

Abstract
Cyclin-dependent kinase (CDK) 4 and CDK6 (CDK4/6) are critical mediators of cellular transition into S phase, and are important for the initiation, growth, and survival of many cancer types. Pharmacological inhibitors of CDK4/6 have rapidly become a new standard of care for patients with advanced hormone receptor-positive breast cancer. As expected, CDK4/6 inhibitors arrest sensitive tumour cells in the G1 phase of the cell cycle. However, the effects of CDK4/6 inhibition are far more wide-reaching. New insights into their mechanisms of action have triggered identification of new therapeutic opportunities including the development of novel combination regimens, expanded application to a broader range of cancers, and use as supportive care to ameliorate the toxicity of other therapies. Exploring these new opportunities in the clinic is an urgent priority, which in many cases has not been adequately addressed. Here, we provide a framework for conceptualising the activity of CDK4/6 inhibitors in cancer and explain how this framework might shape the future clinical development of these agents. We also discuss the biologic underpinnings of CDK4/6 inhibitor resistance, an increasingly common challenge in clinical oncology.
Introduction
For a mammalian cell to divide, it must pass through the series of well-orchestrated phases known collectively as the cell cycle. Progression through the cell cycle in normal cells is tightly regulated by both proliferative and anti-proliferative forces. Cellular division is triggered by mitogenic signals, increasing the levels of cyclin proteins that bind and activate cyclin-dependent kinases (CDKs). This activity is balanced by inhibitory proliferative checkpoints, which prevent inappropriate cell division. Cancer is classically envisaged as a disease in which this balance is disturbed to favour cell division driven by excessive mitogenic signalling, a failure of inhibitory checkpoints, or both. Given this, the development of inhibitors of the CDKs that regulate the cell cycle (particularly CDKs 1, 2, 4 and 6) has been a longstanding aspiration of cancer researchers1.
The development of a safe and effective small molecule CDK inhibitor proved difficult for many years. The first compounds tested (exemplified by flavopiridol) were potent inhibitors of numerous CDKs, rendering them prohibitively toxic and suggesting the need for more selective agents1,2. Unfortunately, creation of these selective inhibitors has, until recently, presented a medicinal chemistry challenge3. Notwithstanding this technical obstacle, the biological complexities of cell cycle regulation in cancer also raise the concern that inhibition of a single CDK might be ineffective in many cases. First, the human cyclin–CDK network is diverse, exhibiting redundancy and plasticity4. This means that inhibition of one or more cyclins or CDKs can be compensated by an increase in the activity of others5–8. Second, cancer cells can harbour genetic aberrations – often in key cell cycle genes – that alter their dependencies on specific CDKs9. Third, tumours originating from distinct cellular lineages use different interphase CDKs as their primary driver of proliferation10–12.
Considering these obstacles, the success story of selective CDK4/6 inhibitors in modern oncology is remarkable. In 2004, the chemical structure of the first CDK4/6 inhibitor – palbociclib – was published, and within eleven years, this agent received accelerated approval from the United States Food and Drug Administration (FDA) as a treatment for breast cancer13,14. Since then, two other agents (ribociclib and abemaciclib) have also entered mainstream breast oncology practice, and all three agents have proven to be well tolerated by patients14. This success has triggered widespread preclinical, translational and clinical research efforts exploring the CDK4/6 pathway across many cancer types, and the research community is now at a crossroads. Behind us lies the clinical success of CDK4/6 inhibitors as treatment for hormone receptor (HR)-positive breast cancer. Ahead of us lies a plethora of preclinical data suggesting other indications where clinical testing of these drugs is warranted. Given this, a detailed Review of CDK4/6 inhibition in cancer is timely and may help focus research efforts as we strive to fully exploit the promise of these drugs. Here, we focus our discussion on emerging concepts in the CDK4/6 field, including a detailed discussion of the biological mechanisms by which CDK4/6 inhibitors exert their effects in cancer. We then suggest how knowledge of these mechanisms will inform the rational development of CDK4/6 inhibitor-based therapeutic approaches in years to come.
The CDK4/6 pathway in cancer
Regulation of S phase entry by CDK4/6
The human cyclin–CDK network comprises over 20 CDKs and up to 30 distinct cyclin proteins15. CDK4 and CDK6 bind to the D-type cyclins (cyclin D1, cyclin D2 and cyclin D3) and are specifically implicated in driving cellular transition from the G1 phase to the S phase of the cell cycle, when DNA synthesis occurs16–18. According to the classical model, the G1 to S transition is driven by mitogenic signalling from extracellular growth factors, which increase cyclin D levels19–22. Cyclin D-bound CDK4/6 then phosphorylates the tumour suppressor protein RB, as well as the RB-like ‘pocket proteins’ p107 and p13023–25. The canonical function of hypo-phosphorylated RB is to bind to and thus sequester activity of the E2F transcription factors, thereby restraining cellular entry into S phase. RB enacts this repression by (i) binding to and blocking the E2F transactivation domain26–29; and (ii) recruiting chromatin modifiers to E2F-bound promoters, which enforce a repressive chromatin environment at these sites30–32. RB phosphorylation by CDK4/6 leads to the release of E2F transcription factors, increasing transcription of the E2F target genes cyclin E1 (CCNE1) and cyclin E2 (CCNE2). E-type cyclins then bind and activate CDK2, leading to RB hyperphosphorylation and phosphorylation of numerous other proteins, which collectively drive an irreversible commitment to S phase33,34 (Figure 1a).
Figure 1. The CDK4 and CDK6 pathway in cancer.
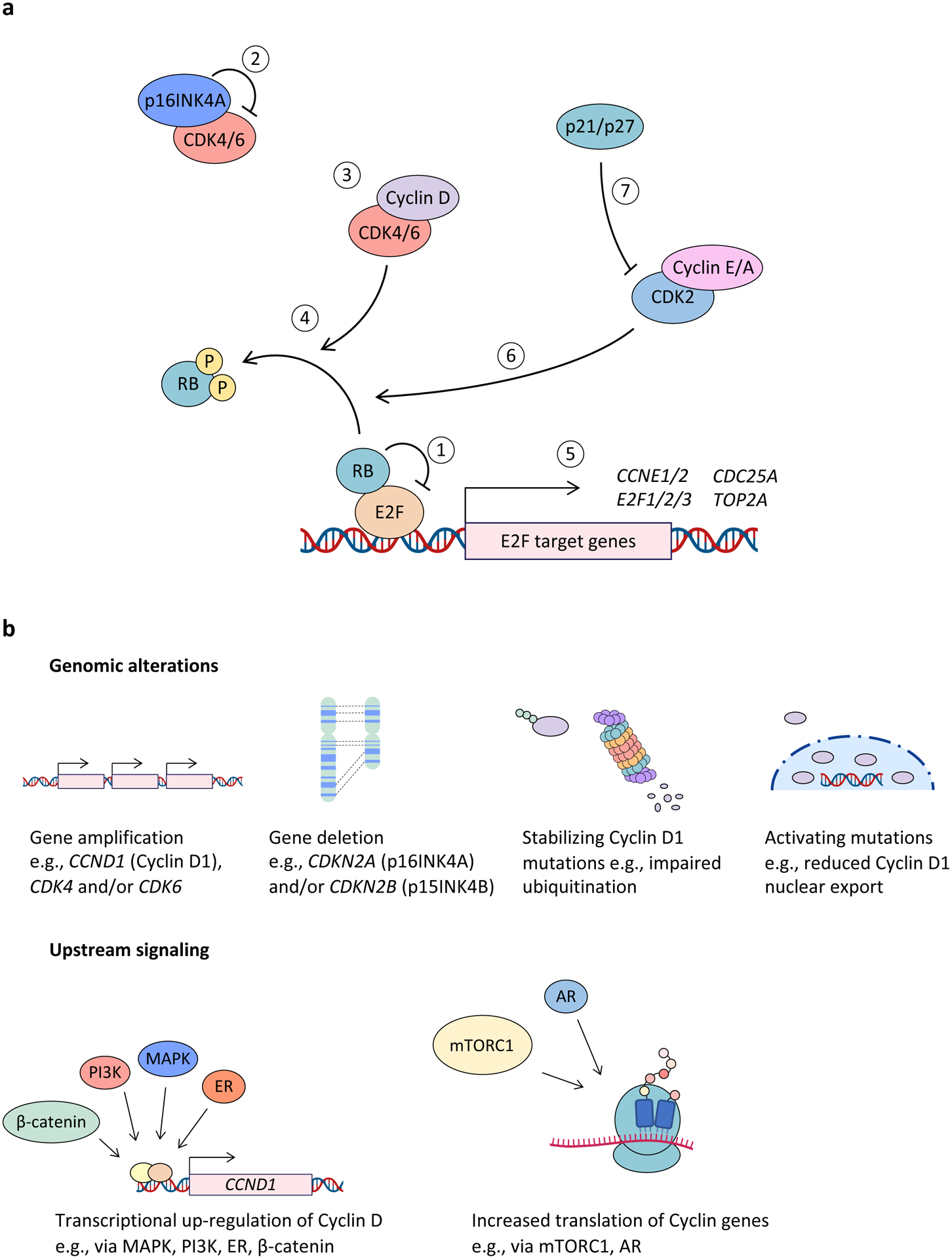
a, Hypophosphorylated RB binds to and represses E2F family transcription factors (1). Negative regulation of cyclin-dependent kinase 4 (CDK4) and CDK6 (CDK4/6) activity is mediated primarily by the INK4 family of cell cycle inhibitors (INK4B (p15; encoded by cyclin-dependent kinase inhibitor 2B (CDKN2B)), INK4A (p16; encoded by CDKN2A), INK4C (p18), and INK4D (p19)), which bind to monomeric CDK4 and CDK6 to form inactive binary complexes (2). Mitogen or growth factor stimulation drives cyclin D up-regulation, leading to CDK4/6 activation (3). RB phosphorylation by cyclin D–CDK4/6 complexes promotes dissociation of RB–E2F binding (4). This in turn allows for E2F-mediated expression of genes required for cell cycle progression (5), which leads to progression through G1 phase and into S-phase. RB phosphorylation by cyclin E–CDK2 and cyclin A–CDK2 complexes (6) also promotes RB–E2F dissociation to drive progression into S phase. On the other hand, WAF1 and KIP family proteins, such as p21 (WAF1), p27 (KIP1) and p57 (KIP2), inhibit CDK2 and are important for inducing cell cycle arrest (7). Of note, p21 and p27 (p21/p27) have been shown to inhibit CDK4/6 activity in some instances and in other instances to stabilize cyclin D–CDK4/6 and thereby form an active trimeric holoenzyme. b, Major mechanisms responsible for dysregulated CDK4/6 activity in cancer include genomic alterations as well as activation of upstream signalling pathways that may up-regulate this pathway at the transcriptional, translational and post-translational levels. AR, androgen receptor; CCN, cyclin; ER, oestrogen receptor; mTORC1, mTOR complex 1.
In addition to the various mechanisms that control the expression, nuclear export, and degradation of D-type cyclins (discussed throughout this Review), CDK4/6 activity is also regulated by the INK4 (INK4B (p15), INK4A (p16), INK4C (p18) and INK4D (p19)) and WAF1 and KIP (p21 (WAF1), p27 (KIP1) and p57 (KIP2)) cyclin-dependent kinase inhibitor protein families. INK4 proteins bind to and inhibit monomeric CDK4 or CDK6 by (i) displacing CDK4 or CDK6 from the heat shock protein 90 (HSP90) co-chaperone CDC37, disrupting their correct folding and assembly; and (ii) inducing a conformational change in CDK4 or CDK6 that weakens associations with cyclin D35–38. WAF1 and KIP proteins play a more nuanced role. On one hand, they bind to and inhibit a broad range of fully assembled cyclin–CDK complexes (including cyclin D-CDK4) to restrain cell cycle progression39–41. However, WAF1 and KIP proteins (particularly p21 and p27) have also been reported to promote S phase progression by forming stable complexes with CDK4/6 and cyclin D, which (i) stimulate CDK4/6 enzymatic function42,43; and (ii) act as reservoirs that sequester p21 and/or p27 and thus allow unrestrained activity of CDK244. Importantly, several in vitro and transgenic animal studies have called into question the essentiality of p21 and/or p27 for CDK4/6 enzymatic activity45–48.
The role of cyclin E–CDK2 in S phase regulation
Although the classical model described above suggests that CDK4/6-mediated RB phosphorylation is a prerequisite for CDK2 activity and thus S phase entry, this linear concept is overly simplistic. Indeed, either CDK4/6 or CDK2 alone could drive proliferation in certain circumstances. For example, certain RB-proficient cancer cells can proliferate in the absence of CDK249. Similarly, mammalian cells lacking both CDK4 and CDK6 proteins can proliferate due to the formation of atypical cyclin D–CDK2 complexes that retain the capacity to phosphorylate RB5. As is discussed throughout this Review, the redundancy between CDK4/6 and CDK2 is critical when considering the variable responses of RB-proficient cancers to inhibition of CDK4/6.
Hyperactivity of CDK4/6 kinases in cancer
Alterations predicted to drive hyperactivity of the cyclin D–CDK4/6 axis are common in human cancers (Figure 1b and Supplementary Table S1). Amplifications of CCND1, CDK4, and CDK6 are reported in a variety of tumour types, as is homozygous deletion of CDKN2A (which encodes INK4A). In addition, a series of rarer genetic events – described as ‘D-cyclin activating features’ (DCAF)50 – can markedly enhance cancer cell dependence on CDK4/6. These include the classical t11:14 translocation of mantle cell lymphoma (juxtaposing CCND1 with the immunoglobulin heavy chain (IGH) locus)51, focal amplifications of CCND2 and CCND350, loss of and/or mutations in the 3’-untranslated region (3’-UTR) of cyclin D genes52, and expression of the Kaposi’s sarcoma-associated herpesvirus v-cyclin53. Finally, mutations in cyclin D genes that prevent either ubiquitination and degradation or nuclear export of cyclin D have been reported in a variety of tumour types50,54–58.
Equally important drivers of CDK4/6 hyperactivity in cancer are the ubiquitous mechanisms underlying cyclin D upregulation in the absence of genetic alterations in cyclin D–CDK4/6 pathway genes themselves. The majority of these are underpinned by inappropriately high levels of mitogenic signalling, which disrupts the finely tuned rise and fall of cyclin D levels typically seen during G1 and S phase59,60. Increased MAPK pathway signalling (through mutation or other means) directly drives CCND1 transcription21, whereas PI3K–AKT signalling increases cyclin D through various mechanisms including derepression of cyclin D gene transcription61, increased mRNA translation62, and reduced nuclear export and protein degradation22. Furthermore, mTOR complex 1 (mTORC1) (downstream of both PI3K–AKT and MAPK pathways) can specifically increase cyclin D mRNA translation63,64. Numerous other signalling pathways can also increase CDK4/6 activity in cancer cells, through diverse mechanisms. For example, (i) steroid hormone signalling through the estrogen receptor (ER) increases CCND1 transcription in estrogen-sensitive cancers such as breast carcinoma65, and signalling through the androgen receptor (AR) in prostate cancer drives increased cyclin D mRNA translation via mTORC166; (ii) β-catenin activates transcription from the CCND1 promoter67; (iii) CCND2 is a direct target gene of MYC68; (iv) signalling through the Notch intracellular domain can drive CCND1 and CCND3 transcription69,70; and (v) Hippo pathway members can increase transcription of both CCND1 and CDK671–73.
Insights from transgenic mouse models
Transgenic animal models have offered fundamental insights into the biological processes regulated by the cyclin D–CDK4/6 axis within tumours. Many of these come from studies of mammary and lymphoid malignancies, stemming from the critical role of cyclin D1–CDK4 and cyclin D3–CDK6 in the development of these organs, respectively5,70,74–76. In the mammary gland, cyclin D1 is a weak oncogene and its overexpression is sufficient to induce mammary carcinogenesis77, a phenomenon underpinned by factors including CDK4/6-dependent stimulation of mammary epithelial cell proliferation78, suppression of oncogene-induced senescence79, and a number of less well-characterised kinase-independent functions of cyclin D180. Cyclin D1–CDK4 is required for the development of mammary carcinomas driven by upstream oncogenes such as Neu (also known as Erbb2) and HRAS, but not others such as Myc or Wnt111,81. Analogous to this, cyclin D3 is required for the development of Notch1 and Lck-driven T cell malignancies70.
Separate to their role in tumorigenesis, CDK4 and CDK6 can promote the sustained growth of established tumours in seemingly distinct and tissue-specific ways. Genetic ablation of cyclin D1 in established mammary tumours leads to cell cycle arrest and cellular senescence, consistent with the role of hypophosphorylated RB as a mediator of senescence12,82. In contrast, cyclin D3 ablation triggers leukaemia cell apoptosis in T cell acute lymphoblastic leukaemia, mediated by accumulation of reactive oxygen species (ROS) as a consequence of inhibited CDK6-mediated phosphorylation of key metabolic enzymes (i.e., an RB-independent process)12,76,83. These examples demonstrate that the precise mechanism by which cyclin D–CDK4/6 complexes sustain tumour growth can differ between tumours depending on which cyclin D–CDK pairing they employ, and that this in turn is often linked to tissue of origin. Collectively, this incontrovertible in vivo genetic evidence coupled with the frequency of cyclin D–CDK4/6 axis activation in cancer have provided a strong rationale for the development of pharmacological inhibitors of CDK4 and CDK6.
