

Abstract
Cardiomyopathies are associated with arrhythmias and cardiac ion channel downregulation. This downregulation is arrhythmogenic. Paradoxically, antiarrhythmic therapies are based on ion channel-blocking drugs that further downregulate these channels and exhibit pro-arrhythmia risk. Recent studies have shown that inhibition of the protein kinase RNA-like ER kinase (PERK) arm of the unfolded protein response prevents select cardiac ion channel downregulation and plays a protective role against arrhythmias. Prevention of ion channel downregulation represents as a novel therapeutic strategy to treat arrhythmias in myocardial infarction and heart failure.
Keywords: cardiac ion channel, myocardial infarction, mRNA degradation, therapy, heart failure
Downregulation of ion channels is a fundamental mechanism causing arrhythmic risk in cardiomyopathy.
Heart failure (HF) is associated with sudden cardiac death and characterized by arrhythmogenic electrical remodeling, which includes downregulations of many cardiac ion channels and transporters that contribute to action potential (AP) (see Glossary) prolongation, QT prolongation on the surface electrocardiogram (ECG), and increased arrhythmic risk [1–4]. Cardiac ion channels are often downregulated in both ischemic and nonischemic cardiomyopathies (Table 1, also reviewed in [5, 6]). Decreased cardiac Na+ channel (Nav1.5) protein and current (INa) in HF cause a decreased upstroke velocity (dV/dtmax) of the AP [2, 7–9], which contributes to slow conduction, a prerequisite for reentrant arrhythmias, and jeopardizes impulse propagation in heart tissue. Human and animal studies reveal downregulations of multiple K+ channel currents in cardiomyopathy (Table 1) [4, 10]. These current reductions have been linked to reduced transcription, translation, and expression of the corresponding channel subunits [3, 4, 10]. The loss of K+ channel repolarizing current causes AP duration (APD) prolongation and its corollary QT prolongation. Prolonged QT interval is also associated with another basic mechanism of arrhythmia known as triggered activity that contributes to polymorphic ventricular tachycardia known as Torsades de Pointes [4].
Table 1.
Cardiac ion channels are reported to be downregulated in cardiomyopathies of human and animal models at mRNA, protein, and current levels.
| Channels | Decreased mRNA | Decreased Protein | Decreased Currents | Effects on AP and ECGs |
|---|---|---|---|---|
| SCN5A/Nav1.5/INa | HF [2, 10] MI [15] |
HF [1] MI [15] |
HF [1, 2, 5, 9] MI [8, 15] AF [69] |
Slow conduction, repolarization abnormalities, ventricular arrhythmias, sustained and isolated conduction defects, promoted re-entrant arrhythmias [1, 15] |
| CACNA1C/Cav1.2/IcaL | HF [10, 70], MI [15], AF [71] |
HF [72], MI [15], AF [71] |
HF[70, 73] MI [5, 15] |
APD prolongation, QT prolongation, arrhythmias, shorter atrial effective refractory period [15, 71, 73] |
| KCND3/Kv4.3/Ito | HF [2–4, 10], MI [15], AF [71, 74] |
HF [3, 4], MI [15], AF [71, 74] |
HF [3, 75, 76] MI [5, 15] |
APD prolongation, QT prolongation, repolarization abnormalities such as EADs/DADs, triggered arrhythmias, nonexcitable gap re-entry, shorter atrial effective refractory period [3, 15, 71, 75, 76] |
| KCNQ1/KVLQT1/Iks | HF [4] AF [71] |
HF [4] | HF [4, 5, 75–77] | APD prolongation, EADs, QT prolongation and/or shortening, ventricular tachyarrhythmias, sudden death [4, 76] |
| KCNH2/hERG/Ikr | HF [4], AF [71] |
HF [4], AF [71] |
HF [4, 73], MI [5] |
APD prolongation, QT prolongation and/or shortening, ventricular tachyarrhythmias, sudden death, shorter atrial effective refractory period [4] [71] |
| KCNJ2/Kir2.1/I k1 | HF [10], MI [15] |
HF [10], MI [15] |
HF [5, 10, 75], MI [5, 15] |
APD prolongation, QT prolongation, arrhythmias [15] |
| KCNJ3/Kir3.1/IKACh | HF [10], AF [71] |
AF [71, 74] | Shorter atrial effective refractory period [71] | |
| KCNJ5/Kir3.4/IKACh | HF [10], AF [71, 74, 78] |
AF [78] | Shorter APD, shorter atrial effective refractory period [71, 78] | |
| KCNE1/minK | HF [4] | HF [4], AF [71] |
APD prolongation, QT prolongation and/or shortening, ventricular tachyarrhythmias, sudden death, shorter atrial effective refractory period [4, 71] | |
| KCNA5/Kv1.5/IKur | HF [10], MI [15] |
MI [15], AF [71, 74] |
MI [15] | APD prolongation, QT prolongation, arrhythmias, shorter atrial effective refractory period [15, 71] |
| KCNJ11/Kir6.2/IKATP | HF [10], AF [71, 74] |
AF [79] | Shorter atrial effective refractory period [71] | |
| SLC8A1/NCX/INCX | HF [10, 70] | HF [70] | ||
| HCN2/HCN4/HCN/If | HF [80] | HF [80] | HF [77] | Increased cycle length, reduced diastolic depolarization rate, increased sinus node recovery time [77, 80] |
AP=action potential; APD=action potential duration; AF=atrial fibrillation; DADs=delayed afterdepolarizations; EADs-early afterdepolarizations; ECG=electrocardiogram; HF=heart failure; MI=myocardial infarction.
