

Abstract
Spinal muscular atrophy, X-linked 2 (SMAX2) is a rare type of spinal muscular atrophy characterized by muscle weakness, hypotonia, areflexia, myopathic face, tongue fibrillations, contractures, bone fractures, and cryptorchidism. Variants of the UBA1 gene lead to SMAX2. The UBA1 gene encodes a protein that activates the ubiquitin pathway which is responsible for protein degradation. Here, we describe a family presenting with hypotonia, muscle weakness, areflexia, contractures, weak cry, in association with other anomalies including myopathic face, scoliosis, tongue fibrillations, and cryptorchidism. Molecular analysis in 2 patients revealed a hemizygous pathogenic variant in the UBA1 gene (NM_153280.3, NP_695012.1: c.1731C>T [p.Asn577Asn]) inherited from their carrier mothers. Our study presents the first patients from Turkey, widening the phenotypic spectrum of SMAX2 by pectus carinatum, medullary sponge kidney, and frontal cyst.
Keywords: SMAX2, UBA1, UBE1, X-linked spinal muscular atrophy
Established Facts
UBA1 gene hemizygous variations are associated with X-linked spinal muscular atrophy 2 (SMAX2).
SMAX2 is a very rare muscular atrophy phenotype.
A synonymous UBA1 gene variant (c.1731C>T) has been described previously.
Novel Insights
Here, we describe clinical features of a Turkish family extending the phenotype of SMAX2 to pectus carinatum, frontal cyst, and medullary sponge kidney.
To our knowledge, this is the first case of SMAX2 from Turkey.
Introduction
Autosomal recessive spinal muscular atrophy (SMA) is a hereditary neuromuscular disorder characterized by degeneration of lower motor neurons in the anterior horn of the spinal cord, leading to progressive symmetrical proximal muscle weakness, atrophy, hypotonia, respiratory insufficiency, areflexia, and delayed motor milestones [Crawford and Pardo, 1996]. It is caused by mostly homozygous deletions and rarely homozygous variants in the SMN1 (survival of motor neuron 1) gene which is located on chromosome 5q13 [Lefebvre et al., 1995]. This disease is categorized into 4 types (SMA I, SMA II, SMA III, and SMA IV) based on the age of onset and clinical severity.
X-linked SMA is mainly divided into 2 categories: X-linked spinal and bulbar muscular atrophy (SBMA, SMAX1) and X-linked spinal muscular atrophy type 2 (SMAX2). SMAX1, also referred to as Kennedy disease, results from an AG repeat expansion in exon 1 of the coding region of the androgen receptor [MacLean et al., 1996]. SMAX2 (MIM 301830) is an extremely rare neuromuscular disorder characterized by muscle weakness, delayed motor milestones, areflexia, hypotonia, respiratory distress, as well as bone fractures and contractures [Greenberg et al., 1988]. The facial features include a myopathic face, high-arched palate, and tent-shaped open mouth. Other associated features are cryptorchidism, micrognathia, kyphosis, scoliosis, and normal intelligence. It is usually progressive, and most patients die in the first few years of life due to respiratory failure [Greenberg et al., 1988].
This disease is caused by hemizygous variants in the UBA1 (ubiquitin-like modifier-activating enzyme 1) gene (previously referred to as UBE1) which encodes a 1,058-amino-acid protein that activates the ubiquitin-proteasome pathway (UPP) which is responsible for regulation of protein degradation [Ramser et al., 2008]. Liu and Pfleger [2013] showed that homozygous variants in the Drosophila Uba1 gene lead to a shorter lifespan and severe motor impairment. SMAX2 was first reported as “X-linked infantile spinal muscular atrophy” by Greenberg et al. [1988]. The prevalence is unknown. To date, 9 molecularly confirmed families have been reported [Greenberg et al., 1988; Ramser et al., 2008; Dlamini et al., 2013; Jedrzejowska et al., 2015; Shaughnessy et al., 2020; Wang et al., 2020].
Here, we present 3 male members of a family from Southeastern Anatolia, Turkey, with contractures, weakness, fractures, cryptorchidism, myopathic face, hypotonia, and areflexia. Two of them were confirmed by molecular genetic analysis which revealed a synonymous variant (c.1731C>T; p.Asn577Asn) that was previously reported in 4 SMAX2 families [Greenberg et al., 1988; Ramser et al., 2008; Jedrzejowska et al., 2015].