Selective CDK4/6 inhibitors
Generation of currently approved CDK4/6 inhibitors
First- and second-generation CDK inhibitors developed over the last three decades have almost universally failed clinical development. In most instances, these compounds inhibited both interphase CDKs as well as CDKs regulating RNA polymerase II-mediated transcriptional activity (CDK7 and CDK9), and this lack of selectivity resulted in significant clinical toxicity. Furthermore, patient selection was not driven by specific tumour types or molecular biomarkers.
Interest in inhibitors of cell cycle CDKs was revitalised by the development of highly specific, potent ATP-competitive inhibitors of CDK4 and CDK6. A critical step in this development was optimisation of the pyrido[2,3-d]pyrimidine scaffold by including a methyl substituent at the C-5 position, which was sufficient to confer excellent selectivity for CDK4 and CDK684. This breakthrough led the biopharmaceutical company Pfizer to develop palbociclib (PD-0332991)13, and Novartis scientists to develop ribociclib using a similar scaffold85. Meanwhile, medicinal chemists at Lilly utilised the 6-pyrimidine benzimidazole core to develop abemaciclib86,87. All three of these agents are administered orally and are currently approved as therapy for advanced HR-positive breast cancer. A fourth recently approved inhibitor – trilaciclib – is based on a tricyclic lactam scaffold, given via intravenous injection, and approved for the reduction of chemotherapy-induced bone marrow suppression in patients with small-cell lung cancer (SCLC)88,89. At the time of writing, many other selective CDK4/6 inhibitors are in various stages of clinical development, and whilst a comprehensive discussion of these molecules lies outside the scope of our Review, they are listed in Table 1.
Table 1.
CDK4 and CDK6 inhibitors approved or under clinical development.
| Agent | Company | Selectivity (IC50) | Clinical development |
|---|---|---|---|
| Approved | |||
Palbociclib |
Pfizer | CDK4: 11 nM CDK6: 16 nM |
Approved for HR+, HER2− advanced breast cancer in combination with hormonal therapy |
| Abemaciclib |
Eli Lilly | CDK4: 2 nM CDK6: 10 nM |
Approved for HR+, HER2− advanced breast cancer in combination with hormonal therapy Approved as monotherapy for advanced HR+, HER2− breast cancer Approved as adjuvant therapy for high-risk, early-stage HR+, HER2− breast cancer in combination with hormonal therapy |
Ribociclib |
Novartis | CDK4: 10 nM CDK6: 39 nM |
Approved for HR+, HER2− advanced breast cancer in combination with hormonal therapy |
Trilaciclib
|
G1 Therapeutics | CDK4: 1 nM CDK6:4 nM |
Approved to reduce chemotherapy-induced bone marrow suppression in patients with extensive-stage SCLC. |
| Phase III | |||
| Dalpiciclib (SHR6390) | Jiangsu Hengrui Medicine | CDK4: 12 nM CDK6: 10 nM |
Phase III for HR+, HER2− breast cancer in combination with hormonal therapy; phase I/II for multiple tumor types in combination with hormone, targeted or immune therapy |
| Phase II | |||
| PF-06873600 | Pfizer | CDK2: 0.09 nM (Ki) CDK4: 0.13 nM (Ki) CDK6: 0.16 nM (Ki) |
Phase II for HR+, HER2− metastatic breast cancer, TNBC and gynecological cancers in combination with hormonal therapy |
| Phase I/II | |||
| Lerociclib (G1T38) | G1 Therapeutics | CDK4: 1 nM CDK6: 2 nM CDK9: 28 nM |
Phase I/II for HR+, HER2− metastatic breast cancer in combination with fulvestrant (SERD); phase I/II for EGFR-mutant metastatic NSCLC in combination with osimertinib (EGFR inhibitor) |
| Birociclib (XZP-3287) | Jilin Sihuan Pharmaceutical/Xuanzhu Pharma | Not available | Phase I/II for HR+, HER2− advanced breast cancer |
| BPI-1178 | Beta Pharma | Not available | Phase I/II for advanced solid tumors, and for HR+, HER2− advanced breast cancer in combination with hormone therapy |
| FCN-437C | Fochon Pharmaceuticals | Not available | Phase I/II for HR+, HER2− advanced breast cancer in combination with letrozole (aromatase inhibitor) |
| TQB3616 | Chia Tai Pharmaceutical Group | Not available | Phase I/II for HR+, HER2− advanced breast cancer in combination with fulvestrant, and for advanced lung cancer in combination with anlotinib (VEGFR inhibitor) or standard chemotherapy |
| Phase I | |||
| AMG-925 (FLX925) | Amgen | CDK4: 1.5 nM CDK6: 8 nM FLT3: 2.4 nM |
Phase I/Ib for relapsed or refractory AML |
| R547 | Hoffmann-La Roche | CDK1: 2 nM (Ki) CDK2: 3 nM (Ki) CDK4:1 nM (Ki) |
Phase I for advanced solid cancers |
| BPI-16350 | Betta Pharmaceuticals | Not available | Phase I for advanced solid cancers |
| CS3002 | CStone Pharmaceuticals | Not available | Phase I for advanced solid cancers |
| HS-10342 | Jiangsu Hansoh Pharmaceutical | Not available | Phase I for advanced solid cancers |
| PF-06842874 | Pfizer | Not available | Phase I in healthy participants |
| TY-302 | TYK Medicines | Not available | Phase I for advanced solid cancers, and for HR+, HER2− breast advanced cancer in combination with tamoxifen |
AML, acute myeloid leukaemia; CDK, cyclin-dependent kinase; EGFR, epidermal growth factor receptor; FLT3, FMS-like tyrosine kinase 3; HER2, human epidermal growth factor receptor 2; HR, hormone receptor; SCLC, small-cell lung cancer; SERD, selective estrogen receptor degrader; TNBC, triple-negative breast cancer; VEGFR, vascular endothelial growth factor receptor.
Mechanism of action and inhibitory spectra
Remarkably, the precise molecular mechanisms by which currently approved CDK4/6 inhibitors suppress cellular proliferation remains an open question, with recent provocative studies positing that we might not fully understand whether these drugs directly inhibit CDK4/6 in cells, and if they do, to what extent. Each compound interacts with the ATP-binding pocket of CDK4 and CDK6, which has been presumed to result in competitive inhibition of active CDK4/6 kinases. However, whilst active CDK4/6 in living cells often exists in a trimer with cyclin D and p21 and/or p2742, the in vitro potency of each compound was determined using cyclin D–CDK4/6 dimers13,86,88. Intriguingly, palbociclib, ribociclib, and abemaciclib do not appear to inhibit active trimeric complexes in vitro, calling into question whether they directly inhibit active CDK4/6 in cells. One alternate model is that CDK4/6 inhibitors bind and sequester monomeric (inactive) CDK4/6, thereby preventing assembly of holoenzyme trimers, which in turn frees p21 to inhibit CDK243. The concept that CDK4/6 inhibitors exert their effects primarily through indirect CDK2 inhibition is intriguing, remains a somewhat open question48, and would be strengthened by demonstrating that CDK4/6 inhibitor-sensitive cancer cells are sensitive to genetic disruption of CDK2 kinase function, even more so than to disruption of CDK4 and CDK6 kinases. Another layer of complexity arises from the fact that CDK6 exists in both thermounstable (HSP90-CDC37 bound) and thermostable (HSP90-CDC37 unbound) forms, the latter exhibiting resistance to existing pharmacological CDK4/6 inhibitors90. Better understanding of the relevance of these findings to cancer therapy are urgently needed, as they have major implications for understanding drug activity and resistance mechanisms.
In vitro kinase and chemo-proteomic studies have suggested that the approved CDK4/6 inhibitors do have distinct non-CDK4/6 kinase targets86,91,92. This is particularly true for abemaciclib, which has been reported to inhibit CDK9, PIM1, homeodomain-interacting protein kinase 2 (HIPK2), dual specificity tyrosine-phosphorylation-regulated kinase 2 (DYRK2), glycogen synthase kinase-3β (GSK3β), and even CDK2 in vitro86,93. Although these targets have been proposed to explain the distinct toxicity and efficacy profiles of abemaciclib, the extent to which they are inhibited in tumour cells by physiological drug concentrations remains unclear50,94–96. Moreover, there is a strong correlation between sensitivity to palbociclib and abemaciclib across large panels of cancer cell lines, suggesting that they act by inhibiting the same pathway(s)50. Finally, and notwithstanding the question of whether CDK4/6 inhibitors directly inhibit CDK4/6, the relative potency of each compound for CDK4 versus CDK6 (Table 1) has been proposed as a determinant of its efficacy or toxicity. However, this notion remains speculative at present.
Clinical development in breast cancer
Clinical trial participants with a wide variety of tumour types have received oral CDK4/6 inhibitor therapy, and an exhaustive review of the related literature lies outside the scope of this Review. However, it is noteworthy that some of the earliest reported clinical data demonstrated anti-proliferative and clinical activity of palbociclib in a cohort of 17 patients with mantle cell lymphoma97. At present, these agents are only approved for the treatment of advanced HR-positive breast cancer. Early development of the compounds for breast cancer was triggered by seminal preclinical studies using breast cancer cell lines. Finn et al.98 treated a panel of 47 human breast cancer or immortalised mammary cell lines and, consistent with findings from transgenic mice described above, demonstrated that luminal and human epidermal receptor 2 (HER2)-amplified cancer cells were the most sensitive to CDK4/6 inhibition. Synergy was also observed between CDK4/6 inhibitors and other common breast cancer therapies (anti-estrogen therapy in ER-positive cells and the monoclonal antibody trastuzumab targeting HER2 in HER2-amplified cells)98.
The ensuing clinical trials have cemented a role for CDK4/6 inhibitors as a mainstay of therapy for advanced HR-positive disease and have been complemented by several neoadjuvant therapy trials, which have added mechanistic insights. Fundamental lessons from these trials include: (i) CDK4/6 inhibitor monotherapy demonstrates anti-proliferative and clinical activity in HR-positive breast cancer99–103; (ii) CDK4/6 inhibition can overcome endocrine therapy (ET) resistance, and these two agents act synergistically to improve progression-free and overall survival when compared to ET alone101,102,104–112; and (iii) these benefits extend to both pre- and post-menopausal women106,113,114. Clinical trials also demonstrate that the distinct toxicity profiles of these agents influence their schedule of administration. Palbociclib and ribociclib induce higher rates of grade 3 or 4 haematological toxicity (presumably an on-target effect of CDK6 inhibition), which necessitate intermittent dose interruptions (typically given as a 21 days on, 7 days off cycle). Haematological toxicity is seen with abemaciclib as well, but it is less frequent, which allows for the drug to be dosed continuously. The extent to which these distinctions account for differences in clinical activity, most notably the higher response rate with abemaciclib monotherapy103, remains a subject of conjecture.
A mechanistic framework for CDK4/6 inhibition
Although preclinical studies suggest a wide range of potential applications and indications for CDK4/6 inhibitors, they are currently only approved to treat two cancer types (Table 1). Successful prioritisation of areas for future clinical development will require a detailed understanding of the mechanisms by which CDK4/6 inhibitors exert their effects in cancer. Here, we provide a mechanistic framework to facilitate this, with six features enumerating the variety of cellular and molecular processes that ultimately dictate phenotypic responses to CDK4/6 inhibition.
Downregulation of E2F target gene expression
The best characterised consequence of CDK4/6 inhibitor-mediated inhibition of E2F transcriptional activity is RB-dependent proliferative arrest (Figure 2a), which has been documented in preclinical and clinical studies12,13,86,97,98,100,104,115–119. This cytostatic effect would be expected to stabilise tumour growth at best and cannot alone explain the objective clinical responses (i.e., tumour shrinkage) seen in CDK4/6 inhibitor monotherapy trials97,99,103. Importantly, E2F targets regulate processes other than S phase entry, including replication origin licensing, DNA repair, DNA methylation and chromatin condensation, cellular metabolism, cellular differentiation, and apoptosis120–127, and CDK4/6 inhibition can also impact these processes in RB-proficient cells in a manner that influences phenotypic responses to treatment128–132.
Figure 2. A conceptual framework to understand the effects of CDK4 and CDK6 inhibitors in cancer.
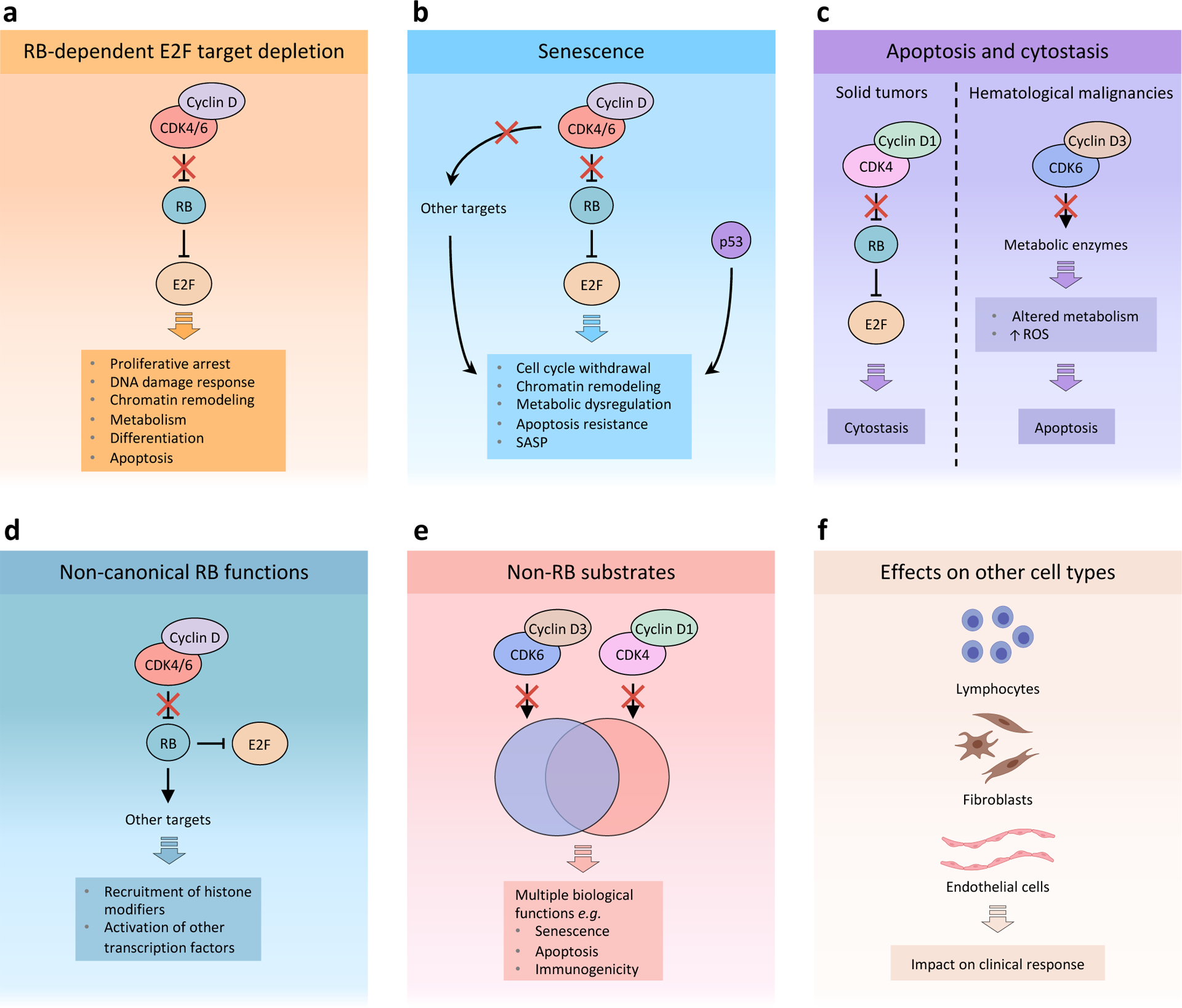
Response to cyclin-dependent kinase 4 (CDK4) and CDK6 (CDK4/6) inhibitors depends on multiple factors that affect the final outcome, including context-dependent tumour cell-intrinsic and -extrinsic features. a, RB-dependent E2F target depletion: Blockade of RB phosphorylation by cyclin D–CDK4/6 complexes leads to sustained binding to and inhibition of E2F by RB. A major consequence of E2F inactivation by RB is proliferative arrest. Moreover, E2F functions in various additional processes that are affected by CDK4/6 inhibition, including DNA damage response, chromatin remodelling, metabolism, differentiation and apoptosis. b, Senescence: CDK4/6 inhibitors may induce an RB-dependent senescent phenotype characterized by cell cycle withdrawal, chromatin remodelling, metabolic dysregulation, resistance to apoptosis and a senescence-associated secretory phenotype (SASP). Downregulation of other CDK4/6 targets, such as forkhead box protein M1 (FOXM1) and DNA methyltransferase 1 (DNMT1), is thought to enhance the senescent phenotype. In addition, p53 plays distinct and overlapping roles with RB in inducing senescence. Current evidence suggests that loss of p53 function may significantly affect senescence induced by CDK4/6 inhibitors, although additional studies have shown that CDK4/6 inactivation can still induce senescent phenotypes in the absence of wild type p53. c, Apoptosis and cytostasis: Induction of apoptosis or cytostasis appears to be cell type-dependent and may depend on differences in signalling between CDK4 and CDK6. In haematological malignancies, inhibition of CDK6–cyclin D3 complexes may lead to an RB-independent state of metabolic dysregulation that results in apoptosis. On the other hand, solid tumours such as breast cancer often depend primarily on CDK4–cyclin D1 activity for proliferation. In this case, CDK4/6 monotherapy predominantly induces RB-dependent proliferative arrest as a result of sustained E2F inhibition. d, Non-canonical RB functions: In addition to inhibiting E2F, RB has been shown to exert other functions, such as mediating chromatin remodelling and activation of other transcription factors. e, Non-RB substrates: In addition to phosphorylating and inhibiting RB, CDK4 and CDK6 phosphorylate a set of unique and overlapping targets that play important functions in numerous biological processes, including senescence, apoptosis and immunogenicity. f, CDK4/6 inhibitors have been shown to exert direct effects on multiple cell types normally present within the tumour microenvironment, including lymphocytes, fibroblasts and endothelial cells. Several studies have shown CDK4/6 inhibitors can directly and indirectly promote anti-tumour T lymphocyte effector function and inhibit immunosuppressive regulatory T (Treg) cells. CDK4/6 inhibitors also induce senescence phenotypes in fibroblasts, which could potentially impair response to therapy by promoting tumour growth, and cell cycle arrest in endothelial cells, which has the potential to impact on clinical response. ROS, reactive oxygen species.