Current anti-arrhythmic medications are ion channel-blocking drugs such as Vaughan Williams class Ia (such as quinidine and procainamide) and Ic agents (such as flecainide and propafenone) that block Nav1.5, and class III agents that block K+ channels (such as amiodarone and sotalol). These agents further inhibit ion channel activity and evoke significant proarrhythmia risk that is worsened by the presence of cardiomyopathy (see Clinician’s corner).
Text Boxes:
Clinician’s corner
All current anti-arrhythmic drugs block ion channels.
All current anti-arrhythmic drugs can induce arrhythmic, especially in cardiomyopathies, a phenomenon called proarrhythmia.
Most ion channels are downregulated in cardiomyopathies, and raising ion channel levels is antiarrhythmic without proarrhythmic risk.
Therefore, it seems likely that the proarrhythmia seen with ion channel blocking drugs is inherent to the mechanism of action and that new strategies to prevent the downregulation of ion channels may be more effective and less risky than current medications.
It is well established that lower cardiac contractile function is inversely associated with arrhythmic risk. Moreover, the unfolded protein response (UPR) is activated in cardiomyopathic states with low contractile function, ion channels are downregulated in cardiomyopathy, and ion channel downregulation can contribute to arrhythmic risk [11–13]. This led us to the idea that UPR may contribute to ion channel downregulation and arrhythmic risk in cardiomyopathy. Recent studies have shown that preventing the cardiomyopathy-induced ion channel downregulations has the advantage of reducing arrhythmia free from the proarrhythmic effects of the ion channel blocking drugs [2, 7, 14–16]. These observations explain the correlation of drug-induced proarrhythmic risk with cardiomyopathic severity and suggest a new therapeutic strategy for antiarrhythmic therapy by preventing the ion channel downregulations associated with HF.
The UPR is one mechanism contributing to ion channel downregulation in cardiomyopathy
Various cardiovascular diseases such as HF [2, 11, 12], myocardial infarction (MI) [17, 18], ischemia/reperfusion [19], dilated cardiomyopathy [20], and hypertension [21] have been associated with the UPR of the endoplasmic reticulum (ER) or sarcoplasmic reticulum [11–13]. The ER is the location for transmembrane protein translation, folding and assembling before trafficking to the plasma membrane, which are crucial for cardiac ion channels and transporter activity. Classically, the UPR is a mechanism that responds to ER protein overload by activation of protein refolding, protein degradation, and inhibition of nascent protein translation.
Three independent arms of UPR have different effects on cardiac proteins and arrhythmia
The UPR signaling pathway has at least three main effectors: protein kinase RNA-like ER kinase (PERK), inositol-requiring enzyme-1 (IRE1), and activating transcription factor-6α (ATF6α). When unfolded/misfolded proteins accumulate in the ER lumen, glucose-regulated protein/78 kDa (Grp78) dissociates from PERK, IRE1, and ATF6α, leading to their activation. Activation of these central UPR effectors initiate complicated signaling cascades to increase gene expression and translation of ER chaperones such as Grp78, which restore the protein folding capacity. On the other hand, activated UPR inhibits protein expression of most other proteins by enhancing mRNA degradation, inhibiting protein translation, and accelerating protein degradation. Under mild ER stress, the UPR acts as an adaptive mechanism to improve ER protein refolding, degrade misfolded proteins, and reestablish the ER homeostasis. Nevertheless, under severe ER stress, the UPR, especially the PERK and IRE1 arms, can shut down the synthesis of many important proteins, leading to cell apoptosis [22]. Cardiomyocyte apoptosis may, itself, contribute to the electrical and structural inhomogeneities known to worsen arrhythmias.
The signals for UPR activation during cardiomyopathy are a matter of speculation, but the PERK arm of UPR is activated in human failing hearts, at least in part, because of abnormal SCN5A (encoding Nav1.5 α subunit) mRNA splicing that results in increased truncated, nonfunctional Nav1.5 proteins trapped in the ER [2]. Since activated PERK can decrease the full-length SCN5A/Nav1.5 expression, this suggests a vicious cycle of abnormal SCN5A splice variants activating the UPR, which then degrades more SCN5A ultimately resulting in a feed forward reduction in INa.
The PERK arm of the UPR.
The PERK arm mainly plays detrimental roles in HF and MI by downregulation of multiple cardiac ion channels via increasing mRNA degradation [2, 15], which causes electrical remodeling and contributes to increased arrhythmic risk (Figure 1). For example, PERK activation in HF and MI inhibits Nav1.5 [2, 15]. PERK also downregulates multiple K+ channels [2, 15, 23]. PERK inhibition has shown protective effects against MI [15, 17, 18, 24] and decreased ventricular arrhythmias in MI [15].
Figure 1.