Case Presentations
Case 1 (IV-1)
The 27-month-old boy presenting muscle weakness was referred to our genetics department. He is the first child of healthy Turkish parents. The boy was born at 38 weeks of gestation by cesarean delivery. Intrauterine growth retardation (IUGR) was noted. He developed respiratory distress during the neonatal period. The boy had 4 extremity contractures which resolved at 24 months, sensorineural polyneuropathy, tongue fasciculations, pectus carinatum, weak cry and suck, myopathic face, and muscle weakness. A muscle biopsy at the age of 27 months showed nonspecific myopathic and dystrophic changes. SMN1 gene deletion and duplication analysis by multiplex ligation-dependent probe amplification (MLPA) and array comparative genomic hybridization (aCGH) genetic tests was planned first. MLPA analysis showed 2 copies of the SMN1 gene. aCGH showed an 819-kb duplication of the Yp11.2 region with the following coordinates: chrY:7,821,883_8,640,857 (build GRCh37, hg19). Because this region did not contain the OMIM gene, it was considered a benign CNV. Expansion of CTG-triplet repeats associated with myotonic dystrophy and variants of the MTM1 gene related to X-linked myotubular myopathy were excluded by investigation. The patient died of respiratory failure at the age of 30 months, shortly after his examination in the intensive care unit. Thereafter, this family was referred to our department after the birth of another affected boy (Case 2, IV-3).
Case 2 (IV-3)
The index patient, a 3-month-old boy, was referred to the medical genetics department because of hypotonia, contracture, and bone fractures. He is a younger brother of Case 1. He is the second child of nonconsanguineous parents, whose mother (gravida 3, parite 2, abort 1) and father are 30 and 32 years old, respectively. Approximately 15 male members of the proband's family showed a similar phenotype (Fig. 1). His brother (case IV-1) died at the age of 2.5 years due to respiratory failure with similar clinical findings. The boy was born by cesarean section in the 39th gestational week. He had neuromotor developmental delay. He was not able to hold his head steady. At birth, a bone fracture occurred in the right humerus. He had contractures in all 4 extremities, predominantly in the left hand. During the clinical examination, small fontanelle, myopathic face with tent-shaped open mouth, scoliosis, hypotonia, weak cry, weak suck, thick eyebrows, broad forehead, downslanting palpebral fissures, broad nasal bridge, long philtrum, retrognathia, pectus excavatum, and bilateral undescended testes were described (Fig. 2, 3, 4). His growth parameters were a weight of 4.5 kg (<3rd centile, −2.19 SD), height of 55 cm (<3rd centile, −2.34 SD), and head circumference of 38 cm (<3rd centile, −2.29 SD). Results of laboratory analyses including creatine kinase (CK) and lactate were in the normal range. Cranial tomography revealed a frontal cystic lesion. Polyneuropathy was detected by neurophysiological studies performed on the patient. In the follow-up, at 18 months of age, weight, height, and head circumference were 8.5 kg (<3rd centile, −2.15 SD), 72 cm (<3rd centile, −2.67 SD), and 45 cm (<3rd centile, −2.17 SD), respectively. The karyotype and aCGH analysis were normal, 46,XY and arr(X,Y)×1,(1–22)×2, respectively. Since we could not diagnose our patient from these test results, we decided to perform whole-exome sequencing (WES). WES was planned in the index patient because of absence of a prediagnosis in the family. WES revealed a hemizygous variant, NM_153280.3, NP_695012.1: c.1731C>T (p.Asn577=), in exon 15 of the UBA1 gene on chromosome X in the index patient. This variant causes SMAX2, infantile. The synonymous variant is located in the catalytic active adenylation domain (ADD) that is a hotspot for SMAX2 of UBA1. It was classified as “pathogenic” according to ACMG variant classification guidelines: Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product (PS3_Strong). Located in a mutational hotspot and well-established functional domain without benign variation (PM1_Moderate). The variant has not been found in gnomAD exomes (PM2_Moderate). Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease (PP1_Supporting). ClinVar databases classified this variant as pathogenic (rs80356547) with 2 submissions, 1 publication (PP5_Supporting). SIFT (Sorting Intolerant From Tolerant) and PROVEAN (Protein Variation Effect Analyzer) databases predicted tolerated and neutral variant, respectively. Mutation Taster database classified this variant as disease-causing. The Deleterious Annotation of genetic variants using Neural Networks (DANN) score of this variant is 0.9282.
