

Abstract
The histone chaperone FACT occupies transcribed regions where it plays prominent roles in maintaining chromatin integrity and preserving epigenetic information. How it is targeted to transcribed regions, however, remains unclear. Proposed models include docking on the RNA polymerase II (RNAPII) C-terminal domain (CTD), recruitment by elongation factors, recognition of modified histone tails and binding partially disassembled nucleosomes. Here, we systematically test these and other scenarios in Saccharomyces cerevisiae and find that FACT binds transcribed chromatin, not RNAPII. Through a combination of high-resolution genome-wide mapping, single-molecule tracking and mathematical modeling, we propose that FACT recognizes the +1 nucleosome, as it is partially unwrapped by the engaging RNAPII, and spreads to downstream nucleosomes aided by the chromatin remodeler Chd1. Our work clarifies how FACT interacts with genes, suggests a processive mechanism for FACT function, and provides a framework to further dissect the molecular mechanisms of transcription-coupled histone chaperoning.
Keywords: Histone chaperone, FACT, Spt16, Pob3, Chd1, RNA polymerase II, nucleosome unwrapping
eTOC blurb
Preserving nucleosomes during transcription elongation requires histone chaperone FACT and chromatin remodeler Chd1, but their interplay is unclear. Here, Jeronimo et al. show that nucleosomal DNA unraveling by RNAPII drives the recruitment of FACT, while the remodeling activity of Chd1 promotes FACT movement along genes, enabling a processive mechanism.
Graphical Abstract
Introduction
Nucleosomes, the building blocks of chromatin, allow for DNA compaction and the regulation of protein-DNA interactions. Nucleosomes also represent roadblocks to molecular machines that translocate along DNA. For instance, during transcription elongation, RNA polymerase II (RNAPII) encounters successions of nucleosomes, which must unfold for the DNA template to feed the polymerase active site. Importantly, these nucleosomes have to reassemble in the wake of the polymerase to maintain chromatin integrity (Lai and Pugh, 2017; Teves et al., 2014; Venkatesh and Workman, 2015). This delicate process is controlled by several chromatin regulators including histone methyltransferases, histone deacetylases, chromatin remodelers and histone chaperones (Lai and Pugh, 2017; Venkatesh and Workman, 2015). Among those, histone chaperones FACT and Spt6 play master roles during transcription elongation as they can mediate both nucleosome disassembly and assembly.
To assist RNAPII during transcription elongation through nucleosomes, chromatin regulators need to be recruited to sites of transcription. Most of these factors are recruited via their interaction with RNAPII (Carrozza et al., 2005; Drouin et al., 2010; Govind et al., 2010; Krogan et al., 2003; Sdano et al., 2017). How FACT makes its way to actively transcribed regions, however, remains unclear. An interaction with RNAPII or an associated protein would provide a simple way to recruit the histone chaperone. Accordingly, several proteins have been proposed to recruit FACT to transcribing RNAPII (Adelman et al., 2006; Carvalho et al., 2013; John et al., 2000; Krogan et al., 2002; Kwon et al., 2010; Mason and Struhl, 2003; Sen et al., 2017), but the relative contribution of these factors to FACT recruitment has never been systematically analyzed. Alternatively, FACT may recognize transcribed chromatin rather than the elongation complex itself. Such a model would explain why FACT is recruited to genes transcribed by RNAPI and III in addition to RNAPII (Birch et al., 2009; Tessarz et al., 2014). Recruitment through transcribed chromatin is supported by the recent analysis of the DNA fragments associated with FACT after micrococcal nuclease (MNase) treatment (Martin et al., 2018) but the biochemical nature of such “transcription-modified nucleosomes” recognized by FACT remains to be determined. Previous studies suggested that histone acetylation, methylation or ubiquitylation contribute to FACT recruitment (Carvalho et al., 2013; Fleming et al., 2008; Murawska et al., 2020; Pathak et al., 2018), potentially providing a molecular mechanism for how transcription –by co-transcriptional histone modifications– promotes the recruitment of FACT. Alternatively, in vitro studies showing increased affinity of FACT for disrupted nucleosomes (Farnung et al., 2021; Formosa et al., 2001; Liu et al., 2020; McCullough et al., 2018; Nesher et al., 2018; Ruone et al., 2003; Tsunaka et al., 2016; Wang et al., 2018) suggest that partial loss of histone-DNA contacts, generated by RNAPII or some associated factors, may allow FACT binding.
Here, we systematically tested many possible recruitment mechanisms, including those proposed previously, and concluded that FACT binds transcribed chromatin rather than RNAPII or elongation factors. We, however, found no evidence for a role of histone modifications in FACT recruitment. Instead, our data support a model where partial unwrapping of the first nucleosome, induced by engaging RNAPII, exposes a FACT interacting surface on the nucleosome and triggers its recruitment. Once docked on the +1 nucleosome, FACT appears to spread to more downstream nucleosomes with the help of the chromatin remodeler Chd1, suggesting that FACT works processively along genes during their transcription.
Results
FACT is recruited after transcription initiation in an RNAPII CTD phosphorylation-independent manner
To start investigating how FACT is recruited to chromatin, we compared the distribution of FACT to that of RNAPII and nucleosomes at high-resolution. We performed chromatin immunoprecipitation (ChIP), followed by high-resolution tiling arrays (ChIP-chip) and sequencing exonuclease-treated ChIP (ChIP-exo) of FACT subunits (Spt16 and Pob3) and RNAPII (Rpb3) in wild type (WT) cells (Figures 1 and S1). In agreement with previous work (Martin et al., 2018; Mason and Struhl, 2003; Pathak et al., 2018; Vinayachandran et al., 2018), we found that both Spt16 and Pob3 occupy the transcribed region of active genes (Figures 1A and S1A). Both FACT subunits correlated with RNAPII occupancy (Figures 1A, 1B and S1B). Moreover, shutting down transcription using the rpb1–1 allele led to a nearly complete loss of FACT occupancy genome-wide (Figure 1C). A comparison of the data for RNAPII and Spt16/Pob3 indicates that FACT accumulates downstream of RNAPII (Figures 1A and S1A). Indeed, FACT occupancy peaks at each nucleosome dyad within transcribed regions while RNAPII occupancy builds up upstream of the +1 nucleosome and has a more uniform distribution further downstream (Figure 1A, metagenes on the left). Thus, genome-wide FACT occupancy correlates with —and depends on— transcription, but does not precisely correlate with where transcription initiates. This observation, which corroborates ChIP-exo, ChIP-seq, and MNase-ChIP-seq data by others (Martin et al., 2018; Vinayachandran et al., 2018), suggests that FACT “joins” RNAPII post-initiation.
Figure 1:

FACT occupies actively transcribed regions and is recruited after initiation in a CTD phosphorylation-independent manner. A) Metagene and heatmap representations of RNAPII (Rpb3) and both FACT subunits (Spt16, Pob3) occupancy (reads per million (rpm)), as determined by ChIP-exo, in WT cells. Metagenes for all (n = 5,456), the most transcribed (Top genes; n = 264) and the least transcribed (Bottom genes; n = 2,792) genes are shown. The average enrichment level of nucleosomes (Jiang and Pugh, 2009) is shown in grey. Genes in the heatmaps were ordered by decreasing RNAPII occupancy (n = 5,456). Data are aligned on the dyad position of +1 nucleosomes, as determined in (Chereji et al., 2018). B) Pearson correlation matrix of RNAPII, Spt16 and Pob3 occupancy, as determined by ChIP-exo. Pearson correlations were calculated using all of the data points covering ORFs that are longer than 1 kb (n = 3,503). C) Metagenes of RNAPII and Spt16 occupancy over highly (n = 85), mildly (n = 190) and lowly (n = 3,241) transcribed genes, as determined by ChIP-chip, relative to Input, in WT and rpb1–1 cells after an 80 min heat-shock at 37°C. D) Metagenes of RNAPII and Spt16 occupancy over transcribed genes (n = 275), as determined by ChIP-chip, relative to Input, in WT and kin28 ATP analog-sensitive (kin28as) cells, both treated 15 min with 1NAPP1. E) Metagenes of RNAPII and Spt16 occupancy over transcribed genes (n = 355), as determined by ChIP-chip, relative to Input, in cells ectopically expressing the indicated CTD versions of RPB1 following nuclear depletion of the endogenous Rpb1 protein by anchor-away. Data for RNAPII in panels D and E (except for the S2A mutant) are from (Jeronimo and Robert, 2014). NCP, nucleosome core particle. TSS, transcription start site. See also Figure S1.