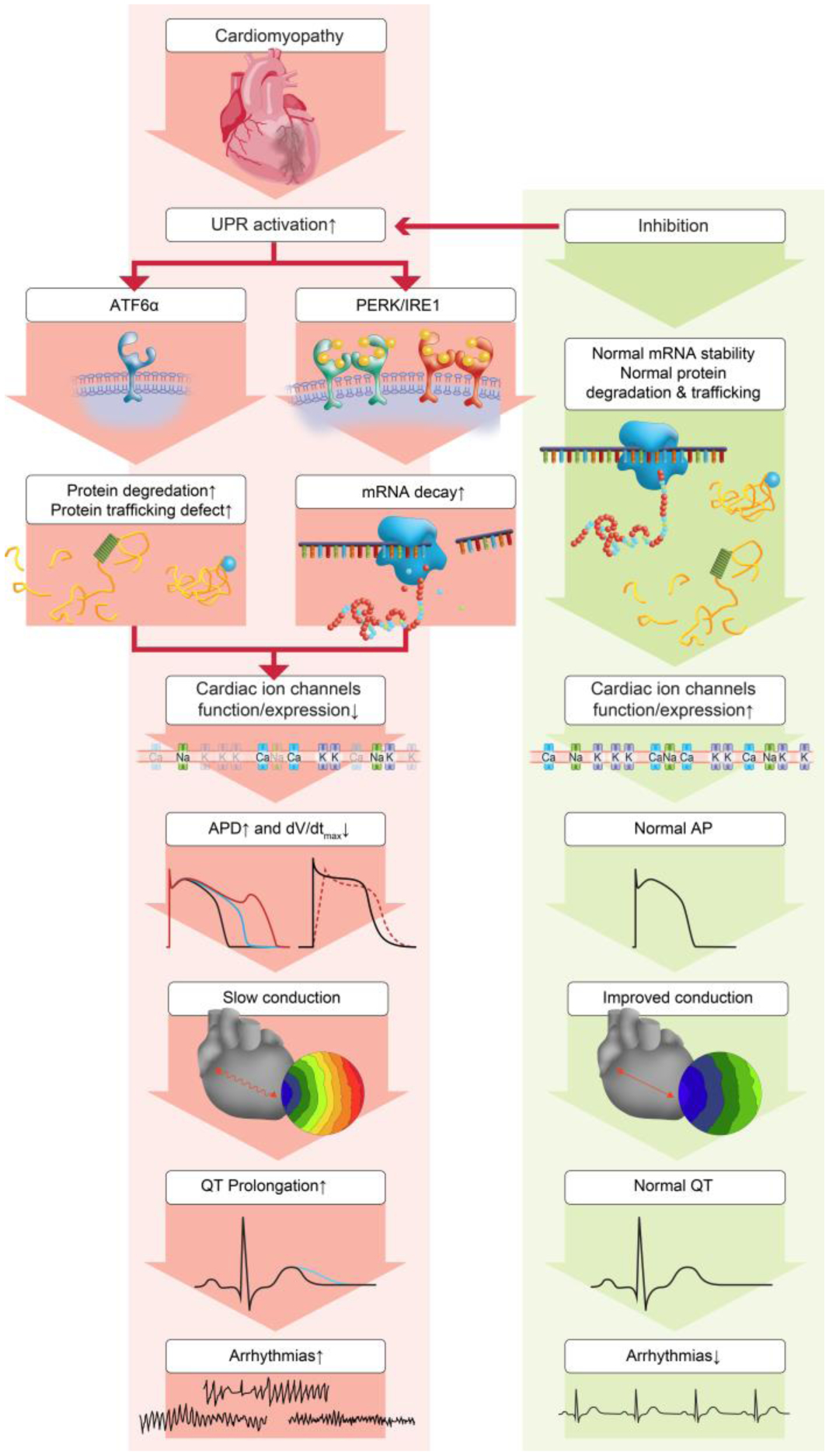
The adverse effects of the UPR on cardiac electrophysiology. Inhibition of these effects may reduce arrhythmic risk in cardiomyopathies. The PERK and IRE1 arm downregulate multiple cardiac ion channels at the mRNA level, and ATF6α increases hERG protein degradation and causes trafficking defects. These deleterious effects alter cardiomyocyte action potential (APD prolongation and dV/dtmax reduction) and prolong QT intervals of the ECG, leading to increased arrhythmic risks. UPR inhibition can normalize channel alterations, raise channel functions and improve arrhythmias in cardiomyopathies.
The IRE1 arm of the UPR.
The IRE1 arm is the most conserved UPR arm that exists from yeast to mammalians. Its activation has been reported in human failing heart [25, 26] and animal models of MI [15]. Inhibition of IRE1 under physiological conditions downregulates some channels and prolongs the APD [23], suggesting that the IRE1 arm contributes to maintaining the expression levels of channels. Nevertheless, when cardiomyocytes are under ER stress and the APD is prolonged, inhibition of the IRE1 arm increases certain channel expressions and shortens the APD [23], indicating that IRE1 downregulates certain channels under ER stress and plays a detrimental role (Figure 1).
The ATF6α arm of the UPR.