Fig. 1.

Pedigree of the family. The index patient (IV-3) is indicated by an arrow. Patients IV-3 and IV-4, and the patients' mothers (III-3 and III-12) were molecularly confirmed. In the pedigree, the mothers II-3, II-4, II-5, II-6, II-7, and III-17 should be obligate carriers, but were not molecularly confirmed.
Fig. 2.
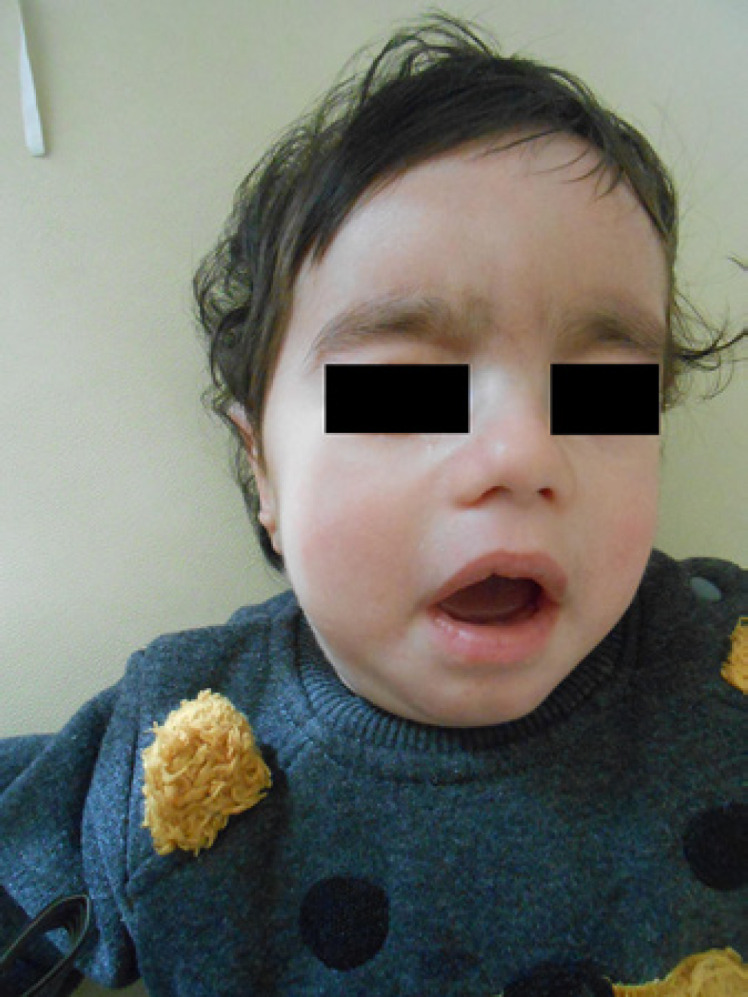
Index patient IV-3.
Fig. 3.

Index patient IV-3 with pectus excavatum.
Fig. 4.

Radiography of the index patient.
Index patient IV-3 died of respiratory failure at the age of 29 months in November 2021.
Case 3 (IV-4)
Case IV-4, a 1-year-old boy, presented with bone fractures and contractures. He was born by cesarean section in the 39th gestational week with IUGR, bone fractures, and oligohydramnios. He was hospitalized for 20 days for IUGR, bone fractures, and feeding difficulties. He was diagnosed with clinical osteogenesis imperfecta (OI) because he had 8 bone fractures after birth, but he did not have blue sclera, dentinogenesis imperfecta, joint laxity associated with OI. During the clinical examination, contractures, myopathic face with tent-shaped open mouth, hypotonia, weak cry, weak suck, high-arched palate, and pectus excavatum were described. His development was globally delayed. He was not able to sit and walk unsupported. He suffered from respiratory distress at the age of 2 months. Because of frequent trials of intubation-extubation, tracheostomy was performed. Severe sensory polyneuropathy was detected by electromyography in the patient. The X-ray revealed scoliosis, and abdominal ultrasonography showed right medullary sponge kidney. He is alive at 37 months. Chromosome analysis revealed a 46,XY karyotype. Thereafter, analysis of COL1A1 and COL1A2 genes was planned due to the prediagnosis of OI in the patient. No pathogenic variant was detected in these genes. However, patient IV-4 was hemizygous for the UBA1 gene variant.
