to determine the corresponding serum levels of protein.
The corresponding maximum serum level of a 0.5 mg/kg dose in mice was approximately 4 μg/mL (Figure S2). Maximum levels were observed 2 days after subcutaneous injection, and detectable levels of c7D11 persisted for at least 15 days. When 50 μg was administered (2.5 mg/kg), serum levels peaked at 27 μg/mL 4 days after injection. As was the case for both doses of antibody, there were gaps in our bleed schedule, and the highest levels of antibody may have been missed. The same stipulation must be made for calculating the 50% clearance of c7D11 from circulation. For 0.5 and 2.5 mg/kg, the half-life of the antibody was between days 10 and 15 or 7 and 10, respectively. The timing (maximum observed serum levels) and the recovery (63% and 59% for 50 and 10 μg groups) observed in our study are similar to reports by others.23
The utilization of disposable syringe jet injectors (DSJIs), such as the PharmaJet Stratis, for delivery of DNA vaccines has been shown to increase the immune response to an encoded target antigen relative to needle injection.24 Although not statistically significant, we observed an approximate 2-fold advantage of PharmaJet injection over the first three sampling points by a head-to-head comparison using a single intramuscular needle or jet injection of the c7D11 mRNA construct. We found that jet injection increased the maximum attained ELISA titer by 2-fold (Figure 2). These data, along with others,25 emboldened our choice to use the PharmaJet injector to help maximize our delivery of the RNA and, in turn, increase serum levels. Rabbits were chosen because of their larger size and muscle mass to accommodate multiple DSJIs.
With the lipid nanoparticle (LNP) encapsulated mRNA constructs we were reaching antibody levels as high as 9.1 μg/mL when rabbits were given 4 mg (approximately 0.9 mg/kg), up to 3.5 μg/mL after a 2 mg dose (0.67 mg/kg), and 0.6 μg/mL after a 1 mg dose (0.28 mg/kg). Our other two constructs produced relatively similar amounts of product (between 200 and 600 ng/mL per milligram). In comparison with our results, others have shown levels of chikungunya mAb in nonhuman primates using 3.0 mg/mL mRNA delivered via intravenous infusion of 16.2 and 28 μg/mL after boosting.26 Comparing doses, we averaged a circulating concentration of antibody that was approximately 6.8-fold less (2.4 μg/mL versus 16.2 μg/mL), while giving the animals 3.3-fold less construct and using the less invasive injection route. The slight differences most likely can be attributed to our utilization of nonhomologous antibody relative to the host and/or administration via intramuscular injection as opposed to intravenous infusion. Even if small differences could still be detected after using a homologous combination of host and antibody, the real-world practicality of intramuscular injection versus intravenous infusion might outweigh that small difference.
We could show from in vitro data that, when different antibodies were generated from mRNAs, the levels of functional antibodies were lower when the different antibodies were administered simultaneously compared with applying only one antibody. Therefore, it was obvious that the combination of all three constructs administered in vivo from “one syringe” would lead to inefficient production of functional antibody. Therefore, these studies were not attempted. Administering mRNA constructs via separate intramuscular injections afforded the opportunity to attempt (and succeed at) concomitantly producing three mAbs from three separate mRNA constructs. When conducting the co-administration experiments, we found that similar antibody levels were obtained whether injected with only the single construct or in combination with the other two constructs, with the exception of our c8A construct (Figure 2). For c8A, there was an approximate 2-fold difference on days 3 and 5 that was statistically significant. Our data suggest that the lower levels of c8A when administered concomitant with c6C and c7D11 were likely not related to weight, as the doses administered per kilogram were very similar, but we cannot conclude whether the other constructs were interfering with our c8A construct. For instance, it is possible that we underestimated variation between rabbits and/or administration of the constructs. In either case, more testing is needed.