Activation of the ATF6α arm has been observed in human HF [27] and animal models of MI [15]. Activation of the ATF6α arm has be reported to show both protective [28, 29] and detrimental effects [20, 30, 31]. ATF6α is essential as an adaptive responder to optimize protein folding, secretion, and degradation, protecting cells from chronic ER stress. When activated, it enhances the gene expression of UPR chaperones and protein disulfide isomerase, induces antioxidant gene expression [29], and promotes ER-associated misfolded protein degradation [32], all of which are likely to alleviate ER stress. ATF6α is activated in ischemia but inactivated upon reperfusion, and overexpression of ATF6α protects the heart from ischemia [28, 29]. Different effects of ATF6α on arrhythmic risks have been reported. A positive effect of ATF6α is reported on AF, where a decreased expression level of ATF6 is associated with AF susceptibility [33]. A negative effect of ATF6α is reported in high glucose treated cells, where ATF6α activation is associated with defected hERG trafficking and decreased hERG protein expression and channel current IKr [31] (Figure 1). This can lead to APD prolongation, QT prolongation, increased arrhythmias and Long QT syndrome.
The three independent arms of UPR have different effects on cardiac proteins and arrhythmia (Figure 1). Only the effect of inhibition of the PERK arm on arrhythmic risk has been tested directly and the effect was determined immediately after MI. As noted above, when cardiomyocytes are under ER stress and the APD is prolonged, inhibition of the IRE1 arm increases certain channel expressions and shortens the APD [23], indicating that IRE1 downregulates certain channels under ER stress and may play a detrimental role to prolong APD. Although not tested directly, this would imply that IRE1 inhibition might be anti-arrhythmic in pathological conditions. Activation of the ATF6α arm has be reported to show both protective [33] and detrimental effects [31] on arrhythmias.
There is little literature on the effects of altering the UPR in conditions other than MI. Cardiac-specific overexpression or depletion of UPR effectors has been performed in studies on mouse models of ischemia/reperfusion and transverse aortic constriction (TAC) (reviewed in [34]). Depletion of the PERK arm (PERK and CHOP) in TAC models shows mainly protection against pressure overload-induced HF [35, 36]. Overexpression of PERK or ATF4 also increases autophagy and causes increased cardiac atrophy [37]. Overexpression of ATF4 in atrial cells increases cytotoxicity and apoptosis [38]. Overexpression of IRE1, sXBP1, or ATF6α mainly presents protection against ischemia reperfusion-induced apoptosis and infarction [28, 29, 39]. The effect of these alterations on arrhythmias remains to be determined.
UPR regulates a set of channels important for heart rhythm
Although most channels are downregulated in HF, the UPR arms are selective in regulating ion channels (Table 2). Studies show that PERK regulates human Nav1.5, the rapidly inactivating K+ channel (Kv4.3, conducting Ito), hERG, and the slowly inactivating K+ channel (KvLQT1 or Kv7.1, conducting IKs) [2, 23], and Nav1.5/Kv4.3/the voltage-gated K+ channel, shaker-subfamily member 5 (Kv1.5, conducting IKur) in mice [15]. PERK also shows activation of ryanodine receptor 2 channel activity that leads to Ca2+ leaks, causing early and delayed afterdepolarizations (EADs and DADs) in rats [40]. The IRE1 arm modulates human Nav1.5/hERG/KvLQT1/the L-types of Ca2+ channels (Cav1.2 conducting ICaL) [23] and Nav1.5/Kv1.5/the inward rectifying K+ channel (Kir2.1, conducting IK1) in mice (unpublished data). ATF6α seems to regulate hERG channel trafficking [31, 41, 42] and its regulation on other channels are unknown. The mechanisms underlying this selectivity are still unknown, but selective UPR-mediated ubiquitination has been suggested as one possibility [43, 44]. Despite this selectivity, inhibition of the PERK arm alone is antiarrhythmic [2, 15], suggesting that preventing downregulation of a subset of channels reduced in HF is sufficient to prevent arrhythmias and implying that select channels may be central to acquired arrhythmogenesis.
Table 2.
Selective regulation of the three UPR arms on cardiac ion channels.
We suggest that a downregulation of ion channel protein increases the risk of arrhythmias in cardiomyopathy. This idea is consistent with many inherited arrhythmic syndromes, such as Brugada Syndrome and most Long QT Syndromes wherein ion channel activity is downregulated. It is of course true that ion channel overactivity can increase the risk of arrhythmias in some cases, for example Long QT syndrome type 3, and overactivity participates directly in the maintenance of arrhythmias. Therefore, any antiarrhythmic therapy would have to consider the effects of increasing ion channel currents beyond normal levels. Nevertheless, by inhibiting mechanisms of ion channel downregulation activated in pathological circumstances, we have not seen an overactivity of ion channels or an increase in arrhythmic risk, and this approach may be safer than a direct upregulation of a particular ion channel.
UPR works mostly at the mRNA level to regulate arrhythmias.
While classically, PERK is thought to reduce protein levels directly by inhibiting translation, data suggest that a fundamental mechanism of UPR activity is to regulate mRNA abundance levels (Figure 1) [15, 23]. The mechanism of this effect is unknown, but phosphorylation of PERK-eIF2α in the UPR activation can regulate the nonsense-mediated RNA decay pathway, which is well known to play a role in RNA quality regulation and rapidly degrade mRNA [45]. PERK regulates the expression of noncoding RNAs such as the miR-424(322)-503 cluster and miR-483, which further affect the mRNA levels of their respective targets [46, 47].