Complete pre-initiation complexes (PICs) are too transient to be detected by ChIP in WT cells (Jeronimo and Robert, 2014; Wong et al., 2014) so that FACT may be a transient component of PICs that escapes detection in WT cells. To test this possibility, we performed RNAPII and FACT ChIP-chip experiments under two conditions that stabilize PICs: inhibition of TFIIH kinase (Kin28) and mutation of serine 5 of the RNAPII CTD. Both conditions led to the accumulation of RNAPII in the promoter region as expected (Jeronimo and Robert, 2014; Wong et al., 2014) (Figures 1D and 1E). In contrast, while FACT occupancy is decreased on gene bodies (reflecting reduced RNAPII occupancy) it does not shift upstream (Figures 1D and 1E). These results do not support a direct role for FACT in transcription initiation genome-wide, although we cannot exclude roles at specific genes.
FACT has been proposed to be recruited to the elongating RNAPII via CTD phosphorylation by TFIIH (Mason and Struhl, 2003). The finding that Spt16 occupancy is unaffected by either Kin28 inhibition or CTD-S5A mutations (Figures 1D and 1E), does not support such a model. Of note, we also observed no FACT occupancy defects behind those solely explained by an effect on RNAPII occupancy in cells expressing CTD-S2A or CTD-S7A mutants (Figure 1E), nor in various CTD kinase mutants (Figure S1C). We, therefore, conclude that FACT is unlikely to be recruited to genes via the phosphorylated CTD.
FACT distribution is uncoupled from RNAPII in CHD1 mutants
Some evidence suggested that FACT may be recruited to the elongating RNAPII via interactions with PAF1C (Adelman et al., 2006), or the N-terminal domain of the capping enzyme Cet1 (Sen et al., 2017). We, therefore, tested FACT and RNAPII occupancy in PAF1C and cet1ΔN mutants. None of these mutations alters FACT localization (Figures S2A and S2B). We conclude that neither the PAF1C nor the N-terminal domain of Cet1 are required to target FACT to transcribed regions.
Our data are consistent with FACT occupancy on transcribed genes simply reflecting the presence of transcription, without a requirement for a recruiting factor. But what makes transcribed chromatin recognized by FACT? One possibility is that transcription generates a specific histone modification pattern that is recognized by FACT. Several studies provided evidence for such a scenario (Fleming et al., 2008; John et al., 2000; Murawska et al., 2020; Pathak et al., 2018; Smart et al., 2009). We therefore profiled FACT and RNAPII occupancy in sas3Δ, gcn5Δ, set2Δ, bre1Δ, ubp8Δ, and ubp10Δ cells. As shown in Figure S2C, however, none of these mutants altered FACT occupancy. We conclude that histone modifications do not contribute significantly to FACT recruitment during transcription, although we cannot rule out the possibility that a modification we have not tested may contribute.
Previous work showed that FACT physically interacts with the ATP-dependent chromatin remodeler Chd1 (Farnung et al., 2021; Farnung et al., 2017; Krogan et al., 2006; Krogan et al., 2002; Krogan et al., 2004; Simic et al., 2003). To investigate the possible role of Chd1 in FACT recruitment, we performed ChIP-chip, ChIP-exo and ChIP-qPCR of FACT in chd1Δ cells. Surprisingly, the deletion of CHD1 had a profound effect on FACT distribution. In chd1Δ cells, FACT occupancy increased in the 5’ end and decreased in the 3’ end of genes (Figures 2A–C and S2D). These data were observed in two different genetic backgrounds (W303 and BY4741; Figure S2D) and in cells where Chd1 is acutely depleted from the nucleus by anchor-away (Haruki et al., 2008) (Figure S2E), but were not observed in mutants for the two other yeast chromatin remodelers involved in nucleosome sliding, Isw1 and Isw2 (Figure S2F). Importantly, this change in FACT occupancy occurred without a change in RNAPII occupancy (Figures 2AC and S2D–E) and correlates with transcription (Figure S2G). Hence, FACT distribution along genes is uncoupled from RNAPII in CHD1 mutants, providing direct evidence that FACT interacts with chromatin rather than with RNAPII. This conclusion is further supported by profiles of FACT occupancy in spt61004, a mutant of the Spt6 histone chaperone that affects histone occupancy in the 5’ end of genes more so than in the 3’ end (Jeronimo et al., 2019; Jeronimo et al., 2015). Indeed, in spt6–1004 cells, while RNAPII occupancy is reduced uniformly along genes, FACT shows a biased decrease in the 5’ end of genes, mirroring the occupancy defect of histones in this mutant (Figure S2H).
Figure 2:

FACT, but not RNAPII, accumulates at 5’ nucleosomes in CHD1 mutants. A) Metagene representation of RNAPII (Rpb3) and FACT (Spt16, Pob3) occupancy over genes (n = 5,456), as determined by ChIP-exo, in WT and chd1Δ cells. The average enrichment level of nucleosomes (Jiang and Pugh, 2009) is shown in grey. Data are aligned on the dyad position of +1 nucleosomes (Chereji et al., 2018). B) Same as panel A but for the 10% most transcribed genes, as determined by RNAPII ChIPexo (n = 264). C) RNAPII (Rpb3–3Flag), FACT (Spt16–6HA) and RNAPII-normalized FACT occupancy over the 5’ and 3’ regions of the PMA1 and TEF1 ORFs, as determined by ChIP-qPCR, in WT and chd1Δ cells. Error bars represent standard deviation from four biological replicates. P values are from Student’s ttest. D) Co-immunoprecipitation experiments of Spt16 and Chd1 in WT, chd1-K407R and chd1ΔN cells expressing an HA-tagged version of Chd1. Western blots representative of four biological replicates. E) Metagene representation of Chd1, RNAPII, and Spt16 occupancy over transcribed genes (n = 295), as determined by ChIP-chip, relative to Input, in WT, chd1Δ, chd1ΔN and chd1-K407R cells. F) Scatter plots of the ORFs occupancy (n = 5,817) of Spt16 versus RNAPII in WT, chd1Δ, chd1ΔN and chd1-K407R cells. See also Figure S2.
To determine whether Chd1 affects FACT distribution through protein-protein interactions or its effect on chromatin, we tested a mutant in the helicase/ATPase domain (chd1-K407R) predicted to prevent Chd1 from remodeling nucleosomes (Simic et al., 2003), and a mutant where Chd1 is N-terminally truncated by 117 amino acids preventing the interaction with FACT in vitro (Farnung et al., 2021). Co-immunoprecipitation experiments confirmed that the chd1ΔN, but not the chd1-K407R, mutant decreased the interaction between FACT and Chd1 in extracts, although most of the interaction appears to depend on nucleic acids (Figure 2D). Interestingly, the chd1-K407R mutant phenocopied the effect of chd1Δ on FACT distribution while deleting the N-terminal region of Chd1 had no effect (Figure 2E). This result strongly suggests that Chd1 impacts FACT distribution via its effect on chromatin rather than by directly recruiting the histone chaperone. It also suggests that the direct interaction between FACT and Chd1 does not play a measurable role in the recruitment or distribution of FACT along genes. We noted, however, that even in chd1Δ and chd1-K407R cells, FACT occupancy correlated with transcription (Figure 2F), demonstrating that FACT recruitment is transcription-dependent even in that context.
