

Abstract
Although it is well established that p53-mediated tumor suppression mainly acts through its ability in transcriptional regulation, the molecular mechanisms of this regulation are not completely understood. Among a number of regulatory modes, acetylation of p53 attracts great interests. p53 was one of the first non-histone proteins found to be functionally regulated by acetylation and deacetylation, and subsequent work has established that reversible acetylation is a general mechanism for regulation of non-histone proteins. Unlike other types of post-translational modifications occurred during stress responses, the role of p53 acetylation has been recently validated in vivo by using the knockin mice with both acetylation-defective and acetylation-mimicking p53 mutants. Here, we review the role of acetylation in p53-mediated activities, with a focus on which specific acetylation sites are critical for p53-dependent transcription regulation during tumor suppression and how acetylation of p53 recruits specific “readers” to execute its promoter-specific regulation of different targets. We also discuss the role of p53 acetylation in differentially regulating its classic activities in cell cycle arrest, senescence and apoptosis as well as newly identified unconventional functions such as cell metabolism and ferroptosis.
Introduction
The prevalence of TP53 gene mutation in human cancers suggests an important function of p53 in tumor suppression (1). Evidence from both in vitro and in vivo experiments has established the pivotal role of p53 in cell fate decisions and tumor suppression (2, 3). In response to various genotoxic insults and cellular stresses, such as DNA damage or oncogene activation, p53 plays important roles in regulating many biological processes such as cell cycle arrest, apoptosis, cellular senescence, genome stability/DNA repair, metabolic reprogramming, ferroptosis, metastasis, stemness maintenance etc. (2). p53 functions as a homo-tetrameric and sequence-specific transcriptional factor that can induce or repress expression of hundreds of target genes, leading to various cellular processes and consequences. Although transcription-independent roles have also been ascribed to p53, transcriptional regulation of p53 is essential for its tumor suppressor function (3). Given its significant role in cell fate decisions, p53 activity must be tightly controlled to prevent tumorigenesis as well as keep normal cells alive. Reversible posttranslational modifications (PTM) represent an efficient mode of regulation that allows quick responses at the protein level (4). p53 function is delicately orchestrated by a comprehensive network of modifications, including phosphorylation, acetylation, ubiquitination, and methylation, among others (4). Since the initial characterization of p53 acetylation in 1997 (ref. 5), devoted efforts from research groups worldwide have established acetylation as one of the hallmarks of p53 activation. The importance of p53 acetylation for p53 activation and function has been demonstrated by a variety of in vitro assays and in vivo experiments using genetically engineered mouse models (6, 7). Although substantial progress has been made in understanding of p53 acetylation, the answers to some of the important questions remain elusive. This review summarizes what is known of p53 acetylation, including the initial discovery, acetylation sites on p53, enzymes involved, functional consequences in p53 transcriptional activity, overall physiological significance or biological outcomes, and molecular mechanisms. There have been other excellent reviews written previously on similar topics (4, 8, 9). This review focuses on the recent progress and refining our understanding of p53 acetylation.
Concept establishment: p53 acetylation as a model for studying non-histone protein acetylation
Posttranslational lysine acetylation was first discovered on histones by Vincent Allfrey and colleagues in 1964 (ref. 10). Acetylation of lysines on N-terminal tails of histones can neutralize their positive charge, causing decondensation of chromatin and inducing changes in gene expression patterns, which represents a critical node of epigenetic regulation (11). In the following decades, lysine acetylation was also discovered on non-histone proteins, like HMG-1 (ref. 12) and tubulin (13). However, acetylation of non-histone protein did not receive much attention until the discovery of p53 acetylation in 1997 (ref. 5). The initial observation that CBP/p300 can bind and function in synergy with p53 to promote its transcriptional activity (14) led to the discovery that human p53 can be acetylated by CBP/p300 at multiple lysine residues (K370, K372, K373, K381, K382) within the C-terminal regulatory domain (5). Acetylation of p53 by p300/CBP activates expression of its target genes, which represented a previously unknown mechanism for p53 activation (5). Over the past 25 years, together with other reseach groups, we have used biochemical analyses combined with genetically engineered mouse models to identify acetylation sites on p53 and determine the roles of lysine acetylation in regulating p53-mediated trasncription and tumor suppression. We established that site-specific acetylation plays a critical role in promoter-specific regulation of p53 targets, resulting in distinct biological outcomes upon stresses (6, 15–19). Significantly, we have shown that acidic domain containing proteins act as a new class of readers for unacetylated p53 C-terminal domain and are critically involved in controlling acetylation-mediated transactivation (20, 21). In contrast, Dicer is a reader for unacetylated DBD of p53 regulating p21 and Puma transcription (22). Since the characterization of p53 C-terminal acetylation, other non-histone proteins have also been identified as bona fide substrates for acetyl transferases (23, 24). Proteins that can be acetylated are involved in diverse cellular processes such as transcription, gene splicing, nuclear transport, cell cycle, metabolism and more. For example, several components of the p53 pathway are acetylated, including Daxx, PML, PTEN, and HAUSP (23). There are other acetylated transcription factors such as Yin Yang 1, STAT3, c-MYC, and HIF-1α. Other acetylated proteins include nuclear receptors like androgen receptor and estrogen receptor, nuclear import factors like Importin-α, DNA repair pathway proteins like Ku70, molecular chaperones like Hsp90, and even viral proteins like E1A, L-HDAg and S-HDAg (24, 25). These result show that the scope of lysine acetylation has expanded from epigenetic events to virtually all nuclear processes and many cytoplasmic functions. The discovery of p53 acetylation and continuous studies in this field have not only established the roles of acetylation-mediated regulation of p53, but also laid the foundation for acetylation as a general mechanism for regulation of non-histone proteins, which goes beyond p53 and cancer biology.
p53 acetylation: sites and enzymes
p53 protein harbors six major domains, namely two intrinsically disordered N-terminal transactivation domains (TADs), a proline-rich domain (PRD), a central deoxyribonucleic acid (DNA)-binding domain (DBD) followed by a tetramerization domain (TD), and an intrinsically disordered C-terminal regulatory domain (CTD) (26) (Figure 1). Acetylation of lysine residues in human p53 can be readily detected in its C-terminal domain (K370, K372, K373, K381, K382), the central DNA binding domain (K101, K120, K139, and K164), and to some extent in lysines between those two domains (K305, K319, K320 and K357). The corresponding sites in mice have also been denoted in Figure 1 (lower panel). Six acetyltransferases, which can be classified into two different groups, p300/CBP/PCAF or Tip60/MOF/MOZ, have been identified as enzymes that acetylate p53 with distinct site specificity. Initial work revealed that five highly conserved C-terminal lysine residues in human p53 (K370, K372, K373, K381, K382) are acetylated by p300 and CBP (5). p300 and CBP have a lot of similarities, in particular they share approximately 91% homology in their histone acetyltransferase (HAT) domains (8). Both p300 and CBP can directly bind to p53 (ref. 5, 14, 27–32) and acetylate the aforementioned five C-terminal sites. Another histone acetyltransferase, PCAF (p300-CBP associated factor), was shown to acetylate p53 at lysine 320 (K320) in the tetramerization domain (33–35). In addition, the DNA binding domain also harbors multiple lysine sites that can be acetylated. K120 is a unique lysine because only the three members of the MYST HAT family, including Tip60 (HIV-1 tat-interacting protein, 60 kilodalton), MOF (males absent on the first) and MOZ (monocytic leukemia zinc finger), but not by CBP/p300 or PCAF can acetylate this site (15, 36–38). The other three acetylation sites (K101, K139 and K164) on the DBD of p53 can all be acetylated by CBP/p300 (ref. 6, 18, 19). Recently, Tip60 was shown to also acetylate K139 (ref. 19) and MOZ also promotes p53 acetylation at K382 (ref. 38). Three additional acetylation sites (K305, K319 and K357) of p53 localized between DBD and CTD were also identified. K305 is acetylated by CBP/p300, while the enzyme catalyzing K319 and K357 acetylation remain unknown (39, 40). Because the significance of these three acetylation sites is not clear, we will not discuss them further.
Figure 1.
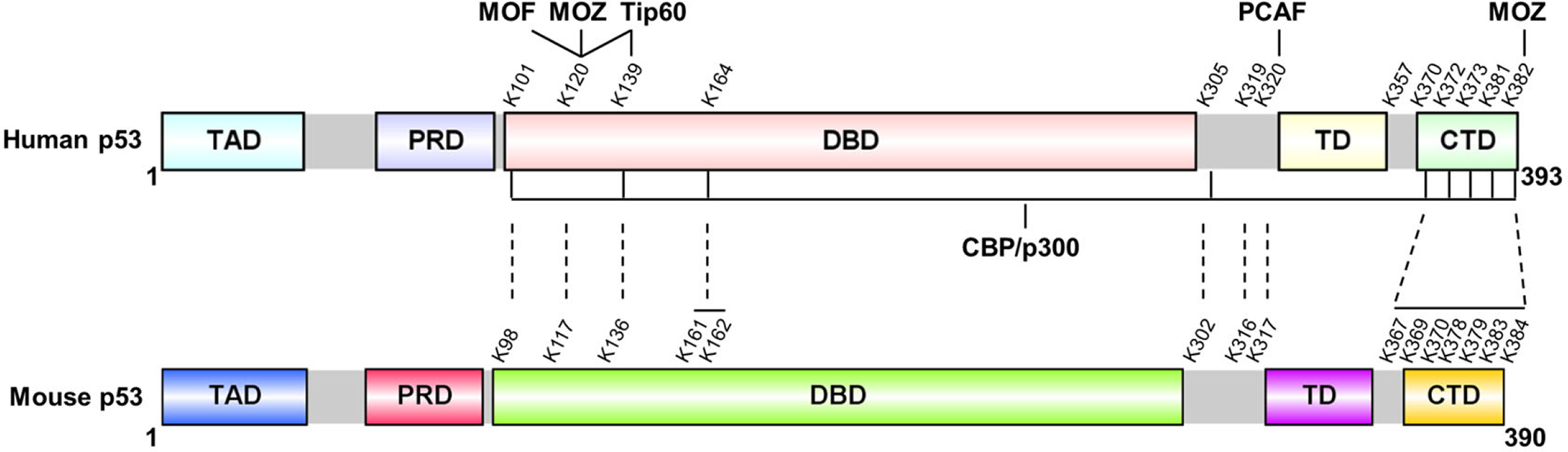
Overview of p53 acetylation sites, enzymes involved, and functional significance.
