

Abstract
Corticotropin-releasing factor (CRF) signaling in the central nucleus of the amygdala (CeA) plays a critical role in rodent models of excessive alcohol drinking. However, the source of CRF acting in the CeA during alcohol withdrawal remains to be identified. In the present study, we hypothesized that CeA CRF interneurons may represent a behaviorally relevant source of CRF to the CeA increasing motivation for alcohol via negative reinforcement. We first observed that Crh mRNA expression in the anterior part of the mouse CeA correlates positively with alcohol intake in C57BL/6J males with a history of chronic binge drinking followed by abstinence and increases upon exposure to chronic intermittent ethanol (CIE) vapor inhalation. We then found that chemogenetic activation of CeA CRF neurons in Crh-IRES-Cre mouse brain slices increases gamma-aminobutyric acid (GABA) release in the medial CeA, in part via CRF1 receptor activation. While chemogenetic stimulation exacerbated novelty-induced feeding suppression (NSF) in alcohol-naïve mice, thereby mimicking the effect of withdrawal from CIE, it had no effect on voluntary alcohol consumption, following either acute or chronic manipulation. Furthermore, chemogenetic inhibition of CeA CRF neurons did not affect alcohol consumption or NSF in chronic alcohol drinkers exposed to air or CIE. Altogether, these findings indicate that CeA CRF neurons produce local release of GABA and CRF and promote hyponeophagia in naïve mice, but do not drive alcohol intake escalation or negative affect in CIE-withdrawn mice. The latter result contrasts with previous findings in rats and demonstrates species specificity of CRF circuit engagement in alcohol dependence.
Introduction
Alcohol use disorders (AUDs) represent a spectrum of pathological patterns of alcoholic beverage consumption that affect 6.2% of the adult population in the United States of America1 and more than 100 million people worldwide2. AUDs are not only characterized by the intake of excessive amounts of alcohol but also by the emergence of a negative emotional state (e.g., anxiety, irritability) upon withdrawal. While the motivation to drink alcohol is initially driven by the desire to experience the pleasurable effects of intoxication (positive reinforcement), it becomes progressively driven by the need for relief from the negative emotional state associated with withdrawal (negative reinforcement)3. Excessive alcohol drinking (i.e., beyond intoxication threshold) and negative affect during withdrawal can be triggered in several mouse and rat models, which has enabled the dissection of underlying neurobiological mechanisms4–8.
Multiple pharmacological and genetic studies have highlighted that recruitment of the extrahypothalamic corticotropin-releasing factor (CRF, encoded by the Crh gene) system plays a pivotal role in the emergence of negative reinforcement in alcohol-withdrawn animals, whereby activation of CRF type 1 receptors (CRF1) contributes to the excessive drinking, heightened anxiety, and stress sensitization characterizing alcohol dependence3, 9–12. In particular, CRF is released in the central nucleus of the amygdala (CeA) during alcohol withdrawal13 and blockade of CRF1 signaling in the CeA reduces the anxiogenic-like effect of alcohol withdrawal14, 15, dependence-induced increase in alcohol self-administration16, 17, as well as heavy binge drinking18. CRF1 in the CeA also mediates the elevation of gamma-aminobutyric acid (GABA) dialysate induced by alcohol dependence19 and CRF1 overexpression in the CeA potentiates stress-induced reinstatement of alcohol seeking20.
Despite this wealth of converging evidence, the specific neurons responsible for the release of CRF in the CeA during alcohol withdrawal have not been identified. In the present study, we tested the hypothesis that these neurons are intrinsic to the CeA rather than arising from an extrinsic source. This hypothesis was supported by evidence that Crh is upregulated in the CeA of alcohol-dependent and post-dependent rats19, 21, 22, along with our own findings (reported here) that Crh expression in the anterior part of the CeA correlates with alcohol intake in mice with a history of chronic alcohol drinking followed by abstinence and increases upon exposure to chronic intermittent ethanol (CIE) vapor inhalation. Importantly, psychological stress also upregulates Crh in the CeA23–25 and CRF synthesis in the CeA plays a critical role in mediating anxiety-like behavior26, 27. In addition, CRF1-expressing neurons in the CeA receive direct input from local interneurons28 and CeA CRF neurons provide both local inhibitory GABA and excitatory CRF1-mediated signals to other CeA neurons in Crh-Cre rats29, which supports the notion that CRF can be released from intrinsic CeA neurons. Furthermore, studies in Crh-Cre rats have recently shown that chemogenetic stimulation of CeA CRF neurons elicits anxiety-like behavior30, 31, while their optogenetic inhibition reduces escalated alcohol self-administration in alcohol-dependent rats32. In the mouse, CeA CRF neurons are more active than their non-CRF neighbors during repeated alcohol binge drinking33.
According to the hypothesis that CeA CRF neurons can release CRF locally, we predicted that chemogenetic stimulation of mouse CeA CRF neurons would replicate cellular and behavioral hallmarks of alcohol dependence. While this manipulation produced local CRF1-mediated GABA release and elicited signs of negative affect resembling those seen during alcohol withdrawal, it did not increase voluntary alcohol consumption. Furthermore, their chemogenetic inhibition did not reverse ethanol intake escalation in alcohol-dependent mice. Accordingly, our data suggest that contrary to reports in rats, CeA CRF neurons are neither sufficient nor necessary to drive excessive alcohol drinking in mice.
Materials and Methods
Animals
C57BL/6J males were purchased from the Jackson Laboratories (stock #000664) or from Scripps Research rodent breeding colony. Crh-IRES-Cre breeders were obtained from The Jackson Laboratory (B6(Cg)Crhtm1(cre)Zjh/J, stock # 012704,34). All Crh-IRES-Cre mice used for experimentation were heterozygous males. Additional details are provided in the Supplement. All procedures adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Scripps Research.
Viral vectors
Adeno-associated viral serotype 2 (AAV2) vectors encoding the hM3Dq (excitatory) or hM4Di (inhibitory) designer receptors35 fused to the red fluorescent protein mCherry, under the control of the human synapsin promoter and in a Cre-dependent manner36, were obtained from the Vector Core at the University of North Carolina (UNC) at Chapel Hill or from Addgene. Vectors were injected bilaterally into the anterior part of the CeA (AP −0.9 mm from bregma, ML ± 2.8–3.0 from the midline, DV −4.5 mm from the skull). Additional details are provided in the Supplement.
Experimental cohorts
In situ hybridization data were collected from two cohorts of C57BL/6J mice subjected to chronic alcohol drinking and alcohol vapor inhalation, respectively. For chemogenetic stimulation, a first cohort of Crh-IRES-Cre mice was used for electrophysiological recordings. A second cohort was tested in the elevated plus-maze (EPM), social approach, and novelty-suppressed feeding (NSF) assays, with at least one week between tests; these mice were then single-housed and tested for alcohol drinking. A third cohort was tested for digging and marble burying, and their brains were used to quantify c-Fos induction one week later. A fourth cohort was used to confirm the phenotype observed in the NSF test and included negative control mice; these mice were also tested for locomotor activity and fasting-refeeding in the home cage. For chemogenetic inhibition, electrophysiological validation was conducted in a first cohort of Crh-IRES-Cre mice and alcohol dependence was induced in a second cohort. The effect of CP376395 on alcohol drinking was tested in a separate cohort of C57BL/6J mice exposed to air or CIE.
Detailed methods for in situ hybridization, electrophysiology, immunohistochemistry, alcohol exposure, and behavioral testing are provided in the Supplement.
Statistical analysis
Data analysis was performed in Statistica 13.3 (TIBCO Software Inc.). The correlation of Crh in situ hybridization signal with ethanol intake was evaluated using Pearson’s r. The effect of alcohol vapor inhalation on Crh signal was analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s comparisons to the Air group. The effect of clozapine-N-oxide (CNO) on c-Fos counts, firing rate, and sIPSC parameters was analyzed using paired two-tailed Student’s t-tests. The effect of CNO combined with R121919 was analyzed by repeated measures (RM) ANOVA and Pearson’s correlation. CNO-responsive cells were identified by comparing the distribution of inter-event intervals at baseline and after CNO application using a Kolmogorov-Smirnov test. In hM3Dq-expressing mice, one-way RM-ANOVAs were used to analyze the effect of CNO on locomotor activity and ethanol intake (acute regimen). The effects of CNO on ethanol intake (chronic regimen), social approach, hyponeophagia, and refeeding were analyzed by two-way RM-ANOVA. Other behavioral endpoints were analyzed using unpaired two-tailed Student’s t-tests. Behavioral measures obtained in hM4Di-expressing mice were analyzed by two-way RM-ANOVA. Tukey posthoc tests were conducted when relevant. The effect of CP376395 was analyzed by two-way RM-ANOVA followed by Dunnett’s comparisons to vehicle. Data are shown as mean ± s.e.m. on the graphs.
Results
Crh expression in the anterior part of the CeA correlates with ethanol intake and increases upon CIE
As described previously using CRF immunostaining and a GFP reporter strain, CRF expression varies along the rostro-caudal extent of the mouse CeA37, 38. We confirmed this observation using chromogenic in situ hybridization to label Crh mRNA (Fig. 1A), as can also be seen in the Allen Mouse Brain Atlas (https://mouse.brain-map.org, experiment 292). At the most anterior level (≈ −0.8 mm from bregma), a V-shaped cluster of CRF neurons is found at the transition between the interstitial nucleus of the posterior limb of the anterior commissure (IPAC) and the CeA. At midlevel (≈ −1.2 mm from bregma), CRF neurons are more sparsely distributed, mostly within the lateral aspect of the CeA, and to a smaller extent within the medial CeA. At a more posterior level (≈ −1.6 mm from bregma), CRF neurons are tightly packed within the lateral CeA, with again a few cell bodies found in the medial CeA.
Figure 1. Crh mRNA levels in the anterior CeA increase upon chronic intermittent alcohol exposure.