The accumulation of FACT at the 5’ end of genes when Chd1 or its remodeling activity is disrupted suggests an unexpected mechanism in which FACT is recruited to the 5’ end of genes in a transcription-dependent manner, and spreads through the gene body via a mechanism that requires the remodeling activity of Chd1. This mechanism is explored in the next sections.
Single-molecule tracking of FACT suggests a processive role during transcription by RNAPII
To acquire kinetic information about FACT-chromatin interactions in live cells, we turned to single-molecule tracking (SMT) (Lionnet and Wu, 2021). We tagged Spt16 with the HaloTag (Los et al., 2008) and labeled mid-log growing cells with Janelia Fluor 552 (JF552) (Zheng et al., 2019). We imaged single molecules at 10 ms resolution and tracked their positions to establish 2D projections of trajectories through nuclear space (Kim et al., 2021; Nguyen et al., 2020). From these “fast-tracking” movies, we calculated the mean-squared displacement (MSD) for each trajectory to extract the molecule’s apparent diffusion coefficient (D). The log D distributions showed values within the technical dynamic range demonstrated by Halo-H2B and nucleus-localized HaloTag as standards for chromatin-bound and chromatin-free behaviors, respectively (Figure 3A). Gaussian fitting resolved two dynamically distinct populations for Spt16-Halo, representing chromatin-bound and free FACT populations (Figure 3B, grey). To achieve more robust quantitation of the average D and fraction (F) of molecules comprising each population, we performed two-state kinetic modeling of single displacements (Hansen et al., 2018) (Figure 3B, Spot-On, grey). These experiments revealed the fraction of chromatin-bound molecules (Fbound) as about one-third (32.5%) in WT cells.
Figure 3:

Single-molecule tracking revealed live-cell dynamics of FACT. A) Diffusion coefficient (D) histograms and corresponding two-Gaussian fits for biological controls, Halo-H2B (brown) and nucleus-localized HaloTag (pink, only fit shown). The majority of histone H2B molecules comprise a “chromatin-bound” population with low average diffusivity, whereas nuclear HaloTag molecules display predominantly fast, “chromatin-free” population. B) Fast-tracking results for Spt16-Halo in WT (grey) and in rpb1-AA cells treated with rapamycin (blue) (DMSO control shown in Figure S3C). (Left) Normalized histograms of log10D of single trajectories and corresponding two-Gaussian fits (solid grey line: sum of two Gaussians; dashed lines: individual Gaussian curves representing chromatin-bound and free subpopulations). The histograms represent combined data from two biological replicates. (Right) Chromatin-bound fractions from Spot-On kinetic modeling of single displacements. Error bars represent standard deviations from two biological replicates. C) Log-log survival-probability curves (1-CDF) for Spt16-Halo (black) and Halo-H2B (grey) from apparent dwell times of single-molecule chromatin-binding events captured by slow-tracking. Data from two biological replicates (circles, with grey shades representing 95% confidence intervals obtained by bootstrapping) were fitted with a doubleexponential decay function (solid lines). D) Log-log survival-probability curves for Spt16-Halo from WT (black) and rpb1-AA (blue) cells, treated with rapamycin (DMSO control shown in Figure S3D). The data (solid lines) are from two biological replicates, with 95% confident intervals shown as grey shades. E) Fast-tracking results for Spt16-Halo in chd1Δ cells (red). See legend of Panel B for details. F) Log-log survival-probability curves for Spt16 from WT (black) and chd1Δ (red) cells from apparent dwell times of single-molecule chromatin-binding events. The data are from two biological replicates. 95% confident intervals are shown as grey shades. Embedded pie charts show the relative fractions of stable and transient binding events, with values for the former indicated. G) Corrected residence times (τsb, left panel) and global fractions (Ceq, right panel) of stable chromatin binding by FACT in WT and chd1Δ cells. Error bars for the residence times are standard deviations of time-lapse results. Ceq errors were propagated using Fb and fsb errors. Ceq result for WT had negligible ~0 error. Errors for τsb H2B and free Halo data are from (Kim et al., 2021). D, diffusion coefficient; CDF, cumulative distribution function; Rapa, rapamycin; Ceq, overall fraction of stably-bound molecules. See also Figure S3.
To determine the residence time of FACT on chromatin, we imaged Spt16-Halo at 250 ms-frame rate, which enables efficient blurring of fast-moving molecules and thus selective tracking of chromatin-binding events (Chen et al., 2014). From these “slow-tracking” experiments, we computed the survival probability curve from the apparent dwell times of several thousand binding events. These curves fitted a double-exponential decay behavior (Figure 3C), indicating the predominance of transient binding (tb), while a small fraction (10%) exhibited more stable binding (sb) with an apparent dissociation rate (ksb = 0.23 s−1) similar to that observed for H2B (ksb = 0.23 s−1) (Figure S3A). Because H2B decay kinetics represented the technical limit primarily imposed by photobleaching, we sought to reduce the effective laser exposure of the sample by alternating 250 ms laser-on and 250 ms or 500 ms laser-off periods. This time-lapse (TL) approach (Gebhardt et al., 2013) extended the technical limit of H2B (ksb = 0.16–0.21 s−1) beyond the apparent dissociation rate of Spt16 (ksb = 0.21–0.25 s−1) (Figures S3A and S3B). We then corrected ksb obtained for Spt16 using the value for H2B (Hansen et al., 2017; Nguyen et al., 2020) and computed the residence time for stable binding (τsb = 1/ ksb). Results from both TL regimes indicated a FACT residence time of 20–25 s for stable binding (τsb), and of 1 s for transient binding (τtb) (Figure S3A).
Remarkably, the dissociation kinetics of Spt16 is highly similar to that observed for RNAPII in live cells (RNAPII τsb = 20–23 s) (Nguyen et al., 2020). This characteristic is unique among all the factors we have tested, which includes the entire PIC (Nguyen et al., 2020) and six chromatin- remodeling complexes (Kim et al., 2021). These results suggest a functional connection between FACT and RNAPII on chromatin in vivo. To validate this relationship, we used the anchor-away approach (Haruki et al., 2008) to deplete RNAPII from the nuclei (rpb1-AA) (Nguyen et al., 2020). In fast-tracking mode, this led to a drastic reduction of the chromatin-bound fraction (Figures 3B, blue and S3C), consistent with our ChIP experiments (Figure 1C). Accordingly, slow-tracking analyses showed that RNAPII depletion caused ~70% reduction in observable binding events per nucleus (Figure S3A), which exhibited much faster dissociation (residence time between 0.24 and 2.9 s) compared to WT kinetics (Figures 3D and S3D). These results indicate a strong dependence on RNAPII for chromatin association by FACT. Considering the similar residence time (20–25 s) between RNAPII and FACT, and because this time is sufficient for the continuous transcription of an average yeast gene (~1 kb) given previously measured transcription rates (~2 kb/min) (Mason and Struhl, 2005), and much longer than the estimated time for RNAPII to transcribe through a nucleosome (~5 s), our SMT data support a processive mechanism for FACT function during transcription (see Discussion).