From the compilation of our data, we decided not to move forward with performing efficacy testing using an infection model. We were near the target serum concentration that, based on our mouse model data, would provide only limited benefit with c7D11 alone. The maximum antibody levels we obtained using mRNA were found by administering 4,000 μg; that is, four injections with 500 μg/injection. It is possible that, by administering a treatment regimen using all three constructs (Figure 2), the combination would prove more efficacious than c7D11 alone. Unfortunately, our study did not include combinatorial efficacy testing of the antibodies, and this would be required before experiments in rabbits. The idea of testing the disease efficacy of mAb combinations becomes even more complex given our knowledge of the differing efficacy of c6C between two vaccinia virus strains based on subpopulations of EVs.22 More specifically, in vitro testing of rabbitpox virus may reveal that both VacV-WR and VacV-IHDJ animal models would be required to sufficiently bridge the gap between our data and the data required to move forward. Given that rabbitpox virus produces quantities of EVs that are similar to VacV-IHDJ, it is conceivable that the sensitivities to c6C are similar to those to VacV-IHDJ and, consequently, may not provide much benefit in the rabbit/rabbitpox virus model.
Having weights comparable to those of 2- to 3-year-old nonhuman primates, rabbits provide a good, nonrodent model system for assessing nucleic acid therapies. This was an important factor for these studies, as the progress and capability of the technology would be more rigorously evaluated in a larger model. More specifically, mRNA technology administered to smaller animals (rodents) has been shown to produce high and/or protective levels protein (antibody).26, 27, 28, 29 Not surprisingly, there are some drawbacks to utilizing rabbits as a model. Since the hindleg muscles are the only appropriate muscle to perform intramuscular jet injection, there are constraints to the number of, and frequency of, mRNA administrations (boosting) in rabbits. For our studies, four injections of 0.5 mL per injection was the maximum we could administer at one time. Healing of the muscle is also necessary before reinjection. Therefore, it was not possible to increase the frequency of injections to potentially improve antibody levels. In larger (more appropriate) hosts, this may be explored.
We found that readministration of 2 mg of the c7D11 mRNA construct 4 weeks post injection provided little to no additional systemic effector antibody (c7D11) (Figure S3). We explored immunological responses, in the form of anti-drug antibody (ADA) responses, as a possible explanation for the reduced titers following boost (Table S5). Although the results of the boost can be explained somewhat by a recall response and ADA targeting and clearing the heterologous antibody, the variability of results after the initial injection cannot (Figure S3).
The magnitude of mAbs after the “boost,” albeit much lower, was relative to the initial injection (with the exception of rabbit 2000(a) (Figure S3), which did not have detectable levels of mAb), suggesting some other inherent factor of the rabbit (e.g., anatomical/physiological, innate, or cell-mediated response) or some other variable(s) occurring during the primary injection may also be important. In fact, others have shown that modifying the bases of an mRNA construct can reduce activation of the innate immune system (e.g., Toll-like receptors).27,29,30 Although the unmodified nature of our constructs could play a role in the magnitude of the ADA response, studies have shown that LNPs themselves can contribute to an immune response.31 Future research should tease out the impact of our specific LNP formulation and the mRNA constructs for stimulating the innate immune effector molecules. In those experiments, it would be necessary to use mAbs homologous to the injected animal (e.g., rabbit mAbs in rabbits).
Intravenous infusions might mitigate logistical issues with intramuscular injection of rabbit muscles and may also increase circulating levels of effector antibody; but again, infusions for nonemergency indications are not a pragmatic delivery method for humans. Another issue is xenogeneic response to our antibody. To pursue an increase in antibody levels, making a host-specific chimera (e.g., rabbit/mouse chimeric mAb) would seem logical, as this would decrease the immunogenicity of the mAb in rabbits and possibly allow less frequent, but constant, boosts until serum target levels were reached. Again, this would be a difficult process to bridge to the target product, a humanized or human chimeric antibody. A more straightforward approach would be to test in nonhuman primates, but this approach is still not perfect. That is, the overall lack of availability and increased resources required for nonhuman primates make them a poor choice for the exploratory and multi-iterative evaluation necessary to optimize a technology. The predictive ability of nonhuman primate tolerability is also questionable.32
As the COVID-19 pandemic grips the globe, there is an urgency to quickly design, develop, and license safe and effective countermeasures. Nucleic acid platforms are perfectly suited to such unforeseen events. Although success will be gauged over time, the rapid generation and evaluation of the now FDA-authorized mRNA vaccines are impressive. The next step might be to use nucleic acid technology to test and license prophylactic or therapeutic effector molecules, such as antibodies. As evidenced by SARS-CoV-2, a responsive platform is likely required to stay ahead of a constantly and rapidly evolving agent. As quickly as the technology is progressing in terms of delivery and stability, the threshold toward such a therapeutic may be close. Our data contribute to establishing the feasibility of producing a nucleic acid-based therapy that may require launching multiple mAbs. It is possible that a single mAb may be sufficient to convey the desired benefit, putting the technology even closer to that threshold, but to avoid resistance, increase efficiency, or address a multifaceted/complex disease, the use of multiple mAbs may be necessary.