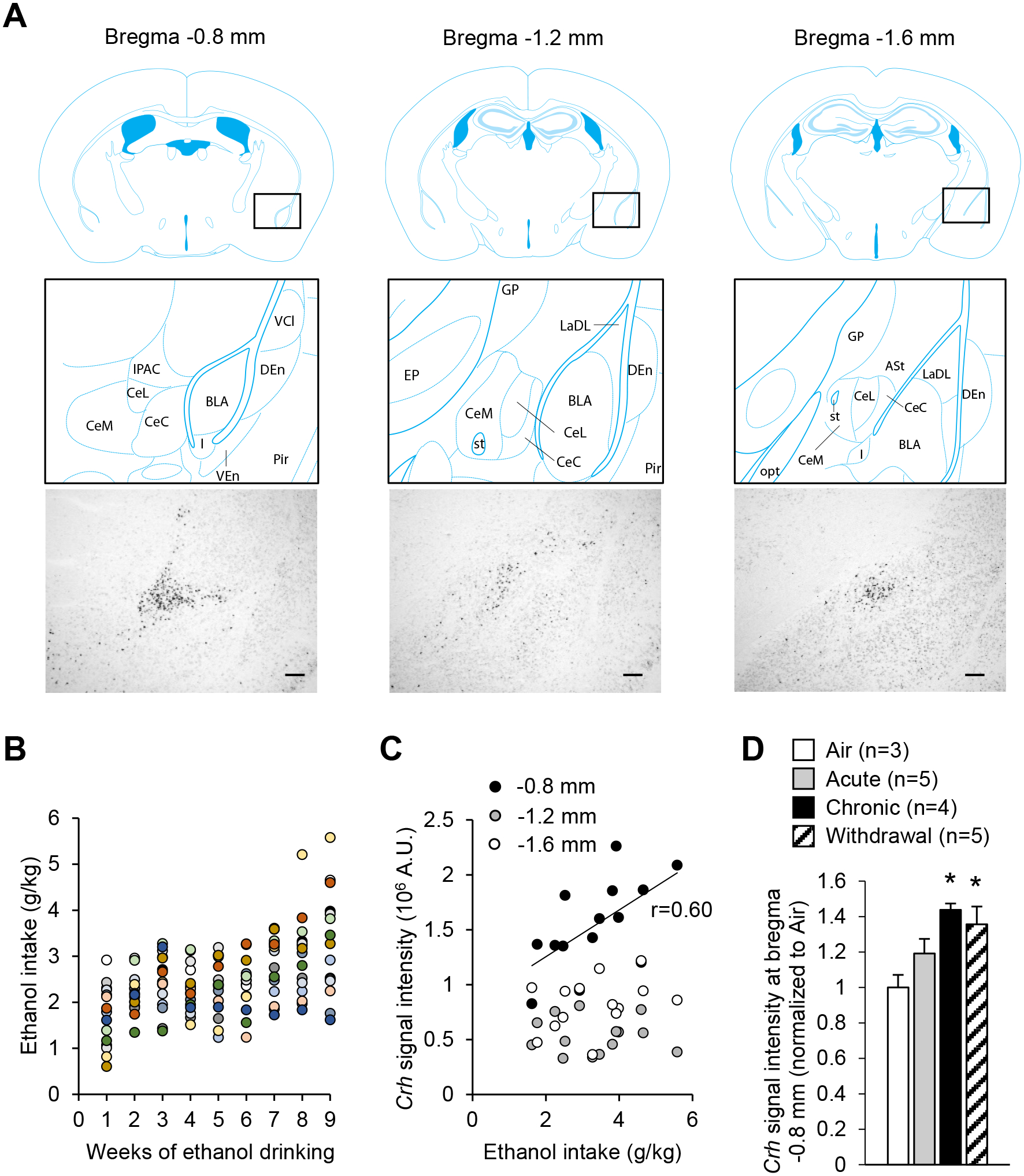
A. Representative images of Crh mRNA distribution at three antero-posterior levels of the mouse CeA (scale bars = 200 μm) and corresponding brain atlas diagrams highlighting a V-shaped cluster of CRF neurons at the junction of the anterior CeL and IPAC (left panels), and scattered CRF neurons at more posterior levels of the CeL (middle and right panels). Brain atlas diagrams were reproduced from85. BLA, basolateral amygdaloid nucleus; CeC, capsular part of the CeA; CeL, lateral division of the CeA; CeM, medial division of the CeA; DEn, dorsal endopiriform claustrum; EP, entopeduncular nucleus; IPAC, interstitial nucleus of the posterior limb of the anterior commissure; GP, globus pallidus; I, intercalated nuclei of the amygdala; LaDL, lateral amygdaloid nucleus, dorsolateral part; opt, optic tract; Pir, piriform cortex; st, stria terminalis; VCl, ventral part of claustrum; VEn, ventral endopiriform claustrum. B. Average weekly ethanol intake in a cohort of mice subjected to 2-h two-bottle choice (ethanol 15% v:v vs water) sessions five days per week for nine weeks. Each color represents an individual mouse. C. Crh chromogenic in situ hybridization signal density at three antero-posterior levels of the CeA as a function of ethanol intake during the last week. There was a significant correlation at the most anterior level (≈ bregma −0.8 mm, p<0.05), but not at more posterior levels. D. Crh chromogenic in situ hybridization signal density at the most anterior level of the CeA in mice exposed to a single 16-h bout of alcohol vapor inhalation (Acute), eight bouts (Chronic), or eight bouts followed by three days of withdrawal (Withdrawal). Data are shown as mean ± s.e.m. of Crh signal normalized to Air controls. Data were analyzed by one-way ANOVA; *, p<0.05 vs. Air, Dunnett’s posthoc test.
Semi-quantitative measures of Crh expression at these three antero-posterior levels were obtained in C57BL/6J males subjected to chronic alcohol binge drinking for nine weeks and euthanized four weeks after their last drinking session. Average ethanol intake during the last drinking week ranged between 1.6 and 5.8 g/kg per 2-h session (Fig. 1B). Crh expression in the most anterior part of the CeA (bregma −0.8 mm, including IPAC) correlated positively with the average ethanol intake measured during the last week of drinking (r=0.60, p=0.02, Fig. 1C). No significant correlation was observed at more posterior levels of the CeA (bregma −1.2 mm: r=0.13, p=0.65; bregma −1.6 mm: r=0.33, p=0.23; Fig. 1C).
Additional analyses were conducted to determine whether Crh expression in the anterior CeA responded to alcohol intoxication or withdrawal. C57BL/6J males were exposed to a single bout (Acute) or eight bouts of 16 h of ethanol vapor inhalation. Mice subjected to eight bouts were euthanized either immediately after vapor exposure (Chronic) or following 72 h withdrawal (Withdrawal). Crh expression at bregma −0.8 mm was significantly higher in Chronic and Withdrawal mice (Fig. 1D; main effect of group: F3,13=4.5, p=0.022; posthoc comparisons to Air group: p=0.014 and p=0.035, respectively).
Based on these results, the anterior level of the CeA was targeted for viral vector infusions in subsequent chemogenetic experiments.
Validation of chemogenetic approach to stimulate CeA CRF neurons
We used Cre-dependent expression of the excitatory designer receptor hM3Dq in Crh-IRES-Cre males to analyze the effects of CeA CRF neuron stimulation on CRF release, alcohol drinking and affective behaviors. We first assessed the specificity of Cre activity distribution in the CeA of Crh-IRES-Cre mice and verified that CNO activated hM3Dq-expressing cells both in vivo and in brain slices.
We analyzed the overlap between Crh and tdTomato mRNAs in CeA sections of Crh-IRES-Cre;Ai9 mice by double in situ hybridization (Fig. 2A). We found that 86.3 ± 4.9 % of tdTomato+ neurons expressed Crh (a measure of reporter fidelity), while 64.0 ± 5.4 % of Crh+ neurons expressed tdTomato (a measure of reporter penetrance). Consistent with these results, in Crh-IRES-Cre mice injected with an AAV2-hSyn-DIO-hM3Dq-mCherry vector in the CeA, mCherry-immunolabeled cell bodies showed the same distribution as Crh mRNA (Fig. 2B, compare with Fig. 1A left panel and Fig. 2A). Mice were perfused 90 min following i.p. administration of either vehicle or CNO (5 mg/kg) and their brains were processed for immunohistochemistry. mCherry immunoreactivity was used to label hM3Dq-expressing cells and c-Fos immunoreactivity was used as a marker of cellular activation (Fig. 2C–D). As expected, CNO increased the number of c-Fos+ cells selectively in the subpopulation expressing hM3Dq (t11=4.1, p=0.002), but it did not affect the number of c-Fos+ cells among mCherry-negative cells (t11=−0.1, p=0.91; Fig. 2D, left). Upon CNO administration, the proportion of active (c-Fos+) hM3Dq-expressing cells increased about 4-fold (t11=6.3, p<0.0001; Fig. 2D, right).
Figure 2. Validation of Cre activity and chemogenetic stimulation in CeA CRF neurons of Crh-IRES-Cre mice.
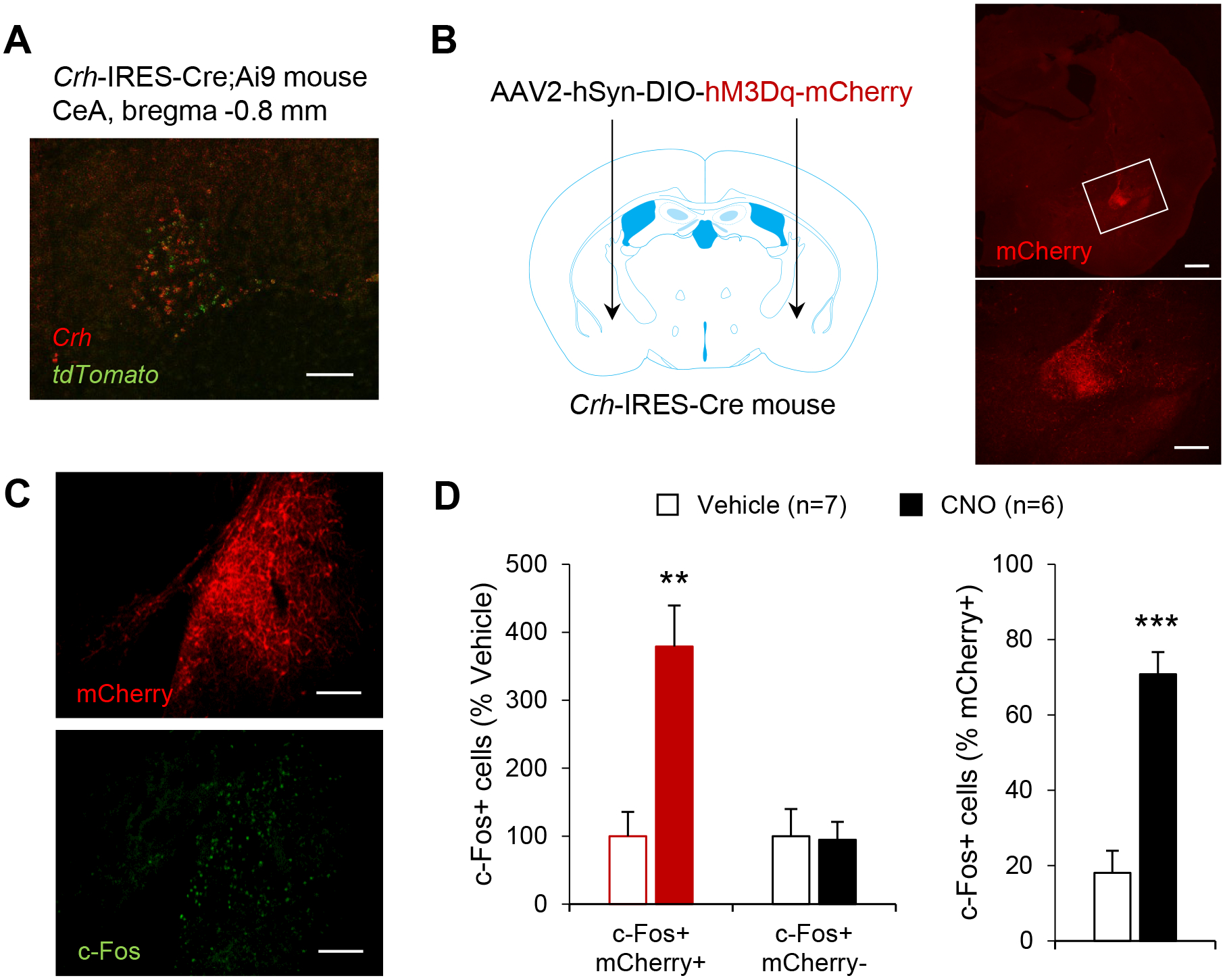
A. Distribution of Crh (red) and tdTomato (green) mRNAs in the CeA of Crh-IRES-Cre;Ai9 mice, as visualized by double fluorescent in situ hybridization (scale bar = 100 μm). B. mCherry immunolabeling recapitulates the V-shaped pattern of Crh expression in the CeA of Crh-IRES-Cre mice injected with a Cre-dependent AAV vector encoding hM3Dq-mCherry. The area framed in the top picture (scale bar = 500 μm) is shown at higher magnification in the bottom picture (scale bar = 100 μm). C. Double immunostaining of mCherry (red) and c-Fos (green) was used to evaluate neuronal activation in the CeA following i.p. injection of vehicle (n=7) or CNO 5 mg/kg (n=6) in Crh-IRES-Cre mice injected with AAV2-hSyn-DIO-hM3Dq-mCherry in the CeA (scale bars = 100 μm). D. Chemogenetic stimulation of CeA CRF neurons increased c-Fos expression selectively in mCherry-positive CeA neurons. Data are shown as mean ± s.e.m. of the number of c-Fos+ cells expressed as percentage of Vehicle values (left graph) or percentage of mCherry+ cells (right graph). Data were analyzed by unpaired t-test: **, p<0.01; ***, p<0.001.