Domain structure of human or mouse p53 is shown. Multiple lysine residues on p53 are targeted for acetylation by six different acetyltransferases: p300/CBP, PCAF, Tip60, MOF, and MOZ. TAD, transactivation domain; PRD, proline-rich domain; DBD, DNA binding domain; TD, tetramerization domain; CTD, C-terminal domain. The figure is modeled from a published review (8).
Functional significance of p53 acetylation underlined by genetically engineered mouse model
To elucidate the function of p53 acetylation, early studies were performed to reduce p53 acetylation either enzymatically or by using acetylation deficient p53 mutant in cell culture-based assays. Results from these experiments have demonstrated that p53 acetylation is critical for p53 activation, even though acetylation at different sites may have some functional redundancy. For example, HDAC1 complex-mediated deacetylation of p53 strongly represses p53-dependent transcriptional activation, cell growth arrest, and apoptosis (41). SIRT1 was reported to bind and deacetylate p53 and repress p53-dependent apoptosis in response to DNA damage and oxidative stress (42, 43). Consistently, mutating K120, K164 and all six C-terminal lysine residues to arginine (p53-8KR) completely abrogated p53-dependent transactivation of p21 and the ability to induce cell growth arrest, suggesting that acetylation is indispensable for p53 activation (6). Nevertheless, the questions whether p53 acetylation is important for p53-mediated tumor suppressor function and what the precise role of p53 acetylation is under physiological conditions were not answered. To address these questions, knock-in mice in which homologous lysine residue(s) in p53 are substituted by arginine (acetylation-deficient K to R mutant) or glutamine (acetylation-mimic K to Q mutant) have been generated (Figure 2). The initial study showed that acetylation of p53 C-terminal lysine residue at K370, K372, K373, K381 and K382 activates p53 and boosts the expression of p53 target genes. Feng et al. generated the C-terminal acetylation-deficient p53-6KR (K367R+K369R+K370R+K378R+K379R+K383R; identical to the human p53-6KR mutant) knock-in mice. Studies using p53-6KR mice showed that p53-dependent gene expression in embryonic stem cells and thymocytes is impaired in a promoter-specific manner after DNA damage, supporting the notion that C-terminal acetylation facilitates p53 dependent transactivation in response to DNA damage (44). Moreover, p53-6KR thymocytes are slightly more resistant to p53-dependent apoptosis compared with the control thymocytes after ironizing radiation. However, this effect is cell type-specific, limiting to embryonic stem cells and thymocytes, but not in embryonic fibroblasts. Similarly, p53-7KR (K367R+K369R+K370R+K378R+K379R+K383R+K384R) knock-in mice were generated because mouse p53 has one additional lysine at the C-terminus. Again, there is no substantial differences in p53 mediated gene expression, cell cycle arrest or apoptosis using the mouse embryonic fibroblasts (MEFs) derived from mouse (45). p53-7KR knock-in mice develop normally and exhibit no increased susceptibility to cancer. Notably, in both studies using p53-6KR or p53-7KR mice, normal p53 stabilization was observed in embryonic stem cells, MEFs, and thymocytes both before and after DNA damage, indicating that ubiquitination at the C-terminal lysine residues is not required for p53 degradation and the remaining lysine residues in p53 are sufficient for p53 degradation. These data suggested that acetylation of C-terminal lysine residues only has modest effects on p53 transactivation. Interestingly, Simeonova et al. showed that homozygous mutant mice expressing p53Δ31, a truncated p53 lacking the C-terminal 31 amino acids, exhibit increased p53 activity and suffer from aplastic anemia and pulmonary fibrosis (46). Hamard et al. found that the CTD regulates gene expression via multiple mechanisms, depending on the tissues and target genes (47). Both studies demonstrate that the deletion of p53 CTD in mice results in p53 activation and premature lethality (46, 47), supporting the notion that the CTD acts as a negative regulatory domain for p53 (ref. 48–50). To further investigate the in vivo functions of CTD acetylation, a p53-7KQ (K to Q mutations at K367, K369, K370, K378, K379, K383 and K384) knock-in mouse was recently generated to mimic constitutive acetylation of the p53 C-terminus (20). p537KQ/7KQ mice were perinatal lethal due to developmental defects in the brain and their inability to obtain nourishment (20), yet this lethality was averted in p537KQ/− mice, with normal postnatal development (51). Nevertheless, p537KQ/− mice died prematurely due to anemia and hematopoiesis failure. Interestingly, increased expression of p53 targets such as p21, Puma, Noxa, Gadd45, and Ccng1 was observed in various tissues of the acetylation-mimicking p53 mutant without obvious increase of p53 levels. In the well-established pancreatic ductal adenocarcinoma mouse model (PDAC), expression of this acetylation-mimicking p53 mutant protein effectively suppressed K-Ras-induced PDAC tumor development in the absence of robust p53 stabilization. This study showed that CTD acetylation can result in tumor suppression, in the absence of p53 stabilization (51, 52). While both K to R and K to Q mutations block p53 major modifications on those lysine residues, such as ubiquitination, methylation, and neddylation, K to Q mutation could specifically mimic the acetylation status of lysine. The hyper-activation of p53-7KQ may reflect the net outcome of p53 CTD acetylation.
Figure 2.
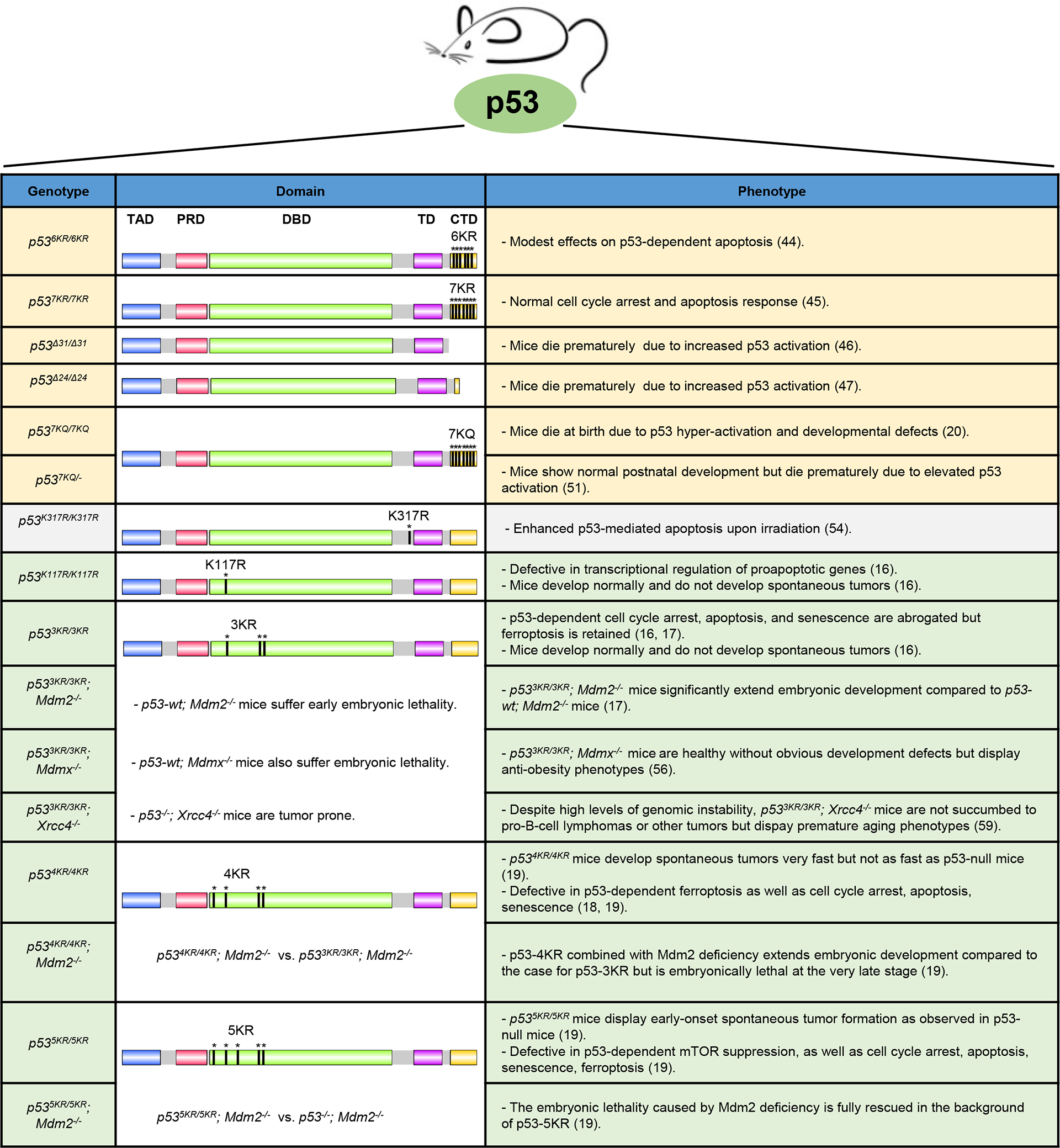
Summary of all the p53-mutant mouse models related to acetylation-mediated regulation as discussed in this review.