In the segregation analysis, this variant was detected in the patients' mothers (III-3 and III-12). In the pedigree, the mothers II-3, II-4, II-5, II-6, II-7, and III-17 should be obligate carriers but were not molecularly confirmed (Fig. 1).
Discussion
SMAX2 was first reported as “X-linked infantile spinal muscular atrophy” in 1988 [Greenberg et al., 1988]. Kobayashi et al. [1995] described that a gene for X-linked infantile SMA is located in chromosome Xp11.3q11.2. Dressman et al. [2007] studied 7 new families and narrowed the X-linked SMA locus to Xp11.3q11.1. Ramser et al. [2008] described that hemizygous variants in the UBA1 gene located on human chromosome X are responsible for SMAX2. The prevalence of SMAX2 is unknown. To date, in only 9 families the diagnosis of SMAX2 has been confirmed by molecular genetic testing, although many cases of SMA with contractures and fractures have been reported [Greenberg et al., 1988; Ramser et al., 2008; Dlamini et al., 2013; Jedrzejowska et al., 2015; Shaughnessy et al., 2020; Wang et al., 2020]. In addition, Kosmicki et al. [2017] demonstrated a missense variant c.199A>G (p.Met67Val) in a male patient with neurodevelopmental disorder, and Peng et al. [2018] described a c.2501C>T (p.Pro834Leu) hemizygous missense variant in the UBA1 gene that was associated with West syndrome. While all reported variants associated with SMAX2 were localized in exon 15 of the UBA1 gene, these variants were detected in exons 4 and 21, respectively.
Clinical features in our patients were compared with previously reported cases of SMAX2 with molecular diagnosis (Tables 1 and 2). The classical features of SMAX2 were found in the family, such as hypotonia, muscle weakness, myopathic facies, contractures, bone fractures, areflexia, and respiratory failure. The patients also presented less common clinical findings such as oligohydramnios, cryptorchidism, and scoliosis. We compared our patients of the family with previously reported cases, and the constant clinical finding of SMAX present in all cases is areflexia. The dysmorphic features found in most cases are myopathic face, open tent-shaped mouth, and high-arched palate. Our patient (IV-3) showed the same dysmorphic features, in addition to thick eyebrows, broad forehead, downslanting palpebral fissures, broad nasal bridge, long philtrum, and retrognathia (Fig. 2), which were not reported as typical dysmorphic facial features for this syndrome. The majority of the cases previously reported had areflexia, progressive muscle weakness, and hypotonia with early onset. Five patients reported by Wang et al. [2020] differed from the other patients by a long life span, dysarthria, mild muscle weakness, late-onset age, no bone fractures, contractures, respiratory distress, and no progression. Neuropathy was detected in approximately 33% of previously reported cases. Although neurological features constitute most of the clinical findings of SMAX2, electromyography is not frequently performed. Skeletal manifestations include bone fractures, contractures, scoliosis/kyphosis, and pectus abnormalities. Although bone fractures appear to be the most characteristic finding of SMAX2, they have been reported in 33% of patients with SMAX. Three patients reported by Greenberg et al. [1988] and 1 patient reported by Jedrzejowska et al. [2015] had bone fractures, while most of the patients reported had none. The 2 patients from our family presented here had bone fractures. Furthermore, clinical OI was suspected in patient IV-4 due to multiple bone fractures. Although the pathogenesis of bone fractures is more complex, it is thought that the fractures are due to decreased fetal movement impairing bone mineralization. Contractures have been reported in 6 out of the 12 cases [Greenberg et al., 1988; Dlamini et al., 2013; Jedrzejowska et al., 2015]. While contractures were present in 3 of our patients, the contracture of patient 1 resolved after 18 months. Three previous studies reported the existence of pectus abnormalities including pectus excavatum and bell-shaped chest in patients with SMAX2 [Dlamini et al., 2013; Jedrzejowska et al., 2015; Shaughnessy et al., 2020]. All of our cases had pectus anomalies, but the pectus carinatum in patient 1 was not reported previously. Although the exact etiology of pectus excavatum and pectus carinatum is unknown, it is suggested that