In another cohort of Crh-IRES-Cre mice injected in the CeA with a Cre-dependent vector encoding hM3Dq-mCherry, action potentials were recorded from mCherry-positive (i.e., hM3Dq-expressing) CeA neurons (Fig. 3A–B). As expected, CNO (500 nM) application significantly increased their firing rate (t7=−6.5, p=0.0003; Fig. 3B).
Figure 3. Chemogenetic stimulation of CeA CRF neurons increases GABA release onto medial CeA neurons in a CRF1-dependent manner.
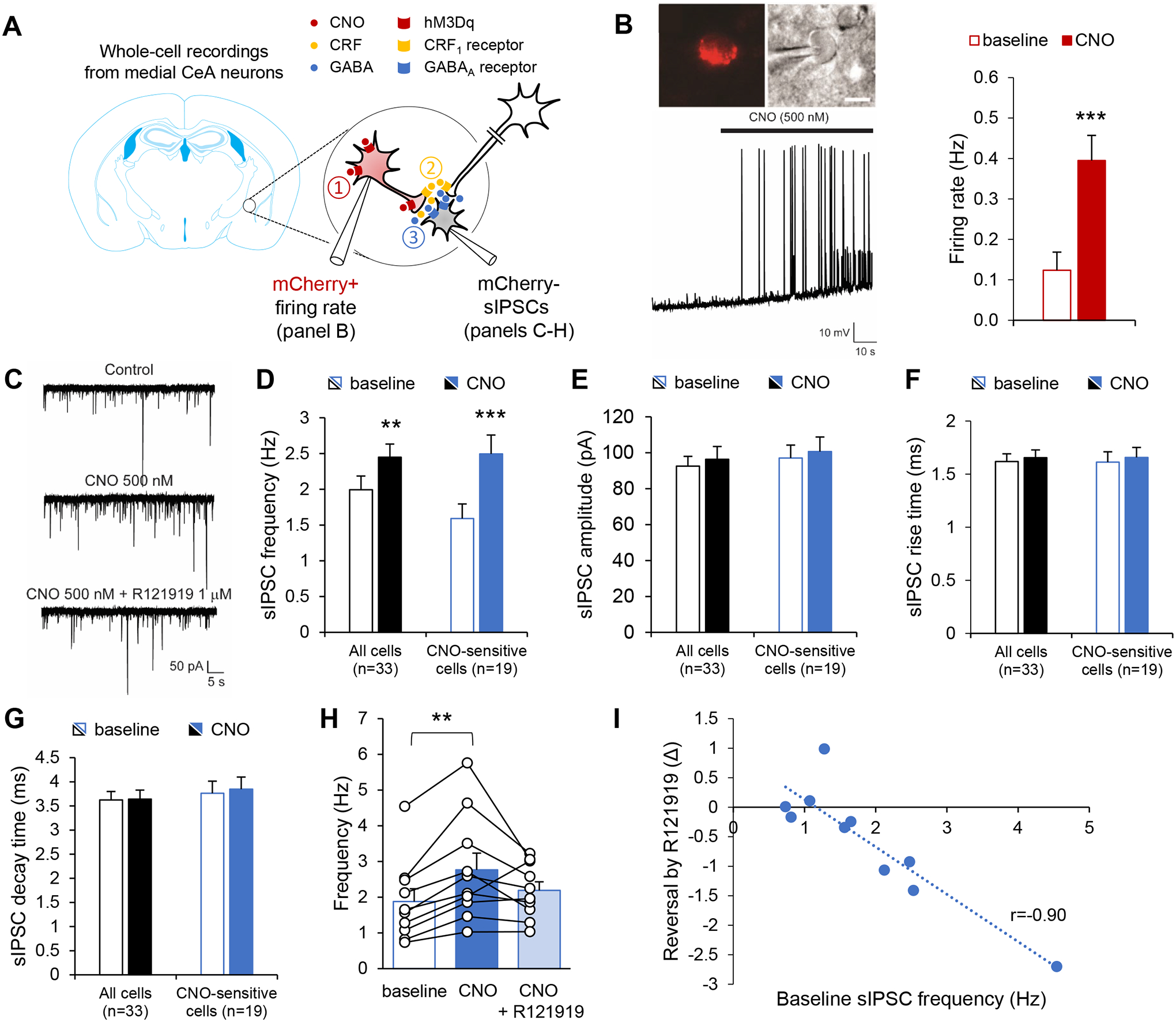
A. Crh-IRES-Cre mice were injected in the anterior CeA with a Cre-dependent AAV vector encoding hM3Dq-mCherry. Whole-cell recordings were then obtained from mCherry+ neurons (panel B) or mCherry− neurons (panels C-I) to test the hypothesis that chemogenetic stimulation of CeA CRF neurons (red, ①) triggers the release of GABA (blue) and CRF (yellow) in the medial CeA (②), which may in turn stimulate GABA release (③) onto non-CRF neurons (grey) via the activation of CRF1 receptors located on intrinsic or extrinsic GABAergic presynaptic terminals39. B. Firing rates were recorded from CeA mCherry+ neurons. Top: red fluorescence and differential interference contrast images of patched CeA neuron. Bottom: representative current-clamp trace before and during CNO application (500 nM). Right: Firing rates are shown as mean ± s.e.m. (n=8 neurons). Data were analyzed by paired t-test: ***, p<0.001. C-I. Spontaneous inhibitory postsynaptic currents (sIPSCs) were recorded from medial CeA mCherry− neurons. C. Representative traces before and during CNO application (500 nM) followed by subsequent R121919 co-application (1 μM). CNO increased sIPSC frequency (D), but did not affect sIPSC amplitude (E), rise time (F) or decay time (G). Data are shown as mean ± s.e.m. for the whole set of recorded neurons (n=33, black bars), as well as for the subset of neurons whose sIPSC frequency was significantly increased by CNO (n=19, blue bars). Data were analyzed by paired t-test: **, p<0.01; ***, p<0.001. H-I. In a subset of CNO-sensitive cells (n=10), the CRF1 antagonist R121919 was co-applied following CNO alone. H. sIPSC frequencies are shown as mean ± s.e.m. and were analyzed by one-way ANOVA; **, p<0.01, Tukey posthoc test. I. The effect of R121919 (expressed as sIPSC frequency difference before and after addition of R121919) was correlated with baseline sIPSC frequency; p<0.001.
Stimulation of CeA CRF neurons increases local GABAergic transmission via activation of CRF1 receptors
We first evaluated the possibility that CeA CRF neurons release CRF in the medial CeA, where both CRF and ethanol are known to increase GABA release via the activation of CRF1 receptors39. Voltage-clamp recordings of spontaneous inhibitory postsynaptic currents (sIPSC) were obtained from mCherry-negative neurons (n=33 neurons from 11 mice) located in the medial CeA before and during application of CNO (500 nM). Overall, CNO produced a significant increase in sIPSC frequency (Fig. 3C–D, t32=−3.5, p=0.001), but had no effect on amplitude (Fig. 3E, t32=−1.5, p=0.16), rise time (Fig. 3F, t32=−0.8, p=0.45) or decay time (Fig. 3G, t32=−0.2, p=0.83). However, the response to CNO was variable between cells. Within-cell analysis of the cumulative distribution of inter-event intervals indicated that sIPSC frequency was significantly increased in 19 cells (blue bars in Fig. 3D–G), reduced in 3 cells, and unchanged in the remaining 11 cells (p>0.05). In the subset of neurons that responded with increased sIPSC frequency (t18=−7.6, p<0.0001), there was still no effect of CNO on sIPSC amplitude (t18=−1.5, p=0.15), rise time (t18=−0.6, p=0.53) and decay time (t18=−0.7, p=0.52).
In a subset of CNO-sensitive neurons (n=10), the CRF1 antagonist R121919 (1 μM) was applied with CNO to determine whether the effect of CNO on sIPSC frequency was in part mediated by CRF1 activation (see Fig. 3A for schematic representation of tested hypothesis). There was a significant main effect of treatment (Fig. 3H, F2,18=6.1, p=0.01) reflecting the increase in sIPSC frequency induced by CNO and partial reversal of the effect of CNO by R121919. Posthoc analysis revealed a significant difference between baseline and CNO (p=0.008). Furthermore, R121919 exerted a stronger inhibition in cells with higher sIPSC frequency, as indicated by a significant correlation between the reversal effect of R121919 (expressed as frequency change after addition of R121919) and baseline frequency (Fig. 3I, r=−0.90, p<0.001).
Altogether, these data indicate that chemogenetic stimulation of mouse CeA CRF neurons increases GABA release onto local medial CeA neurons and that this effect is at least partially mediated by CRF1 activation.
Stimulation of CeA CRF neurons does not affect alcohol drinking
These results, combined with the fact that CRF1 signaling in the CeA drives excessive alcohol intake in rodent models of binge-like drinking and dependence16–18, led us to hypothesize that chemogenetic stimulation of CeA CRF neurons (Fig. 4A) may promote voluntary alcohol consumption. To test this hypothesis, the effect of CNO on ethanol intake during 2-h ethanol (15% v:v)/water two-bottle choice (2BC) drinking sessions was tested under different conditions (see Fig. 4B for experimental timeline).
Figure 4. Chemogenetic stimulation of CeA CRF neurons does not affect alcohol drinking.