It was reported that acetylation of K320 by PCAF upon DNA damage favors cell survival by increasing p53-mediated activation of genes causing cell cycle arrest, such as p21, and suppressing expression of pro-apoptotic genes such as Noxa (53). Consistently, mice with a K317R (equivalent to K320R in humans) mutation in p53 displayed increased expression of pro-apoptotic target genes and enhanced p53-mediated apoptosis upon irradiation (54). These data support a pro-survival role of p53 K320 acetylation by suppressing apoptosis and allowing the cells with time to repair DNA and resume proliferation once the cellular damage is repaired. This is an example that p53 acetylation dictates promoter specificity to selectively activate target genes. This model of regulation is particularly prominent for acetylation of p53 DNA binding domain.
Acetylation of lysine residues within the DNA binding domain (DBD) hardly affects p53 stability or DNA binding but plays a critical role in promoter-specific regulation of p53 target genes. Two groups independently showed that Tip60 acetylates p53 at K120 to selectively induce the expression of proapoptotic genes Puma and Bax, but not cell cycle arrest genes p21 and others like Mdm2 (ref. 15, 36). In contrast, K164 acetylation is responsible for the induction of cell cycle arrest by activating p21 expression (6). Notably, the studies using a mouse model of p53 DBD acetylation deficient mutant mice have revealed unique insights in acetylation mediated gene regulation and tumor suppressor function by p53 (Figure 3). The first acetylation-deficient mouse was p53-K117R, where lysine 117 in murine p53 (corresponding to human K120) was replaced with an arginine. K117R mutation has no effect on p53 stabilization but completely abolishes p53-mediated apoptosis, yet the mouse did not develop spontaneous cancer, suggesting that apoptosis is dispensable for tumor suppression by p53 (ref. 16). In fact, p53-dependent cell cycle arrest and senescence remain intact in p53K117R/K117R MEFs. In a subsequent mouse model, the acetylation sites K117, K161, and K162 were all replaced with arginine residues. This p53-3KR mouse lost the ability to transactivate p21 and was unable to induce cell cycle arrest or senescence, in addition to apoptosis. Surprisingly, these mice were also not susceptible to spontaneous tumor development (16). These results suggest that the ability of p53 to induce growth arrest/senescence and apoptosis may be dispensable, in certain tissues and tumor types, to suppress spontaneous tumor development. Strikingly, p53-3KR was shown to still be able to induce ferroptosis by regulating genes involved in oxidative stress, such as Gls2 and Slc7a11 (ref. 16, 17). Moreover, it had been known that the Mdm2 deficient mouse was embryonically lethal, and that this could be rescued by the knockout of p53 (ref. 55). Surprisingly, the p53-3KR mutant, which is largely defective in transcription except for ferroptosis genes, was also embryonic lethal in the Mdm2 knockout background, although p53-3KR mutant extended the embryonic development (17). Notably, p53-3KR protein levels dramatically increased in the absence of Mdm2, demonstrating again that Mdm2 is critical for p53 stability (17). Unlike p533KR/3KR; Mdm2−/− mice, p533KR/3KR; Mdmx−/− mice are healthy but show anti-obesity phenotypes without obvious developmental defects (56). Notably, the levels of p53 protein are only slightly increased and can be further induced upon DNA damage in p533KR/3KR; Mdmx−/− mice, suggesting that Mdmx (also called Mdm4) is only partially required for p53 degradation in vivo. The anti-obesity phenotypes in p533KR/3K; Mdmx−/− mice are caused by activation of lipid oxidation and thermogenic programs in adipose tissues (56). These results demonstrate the specific effects of the p53/Mdmx axis in lipid metabolism and adipose tissue remodeling, other than commonly expected tumor suppression pathway. To identify the mechanism responsible for regulation of ferroptosis by p53, we have demonstrated that acetylation at K101 of human p53 (lysine K98 for mouse p53) is critical for the regulation of the metabolic and ferroptosis-related genes, such as TIGAR and SLC7A11 (ref. 18). While the loss of mouse p53 K98 acetylation (p53-K98R) alone has very modest effects on p53-mediated transactivation, simultaneous mutations at four acetylation sites (p53-4KR: K98R+K117R+K161R+K162R) completely abolish its ability to regulate metabolic targets such as TIGAR and SLC7A11 (ref. 18). Defective in conventional p53 responses and ferroptosis, p53-4KR mice develop tumor with time but are still resistant to early-onset tumorigenesis. p53-4KR combined with mdm2 deficiency is still embryonically lethal, although mdm2 knockout embryos are partially rescued by p53-4KR compared to p53-3KR. More recently, K139 of human p53 (K136 of mouse p53) was identified as a novel acetylation site that is important for p53 mediated mTOR suppression (19). While the acetylation-defective mutant p53-4KR retains the ability to inhibit mTOR activity through activating cellular targets such as SENS2 and DITT4, this activity is completely abolished in p53-5KR (K136R+K98R+K117R+K161R+K162R). Simultaneous mutations at all five acetylation sites (p53-5KR) diminished its remaining tumor suppression function. Moreover, the embryonic lethality caused by the deficiency of Mdm2 was fully rescued in the background of p535KR/5KR, but not in p534KR/4KR background. Notably, the early-onset tumor formation observed in p535KR/5KR was similar to that in p53-null mice and could be suppressed upon the treatment of the mTOR inhibitor. This study suggests that p53-mediated mTOR regulation acts as a previously unanticipated checkpoint in both embryonic development and tumor suppression, independent of cell cycle arrest, senescence, apoptosis, and ferroptosis (Figure 3).
Figure 3.
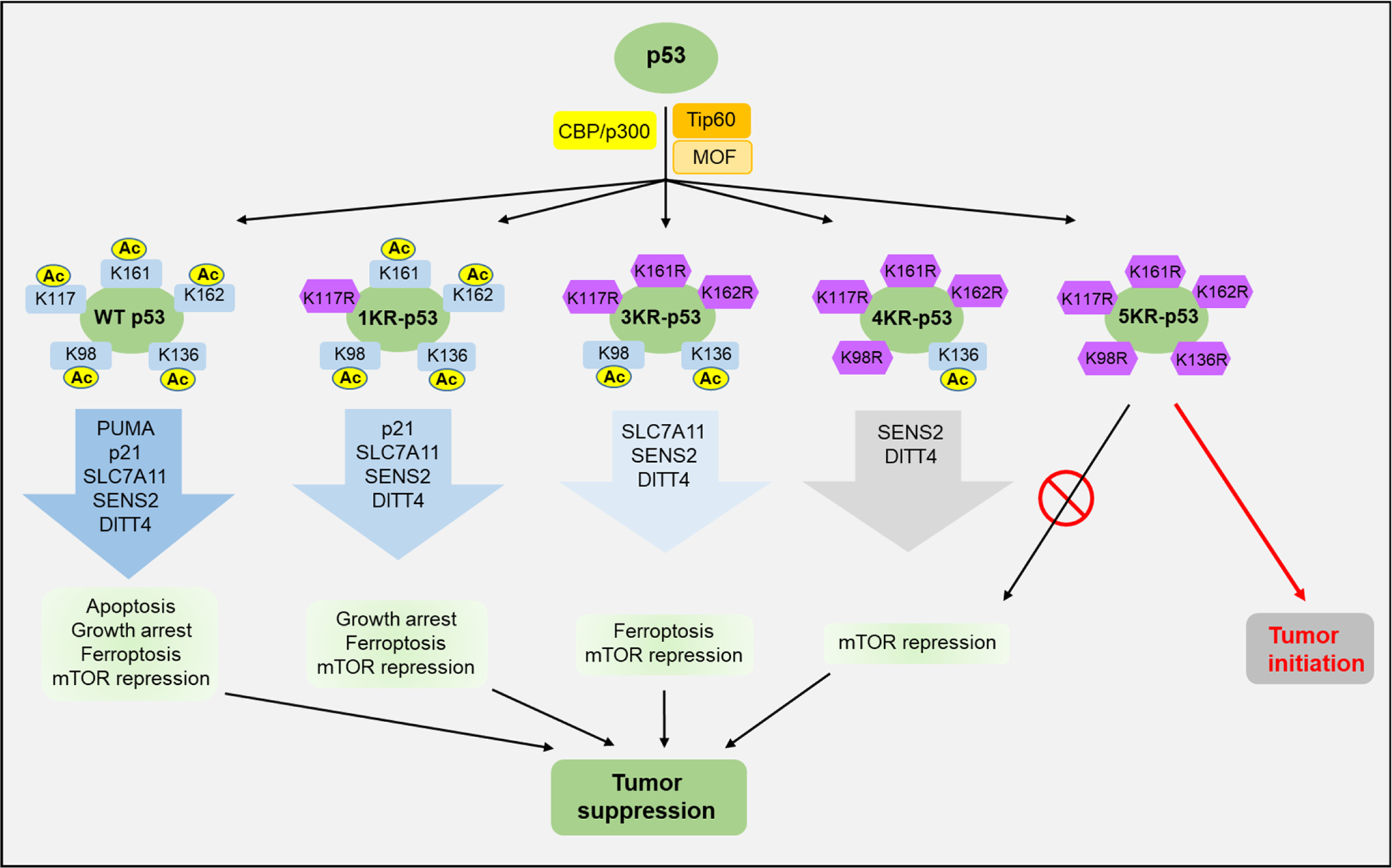
Graphical presentation of acetylation mediated regulation of p53 tumor suppressive functions, as revealed in mouse models.