disruption of chondrogenesis and collagen metabolism plays an important role in the pathogenesis of these chest deformities [Fokin et al., 2009; Rea and Sezen, 2020]. The reason why pectus excavatum and pectus carinatum, 2 opposite phenotypes, are seen in the same family may be that the pathogenesis of both pectus deformities is similar. Genitourinary manifestations were observed in ∼25% of all reported patients (Table 2). The 2 patients from our family presented here had bilateral cryptorchidism. Additionally, case 3 (IV-4) had a medullary sponge kidney which was not reported previously. Other reported genitourinary anomalies include absence of right testis [Dlamini et al., 2013]. Other common features include decreased fetal movement, feeding difficulties, weak cry, and tongue fasciculations (Table 2). Many reported cases (40%) had a short lifespan [Greenberg et al., 1988; Dlamini et al., 2013; Jedrzejowska et al., 2015]. Among our patients, patient IV-4 reached the age of 37 months, the other patients (IV-1 and IV-3) died at the age of 30 and 29 months, respectively. Five patients from a family reported by Wang et al. [2020] differ from the others in long lifespan, ranging from 5 to 86 years. Four of the 5 patients with previously reported c.1731C>T variant died within 2 years of age, suggesting that this variant may be associated with a poor prognosis [Greenberg et al., 1988; Jedrzejowska et al., 2015]. SMAX2 is caused by variants in UBA1 which is located in chromosome Xp11.3 and escapes X inactivation [Goto and Kimura, 2009]. The UBA1 gene encodes a highly conserved UBA1 protein that activates the ubiquitin pathway which is responsible for protein degradation. A study by Liu and Pfleger [2013] showed that loss of the Uba1 gene, which is essential for neuron development in Drosophila causes motor impairment and short lifespan. They suggested that women who carry a mutant copy of UBA1 may have a shortened life expectancy. Recent studies have also detected low levels of UBA1 in mouse models of SMN1-dependent SMA, suggesting that UBA1 may play a role in the pathogenesis of SMA [Wishart et al., 2014]. The study in mice with SMA1-dependent SMA showed that AAV9-UBA1 gene therapy improved body weight, lifespan, and motor performance in mice [Powis et al., 2016]. To date, only 6 variants of UBA1 have been associated with XL-SMA, and all of them are located in exon 15, which encodes the active adenylation domain (5 missense variants: c.1617G>T [p.Met539Ile]; c.1617G>A [p.Met539Ile]; c.1639A>G [p.Ser547Gly]; c.1670A>T [p.Glu557Val]; c.1681G>A [p.Asp561Asn] and a synonymous variant: c.1731C>T [p.Asn577Asn]). The missense variant c.1617G>A was also found in 3 individuals in a family described by Wang et al. [2020]. These patients were milder affected when compared to other molecularly confirmed patients. In this study, we reported the synonymous variant c.1731C>T (p.Asn577Asn) in UBA1. This variant has been already reported in 4 SMAX2 families [Greenberg et al., 1988; Ramser et al., 2008; Jedrzejowska et al., 2015]. Ramser et al. [2008] demonstrated that this synonymous variant reduced the UBA1 gene expression by altering the methylation of exon 15. They suggested that this synonymous change in exon 15 of the UBA1 gene partially reduced the methylation of this region, allowing the binding of proteins that block the specific transcription enhancer element, resulting in reduced transcription. Likewise, Lenski et al. [2015] suggested that the c.1731C>T variant alters the pre-mRNA process by affecting exonic splicing enhancers and splicing regulatory proteins. The management of SMAX2 should be multidisciplinary to meet the specific needs of affected cases. Children with this disorder frequently have hypotonia, muscle weakness, feeding difficulties, and inadequate sucking ability. They also commonly show respiratory distress, failure to thrive, and a short lifespan. For this reason, regular respiratory and swallowing assessments are necessary to prevent these patients from complications. In some cases, tracheostomy would be an influential solution for patients with significant respiratory distress. Furthermore, neurological, genitourinary, and orthopedic evaluation is essential for affected children. The prognosis of these patients is poor and many patients die, mainly due to respiratory distress and recurrent infections.
Table 1.