Cellular senescence
CDK4/6 inhibitors can induce a ‘senescence-like’ state in cancer cells (Figure 2b), evidenced by cellular enlargement and increased β-galactosidase activity (a recognized marker of cellular senescence)12,133,134. Early studies showed that this phenomenon is mediated by active RB133,135, which is not surprising given the role of RB as a principal mediator of the cellular senescence program82,136. The senescent phenotype might also be augmented by reduced activity of the direct CDK4/6 substrates forkhead box protein M1 (FOXM1) and DNA methyltransferase 1 (DNMT1)79,137. However, FOXM1 and DNMT1 are also E2F targets, and there is currently no evidence that inhibition of the phosphorylation of FOXM1 and DNMT1 without downregulation of other E2F targets (e.g., after CDK4/6 inhibitor treatment of RB-deficient cancer cells) is sufficient to induce senescence137,138.
The phenotypic hallmarks of classical cellular senescence include cell cycle withdrawal, chromatin remodelling, metabolic dysregulation, apoptosis resistance, and secretion of growth factors and cytokines (known as the senescence-associated secretory phenotype (SASP))139. These phenomena were characterised in models of DNA damage-induced senescence in fibroblasts, and it cannot be assumed that they are recapitulated in cancer cells treated with CDK4/6 inhibitors. Unlike classical senescence in benign cells, CDK4/6 inhibitor-induced senescence is distinct in that (i) the initiating mechanism is RB activation rather than DNA damage, which activates both RB and p53140,141; (ii) it occurs in cancer cells that harbour DNA mutations and have therefore presumably acquired senescence escape mechanisms.
Notwithstanding this, certain aspects of classical senescence have been described in CDK4/6 inhibitor-treated cancer cells. Watt and colleagues142 reported chromatin remodelling in cell-based models and clinical specimens of breast cancer treated with CDK4/6 inhibitors, characterised by widespread enhancer activation akin to that seen in senescent fibroblasts143. The transcriptional activity mediated by newly activated enhancers was directly implicated in mediating apoptotic evasion and enhanced cellular immunogenicity, both known features of classical senescence, and was principally driven by the AP-1 transcription factors, which have also been implicated as master regulators of the classical senescence phenotype142,144. Notably, these new enhancers also provoked a more differentiated cellular phenotype, supporting the mechanistic link between RB activation and cellular differentiation in cancer142,145.
CDK4/6 inhibition, either alone or in combination with other agents that heighten the senescent phenotype, can also induce a SASP, presumably attributable to the enhancer remodelling described above135,146,147. The SASP factors released can include immunomodulatory proteins that mediate surveillance by components of the innate immune system146, pro-angiogenic factors and matrix metalloproteinases that facilitate tumour angiogenesis147, growth factors that can drive autocrine tumour cell proliferation148, and classical SASP factors described in fibroblast models135. SASP factors secreted by CDK4/6 inhibitor-treated cells are likely to vary by tumour lineage, and a more comprehensive portrait of these is required given the major impact they can have on the tumour microenvironment.
Although p53 plays a critical role in classical senescence, its role in CDK4/6 inhibitor-mediated senescence remains unclear. On one hand, genetic and pharmacological CDK4/6 inhibition can induce phenotypic features of senescence in malignant cells that lack functional p53133,135,149,150. Conversely, transgenic mouse models of KRAS-driven lung carcinoma have shown that genetic inhibition of CDK4 kinase activity can only induce tumour cell senescence if p53 function is intact151. Furthermore, TP53 mutations emerged as the strongest genomic predictor of CDK4/6 inhibitor resistance in a panel of 560 cancer cell lines, suggesting that p53 function contributes to drug-induced proliferative arrest50. RB and p53 have partially overlapping but distinct functions in classical senescence, and it is conceivable that both p53 wild-type and mutant cancer cells can display features of senescence in response to CDK4/6 inhibition, but that the two senescent phenotypes are qualitatively different.
Cellular apoptosis and cytostasis
Pharmacological CDK4/6 inhibition can also directly induce tumour cell apoptosis. As described above, the evidence for this is strongest in haematological malignancies that are primarily driven by cyclin D3–CDK6 activity12,76,83,152. In contrast, the dominant response to CDK4/6 inhibitor monotherapy in solid tumours that are thought to be primarily driven by Cyclin D1–CDK4 activity (e.g. in breast cancer and non-small-cell lung cancer) is cytostasis12,153 (Figure 2c).
Non-canonical functions of hypo-phosphorylated RB
Hypo-phosphorylated RB has non-canonical functions, including the recruitment of histone modifying enzymes to DNA, and the stimulation of transcriptional activity of factors such as JUN and the glucocorticoid receptor154–156 (Figure 2d). It is conceivable that these functions could also be triggered by CDK4/6 inhibition in RB-proficient cells, as has been demonstrated for the case of JUN and other AP-1 factors142.
Non-RB substrates
CDK4 and CDK6 bind and phosphorylate numerous non-RB substrates. In brief, they include a variety of transcription factors79,157–159, proteins regulating oncogene expression152, DNMTs121, metabolic enzymes83, ubiquitin ligase adaptor proteins and deubiquitinases160,161, suppressors of oncogenic kinase signalling153,162,163, and others79 (Figure 2e). Inhibiting the phosphorylation of these proteins with CDK4/6 inhibitors can impact diverse biological processes including senescence and lysosome biogenesis79,137,159, apoptosis83, tumour cell metastasis160, tumour cell immunogenicity161, and T lymphocyte activation158, but we lack the information needed to dissect the relative contributions of these effects in any given tumour context. Importantly, (i) hypo-phosphorylation of these substrates by CDK4/6 inhibitors is expected to impact both RB-proficient and RB-deficient cells; and (ii) although the network of CDK4/6 substrates is not fully characterized, several non-RB substrates are unique to either CDK4 or CDK679,83,152.
Effects on stromal cell types
Although most CDK4/6 inhibitor studies have focused on the treatment of cancer cell lines in vitro, other cell types, including lymphocytes130,158,164, fibroblasts140, and endothelial cells165 can respond to CDK4/6 inhibitors in ways that can meaningfully impact response to therapy (Figure 2f). In particular, CDK4/6 inhibitors have been shown to directly stimulate CD8+ T cell effector function while repressing T regulatory (Treg) cell proliferation in vitro and in vivo, thereby promoting anti-tumour responses130,158,164. The reported impact of CDK4/6 inhibition on fibroblasts, and the consequences for tumour behaviour, are mixed. On one hand, treatment of normal mouse embryonic fibroblasts (MEFs) with palbociclib induced senescence in vitro, and co-injection of palbociclib-treated MEFs with tumour cells into immunocompetent mice enhanced tumour growth in certain models of melanoma with WT Braf and Nras140. Conversely, a recent study showed that although abemaciclib-treated fibroblasts do exhibit as SASP, this is not sufficient to stimulate cancer cell proliferation166. Likewise, treatment of endothelial cells with palbociclib in vitro results in cell cycle arrest, but the consequences of this effect in cancer was not explored165.
Designing rational combination therapies
The key to improving the utility of CDK4/6 inhibitor therapy for a broad range of cancer types will be the identification of effective and tolerable combination therapy regimens. By changing tumour cell and stromal cell phenotypes so profoundly, CDK4/6 inhibitors invoke new cancer phenotypes, dependencies, and vulnerabilities that can be exploited through the addition of additional therapeutic agents167. Currently, combination therapies with CDK4/6 inhibitors are being evaluated in a vast number of clinical trials encompassing numerous and varied pharmacological agents in multiple cancer types (Supplementary Table S2). In this section, we discuss novel combination therapy approaches and the biological rationale for their development.
Endocrine therapy in breast cancer
Preclinical and clinical studies clearly demonstrate synergy between ET and CDK4/6 inhibitors in luminal breast cancer, which underpins the remarkable efficacy of this combination8,98,102,168–170. The ER and cyclin D1–CDK4 interact at several levels: (i) CCND1 is a direct ER target gene65; (ii) ER directs an estrogen-independent, E2F-driven transcriptional signature that facilitates tumour cell proliferation171; and (iii) cyclin D1 can directly bind ER and promote expression of ER target genes in a CDK-independent fashion172. How these interactions give rise to synergistic interactions between ET and CDK4/6 inhibitors is not clear, but likely relates to heightened inhibition of RB phosphorylation170.
Combining oncogenic kinase and CDK4/6 inhibitors
A plethora of preclinical data demonstrates cooperativity between CDK4/6 inhibitors and inhibitors of mitogenic signaling pathways in vitro and in vivo, with studies having been performed in dozens of tumour types. Here, we outline the major principles that have emerged from this literature.
Synergy between CDK4/6 inhibition and inhibition of other oncogenic kinases (e.g. those in the PI3K–AKT signalling pathway, the MAPK pathway, and upstream receptor tyrosine kinases such as epidermal growth factor receptor (EGFR), HER2, fibroblast growth factor receptors (FGFRs) and insulin-like growth factor 1 receptor (IGF1R)) is bi-directional. On one hand, persistent CDK4/6 enzymatic activity can mediate resistance to inhibition of oncogenic kinases153,173–177, and in breast cancer specifically, this is related to a failure of upstream kinase inhibitors to reduce cellular levels of cyclin D1153,173. Conversely, uninhibited hyperactivity of mitogenic pathways can mediate resistance to CDK4/6 inhibitors through a variety of mechanisms discussed below8,178–181. These two phenomena often co-occur, underpinning potent synergy between CDK4/6 inhibitors and inhibitors of upstream kinases known to be active in a particular tumour.
CDK4/6 inhibition often induces adaptive rewiring of kinase circuits in cancer cells (Figure 3a), enhancing their dependence on other signalling pathways. This has been demonstrated in a variety of cell-based and animal models: for example, CDK4/6 inhibition increases (i) phosphorylation and activity of HER receptors in HER2-positive breast cancer153,182; (ii) PI3K–AKT pathway activity in numerous breast cancer models8,170,173,180,182; (iii) IGF1R pathway activity in models of Ewing’s sarcoma and pancreatic carcinoma177,181; (iv) MAPK pathway activity in models of prostate carcinoma183; and (v) FGFR1 activity in models of KRAS-mutant lung cancer148. Several mechanisms underlie this adaptive rewiring. First, CDK4/6 can directly stimulate mTORC1 activity by binding and phosphorylating the tuberous sclerosis 2 (TSC2) tumour suppressor162,163,184, and partial inhibition of mTORC1 by CDK4/6 inhibitors can relieve feedback inhibition on upstream receptor tyrosine kinases153,185. Second, hypo-phosphorylation of RB by CDK4/6 inhibitors directly facilitates access of AKT to SIN1, a member of the mTORC2 complex, ultimately increasing AKT phosphorylation by mTORC2186. Third, CDK4/6 inhibitors can induce secretion of growth factors from tumour cells (possibly as part of the SASP), which stimulate signaling through receptor tyrosine kinases in an autocrine manner148,187.
Figure 3. Effect of CDK4 and CDK6 inhibition upon tumour cell-intrinsic and -extrinsic signalling pathways.