UPR and oxidative stress.
ER stress and oxidative stress are correlated and can be induced by each other [21, 48]. Oxidative stress has been reported to cause arrhythmias and downregulate ion channels. Therefore, part of the UPR inhibition effect may be because of reduced oxidative stress. Mitochondrial oxidative stress downregulates Nav1.5 at the post-translational level by affecting single channel current of Nav1.5 without altering the channel protein expression [49–51]. NADPH oxidase-induced oxidative stress decreases Kv4.3 mRNA and protein levels, suggesting a modulation at the transcriptional level [52]. Inhibition of oxidative stress may allow for reversal of a post-translational regulation of Na+-Ca2+ exchanger (NCX) [53], and decreased ICaL and IK1 [54–56].
Future directions: Implementing UPR inhibition for arrhythmias.
Possible treatments to inhibit UPR in cardiomyopathy include applications of specific inhibitors and genetic knockdown of the UPR arms. The specificity of UPR inhibitors is important to ensure that only the harmful arm(s), for example the PERK arm in MI, is inhibited, while the other arms are still activated to help suppress the ER stress. Reported PERK inhibitors include GSK2606414, apelin-13, panax quinquefolium saponin, and atorvastatin [15, 17, 18, 24, 57]. There are also specific inhibitors for IRE1 (such as 4μ8c, STF-083010, MKC-3946) [58–60] and for ATF6α (such as 4-(2-aminoethyl) benzenesulfonyl fluoride) [61]. Possible drawbacks of UPR inhibition could be sustained ER stress and inhibition of UPR regulation on maintaining important basal cellular functions, such as pancreatic β cell function, which can be impaired by PERK inhibition with GSK2656157 or genetic knockout with decreased proinsulin and insulin levels [62]. Nevertheless, ISRIB, which acts on eIF2α (downstream of PERK) by reversing its phosphorylation, shows no pancreatic toxicity [63]. It may be that drugs need to be used transiently and specific timing still needs to be investigated. Genetic knockdown of the harmful arms has an advantage of only targeting the heart (for example using AAV9-based virus infection) and therefore maintain the UPR function in other organs at the same time. Genetic inhibition also avoids possible side effects of chemical inhibitors. Aside from GSK2606414 in mouse MI, the effects of drugs on arrhythmic risk in cardiomyopathy remain to be determined.
Another approach to implementing modulation of the UPR for treating arrhythmias is the use of chemical protein chaperones. Chemical chaperones, such as taurine-conjugated ursodeoxycholic acid, 4-phenylbutyric acid (4-PBA) and 4-PBA analogs (2-POAA-OMe, 2-POAA-NO2, and 2-NOAA) exhibit significant effects on reducing cardiac damage by suppressing the ER stress in hypertension and obesity-induced cardiac hypertrophy [64–66], but it remains to be seen if these agents will have similar effects to inhibiting the main UPR effectors.
Concluding Remarks
Preventing downregulation of a set of ion channels by inhibiting the PERK arm of the UPR has been shown to prevent arrhythmias during cardiomyopathy [2, 15]. The concept that preventing ion channel downregulation by pathogenic processes active in cardiomyopathy can reduce arrhythmic risk has proven to extend beyond just the effects of the UPR. In addition to inhibiting the UPR, reducing oxidative stress [7, 50], minimizing metabolic stress [49], preventing pathogenic mRNA splicing by RBM25/LUC7L3 [67], inhibiting the effects of miR-448 [16], and upregulating the mRNA stabilizing protein, HuR, [14, 68] have been shown to reduce arrhythmic risk in cardiomyopathy without showing new proarrhythmic risk. Nevertheless, there are a number of outstanding questions and limitations of our current knowledge that would need to be addressed to apply these concepts to patients (see Outstanding questions and limitations). In summary, PERK inhibition in cardiomyopathy may represent a novel antiarrhythmic strategy, and the success of this strategy may point to a larger conceptual idea that preventing ion channel dysregulation rather than blocking ion channels is the future of antiarrhythmic therapy.
Outstanding questions and limitations.
Why is the PERK effect on ion channels mostly at the RNA level?
Why, if PERK regulates only a subset of channels, can PERK inhibition be anti-arrhythmic?
Does PERK inhibition have similar effects in all forms of cardiomyopathy, and can PERK be inhibited without affecting contractile function, which also is correlated with arrhythmic risk?
Does the antiarrhythmic effect of UPR inhibition vary by disease state, type of arrhythmia, heart chamber, gender, age, and other noncardiac, concurrent diseases?
The UPR is a ubiquitous process that has salutary and deleterious effects in many organs. Any therapeutic targeting of the UPR for arrhythmic risk would need to consider off-target effects on other organs or be directed solely to the heart.
When identified, addressing the upstream activators of the UPR may be a more effective antiarrhythmic strategy with less off-target effects than downstream strategies that address the effects of UPR activation.
Supplementary Material
Highlights.
Cardiac ion channels are often downregulated in cardiomyopathies and this downregulation contributes to lethal arrhythmias.
Current treatments for arrhythmias with ion channel blocking drugs are pro-arrhythmic.
Activation of the UPR in cardiomyopathies contributes to the downregulation of cardiac ion channels
Inhibiting the PERK arm of UPR can prevent ion channel downregulation and is antiarrhythmic.