Summary of clinical characteristics of cases with SMAX2 compared to our family
| Case | Variant | Ethnicity | Clinical features |
Death | References | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| contractures | bone fractures | hypotonia | muscle weakness | scoliosis | respiratory distress | weak cry | areflexia | decreased fetal movement | weak suck | pectus abnormality | myopathic face | tongue fasciculations | elevated creatine kinase | neuropathy | cryptorchidism | |||||
| 1 | NP | Turkey | + | − | + | + | + | + | + | + | + | + | + | + | + | − | + | − | 30 mo | Our case 1 |
|
| ||||||||||||||||||||
| 2 | c.1731C>T, p.Asn577Asn | Turkey | + | + | + | + | + | + | + | + | − | + | + | + | + | − | + | + | Alive at age 26 mo | Our case 2 |
|
| ||||||||||||||||||||
| 3 | c.1731C>T, p.Asn577Asn | Turkey | + | + | + | + | + | + | − | + | + | + | + | + | + | − | + | + | Alive at age 37 mo | Our case 3 |
|
| ||||||||||||||||||||
| 4 | c.1681G>A, p.Asp561Asn | reland | − | − | + | + | NR | + | + | + | − | − | + | + | + | + | + | − | Alive at age 3.5 mo | Shaughnessy et al., 2020–2 |
|
| ||||||||||||||||||||
| 5 | c.1617G>A, p.Met539lle | China | − | − | − | + | NR | − | NR | + | NR | + | NR | NR | NR | NR | NP | NR | Alive at age 86 years | Wang et al., 2020 |
|
| ||||||||||||||||||||
| 6 | c.1617G>A, p.Met539lle | China | − | − | − | − | NR | − | NR | + | NR | + | NR | NR | NR | NR | NP | NR | Alive at age 60 years | Wang et al., 2020 |
|
| ||||||||||||||||||||
| 7 | c.1617G>A, p.Met539lle | China | − | − | − | + | NR | − | NR | + | NR | + | NR | NR | NR | NR | + | NR | Alive at age 5 years | Wang et al., 2020 |
|
| ||||||||||||||||||||
| 8 | NP | China | − | − | − | − | NR | − | NR | + | NR | + | NR | NR | NR | NR | NP | NR | Alive at age 68 years | Wang et al., 2020 |
|
| ||||||||||||||||||||
| 9 | NP | China | − | − | − | − | NR | − | NR | + | NR | + | NR | NR | NR | NR | NP | NR | Alive at age 65 years | Wang et al., 2020 |
|
| ||||||||||||||||||||
| 10 | c.1731C>T, p.Asn577Asn | Poland | + | + | + | + | NR | + | + | + | + | + | + | + | + | − | NP | + | 5.5 mo | Jedrzejowska et al., 2015 |
|
| ||||||||||||||||||||
| 11 | c.1670A>T, p.Glu557Val | Caucasus | + | − | + | + | + | + | + | + | + | + | + | + | + | + | + | + | 4 mo | Dlamini et al., 2013 |
|
| ||||||||||||||||||||
| 12 | c.1731C>T, p.Asn577Asn | North America | + | + | + | + | NR | NR | NR | + | − | NR | NR | NR | + | NR | NP | + | 18 mo | Greenberg et al., 1988 Variant analysis: Ramser et al., 2008 |
|
| ||||||||||||||||||||
| 13 | c.1731C>T, p.Asn577Asn | North America | + | + | + | + | NR | + | NR | + | − | NR | NR | NR | + | NR | NR | NR | Alive at age 13 years [Kobayashi et al., 1995] | Greenberg et al., 1988 |
|
| ||||||||||||||||||||
| 14 | c.1731C>T, p.Asn577Asn | North America | + | − | + | NR | NR | NR | NR | + | + | NR | NR | NR | NR | NR | + | NR | 5 mo | Greenberg et al., 1988 |
|
| ||||||||||||||||||||
| 15 | c.1731C>T, p.Asn577Asn | North America | + | + | + | NR | NR | + | NR | + | + | NR | NR | NR | NR | NR | NR | NR | 11 mo | Greenberg et al., 1988 |
NR, not reported; NP, not performed; mo, months.
Table 2.