Materials and methods
RNA constructs
The design and synthesis of mRNA sequences have been described previously.33 In brief, mRNAs encoding mAbs contained a 5′ cap structure (Cap1), 5′ UTR,33 open reading frame, 3′ UTR,33 and poly(A) sequence followed by an C30 stretch followed by a histone stem-loop sequence.34 Sequences were codon optimized (GC enrichment) for human use and did not include chemically modified bases (Figure 1A).33
RNA formulation
LNP formulation was conducted at Arcturus Therapeutics (San Diego, CA, USA) and has been described previously.35 RNAs encoding mAbs were mixed at a molar ratio of approximately 1.5 (heavy- over light-chain-encoding mRNA for all antibodies). For injections, mRNA-LNP was diluted in Lunar bBuffer.35,36
Cells and viruses
BSC-1 cells (CCL-26) were maintained in EMEM with 5% heat-inactivated fetal bovine serum (FBS) (Gibco) and utilized for PRNT. Vaccinia IHDJ was obtained from Dr. Alan Schmaljohn and vaccinia WR from the ATCC (VR-119). BHK cells were cultured in MEM containing 10% heat-inactivated fetal calf serum (FCS) and nonessential amino acids.
Monoclonal antibodies
Characterization of the unmodified mAbs has previously been reported.18,20,21 Human-murine c7D11 and human-chimpanzee c6C and c8A chimeric antibodies were produced by BioFactura, Inc., and have been described elsewhere.22,37
Cell transfections
For in vitro transfection of cells, RNAs were complexed with Lipofectamine 2000 (Life Technologies, Darmstadt, Germany) at 1.5 μL Lipofectamine/μg of mRNA for BHK and transfected into cells according to the manufacturer’s instructions. To analyze the expression of different mAbs, 400,000 BHK cells were seeded 1 day before transfection in six-well plates. Cells were transfected with 5 μg of antibody-encoding RNAs. Approximately 2 h after transfection, the transfection mix was replaced by 1.5 mL of serum-free FreeStyle 293 medium (Thermo Scientific, cat. no. 12338018). Cells were grown for approximately 48 h, and supernatants were harvested, spun down to remove cell debris, and used for analyses. To analyze different heavy-to-light-chain ratios, 10,000 BHK cells were seeded 1 day before transfection in 96-well plates. Cells were transfected with 100 ng of antibody-encoding RNAs. Approximately 2 h after transfection, the transfection mixes were replaced with 200 μL FreeStyle 293 medium (Thermo Scientific, cat. no. 12338018). Cells were grown for approximately 48 h, and supernatants were harvested and used for analyses. Antibody titers were measured by IgG-specific ELISA as described below.
Western blot analysis
For all experiments, pooled triplicates of equal amounts of lysates or supernatants were loaded. For all western blots depicted in this study, 12% SDS Tris-glycine gels were used. Proteins were transferred to a nitrocellulose membrane (Odyssey nitrocellulose membrane, 0.22 μm; Li-COR Biosciences, cat. no. 926-31092) and afterward blocked in 5% skimmed milk in TBST buffer (TBS containing 0.1% Tween 20; Sigma, cat. no. P2287). Membranes were first incubated with rabbit anti-a/b-tubulin 1:1,000 (New England Biolabs, cat. no. 2148S) in 0.5% skimmed milk in TBST for 1 h. After three washes (10 min each) in TBST, both a secondary antibody against rabbit (goat anti-rabbit IgG [H + L] IRDye 680RD; Li-COR Biosciences, cat. no. 926-68071) and an antibody to detect human antibodies (goat anti-human IgG [H + L] IRDye 800CW; Li-COR Biosciences, cat. no. 926-32232) were incubated at 1:15,000 in 0.5% skimmed milk in TBST for 1 h or, in case of HepG2 supernatants, overnight. Immediately before band detection, all membranes were washed three times each for 10 min in TBST and stored in TBS lacking Tween 20 until analysis. Protein detection and image processing were carried out in an Odyssey CLx imaging system and LI-COR’s Image Studio version 5.2.5 according to the manufacturer’s recommendations.