Preventing ion channel downregulation can reduce arrhythmic risk with no proarrhythmic potential and represents a new treatment paradigm.
Acknowledgement:
This work is supported by R01 HL104025 (SCD) and Rhode Island Foundation grant 20154145 (ML).
Glossary:
- AP
action potential. The cardiac AP is generated by the opening and closing of cardiac ion channels in the cardiomyocyte plasma membrane. Nav1.5 governs the initial AP depolarization. Cardiac K+ and Ca2+ channels determine the characteristic plateau of the AP. Inward rectifying K+ channels set the resting membrane potential. The AP underlies the electrical activity measured by the electrocardiogram (ECG).
- Arrhythmia
improper beating of the heart, either irregular, too fast, or too slow. In the context of this manuscript, the term refers to potentially lethal rapid heartbeats from the ventricles, known as ventricular tachycardia.
- ATF6α
activating transcription factor 6α, one of the three arms of the UPR. ATF6α is activated by Grp78 dissociation. Activated ATF6α translocates to the Golgi apparatus, where the N-terminus of ATF6α is cleaved and then translocates to the nucleus to enhance the gene expression of UPR chaperones and components of ER associated protein degradation.
- IRE1
inositol-requiring ER-to-nucleus signal kinase 1, one of the three arms of the UPR. IRE1 is activated by Grp78 dissociation and auto-phosphorylation. Phosphorylated IRE1 induces X-box binding protein 1 (XBP1) splicing. Spliced XBP1 degrades mRNA, enhances ER-associated protein degradation, and upregulates gene expression of ER chaperones.
- PERK
protein kinase-like ER kinase, one of the three arms of the UPR. PERK is activated by Grp78 dissociation auto-phosphorylation. Phosphorylated PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), which inhibits ribosomal-mRNA interaction and causes subsequent mRNA degradation. This leads to reduction of protein translation. Phosphorylated eIF2α also enhances gene expression of activating transcription factor 4 (ATF4), which, in turn, increases the gene expression of chaperone proteins involved in protein folding.
- QT prolongation
a QT interval measures the length of time between the start of the Q-wave and the end of the T-wave of the ECG. This interval represents the time it takes for the lower chambers or ventricles depolarize and then repolarize. QT prolongation is associated with arrhythmia and sudden cardiac death.
- UPR
the unfolded protein response, a mechanism that responds to ER protein overload by activation of protein refolding, protein degradation, and inhibition of nascent protein translation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors have declared that no conflict of interest exists
References
- 1.Zicha S et al. (2004) Post-transcriptional alterations in the expression of cardiac Na+ channel subunits in chronic heart failure. J Mol Cell Cardiol 37, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao G et al. (2013) Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ Arrhythm Electrophysiol. 6, 1018–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaab S et al. (1998) Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation 98, 1383–1393. [DOI] [PubMed] [Google Scholar]
- 4.Tsuji Y et al. (2006) Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia. Circulation 113, 345–355. [DOI] [PubMed] [Google Scholar]
- 5.Nattel S et al. (2007) Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 87, 425–456. [DOI] [PubMed] [Google Scholar]
- 6.Voigt N and Dobrev D (2011) Ion channel remodelling in atrial fibrillation. European Cardiology 7, 97–103. [Google Scholar]
- 7.Liu M et al. (2013) Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol 54, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pu J and Boyden PA (1997) Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart: a possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res 81, 110–119. [DOI] [PubMed] [Google Scholar]
- 9.Valdivia CR et al. (2005) Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol 38, 475–483. [DOI] [PubMed] [Google Scholar]
- 10.Borlak J and Thum T (2003) Hallmarks of ion channel gene expression in end-stage heart failure. FASEB J 17, 1592–1608. [DOI] [PubMed] [Google Scholar]
- 11.Liu M and Dudley SC Jr. (2014) Targeting the unfolded protein response in heart diseases. Expert Opin Ther Targets 18, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M and Dudley SC Jr. (2016) Role for the unfolded protein response in heart disease and cardiac arrhythmias. Int J Mol Sci 17, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu M and Dudley SC Jr. (2018) The role of the unfolded protein response in arrhythmias. Channels (Austin) 12, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou A et al. (2018) HuR-mediated SCN5A messenger RNA stability reduces arrhythmic risk in heart failure. Heart Rhythm 15, 1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu M et al. (2021) Inhibition of the unfolded protein response reduces arrhythmia risk after myocardial infarction. J Clin Invest 131, e147836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang GJ et al. (2020) MIR448 antagomir reduces arrhythmic risk after myocardial infarction by upregulating the cardiac sodium channel. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu M et al. (2013) Panax quinquefolium saponin attenuates ventricular remodeling after acute myocardial infarction by inhibiting chop-mediated apoptosis. Shock 40, 339–344. [DOI] [PubMed] [Google Scholar]
- 18.Shi ZY et al. (2016) Cortistatin improves cardiac function after acute myocardial infarction in rats by suppressing myocardial apoptosis and endoplasmic reticulum stress. J Cardiovasc Pharmacol Ther 22, 83–93. [DOI] [PubMed] [Google Scholar]