Clinical findings in SMAX2 cases
| Clinical findings | Previously reported cases | P1 | P2 | P3 | Total |
|---|---|---|---|---|---|
| Prenatal complications | |||||
| Decreased fetal movement | 4 of 12 | + | − | + | 6 of 15 (40%) |
| Oligohydramnios | 1 of 12 | − | − | + | 2 of 15 (13%) |
| Breech presentation | 2 of 12 | − | − | − | 2 of 15 (13%) |
| Neonatal complications | |||||
| Respiratory distress | 5 of 12 | + | + | + | 8 of 15 (53%) |
| Weak cry | 3 of 12 | + | + | + | 6 of 15 (40%) |
| Weak suck/feeding difficulties | 7 of 12 | + | + | + | 10 of 15 (66%) |
| Neurological abnormalities | |||||
| Muscle weakness | 7 of 12 | + | + | + | 10 of 15 (66%) |
| Hypotonia | 7 of 12 | + | + | + | 10 of 15 (66%) |
| Areflexia | 12 of 12 | + | + | + | 15 of 15 (100%) |
| Neuropathy | 4 of 12 | + | + | + | 7 of 15 (46%) |
| Tongue fasciculations | 5 of 12 | + | + | + | 8 of 15 (53%) |
| Skeletal abnormalities | |||||
| Bone fracture | 4 of 12 | − | + | + | 6 of 15 (40%) |
| Contracture | 6 of 12 | + | + | + | 9 of 15 (60%) |
| Scoliosis/kyphosis | 1 of 12 | + | + | + | 4 of 15 (27%) |
| Pectus abnormalities | 3 of 12 | + | + | + | 6 of 15 (40%) |
| Genitourinary abnormalities | |||||
| Cryptorchidism | 3 of 12 | − | + | + | 5 of 15 (33%) |
| Medullary sponge kidney | 0 of 12 | − | − | + | 1 of 15 (6%) |
| Dysmorphic features | |||||
| Myopathic face | 3 of 12 | + | + | + | 6 of 15 (40%) |
| High-arched palate | 5 of 12 | − | + | + | 7 of 15 (46%) |
| Laboratory abnormalities | |||||
| Elevated CK | 2 of 12 | − | − | − | 2 of 15 (13%) |
| Early death | 5 of 12 | + | − | − | 6 of 15 (40%) |
| Additional findings identified in single individuals | Laryngomalacia Patent foramen ovale Absence of right testis Degenerative cerebellar abnormalities Macrocephaly Potter-like face Cranial sclerosis | Frontal cyst Medullary sponge kidney Pectus carinatum |
Data from previously reported studies. P, patient; CK, creatine kinase.
In conclusion, we present 3 male members of a family from Turkey with contractures, muscle weakness, fractures, cryptorchidism, myopathic face, hypotonia, and areflexia. Novel findings include frontal cyst on computed tomography brain imaging, medullary sponge kidney on abdominal ultrasonography, and pectus carinatum. This case report may help to understand the relationship between phenotype and genotype in SMAX2 syndrome.
Statements of Ethics
All procedures performed in the study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Written informed consent was obtained from the parents/legal guardians of the patients for publication of the details of their medical case and any accompanying images.
Conflict of Interest Statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Funding Sources
No financial support was received from any source.
Author Contributions
H.B. and Ö.Ö. designed the study; Ö.Ö. and B.E.Ç. examined the patients and families; H.B. and Ö.Ö. wrote the manuscript. All authors read and approved the final manuscript.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.
Acknowledgments
We thank all clinicians for their participation in this study and Gaziantep Dr. Ersin Arslan Training and Research Hospital Genetics Laboratory doctors for genetic analysis.