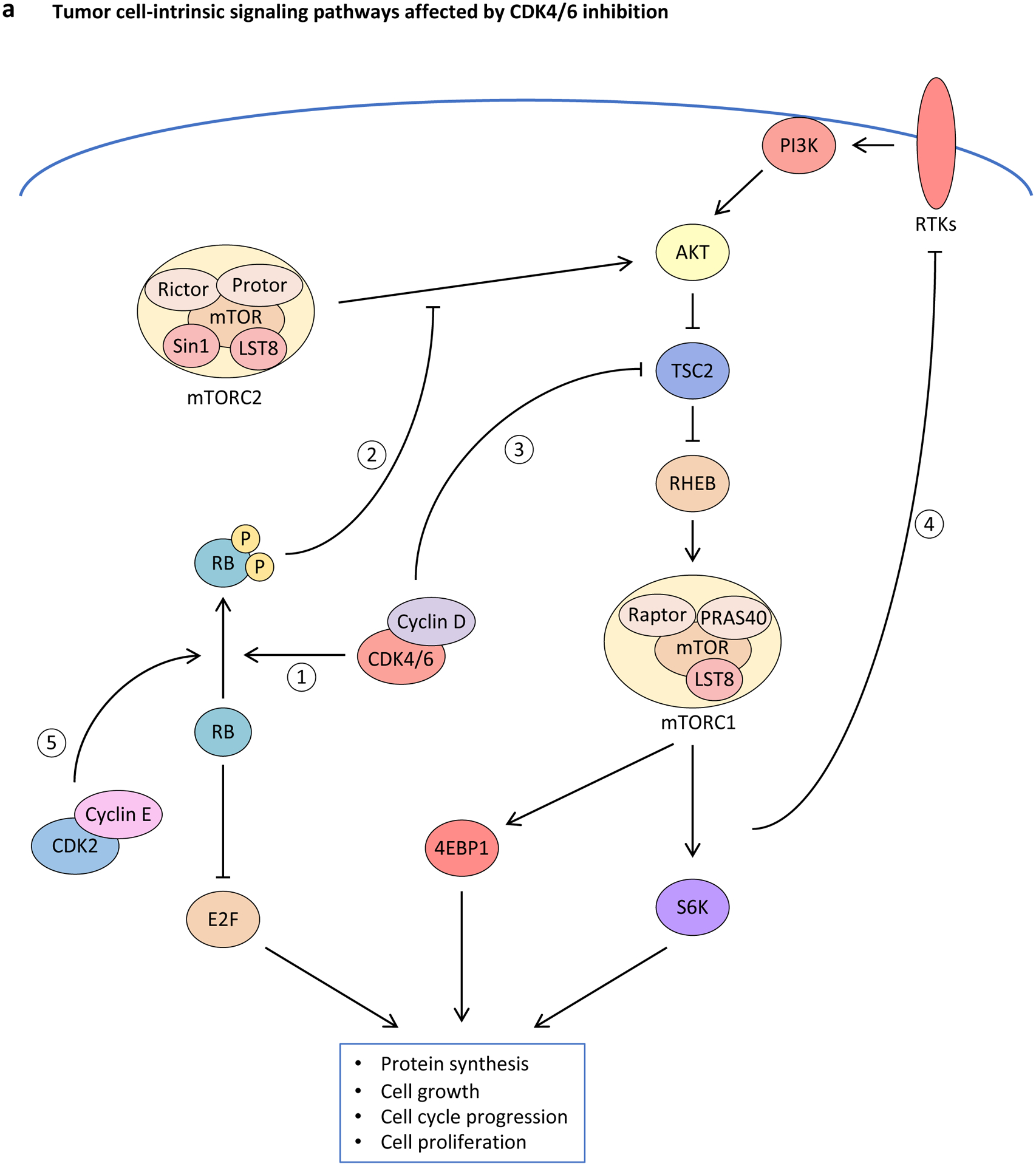
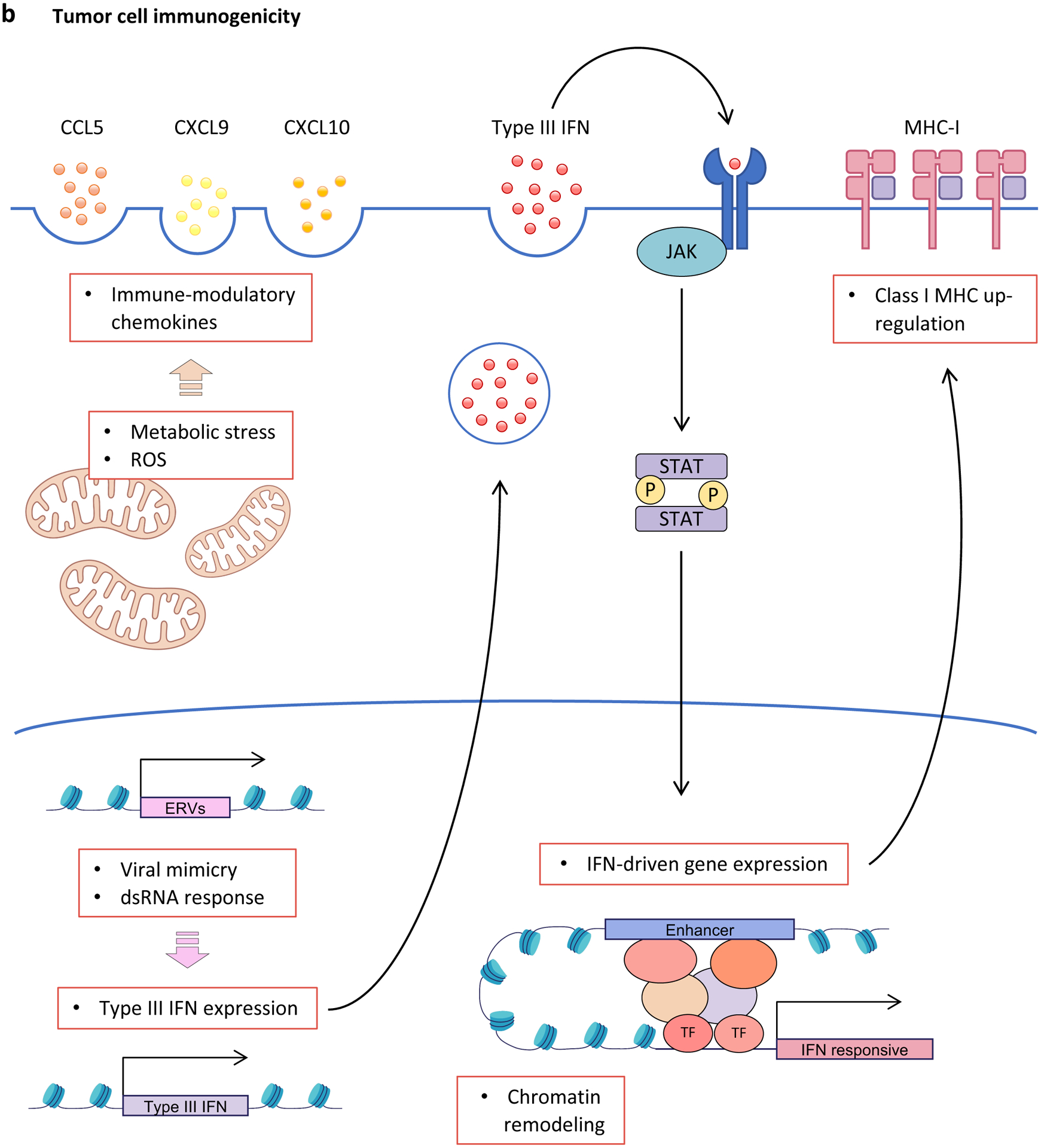
a, Inhibition of cyclin-dependent kinase 4 (CDK4) and CDK6 (CDK4/6) activity induces rewiring of multiple interconnected kinase circuits in tumour cells. First, blocking CDK4/6 inhibits RB phosphorylation (1), thereby preventing E2F-mediated cell cycle progression. However, multiple mechanisms may counteract these effects. For example, decreased phosphorylated-RB levels also lead to enhanced AKT phosphorylation by mTORC complex 2 (mTORC2) (2), which can stimulate cell survival mechanisms. Similarly, decreased CDK4/6 activity can down-regulate mTORC1 signalling via enhanced tuberous sclerosis 2 (TSC2) activity (3), which may lead to loss of negative-feedback regulation of receptor tyrosine kinase (RTK) signalling (4), resulting in up-regulated upstream PI3K–AKT pathway activity. In addition, compensatory CDK2-mediated RB phosphorylation (5) can stimulate cell cycle progression in the absence of CDK4/6 signalling. b, CDK4/6 inhibitors promote tumour cell immunogenicity via multiple mechanisms: (i) Metabolic stress and increased production of reactive oxygen species (ROS) up-regulate secretion of inflammatory chemokines CC-chemokine ligand 5 (CCL5), CXC-chemokine ligand 9 (CXCL9) and CXCL10; (ii) hypomethylation and therefore expression of endogenous retroviruses (ERVs) induces a double-stranded RNA (dsRNA) response, which leads to increased expression and secretion of type III interferon (IFN), activation of Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signalling, enhanced IFN-driven gene expression, and up-regulation of major histocompatibility complex (MHC) class I expression; and (iii) chromatin remodelling facilitates IFN-mediated expression of interferon-responsive genes. 4EBP1; eukaryotic translation initiation factor 4E binding protein 1; IFNLR1, interferon λ receptor 1; IL-10RB, interleukin-10 receptor subunit β; S6K, S6 kinase; TF, transcription factor.
In many instances, these adaptive responses are sufficient to drive acquired CDK4/6 inhibitor resistance, explaining the efficacy of co-targeting CDK4/6 and other mitogenic pathways and possibly the failure of CDK4/6 inhibitor monotherapy in some clinical trials188. A common theme is that upstream mitogenic signalling enhances the activity of CDK2, facilitating RB phosphorylation and S phase entry despite ongoing CDK4/6 blockade8,145,148,189,190. Several mechanisms for stimulation of mitogenic signaling-driven increase in CDK2 activity have been proposed, including (i) increased formation of atypical, enzymatically active cyclin D1–CDK2 complexes due to elevated cyclin D1 levels8,148; (ii) sequestration of p21 and/or p27 in cyclin D1–CDK4/6 complexes190,191; and (iii) direct downregulation of p27189. A second possibility is that increased upstream mitogenic signalling increases the activity of mTORC1, another regulator of S phase entry192, which drives proliferation in the face of sustained CDK4/6 inhibition193,194. Consistent with this, several studies have demonstrated synergistic efficacy from combined CDK4/6 and mTOR inhibition148,190,195,196. Finally, these two mechanisms could be connected, given that mTORC1 can directly enhance cyclin D1 and/or cyclin E1 translation190.
Cellular responses to combined inhibition of CDK4/6 and other oncogenic kinases can include cytostasis, apoptosis, or both. In cases where a combination therapy induces cytostasis, it is typically accompanied by greater RB hypo-phosphorylation than seen with either agent alone, accompanied by increased downregulation of E2F targets, and heightened senescence – all of which could be linked to suppression of CDK4/6 together with either CDK2 or mTORC1135,153,173–175,177,197–200. Frank apoptosis has also been observed in several in vitro and in vivo models8,176,180,196,201,202. However, the underlying molecular determinants of both of these two phenotypes are not well understood.
Immunological effects of CDK4/6 inhibition
Several preclinical studies have converged upon the notion that CDK4/6 inhibitors can stimulate anti-tumour immune responses through tumour cell-intrinsic and -extrinsic mechanisms. Although this was initially considered counterintuitive given concerns that CDK6 inhibition would dampen immune function by preventing T lymphocyte expansion130,203, it is now clear that these drugs induce an ‘inflamed’ tumour microenvironment, and that CD8+ and/or CD4+ T lymphocytes partially mediate therapeutic responses130,158,164,204–206.
The association between CDK4/6 hyperactivity and impaired immunogenicity in tumour cells is supported by clinical data from multiple tumour types. Tumour cells harbouring amplifications in CCND1 (multiple tumour types) or CDK4 (melanoma) express lower levels of major histocompatibility complex (MHC) class I genes and demonstrate resistance to immune checkpoint inhibitors (ICIs)130,207,208. Similarly, a tumour cell-specific gene expression signature reflecting CDK4/6 activity is associated with worse ICI response in patients with melanoma205. Finally, pharmacological CDK4/6 inhibition upregulates interferon-driven gene expression programs in early-stage luminal breast cancers101,130.
CDK4/6 inhibition enhances tumour cell immunogenicity through a variety of RB-dependent mechanisms (Figure 3b). First, these agents can induce a viral mimicry response in luminal breast cancer cells, characterised by activation of an interferon-driven gene expression program that fosters secretion of lymphokines and enhances antigen presentation to CD8+ T cells via MHC class I130,204. Second, drug-induced chromatin remodelling activates a subset of enhancers specifically predicted to regulate interferon-driven genes142. Third, CDK4/6 inhibitors can induce metabolic stress in tumour cells, which drives secretion of immunostimulatory chemokines206. Each of these mechanisms is likely linked to underlying therapy-induced senescence, and similar findings have been reported in mouse models of lung and pancreatic carcinomas induced into a senescence-like state by combined CDK4/6 and MEK inhibition146,209.
T lymphocyte activation is also a common feature of CDK4/6 inhibitor-treated solid cancers. Whilst this might in part relate to tumour cell secretion of chemoattractant cytokines, these agents can also directly stimulate T cell function. A consistent observation across studies has been a relative depletion of immunosuppressive Treg cells (Figure 4a), resulting in an increase in the CD8+:Treg cell ratio that favours immune activation130,164,206,210. This is largely explained by an increased sensitivity of Treg cells to the anti-proliferative effect of CDK4/6 inhibitors130. T lymphocytes from CDK4/6 inhibitor-treated mouse models of mammary and lung carcinoma models also show increased effector activity, reduced expression of numerous exhaustion markers, and increased expression of stimulatory receptors130,158,206 – related in part to direct stimulation of T cell effector function by the CDK6 substrates nuclear factor of activated T-cells 2 (NFAT2) and NFAT4158 (Figure 4b). Finally, recent preclinical and clinical data have described RB-dependent and independent mechanisms by which CDK4/6 inhibition promotes CD8+ T cell memory differentiation (Figure 4b), supporting the prior preclinical observation that these agents protect against tumour re-challenge and might confer long-lasting protective immunity130,211,212.
Figure 4. CDK4 and CDK6 inhibitors exert differential effects in distinct immune cell populations.
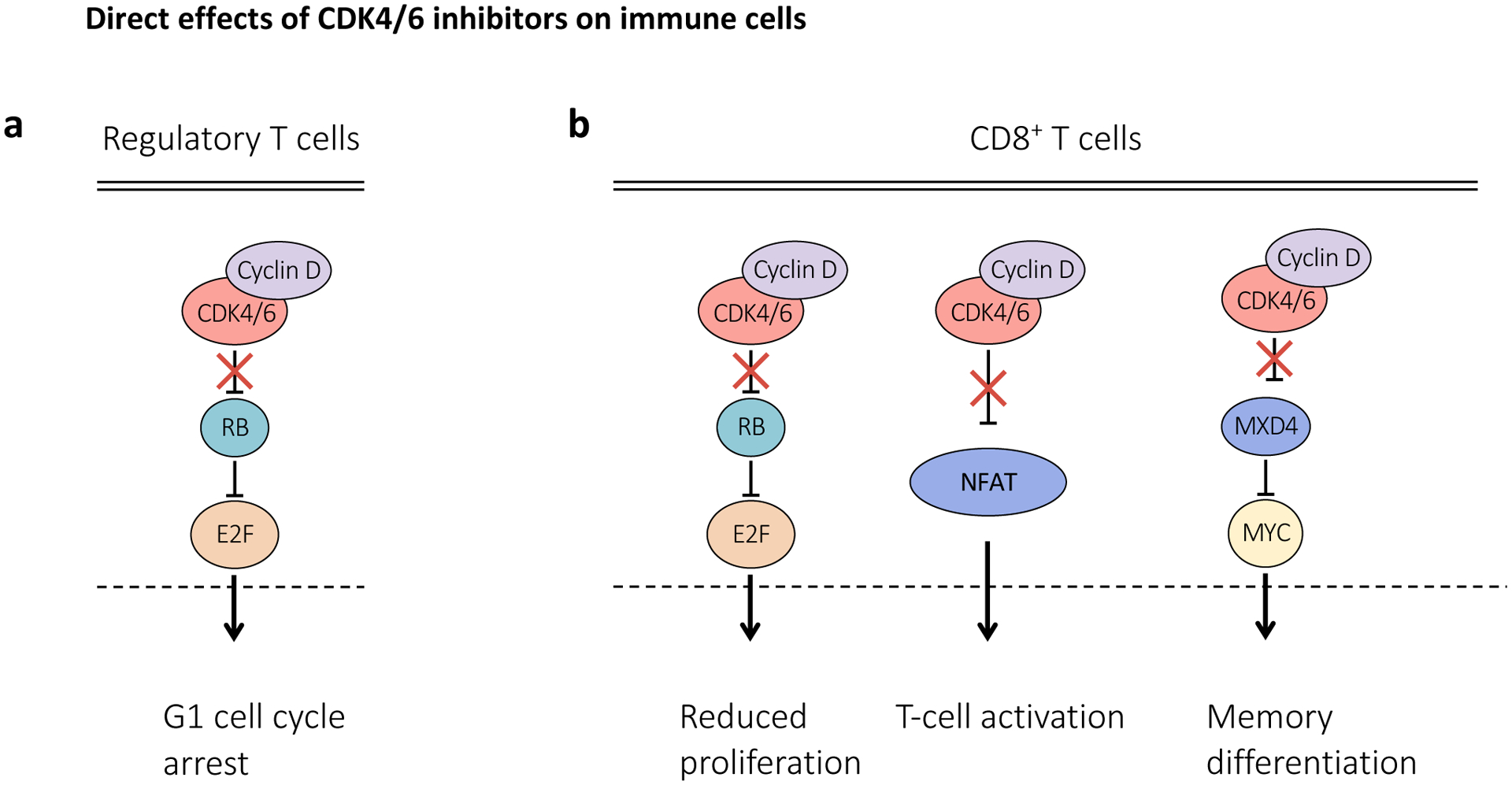
a, Upon treatment with cyclin-dependent kinase (CDK4) and CDK6 (CDK4/6) inhibitors, regulatory T (Treg) cells preferentially undergo cell cycle arrest, likely due to increased reliance on CDK4/6 signalling for cell cycle progression. b, CDK4/6 inhibition enhances CD8+ T-cell activation and induction of effector function via upregulation of nuclear factor of activated T-cells (NFAT) signalling. In addition, CDK4/6 inhibitors have been shown to promote memory T-cell differentiation through RB-dependent and RB-independent (increased MXD4 gene expression) mechanisms. MXD4, MAX dimerization protein 4.
A key challenge now is to leverage the above observations to develop new clinically effective therapies. Several studies, even one suggesting that CDK4/6 inhibitors might foster immune evasion161, have demonstrated synergy between CDK4/6 inhibition and immunotherapies (both ICIs and agonists of stimulatory receptors)130,158,204–206. Clinical development will require identification of ideal immunotherapy partners and tumour types for this approach. Notably, metastatic luminal breast cancer is notoriously unresponsive to immune-directed strategies, and it will be important to direct efforts towards other, more immunogenic tumour types.
A noteworthy example supporting clinical development of CDK4/6 inhibitors as immunostimulatory agents is a randomised trial demonstrating that the addition of trilaciclib to chemotherapy improved overall survival of patients with advanced triple-negative breast cancer213. Biomarker studies from this trial suggested that this unexpected result might have been driven by trilaciclib-mediated immune-stimulation, and a larger confirmatory trial is underway (NCT04799249)214.
Converting senescence to cell death
Like classically senescent cells, cancer cells that have entered CDK4/6 inhibitor-induced senescence show relative resistance to apoptotic insults in vitro130,135,139,142. This phenomenon is supported by the clinical observation that the addition of palbociclib to ET suppresses indices of apoptosis in luminal breast cancers105. Efforts are now underway to improve tumour eradication through the use of senolytics, agents that selectively kill senescent cells. This has been described as a ‘one-two punch’ approach, rendering a cancer cell senescent with a CDK4/6 inhibitor before killing it with a senolytic.
To date, anti-apoptotic BH3-family proteins have been implicated as the primary mediators of CDK4/6 inhibitor-induced apoptotic evasion. In luminal breast cancer cells, CDK4/6 inhibition induces the RB-dependent commissioning of a superenhancer spanning the BCL2L1 locus, increasing BCL2L1 expression and cellular levels of BCL-XL142. This in turn primes tumour cells away from apoptosis at the level of the inner mitochondrial membrane. Consistent with this, CDK4/6 inhibitor-pretreated tumour cells can be re-primed towards their apoptotic threshold and, in some instances, killed by agents that selectively inhibit BCL-XL142. Similarly, selective BCL-2 or dual BCL-2 and BCL-XL inhibitors also show senolytic activity when given after CDK4/6 inhibitor therapy182,210,215–217, and trials combining CDK4/6 and BCL-2 inhibitors in breast cancer are underway (e.g. NCT03900884)214. Although BCL-XL inhibitors induce significant thrombocytopenia, which has limited their clinical development, novel formulations that selectively target senescent cells are currently under development215.