To measure the impact of Chd1 on FACT dynamics, we imaged Spt16 in chd1Δ cells. Results from fast-tracking experiments indicated no significant differences in the global dynamics of FACT in the absence of Chd1 (Fbound = 33.9% in chd1Δ compared to 32.5% in WT) (Figure 3E). In contrast, slow-tracking experiments (continuous exposure and 250 ms/500 ms TL) revealed a measurable effect of CHD1 deletion on chromatin binding by FACT (Figures 3F, 3G, S3A, S3E and S3F). Compared to its WT behavior, stable binding by Spt16 in chd1Δ cells exhibited shorter residence time (τsb = 14–17 s; compared to 20–25 s in WT) (Figures 3G and S3A). Notably, among all events detected by slow-tracking, the fraction of stable binding (fsb) was higher in chd1Δ (17%) compared to WT (10%) (Figure 3F). Based on the Fbound values from fast-tracking (Figure 3E), which correspond to all stable and transient binding occurring globally, we estimated the overall fraction of all FACT molecules stably engaged with chromatin (Ceq = Fbound x fsb) as 3% in WT and 6% in chd1Δ cells (Figure 3G). Considering that there are ~15,000–42,000 FACT molecules per cell, these Ceq values suggest that about 450–1,500 molecules are stably bound to chromatin at any given time in WT cells (Ho et al., 2018; McCullough et al., 2015), consistent with estimates of elongating RNAPII molecules (690–1,380 molecules) (Pelechano et al., 2010). Notably, our calculations indicate an increase in stably-bound FACT with reduced residence time in chd1Δ cells. To further understand these differences, we applied our fast- and slow-tracking results to derive the average search time τsearch, which represents the average time between two specific target-binding events by a single FACT molecule (Chen et al., 2014; Lionnet and Wu, 2021; Nguyen et al., 2020), as 163 s in WT and 76 s in chd1Δ cells (Figure S3A). Thus, FACT appears to find chromatin targets more efficiently in the absence of Chd1. Assuming similar chromatin target and protein abundances in WT and chd1Δ cells, higher search efficiency indicates faster “on” rate (inversely proportional to τsearch) (Lionnet and Wu, 2021), consistent with a global increase in stably bound FACT. These insights from SMT experiments provide key information for our understanding of the role of Chd1 in FACT-chromatin interactions during transcription as detailed below.
FACT binds disorganized nucleosomal particles on transcribed genes
Because all our data point to recognition of altered nucleosomes produced by transcription, and, perhaps, by Chd1, as key to determining where FACT binds, we investigated the structure of the nucleosomes bound by FACT in vivo. We performed MNase-ChIP-seq of FACT in both WT and chd1Δ cells and scrutinized the size of the mapped DNA fragments. As expected, the Input material contains mainly mononucleosomal-size DNA (150 bp), with some dinucleosomes (300 bp) (Figure 4A, grey shades). Interestingly, however, the Spt16 ChIPs in both WT and chd1Δ cells were enriched for DNA fragments of subnucleosomal (<150 bp) and internucleosomal (150–300 bp) sizes (Figure 4A, blue traces). The enrichment of these DNA fragments relative to mononucleosomes is even more pronounced when looking only at fragments aligning on the most highly transcribed genes (Figure 4A, right panel). Similar results were obtained with Pob3 (Figure S4A).
Figure 4:

FACT binds disorganized nucleosomal particles on transcribed genes. A) Distribution plots of the size of DNA fragments recovered from Input and FACT (Spt16) MNase-ChIP-seq samples from WT and chd1Δ cells for all (n = 5,796) and the top 10% transcribed (n = 580) genes. B-C) Heatmaps representation of Input and Spt16 average occupancy (rpm) (B) and of Input-subtracted Spt16 average occupancy (rpm) (C) on gene body (n = 5,796, sorted by decreasing RNAPII occupancy on the y-axis) from WT and chd1Δ cells, as determined by MNase-ChIP-seq, computed with DNA fragments from different sizes (x-axis). Mono-, di- and tri-nucleosome-sized DNA fragments are indicated. See also Figure S4.
To systematically investigate the relationship between DNA fragment size associated with FACT and transcription, we generated heatmaps of the average fragment density over genes sorted by decreasing RNAPII occupancy (y-axis) for DNA fragments from different sizes (x-axis). Figures 4B and 4C clearly show that the transcription-dependent signal in the Spt16 ChIP emanates from the internucleosomal size (150–300 bp and 300–450 bp) and subnucleosomal size (<150 bp) fragments, while the mononucleosomal or dinucleosomal size fragments, despite being more abundant in the sample, do not preferentially map to transcribed genes (similar results were obtained for Pob3, see Figure S4B). Hence, transcription-dependent FACT occupancy is reflected in the internucleosomal and subnucleosomal size fragments in these datasets. Note that these fragments are also observed in histone H4 (Figures S4C and S4D) and RNAPII (Figures S4E and S4F) MNase-ChIP-seq experiments. Taken together, these results show that FACT, together with RNAPII and most likely other factors, associates with disorganized nucleosomes present in transcribed regions.
FACT spreads inside the gene body from the +1 nucleosome in a Chd1-dependent manner
To determine where FACT-associated DNA fragments map relative to genes, we generated two-dimensional occupancy (2DO) plots (Chereji et al., 2018; Henikoff et al., 2011) of the center of Input- and Spt16-associated fragments relative to the dyad of the +1 nucleosome in both WT and chd1Δ cells (Figures 5A and 5B; see Figures S5A and S5B for similar experiments performed with Pob3 and Figures S5D and S5E for H4 and RNAPII additional controls). As expected, the Input samples are dominated by nucleosomal size fragments aligning with nucleosome dyads, with some dinucleosomal size fragments aligning between nucleosomal dyads (Figure 5A, top left panel). In addition to these dominant features, the Spt16 ChIPs from WT cells contain additional signals (notably between 150 bp and 300 bp) (Figure 5A, top right panel). Because we showed that these internucleosomal size fragments are those relevant for transcription-dependent FACT occupancy (see Figure 4), our subsequent analyses will focus on those fragments. Interestingly, the plot revealed that the center of these FACT-enriched fragments covers the entire range of positions, forming an “inverted-v” linking the mononucleosomal and dinucleosomal signals. Fragments forming the upward diagonal of the inverted-v share a common 5’ extremity aligning with the upstream edge of their corresponding nucleosome but are increasingly longer from their 3’ end ascending the diagonal (Figure 5C, left panel, fragments a-d). These fragments therefore represent nucleosomal particles with extended MNase protection on the side distal to the transcription start site (TSS-distal). Conversely, the fragments along the downward diagonal (Figure 5C, right panel, fragments d-g) represent nucleosomal particles with extended MNase protection on the TSS-proximal side. In line with the analyses shown in Figure 4, these internucleosomal size fragments are more prominent on the most highly transcribed genes (Figure 5A). We surmise those fragments represent intermediate states of FACT —likely together with other factors— dynamically interacting with both sides of nucleosomes as they are being transcribed.
Figure 5:

FACT spreads inside the gene body from the +1 nucleosome in a Chd1-dependent manner. A) 2DO plots of the coverage of the sequenced fragment mid-points (nucleosome dyads) in Input and FACT (Spt16) ChIP from MNase-digested chromatin from WT cells, relative to the +1 nucleosome dyads, on all (n = 5,796) and on the most transcribed (n = 580) genes. On the right of each heatmap is the distribution of the fragment sizes. B) Same as panel A but for chd1Δ cells. C) A graphical representation of the fragments aligning on the inverted-v’s observed in 2DO plots from FACT ChIP samples. The blue diagonal lines highlight the upward and downward trajectories of the fragment mid-points and the red bars depict examples of fragments at different positions along the upward (fragments “a” through “d” ) and the downward (fragments “d” through “g”) diagonals. D) A working model for FACT spreading down a gene based on the MNase-ChIP-seq data in WT and chd1Δ cells from panel A. Fragments are considered as different snapshots of a dynamic process (y-axis speculatively denoted as “Time”) and were ordered as they appear when walking through the inverted-v zig-zag. Note the under-represented fragments and the progressive loss of signal in chd1Δ. See also Figure S5.