References
- Crawford TO, Pardo CA. The Neurobiology of Childhood Spinal Muscular Atrophy. Neurobiol Dis. 1996;3((2)):97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Dlamini N, Josifova DJ, Paine SM, Wraige E, Pitt M, Murphy AJ, et al. Clinical and neuropathological features of X-linked spinal muscular atrophy (SMAX2) associated with a novel mutation in the UBA1 gene. Neuromuscul Disord. 2013;23((5)):391–8. doi: 10.1016/j.nmd.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Dressman D, Ahearn ME, Yariz KO, Basterrecha H, Martínez F, Palau F, et al. X-linked infantile spinal muscular atrophy: clinical definition and molecular mapping. Genet Med. 2007;9((1)):52–60. doi: 10.1097/gim.0b013e31802d8353. [DOI] [PubMed] [Google Scholar]
- Fokin AA, Steuerwald NM, Ahrens WA, Allen KE. Anatomical, histologic, and genetic characteristics of congenital chest wall deformities. Semin Thorac Cardiovasc Surg. 2009;21((1)):44–57. doi: 10.1053/j.semtcvs.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Goto Y, Kimura H. Inactive X Chromosome-Specific Histone H3 Modifications and CpG Hypomethylation Flank a Chromatin Boundary between an X-Inactivated and an Escape Gene. Nucleic Acids Res. 2009;37((22)):7416–28. doi: 10.1093/nar/gkp860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg F, Fenolio KR, Hejtmancik JF, Armstrong D, Willis JK, Shapira E, et al. X-linked infantile spinal muscular atrophy. Am J Dis Child. 1988;142:217–9. doi: 10.1001/archpedi.1988.02150020119045. [DOI] [PubMed] [Google Scholar]
- Jedrzejowska M, Jakubowska-Pietkiewicz E, Kostera-Pruszczyk A. X-Linked Spinal Muscular Atrophy (SMAX2) Caused by de novo c.1731C>T Substitution in the UBA1 Gene. Neuromuscul Disord. 2015;25((8)):661–6. doi: 10.1016/j.nmd.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Baumbach L, Matise TC, Schiavi A, Greenberg F, Hoffman EP. A gene for a severe lethal form of X-linked arthrogryposis (X-linked infantile spinal muscular atrophy) maps to human chromosome Xp11.3-q11.2. Hum Mol Genet. 1995;4((7)):1213–6. doi: 10.1093/hmg/4.7.1213. [DOI] [PubMed] [Google Scholar]
- Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M, et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet. 2017;49((4)):504–10. doi: 10.1038/ng.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80((1)):155–65. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lenski C, Erkelenz S, Ramser J, Widera M, Peter J, Schaal, et al. Synonymous mutations in the X-linked disease genes UBA1 and HADH2 affect binding of the splicing regulatory proteins SRSF2, SRSF6 and hnRNP F/H. JSM Genet Genomics. 2015;2((1)):1007. [Google Scholar]
- Liu HY, Pfleger CM. Mutation in E1, the ubiquitin activating enzyme, reduces Drosophila lifespan and results in motor impairment. PLoS One. 2013;8((1)):e32835. doi: 10.1371/journal.pone.0032835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean HE, Warne GL, Zajac JD. Spinal and bulbar muscular atrophy: androgen receptor dysfunction caused by a trinucleotide repeat expansion. J Neurol Sci. 1996;135((2)):149–57. doi: 10.1016/0022-510x(95)00284-9. [DOI] [PubMed] [Google Scholar]
- Peng J, Wang Y, He F, Chen C, Wu LW, Yang LF, et al. Novel West syndrome candidate genes in a Chinese cohort. CNS Neurosci Ther. 2018;24((12)):1196–206. doi: 10.1111/cns.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis RA, Karyka E, Boyd P, Côme J, Jones RA, Zheng Y, et al. Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight. 2016;1((11)):e87908–16. doi: 10.1172/jci.insight.87908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramser J, Ahearn ME, Lenski C, Yariz KO, Hellebrand H, von Rhein M, et al. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am J Hum Genet. 2008;82((1)):188–93. doi: 10.1016/j.ajhg.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea G, Sezen CB. StatPearls[Internet]. Treasure Island. StatPearls Publishing; 2020. Chest Wall Deformities. [PubMed] [Google Scholar]
- Shaughnessy N, Forman EB, O'Rourke D, Lynch SA, Lynch B. X-linked infantile spinal muscular atrophy (SMAX2) caused by novel c.1681G>A substitution in the UBA1 gene, expanding the phenotype. Neuromuscul Disord. 2020;30((1)):35–7. doi: 10.1016/j.nmd.2019.11.004. [DOI] [PubMed] [Google Scholar]
- Wang XH, Zhang LM, Yang X, Zhou SZ. A Pathogenic Missense Variant (c.1617G>A, p.Met539Ile) in UBA1 Causing Infantile X-Linked Spinal Muscular Atrophy (SMAX2) Front Pediatr. 2020;8:64. doi: 10.3389/fped.2020.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart TM, Mutsaers CA, Riessland M, Reimer MM, Hunter G, Hannam ML, et al. Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy. J Clin Invest. 2014;124((4)):1821–34. doi: 10.1172/JCI71318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.