Wild type p53 retains all tumor suppressive functions; 1KR is unable to induce apoptosis; 3KR is defective in apoptosis and cell cycle arrest; 4KR is not capable of undergoing apoptosis, cell cycle arrest and ferroptosis; 5KR further loses the ability to repress mTOR pathway.
In summary, in vivo studies using genetically engineered mice validated the physiological significance of p53 acetylation for its function in embryonic development and tumor suppression (19) (Figure 2). On one hand, C-terminal acetylation fine-tunes p53 function by relieving the inhibitory effects of the CTD; one the other hand, acetylation in the DNA binding domain determines the target selectivity and leads to diverged biological outcomes (Figure 4). Likely, acetylation in these domains works in concert to promote gene expression in a timely and tissue specific manner. Through site-by-site analysis of acetylation at individual lysine residues within p53 DBD, the downstream responses required for p53’s anti-tumor function can be revealed sequentially, which not only aid our understanding of p53, but also provide a path for studying acetylation of non-histone proteins.
Figure 4.
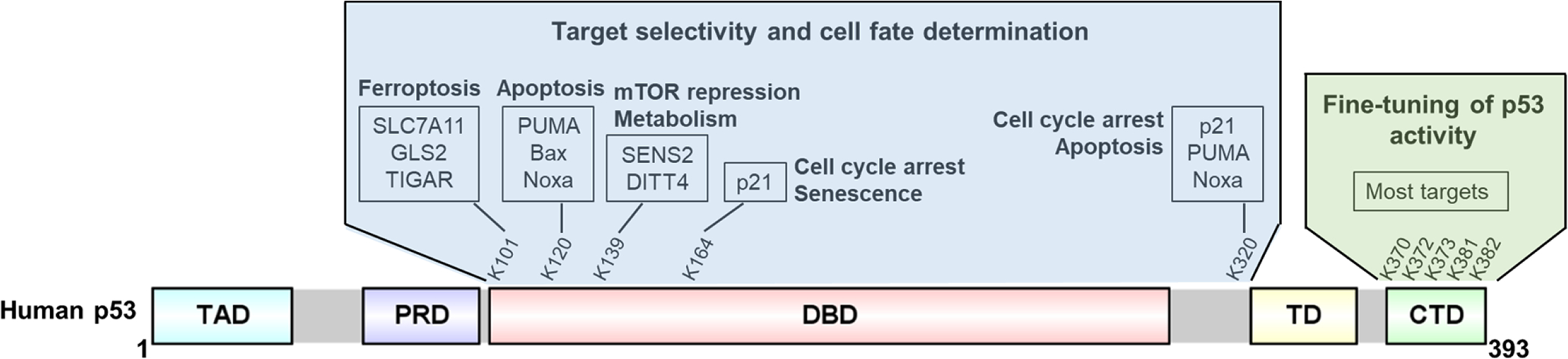
Distinct functional consequences of p53 acetylation in DBD and CTD.
Acetylation in p53 C-terminus fine-tunes expression of major p53 targets, while acetylation in the DNA binding domain plus at K320 determines the target selectivity, leading to divergent biological outcomes.
Ferroptosis: an unconventional but important aspect of p53 tumor suppressive function unveiled by p53-3KR
Well characterized biological outcomes of p53 activation — cell cycle arrest, senescence, and apoptosis, had been long believed to be essential for p53-mediated tumor suppression. However, this notion has been challenged by multiple lines of evidence. p53-3KR mice are able to suppress spontaneous tumor development in the absence of cell cycle arrest, senescence and apoptosis (16). While the transactivation-dead p5325,26,53,54 TAD1/2 mutant underlines the importance of transcriptional activation for p53-mediated tumor suppression, the p5325,26 TAD1 mutant, which has lost the capacity to drive cell cycle arrest or apoptosis, is still completely functional as an effective tumor suppressor in mice (57). Moreover, a study shows that p21−/−; Puma−/−; Noxa−/− mutant mice, which display defective cell cycle arrest and apoptosis responses, are not prone to spontaneous tumor development (58). Finally, the embryonic lethality caused by the deficiency of Xrcc4, a key DNA double strand break repair factor, can be fully rescued in the p533KR/3KR background. Notably, despite high levels of genomic instability, p533KR/3KR; Xrcc4−/− mice, unlike p53−/−; Xrcc4−/− mice, are not succumbed to pro-B-cell lymphomas. Nevertheless, p533KR/3KR; Xrcc4−/− mice display aging-like phenotypes (59) (Figure 2). Collectively, these studies challenge the significance of the p53 responses to DNA damage, such as cell cycle arrest, senescence, and apoptosis for its tumor suppression activity, and force us to rethink the mechanisms underlying p53-mediated tumor suppression.
Earlier studies suggested the implication of p53-regulated metabolic processes in tumor suppression by identifying a number of metabolic targets of p53 such as TIGAR, GLS2 and SCO2, which limit glycolysis and the levels of reactive oxygen species (ROS) (1). One of the research focuses was to investigate whether p53 has the ability to suppress ROS, which in turn limits oxidative stress and DNA damage (60). Interestingly, while defective for the three conventional p53 functions, p53-3KR mice are still capable of regulating the expression of multiple metabolic p53 target genes including GLS2, TIGAR and GLUT3 (ref. 16). Among these p53 metabolic target genes, TIGAR, a protein with bisphosphatase activity, plays a critical role in limiting cellular ROS and is therefore deemed as a promising anti-tumor effector of p53 (ref. 61, 62). However, loss of Tigar in mice leads to increased survival and slowed tumor development in intestinal and lymphoma models, suggesting a benefiting role of Tigar for tumor growth (63). Recently, it was shown that ROS regulation by TIGAR supports premalignant pancreas tumor development, and TIGAR and ROS levels are dynamically regulated throughout tumor progression (64). These divergent research results urges us to reconsider the role of p53 in ROS and tumorigenesis (65).
Strikingly, we found p53-3KR was capable of inducing ferroptosis through direct transcriptional repression of SLC7A11, a key component of the cystine-glutamate antiporter (the xCT system) (17). Ferroptosis is a form of programmed cell death that results from the accumulation of lipid peroxides (66). The primary cellular mechanism of protection against ferroptosis is mediated by glutathione peroxidase 4 (GPX4), a glutathione (GSH)-dependent hydroperoxidase that converts lipid peroxides into non-toxic lipid alcohols at the expense of reduced glutathione (67). Decreases in GSH biogenesis or inactivation of GPX4 can lead to the accumulation of lipid peroxides, resulting in ferroptotic cell death (66). By repressing SLC7A11 expression, p53 limits cystine uptake and reduces the biogenesis of intracellular glutathione (GSH), the primary cellular antioxidant, thus diminishing GPX4-mediated suppression of ferroptosis. Unlike p53 knockout cells, p53-3KR cells retain the ability to undergo ferroptosis, which is consistent with the fact that p53-3KR mice are not susceptible to early-onset tumorigenesis (16). Moreover, although the p53-3KR mutant is defective in transcription for proapoptotic genes but not for ferroptosis genes, p53-3KR embryos in the MDM2 knockout background die during embryonic development, suggesting other type(s) of cell death cause the lethality (55). Interestingly, treating embryos with ferrostatin-1, which inhibits ferroptosis, extended the embryonic development (17). In a xenograft mouse model, p53-3KR has been shown to efficiently inhibit tumor growth, which was restored by overexpression of SCL7A11. p53-4KR, unable to induce ferroptosis, is severely impaired in suppressing tumor growth in mouse xenograft models (18). Of note, the p5325,26,53,54 transactivation-dead mutant is unable to inhibit SLC7A11 or promote ferroptosis (68). Moreover, the P47S variant of p53, with impaired ability to downregulate SLC7A11, is defective in promoting ferroptosis and repressing tumor development (69, 70). These studies indicate that p53-mediated transcriptional repression of SLC7A11 is critical for inducing ferroptosis and tumor suppression. Considering these observations and previous studies on antioxidative roles of p53, an intriguing mode can be proposed, in which p53 can dynamically regulate the accumulation of ROS. In response to low or basal ROS levels, p53 may prevent cells from accumulating lethal levels of ROS while also allowing survival and repair of moderate oxidative damage. However, in response to higher or unmanageable ROS levels, p53 may instead facilitate the removal of unsalvageable cancer cells through ferroptotic cell death (68).