Anti-human IgG ELISA
Goat anti-human IgG (1 mg/mL; SouthernBiotech, cat. no. 2044-01) was diluted 1:1,000 in coating buffer (15 mM Na2CO3, 15 mM NaHCO3, and 0.02% NaN3 [pH 9.6]) and used to coat Nunc MaxiSorp flat bottom 96-well plates (Thermo Fisher) with 100 μL for 4 h at 37°C. After being coated, the wells were washed three times (PBS [pH 7.4] and 0.05% Tween 20) and blocked overnight in 200 μL blocking buffer (PBS, 0.05% Tween 20, and 1% BSA) at 4°C. The respective recombinant antibodies c7D11, c8A, and c6C (manufactured by BioFactura, Inc.) were diluted in blocking buffer to 100 ng/mL. Starting with this solution, a serial dilution was prepared for generating a standard curve. Samples were diluted appropriately in blocking buffer (PBS, 0.05% Tween 20, and 1% BSA) to allow for quantification. All further incubations were carried out at room temperature. Diluted supernatants or sera were added to the coated wells and incubated for 2 h. The solution was discarded and the wells were washed three times. Detection antibody (goat anti-human IgG biotin; Dianova, cat. no. 109065088) was diluted 1:20,000 in blocking buffer, and 100 μL was added to the wells and incubated for 60–90 min. The solution was discarded and the wells were washed three times. Horseradish peroxidase (HRP)-streptavidin (BD Pharmingen, cat. no. 554066) was diluted 1:1,000 in blocking buffer, and 100 μL was added to the wells and incubated for 30 min. The HRP solution was discarded and the wells were washed four times. One hundred microliters of tetramethylbenzidine (TMB; Thermo Scientific, cat. no. 34028) substrate was added and the reaction was stopped by using 100 μL of 20% sulfuric acid. For the detection of antibodies, an ELISA was carried out as described above. BHK cells were transfected with mRNA encoding the different antibodies as described above. After 72 h, supernatants were collected and antibody was purified using protein A beads from Pierce (Thermo Scientific, cat. no. 44667) according to the manufacturer’s instructions. Wells were then coated with 100 μL of antibodies at 1 μg/mL. Mouse anti-human IgG (H + L) cross-adsorbed secondary antibody (Thermo Scientific, cat. no. 31135) was used as an internal standard. Goat anti-mouse IgG (H + L) secondary antibody, biotin (Thermo Scientific, cat. no. 31800), was used at 1:50,000 for detection. Absorbance at 450 nm was measured in a plate reader (Hidex Chameleon Model 425-156 or BertholdTech TriStar2 Model LB 942).
Immunogen-specific quantitative ELISA
Three immunogen-specific ELISAs were performed as previously described.22 Antibodies c7D11, c8A, and c6C targeting poxviral proteins L1, B5, and A33, respectively, were provided by BioFactura, Inc. Normal mouse and rabbit sera were utilized to match the proper matrix of the samples being tested and were purchased from BioIVT.
Mature virion neutralization assay
Neutralization assays were performed similar to those previously described.37 Vaccinia virus was incubated with serial 5-fold dilutions of test samples and subsequently titrated on BSC-1 cells with a 1.5% methylcellulose overlay. Plates were stained after 4 days with crystal violet and plaques enumerated.
Extracellular virus neutralization assay
Extracellular enveloped vaccinia virus was prepared, and neutralization was performed as described elsewhere.22 Briefly, serial dilutions were made of the samples and controls, mixed with EVs in the presence or absence of 5% (final) human complement and an anti-MV neutralizing antibody (10F5), incubated, and plaque titrated on BSC-1 cells using a 1.5% methylcellulose overlay. The cells were stained after 4 days with crystal violet and plaques enumerated.