Targeting tumour metabolism and autophagy
An additional function of the cyclin D1–CDK4 axis in mammary epithelial cells is the suppression of cellular autophagy218. Indeed, both genetic and pharmacological studies have shown that CDK4/6 pathway inhibition can induce autophagy in normal and malignant luminal mammary cells218,219. Similar observations have been made in RB-proficient tumour cells, where senescence and autophagy accompany one another in response to CDK4/6 inhibition137. These findings have prompted researchers to combine CDK4/6 inhibitors with inhibitors of autophagy (e.g., hydroxychloroquine and bafilomycin). Interestingly, such combinations demonstrate synergistic anti-tumour activity, manifest by increases in both senescence and apoptosis. Notably, drugs such as hydroxychloroquine can also promote release of CDK4/6 inhibitors from lysosomes in which they are sequestered, which may in part explain these observations220.
The impact of CDK4/6 inhibitors on tumour cell metabolism is an understudied topic, especially considering the profound metabolic derangements reported in senescent cells139. One study demonstrated RB-dependent metabolic reprogramming in pancreatic carcinoma cells in response to CDK4/6 inhibition, which was characterised by increased mitochondrial mass and consumption of glucose and glutamine221. Although this might explain the reported sensitivity of CDK4/6 inhibitor-treated cancer cells to glutaminase inhibition222, the underlying biological mechanisms require further study before their clinical applicability is determined.
Combined inhibition of CDK4/6 and epigenetic proteins
As the impact of CDK4/6 inhibitors on the cancer cell epigenome is increasingly understood, so too is interest in determining how this might be leveraged for therapeutic benefit. At the time of writing, promising activity has been reported in preclinical studies combining CDK4/6 inhibitors with drugs targeting epigenetic readers and writers that drive transcriptional activity (e.g., bromodomain-containing protein 4 (BRD4) and p300 and CREB binding protein (CBP))32,223,224, but information is limited.
Novel applications and future challenges
Combination with cytotoxic therapy
CDK4/6 inhibitors arrest RB-proficient cancer cells in G1, raising concerns that they might antagonise the effects of cytotoxic chemotherapy drugs, which typically exert their effects during S phase or G2/M. Several preclinical studies confirmed this notion, demonstrating that CDK4/6 inhibitor-induced proliferative arrest diminishes the efficacy of chemotherapy administered shortly thereafter119,129,225,226. Similar antagonism has been reported when administering CDK4/6 inhibitors prior to external beam radiation therapy227.
More recent studies, however, have challenged the idea that CDK4/6 inhibitors should not be combined with cytotoxic therapies by employing alternative dosing schedules that enhance efficacy in RB-proficient tumours. For example, administering microtubule stabilisers or DNA damaging agents prior to CDK4/6 inhibition was shown highly effective in in vitro and in vivo models of pancreatic carcinoma131. These findings could be explained by impaired recovery from chemotherapy-induced DNA damage due to suppressed expression of E2F target genes whose protein products mediate DNA repair by homologous recombination129,131 (Figure 5a). RB-independent effects of CDK4/6 inhibitors (e.g., inhibited phosphorylation of CDK4/6 substrates FOXM1 or FOXO3) might also be sufficient to impair DNA repair after chemotherapy. Similar findings have been reported when CDK4/6 inhibitors were given after radiotherapy in a variety of different tumour models228–230.
Figure 5. Novel approaches for the use of CDK4 and CDK6 inhibitors.
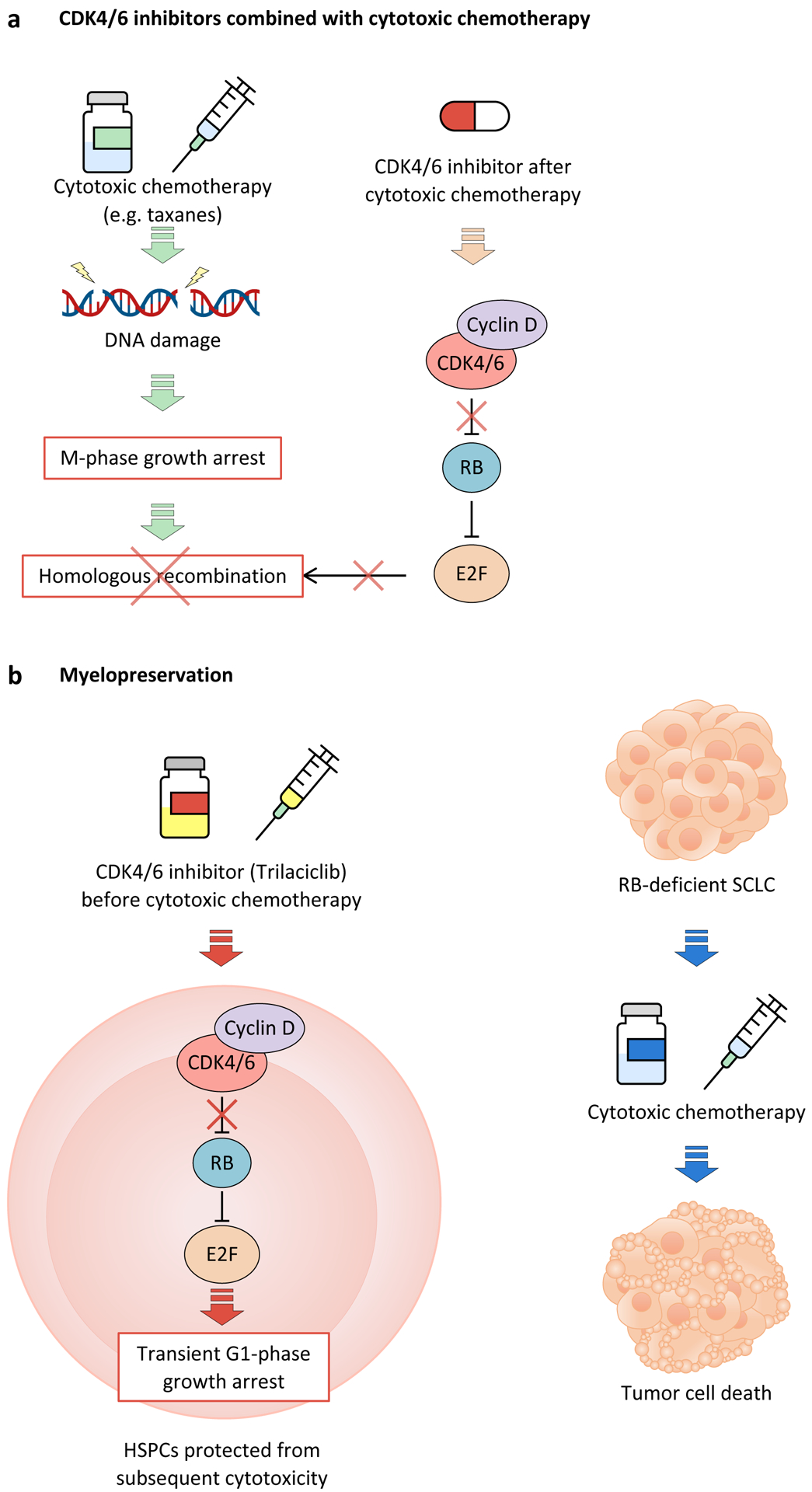
a, Administration of cyclin-dependent kinase 4 (CDK4) and CDK6 (CDK4/6) inhibitors after cytotoxic chemotherapy has been shown to enhance response by preventing E2F-mediated DNA damage repair after growth arrest during M-phase. Conversely, treatment with CDK4/6 inhibitors prior to chemotherapy would induce G1 cell cycle arrest, thus preventing chemotherapy-induced cytotoxicity that occurs during DNA replication or chromosome segregation. b, The CDK4/6 inhibitor trilaciclib can be used to protect hematopoietic stem and progenitor cells (HSPCs) from the cytotoxic effects of chemotherapy by inducing transient cell cycle arrest prior to treatment with chemotherapy. In this case, the target population would be patients with cancer types that are typically RB-deficient, such as small-cell-lung cancer (SCLC), so as to not interfere with cytotoxicity against tumour cells.
CDK4/6 inhibitors for supportive care
The idea that CDK4/6 inhibitors could protect normal tissue from the harmful effects of cytotoxic therapy was first proposed in 2010. Preclinical modelling demonstrated that pre-treatment of solid tumour-bearing mice with palbociclib reduces the multi-lineage haematological toxicity of total body irradiation or carboplatin chemotherapy226,227. The primary mechanism underlying this myelopreservation is an RB-dependent G1 arrest in haematopoietic stem and progenitor cells (HSPCs), reducing their vulnerability to DNA damaging agents. These findings prompted the development of trilaciclib as a myelopreservative agent. Intravenous trilaciclib therapy induces a rapid but transient G1 arrest in HSPCs and is now FDA-approved to reduce haematological toxicity for patients receiving cytotoxic chemotherapy for SCLC231 (Figure 5b). Of note, SCLCs are most commonly RB-deficient, thereby mitigating the risk of CDK4/6 inhibitor-chemotherapy antagonism in tumour cells.
The approval of trilaciclib will open new avenues for CDK4/6 inhibitors in the field of oncology supportive care, particularly for RB-deficient tumours. Indeed, preclinical studies have shown that palbociclib or ribociclib can protect against radiation-induced intestinal injury232, minimise hair follicle damage after taxane chemotherapy233, and mitigate acute kidney injury induced by cisplatin and other nephrotoxins234,235.
Prevention of cancer relapse
Oral CDK4/6 inhibitors are only approved for the treatment of metastatic malignancy, and a critical question is whether they can be successfully used in the adjuvant setting to prevent cancer relapse. Three clinical trials of high-risk, early-stage luminal breast cancer have provided contrasting results. The MonarchE trial demonstrated a significant improvement in disease-free survival when abemaciclib is added to endocrine therapy in patients with high-risk HR-positive early breast cancer. Conversely, the PALLAS and PENELOPE trials failed to show similar improvements with palbociclib, albeit in slightly different patient populations. The reasons for these differences are not clear, and it is hoped that longer follow-up of these and other studies will provide clarity on the role of CDK4/6 inhibitors in this setting236–238. From a biological standpoint, the unanswered question is what impact CDK4/6 inhibitors have on micrometastatic dormant tumour cells. In one preclinical study, a short course of CDK4/6 inhibitor therapy delayed, but did not prevent, recurrence of HER2-driven mammary carcinomas153, and it remains an open question in the clinical arena whether a cytostatic therapy can do any more than delay an inevitable relapse.
Selective CDK4 and CDK6 degradation
Novel technologies such as proteolysis targeting chimeras (PROTACs) have enabled synthesis of compounds that degrade CDK4/6 rather than inhibit their enzymatic activity. Although most currently available CDK4/6 degraders are based on the structure of approved CDK4/6 inhibitors, they show variable selectivity for degradation of CDK4, CDK6, or both90,239–243. Given this, these drugs are appealing for indications where selective inhibition of either CDK4 or CDK6 is desired. They also enable inhibition of kinase-independent effects of CDK4/6, which could offer further additional therapeutic potential244, and the ability to ‘dial-in’ simultaneous degradation of other important cancer drivers241. Several CDK4/6 degraders inhibit RB phosphorylation and induce G1 arrest as well, if not more potently than, traditional inhibitors90. Of note, however, current evidence suggests that the kinase-inhibitor resistant, thermostable CDK6 is also resistant to CDK6 degradation90.
De novo and acquired resistance
Despite the success of CDK4/6 inhibitors, therapeutic failures and acquired resistance represent major barriers to their more widespread implementation. In the case of advanced endocrine-sensitive breast cancer, most patients derive clinical benefit from CDK4/6 inhibitor therapy before eventually experiencing disease progression107,109,245. For other cancer types, a significant gap remains between the promise CDK4/6 inhibitors have shown in laboratory studies and their apparent clinical efficacy. The cellular and molecular mechanisms reported to drive CDK4/6 inhibitor resistance have been comprehensively reviewed elsewhere, and so we discuss them only at a high-level here246.
Many of the tumour cell-intrinsic resistance mechanisms ultimately alter or ‘rewire’ cellular CDK dependence. First, genomic alterations conferring loss of RB function (mutations and/or deletions in RB1) are, not surprisingly, associated with de novo and acquired resistance to CDK4/6 inhibitors in breast cancer72,247–250. Second, various resistance mechanisms faciliate CDK2-mediated S phase entry despite sutained inhibition of CDK4/6. These include amplification or heightened expression of cyclin E genes8,190,250,251, the formation of atypical cyclin D–CDK2 complexes that can phosphorylate RB5,8, and increased translation of cyclin E proteins as a result of heightened mTOR activity190. The importance of CDK2 as a pharmacological target in de novo and acquired CDK4/6 inhibitor resistance is underscored by the recent development of selective inhibitors targeting either CDK2252,253 or all interphase CDKs (i.e. CDK2/4/6 inhibitors)254. Third, a diverse series of mechanisms that increase CDK6 levels – including CDK6 amplification, upstream mutations (e.g. in FAT1) that increase CDK6 expression, and transforming growth factor β (TGFβ) pathway suppression – have all been linked to CDK4/6 inhibitor resistance72,255–257. How increased levels of wild-type CDK6 itself mediates CDK4/6 inhibitor resistance is a fascinating question, given that these agents are potent CDK6 inhibitors. A possible explanation is that CDK6 induces expression of INK4C, resulting in formation of enzymatically active INK4C–cyclin D–CDK4/6 complexes, which are not bound by CDK4/6 inhibitors257. Importantly, these complexes remain sensitive to CDK4/6 degraders257.
Given the inherent plasticity of the cell cycle machinery, it is also possible that cancer cells might develop mechanisms to proliferate even in the face of combined inhibition of all interphase CDKs. For example, mammalian cells lacking CDK2, CDK3, CDK4, and CDK6 can proliferate using CDK1, and it remains to be seen whether cancer cells treated with combined CDK2/4/6 inhibitors, for example, might co-opt this mechanism7. A potential strategy to overcome this would be pharmacological inhibition of CDK7, which has dual roles as a CDK-activating kinase (phosphorylating CDK1, CDK2, CDK4 and CDK6) and a mediator of RNA polymerase II-mediated transcription258,259. Indeed, selective CDK7 inhibitors are in clinical development and have shown promise in initial trials of CDK4/6 inhibitor-resistant luminal breast cancer260.
Laboratory studies in several cancer types have also uncovered numerous other mechanisms underlying acquired CDK4/6 inhibitor resistance. Some of these are tumour-cell intrinsic and include drug-induced activation of upstream mitogenic signalling pathways (described earlier), drug sequestration in the tumour cell lysosomal compartment220,261, and changes in chromatin modifiers that faciliate re-expression of E2F target genes32,262. In addition, therapy-induced stromal changes, such as fibroblast senescence, have also been implicated to drive resumption of tumour cell proliferation despite CDK4/6 inhibition140. However, none of these mechanisms have been looked for or identified in clinical specimens of CDK4/6 inhibitor-resistant cancers.
A number of important yet unanswered questions face the CDK4/6 inhibitor resistance field. First, it is not clear how common genomic alterations found in clinical specimens of CDK4/6 inhibitor-resistant breast cancer (i.e. mutations and copy number alterations) actually drive resistance, as opposed to simply co-occur at the time of resistance. Estimates of the frequency of newly emergent mutations in CDK4/6 inhibitor-resistant breast cancers vary markedly, and resolving this issue is important given the plethora of non-genetic mechanisms reported and the tendency for clinical investigators to rely on DNA sequencing when studying drug resistant cancers248,250. Second, the extent to which preclinical mechanisms of breast cancer resistance to CDK4/6 inhibitor monotherapy can be extrapolated to the clinic, where patients are treated with combined CDK4/6 inhibition and endocrine therapy, is not known. For this reason, studies focusing on resistance to combination therapy are needed, as is a greater understanding of how these two treatments interact. Finally, it is likely that resistance mechanisms identified in breast cancer are not universally applicable to other cancer types, and lineage- and mutation-specific drivers of cell cycle progression further complicate progress in the field.
Conclusions and future perspectives
A significant amount of data regarding the use of CDK4/6 inhibitors in cancer has accumulated in recent years. Important studies in preclinical models have shed light into the cellular and molecular mechanisms underlying the basis for their success in the clinic. Validating these findings in clinical settings will be an important step in guiding the design of further trials based on preclinical results, especially as there is still a disconnect between preclinical results and clinical development strategies. Addressing this concern will also help identify and prioritize specific areas for further development. Indeed, the large number of clinical trials currently evaluating combinations of CDK4/6 inhibitors with other agents – both FDA-approved and those under development – underscores the importance of this challenge. Moreover, molecular pathways and synergies described in vitro may not be recapitulated in in vivo settings due to the impact of different microenvironments and other variables, such as genetic and epigenetic alterations. Another important consideration will be the potential toxicity of combination therapies, which could severely limit clinical success, as has already been observed in recent trials of combination CDK4/6 and PI3K or mTOR inhibitors. Critically, it will be important to consider the diverse determinants of CDK4/6 inhibitor response, as described by our conceptual framework, when designing future clinical trials with these agents.