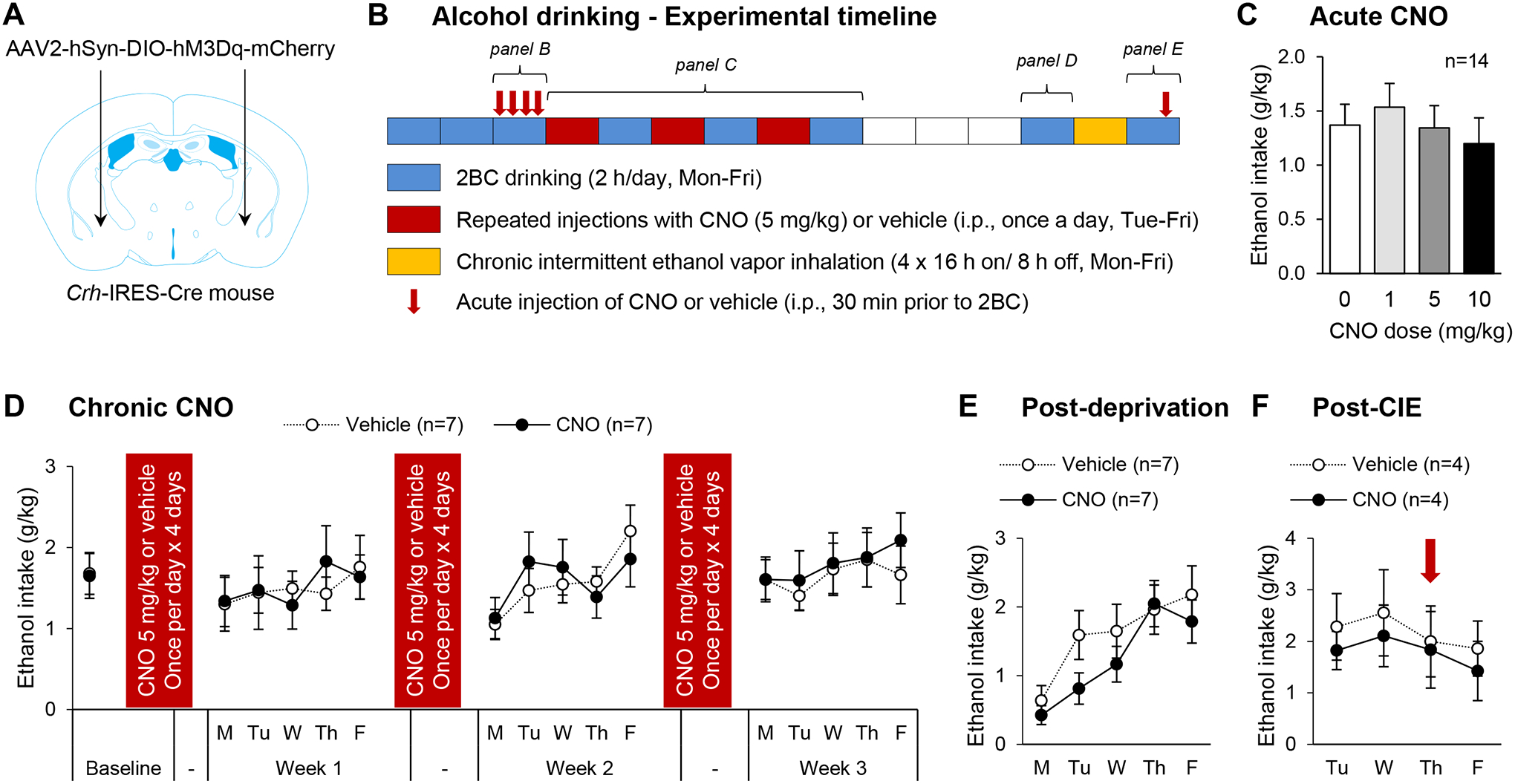
A. Crh-IRES-Cre male mice were injected with a Cre-dependent hM3Dq-encoding vector in the anterior CeA and were tested for voluntary ethanol intake under different experimental conditions. B. Experimental timeline. Each box represents one week, the color code indicates the experimental procedure conducted during that week. Two-bottle choice (2BC) drinking sessions were conducted Mon-Fri, starting at the beginning of the dark phase and lasting 2 h (blue boxes). Mice were given ten baselining sessions prior to testing the acute effect of CNO (0, 1, 5 and 10 mg/kg, i.p., 30-min pretreatment) according to a within-subject Latin-square design over four consecutive days (red arrows; data shown in panel C). An additional 2BC session without pretreatment was conducted and mice were then split in two groups exhibiting equivalent baseline ethanol intake, which were repeatedly injected with either CNO (5 mg/kg) or vehicle. Weeks of CNO or vehicle administration (once per day, Tue-Fri; red boxes) were alternated with weeks of 2BC drinking sessions (Mon-Fri, as described above; blue boxes; data shown in panel D) for a total of 3 rounds. Mice were then given a 3-week ethanol deprivation period (white boxes), after which 2BC sessions were resumed for a week (blue box; data shown in panel E). Next, the mice were exposed to four cycles of chronic intermittent ethanol exposure (16-h ethanol vapor inhalation followed by 8-h air inhalation, Mon-Fri; yellow box). The mice were then returned to their home cages and 2BC sessions resumed four days later (Tue-Fri; blue box; data shown in panel F). On the third session (Thu), CNO (5 mg/kg) or vehicle was administered 30 min prior to the session (red arrow). C-F. Ethanol intake is expressed in g ethanol per kg body weight in 2-h session. Data are shown as mean ± s.e.m. Number of mice per group is shown in the legend of each graph.
Acute CNO (1, 5 and 10 mg/kg) administration (30 min prior to drinking session) did not affect ethanol intake (Fig. 4C, F3,39=1.6, p=0.21). We then attempted to replicate the pattern of CRF release in the CeA elicited by chronic intermittent ethanol (CIE) exposure. In the CIE-2BC model, weeks of 2BC (Mon-Fri) are alternated with weeks of CIE consisting of four 16-h periods of ethanol vapor inhalation separated by 8-h periods of air inhalation (Mon-Fri)4. Extracellular CRF levels are known to gradually increase in the amygdala of rats withdrawn from chronic alcohol exposure starting about 6 h after the onset of withdrawal13. Accordingly, to replicate the predicted pattern of CRF release experienced by CIE-exposed mice, we injected CNO (5 mg/kg) once per day (Tue-Fri) and conducted 2BC sessions on alternate weeks (Fig. 4B). In the CIE-2BC model, CIE-exposed mice typically start increasing their voluntary ethanol intake after 1–3 weeks of CIE exposure4, 40. However, alternating weeks of repeated CNO administration with weeks of 2BC for three rounds did not affect ethanol intake compared to mice repeatedly injected with vehicle (Fig. 4D). A two-way ANOVA of weekly average intakes revealed no effect of time (F3,36=1.4, p=0.26), no effect of treatment (F1,36=0.01, p=0.91) and no time × treatment interaction (F3,36=0.3, p=0.86). We examined whether repeated CNO administration may produce a delayed effect but there was again no effect of treatment on ethanol intake following 3 weeks of deprivation (Fig. 4E, t12=−1.0, p=0.33). Finally, we exposed the mice to a single week of CIE, which was not sufficient to alter ethanol intake in subsequent 2BC sessions (Tue-Fri). We then tested the ability of acute CNO (5 mg/kg) administration to sensitize the mice and increase their post-vapor ethanol intake. There was again no significant effect of CNO (Fig. 4F, t6=−0.2, p=0.87).
In conclusion, chemogenetic stimulation of mouse CeA CRF neurons is not sufficient to increase ethanol drinking following acute or chronic administration, or in combination with CIE exposure.
Stimulation of CeA CRF neurons exacerbates hyponeophagia
We then examined whether chemogenetic stimulation of CeA CRF neurons (Fig. 5A) may elicit anxiety-like behavior. CNO (1 or 5 mg/kg) did not affect locomotor activity in a familiar environment (Fig. 5B, F2,16=0.9, p=0.44). The dose of 5 mg/kg was used in all other tests. In the digging assay (Fig. 5C), CNO tended to increase the total duration of digging (t10=−2.0, p=0.07), but had no effect on marble burying (t10=−0.5, p=0.62). In the EPM (Fig. 5D), CNO had no effect on the total distance traveled (t10=0.1, p=0.94), the number of entries on open arms (t10=0.4, p=0.73) or the time spent on open arms (t10=−1.5, p=0.16). In the social approach test (Fig. 5E), there was a significant main effect of compartment (F2,24=29.9, p<0.0001), reflecting a preference for the chamber containing a stranger mouse compared to both the center and empty cup compartments (p<0.0001 for both posthoc comparisons). CNO did not alter this preference, as reflected by a lack of treatment effect (F1,12=1.5, p=0.24) and compartment × treatment interaction (F2,24=0.2, p=0.80). There was also no effect of CNO on the number of transitions between compartments (t12=0.6, p=0.56), in agreement with the lack of effect of CNO on locomotor activity.
Figure 5. Chemogenetic stimulation of CeA CRF neurons exacerbates hyponeophagia without altering other affective responses nor appetite.