Rabbits and injections
Rabbit experiments were performed by a contract facility. New Zealand White rabbits (Oryctolagus cuniculus) were purchased from Charles River Laboratories. Rabbits were acclimated for at least 5 days and clinically cleared before enrollment in the study. The rabbits were anesthetized before the hindleg(s) was clipped and subsequently administered an intramuscular injection(s) of the test material. Up to four injections were given per rabbit (two per hindleg), and all injections were given within a relatively short time frame of <2 min. The injections were performed utilizing a PharmaJet Stratis device in a volume of 0.5 mL. Blood was collected via marginal ear veins. Individual constructs were administered to the same leg of each rabbit for experiments administering two different mRNA constructs.
Research was conducted under an IACUC-approved protocol in compliance with the Animal Welfare Act, PHS Policy, and other federal statutes and regulations relating to animals and experiments involving animals. The facility where this research was conducted is accredited by AAALAC International and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 2011.
Statistics
GraphPad Prism was used to construct graphs, associated descriptive statistics, and two-way ANOVA with multiple comparisons (Sidak’s multiple comparisons).
Acknowledgments
We would like to thank USAMRIID’s Veterinary Medicine Division for supporting our efforts. In addition, we would like to thank Dr. Darryl Sampey, BioFactura, for providing antibody sequence information and control antibodies used in this study. The Defense Advanced Research Projects Agency (DD1144 A629-ISA) funded this research. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the US Army.
Author contributions
E.M.M. designed and performed experiments, analyzed data, prepared the draft manuscript and figures, and edited the manuscript. C.T.-S. designed and performed experiments, analyzed data, prepared figures, and edited the manuscript. P.B. designed experiments, analyzed data, and edited the manuscript. J.W.H. designed experiments, analyzed data, and edited the manuscript.
Declaration of interests
P.B. and C.T.-S. are employed by CureVac, a company that has licensing rights related to the mRNA utilized in these studies. J.W.H. is an inventor on a patent related to mAb c7D11 and antibody therapies generated using L1, A33, and B5 as antigens. These interests do not alter the authors’ adherence to policies on sharing data and materials.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.05.025.
Supplemental information
References
- 1.Golden J.W., Hooper J.W. The strategic use of novel smallpox vaccines in the post-eradication world. Expert Rev. Vaccin. 2011;10:1021–1035. doi: 10.1586/erv.11.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kempe C.H. Studies smallpox and complications of smallpox vaccination. Pediatrics. 1960;26:176–189. doi: 10.1542/peds.26.2.176. [DOI] [PubMed] [Google Scholar]
- 3.Sharp J.C., Fletcher W.B. Experience of anti-vaccinia immunoglobulin in the United Kingdom. Lancet. 1973;301:656–659. doi: 10.1016/s0140-6736(73)92215-0. [DOI] [PubMed] [Google Scholar]
- 4.Xiao Y., Isaacs S.N. Therapeutic vaccines and antibodies for treatment of orthopoxvirus infections. Viruses. 2010;2:2381–2403. doi: 10.3390/v2102381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golden J.W., Zaitseva M., Kapnick S., Fisher R.W., Mikolajczyk M.G., Ballantyne J., Golding H., Hooper J.W. Polyclonal antibody cocktails generated using DNA vaccine technology protect in murine models of orthopoxvirus disease. Virol. J. 2011;8:441. doi: 10.1186/1743-422X-8-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lustig S., Fogg C., Whitbeck J.C., Eisenberg R.J., Cohen G.H., Moss B. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 2005;79:13454–13462. doi: 10.1128/JVI.79.21.13454-13462.