The importance of SLC7A11-mediated ferroptosis in tumor suppression was highlighted by a study using PDAC mouse model in which the tumor growth was inhibited after deletion of Slc7a11 (ref. 71). By screening for potential contributors to p53/SCL7A11-meidated ferroptosis, we identified a new pathway involving ALXO12 (ref. 72). ALOX12 is a member of the ALOX arachidonate lipoxygenase family. SLC7A11 binds and sequesters ALOX12 from its substrate, polyunsaturated fatty acids (PUFAs). Inhibition of SCL7A11 expression by p53 allows the release of ALOX12 and results in increased ALOX12 activity, which subsequently oxidizes PUFAs and causes ferroptosis. ALOX12 inactivation diminishes p53-mediated ferroptosis induced by reactive oxygen species stress and abrogates p53-dependent inhibition of tumor growth in xenograft models. Loss of one Alox12 allele is sufficient to accelerate tumorigenesis in Eμ-Myc lymphoma models. Early studies showed that the ferroptotic responses induced by either erastin or GPX4 inhibitors are dependent on acyl-CoA synthetase long-chain family member 4 (ACSL4), an enzyme that promotes biosynthesis of unsaturated phospholipids, the main substrates for lipid peroxidation (73, 74). Notably, ACSL4 is shown to be dispensable for p53/ALOX12-mediated ferroptosis, suggesting the p53/SCL7A11/ALOX12 axis is independent of the p53/SCL7A11/GPX4 pathway. Conversely, ALOX12 is also dispensable for ferroptosis induced by erastin or GPX4 inhibitors. p53 has also been shown to promote ferroptosis through regulating other metabolic targets and pathways, such as GLS2 (glutaminolysis), SAT1 (polyamine catabolism), and ferredoxin reductase (unknown mechanism) (75–77). In summary, p53 has a crucial role in regulating ferroptosis and p53-mediated ferroptosis is important for p53 tumor suppressor function, reviewed elsewhere (78).
p53 acetylation and stabilization
p53 is maintained at low levels under normal homeostasis conditions by MDM2-catalized polyubiquitination, and subsequent proteasome-mediated degradation. Upon DNA damage, p53 levels increase dramatically for its activation. Early studies showed that p53 C-terminal acetylation at K370, K372, K373, K381 and K382 boost p53 activation through several mechanisms including promoting p53 protein stabilization (79, 80). The C-terminal lysine residues of p53 can undergo acetylation, as well as MDM2-mediated polyubiquitination (81, 82). Because acetylation and ubiquitylation occur on the same sites, they are mutually exclusive events and represent competing mechanisms for stabilization and activation (79). Consistent with this notion, the acetylation levels of p53 are significantly enhanced in vivo in response to almost every type of stress, which correlates with stress-induced stabilization (82). Conversely, MDM2 can reduce acetylation of p53 by inhibiting CBP/p300 (ref. 82, 83) or by recruiting HDAC1 to deacetylate p53, thus allowing MDM2 to ubiquitinate p53 and reduce p53 levels (84). Although in vitro studies showed that the C-terminal lysine residues are the major sites for ubiquitination, both the levels and the activity of p53 were not significantly altered in the p53 knock-in mice, in which the C-terminal lysine residues were replaced with arginine (44, 45). These observations suggest that the C-terminal lysines are not the only residues ubiquitinated by MDM2 and alternative lysine residues are utilized for MDM2-mediated degradation. Indeed, p53 acetylation was shown to inhibit MDM2 binding and p53 ubiquitination not only at acetylated lysine residues but also at unacetylated residues, which represents a unique mechanism in addition to the site competition (79). More recently, we generated acetylation-mimicking p53-7KQ mouse model and demonstrated that mimicking p53 CTD acetylation can induce transcriptional activation and tumor suppression in vivo, in the absence of robust p53 stabilization. This study provides evidence that CTD acetylation can induce p53 transcriptional activation and tumor suppression through stabilization-independent mechanisms (51, 52).
Mechanistic insights into p53 acetylation mediated actions: molecular readers for p53 acetylation
Given the significant role of p53 CTD acetylation in p53 transactivation, independent of mediating p53 stabilization (51), the underlining mechanisms need to be elucidated. As p53-3KR or p53-4KR does not affect p53 stability or DNA binding (16, 18), what are the mechanisms that determine promoter-specific regulation and lead to distinct biological outcomes? Since the lysine sites, acetyltransferases (acetylation writers) and deacetylases (acetylation erasers) of p53 acetylation have been well characterized and extensively studied; the missing piece for understanding p53 acetylation-mediated actions could be the functional molecular readers that specifically “read” p53 acetylation status. Bromodomains, a protein domain of approximately 110 amino acids, has been shown to “read” histone N-terminal lysine acetylation (85). Recognition of histone lysine acetylation by bromodomains can translate the signals carried by acetylated lysine residues into various phenotypes (85). Notably, bromodomains display higher affinity preferentially for regions where multiple acetylation sites exist in proximity, which is present in p53 C-terminus. Indeed, bromodomain-containing proteins such as CBP/p300 were shown to recognize acetylation in p53 CTD (Figure 5.A). DNA damage induced p53 acetylation at K382 and K320 promotes recruitment of its coactivators, p300/CBP and TRRAP, to the CDKN1A promoter and increases histone acetylation (86), which is required for the activation of p53-induced cell cycle arrest after UV exposure (87) (Figure 5.A). Likewise, TAF1, the largest subunit of the transcription initiation factor TATA-box binding protein (TFIID), recognizes acetylation at K373 and K382 of p53 through its bromodomains after UV damage, which is important for TAF1 recruitment to CDKN1A promoter (87) (Figure 5.A). A recent study showed that Polybromo 1 (PBRM1, also known as BAF180), a component of SWI/SNF chromatin remodeler complex, acts as a reader for p53 acetylation at lysine 382 through its bromodomain 4 (BD4) to achieve full activation of p53 targets such as CDKN1A upon DNA damage (88) (Figure 5.A).
Figure 5.
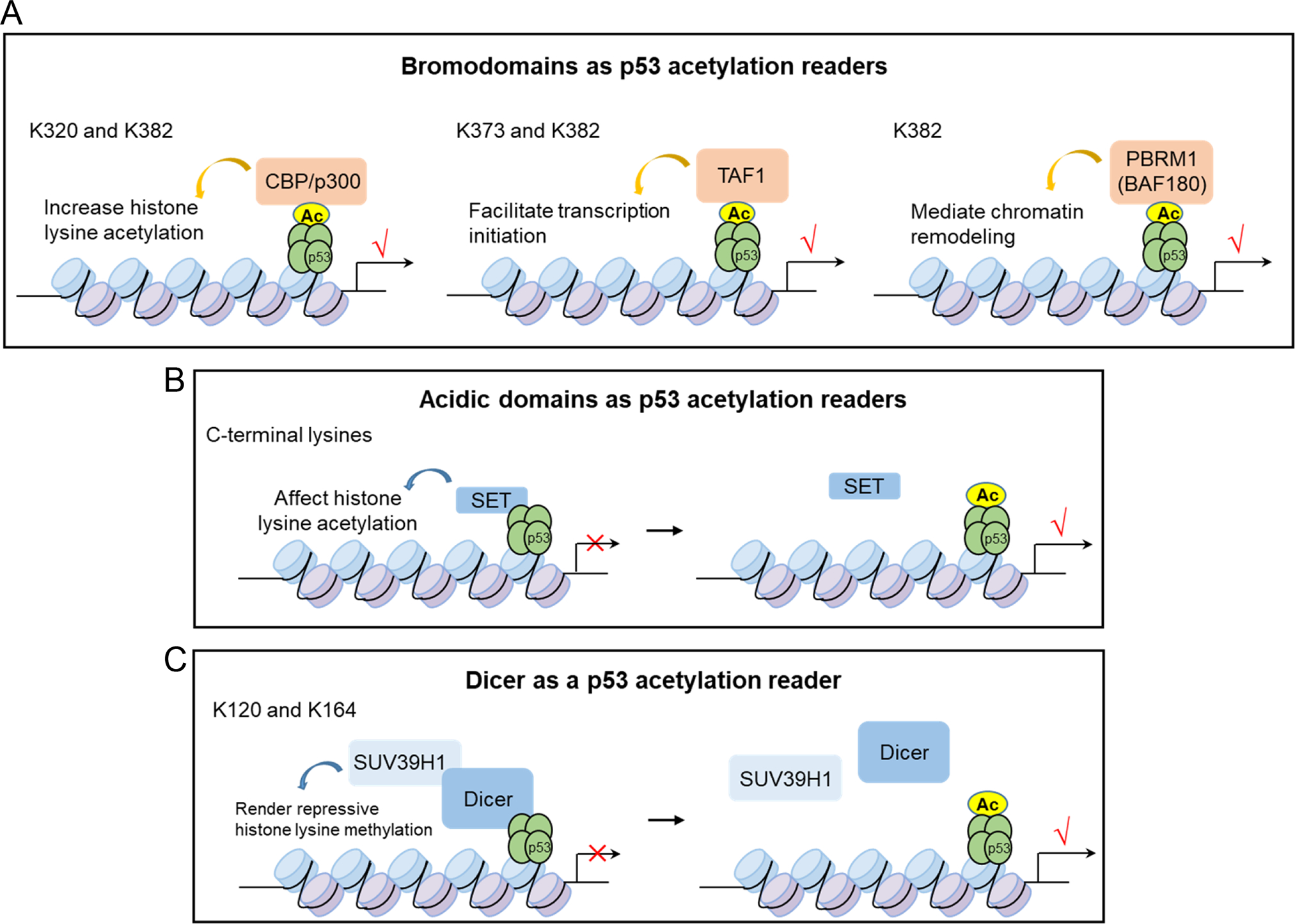
Molecular readers for p53 acetylation.
(A) Bromodomains as p53 acetylation readers. p53 acetylation at specific lysine sites can be recognized by bromodomain-containing proteins such as CBP/p300, TAF1, or PBRM1, which promotes their recruitment to chromatin and activates transcription through different mechanisms.
(B) Acidic domains as p53 acetylation readers. Acidic domain-containing proteins, such as SET, bind unacetylated p53 CTD and function as transcriptional corepressors. p53 acetylation on CTD can repel these acidic domain corepressors, mediating “anti-repression”.
(C) Dicer as a p53 acetylation reader. Dicer binds unacetylated p53 DBD and represses the transcription of p21 and Puma by recruiting SUV39H1 to render repressive histone methylation. p53 acetylation at K120 and K164 dislodges Dicer, thus activating p21 and Puma transcription.