Supplementary Material
Acknowledgements
SG is a Snow Fellow of the Snow Medical Research Foundation. SG receives also research funding support from the National Health and Medical Research Council of Australia (Investigator Grant GNT1177357), the US National Institutes of Health (P50 CA165962-06A1), and The Mark Foundation (ASPIRE award). JJZ acknowledges funding from the Breast Cancer Research Foundation (BCRF), DOD CDMRP W81XWH-18-1-0491, National Institutes of Health (NIH)/National Cancer Institute (NCI) CA168504,CA165962, CA203655 and R35 CA210057.
Competing interests
SG has received funding to support laboratory research from Eli Lilly and G1 Therapeutics. SG has served as a paid advisor to Eli Lilly, G1 Therapeutics and Pfizer. JSB is a scientific consultant for Geode Therapeutics. JJZ is a co-founder and board director of Crimson Biotech and Geode Therapeutics.
Glossary
- Mitogenic signals
Signals arising from small extracellular proteins or peptides that induce a cell to begin cell division.
- Interphase
The portion of the cell cycle including G1, S and G2 phases, but excluding M phase; interphase begins at the end of one mitotic division and ends at the beginning of the next mitotic division.
- Endocrine therapy (ET)
A therapy that alters the effect of sex steroid hormones in cancer cells; in breast cancer, ET is used to block the effect of oestrogen in hormone receptor-positive breast tumours.
- Lymphokines
A type of cytokine (i.e., a small secreted protein with autocrine, paracrine and/or endocrine functions) produced and secreted by lymphocytes.
- Thrombocytopenia
Low blood count of platelets (thrombocytes), a type of blood cell important for clotting.
- Homologous recombination
The exchange of genetic material between homologous chromosomes; an important mechanism used by cells to repair deleterious DNA damages such as double strand breaks and collapsed replication forks.
References
- 1.Asghar U, Witkiewicz AK, Turner NC & Knudsen ES The history and future of targeting cyclin-dependent kinases in cancer therapy. Nature reviews. Drug discovery 14, 130–146, doi: 10.1038/nrd4504 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Losiewicz MD, Carlson BA, Kaur G, Sausville EA & Worland PJ Potent inhibition of CDC2 kinase activity by the flavonoid L86–8275. Biochem Biophys Res Commun 201, 589–595, doi: 10.1006/bbrc.1994.1742 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Sanchez-Martinez C, Lallena MJ, Sanfeliciano SG & de Dios A Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorg Med Chem Lett 29, 126637, doi: 10.1016/j.bmcl.2019.126637 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Malumbres M Cyclin-dependent kinases. Genome biology 15, 122, doi: 10.1186/gb4184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malumbres M et al. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 118, 493–504 (2004). [DOI] [PubMed] [Google Scholar]; Using CDK4/CDK6 double knockout mice, this study established that mammalian cells can proliferate in the absence of both CDK4 and CDK6, demonstrating a plasticity of cyclin-CDK networks that has implications for CDK4/6 inhibitor resistance.
- 6.Xiong Y, Zhang H & Beach D D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell 71, 505–514, doi: 10.1016/0092-8674(92)90518-h (1992). [DOI] [PubMed] [Google Scholar]
- 7.Santamaria D et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815, doi: 10.1038/nature06046 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Herrera-Abreu MT et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer research 76, 2301–2313, doi: 10.1158/0008-5472.CAN-15-0728 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Leary B, Finn RS & Turner NC Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13, 417–430, doi: 10.1038/nrclinonc.2016.26 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Puyol M et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer cell 18, 63–73, doi: 10.1016/j.ccr.2010.05.025 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Yu Q et al. Requirement for CDK4 kinase function in breast cancer. Cancer cell 9, 23–32, doi: 10.1016/j.ccr.2005.12.012 (2006). [DOI] [PubMed] [Google Scholar]; This research established that cyclin D1-associated CDK4 is necessary for the formation and continued proliferation of mammary carcinomas driven by ErbB2.
- 12.Choi YJ et al. The requirement for cyclin D function in tumor maintenance. Cancer cell 22, 438–451, doi: 10.1016/j.ccr.2012.09.015 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provided in vivo evidence that CDK4/6 inhibitors induce a senescence-like phenotype in mammary carcinomas, but cellular apoptosis in T-cell leukemias.
- 13.Fry DW et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Molecular cancer therapeutics 3, 1427–1438 (2004). [PubMed] [Google Scholar]
- 14.Spring LM et al. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet 395, 817–827, doi: 10.1016/S0140-6736(20)30165-3 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Alonso D & Malumbres M Mammalian cell cycle cyclins. Semin Cell Dev Biol 107, 28–35, doi: 10.1016/j.semcdb.2020.03.009 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Xiong Y, Connolly T, Futcher B & Beach D Human D-type cyclin. Cell 65, 691–699 (1991). [DOI] [PubMed] [Google Scholar]
- 17.Lew DJ, Dulic V & Reed SI Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell 66, 1197–1206, doi: 10.1016/0092-8674(91)90042-w (1991). [DOI] [PubMed] [Google Scholar]
- 18.Matsushime H, Roussel MF, Ashmun RA & Sherr CJ Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell 65, 701–713, doi: 10.1016/0092-8674(91)90101-4 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Pardee AB A restriction point for control of normal animal cell proliferation. Proceedings of the National Academy of Sciences of the United States of America 71, 1286–1290, doi: 10.1073/pnas.71.4.1286 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aktas H, Cai H & Cooper GM Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Molecular and cellular biology 17, 3850–3857, doi: 10.1128/MCB.17.7.3850 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peeper DS et al. Ras signalling linked to the cell-cycle machinery by the retinoblastoma protein. Nature 386, 177–181, doi: 10.1038/386177a0 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Diehl JA, Cheng M, Roussel MF & Sherr CJ Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes & development 12, 3499–3511 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsushime H et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell 71, 323–334 (1992). [DOI] [PubMed] [Google Scholar]
- 24.Kato J, Matsushime H, Hiebert SW, Ewen ME & Sherr CJ Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes & development 7, 331–342 (1993). [DOI] [PubMed] [Google Scholar]
- 25.Meyerson M & Harlow E Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Molecular and cellular biology 14, 2077–2086, doi: 10.1128/mcb.14.3.2077-2086.1994 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weintraub SJ, Prater CA & Dean DC Retinoblastoma protein switches the E2F site from positive to negative element. Nature 358, 259–261, doi: 10.1038/358259a0 (1992). [DOI] [PubMed] [Google Scholar]
- 27.Weintraub SJ et al. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature 375, 812–815, doi: 10.1038/375812a0 (1995). [DOI] [PubMed] [Google Scholar]
- 28.Hiebert SW, Chellappan SP, Horowitz JM & Nevins JR The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes & development 6, 177–185 (1992). [DOI] [PubMed] [Google Scholar]
- 29.Sellers WR, Rodgers JW & Kaelin WG Jr. A potent transrepression domain in the retinoblastoma protein induces a cell cycle arrest when bound to E2F sites. Proceedings of the National Academy of Sciences of the United States of America 92, 11544–11548, doi: 10.1073/pnas.92.25.11544 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chicas A et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proceedings of the National Academy of Sciences of the United States of America 109, 8971–8976, doi: 10.1073/pnas.1119836109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo RX, Postigo AA & Dean DC Rb interacts with histone deacetylase to repress transcription. Cell 92, 463–473, doi: 10.1016/s0092-8674(00)80940-x (1998). [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y et al. HDAC5 Loss Impairs RB Repression of Pro-Oncogenic Genes and Confers CDK4/6 Inhibitor Resistance in Cancer. Cancer research 81, 1486–1499, doi: 10.1158/0008-5472.CAN-20-2828 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Harbour JW, Luo RX, Dei Santi A, Postigo AA & Dean DC Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98, 859–869 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Goodrich DW, Wang NP, Qian YW, Lee EY & Lee WH The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell 67, 293–302 (1991). [DOI] [PubMed] [Google Scholar]
- 35.Serrano M, Hannon GJ & Beach D A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366, 704–707, doi: 10.1038/366704a0 (1993). [DOI] [PubMed] [Google Scholar]
- 36.Stepanova L, Leng X, Parker SB & Harper JW Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes & development 10, 1491–1502, doi: 10.1101/gad.10.12.1491 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Zhao Q, Boschelli F, Caplan AJ & Arndt KT Identification of a conserved sequence motif that promotes Cdc37 and cyclin D1 binding to Cdk4. J Biol Chem 279, 12560–12564, doi: 10.1074/jbc.M308242200 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Lamphere L et al. Interaction between Cdc37 and Cdk4 in human cells. Oncogene 14, 1999–2004, doi: 10.1038/sj.onc.1201036 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Harper JW, Adami GR, Wei N, Keyomarsi K & Elledge SJ The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816, doi: 10.1016/0092-8674(93)90499-g (1993). [DOI] [PubMed] [Google Scholar]
- 40.Xiong Y et al. p21 is a universal inhibitor of cyclin kinases. Nature 366, 701–704, doi: 10.1038/366701a0 (1993). [DOI] [PubMed] [Google Scholar]
- 41.Toyoshima H & Hunter T p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78, 67–74, doi: 10.1016/0092-8674(94)90573-8 (1994). [DOI] [PubMed] [Google Scholar]
- 42.LaBaer J et al. New functional activities for the p21 family of CDK inhibitors. Genes & development 11, 847–862 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Guiley KZ et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 366, doi: 10.1126/science.aaw2106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study proposes an alternate hypothesis for CDK4/6 inhibitor mechanism of action, positing that these agents do not directly inhibit the CDK4 kinase, but rather sequester monomeric CDK4, thereby freeing p21 to bind and inhibit CDK2.
- 44.McConnell BB, Gregory FJ, Stott FJ, Hara E & Peters G Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Molecular and cellular biology 19, 1981–1989, doi: 10.1128/MCB.19.3.1981 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bagui TK, Mohapatra S, Haura E & Pledger WJ P27Kip1 and p21Cip1 are not required for the formation of active D cyclin-cdk4 complexes. Molecular and cellular biology 23, 7285–7290, doi: 10.1128/MCB.23.20.7285-7290.2003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sugimoto M et al. Activation of cyclin D1-kinase in murine fibroblasts lacking both p21(Cip1) and p27(Kip1). Oncogene 21, 8067–8074, doi: 10.1038/sj.onc.1206019 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Cerqueira A et al. Genetic characterization of the role of the Cip/Kip family of proteins as cyclin-dependent kinase inhibitors and assembly factors. Molecular and cellular biology 34, 1452–1459, doi: 10.1128/MCB.01163-13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pennycook BR & Barr AR Palbociclib-mediated cell cycle arrest can occur in the absence of the CDK inhibitors p21 and p27. Open Biol 11, 210125, doi: 10.1098/rsob.210125 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; Using a series of genetic manipulations in cell-based models, this study demonstrated that p21 and p27 are not required for cells to enter or remain in palbociclib-mediated G1 arrest (providing an alternate hypothesis to that presented in reference 43).
- 49.Tetsu O & McCormick F Proliferation of cancer cells despite CDK2 inhibition. Cancer cell 3, 233–245, doi: 10.1016/s1535-6108(03)00053-9 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Gong X et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer cell 32, 761–776 e766, doi: 10.1016/j.ccell.2017.11.006 (2017). [DOI] [PubMed] [Google Scholar]; This study provides a reference dataset for the sensitivity of human cancer cell lines to CDK4/6 inhibitors (abemaciclib, n = 560; palbociclib, n = 492), and highlights a series of genomic alterations that are associated with sensitivity or resistance to these agents.
- 51.Seto M et al. Gene rearrangement and overexpression of PRAD1 in lymphoid malignancy with t(11;14)(q13;q32) translocation. Oncogene 7, 1401–1406 (1992). [PubMed] [Google Scholar]
- 52.Wiestner A et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 109, 4599–4606, doi: 10.1182/blood-2006-08-039859 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li M et al. Kaposi’s sarcoma-associated herpesvirus encodes a functional cyclin. J Virol 71, 1984–1991, doi: 10.1128/JVI.71.3.1984-1991.1997 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santra MK, Wajapeyee N & Green MR F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 459, 722–725, doi: 10.1038/nature08011 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simoneschi D et al. CRL4(AMBRA1) is a master regulator of D-type cyclins. Nature 592, 789–793, doi: 10.1038/s41586-021-03445-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moreno-Bueno G et al. Cyclin D1 gene (CCND1) mutations in endometrial cancer. Oncogene 22, 6115–6118, doi: 10.1038/sj.onc.1206868 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Barbash O et al. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer cell 14, 68–78, doi: 10.1016/j.ccr.2008.05.017 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benzeno S et al. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene 25, 6291–6303, doi: 10.1038/sj.onc.1209644 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Jones SM & Kazlauskas A Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nature cell biology 3, 165–172, doi: 10.1038/35055073 (2001). [DOI] [PubMed] [Google Scholar]
- 60.Choi YJ & Anders L Signaling through cyclin D-dependent kinases. Oncogene 33, 1890–1903, doi: 10.1038/onc.2013.137 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Schmidt M et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Molecular and cellular biology 22, 7842–7852, doi: 10.1128/MCB.22.22.7842-7852.2002 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muise-Helmericks RC et al. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem 273, 29864–29872, doi: 10.1074/jbc.273.45.29864 (1998). [DOI] [PubMed] [Google Scholar]
- 63.Averous J, Fonseca BD & Proud CG Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene 27, 1106–1113, doi: 10.1038/sj.onc.1210715 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Rosenwald IB, Lazaris-Karatzas A, Sonenberg N & Schmidt EV Elevated levels of cyclin D1 protein in response to increased expression of eukaryotic initiation factor 4E. Molecular and cellular biology 13, 7358–7363, doi: 10.1128/mcb.13.12.7358-7363.1993 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prall OW, Sarcevic B, Musgrove EA, Watts CK & Sutherland RL Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem 272, 10882–10894, doi: 10.1074/jbc.272.16.10882 (1997). [DOI] [PubMed] [Google Scholar]; This study demonstrated that estradiol (E2) directly stimulates CCND1 expression in human breast cancer cells, clarifying a key mechanism underlying E2-driven mitogenic signaling and strengthening the rationale for using CDK4/6 inhibitors to treat HR-positive breast cancer.
- 66.Xu Y, Chen SY, Ross KN & Balk SP Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer research 66, 7783–7792, doi: 10.1158/0008-5472.Can-05-4472 (2006). [DOI] [PubMed] [Google Scholar]
- 67.Tetsu O & McCormick F Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426, doi: 10.1038/18884 (1999). [DOI] [PubMed] [Google Scholar]
- 68.Bouchard C et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J 18, 5321–5333, doi: 10.1093/emboj/18.19.5321 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ronchini C & Capobianco AJ Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic). Molecular and cellular biology 21, 5925–5934, doi: 10.1128/mcb.21.17.5925-5934.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sicinska E et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer cell 4, 451–461, doi: 10.1016/s1535-6108(03)00301-5 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Mizuno T et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 31, 5117–5122, doi: 10.1038/onc.2012.5 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Li Z et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer cell 34, 893–905 e898, doi: 10.1016/j.ccell.2018.11.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li F et al. YAP1-Mediated CDK6 Activation Confers Radiation Resistance in Esophageal Cancer - Rationale for the Combination of YAP1 and CDK4/6 Inhibitors in Esophageal Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 25, 2264–2277, doi: 10.1158/1078-0432.Ccr-18-1029 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Sicinski P et al. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82, 621–630 (1995). [DOI] [PubMed] [Google Scholar]
- 75.Jeselsohn R et al. Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer cell 17, 65–76, doi: 10.1016/j.ccr.2009.11.024 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sawai CM et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer cell 22, 452–465, doi: 10.1016/j.ccr.2012.09.016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang TC et al. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature 369, 669–671, doi: 10.1038/369669a0 (1994). [DOI] [PubMed] [Google Scholar]
- 78.Musgrove EA, Lee CS, Buckley MF & Sutherland RL Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proceedings of the National Academy of Sciences of the United States of America 91, 8022–8026, doi: 10.1073/pnas.91.17.8022 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anders L et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer cell 20, 620–634, doi: 10.1016/j.ccr.2011.10.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Musgrove EA, Caldon CE, Barraclough J, Stone A & Sutherland RL Cyclin D as a therapeutic target in cancer. Nature reviews. Cancer 11, 558–572, doi: 10.1038/nrc3090 (2011). [DOI] [PubMed] [Google Scholar]
- 81.Yu Q, Geng Y & Sicinski P Specific protection against breast cancers by cyclin D1 ablation. Nature 411, 1017–1021, doi: 10.1038/35082500 (2001). [DOI] [PubMed] [Google Scholar]; This study demonstrated a specific role for cyclin D1 in the initiation of murine mammary tumours driven by Neu or Ras, signifying a potential role for inhibiting cyclin D1 function in breast cancer.