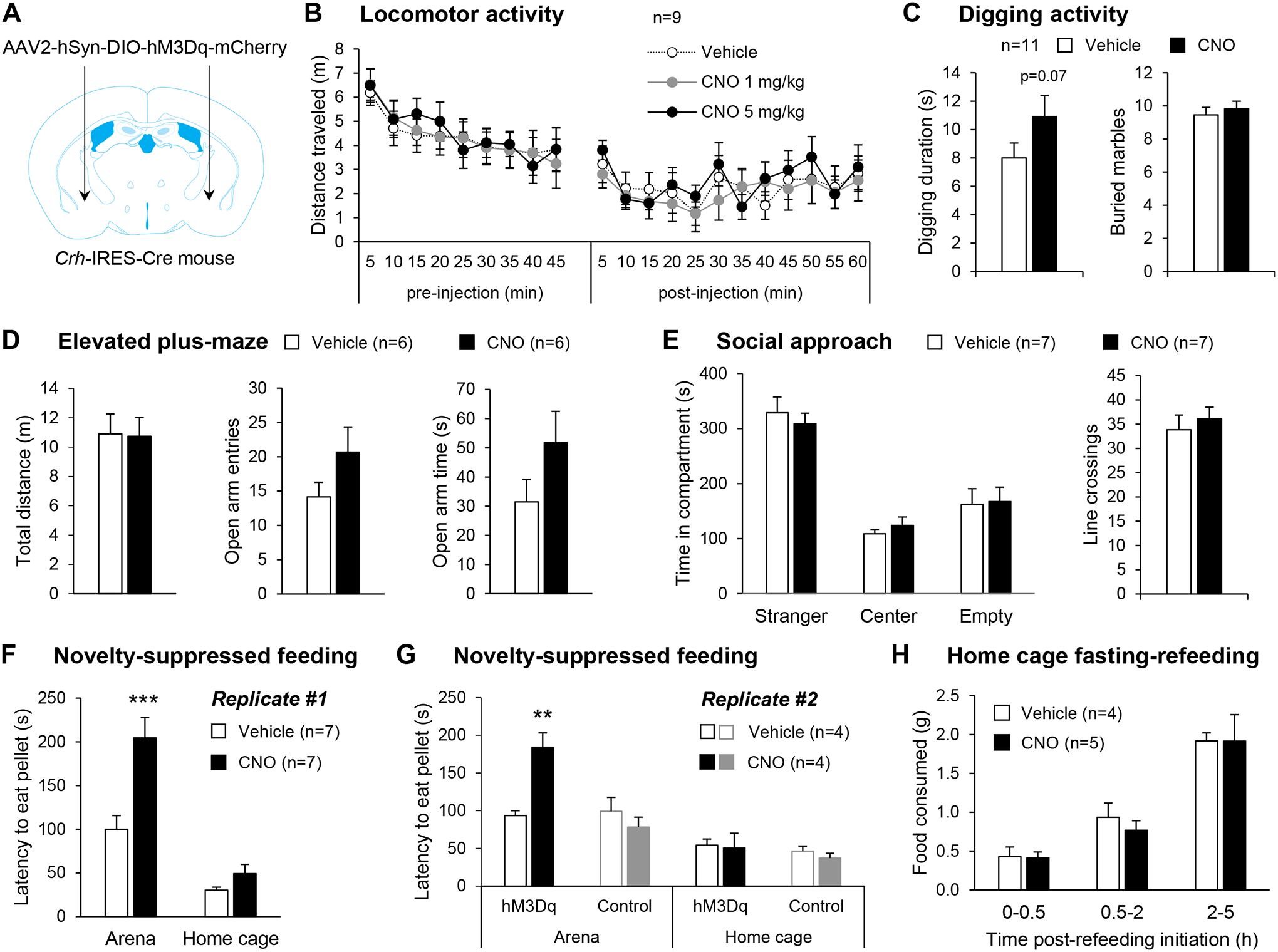
A. Crh-IRES-Cre male mice were injected with a Cre-dependent hM3Dq-encoding vector in the anterior CeA. B. Locomotor activity 45 min prior and 60 min following i.p. injection of vehicle or CNO. C-H. Mice were tested in assays probing affective behavior 30 min after i.p. injection of saline or CNO (5 mg/kg). C. CNO tended to increase digging duration but did not affect marble burying. D. CNO did not affect open arm exploration in the elevated plus maze. E. CNO did not affect the preference for social interaction. F-G. CNO increased the latency to start feeding in an anxiogenic arena, but not in the home cage. This effect of CNO was replicated in an independent cohort and was not observed in control mice with no hM3Dq expression. **, p<0.01; ***, p<0.001, Tukey posthoc test. H. CNO did not alter the amount of food consumed following 24-h food deprivation. Data are shown as mean ± s.e.m. Number of mice per group is shown in the legend of each graph.
In the NSF test (Fig. 5F), it took longer for the food-deprived mice to start eating in the arena than in their home cage (main effect of environment: F1,12=57.1, p<0.0001) and CNO selectively prolonged the arena latency (environment × treatment interaction: F1,12=8.3, p=0.01; posthoc comparison Arena-Vehicle vs Arena-CNO: p=0.0006; Home cage-Vehicle vs Home cage-CNO: p=1.00). We replicated this finding in an independent cohort, which also included control mice (Cre-dependent tdTomato expression, no hM3Dq). In this second replicate (Fig. 5G), CNO again increased the latency to start eating the pellet in the arena but not the home cage in hM3Dq-expressing mice (environment × treatment interaction: F1,6=6.9, p=0.04; posthoc comparison Arena-Vehicle vs Arena-CNO: p=0.007; Home cage-Vehicle vs Home cage-CNO: p=1.00), and it had no effect in Control mice (main effect of environment: F1,6=32.1, p=0.001; main effect of treatment: F1,6=1.0, p=0.36; environment × treatment interaction: F1,6=0.5, p=0.50). To verify that the anxiogenic-like effect of CNO in the NSF test was not related to reduced appetite, we further examined the effect of CNO on feeding in a home cage setting. CNO had no effect on food consumption after 24-h deprivation at any of the time points examined (Fig. 5H, F1,7=0.09, p=0.77).
In summary, chemogenetic stimulation of mouse CeA CRF neurons promotes hyponeophagia without altering appetite. The anxiogenic-like effect of this manipulation is not detected in other assays of anxiety-like behavior.
Chemogenetic inhibition of CeA CRF neurons does not reverse alcohol dependence phenotypes
Although stimulating CeA CRF neurons was not sufficient to increase alcohol drinking, this population might still be necessary to ethanol intake escalation in the CIE-2BC model. We employed chemogenetic inhibition to test this hypothesis. Crh-IRES-Cre mice were injected in the CeA with a Cre-dependent vector encoding hM4Di-mCherry (Fig. 6A). CNO reduced the frequency of events recorded in loose cell attached configuration selectively in hM4Di-expressing cells, thereby validating our approach (Fig. 6B, mCherry+: t5=−6.3, p=0.003; mCherry-: t4=0.5, p=0.68). The effect of CNO on CIE-induced behaviors was then tested (see Fig. 6C for experimental timeline). As expected, withdrawal from CIE increased voluntary alcohol consumption (Fig. 6D, main effect of CIE: F1,18=18.2, p<0.001), digging activity (Fig. 6E, F1,16=4.1, p=0.06), and hyponeophagia (Fig. 6F, F1,14=9.6, p=0.008). However, there was no significant effect of CNO (ethanol intake: F2,36=0.8, p=0.46; digging duration: F1,16=2.4, p=0.14; arena latency: F1,16=0.001, p=0.97) or CIE × CNO interaction (ethanol intake: F2,36=1.7, p=0.19; digging duration: F1,16=0.8, p=0.39; arena latency: F1,16=1.0, p=0.35) on either measure.
Figure 6. Chemogenetic inhibition of CeA CRF neurons does not reverse ethanol intake escalation or affective disturbance in alcohol-dependent mice.
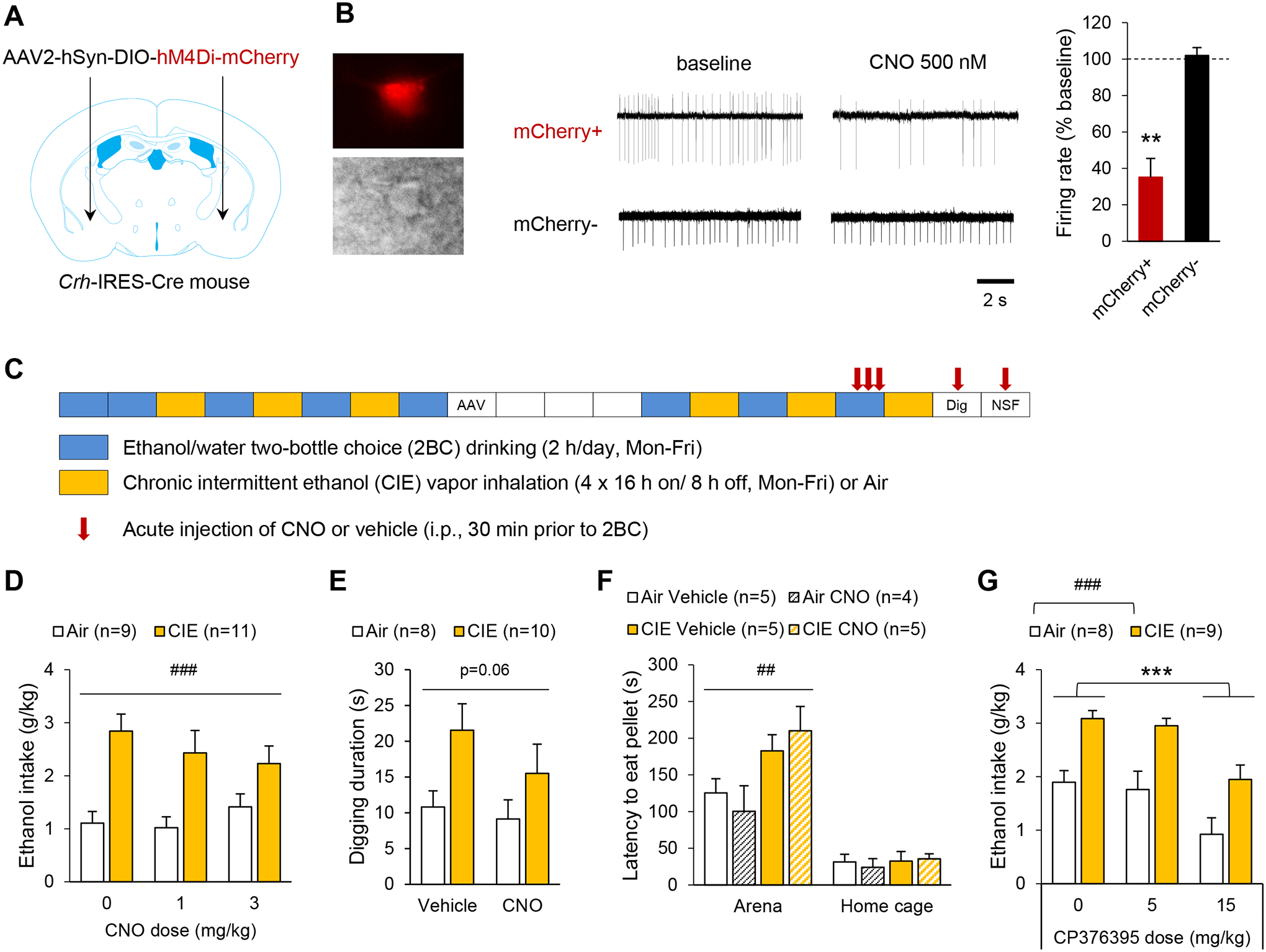
A-F. Crh-IRES-Cre male mice were injected with a Cre-dependent hM4Di-encoding vector in the anterior CeA. B. Firing rates were recorded from CeA mCherry+ and mCherry− neurons. Left: red fluorescence and differential interference contrast images of patched CeA neuron. Middle: representative traces before and during CNO application (500 nM). Right: CNO-induced change in firing rate (mCherry+, n=5; mCherry-, n=4). **, p<0.01, one-sample t-test. C. Experimental timeline for behavioral testing. Each box represents one week. AAV designates the timepoint at which the viral vector was injected, mice recovered for 3 weeks before resuming alcohol drinking sessions. The digging test (Dig) was performed 7 and 10 days after last vapor exposure (within-subject design). The novelty-suppressed feeding (NSF) test was conducted 14 days after last vapor exposure (between-subject design). D-F. Exposure to CIE increased ethanol intake (D), digging activity (E), and hyponeophagia (F). Acute CNO administration had no significant effect on these measures. Data were analyzed by two-way ANOVA (main effect of CIE: ##, p<0.01; ###, p<0.001). G. C57BL/6J mice subjected to the CIE-2BC procedure were injected with the CRF1 antagonist CP376395 30 min prior to 2BC. CP376395 reduced ethanol intake in both Air and CIE mice. Data were analyzed by two-way repeated-measures ANOVA (###, p<0.001, main effect of CIE; ***, p<0.001, Dunnett’s posthoc test). Data are shown as mean ± s.e.m. Number of mice per group is shown in the legend of each graph.