In contrast, we recently identified acidic domain (AD)-containing proteins as a novel class of acetylation reader proteins that recognize the unacetylated CTD of p53, such as SET nuclear proto-oncogene (SET), DDB1 and CUL4 associated factor 1 (VPRBP), death domain associated protein (DAXX), and proline, glutamate and leucine rich protein 1 (PELP1) (20). Whereas the bromodomain-containing HATs such as p300/CBP, induce histone acetylation after binding to promoters, SET binds unacetylated CTD of p53 and represses p53 transactivation by attenuating p300/CBP-dependent acetylation of H3K18 and H3K27 on the promoters of p53 target genes. Loss of SET induces activation of p53 targets including p21, Puma, and Mdm2. The acidic domain of SET possesses a negative charge and favorably binds unacetylated CTD, which is lysine rich and positively charged. Acetylation of p53 C-terminal lysine residues can neutralize its positive charge and lead to p53 activation by blocking the interactions of p53 with SET without affecting p53 stability (Figure 5.B), reminiscent of the “anti-repression” model proposed previously (80). Recently, it was shown that Kaposi Sarcoma (KS)-associated herpesvirus (KSHV) encoded latency-associated nuclear antigen specifically interacts with unacetylated p53 through two tandem acidic domains and represses p53 response to facilitate KSHV latency establishment (89). These studies showed that p53 CTD can serve as a docking site for corepressors, consistent with the notion that the CTD is a negative regulatory domain for p53, which has been proposed in early studies (46, 47). Moreover, this acetylation-regulated interaction model can also be applied to other lysine rich proteins that undergo acetylation, such as histone H3, FOXO1, and Ku70, suggesting this model is widely present in nature (20, 21).
As for acetylation in p53 DBD, we recently revealed an unexpected role for Dicer as a reader of the unacetylated DNA binding domain of p53 in association with transcriptional regulation (22), which explains how p53-3KR loses the ability to regulate p21 and Puma. Dicer suppresses the p53-dependent activation of p21 and Puma but have no obvious effect on Mdm2 or Tigar expression, similar to the observations that the acetylation-defective p53-3KR mutant losses its ability to activate the expression of targets such as p21 and Puma, but not Mdm2 (ref. 16). Mechanically, Dicer was found to directly bind the DNA binding domain of p53 and recruited to its target gene promoters, where Dicer recruits histone-lysine methyltransferase SUV39H1 to elicit transcriptional repression. Further investigation demonstrated that Dicer preferentially binds to unacetylated forms of p53, whereas the interaction between Dicer and p53 is dramatically suppressed by stress-induced p53 acetylation at K120 and K164 (Figure 5.C). Upon loss of Dicer expression, the transcriptional activation of p21 and Puma by mouse p53-3KR is largely restored. In contrast, p53-mediated Mdm2 transcription remains unchanged (22). This study established Dicer as a previously unrecognized transcriptional repressor that serves as a reader for unacetylated p53 DBD and regulates the p53 transcriptional program in a promoter-specific manner. This study partially explains how p53 acetylation controls target selectivity without affecting p53 stability or p53 DNA binding ability. To better understand the mechanisms of p53 regulation by acetylation at other sites, more studies are required to uncover unknown readers and mechanisms.
Crosstalk between acetylation and other modifications of p53
In response to different cellular signals, p53 also undergoes other post-translational modifications, including ubiquitylation, phosphorylation, methylation, SUMOylation, and neddylation. The crosstalk between acetylation and these PTMs in temporal or spatial order potentially forms a finely orchestrated regulatory network (4, 9). Ubiquitination and acetylation antagonize each other at several levels, as discussed above in the “p53 acetylation and stabilization” section. p53 phosphorylation is generally considered to activate p53 and critical in determining p53 function and cell fate (4, 80, 90). Importantly, N-terminal p53 phosphorylation has been shown to promote p53 acetylation. For example, phosphorylation of p53 at N-terminal residues like S15, T18, S20 and S37, and/or multisite phosphorylation of p53 can recruit acetyltransferase p300/CBP to promote p53 acetylation (91–95). Similarly, p53 phosphorylation at S33 and S37 upon UV radiation or IR promotes the binding between p53 and PCAF (35). Consistent with this notion, phosphorylation at sites within p53 N-terminus has been shown to be required for K320 and K382 acetylation (96–98). Moreover, phosphorylation of p53 at S15 and S20 promotes its interaction with MOZ and potentially increases p53 K120 acetylation (99). Additionally, phosphorylation of p53 N-terminus (S15, T18, S20 and S37) blocks MDM2 binding and indirectly favors p53 acetylation, particularly when phosphorylation occurs at multiple sites (93, 95, 100–102). However, phosphorylated sites in the C-terminus of p53 (T377 and S378) were shown to negatively affect acetylation at the nearby sites (103). Phosphorylation-deficient p53 mutant (T377A/S378A) showed increased binding with p300, and greater acetylation at K373 and K382. Taken together, C-terminal phosphorylation antagonizes the acetylation events on p53, which is different from the enhancement of p53 acetylation by N-terminal phosphorylation. Methylation has also been shown to interplay with p53 acetylation in distinct manners, depending on the site of modification. p53 is methylated at K370 by Smyd2, K372 by Set 7/9, and K382 by Set8 (ref. 104–108). Methylation of K370 and K382 represses p53 DNA binding ability and transcriptional activity (105, 106), and K382 methylation was shown to inhibit acetylation on the same site (106). By comparison, methylation at K372 of p53 promotes its acetylation at K373 and K382 in response to various stresses (107), but inhibits K370 methylation by Smyd2 (ref. 105). This is consistent with earlier observations that K372 methylation is essential for efficient p53 transactivation and induction of cell cycle arrest and death (104). In another study, cells from Set7/9 mutant mice, which are unable to methylate p53 K369 (equivalent to human K372), displayed abrogated p53 acetylation at multiple sites, impaired p53-dependent transcriptional activation of p21 and Puma, and greatly reduced induction of cell cycle arrest and apoptosis in response to DNA damage and oncogenic stress (108). However, two independent groups have generated additional Set7/9 knockout mice and demonstrated that methyltransferase have no effect on p53 acetylation (at mouse K379, equivalent to human K382), transcriptional activation or tumor suppression (109, 110). Given the contradictory results from these different mouse studies, the physiological effect of p53 C-terminal methylation on p53 acetylation and activity remains unknown. The function of p53 SUMOylation is poorly understood as reports are often contradicting each other. Nevertheless, early studies showed that SUMOylation promotes p53 recruitment into PML nuclear bodies and transcriptional activation (111–113), whereas another study showed that SUMO-1 modification of K386 had no effect on p53-regulated transcription or subnuclear localization (114). Others showed that SUMOylation of p53 by the SUMO E3 ligase PIASy promotes nuclear export of p53 (ref. 115, 116). In the study by Naidu et al., SUMOylation of both p53 and Tip60 by PIASy was shown to augment p53 K120 acetylation, p53 cytoplasmic accumulation, and induction of Puma-independent autophagic cell death (116). Recent findings support the idea that SUMOylation correlates with impaired transcriptional activity (117). In agreement, Wu et al. found that upon SUMOylation at K386, p53 binds p300 but fails to undergo p300-mediated acetylation at K373 and K382, which is associated with the inability of p53 to bind DNA and promote the transcription of p53 target genes (118). The same study showed that a SUMOylation-deficient p53 mutant has increased the transactivation of endogenous p21 compared to wild-type p53. Interestingly, acetylation restored binding of SUMOylated p53 to DNA, suggesting that acetylation can antagonize the inhibitory effect of SUMOylation on p53 DNA binding. Neddylation of p53 can occur on K370, K372, and K373 by Mdm2 and on K320 and K321 by FBXO11, and in each case it is associated with repression of p53-mediated transcription (119, 120). The effect of p53 neddylation on its acetylation is not well defined but it is hypothesized that Mdm2 and FBX011 may compete with the acetyltransferases for modification of p53 at the same C-terminal lysine sites.
Concluding remarks and prospects
Over past 25 years, we have substantially advanced our understanding towards roles of p53 acetylation in regulating its transcription activity, stress response and tumor suppression function. In light of these studies, many novel tumor suppressive functions mediated by p53 have been revealed and appreciated, such as metabolic reprogramming, ferroptosis, and mTOR repression (60), in addition to the conventional functions of p53 including cell cycle arrest, cellular senescence, and apoptotic cell death. Acetylation controls these p53 mediated responses upon stresses through modulating p53 protein stability, conformation change, transactivation, target specificity, protein interactions and crosstalk with other modifications (16–19). So far, acetylation has been well established as activating p53 and promoting its tumor suppressive function in broad terms. Acetylation of the DBD mainly controls p53 promoter specificity and target selectivity, while acetylation of the CTD fine-tunes p53 activity.
An intriguing but elusive question is how acetylation at specific lysine sites mediate p53 promoter-specific regulation and target gene selectivity. While the p53-5KR is defective in transcription and tumor suppression (19), it remains unclear how much this is due to defects in acetylation or alteration in protein structure and DNA binding ability caused by five point-mutations in the DNA binding domain. Although lysine to arginine mutation would have minimal perturbation on the p53 structure, it remains unknown whether p53-5KR is completely transcription-dead or still retains some transcription activity for certain target genes. Future studies should address these questions by determining whether p53-5KR mutant can still bind p53 targets under physiological conditions. Of note, we previously performed chromatin-immunoprecipitation and found that wild-type mouse p53 and p53-3KR/p53-4KR mutants were all able to recruit to the TIGAR gene promoter, although with a slight reduction in binding affinity with the p53-4KR mutant (18). However, the TIGAR expression in completely abrogated in the presence of the p53-4KR mutant, suggesting that its mild effect on DNA binding could not account for the transcriptional defect observed (18). Clearly, there exists other mechanisms responsible for the promoter-specific regulation of transcription. Recent studies reporting Dicer as a reader of p53 DBD acetylation shed light on this question. Dicer serves as the corepressor of p53-3KR for suppressing p21 and Puma expression. Acetylation at K120 and K164 within p53 DBD can repel Dicer, thus activating the expression of p21 and Puma (22). The mechanisms of promoter selectivity mediated by acetylation at other sites, such as K101, K139 and K320 remain largely unknown. Are there other readers or cofactors yet to be discovered? Are they proteins or RNAs? Long non-coding RNAs (lncRNAs) are important regulators in many biological processes including transcription, epigenetics, and tumorigenicity (121, 122). Unsurprisingly, accumulating evidence suggests that lncRNAs are regulatory components of p53 pathway, either by binding and modulating p53 or by functioning downstream of p53 as its transcriptional targets, or both (123–125). For example, long non-coding RNA MALAT1 was reported to regulate p53 acetylation level by interacting with DBC1 and preventing the association of SIRT1 and DBC1 (ref. 126). MALAT1 enhances the deacetylation activity of SIRT1 and downregulate the p53 acetylation level, therefore suppressing the transactivation of p53 target genes such as p21, Bax, Puma, etc. MALAT1 level can be transcriptionally inhibited by p53, which directly binds to the two p53 binding motifs on the promoter region of MALAT1 (ref. 127), forming an inhibitory feedback loop. These studies hint that lncRNAs could be involved in p53 acetylation-mediated actions, which warrants further investigation.