Interestingly, FACT MNase-ChIP-seq from chd1Δ cells yielded dramatically different patterns (Figures 5B and Figure S5B). First, and in agreement with our ChIP-chip and ChIP-exo data, the signal decreases from 5’ to 3’ confirming that FACT does not distribute evenly along genes in the absence of the chromatin remodeler. Second, DNA fragments along the downward diagonal of the inverted-v (fragments e and f in Figure 5C) are absent; suggesting that in chd1Δ cells, FACT mostly binds nucleosomal particles with extended MNase protection on the TSS-distal side. Putting these observations together, and considering the fact that FACT works as a processive histone chaperone (Figure 3), we hypothesized that the different sized fragments represent intermediate states of a dynamic process by which FACT interacts with nucleosomes from the TSS-proximal to distal sides (Figures 5C and 5D). The loss of the downward inverted-v fragments reflecting binding to the downstream side of the nucleosome in chd1Δ cells suggests that Chd1 is required for the appearance of the later stage intermediates. Also, because FACT’s signal logarithmically decreases at each nucleosome from 5’ to 3’ in chd1Δ cells (Figure S5C), the data suggest that Chd1 allows FACT to translocate from one nucleosome to the next (see Figure 5D).
Mathematical modeling suggests mechanisms for Chd1-dependent spreading of FACT from the +1 nucleosome
Our model, although compatible with our and published data, assumes that the different MNase-protected fragments bound by FACT represent intermediates of a dynamic process. It also assumes that their appearance over time follows their appearance along the inverted-v patterns (see placement of fragments in Figure 5D). A formal validation of this model would require experimental evidence for the time-dependence of the appearance of the MNase-protected fragments. Such experiments, however, are not practically doable, notably because yeast genes are very short and no efficient methods would allow us to synchronize transcription with the sufficient time scale. To challenge our model against alternative ones, we turned to mathematical modeling and built stochastic models with multiple potential mechanisms (Figures 6 and S6). Among the models tested, one (with a core “inchworm-like” mechanism of progression) was able to capture many qualitative features of the inverted-v pattern observed in the FACT MNase-ChIP data and can be summarized as follows. FACT (together with other factors) binds at the +1 nucleosome location; extends from the downstream end in the direction of transcription; reaches full extension when encompassing the +1 and +2 nucleosome locations; transitions to a retracting state; retracts from the upstream end in the direction of transcription; reaches full retraction when encompassing the location of the +2 nucleosome; transitions to an extending state; resumes extension and repeats the extension/retraction process until the end of the gene is reached and the complex dissociates from chromatin (Figure 6A). To reflect the fact that chromatin interactions are dynamic, the model also includes a probability of unbinding at each point in the process. Upon simulations, this model reached a steady-state recapitulating the key features of the observed FACT MNase-ChIP in WT cells (Figure 6B and Movie S1). We tested alternative models, but none was as successful in recapitulating the pattern observed in the FACT MNase-ChIP data (Figures S6A).
Figure 6:

Mathematical modeling supports FACT binding at the +1 nucleosome and traversing the gene with an “inchworm” mechanism and suggests mechanisms for Chd1-dependent spreading. A) Schematic of the inchworm mechanism. B) Simulated protected fragment sizes and locations from the WT model. C) Simulated protected fragment sizes and locations for the same model as in panel A but with a reduced rate of extension and increase unbinding (chd1Δ model 1). D) Simulated protected fragment sizes and locations for the same model as in panel A but with increase unbinding and adding a probability of stalling during extension (chd1Δ model 2). The units of the color bars in panels B-D represent the number of fragments observed in a complete simulation averaged over a 20×20 bp window. E) Top: A schematic representation of the YLR454W gene under the control of the GAL1 promoter and of the ChIP time-course. Bottom: RNAPII (Rpb3–3Flag) and FACT (Spt16–6HA) occupancy, as determined by ChIP-qPCR, over the 5’ and 3’ regions of GAL1-YLR454W at different times after repression by addition of glucose, in WT and chd1Δ cells. The circles represent individual biological replicates (n = 4) and the traces represent their average. See also Figure S6 and Movies S1, S2 and S3.
We then took advantage of our mathematical model to test possible mechanisms for the role of Chd1 by systematically assessing the impact of increasing or decreasing each variable in the model (extension, transition, retraction, unbinding) as well as introducing other variables (see Figures 6C, 6D and S6B). Among those simulations, impairing the extension rate (Figure 6C and Movie S2) or introducing a probability of stalling during the extension step (Figure 6D and Movie S3) both recapitulated all features of the chd1Δ experimental data: i) enhanced signal on the upward diagonal, ii) loss of the downward diagonal and iii) a decrease in intensity towards the 3’ end of the gene. Note that, while not necessary to obtain a chd1Δ-like pattern (Figure S6C), both chd1Δ models include increase unbinding as instructed by the SMT data (Figure 3G). These data suggest that Chd1 promotes FACT spreading by energetically favoring the extension step.
In the course of building and testing our models, we noticed that both conditions mimicking the chd1Δ data systematically led to the appearance of increased signal for fragments greater than 300 bp as a natural consequence of not allowing FACT complexes to overlap (Figure S6D, middle and right panels). These long fragments therefore appear consequent to a “traffic jam” caused by either slowly progressing (model 1) or poorly processive (model 2) FACT molecules. Looking back at the experimental data, we noticed that such signal was also observed in the chd1Δ MNase-ChIP data when displaying fragments up to 800 bp (Figure S6D, left panel). Because these fragments were not used in the construction of the models, their presence in both the simulations and the experimental data represents an internal validation of the models.
Chd1 does not affect the velocity of FACT spreading along genes during transcription
Our mapping, imaging, and modeling data suggest that Chd1 promotes FACT spreading along genes during transcription but cannot discriminate between two molecular mechanisms. Model 1 (Figure 6C and Movie S2) suggests that Chd1 promotes the speed of FACT as it extends the nucleosome footprint on the distal side (exiting the nucleosome). In the second model (Figure 6D and Movie S3), Chd1 contributes to the fraction of FACT that successfully moves from one nucleosome to the next. Regardless the exact biochemical details, these two models are different with respect to the dynamics of FACT during transcription. While the first model predicts that FACT spreading along genes should be slower in chd1Δ cells, the second model rather implies that the fraction of FACT molecules progressing forward (but not their speed) is reduced in chd1Δ cells. We therefore looked at the movement of FACT along a gene during transcription. We used the well-established GAL1-YLR454W system, where the expression of YLR454W (a ~8kb-long gene) is driven by the GAL1 promoter (Figure 6E). Occupancy of FACT and RNAPII at different regions along the gene is assessed by ChIP-qPCR after addition of excess glucose (leading to acute repression). This system enables tracking the last wave of RNAPII (and potentially FACT) moving along the gene (Malik et al., 2017; Mason and Struhl, 2003, 2005; Schwabish and Struhl, 2004; Strasser et al., 2002). Consistent with FACT processively moving along genes with RNAPII (Mason and Struhl, 2003) (Figure 3), we observed FACT moving from 5’ to 3’ at a rate similar to that of RNAPII in WT cells (Figure 6E). Interestingly, deletion of CHD1 did not reduce FACT (or RNAPII) velocity (Figure 6E), arguing against model 1.
In order to solidify this important conclusion, we turned to Drosophila melanogaster cells. First, we showed that the knockdown of Chd1 in Drosophila S2R+ cells (Figure S6E) led to a 5’/3’ imbalance in FACT occupancy, without affecting RNAPII, at the Cam gene (Figure S6F), a gene previously shown to be transcribed and highly occupied by FACT (Tettey et al., 2019). This result demonstrates that the role of Chd1 in FACT distribution is conserved in metazoans. Then, we looked at RNAPII and FACT over the Cam gene after a release from a DRB block, a system used to measure elongation rate in metazoans (Fuchs et al., 2015). Similar to yeast, we observed progression of RNAPII and FACT at a similar rate, and this rate was not affected by the knockdown of Chd1 (Figure S6G). These experiments support model 2 where Chd1 promotes FACT movement by preventing stalling during the extension step of the inchworm process (Figure 6D). Model 2 is also more consistent with the SMT data (Figure 3) than model 1. Indeed, slower progression of FACT (model 1) would have been consistent with an increased FACT residence time in chd1Δ cells, while we observed the opposite (Figure 3G). Moreover, the increased stable chromatin-bound fraction of FACT in chd1Δ (Figures 3F and 3G) may be explained by attempts to load additional FACT molecules on FACT-free RNAPII-engaged nucleosomes inside the gene.