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crickard L., Babas T., Seth S., Silvera P., Koriazova L., Crotty S. Protection of rabbits and immunodeficient mice against lethal poxvirus infections by human monoclonal antibodies. PLoS One. 2012;7:e48706. doi: 10.1371/journal.pone.0048706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaitseva M., Kapnick S.M., Meseda C.A., Shotwell E., King L.R., Manischewitz J., Scott J., Kodihalli S., Merchlinsky M., Nielsen H., et al. Passive immunotherapies protect WRvFire and IHD-J-Luc vaccinia virus-infected mice from lethality by reducing viral loads in the upper respiratory tract and internal organs. J. Virol. 2011;85:9147–9158. doi: 10.1128/JVI.00121-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boulter E.A. Humoral and cellular aspects of the response to infection: Protection against poxviruses. Proc. R. Soc. Med. 1969;62:295–297. doi: 10.1177/003591576906200349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boulter E.A., Zwartouw H.T., Titmuss D.H., Maber H.B. The nature of the immune state produced by inactivated vaccinia virus in rabbits. Am. J. Epidemiol. 1971;94:612–620. doi: 10.1093/oxfordjournals.aje.a121360. [DOI] [PubMed] [Google Scholar]
- 11.Boulter E.A., Appleyard G. Differences between extracellular and intracellular forms of poxvirus and their implications. Prog. Med. Virol. 1973;16:86–108. [PubMed] [Google Scholar]
- 12.Golden J.W., Josleyn M., Mucker E.M., Hung C.F., Loudon P.T., Wu T.C., Hooper J.W. Side-by-side comparison of gene-based smallpox vaccine with MVA in nonhuman primates. PLoS One. 2012;7:e42353. doi: 10.1371/journal.pone.0042353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flingai S., Plummer E.M., Patel A., Shresta S., Mendoza J.M., Broderick K.E., Sardesai N.Y., Muthumani K., Weiner D.B. Protection against dengue disease by synthetic nucleic acid antibody prophylaxis/immunotherapy. Sci. Rep. 2015;5:12616. doi: 10.1038/srep12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muthumani K., Block P., Flingai S., Muruganantham N., Chaaithanya I.K., Tingey C., Wise M., Reuschel E.L., Chung C., Muthumani A., et al. Rapid and long-term immunity elicited by DNA-encoded antibody prophylaxis and DNA vaccination against chikungunya virus. J. Infect. Dis. 2016;214:369–378. doi: 10.1093/infdis/jiw111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muthumani K., Flingai S., Wise M., Tingey C., Ugen K.E., Weiner D.B. Optimized and enhanced DNA plasmid vector based in vivo construction of a neutralizing anti-HIV-1 envelope glycoprotein Fab. Hum. Vaccin. Immunother. 2013;9:2253–2262. doi: 10.4161/hv.26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schnepp B.C., Johnson P.R. Vector-mediated antibody gene transfer for infectious diseases. Adv. Exp. Med. Biol. 2015;848:149–167. doi: 10.1007/978-1-4939-2432-5_8. [DOI] [PubMed] [Google Scholar]
- 17.Mucker E.M., Karmali P.P., Vega J., Kwilas S.A., Wu H., Joselyn M., Ballantyne J., Sampey D., Mukthavaram R., Sullivan E., et al. Lipid nanoparticle formulation increases efficiency of DNA-vectored vaccines/immunoprophylaxis in animals including transchromosomic bovines. Sci. Rep. 2020;10:8764. doi: 10.1038/s41598-020-65059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolffe E.J., Vijaya S., Moss B. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology. 1995;211:53–63. doi: 10.1006/viro.1995.1378. [DOI] [PubMed] [Google Scholar]
- 19.Mucker E.M., Wollen-Roberts S.E., Kimmel A., Shamblin J., Sampey D., Hooper J.W. Intranasal monkeypox marmoset model: prophylactic antibody treatment provides benefit against severe monkeypox virus disease. PLoS Negl. Trop. Dis. 2018;12:e0006581. doi: 10.1371/journal.pntd.0006581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z., Earl P., Americo J., Damon I., Smith S.K., Zhou Y.H., Yu F., Sebrell A., Emerson S., Cohen G., et al. Chimpanzee/human mAbs to vaccinia virus B5 protein neutralize vaccinia and smallpox viruses and protect mice against vaccinia virus. Proc. Natl. Acad. Sci. U S A. 2006;103:1882–1887. doi: 10.1073/pnas.0510598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z., Earl P., Americo J., Damon I., Smith S.K., Yu F., Sebrell A., Emerson S., Cohen G., Eisenberg R.J., et al. Characterization of chimpanzee/human monoclonal antibodies to vaccinia virus A33 glycoprotein and its variola virus homolog in vitro and in a vaccinia virus mouse protection model. J. Virol. 