Another line of research is targeting p53 deacetylases to treat p53-related diseases, such as cancer. Dynamic equilibrium of p53 acetylation and deacetylation provides layers of regulation of p53, which can be potentially targeted. HDAC1 and SIRT1 have been shown to suppress p53 acetylation level, transactivation activity and anti-tumor responses. Other HDACs (2, 3, 6 and 8) and SIRT3 have also been reported to deacetylate p53 and inhibit its activity, although more studies are needed to establish those interactions and their biological significance (8). However, research on targeting deacetylating enzymes using small-molecule compounds to activate p53 has issues such as poor specificity (8). Since HDAC family consists of a number of members bearing similar structures, a small-molecule compound usually targets not only HDAC1 but also other HDACs. In addition, given each HDAC has many substrates, inhibiting the HDAC may cause a global epigenetic state and gene expression change. Likewise, targeting Mdm2/Mdmx-p53 interaction to increase p53 level for cancer treatment has not been clinically successful due to various issues (128). Maybe it’s time to think outside of the box, such as targeting other p53 interacting proteins or RNAs. As more novel acetylation readers are uncovered in the future, hopefully, we will find proteins or RNAs with specific regulations in p53 signaling, which could provide more specific targets for treating disease with better outcomes. For instance, combined with p53 acetylation pathway, targeting lncRNA using antisense oligo (ASO) with higher specificity may be promising in context-specific cancer therapy (129–131).
Supplementary Material
Acknowledgements
This work was supported by the National Cancer Institute of the National Institutes of Health under Award R35CA253059, RO1CA258390 and R01CA254970 to W.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Ethics declarations
Conflict of interest
O. Tavana is currently an employee of AstraZeneca and has stock ownership in AstraZeneca. All other authors declare that they have no conflict of interest.
References
- 1.Boutelle AM, Attardi LD. p53 and Tumor Suppression: It Takes a Network. Trends Cell Biol. 2021;31(4):298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mello SS, Attardi LD. Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol. 2018;51:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaiser AM, Attardi LD. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018;25(1):93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y, Tavana O, Gu W. p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol. 2019;11(7):564–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90(4):595–606. [DOI] [PubMed] [Google Scholar]
- 6.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133(4):612–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnoud T, Indeglia A, Murphy ME. Shifting the paradigms for tumor suppression: lessons from the p53 field. Oncogene. 2021;40(25):4281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reed SM, Quelle DE. p53 Acetylation: Regulation and Consequences. Cancers (Basel). 2014;7(1):30–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen J, Wang D. Deciphering the PTM codes of the tumor suppressor p53. J Mol Cell Biol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A. 1964;51:786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nitsch S, Zorro Shahidian L, Schneider R. Histone acylations and chromatin dynamics: concepts, challenges, and links to metabolism. EMBO Rep. 2021;22(7):e52774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sterner R, Vidali G, Allfrey VG. Studies of acetylation and deacetylation in high mobility group proteins. Identification of the sites of acetylation in HMG-1. J Biol Chem. 1979;254(22):11577–83. [PubMed] [Google Scholar]
- 13.L’Hernault SW, Rosenbaum JL. Chlamydomonas alpha-tubulin is posttranslationally modified by acetylation on the epsilon-amino group of a lysine. Biochemistry. 1985;24(2):473–8. [DOI] [PubMed] [Google Scholar]
- 14.Gu W, Shi XL, Roeder RG. Synergistic activation of transcription by CBP and p53. Nature. 1997;387(6635):819–23. [DOI] [PubMed] [Google Scholar]
- 15.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24(6):827–39. [DOI] [PubMed] [Google Scholar]
- 16.Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149(6):1269–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016;17(2):366–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kon N, Ou Y, Wang SJ, Li H, Rustgi AK, Gu W. Corrigendum: mTOR inhibition acts as an unexpected checkpoint in p53-mediated tumor suppression. Genes Dev. 2021;35(3–4):300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang D, Kon N, Lasso G, Jiang L, Leng W, Zhu WG, et al. Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature. 2016;538(7623):118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang D, Kon N, Tavana O, Gu W. The “readers” of unacetylated p53 represent a new class of acidic domain proteins. Nucleus. 2017;8(4):360–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X, Wang X, Li Z, Duan S, Li H, Jin J, et al. An unexpected role for Dicer as a reader of the unacetylated DNA binding domain of p53 in transcriptional regulation. Sci Adv. 2021;7(44):eabi6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–40. [DOI] [PubMed] [Google Scholar]
- 24.Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. 2019;20(3):156–74. [DOI] [PubMed] [Google Scholar]
- 25.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16(4):258–64. [DOI] [PubMed] [Google Scholar]
- 26.Joerger AC, Fersht AR. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu Rev Biochem. 2016;85:375–404. [DOI] [PubMed] [Google Scholar]
- 27.Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89(7):1175–84. [DOI] [PubMed] [Google Scholar]
- 28.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387(6635):823–7. [DOI] [PubMed] [Google Scholar]
- 29.Scolnick DM, Chehab NH, Stavridi ES, Lien MC, Caruso L, Moran E, et al. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res. 1997;57(17):3693–6. [PubMed] [Google Scholar]
- 30.Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao ZX, et al. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2(4):405–15. [DOI] [PubMed] [Google Scholar]
- 31.Wadgaonkar R, Phelps KM, Haque Z, Williams AJ, Silverman ES, Collins T. CREB-binding protein is a nuclear integrator of nuclear factor-kappaB and p53 signaling. J Biol Chem. 1999;274(4):1879–82. [DOI] [PubMed] [Google Scholar]
- 32.Buschmann T, Adler V, Matusevich E, Fuchs SY, Ronai Z. p53 phosphorylation and association with murine double minute 2, c-Jun NH2-terminal kinase, p14ARF, and p300/CBP during the cell cycle and after exposure to ultraviolet irradiation. Cancer Res. 2000;60(4):896–900. [PubMed] [Google Scholar]
- 33.Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382(6589):319–24. [DOI] [PubMed] [Google Scholar]
- 34.Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19(2):1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12(18):2831–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24(6):841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Wu L, Corsa CA, Kunkel S, Dou Y. Two mammalian MOF complexes regulate transcription activation by distinct mechanisms. Mol Cell. 2009;36(2):290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rokudai S, Laptenko O, Arnal SM, Taya Y, Kitabayashi I, Prives C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc Natl Acad Sci U S A. 2013;110(10):3895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang YH, Tsay YG, Tan BC, Lo WY, Lee SC. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J Biol Chem. 2003;278(28):25568–76. [DOI] [PubMed] [Google Scholar]
- 40.Joubel A, Chalkley RJ, Medzihradszky KF, Hondermarck H, Burlingame AL. Identification of new p53 acetylation sites in COS-1 cells. Mol Cell Proteomics. 2009;8(6):1167–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408(6810):377–81. [DOI] [PubMed] [Google Scholar]
- 42.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107(2):137–48. [DOI] [PubMed] [Google Scholar]
- 43.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107(2):149–59. [DOI] [PubMed] [Google Scholar]
- 44.Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005;25(13):5389–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krummel KA, Lee CJ, Toledo F, Wahl GM. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci U S A. 2005;102(29):10188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simeonova I, Jaber S, Draskovic I, Bardot B, Fang M, Bouarich-Bourimi R, et al. Mutant mice lacking the p53 C-terminal domain model telomere syndromes. Cell Rep. 2013;3(6):2046–58. [DOI] [PubMed] [Google Scholar]
- 47.Hamard PJ, Barthelery N, Hogstad B, Mungamuri SK, Tonnessen CA, Carvajal LA, et al. The C terminus of p53 regulates gene expression by multiple mechanisms in a target- and tissue-specific manner in vivo. Genes Dev. 2013;27(17):1868–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell. 1992;71(5):875–86. [DOI] [PubMed] [Google Scholar]
- 49.Hupp TR, Sparks A, Lane DP. Small peptides activate the latent sequence-specific DNA binding function of p53. Cell. 1995;83(2):237–45. [DOI] [PubMed] [Google Scholar]
- 50.Jayaraman J, Prives C. Activation of p53 sequence-specific DNA binding by short single strands of DNA requires the p53 C-terminus. Cell. 1995;81(7):1021–9. [DOI] [PubMed] [Google Scholar]
- 51.Kon N, Churchill M, Li H, Mukherjee S, Manfredi JJ, Gu W. Robust p53 Stabilization Is Dispensable for Its Activation and Tumor Suppressor Function. Cancer Res. 2021;81(4):935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kon N, Gu W. p53 activation vs. stabilization: an acetylation tale from the C-terminal tail. Oncoscience. 2021;8:58–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173(4):533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T, et al. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol Cell Biol. 2006;26(18):6859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378(6553):203–6. [DOI] [PubMed] [Google Scholar]
- 56.Kon N, Wang D, Li T, Jiang L, Qiang L, Gu W. Inhibition of Mdmx (Mdm4) in vivo induces anti-obesity effects. Oncotarget. 2018;9(7):7282–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145(4):571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, et al. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013;3(5):1339–45. [DOI] [PubMed] [Google Scholar]