FACT recognizes partially unwrapped +1 nucleosomes induced by RNAPII
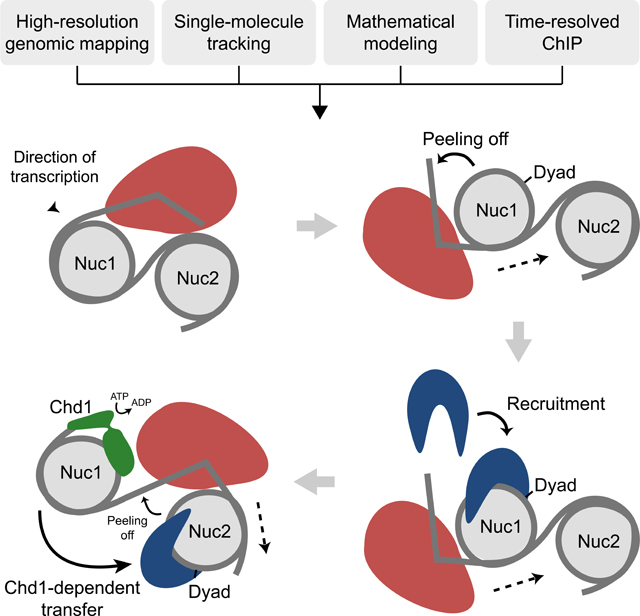
While the data presented above provide a mechanism to explain how FACT spreads down a gene, it does not explain how it is initially recruited to the +1 nucleosome of active genes. We addressed this question by analyzing the subnucleosomal-size DNA fragments from FACT MNase-ChIP-seq experiments. We mapped the center of DNA fragments of different sizes on the dyad of the +1 nucleosome. As previously shown in Drosophila cells (Ramachandran et al., 2017), the distribution of subnucleosomal-size DNA fragments from MNase-seq is bimodal and can be fit to two Gaussians, representing two populations of nucleosomes partially unwrapped from one end or the other (see Figure 7A for a graphical representation). Hence, guided by this previous work (Ramachandran et al., 2017), we mapped the distributions of fragments of 90 bp, 103 bp, and 125 bp around the dyad of the +1 nucleosome both in WT and chd1Δ cells. The 90 bp and 103 bp fragments informing on asymmetrically unwrapped nucleosomal particles and the 125 bp fragments reflecting symmetrically unwrapped particles (Ramachandran et al., 2017). The Input material of WT cells revealed that the proportion of +1 nucleosomes partially unwrapped from the TSS-proximal side is similar to those unwrapped from the TSS-distal side, suggesting asymmetric nucleosome breathing (Figures 7B and S7A, 103 bp and 90 bp fragments plots for Input). As previously described (Ramachandran et al., 2017), symmetrical unwrapping is also observed, as shown through the analysis of the 125 bp fragments (Figure S7A). Also, note that similar observations were made when looking at the +2 nucleosome (Figure S7B). Subnucleosomal-size DNA fragments from the FACT ChIPs, however, were heavily biased for asymmetrically unwrapping from the TSS-proximal side (Figures 7B and S7A, 103 bp and 90 bp fragments plots) demonstrating that FACT preferentially binds to +1 nucleosomes with TSS-proximal DNA contact loss. Interestingly, similar results were observed in chd1Δ cells, further supporting the idea that the chromatin remodeler is involved in post-recruitment steps (FACT spreading) rather than during the initial recruitment at the +1 nucleosome. Hence, we propose that transcription-induced unwrapping of the TSS-proximal side of the +1 nucleosome triggers FACT recruitment.
Figure 7:

FACT recognizes +1 nucleosomes, asymmetrically unwrapped from their TSS-proximal side. A) Graphical explanation for the interpretation of the distribution plots shown in panel B. The graphs show the distribution of the center of subnucleosomal size fragments. A distribution centered at “0” (grey trace) indicates symmetrically unwrapped nucleosomal particles, whereas a distribution centered to the left (tan) or the right (green) indicates nucleosomal particles asymmetrically unwrapped on their TSSdistal and TSS-proximal side, respectively. We used 90 bp, 103 bp, and 125 bp fragments as per (Ramachandran et al., 2017). B) Distribution of 103 bp (+/− 5 bp) fragment centers, from FACT (Spt16 and Pob3) MNase-ChIP-seq experiments (and their Inputs) from WT and chd1Δ cells, plotted relative to the position of +1 nucleosome dyads on all genes (n = 5,796). The data (grey) were fitted to a doubleGaussian (dotted). The tan and green traces show the two individual Gaussians representing asymmetrically unwrapped on their TSS-distal and TSS-proximal side, respectively as depicted in panel A. C) Full recruitment of FACT requires Spt4/Spt5. Left: Metagene of Spt16 and RNAPII (Rpb3) occupancy over transcribed genes, as determined by ChIP-chip, relative to Input, in WT and spt4Δ cells. Data are shown for experiments performed in two genetic backgrounds (BY4741, n = 275 and W303, n = 295). Right: A box plot of the average RNAPII and Spt16 occupancy in WT and spt4Δ cells for transcribed genes from the BY4741 experiment. D) A cartoon representation of the proposed mechanism for FACT recruitment and spreading along transcribed genes. See also Figure S7.
Further support for that model came from our FACT ChIP-chip experiments in spt4Δ cells. Spt4/Spt5 is an elongation factor that interacts with FACT (Foltman et al., 2013; Han et al., 2010; Lindstrom et al., 2003). Detailed in vitro analyses showed that Spt4/5 stabilizes a partially unwrapped state of the nucleosome during transcription in vitro (Crickard et al., 2017). In another study, cryo-EM revealed that Spt4/5 prevents DNA from reassociation with histones when RNAPII is stalled near the dyad (Ehara et al., 2019). Together, these two studies support a role for Spt4/5 in assisting RNAPII negotiating with nucleosomes by preventing undesirable DNA-histone interactions that may cripple the progression of the polymerase through nucleosomes. Consistent with these in vitro experiments and with in vivo data in Schizosaccharomyces pombe (Shetty et al., 2017), our ChIP-chip analyses of RNAPII in spt4Δ cells revealed an accumulation at the 5’ end of genes (Figure 7C). Interestingly, FACT occupancy is decreased in spt4Δ cells (Figure 7C). This observation contrasts with other mutants we have tested (see above), where variations in RNAPII occupancy are generally mirrored by FACT. The decreased FACT recruitment in a context where RNAPII is accumulated rather suggests that Spt4/5 contributes to FACT recruitment. In light of the in vitro data mentioned above (Crickard et al., 2017), we propose that Spt4/5 contributes to the unwrapping (by preventing unsuitable rewrapping) of the +1 nucleosome, hence contributing to the proper recruitment of FACT.
DISCUSSION
Why do we propose a processive model for FACT? In principle, a different FACT molecule may assist RNAPII at each nucleosome. Such a distributive model would be consistent with the fact that FACT is highly abundant (15,000–42,000 molecules per cell, relative to ~70,000 nucleosomes per cell) (Formosa and Winston, 2020). However, two key findings —derived from our SMT data— rather support a model where a single FACT molecule tracks along the entire gene with RNAPII (processive model). First, the residence time of FACT and RNAPII are very similar (20–25 s on average) and consistent with both RNAPII and FACT persisting on genes from start to finish, considering the speed of transcription elongation and the size of yeast genes. We note that RNAPII takes about 5 s to transcribe through a nucleosome, a time that resembles the residence time of chromatin remodelers (Kim et al., 2021) but much shorter than the observed residence time of FACT. Second, despite FACT being very abundant, the number of FACT molecules stably associated with chromatin at a given time is estimated at 450–1,500 (3% of the total number of FACT molecules), similar to the number of elongating RNAPII molecules (690–1,380) (Pelechano et al., 2010). Hence, both the residence time and the number of bound molecules are similar between FACT and RNAPII, consistent with a processive model. To accommodate a distributive model, the FACT residence time would have to be considerably shorter to explain the relatively low number of chromatin-bound FACT molecules.