2007;81:8989–8995. doi: 10.1128/JVI.00906-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mucker E.M., Lindquist M., Hooper J.W. Particle-specific neutralizing activity of a monoclonal antibody targeting the poxvirus A33 protein reveals differences between cell associated and extracellular enveloped virions. Virology. 2020;544:42–54. doi: 10.1016/j.virol.2020.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Wang W., Wang E.Q., Balthasar J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008;84:548–558. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- 24.Kwilas S., Kishimori J.M., Josleyn M., Jerke K., Ballantyne J., Royals M., Hooper J.W. A hantavirus pulmonary syndrome (HPS) DNA vaccine delivered using a spring-powered jet injector elicits a potent neutralizing antibody response in rabbits and nonhuman primates. Curr. Gene Ther. 2014;14:200–210. doi: 10.2174/1566523214666140522122633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alberer M., Gnad-Vogt U., Hong H.S., Mehr K.T., Backert L., Finak G., Gottardo R., Bica M.A., Garofano A., Koch S.D., et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet. 2017;390:1511–1520. doi: 10.1016/S0140-6736(17)31665-3. [DOI] [PubMed] [Google Scholar]
- 26.Kose N., Fox J.M., Sapparapu G., Bombardi R., Tennekoon R.N., de Silva A.D., Elbashir S.M., Theisen M.A., Humphris-Narayanan E., Ciaramella G., et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 2019;4:eaaw6647. doi: 10.1126/sciimmunol.aaw6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pardi N., Hogan M.J., Pelc R.S., Muramatsu H., Andersen H., DeMaso C.R., Dowd K.A., Sutherland L.L., Scearce R.M., Parks R., et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. 2017;543:248–251. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pardi N., Tuyishime S., Muramatsu H., Kariko K., Mui B.L., Tam Y.K., Madden T.D., Hope M.J., Weissman D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control Release. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pardi N., Secreto A.J., Shan X., Debonera F., Glover J., Yi Y., Muramatsu H., Ni H., Mui B.L., Tam Y.K., et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017;8:14630. doi: 10.1038/ncomms14630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kariko K., Buckstein M., Ni H., Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Alameh M.G., Tombacz I., Bettini E., Lederer K., Sittplangkoon C., Wilmore J.R., Gaudette B.T., Soliman O.Y., Pine M., Hicks P., et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity. 2021;54:2877–2892.e7. doi: 10.1016/j.immuni.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Meer P.J., Kooijman M., Brinks V., Gispen-de Wied C.C., Silva-Lima B., Moors E.H., Schellekens H. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. MAbs. 2013;5:810–816. doi: 10.4161/mabs.25234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thess A., Grund S., Mui B.L., Hope M.J., Baumhof P., Fotin-Mleczek M., Schlake T. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 2015;23:1456–1464. doi: 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thran M., Mukherjee J., Ponisch M., Fiedler K., Thess A., Mui B.L., Hope M.J., Tam Y.K., Horscroft N., Heidenreich R., et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 2017;9:1434–1447. doi: 10.15252/emmm.201707678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramaswamy S., Tonnu N., Tachikawa K., Limphong P., Vega J.B., Karmali P.P., Chivukula P., Verma I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. U S A. 2017;114:E1941–E1950. doi: 10.1073/pnas.1619653114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yaghi N.K., Wei J., Hashimoto Y., Kong L.Y., Gabrusiewicz K., Nduom E.K., Ling X., Huang N., Zhou S., Kerrigan B.C.P., et al. Immune modulatory nanoparticle therapeutics for intracerebral glioma. Neuro Oncol. 2017;19:372–382. doi: 10.1093/neuonc/now198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mucker E.M., Chapman J., Huzella L.M., Huggins J.W., Shamblin J., Robinson C.G., Hensley L.E. Susceptibility of marmosets (Callithrix jacchus) to monkeypox virus: a low dose prospective model for monkeypox and smallpox disease. PLoS One. 2015;10:e0131742. doi: 10.1371/journal.pone.0131742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.