- 19.Hou JY et al. (2016) Protective effect of hyperoside on cardiac ischemia reperfusion injury through inhibition of ER stress and activation of Nrf2 signaling. Asian Pac J Trop Med 9, 76–80. [DOI] [PubMed] [Google Scholar]
- 20.Hu J et al. (2019) Inhibition of CACNA1H attenuates doxorubicin-induced acute cardiotoxicity by affecting endoplasmic reticulum stress. Biomed Pharmacother 120, 109475. [DOI] [PubMed] [Google Scholar]
- 21.Santos C et al. (2014) Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: potential role in hypertension. Antioxid Redox Signal 20, 121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hetz C et al. (2015) Proteostasis control by the unfolded protein response. Nat Cell Biol 17, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M et al. (2018) Activation of the unfolded protein response downregulates cardiac ion channels in human induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 117, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song XJ et al. (2011) Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating ER stress response. Int J Med Sci 8, 564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawada T et al. (2010) X-box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE-like element in cardiomyocytes. J Mol Cell Cardiol 48, 1280–1289. [DOI] [PubMed] [Google Scholar]
- 26.Jensen BC et al. (2017) Upregulation of autophagy genes and the unfolded protein response in human heart failure. Int J Clin Exp Med 10, 1051–1058. [PMC free article] [PubMed] [Google Scholar]
- 27.Ortega A et al. (2014) Endoplasmic reticulum stress induces different molecular structural alterations in human dilated and ischemic cardiomyopathy. PLoS One 9, e107635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martindale JJ et al. (2006) Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res 98, 1186–1193. [DOI] [PubMed] [Google Scholar]
- 29.Jin J-K et al. (2017) ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res 120, 862–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bektur Aykanat NE et al. (2021) Cardiac hypertrophy caused by hyperthyroidism in rats: the role of ATF-6 and TRPC1 channels. Can J Physiol Pharmacol 99, 1226–1233. [DOI] [PubMed] [Google Scholar]
- 31.Shi YQ et al. (2015) High glucose represses hERG K+ channel expression through trafficking inhibition. Cell Physiol Biochem 37, 284–96. [DOI] [PubMed] [Google Scholar]
- 32.Horimoto S et al. (2013) The unfolded protein response transducer ATF6 represents a novel transmembrane-type endoplasmic reticulum-associated degradation substrate requiring both mannose trimming and SEL1L protein. J Biol Chem 288, 31517–31527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deshmukh A et al. (2015) Left atrial transcriptional changes associated with atrial fibrillation susceptibility and persistence. Circ Arrhythm Electrophysiol 8, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren J et al. (2021) Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol 18, 499–521. [DOI] [PubMed] [Google Scholar]
- 35.Fu HY et al. (2010) Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 122, 361–369. [DOI] [PubMed] [Google Scholar]
- 36.Liu X et al. (2014) Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension 64, 738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanhoutte D et al. (2021) Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Nature Commun 12, 3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freundt JK et al. (2018) The transcription factor ATF4 promotes expression of cell stress genes and cardiomyocyte death in a cellular model of atrial fibrillation. BioMed Res Int 2018, 3694362–3694362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang ZV et al. (2014) Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 156, 1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Z et al. (2014) Protein kianse RNA-like endoplasmic reticulum kinase (PERK)/calcineurin signaling is a novel pathway regulating intracellular calcium accumulation which might be involved in ventricular arrhythmias in diabetic cardiomyopathy. Cell Signal 26, 2591–2600. [DOI] [PubMed] [Google Scholar]
- 41.Feng PF et al. (2019) Intracellular mechanism of rosuvastatin-induced decrease in mature hERG protein expression on membrane. Mol Pharm 16, 1477–1488. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y et al. (2020) Thioridazine induces cardiotoxicity via reactive oxygen species-mediated hERG channel deficiency and L-type calcium channel activation. Oxid Med Cell Longev 2020, 3690123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuki Y et al. (2020) Ribosomal protein S7 ubiquitination during ER stress in yeast is associated with selective mRNA translation and stress outcome. Sci Rep 10, 19669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qu J et al. (2021) The roles of the ubiquitin-proteasome system in the endoplasmic reticulum stress pathway. Int J Mol Sci 22, 1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fatscher T et al. (2015) Mechanism, factors, and physiological role of nonsense-mediated mRNA decay. Cell Mol Life Sci 72, 4523–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta A et al. (2015) PERK regulated miR-424(322)-503 cluster fine-tunes activation of IRE1 and ATF6 during Unfolded Protein Response. Sci Rep 5, 18304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hiramatsu N et al. (2020) PERK-mediated induction of microRNA-483 disrupts cellular ATP homeostasis during the unfolded protein response. J Biol Chem 295, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Safiedeen Z et al. (2017) Temporal cross talk between endoplasmic reticulum and mitochondria regulates oxidative stress and mediates microparticle-induced endothelial dysfunction. Antioxid Redox Signal 26, 15–27. [DOI] [PubMed] [Google Scholar]