FACT recognizing partially unwrapped nucleosomes is consistent with a plethora of in vitro FACT-nucleosome interaction data (Formosa et al., 2001; Liu et al., 2020; McCullough et al., 2018; Nesher et al., 2018; Ruone et al., 2003; Tsunaka et al., 2016; Wang et al., 2018). In vivo, partially unwrapped nucleosomes were detected in transcribed regions in Drosophila (Ramachandran et al., 2017) and yeast (this study). In addition, recent structural and biochemical studies showed DNA unwrapping from the upstream side of RNAPII-engaged nucleosomes (Farnung et al., 2021; Farnung et al., 2018; Kujirai et al., 2018). It is therefore reasonable to think that asymmetrical DNA unwrapping of the +1 nucleosome — triggered by RNAPII— would promote FACT recruitment. Because such partial unwrapping can be achieved multiple ways and in different circumstances in cells, the model proposed here provides an attractive recruitment mechanism for FACT to function in different contexts (reviewed in (Formosa, 2013; Formosa and Winston, 2020; Gurova et al., 2018)).
Our work shows that proper distribution of FACT along genes requires the catalytic activity of Chd1. We propose that, once recruited on chromatin at the 5’ end of genes, FACT moves towards the 3’ end with the help of Chd1; but how does Chd1 remodeling activity promote FACT spreading? Because the velocity of FACT’s progression is not altered —and because neither the distribution nor the speed of RNAPII is affected— in chd1Δ cells, we propose that Chd1 promotes the release of FACT from the post-transcribed nucleosome, a necessary step for its transfer to the next (transcribed) nucleosome (Figure 7D). In the absence of Chd1, we imagine that transcription progresses forward, leaving FACT behind, “stuck” on the post-transcribed nucleosome. In this model, Chd1 therefore promotes the “processivity” of FACT during transcription. Interestingly, while FACT (this study) and RNAPII (Nguyen et al., 2020) have a dwell time consistent with both factors persisting on an average gene from start to finish (~22 s), Chd1 resides on chromatin for much shorter time (7 s) (Kim et al., 2021). These observations suggest that different molecules of Chd1 may take turns in promoting the progression of a single FACT complex. The structures of FACT and Chd1 bound to a nucleosome engaged by RNAPII were recently released (Farnung et al., 2021). Overlapping these structures highlighted a clash between Chd1 and FACT, perhaps providing an insight about how Chd1 may promote FACT eviction. In this study, however, Chd1 was proposed to bind the nucleosome first, and swap with FACT as RNAPII progresses further into the nucleosome. It was also proposed that Chd1 promotes FACT recruitment by physically holding on it via its N-terminal domain. While our work does not dispute this sequence of events, it shows that the N-terminal domain of Chd1 has no measurable impact on FACT occupancy or distribution on genes. Hence, we propose that Chd1 (perhaps in addition to binding to the pre-transcribed nucleosome) binds to the post-transcribed nucleosome (still bound by FACT) when RNAPII is exiting, promoting the transfer of FACT to the downstream nucleosome (already engaged by RNAPII and, perhaps, Chd1). Because all current nucleosome-RNAPII structures depict RNAPII engaging (not exiting) the nucleosome, our findings call for the investigation of structures of FACT bound to more complex substrates (e.g. di-nucleosomes) together with Chd1, RNAPII, and perhaps other elongation factors. Collectively, the work presented here ties previous in vitro information to a relevant in vivo context (transcription) and provides a ground state for the dissection of FACT chaperoning mechanisms, both in vivo and in vitro.
Limitations of the study
By scrutinizing the DNA fragments from MNase-digested chromatin, and aided by mathematical simulations, we were able to formulate models that could be experimentally tested. Translating these abstract models into biochemical ones involves making assumptions. Hence, while our data solidly established that Chd1 affects the “processivity” of FACT along genes rather than its speed, the biochemical details of how this is achieved will require additional work, and our proposal that Chd1 promotes the transfer of FACT from the post-transcribed nucleosome is to some extent hypothetical.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following: Outline to be created by the Production team.
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, François Robert (francois.robert@ircm.qc.ca).
Materials Availability
Yeast strains and plasmids generated in this study are available without restrictions upon request.
Data and Code Availability
The microarray (ChIP-chip) and DNA sequencing (ChIP-exo and MNase-ChIP-seq) data generated during this study are available at the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE155144.
The single-molecule tracking data generated during this study are available in Mendeley Data, DOI: 10.17632/rnf4nc3g6y.1 (https://data.mendeley.com/datasets/rnf4nc3g6y/1).
The in-house scripts used to generate some of the analyses during this study are available at https://github.com/francoisrobertlab.
The code related to the mathematical modeling of the MNase-ChIP-seq data is available at https://github.com/aangel-code/FACT-inchworm-model.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains
S. cerevisiae strains used in this study (all derived from the W303–1A, BY4741 or FY4 genetic background) are described in Table S1. Unless otherwise indicated, cells were grown at 30°C and 200 rpm in YPD (yeast extract-peptone-2% glucose, supplemented with 44 μM adenine) medium as follows. Strains were streaked from glycerol stocks onto 2% agar YPD plates and grown at 30°C for 2–3 days. An isolated colony was then grown overnight in 10 mL of YPD. This preculture was used to inoculate 50 mL of YPD at an OD600 of 0.1 which was grown to an OD600 of 0.7–0.9.
Drosophila cells
S2R+ Drosophila cells were grown at 25°C in Shields and Sang M3 medium (Sigma-Aldrich, S8398), supplemented with bactopeptone and yeast extract, 5 mM KHCO3 and 10% FBS (Wisent, 080–150).
METHOD DETAILS
Yeast strains and plasmids
Genotypes for the yeast strains used in this study are listed in Table S1. 3HA, 6HA, and Halo tags were inserted by homologous recombination of a PCR cassette amplified from the pMPY-3HA (Schneider et al., 1995), pYM15 (Janke et al., 2004), or pBS-SK-Halo-NatMX (Ranjan et al., 2020) plasmid, respectively. The 3Flag tag sequence was amplified from strain YSC001 (a gift from S. Churchman) (Churchman and Weissman, 2011). The chd1-K407R catalytic dead point mutation (Simic et al., 2003), the chd1ΔN and 3HA-chd1ΔN truncations (Farnung et al., 2021), and the FRB-GFP tag in Chd1 were introduced directly in the genome by CRISPR-Cas9 editing as previously described (Collin et al., 2019). Oligonucleotides corresponding to gRNAs targeting the different regions in CHD1 (Table S2) were cloned into the pML107 or pML104 plasmid (Laughery et al., 2015). Donor DNAs containing the desired CHD1 mutations (including silent mutations destroying the protospacer adjacent motif) were generated by PCR (see Table S2 for oligonucleotide sequences). Gene deletions were made by replacing the gene of interest with an auxotrophic or antibiotic resistance marker.
To generate a donor DNA introducing a 6Flag-blasticidin cassette at the Ssrp C-terminus in SR2+ cells, the pKF296 plasmid (Kunzelmann et al., 2016) was modified by replacing the TEV site-2V5 cassette with an AID*−6Flag tag.