Given the lack of effect of chemogenetic stimulation or inhibition of CeA CRF neurons, we sought to verify the involvement of CRF1 signaling in the control of alcohol drinking under our experimental conditions. As expected, systemic administration of the CRF1 antagonist CP376395 dose-dependently reduced ethanol intake during 2-h 2BC sessions in both air- and CIE-exposed C57BL/6J mice (Fig, 6G; main effect of dose: F2,30=20.5, p=0.000002; main effect of CIE: F1,30=17.3, p=0.0008; dose × CIE interaction: F2,30=0.17, p=0.85). Posthoc analysis indicated a significant effect of the 15 mg/kg dose (p=0.00001), but not 5 mg/kg (p=0.69).
In conclusion, chemogenetic inhibition of mouse CeA CRF neurons does not reverse behavioral hallmarks of alcohol dependence in mice, and it does not reduce alcohol drinking or hyponeophagia in moderate drinkers despite pharmacological evidence of a significant role of CRF1 signaling in alcohol intake.
Discussion
Our study first shows that chronic binge-like drinking and repeated alcohol vapor inhalation both elevate Crh expression at the most anterior level of the mouse CeA, suggesting that anterior CeA CRF neurons may be functionally involved in the behavioral consequences of chronic intermittent alcohol exposure. However, these neurons are neither necessary nor sufficient to drive ethanol intake escalation in mice. Their chemogenetic stimulation increases GABA release onto medial CeA neurons and exacerbates hyponeophagia but does not affect voluntary alcohol consumption. Furthermore, their chemogenetic inhibition does not alter ethanol intake or hyponeophagia in alcohol-dependent or non-dependent mice. The latter finding is at odds with the effect of optogenetic manipulation in rats32, which highlights species differences in the function of CeA CRF circuits. The reduction in alcohol intake elicited by CP376395 is consistent with the effects of systemic CRF1 blockade on binge-like drinking previously reported in mice and confirms that CRF1 signaling promotes alcohol consumption under our experimental conditions12, 18, 41–43.
While multiple studies had previously demonstrated Crh upregulation in the CeA of animals withdrawn from chronic alcohol exposure19, 21, 22, 44, most of them did not consider anteroposterior level in their analysis. In rats withdrawn from CIE for 2 h, CRF immunoreactivity selectively increased in the most posterior area of the CeA17. In contrast, we found that the impact of chronic binge drinking on Crh expression was specific to the most anterior level of the CeA – the area that shows the highest density of CRF-producing neurons in mice while most CRF-expressing neurons are located at middle and posterior levels in rats37. Furthermore, CIE also increased Crh expression at this level and the effect persisted after 72 h withdrawal. These observations indicate that the anatomical organization of the CeA CRF system as it relates to alcohol sensitivity differs between rats and mice.
The penetrance of Cre activity in CeA CRF neurons was lower in Crh-IRES-Cre mice (64%) than in Crh-Cre rats (99%), which may also explain differential experimental outcomes in the two species. However, our quantification by in situ hybridization may have been less sensitive than the method used in rats (immunohistochemistry, confocal imaging at 63x and 3D reconstruction)29. Previous characterization of the distribution of Cre activity in the amygdala of Crh-IRES-Cre;Ai14 mice by immunohistochemistry concluded that the pattern “largely recapitulated” the distribution of CRF, although no quantification was presented45. We found that the fidelity of Cre activity for CeA CRF neurons was reasonably high (86% - which may again be an underestimate), confirming that the cellular and behavioral effects of CNO reported in the present study result, for the most part, from the activation of CeA CRF neurons.
There was no effect of CeA CRF neuron stimulation on the overall activity of other CeA neurons (as indexed by c-Fos expression in mCherry-negative CeA neurons). This observation contrasts with the effect of the same manipulation in Crh-Cre rats, where robust c-Fos induction was also detected in non-CRF neurons of the medial and lateral CeA and was mediated by CRF1 activation29. This discrepancy could relate to the differential anatomy of the CRF system, with the dense cluster of CRF neurons at the junction of the IPAC and anterior CeA being unique to mice and possibly exerting a stronger inhibitory control over the remainder of the CeA than the more posterior CRF neurons. Consistent with this hypothesis, the connectivity of CeA CRF neurons is species-specific, as local synapses are predominant in mice46, while long-range projections to the BNST, lateral hypothalamus and parabrachial nucleus are robust in rats30, 32. Alternatively, the discrepancy may stem from a differential excitatory/inhibitory signaling balance between the two species, whereby a higher ratio of GABA to CRF may be released in mice compared to rats, inhibitory neuromodulators may be co-released to a larger extent in mice, or inhibitory signaling may override excitatory signaling via mouse-specific postsynaptic mechanisms. Of note, the GABA release induced by CNO likely reduced the activity of neurons directly contacted by hM3Dq-expressing neurons but c-Fos immunolabeling is not well suited to detect such inhibition owing to low baseline expression of this immediate early gene.
Chemogenetic stimulation of CeA CRF neurons triggered GABA release onto a subset of medial CeA neurons, as reflected by increased sIPSC frequency in response to CNO. This result is consistent with the ability of optogenetic stimulation of CeA CRF terminals to evoke IPSCs in a subset of CeA neurons in Crh-Cre rats and mice29, 46. The CNO insensitivity of some medial CeA mCherry-negative ties in with the notion that all medial CeA cells may not be contacted by CRF (i.e., hM3Dq-expressing) neurons. Combined with the lack of activation of mCherry-negative CeA cell bodies we observed with c-Fos labeling, our data indicate that mouse CeA CRF neurons release GABA from their own terminals (given that CeA CRF neurons are GABAergic47) or possibly stimulate GABA release from extrinsic afferents, but not from local non-CRF interneurons as may be the case in rats (see discussion above). The ability of R121919 to reverse this increase in GABA release in cells with high baseline release is consistent with previous work showing that exogenous CRF acts at presynaptic CRF1 receptors to increase GABA release in the mouse CeA39. We chose to focus on inhibitory transmission as it provides a consistent readout of CRF1 signaling in both the rat and mouse CeA, while CRF effects on excitatory transmission are more complex, at least in rats48–50. Relevant to the present study, although ethanol can increase glutamate release in the mouse and rat CeA and this action requires activation of CRF receptors in both species, CeA CRF neurons do not mediate this effect in the mouse50, 51.
The effect of chemogenetic stimulation of CeA CRF neurons on anxiety-like behavior appears to be species- and assay-dependent. While this manipulation did not affect mouse EPM behavior in the present study, it reduces open arm exploration in rats30, 31. Previous studies in Crh-IRES-Cre mice also showed that chemogenetic activation of CeA CRF neurons increases immobility in the EPM (although this may not necessarily translate into reduced time spent on open arms) and optogenetic activation of the CeA CRF projection to the locus coeruleus reduced exploration of open sections of an elevated zero-maze52, 53. We therefore anticipated that activating CeA CRF neurons would reduce open arm exploration in the EPM, but it had no effect. It is possible that other projections of CeA CRF neurons oppose the anxiogenic-like effect of the coerulear projection, resulting in a lack of EPM phenotype in the present study.
It is important to note, however, that mice and rats can manifest negative affect in different ways, even when undergoing a similar aversive experience. For instance, while it has been repeatedly shown that rats withdrawn from chronic alcohol exposure exhibit anxiety-like behavior in the EPM54–61, a similar phenotype has been difficult to detect in mice using the same assay62. However, negative affect can be captured in alcohol-withdrawn mice using alternative assays. Most relevant to the present study, C57BL/6J males display increased digging activity and exacerbated hyponeophagia, but unaltered light-dark box exploration and social interaction, when tested 3–10 days into withdrawal from CIE8, 63–67. The latter data are consistent with our observation that chemogenetic stimulation of CeA CRF neurons in mice exacerbates hyponeophagia and tends to increase digging activity but has no effect in the EPM and social approach tests. However, chemogenetic inhibition of CeA CRF neurons failed to reverse the exacerbated digging and hyponeophagia of CIE-withdrawn mice, discounting a functional implication of these neurons in withdrawal-induced negative affect. Altogether, our results indicate that mouse CeA CRF neurons have the capacity to drive hyponeophagia but do not contribute to the hyponeophagic phenotype associated with CIE withdrawal. Based on the molecular dissection of the anxiety-like behavior driven by CeA CRF neurons in rats, the NSF phenotype we observed in alcohol-naïve mice may result from the combined release of CRF and dynorphin31. Furthermore, although our electrophysiological results demonstrated local GABA and CRF release upon activation of CeA CRF neurons, it is possible that the effect of CNO in the NSF assay is driven by CeA CRF neurons projecting outside of the CeA. There is, to the best of our knowledge, no experimental approach currently available to target CRF neurons intrinsic to the CeA while sparing CRF neurons projecting to other regions.
We had hypothesized that CeA CRF neurons would not only drive negative affect but also increase alcohol drinking by mimicking negative reinforcement-driven escalation of alcohol self-administration elicited by CIE68. However, neither acute nor chronic chemogenetic stimulation of CeA CRF neurons altered alcohol intake in mice given limited access to alcohol. Furthermore, chemogenetic inhibition failed to reduce ethanol intake in alcohol-dependent or non-dependent mice. This observation contrasts with the ability of optogenetic inhibition of the same neuronal population to selectively reduce escalated alcohol self-administration in CIE-exposed rats32. This discrepancy demonstrates a differential involvement of CeA CRF neurons in the control of ethanol intake in mice vs. rats. Our results, combined with the prior demonstration that intra-CeA CRF1 antagonism reduces alcohol consumption in binge drinking and CIE-exposed mice16, 18, indicate that an extrinsic source of CRF to the CeA promotes alcohol drinking in mice. Future work will aim to elucidate the identity of behaviorally relevant CRF-containing projections to the CeA and their recruitment by CIE exposure.
A recent study demonstrated that a subset of CeA CRF neurons is activated prior to alcohol licks in binge-drinking mice33. The inability of chemogenetic inhibition to reduce the alcohol intake of Air or CIE mice indicates that these neurons may encode the anticipation of drinking but are not required for moderate or excessive consumption. Alternatively, there may be another subset of CeA CRF neurons exerting an opposing influence on alcohol consumption, such that concomitant inhibition of both subsets in our study may have neutralized the role of the pre-lick activated subset.
Aside from fear learning and anxiety, CeA neurons have also been implicated in feeding control. Specifically, lateral CeA PKCδ-positive neurons suppress food intake69. In the NSF test, we found that chemogenetic stimulation of CeA CRF neurons increased the latency of food-deprived mice to start eating in an anxiogenic arena but did not affect their home cage feeding. This observation is consistent with the minimal overlap between mouse CeA populations expressing PKCδ versus CRF46, 70. Interestingly, CRF suppresses feeding when administered in the brain and CRF signaling mediates the anorexigenic effect of several stressors (see71 for review). While CeA CRF neurons can be recruited by stress (72, 73, but see74), our findings indicate that their activity is not sufficient to inhibit feeding.
A general limitation of our study is that mice from each cohort were subjected to multiple behavioral tests and CNO injections. We cannot rule out that potential effects of CNO may have been occluded by prior testing, despite the 1-week interval between tests. However, this concern is mitigated by the robust effect of CNO on hyponeophagia in hM3Dq-expressing mice, which was observed after multiple injections. It is also possible that significant effects of CNO would have been detected in hM4Di-expressing CIE-withdrawn mice if we had used a higher dose of CNO.