- 59.Li T, Liu X, Jiang L, Manfredi J, Zha S, Gu W. Loss of p53-mediated cell-cycle arrest, senescence and apoptosis promotes genomic instability and premature aging. Oncotarget. 2016;7(11):11838–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Y, Gu W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin Cancer Biol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–20. [DOI] [PubMed] [Google Scholar]
- 62.Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci U S A. 2012;109(50):20491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, et al. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev Cell. 2013;25(5):463–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, et al. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell. 2020;37(2):168–82 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Assi M The differential role of reactive oxygen species in early and late stages of cancer. Am J Physiol Regul Integr Comp Physiol. 2017;313(6):R646–R53. [DOI] [PubMed] [Google Scholar]
- 66.Stockwell BR, Jiang X, Gu W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020;30(6):478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26(3):165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang L, Hickman JH, Wang SJ, Gu W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle. 2015;14(18):2881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016;30(8):918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leu JI, Murphy ME, George DL. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci U S A. 2019;116(17):8390–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368(6486):85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59(2):298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113(44):E6806–E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y, Qian Y, Zhang J, Yan W, Jung YS, Chen M, et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017;31(12):1243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu Y, Gu W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem. 2002;277(52):50607–11. [DOI] [PubMed] [Google Scholar]
- 80.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137(4):609–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol. 2000;20(22):8458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, et al. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001;20(6):1331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kobet E, Zeng X, Zhu Y, Keller D, Lu H. MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc Natl Acad Sci U S A. 2000;97(23):12547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, et al. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21(22):6236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fujisawa T, Filippakopoulos P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol. 2017;18(4):246–62. [DOI] [PubMed] [Google Scholar]
- 86.Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8(6):1243–54. [DOI] [PubMed] [Google Scholar]
- 87.Li AG, Piluso LG, Cai X, Gadd BJ, Ladurner AG, Liu X. An acetylation switch in p53 mediates holo-TFIID recruitment. Mol Cell. 2007;28(3):408–21. [DOI] [PubMed] [Google Scholar]
- 88.Cai W, Su L, Liao L, Liu ZZ, Langbein L, Dulaimi E, et al. PBRM1 acts as a p53 lysine-acetylation reader to suppress renal tumor growth. Nat Commun. 2019;10(1):5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Juillard F, de Miranda MP, Li S, Franco A, Seixas AF, Liu B, et al. KSHV LANA acetylation-selective acidic domain reader sequence mediates virus persistence. Proc Natl Acad Sci U S A. 2020;117(36):22443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1(6):a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R, Brady JN. Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem. 1998;273(49):33048–53. [DOI] [PubMed] [Google Scholar]
- 92.Kar S, Sakaguchi K, Shimohigashi Y, Samaddar S, Banerjee R, Basu G, et al. Effect of phosphorylation on the structure and fold of transactivation domain of p53. J Biol Chem. 2002;277(18):15579–85. [DOI] [PubMed] [Google Scholar]
- 93.Teufel DP, Bycroft M, Fersht AR. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene. 2009;28(20):2112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jenkins LM, Yamaguchi H, Hayashi R, Cherry S, Tropea JE, Miller M, et al. Two distinct motifs within the p53 transactivation domain bind to the Taz2 domain of p300 and are differentially affected by phosphorylation. Biochemistry. 2009;48(6):1244–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee CW, Ferreon JC, Ferreon AC, Arai M, Wright PE. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc Natl Acad Sci U S A. 2010;107(45):19290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saito S, Goodarzi AA, Higashimoto Y, Noda Y, Lees-Miller SP, Appella E, et al. ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J Biol Chem. 2002;277(15):12491–4. [DOI] [PubMed] [Google Scholar]
- 97.Hofmann TG, Moller A, Sirma H, Zentgraf H, Taya Y, Droge W, et al. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol. 2002;4(1):1–10. [DOI] [PubMed] [Google Scholar]
- 98.Puca R, Nardinocchi L, Sacchi A, Rechavi G, Givol D, D’Orazi G. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol Cancer. 2009;8:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rokudai S, Aikawa Y, Tagata Y, Tsuchida N, Taya Y, Kitabayashi I. Monocytic leukemia zinc finger (MOZ) interacts with p53 to induce p21 expression and cell-cycle arrest. J Biol Chem. 2009;284(1):237–44. [DOI] [PubMed] [Google Scholar]
- 100.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91(3):325–34. [DOI] [PubMed] [Google Scholar]
- 101.Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000;14(3):278–88. [PMC free article] [PubMed] [Google Scholar]
- 102.Ferreon JC, Lee CW, Arai M, Martinez-Yamout MA, Dyson HJ, Wright PE. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc Natl Acad Sci U S A. 2009;106(16):6591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ou YH, Chung PH, Sun TP, Shieh SY. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol Biol Cell. 2005;16(4):1684–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432(7015):353–60. [DOI] [PubMed] [Google Scholar]
- 105.Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444(7119):629–32. [DOI] [PubMed] [Google Scholar]
- 106.Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell. 2007;27(4):636–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ivanov GS, Ivanova T, Kurash J, Ivanov A, Chuikov S, Gizatullin F, et al. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol. 2007;27(19):6756–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, et al. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol Cell. 2008;29(3):392–400. [DOI] [PubMed] [Google Scholar]
- 109.Lehnertz B, Rogalski JC, Schulze FM, Yi L, Lin S, Kast J, et al. p53-dependent transcription and tumor suppression are not affected in Set7/9-deficient mice. Mol Cell. 2011;43(4):673–80. [DOI] [PubMed] [Google Scholar]
- 110.Campaner S, Spreafico F, Burgold T, Doni M, Rosato U, Amati B, et al. The methyltransferase Set7/9 (Setd7) is dispensable for the p53-mediated DNA damage response in vivo. Mol Cell. 2011;43(4):681–8. [DOI] [PubMed] [Google Scholar]
- 111.Rodriguez MS, Desterro JM, Lain S, Midgley CA, Lane DP, Hay RT. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999;18(22):6455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gostissa M, Hengstermann A, Fogal V, Sandy P, Schwarz SE, Scheffner M, et al. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999;18(22):6462–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000;19(22):6185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kwek SS, Derry J, Tyner AL, Shen Z, Gudkov AV. Functional analysis and intracellular localization of p53 modified by SUMO-1. Oncogene. 2001;20(20):2587–99. [DOI] [PubMed] [Google Scholar]
- 115.Carter S, Bischof O, Dejean A, Vousden KH. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat Cell Biol. 2007;9(4):428–35. [DOI] [PubMed] [Google Scholar]
- 116.Naidu SR, Lakhter AJ, Androphy EJ. PIASy-mediated Tip60 sumoylation regulates p53-induced autophagy. Cell Cycle. 2012;11(14):2717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hay RT. SUMO: a history of modification. Mol Cell. 2005;18(1):1–12. [DOI] [PubMed] [Google Scholar]
- 118.Wu SY, Chiang CM. Crosstalk between sumoylation and acetylation regulates p53-dependent chromatin transcription and DNA binding. EMBO J. 2009;28(9):1246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118(1):83–97. [DOI] [PubMed] [Google Scholar]
- 120.Abida WM, Nikolaev A, Zhao W, Zhang W, Gu W. FBXO11 promotes the Neddylation of p53 and inhibits its transcriptional activity. J Biol Chem. 2007;282(3):1797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. [DOI] [PubMed] [Google Scholar]
- 122.Statello L, Guo CJ, Chen LL, Huarte M. Author Correction: Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22(2):159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang A, Xu M, Mo YY. Role of the lncRNA-p53 regulatory network in cancer. J Mol Cell Biol. 2014;6(3):181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chaudhary R, Lal A. Long noncoding RNAs in the p53 network. Wiley Interdiscip Rev RNA. 2017;8(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lin T, Hou PF, Meng S, Chen F, Jiang T, Li ML, et al. Emerging Roles of p53 Related lncRNAs in Cancer Progression: A Systematic Review. Int J Biol Sci. 2019;15(6):1287–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen R, Liu Y, Zhuang H, Yang B, Hei K, Xiao M, et al. Corrigendum to article “Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation”. Nucleic Acids Res. 2021;49(7):4199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ma XY, Wang JH, Wang JL, Ma CX, Wang XC, Liu FS. Malat1 as an evolutionarily conserved lncRNA, plays a positive role in regulating proliferation and maintaining undifferentiated status of early-stage hematopoietic cells. BMC Genomics. 2015;16:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jiang L, Zawacka-Pankau J. The p53/MDM2/MDMX-targeted therapies-a clinical synopsis. Cell Death Dis. 2020;11(4):237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Amodio N, Raimondi L, Juli G, Stamato MA, Caracciolo D, Tagliaferri P, et al. MALAT1: a druggable long non-coding RNA for targeted anti-cancer approaches. J Hematol Oncol. 2018;11(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Slack FJ, Chinnaiyan AM. The Role of Non-coding RNAs in Oncology. Cell. 2019;179(5):1033–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jiang MC, Ni JJ, Cui WY, Wang BY, Zhuo W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am J Cancer Res. 2019;9(7):1354–66. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.