- 82.Narita M et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716 (2003). [DOI] [PubMed] [Google Scholar]
- 83.Wang H et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 546, 426–430, doi: 10.1038/nature22797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.VanderWel SN et al. Pyrido[2,3-d]pyrimidin-7-ones as specific inhibitors of cyclin-dependent kinase 4. J Med Chem 48, 2371–2387, doi: 10.1021/jm049355+ (2005). [DOI] [PubMed] [Google Scholar]
- 85.Besong G et al. Pyrrolopyrimidine compounds and their uses. USA patent (2010). [Google Scholar]
- 86.Gelbert LM et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs 32, 825–837, doi: 10.1007/s10637-014-0120-7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sanchez-Martinez C, Gelbert LM, Lallena MJ & de Dios A Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg Med Chem Lett 25, 3420–3435, doi: 10.1016/j.bmcl.2015.05.100 (2015). [DOI] [PubMed] [Google Scholar]
- 88.Bisi JE, Sorrentino JA, Roberts PJ, Tavares FX & Strum JC Preclinical Characterization of G1T28: A Novel CDK4/6 Inhibitor for Reduction of Chemotherapy-Induced Myelosuppression. Molecular cancer therapeutics 15, 783–793, doi: 10.1158/1535-7163.Mct-15-0775 (2016). [DOI] [PubMed] [Google Scholar]
- 89.He S et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Science translational medicine 9, doi: 10.1126/scitranslmed.aal3986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated that trilaciclib induces transient inhibition of hematopoietic stem cells in mice and humans, providing a rationale for its use as a myelopreservative in patients receiving cytotoxic chemotherapy.
- 90.Wu X et al. Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nature Cancer 2, 429–443, doi: 10.1038/s43018-021-00174-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sumi NJ, Kuenzi BM, Knezevic CE, Remsing Rix LL & Rix U Chemoproteomics Reveals Novel Protein and Lipid Kinase Targets of Clinical CDK4/6 Inhibitors in Lung Cancer. ACS Chem Biol 10, 2680–2686, doi: 10.1021/acschembio.5b00368 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hafner M et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem Biol 26, 1067–1080.e1068, doi: 10.1016/j.chembiol.2019.05.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Litchfield LM et al. Combined inhibition of PIM and CDK4/6 suppresses both mTOR signaling and Rb phosphorylation and potentiates PI3K inhibition in cancer cells. Oncotarget 11, 1478–1492, doi: 10.18632/oncotarget.27539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Torres-Guzmán R et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 8, 69493–69507, doi: 10.18632/oncotarget.17778 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cousins EM et al. Competitive Kinase Enrichment Proteomics Reveals that Abemaciclib Inhibits GSK3β and Activates WNT Signaling. Mol Cancer Res 16, 333–344, doi: 10.1158/1541-7786.Mcr-17-0468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Knudsen ES, Hutcheson J, Vail P & Witkiewicz AK Biological specificity of CDK4/6 inhibitors: dose response relationship, in vivo signaling, and composite response signature. Oncotarget 8, 43678–43691, doi: 10.18632/oncotarget.18435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Leonard JP et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 119, 4597–4607, doi: 10.1182/blood-2011-10-388298 (2012). [DOI] [PubMed] [Google Scholar]
- 98.Finn RS et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast cancer research : BCR 11, R77, doi: 10.1186/bcr2419 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This preclinical study demonstrated that human breast cancer cell lines representing luminal, HR+ subtypes (both HER2-positive and HER2-negative) are more sensitive to CDK4/6 inhibition than those representing non-luminal or basal subtypes.
- 99.DeMichele A et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clinical cancer research : an official journal of the American Association for Cancer Research 21, 995–1001, doi: 10.1158/1078-0432.CCR-14-2258 (2015). [DOI] [PubMed] [Google Scholar]
- 100.Patnaik A et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer discovery 6, 740–753, doi: 10.1158/2159-8290.CD-16-0095 (2016). [DOI] [PubMed] [Google Scholar]; This phase 1/2 clinical trial demonstrated clinical activity for abemaciclib monotherapy in subsets of patients with breast cancer, non-small cell lung cancer, and melanoma.
- 101.Hurvitz S et al. Treatment with abemaciclib modulates the immune response in gene expression analysis of the neoMONARCH neoadjuvant study of abemaciclib in postmenopausal women with HR+, HER2 negative breast cancer. Proceedings of the 2018 San Antonio Breast Cancer Symposium, San Antonio TX. Cancer Res 79, Abstr nr PD2–10 (2019). [Google Scholar]
- 102.Malorni L et al. Palbociclib as single agent or in combination with the endocrine therapy received before disease progression for estrogen receptor-positive, HER2-negative metastatic breast cancer: TREnd trial. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 29, 1748–1754, doi: 10.1093/annonc/mdy214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dickler MN et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(−) Metastatic Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 23, 5218–5224, doi: 10.1158/1078-0432.CCR-17-0754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ma CX et al. NeoPalAna: Neoadjuvant Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, and Anastrozole for Clinical Stage 2 or 3 Estrogen Receptor-Positive Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 23, 4055–4065, doi: 10.1158/1078-0432.CCR-16-3206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Johnston S et al. Randomized Phase II Study Evaluating Palbociclib in Addition to Letrozole as Neoadjuvant Therapy in Estrogen Receptor-Positive Early Breast Cancer: PALLET Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 37, 178–189, doi: 10.1200/jco.18.01624 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Tripathy D et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. The lancet oncology 19, 904–915, doi: 10.1016/S1470-2045(18)30292-4 (2018). [DOI] [PubMed] [Google Scholar]
- 107.Finn RS et al. Palbociclib and Letrozole in Advanced Breast Cancer. The New England journal of medicine 375, 1925–1936, doi: 10.1056/NEJMoa1607303 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Hortobagyi GN et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. The New England journal of medicine, doi: 10.1056/NEJMoa1609709 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Goetz MP et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 35, 3638–3646, doi: 10.1200/JCO.2017.75.6155 (2017). [DOI] [PubMed] [Google Scholar]; Collectively, references 107–109 present randomized phase 3 clinical trials indicating that the addition of CDK4/6 inhibitors to first-line endocrine therapy significantly prolongs progression-free survival for patients with advanced, HR-positive breast cancer.
- 110.Turner NC et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. The New England journal of medicine 379, 1926–1936, doi: 10.1056/NEJMoa1810527 (2018). [DOI] [PubMed] [Google Scholar]
- 111.Sledge GW Jr. et al. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA oncology 6, 116–124, doi: 10.1001/jamaoncol.2019.4782 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Slamon DJ et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. The New England journal of medicine 382, 514–524, doi: 10.1056/NEJMoa1911149 (2020). [DOI] [PubMed] [Google Scholar]
- 113.Sledge GW Jr. et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 35, 2875–2884, doi: 10.1200/JCO.2017.73.7585 (2017). [DOI] [PubMed] [Google Scholar]
- 114.Turner NC et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. The New England journal of medicine 373, 209–219, doi: 10.1056/NEJMoa1505270 (2015). [DOI] [PubMed] [Google Scholar]
- 115.Toogood PL et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem 48, 2388–2406, doi: 10.1021/jm049354h (2005). [DOI] [PubMed] [Google Scholar]
- 116.Dean JL, Thangavel C, McClendon AK, Reed CA & Knudsen ES Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 29, 4018–4032, doi: 10.1038/onc.2010.154 (2010). [DOI] [PubMed] [Google Scholar]
- 117.Arnedos M et al. Modulation of Rb phosphorylation and antiproliferative response to palbociclib: the preoperative-palbociclib (POP) randomized clinical trial. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 29, 1755–1762, doi: 10.1093/annonc/mdy202 (2018). [DOI] [PubMed] [Google Scholar]
- 118.Infante JR et al. A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 5696–5705, doi: 10.1158/1078-0432.Ccr-16-1248 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Konecny GE et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 17, 1591–1602, doi: 10.1158/1078-0432.Ccr-10-2307 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dimova DK & Dyson NJ The E2F transcriptional network: old acquaintances with new faces. Oncogene 24, 2810–2826, doi: 10.1038/sj.onc.1208612 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Kimura H, Nakamura T, Ogawa T, Tanaka S & Shiota K Transcription of mouse DNA methyltransferase 1 (Dnmt1) is regulated by both E2F-Rb-HDAC-dependent and -independent pathways. Nucleic acids research 31, 3101–3113 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ren B et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes & development 16, 245–256, doi: 10.1101/gad.949802 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Annicotte JS et al. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nature cell biology 11, 1017–1023, doi: 10.1038/ncb1915 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Blanchet E et al. E2F transcription factor-1 regulates oxidative metabolism. Nature cell biology 13, 1146–1152, doi: 10.1038/ncb2309 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tsai KY et al. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell 2, 293–304, doi: 10.1016/s1097-2765(00)80274-9 (1998). [DOI] [PubMed] [Google Scholar]
- 126.Ziebold U, Reza T, Caron A & Lees JA E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes & development 15, 386–391, doi: 10.1101/gad.858801 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Müller H et al. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes & development 15, 267–285, doi: 10.1101/gad.864201 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Crozier L et al. CDK4/6 inhibitors induce replication stress to cause long-term cell cycle withdrawal. EMBO J, e108599, doi: 10.15252/embj.2021108599 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dean JL, McClendon AK & Knudsen ES Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem 287, 29075–29087, doi: 10.1074/jbc.M112.365494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Goel S et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475, doi: 10.1038/nature23465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This was the first study to demonstrate that CDK4/6 inhibitors can stimulate anti-tumour immune responses, underpinning synergy upon dual inhibition of CDK4/6 and inhibitory immune checkpoints.
- 131.Salvador-Barbero B et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer cell 37, 340–353.e346, doi: 10.1016/j.ccell.2020.01.007 (2020). [DOI] [PubMed] [Google Scholar]; This preclinical study demonstrated that the impact of combined CDK4/6 inhibition and cytotoxic chemotherapy is sequence-dependent, providing a rationale for the development of therapeutic regimens in which CDK4/6 inhibitors are administered after anti-mitotic chemotherapy (e.g. taxanes).
- 132.Thangavel C et al. Therapeutic Challenge with a CDK 4/6 Inhibitor Induces an RB-Dependent SMAC-Mediated Apoptotic Response in Non-Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research, doi: 10.1158/1078-0432.CCR-17-2074 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Michaud K et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer research 70, 3228–3238, doi: 10.1158/0008-5472.Can-09-4559 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Thangavel C et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocrine-related cancer 18, 333–345, doi: 10.1530/erc-10-0262 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yoshida A, Lee EK & Diehl JA Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer research 76, 2990–3002, doi: 10.1158/0008-5472.CAN-15-2931 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shay JW, Pereira-Smith OM & Wright WE A role for both RB and p53 in the regulation of human cellular senescence. Experimental cell research 196, 33–39, doi: 10.1016/0014-4827(91)90453-2 (1991). [DOI] [PubMed] [Google Scholar]
- 137.Acevedo M et al. A CDK4/6-Dependent Epigenetic Mechanism Protects Cancer Cells from PML-induced Senescence. Cancer research 76, 3252–3264, doi: 10.1158/0008-5472.CAN-15-2347 (2016). [DOI] [PubMed] [Google Scholar]
- 138.Rubio C et al. CDK4/6 Inhibitor as a Novel Therapeutic Approach for Advanced Bladder Cancer Independently of RB1 Status. Clinical Cancer Research 25, 390–402, doi: 10.1158/1078-0432.Ccr-18-0685 (2019). [DOI] [PubMed] [Google Scholar]
- 139.Gorgoulis V et al. Cellular Senescence: Defining a Path Forward. Cell 179, 813–827, doi: 10.1016/j.cell.2019.10.005 (2019). [DOI] [PubMed] [Google Scholar]
- 140.Guan X et al. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol Cancer Res 15, 237–249, doi: 10.1158/1541-7786.MCR-16-0319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rodier F & Campisi J Four faces of cellular senescence. Journal of Cell Biology 192, 547–556, doi: 10.1083/jcb.201009094 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Watt AC et al. CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nature Cancer 2, 34–48 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study utilized in vitro and in vivo models, as well as clinical specimens, to establish that CDK4/6 inhibitor-induced “senescence” is accompanied by remodelling of cancer cell chromatin, and that the consequent transcriptional changes underpin key effects of these drugs in breast cancer.
- 143.Tasdemir N et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer discovery 6, 612–629, doi: 10.1158/2159-8290.CD-16-0217 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Martinez-Zamudio RI et al. AP-1 imprints a reversible transcriptional programme of senescent cells. Nature cell biology 22, 842–855, doi: 10.1038/s41556-020-0529-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Walter DM et al. RB constrains lineage fidelity and multiple stages of tumour progression and metastasis. Nature, doi: 10.1038/s41586-019-1172-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ruscetti M et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362, 1416–1422, doi: 10.1126/science.aas9090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ruscetti M et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 181, 424–441 e421, doi: 10.1016/j.cell.2020.03.008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Haines E et al. Palbociclib resistance confers dependence on an FGFR-MAP kinase-mTOR-driven pathway in KRAS-mutant non-small cell lung cancer. Oncotarget 9, 31572–31589, doi: 10.18632/oncotarget.25803 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kovatcheva M et al. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zou X et al. Cdk4 disruption renders primary mouse cells resistant to oncogenic transformation, leading to Arf/p53-independent senescence. Genes & development 16, 2923–2934, doi: 10.1101/gad.1033002 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Esteban-Burgos L et al. Tumor regression and resistance mechanisms upon CDK4 and RAF1 inactivation in KRAS/P53 mutant lung adenocarcinomas. Proceedings of the National Academy of Sciences 117, 24415–24426, doi: 10.1073/pnas.2002520117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Uras IZ et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 127, 2890–2902, doi: 10.1182/blood-2015-11-683581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Goel S et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer cell 29, 255–269, doi: 10.1016/j.ccell.2016.02.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study outlined the molecular rationale for combined inhibition of CDK4/6 and upstream mitogenic signals, and also provided the first evidence that abemaciclib can delay the relapse of dormant cancer cells in vivo.
- 154.Dick FA, Goodrich DW, Sage J & Dyson NJ Non-canonical functions of the RB protein in cancer. Nature reviews. Cancer 18, 442–451, doi: 10.1038/s41568-018-0008-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nead MA, Baglia LA, Antinore MJ, Ludlow JW & McCance DJ Rb binds c-Jun and activates transcription. EMBO J 17, 2342–2352, doi: 10.1093/emboj/17.8.2342 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Singh P, Coe J & Hong W A role for retinoblastoma protein in potentiating transcriptional activation by the glucocorticoid receptor. Nature 374, 562–565, doi: 10.1038/374562a0 (1995). [DOI] [PubMed] [Google Scholar]
- 157.Liu L et al. G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nature cell biology 19, 177–188, doi: 10.1038/ncb3474 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Deng J et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer discovery 8, 216–233, doi: 10.1158/2159-8290.CD-17-0915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlighted that CDK4/6 inhibitor-induced immune stimulation is driven, in part, by direct effects on effector T lymphocytes.