Altogether, our results indicate that excitation of CeA CRF neurons is sufficient to produce negative affect but not to increase the motivation to consume alcohol in mice. Furthermore, although CeA neurons are known to be hyperactive during CIE withdrawal in both rats and mice32, 75–77, we found that the CRF-expressing subset does not contribute to the behavioral phenotypes of CIE withdrawal in mice. Their lack of involvement in CIE-induced ethanol intake escalation contrasts with their role in rats and indicates that observations made in one species may not translate to another species. This consideration is relevant to the targeting of CRF signaling for the treatment of alcohol use disorders in humans, as the differential engagement of central amygdala CRF neurons in the control of alcohol consumption in two phylogenetically close species (rats and mice) likely extends to a more distantly related species (humans) and provides a possible explanation for the disappointing results of clinical trials that were motivated in large part by the promises of preclinical studies conducted in rats78, 79. Relevant to the latter point, available evidence points to substantial discrepancies in the anatomical localization and molecular identity of amygdala CRF neurons between humans and rodents. Notably, in the human amygdala, CRH transcripts are enriched in a subpopulation of inhibitory neurons that expresses high levels of CALB2 (calretinin), CCK, and VIP transcripts80. Both CRF- and calretinin-immunoreactive cells are scarce in the human central nucleus81, 82. Moreover, CCK/Cck and VIP/Vip do not co-localize with CRF/Crh in the rodent amygdala37, 83, 84.
Altogether, our findings, combined with the ability of intra-CeA CRF1 antagonism to reduce ethanol intake in C57BL/6J mice, suggest that CRF acting in the mouse CeA to promote alcohol drinking arises from an extrinsic source16, 18. Future work will be needed to identify other mechanisms contributing to alcohol drinking escalation in CIE-exposed mice, including additional neural circuits releasing CRF in the CeA, additional projection targets of CeA CRF neurons, as well as non-CRF signaling pathways that may potentiate the influence of CRF transmission on alcohol drinking.
Supplementary Material
Acknowledgments
We wish to thank Sophia Zhu, Tanvi Shah, and Catherine Lopez for their assistance with brain slicing and immunohistochemistry, and Dr. Michal Bajo for his assistance with electrophysiological recordings. We are also grateful for the support of TSRI Alcohol Research Center Animal Models Core, which conducted blood ethanol concentration analysis for this study.
Funding
This work was supported by the following grants from the National Institutes of Health: AA024198 (CC), AA026685 (CC), AA027636 (CC), AA027372 (CC), AA006420 (CC and MR), AA021491 (MR), AA015566 (MR), AA023002 (MAH), and AA024952 (HS), as well as stipends from University of Benin, Benin City, Nigeria (AO) and Kingsefe Pharmacy, Benin City, Nigeria (AO). These funding sources were not involved in study design, data collection, analysis, interpretation, or decision to publish.
Footnotes
CRediT authorship contribution statement
Max Kreifeldt: Investigation, Methodology, Validation. Melissa A Herman: Conceptualization, Investigation, Formal analysis, Writing – review & editing. Harpreet Sidhu: Investigation, Methodology. Agbonlahor Okhuarobo: Investigation, Validation. Giovana C Macedo: Validation. Roxana Shahryari: Validation. Pauravi J Gandhi: Validation. Marisa Roberto: Conceptualization, Writing – review & editing. Candice Contet: Conceptualization, Funding acquisition, Investigation, Validation, Formal analysis, Supervision, Writing – original draft.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.National Survey on Drug Use and Health: Substance Abuse and Mental Health Services Administration; 2015. [PubMed]
- 2.Collaborators GBDA. Alcohol use and burden for 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 2018; 392(10152): 1015–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koob GF, Le Moal M. Addiction and the brain antireward system. Annual review of psychology 2008; 59: 29–53. [DOI] [PubMed] [Google Scholar]
- 4.Becker HC, Lopez MF. Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res 2004; 28(12): 1829–1838. [DOI] [PubMed] [Google Scholar]
- 5.Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiology & behavior 2005; 84(1): 53–63. [DOI] [PubMed] [Google Scholar]
- 6.Gilpin NW, Richardson HN, Cole M, Koob GF. Vapor inhalation of alcohol in rats. Current protocols in neuroscience / editorial board, Jacqueline N Crawley [et al] 2008; Chapter 9: Unit 9 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holleran KM, Winder DG. Preclinical voluntary drinking models for alcohol abstinence-induced affective disturbances in mice. Genes, brain, and behavior 2017; 16(1): 8–14. [DOI] [PubMed] [Google Scholar]
- 8.Sidhu H, Kreifeldt M, Contet C. Affective Disturbances During Withdrawal from Chronic Intermittent Ethanol Inhalation in C57BL/6J and DBA/2J Male Mice. Alcohol Clin Exp Res 2018; 42(7): 1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends in neurosciences 2007; 30(8): 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koob GF. The role of CRF and CRF-related peptides in the dark side of addiction. Brain research 2010; 1314: 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menzaghi F, Rassnick S, Heinrichs S, Baldwin H, Pich EM, Weiss F et al. The role of corticotropin-releasing factor in the anxiogenic effects of ethanol withdrawal. Annals of the New York Academy of Sciences 1994; 739: 176–184. [DOI] [PubMed] [Google Scholar]
- 12.Chu K, Koob GF, Cole M, Zorrilla EP, Roberts AJ. Dependence-induced increases in ethanol self-administration in mice are blocked by the CRF1 receptor antagonist antalarmin and by CRF1 receptor knockout. Pharmacology, biochemistry, and behavior 2007; 86(4): 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF et al. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci 1995; 15(8): 5439–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rassnick S, Heinrichs SC, Britton KT, Koob GF. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain research 1993; 605(1): 25–32. [DOI] [PubMed] [Google Scholar]
- 15.Huang MM, Overstreet DH, Knapp DJ, Angel R, Wills TA, Navarro M et al. Corticotropin-releasing factor (CRF) sensitization of ethanol withdrawal-induced anxiety-like behavior is brain site specific and mediated by CRF-1 receptors: relation to stress-induced sensitization. The Journal of pharmacology and experimental therapeutics 2010; 332(1): 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finn DA, Snelling C, Fretwell AM, Tanchuck MA, Underwood L, Cole M et al. Increased drinking during withdrawal from intermittent ethanol exposure is blocked by the CRF receptor antagonist D-Phe-CRF(12–41). Alcohol Clin Exp Res 2007; 31(6): 939–949. [DOI] [PubMed] [Google Scholar]
- 17.Funk CK, O’Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci 2006; 26(44): 11324–11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowery-Gionta EG, Navarro M, Li C, Pleil KE, Rinker JA, Cox BR et al. Corticotropin releasing factor signaling in the central amygdala is recruited during binge-like ethanol consumption in C57BL/6J mice. J Neurosci 2012; 32(10): 3405–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M et al. Corticotropin releasing factor-induced amygdala gamma-aminobutyric Acid release plays a key role in alcohol dependence. Biol Psychiatry 2010; 67(9): 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broccoli L, Uhrig S, von Jonquieres G, Schonig K, Bartsch D, Justice NJ et al. Targeted overexpression of CRH receptor subtype 1 in central amygdala neurons: effect on alcohol-seeking behavior. Psychopharmacology (Berl) 2018; 235(6): 1821–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lack AK, Floyd DW, McCool BA. Chronic ethanol ingestion modulates proanxiety factors expressed in rat central amygdala. Alcohol 2005; 36(2): 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sommer WH, Rimondini R, Hansson AC, Hipskind PA, Gehlert DR, Barr CS et al. Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala crhr1 expression following a history of dependence. Biol Psychiatry 2008; 63(2): 139–145. [DOI] [PubMed] [Google Scholar]
- 23.Albeck DS, McKittrick CR, Blanchard DC, Blanchard RJ, Nikulina J, McEwen BS et al. Chronic social stress alters levels of corticotropin-releasing factor and arginine vasopressin mRNA in rat brain. J Neurosci 1997; 17(12): 4895–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makino S, Shibasaki T, Yamauchi N, Nishioka T, Mimoto T, Wakabayashi I et al. Psychological stress increased corticotropin-releasing hormone mRNA and content in the central nucleus of the amygdala but not in the hypothalamic paraventricular nucleus in the rat. Brain research 1999; 850(1–2): 136–143. [DOI] [PubMed] [Google Scholar]
- 25.Boutros N, Der-Avakian A, Kesby JP, Lee S, Markou A, Semenova S. Effects of adolescent alcohol exposure on stress-induced reward deficits, brain CRF, monoamines and glutamate in adult rats. Psychopharmacology (Berl) 2018; 235(3): 737–747. [DOI] [PubMed] [Google Scholar]
- 26.Regev L, Tsoory M, Gil S, Chen A. Site-specific genetic manipulation of amygdala corticotropin-releasing factor reveals its imperative role in mediating behavioral response to challenge. Biol Psychiatry 2012; 71(4): 317–326. [DOI] [PubMed] [Google Scholar]
- 27.Flandreau EI, Ressler KJ, Owens MJ, Nemeroff CB. Chronic overexpression of corticotropin-releasing factor from the central amygdala produces HPA axis hyperactivity and behavioral anxiety associated with gene-expression changes in the hippocampus and paraventricular nucleus of the hypothalamus. Psychoneuroendocrinology 2012; 37(1): 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herman MA, Contet C, Justice NJ, Vale W, Roberto M. Novel subunit-specific tonic GABA currents and differential effects of ethanol in the central amygdala of CRF receptor-1 reporter mice. J Neurosci 2013; 33(8): 3284–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pomrenze MB, Millan EZ, Hopf FW, Keiflin R, Maiya R, Blasio A et al. A Transgenic Rat for Investigating the Anatomy and Function of Corticotrophin Releasing Factor Circuits. Frontiers in neuroscience 2015; 9: 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pomrenze MB, Tovar-Diaz J, Blasio A, Maiya R, Giovanetti SM, Lei K et al. A Corticotropin Releasing Factor Network in the Extended Amygdala for Anxiety. J Neurosci 2019; 39(6): 1030–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pomrenze MB, Giovanetti SM, Maiya R, Gordon AG, Kreeger LJ, Messing RO. Dissecting the Roles of GABA and Neuropeptides from Rat Central Amygdala CRF Neurons in Anxiety and Fear Learning. Cell reports 2019; 29(1): 13–21 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Guglielmo G, Kallupi M, Pomrenze MB, Crawford E, Simpson S, Schweitzer P et al. Inactivation of a CRF-dependent amygdalofugal pathway reverses addiction-like behaviors in alcohol-dependent rats. Nat Commun 2019; 10(1): 1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aroni S, Marino RAM, Girven KS, Irving JM, Cheer JF, Sparta DR. Repeated binge ethanol drinking enhances electrical activity of central amygdala corticotropin releasing factor neurons in vivo. Neuropharmacology 2021; 189: 108527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K et al. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 2011; 71(6): 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 2007; 104(12): 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 2011; 121(4): 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asan E, Yilmazer-Hanke DM, Eliava M, Hantsch M, Lesch KP, Schmitt A. The corticotropin-releasing factor (CRF)-system and monoaminergic afferents in the central amygdala: investigations in different mouse strains and comparison with the rat. Neuroscience 2005; 131(4): 953–967. [DOI] [PubMed] [Google Scholar]