- 49.Liu M et al. (2009) Cardiac Na+ current regulation by pyridine nucleotides. Circ Res 105, 737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu M et al. (2010) Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res 107, 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu M et al. (2017) Role of protein kinase C in metabolic regulation of the cardiac Na+ channel. Heart Rhythm 14, 440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou C et al. (2008) AUF1 is upregulated by angiotensin II to destabilize cardiac Kv4.3 channel mRNA. J Mol Cell Cardiol 45, 832–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu T and O’Rourke B (2013) Regulation of the Na+/Ca2+ exchanger by pyridine nucleotide redox potential in ventricular myocytes. J Biol Chem 288, 31984–31992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gill JS et al. (1995) Free radicals irreversibly decrease Ca2+ currents in isolated guinea-pig ventricular myocytes. Eur J Pharmacol 292, 337–40. [DOI] [PubMed] [Google Scholar]
- 55.Sunagawa T et al. (2014) Cardiac electrophysiological alterations in heart/muscle-specific manganese-superoxide dismutase-deficient mice: prevention by a dietary antioxidant polyphenol. BioMed Res Int 2014, 704291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dong D et al. (2004) Decreases of voltage-dependent K+ currents densities in ventricular myocytes of guinea pigs by chronic oxidant stress. Acta Pharmacol Sin 25, 751–755. [PubMed] [Google Scholar]
- 57.Tao J et al. (2011) Apelin-13 protects the heart against ischemia-reperfusion injury through inhibition of ER-dependent apoptotic pathways in a time-dependent fashion. Am J Physiol Heart Circ Physiol 301, H1471–H1486. [DOI] [PubMed] [Google Scholar]
- 58.Qiu Q et al. (2013) Toll‐like receptor‐mediated IRE1 α activation as a therapeutic target for inflammatory arthritis. The EMBO Journal 32, 2477–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang L et al. (2012) Divergent allosteric control of the IRE1α endoribonuclease using kinase inhibitors. Nat Chem Biol 8, 982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mimura N et al. (2012) Blockade of XBP1 splicing by inhibition of IRE1 is a promising therapeutic option in multiple myeloma. Blood 119, 5772–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toko H et al. (2010) ATF6 is important under both pathological and physiological states in the heart. J Mol Cell Cardiol 49, 113–120. [DOI] [PubMed] [Google Scholar]
- 62.Atkins C et al. (2013) Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 73, 1993–2002. [DOI] [PubMed] [Google Scholar]
- 63.Halliday M et al. (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis 6, e1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kassan M et al. (2012) Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Atertio Thromb Vasc Biol 32, 1652–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ceylan-Isik AF et al. (2011) Endoplasmic reticulum chaperone tauroursodeoxycholic acid alleviates obesity-induced myocardial contractile dysfunction. J Mol Cell Cardiol 50, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Zhang H et al. (2013) Selective, potent blockade of the IRE1 and ATF6 pathways by 4-phenylbutyric acid analogues. Br J Pharmacol 170, 822–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao G et al. (2011) Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure/clinical perspective. Circulation 124, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou A et al. (2018) RNA binding protein, HuR, regulates SCN5A expression through stabilizing MEF2C transcription factor mRNA. J Am Heart Assoc 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gaspo R et al. (1997) Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ Res 81, 1045–52. [DOI] [PubMed] [Google Scholar]
- 70.Yao A et al. (1998) Abnormal myocyte Ca2+ homeostasis in rabbits with pacing-induced heart failure. Am J Physiol Heart Circ Physiol 275, H1441–H1448. [DOI] [PubMed] [Google Scholar]
- 71.Brundel BJ et al. (2001) Ion channel remodeling is related to intraoperative atrial effective refractory periods in patients with paroxysmal and persistent atrial fibrillation. Circulation 103, 684–90. [DOI] [PubMed] [Google Scholar]
- 72.Hong TT et al. (2012) BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm 9, 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsuji Y et al. (2000) Pacing-induced heart failure causes a reduction of delayed rectifier potassium currents along with decreases in calcium and transient outward currents in rabbit ventricle. Cardiovasc Res 48, 300–309. [DOI] [PubMed] [Google Scholar]
- 74.Brundel BJJM et al. (2001) Alterations in potassium channel gene expression in atria of patients with persistent and paroxysmal atrial fibrillation: differential regulation of protein and mRNA levels for K+ channels. J Am Coll Cardiol 37, 926–932. [DOI] [PubMed] [Google Scholar]
- 75.Li GR et al. (2002) Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol 283, H1031–H1041. [DOI] [PubMed] [Google Scholar]
- 76.Li GR et al. (2004) Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm 1, 460–468. [DOI] [PubMed] [Google Scholar]
- 77.Verkerk AO et al. (2003) Ionic remodeling of sinoatrial node cells by heart failure. Circulation 108, 760–766. [DOI] [PubMed] [Google Scholar]
- 78.Dobrev D et al. (2001) Molecular basis of downregulation of G-protein–coupled inward rectifying K+ current (IK,ACh) in chronic human atrial fibrillation: decrease in GIRK4 mRNA correlates with reduced IK,ACh and muscarinic receptor–mediated shortening of action potentials. Circulation 104, 2551–2557. [DOI] [PubMed] [Google Scholar]
- 79.Balana B et al. (2003) Decreased ATP-sensitive K+ current density during chronic human atrial fibrillation. J Mol Cell Cardiol 35, 1399–405. [DOI] [PubMed] [Google Scholar]
- 80.Zicha S et al. (2005) Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc Res 66, 472–481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.