- 159.Yin Q et al. CDK4/6 regulate lysosome biogenesis through TFEB/TFE3. Journal of Cell Biology 219, doi: 10.1083/jcb.201911036 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liu T et al. CDK4/6-dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nat Commun 8, 13923, doi: 10.1038/ncomms13923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhang J et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95, doi: 10.1038/nature25015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Romero-Pozuelo J, Demetriades C, Schroeder P & Teleman AA CycD/Cdk4 and Discontinuities in Dpp Signaling Activate TORC1 in the Drosophila Wing Disc. Dev Cell 42, 376–387 e375, doi: 10.1016/j.devcel.2017.07.019 (2017). [DOI] [PubMed] [Google Scholar]
- 163.Romero-Pozuelo J, Figlia G, Kaya O, Martin-Villalba A & Teleman AA Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep 31, 107504, doi: 10.1016/j.celrep.2020.03.068 (2020). [DOI] [PubMed] [Google Scholar]
- 164.Lai AY et al. CDK4/6 inhibition enhances antitumor efficacy of chemotherapy and immune checkpoint inhibitor combinations in preclinical models and enhances T-cell activation in patients with SCLC receiving chemotherapy. J Immunother Cancer 8, doi: 10.1136/jitc-2020-000847 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fang JS et al. Shear-induced Notch-Cx37-p27 axis arrests endothelial cell cycle to enable arterial specification. Nature Communications 8, 2149, doi: 10.1038/s41467-017-01742-7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang B et al. Pharmacological CDK4/6 inhibition reveals a p53-dependent senescent state with restricted toxicity. EMBO J, e108946, doi: 10.15252/embj.2021108946 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Goel S, DeCristo MJ, McAllister SS & Zhao JJ CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol 28, 911–925, doi: 10.1016/j.tcb.2018.07.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wardell SE et al. Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy–Resistant Breast Cancer. Clinical Cancer Research 21, 5121–5130, doi: 10.1158/1078-0432.Ccr-15-0360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Andreano KJ et al. G1T48, an oral selective estrogen receptor degrader, and the CDK4/6 inhibitor lerociclib inhibit tumor growth in animal models of endocrine-resistant breast cancer. Breast cancer research and treatment 180, 635–646, doi: 10.1007/s10549-020-05575-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.O’Brien N et al. Preclinical Activity of Abemaciclib Alone or in Combination with Antimitotic and Targeted Therapies in Breast Cancer. Molecular cancer therapeutics 17, 897–907, doi: 10.1158/1535-7163.Mct-17-0290 (2018). [DOI] [PubMed] [Google Scholar]
- 171.Miller TW et al. ERα-Dependent E2F Transcription Can Mediate Resistance to Estrogen Deprivation in Human Breast Cancer. Cancer discovery 1, 338–351, doi: 10.1158/2159-8290.Cd-11-0101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zwijsen RM et al. CDK-independent activation of estrogen receptor by cyclin D1. Cell 88, 405–415 (1997). [DOI] [PubMed] [Google Scholar]
- 173.Vora SR et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer cell 26, 136–149, doi: 10.1016/j.ccr.2014.05.020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hallin J et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer discovery 10, 54–71, doi: 10.1158/2159-8290.Cd-19-1167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lou K et al. KRASG12C inhibition produces a driver-limited state revealing collateral dependencies. Science Signaling 12, eaaw9450, doi: 10.1126/scisignal.aaw9450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kwong LN et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nature medicine 18, 1503–1510, doi: 10.1038/nm.2941 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Heilmann AM et al. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer research 74, 3947–3958, doi: 10.1158/0008-5472.CAN-13-2923 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Formisano L et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun 10, 1373, doi: 10.1038/s41467-019-09068-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tong Z et al. Functional genomics identifies predictive markers and clinically actionable resistance mechanisms to CDK4/6 inhibition in bladder cancer. J Exp Clin Cancer Res 38, 322, doi: 10.1186/s13046-019-1322-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jansen VM et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer research 77, 2488–2499, doi: 10.1158/0008-5472.CAN-16-2653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Guenther LM et al. A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clinical Cancer Research 25, 1343–1357, doi: 10.1158/1078-0432.Ccr-18-0372 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhao M et al. Combining neratinib with CDK4/6, mTOR and MEK inhibitors in models of HER2-positive cancer. Clinical Cancer Research, clincanres.3017.2020, doi: 10.1158/1078-0432.Ccr-20-3017 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.de Leeuw R et al. MAPK Reliance via Acquired CDK4/6 Inhibitor Resistance in Cancer. Clinical Cancer Research 24, 4201–4214, doi: 10.1158/1078-0432.Ccr-18-0410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zacharek SJ, Xiong Y & Shumway SD Negative regulation of TSC1-TSC2 by mammalian D-type cyclins. Cancer research 65, 11354–11360, doi: 10.1158/0008-5472.CAN-05-2236 (2005). [DOI] [PubMed] [Google Scholar]
- 185.Chandarlapaty S et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer cell 19, 58–71, doi: 10.1016/j.ccr.2010.10.031 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhang J et al. Inhibition of Rb Phosphorylation Leads to mTORC2-Mediated Activation of Akt. Mol Cell 62, 929–942, doi: 10.1016/j.molcel.2016.04.023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Olmez I et al. Combined c-Met/Trk Inhibition Overcomes Resistance to CDK4/6 Inhibitors in Glioblastoma. Cancer research 78, 4360–4369, doi: 10.1158/0008-5472.Can-17-3124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Goldman JW et al. A Randomized Phase III Study of Abemaciclib Versus Erlotinib in Patients with Stage IV Non-small Cell Lung Cancer With a Detectable KRAS Mutation Who Failed Prior Platinum-Based Therapy: JUNIPER. Front Oncol 10, 578756, doi: 10.3389/fonc.2020.578756 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kumarasamy V, Vail P, Nambiar R, Witkiewicz AK & Knudsen ES Functional Determinants of Cell Cycle Plasticity and Sensitivity to CDK4/6 Inhibition. Cancer research 81, 1347–1360, doi: 10.1158/0008-5472.Can-20-2275 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Knudsen ES et al. Cell cycle plasticity driven by MTOR signaling: integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene 38, 3355–3370, doi: 10.1038/s41388-018-0650-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Vilgelm AE et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Science translational medicine 11, doi: 10.1126/scitranslmed.aav7171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fingar DC et al. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Molecular and cellular biology 24, 200–216 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Teh JLF et al. In vivo E2F reporting reveals efficacious schedules of MEK1/2-CDK4/6 targeting and mTOR-S6 resistance mechanisms. Cancer discovery, doi: 10.1158/2159-8290.CD-17-0699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Romano G et al. A pre-existing rare PIK3CAE545K subpopulation confers clinical resistance to MEK plus CDK4/6 inhibition in NRAS melanoma and is dependent on S6K1 signaling. Cancer discovery, doi: 10.1158/2159-8290.CD-17-0745 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Michaloglou C et al. Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long term growth inhibition in estrogen receptor positive breast cancer. Molecular cancer therapeutics, doi: 10.1158/1535-7163.MCT-17-0537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Olmez I et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clinical cancer research : an official journal of the American Association for Cancer Research 23, 6958–6968, doi: 10.1158/1078-0432.CCR-17-0803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hart LS et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research 23, 1785–1796, doi: 10.1158/1078-0432.CCR-16-1131 (2017). [DOI] [PubMed] [Google Scholar]
- 198.Ziemke EK et al. Sensitivity of KRAS-Mutant Colorectal Cancers to Combination Therapy That Cotargets MEK and CDK4/6. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 405–414, doi: 10.1158/1078-0432.CCR-15-0829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Song X et al. Combined CDK4/6 and Pan-mTOR Inhibition Is Synergistic Against Intrahepatic Cholangiocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 25, 403–413, doi: 10.1158/1078-0432.Ccr-18-0284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhou J et al. Pan-ERBB kinase inhibition augments CDK4/6 inhibitor efficacy in oesophageal squamous cell carcinoma. Gut, gutjnl-2020–323276, doi: 10.1136/gutjnl-2020-323276 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yadav V et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Molecular cancer therapeutics 13, 2253–2263, doi: 10.1158/1535-7163.MCT-14-0257 (2014). [DOI] [PubMed] [Google Scholar]
- 202.Tao Z et al. Coadministration of Trametinib and Palbociclib Radiosensitizes KRAS-Mutant Non-Small Cell Lung Cancers In Vitro and In Vivo. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 122–133, doi: 10.1158/1078-0432.CCR-15-0589 (2016). [DOI] [PubMed] [Google Scholar]
- 203.Sherr CJ A New Cell-Cycle Target in Cancer — Inhibiting Cyclin D–Dependent Kinases 4 and 6. New England Journal of Medicine 375, 1920–1923, doi: 10.1056/NEJMp1612343 (2016). [DOI] [PubMed] [Google Scholar]
- 204.Schaer DA et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep 22, 2978–2994, doi: 10.1016/j.celrep.2018.02.053 (2018). [DOI] [PubMed] [Google Scholar]
- 205.Jerby-Arnon L et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e924, doi: 10.1016/j.cell.2018.09.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Uzhachenko RV et al. Metabolic modulation by CDK4/6 inhibitor promotes chemokine-mediated recruitment of T cells into mammary tumors. Cell Rep 35, 108944, doi: 10.1016/j.celrep.2021.108944 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Litchfield K et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 184, 596–614.e514, doi: 10.1016/j.cell.2021.01.002 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yu J et al. Genetic Aberrations in the CDK4 Pathway Are Associated with Innate Resistance to PD-1 Blockade in Chinese Patients with Non-Cutaneous Melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 25, 6511–6523, doi: 10.1158/1078-0432.Ccr-19-0475 (2019). [DOI] [PubMed] [Google Scholar]
- 209.Knudsen ES et al. Targeting dual signalling pathways in concert with immune checkpoints for the treatment of pancreatic cancer. Gut 70, 127–138, doi: 10.1136/gutjnl-2020-321000 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Whittle JR et al. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor-Positive Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research, doi: 10.1158/1078-0432.CCR-19-1872 (2020). [DOI] [PubMed] [Google Scholar]
- 211.Lelliott EJ et al. CDK4/6 inhibition promotes anti-tumor immunity through the induction of T cell memory. Cancer discovery, candisc.1554.2020, doi: 10.1158/2159-8290.Cd-20-1554 (2021). [DOI] [PubMed] [Google Scholar]
- 212.Heckler M et al. Inhibition of CDK4/6 promotes CD8 T-cell memory formation. Cancer discovery, candisc.1540.2020, doi: 10.1158/2159-8290.Cd-20-1540 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tan AR et al. Trilaciclib plus chemotherapy versus chemotherapy alone in patients with metastatic triple-negative breast cancer: a multicentre, randomised, open-label, phase 2 trial. The lancet oncology 20, 1587–1601, doi: 10.1016/S1470-2045(19)30616-3 (2019). [DOI] [PubMed] [Google Scholar]
- 214.US National Library of Medicine. Clinicaltrials.gov, http://www.clinicaltrials.gov/ct2/show/NCT03900884> (2009). [DOI] [PubMed] [Google Scholar]
- 215.Gonzalez-Gualda E et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 19, e13142, doi: 10.1111/acel.13142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gadsden NJ et al. Palbociclib Renders Human Papilloma Virus-Negative Head and Neck Squamous Cell Carcinoma Vulnerable to the Senolytic Agent Navitoclax. Mol Cancer Res 19, 862–873, doi: 10.1158/1541-7786.Mcr-20-0915 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ji Y et al. Use of ratiometrically designed nanocarrier targeting CDK4/6 and autophagy pathways for effective pancreatic cancer treatment. Nature Communications 11, 4249, doi: 10.1038/s41467-020-17996-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Brown NE et al. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer research 72, 6477–6489, doi: 10.1158/0008-5472.CAN-11-4139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Vijayaraghavan S et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun 8, 15916, doi: 10.1038/ncomms15916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fassl A et al. Increased lysosomal biomass is responsible for the resistance of triple-negative breast cancers to CDK4/6 inhibition. Sci Adv 6, eabb2210, doi: 10.1126/sciadv.abb2210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Franco J, Balaji U, Freinkman E, Witkiewicz AK & Knudsen ES Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep 14, 979–990, doi: 10.1016/j.celrep.2015.12.094 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Tarrado-Castellarnau M et al. De novo MYC addiction as an adaptive response of cancer cells to CDK4/6 inhibition. Mol Syst Biol 13, 940, doi: 10.15252/msb.20167321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ge JY et al. Acquired resistance to combined BET and CDK4/6 inhibition in triple-negative breast cancer. Nature Communications 11, 2350, doi: 10.1038/s41467-020-16170-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Liao S, Maertens O, Cichowski K & Elledge SJ Genetic modifiers of the BRD4-NUT dependency of NUT midline carcinoma uncovers a synergism between BETis and CDK4/6is. Genes & development 32, 1188–1200, doi: 10.1101/gad.315648.118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.McClendon AK et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell cycle 11, 2747–2755, doi: 10.4161/cc.21127 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Roberts PJ et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. Journal of the National Cancer Institute 104, 476–487, doi: 10.1093/jnci/djs002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Johnson SM et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. The Journal of clinical investigation 120, 2528–2536, doi: 10.1172/jci41402 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hashizume R et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neurooncology 18, 1519–1528, doi: 10.1093/neuonc/now106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Barton KL et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS One 8, e77639, doi: 10.1371/journal.pone.0077639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Naz S et al. Abemaciclib, a Selective CDK4/6 Inhibitor, Enhances the Radiosensitivity of Non–Small Cell Lung Cancer In Vitro and In Vivo. Clinical Cancer Research 24, 3994–4005, doi: 10.1158/1078-0432.Ccr-17-3575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Weiss JM et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: a phase Ib/randomized phase II trial. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 30, 1613–1621, doi: 10.1093/annonc/mdz278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wei L et al. Inhibition of CDK4/6 protects against radiation-induced intestinal injury in mice. The Journal of clinical investigation 126, 4076–4087, doi: 10.1172/JCI88410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Purba TS et al. CDK4/6 inhibition mitigates stem cell damage in a novel model for taxane-induced alopecia. EMBO Molecular Medicine 11, e11031, doi: 10.15252/emmm.201911031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Pabla N et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proceedings of the National Academy of Sciences 112, 5231–5236, doi: 10.1073/pnas.1424313112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.DiRocco DP et al. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am J Physiol Renal Physiol 306, F379–388, doi: 10.1152/ajprenal.00475.2013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Mayer EL et al. Palbociclib with adjuvant endocrine therapy in early breast cancer (PALLAS): interim analysis of a multicentre, open-label, randomised, phase 3 study. The lancet oncology 22, 212–222, doi: 10.1016/s1470-2045(20)30642-2 (2021). [DOI] [PubMed] [Google Scholar]
- 237.Johnston SRD et al. Abemaciclib Combined With Endocrine Therapy for the Adjuvant Treatment of HR+, HER2−, Node-Positive, High-Risk, Early Breast Cancer (monarchE). Journal of Clinical Oncology 38, 3987–3998, doi: 10.1200/jco.20.02514 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This randomized phase 3 trial demonstrated that the addition of abemaciclib to endocrine therapy improves disease-free survival for patients with high-risk, HR-positive HER2-negative breast cancer.
- 238.Loibl S et al. Phase III study of palbociclib combined with endocrine therapy (ET) in patients with hormone-receptor-positive (HR+), HER2-negative primary breast cancerand with high relapse risk after neoadjuvant chemotherapy (NACT): First results from PENELOPE-B. Cancer research 81, GS1–02 (2021). [Google Scholar]
- 239.Zhao B & Burgess K PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chemical Communications 55, 2704–2707, doi: 10.1039/C9CC00163H (2019). [DOI] [PubMed] [Google Scholar]
- 240.Brand M et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem Biol 26, 300–306.e309, doi: 10.1016/j.chembiol.2018.11.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Jiang B et al. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew Chem Int Ed Engl 58, 6321–6326, doi: 10.1002/anie.201901336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Anderson NA et al. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg Med Chem Lett 30, 127106, doi: 10.1016/j.bmcl.2020.127106 (2020). [DOI] [PubMed] [Google Scholar]
- 243.Rana S et al. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg Med Chem Lett 29, 1375–1379, doi: 10.1016/j.bmcl.2019.03.035 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.De Dominici M et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 135, 1560–1573, doi: 10.1182/blood.2019003604 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hortobagyi GN et al. in San Antonio Breast Cancer Symposium. PD4–06. [Google Scholar]
- 246.Alvarez-Fernandez M & Malumbres M Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer cell 37, 514–529, doi: 10.1016/j.ccell.2020.03.010 (2020). [DOI] [PubMed] [Google Scholar]
- 247.Condorelli R et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO, doi: 10.1093/annonc/mdx784 (2017). [DOI] [PubMed] [Google Scholar]
- 248.O’Leary B et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer discovery 8, 1390–1403, doi: 10.1158/2159-8290.CD-18-0264 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This circulating biomarker analysis of the PALOMA-3 study describes mutational profiling of HR-positive breast cancers treated with ET +/− palbociclib, performed using paired blood samples taken at baseline and at disease progression.
- 249.Costa C et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer discovery 10, 72–85, doi: 10.1158/2159-8290.CD-18-0830 (2020). [DOI] [PubMed] [Google Scholar]
- 250.Wander SA et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor–Positive Metastatic Breast Cancer. Cancer discovery 10, 1174–1193, doi: 10.1158/2159-8290.Cd-19-1390 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Turner NC et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 37, 1169–1178, doi: 10.1200/jco.18.00925 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Choi YJ et al. Development of a selective CDK2-E inhibitor in CCNE driven cancers. Cancer research 80, Abstr 1279 (2021). [Google Scholar]
- 253.Sabnis RW Novel CDK2 Inhibitors for Treating Cancer. ACS Med Chem Lett 11, 2346–2347, doi: 10.1021/acsmedchemlett.0c00500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Freeman-Cook K et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer cell 39, 1404–1421 e1411, doi: 10.1016/j.ccell.2021.08.009 (2021). [DOI] [PubMed] [Google Scholar]
- 255.Yang C et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36, 2255–2264, doi: 10.1038/onc.2016.379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Cornell L, Wander SA, Visal T, Wagle N & Shapiro GI MicroRNA-Mediated Suppression of the TGF-beta Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance. Cell Rep 26, 2667–2680 e2667, doi: 10.1016/j.celrep.2019.02.023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Li Q et al. INK4 tumor suppressor proteins mediate resistance to CDK4/6 kinase inhibitors. Cancer discovery, doi: 10.1158/2159-8290.CD-20-1726 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that CDK6 can induce CDK4/6 inhibitor resistance by inducing expression of p18, providing a mechanistic hypothesis to explain prior studies implicating CDK6 as a driver of resistance.
- 258.Glover-Cutter K et al. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Molecular and cellular biology 29, 5455–5464, doi: 10.1128/MCB.00637-09 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Larochelle S, Pandur J, Fisher RP, Salz HK & Suter B Cdk7 is essential for mitosis and for in vivo Cdk-activating kinase activity. Genes & development 12, 370–381, doi: 10.1101/gad.12.3.370 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Coombes C et al. in San Antonio Breast Cancer Symposium (San Antonio, 2021). [Google Scholar]
- 261.Llanos S et al. Lysosomal trapping of palbociclib and its functional implications. Oncogene 38, 3886–3902, doi: 10.1038/s41388-019-0695-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Sudhan D et al. in San Antonio Breast Cancer Symposium (San Antonio, 2021). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.