- 38.De Francesco PN, Valdivia S, Cabral A, Reynaldo M, Raingo J, Sakata I et al. Neuroanatomical and functional characterization of CRF neurons of the amygdala using a novel transgenic mouse model. Neuroscience 2015; 289: 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nie Z, Zorrilla EP, Madamba SG, Rice KC, Roberto M, Siggins GR. Presynaptic CRF1 receptors mediate the ethanol enhancement of GABAergic transmission in the mouse central amygdala. TheScientificWorldJournal 2009; 9: 68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kreifeldt M, Le D, Treistman SN, Koob GF, Contet C. BK channel beta1 and beta4 auxiliary subunits exert opposite influences on escalated ethanol drinking in dependent mice. Front Integr Neurosci 2013; 7: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giardino WJ, Ryabinin AE. CRF1 receptor signaling regulates food and fluid intake in the drinking-in-the-dark model of binge alcohol consumption. Alcohol Clin Exp Res 2013; 37(7): 1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sparta DR, Sparrow AM, Lowery EG, Fee JR, Knapp DJ, Thiele TE. Blockade of the corticotropin releasing factor type 1 receptor attenuates elevated ethanol drinking associated with drinking in the dark procedures. Alcohol Clin Exp Res 2008; 32(2): 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hwa LS, Shimamoto A, Kayyali T, Norman KJ, Valentino RJ, DeBold JF et al. Dissociation of mu-opioid receptor and CRF-R1 antagonist effects on escalated ethanol consumption and mPFC serotonin in C57BL/6J mice. Addict Biol 2016; 21(1): 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eisenhardt M, Hansson AC, Spanagel R, Bilbao A. Chronic intermittent ethanol exposure in mice leads to an up-regulation of CRH/CRHR1 signaling. Alcohol Clin Exp Res 2015; 39(4): 752–762. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Molet J, Gunn BG, Ressler K, Baram TZ. Diversity of Reporter Expression Patterns in Transgenic Mouse Lines Targeting Corticotropin-Releasing Hormone-Expressing Neurons. Endocrinology 2015; 156(12): 4769–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanford CA, Soden ME, Baird MA, Miller SM, Schulkin J, Palmiter RD et al. A Central Amygdala CRF Circuit Facilitates Learning about Weak Threats. Neuron 2017; 93(1): 164–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veinante P, Stoeckel ME, Freund-Mercier MJ. GABA- and peptide-immunoreactivities co-localize in the rat central extended amygdala. Neuroreport 1997; 8(13): 2985–2989. [DOI] [PubMed] [Google Scholar]
- 48.Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci 2004; 24(7): 1594–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herman MA, Varodayan FP, Oleata CS, Luu G, Kirson D, Heilig M et al. Glutamatergic transmission in the central nucleus of the amygdala is selectively altered in Marchigian Sardinian alcohol-preferring rats: Alcohol and CRF effects. Neuropharmacology 2016; 102: 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varodayan FP, Correia D, Kirson D, Khom S, Oleata CS, Luu G et al. CRF modulates glutamate transmission in the central amygdala of naive and ethanol-dependent rats. Neuropharmacology 2017; 125: 418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silberman Y, Fetterly TL, Awad EK, Milano EJ, Usdin TB, Winder DG. Ethanol produces corticotropin-releasing factor receptor-dependent enhancement of spontaneous glutamatergic transmission in the mouse central amygdala. Alcohol Clin Exp Res 2015; 39(11): 2154–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP et al. CRH Engagement of the Locus Coeruleus Noradrenergic System Mediates Stress-Induced Anxiety. Neuron 2015; 87(3): 605–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pliota P, Bohm V, Grossl F, Griessner J, Valenti O, Kraitsy K et al. Stress peptides sensitize fear circuitry to promote passive coping. Mol Psychiatry 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT. CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat. Psychopharmacology (Berl) 1991; 103(2): 227–232. [DOI] [PubMed] [Google Scholar]
- 55.Knapp DJ, Duncan GE, Crews FT, Breese GR. Induction of Fos-like proteins and ultrasonic vocalizations during ethanol withdrawal: further evidence for withdrawal-induced anxiety. Alcohol Clin Exp Res 1998; 22(2): 481–493. [PubMed] [Google Scholar]
- 56.Lal H, Prather PL, Rezazadeh SM. Anxiogenic behavior in rats during acute and protracted ethanol withdrawal: reversal by buspirone. Alcohol 1991; 8(6): 467–471. [DOI] [PubMed] [Google Scholar]
- 57.Overstreet DH, Knapp DJ, Breese GR. Modulation of multiple ethanol withdrawal-induced anxiety-like behavior by CRF and CRF1 receptors. Pharmacology, biochemistry, and behavior 2004; 77(2): 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rasmussen DD, Mitton DR, Green J, Puchalski S. Chronic daily ethanol and withdrawal: 2. Behavioral changes during prolonged abstinence. Alcohol Clin Exp Res 2001; 25(7): 999–1005. [PubMed] [Google Scholar]
- 59.Somkuwar SS, Vendruscolo LF, Fannon MJ, Schmeichel BE, Nguyen TB, Guevara J et al. Abstinence from prolonged ethanol exposure affects plasma corticosterone, glucocorticoid receptor signaling and stress-related behaviors. Psychoneuroendocrinology 2017; 84: 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP et al. Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res 2002; 26(10): 1494–1501. [DOI] [PubMed] [Google Scholar]
- 61.Zhao Y, Weiss F, Zorrilla EP. Remission and resurgence of anxiety-like behavior across protracted withdrawal stages in ethanol-dependent rats. Alcohol Clin Exp Res 2007; 31(9): 1505–1515. [DOI] [PubMed] [Google Scholar]
- 62.Okhuarobo A, Bolton JL, Igbe I, Zorrilla EP, Baram TZ, Contet C. A novel mouse model for vulnerability to alcohol dependence induced by early-life adversity. Neurobiol Stress 2020; 13: 100269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jury NJ, DiBerto JF, Kash TL, Holmes A. Sex differences in the behavioral sequelae of chronic ethanol exposure. Alcohol 2017; 58: 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pleil KE, Lowery-Gionta EG, Crowley NA, Li C, Marcinkiewcz CA, Rose JH et al. Effects of chronic ethanol exposure on neuronal function in the prefrontal cortex and extended amygdala. Neuropharmacology 2015; 99: 735–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rose JH, Karkhanis AN, Chen R, Gioia D, Lopez MF, Becker HC et al. Supersensitive Kappa Opioid Receptors Promotes Ethanol Withdrawal-Related Behaviors and Reduce Dopamine Signaling in the Nucleus Accumbens. Int J Neuropsychopharmacol 2016; 19(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCool BA, Chappell AM. Chronic intermittent ethanol inhalation increases ethanol self-administration in both C57BL/6J and DBA/2J mice. Alcohol 2015; 49(2): 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hartmann MC, Haney MM, Smith CG, Kumar V, Rosenwasser AM. Affective Disruption During Forced Ethanol Abstinence in C57BL/6J and C57BL/6NJ Mice. Alcohol Clin Exp Res 2020; 44(10): 2019–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zorrilla EP, Koob GF. Impulsivity Derived From the Dark Side: Neurocircuits That Contribute to Negative Urgency. Front Behav Neurosci 2019; 13: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cai H, Haubensak W, Anthony TE, Anderson DJ. Central amygdala PKC-delta(+) neurons mediate the influence of multiple anorexigenic signals. Nat Neurosci 2014; 17(9): 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCullough KM, Morrison FG, Hartmann J, Carlezon WA Jr., Ressler KJ. Quantified Coexpression Analysis of Central Amygdala Subpopulations. eNeuro 2018; 5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stengel A, Tache Y. CRF and urocortin peptides as modulators of energy balance and feeding behavior during stress. Frontiers in neuroscience 2014; 8: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Honkaniemi J Colocalization of peptide- and tyrosine hydroxylase-like immunoreactivities with Fos-immunoreactive neurons in rat central amygdaloid nucleus after immobilization stress. Brain research 1992; 598(1–2): 107–113. [DOI] [PubMed] [Google Scholar]
- 73.Butler RK, Oliver EM, Sharko AC, Parilla-Carrero J, Kaigler KF, Fadel JR et al. Activation of corticotropin releasing factor-containing neurons in the rat central amygdala and bed nucleus of the stria terminalis following exposure to two different anxiogenic stressors. Behav Brain Res 2016; 304: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crane JW, French KR, Buller KM. Patterns of neuronal activation in the rat brain and spinal cord in response to increasing durations of restraint stress. Stress 2005; 8(3): 199–211. [DOI] [PubMed] [Google Scholar]
- 75.de Guglielmo G, Crawford E, Kim S, Vendruscolo LF, Hope BT, Brennan M et al. Recruitment of a Neuronal Ensemble in the Central Nucleus of the Amygdala Is Required for Alcohol Dependence. J Neurosci 2016; 36(36): 9446–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kimbrough A, Lurie DJ, Collazo A, Kreifeldt M, Sidhu H, Macedo GC et al. Brain-wide functional architecture remodeling by alcohol dependence and abstinence. Proc Natl Acad Sci U S A 2020; 117(4): 2149–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smith RJ, Anderson RI, Haun HL, Mulholland PJ, Griffin WC 3rd, Lopez MF et al. Dynamic c-Fos changes in mouse brain during acute and protracted withdrawal from chronic intermittent ethanol exposure and relapse drinking. Addict Biol 2020; 25(6): e12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pomrenze MB, Fetterly TL, Winder DG, Messing RO. The Corticotropin Releasing Factor Receptor 1 in Alcohol Use Disorder: Still a Valid Drug Target? Alcohol Clin Exp Res 2017; 41(12): 1986–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spierling SR, Zorrilla EP. Don’t stress about CRF: assessing the translational failures of CRF1antagonists. Psychopharmacology (Berl) 2017; 234(9–10): 1467–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tran MN, Maynard KR, Spangler A, Huuki LA, Montgomery KD, Sadashivaiah V et al. Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron 2021; 109(19): 3088–3103 e3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sorvari H, Soininen H, Pitkanen A. Calretinin-immunoreactive cells and fibers in the human amygdaloid complex. J Comp Neurol 1996; 369(2): 188–208. [DOI] [PubMed] [Google Scholar]
- 82.Powers RE, Walker LC, DeSouza EB, Vale WW, Struble RG, Whitehouse PJ et al. Immunohistochemical study of neurons containing corticotropin-releasing factor in Alzheimer’s disease. Synapse 1987; 1(5): 405–410. [DOI] [PubMed] [Google Scholar]
- 83.Roberts GW, Woodhams PL, Polak JM, Crow TJ. Distribution of neuropeptides in the limbic system of the rat: the amygdaloid complex. Neuroscience 1982; 7(1): 99–131. [DOI] [PubMed] [Google Scholar]
- 84.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007; 445(7124): 168–176. [DOI] [PubMed] [Google Scholar]
- 85.Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates, Compact. 3rd edn. Academic Press; 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.