Growth conditions
For shutting off transcription experiments, a strain harboring a temperature-sensitive allele of RNAPII (rpb1–1) was grown to an OD600 of 0.7 and an equal volume of YPD pre-warmed to 49°C was added to the flask, which was immediately transferred to 37°C for additional 80 min. For inhibition of Kin28 activity experiments, ATP analog-sensitive kin28as strains were precultured in yeast nitrogen base (YNB) medium lacking uracil before inoculation in YPD medium. kin28as strains and their controls were then grown to an OD600 of 0.8 and treated with 6 μM of 1-Naphthyl PP1 (1NAPP1; Tocris Bioscience, 3063) for 15 min. Strains expressing the RPB1 CTD WT or CTD mutant plasmids were grown in YNB medium lacking histidine to an OD600 of 0.5 and treated with 1 μg/mL of rapamycin (Bio Basic, R706203) for 90 min to anchor-away the endogenous Rpb1 protein. For the experiments involving the spt6–1004 temperature-sensitive allele, cells were grown to an OD600 of 0.5 and transferred to 37°C for 80 min. For ChIP experiments involving the anchor-away of Chd1, an FRB-GFP-CHD1 (AA-chd1) strain was grown to an OD600 of 0.5 prior to addition of 1 μg/ml of rapamycin (or DMSO as control) for 90 min. For the yeast elongation rate assay, a 300 mL culture of a strain where the YLR454W is under the control of the GAL1 promoter, and where SPT16 and RPB3 are tagged with 6HA and 3Flag, respectively, was grown in yeast extract-peptone supplemented with 2% galactose and 2% raffinose until an OD600 of 0.8. At that point, a 50 mL aliquot was removed (t0) and glucose was added to a final concentration of 4%. Aliquots were taken every 2 min for a total of 8 min. For SMT experiments, cells were grown at 30°C in complete supplement mixture medium in the presence 1 nM (slow-tracking) or 10 nM (fast-tracking) of JF552 to label Spt16-Halo until an OD600 of 0.6–0.8. For SMT experiments involving the anchor-away of Rpb1, 1 μg/mL rapamycin (or DMSO as control) was added to mid-log cultures (OD600~0.6) for 1 hr before harvesting.
DRB release experiments in Drosophila cells were performed by incubating the cells for 1 hr with 100 μM DRB (Sigma-Aldrich, D1916) and replacing with fresh media for the appropriate amount of time of DRB release.
dsRNAi
dsRNA probes for RNAi were generated against mRNAs for lig4, mus308, GFP and Chd1 by PCR amplification with oligonucleotides containing the T7 promoter sequence and S2R+ cDNA as template (Table S2). The PCR products were used as templates for in vitro transcription with T7 polymerase. After RQ1 DNase I treatment (Promega, M610A) for 30 min at 37°C and inactivation for 10 min at 95°C, RNA strands were annealed by heating 20 min at 65°C and slow cooling at room temperature. dsRNA concentration was estimated by agarose gel electrophoresis. dsRNAi was performed by adding 40 μg of dsRNA to 8×106 S2R+ cells and incubating for 4 days, with media and dsRNA replacement after 48 hr. To validate dsRNAi efficiency at the protein level, cells were harvested in S2-RIPA lysis buffer (10 mM Tris-HCl pH 8.0, 140 mM NaCl, 0.1% SDS, 0.1% Na-deoxycholate, 0.5% Sarkosyl, 1% Triton X-100 and protease inhibitor cocktail (1 mM PMSF, 1 mM Benzamidine, 10 μg/mL Aprotinin, 1 μg/mL Leupeptin, 1 μg/mL Pepstatin)). Samples were incubated at 95oC for 5 min, briefly centrifuged and analyzed by SDS-PAGE and western blot using anti-Chd1 rabbit polyclonal (a gift from A. Lusser; 1:2,000 dilution) and anti-α-tubulin mouse monoclonal (Sigma-Aldrich, T5168; 1:25,000 dilution) antibodies. Membranes were incubated with donkey anti-rabbit IRDye 800CW and donkey anti-mouse IRDye 680RD antibodies (LI-COR Biosciences, 926–32213 and 926–68072) according to the manufacturers’ instructions and scanned on the Odyssey infrared imaging system (LI-COR Biosciences). dsRNAi experiments were also validated by RT-qPCR using two biological replicates. Total RNA from S2R+ cells was extracted using RNeasy Mini kit (QIAGEN, 74134). cDNA was reverse transcribed using SuperScript IV Reverse Transcriptase kit (Thermo Fisher Scientific, 18090050). Oligonucleotides against Chd1, RpL15 and RpL32 were used to quantify mRNA levels by qPCR (Table S2).
Tagging of Ssrp1 by CRISPR knockin
PCR amplification of a sgRNA targeting Ssrp in S2R+ cells for 6Flag integration was done using a combination of oligonucleotides for U6 promoter, Ssrp CRISPR, sgRNA scaffold and sgRNA antisense sequences (Table S2). The pRB17 plasmid (Bottcher et al., 2014) was used as a template for the U6 promoter sequence during the PCR reaction. A homology repair (HR) donor to integrate a 6Flag-blasticidin cassette at the Ssrp C-terminus was generated by PCR amplification, using oligonucleotides overlapping 60 bp at either side of the stop codon (Table S2). A modified version of the pKF296 plasmid (Kunzelmann et al., 2016) was used as a template for the HR donor PCR. Both PCR products (sgRNA and HR donor) were purified before transfection using QIAquick PCR purification kit (QIAGEN, 28106). 1.2×105 S2R+ cells bathed 3 days with a combination of dsRNAs against lig4 and mus308 (1 μg/ml each) were seeded in a 96-well plate. A mixture of 50 ng of each purified PCR product and 50 ng pRB14 (plasmid encoding Cas9) (Bottcher et al., 2014) was transfected with FuGENE HD (Promega, E2311) using a 6:1 ratio (FuGENE HD:DNA). Cells were incubated 4 days with the transfection mix, before being transferred in a 24-well plate in media containing 5 μg/mL blasticidin to select for Ssrp1–6Flag cells.
ChIP
Yeast ChIP experiments were performed at least from two independent biological replicates according to (Jeronimo et al., 2015). In brief, yeast cultures were grown in 50 mL of the appropriate medium (see above) to an OD600 of 0.7–0.9 before crosslinking with 1% formaldehyde (Fisher Scientific, BP531–500) at room temperature for 30 min and quenched with 125 mM glycine. Crosslinked cells were collected by centrifugation and washed twice with 1X TBS (20 mM Tris-HCl pH 7.5, 150 mM NaCl). Cell pellets were then resuspended in 700 μL Lysis buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate and protease inhibitor cocktail (1 mM PMSF, 1 mM Benzamidine, 10 μg/mL Aprotinin, 1 μg/mL Leupeptin, 1 μg/mL Pepstatin)). About the same number of OD600 units was used for all samples. Cells were lysed by bead beating and the lysate was sonicated with a Model 100 Sonic dismembrator equipped with a microprobe (Fisher Scientific), 4 × 20 sec at output 7 Watts, with a 1 min break between sonication cycles. Soluble fragmented chromatin was recovered by centrifugation. 600 μL of the chromatin sample was taken per immunoprecipitation (IP) and, when necessary, 6 μL (1%) were saved as an Input sample. The following amounts of antibody per IP were used: anti-Spt16 (a gift from T. Formosa, 1 μL), anti-Pob3 (a gift from T. Formosa, 1 μL), anti-Rpb3 (Neoclone W0012, 3 μL; BioLegend 665004, 1.5 μL), anti-H4 (a gift from A. Verreault, 2 μL), anti-Flag (Sigma-Aldrich F3165, 5 μg), anti-HA (Santa Cruz Biotechnology sc-7392, 15 μL or 30 μL) and rabbit IgG (Millipore 12–370, 2.5 μg). All antibodies have been validated for ChIP (see (Mason and Struhl, 2003) for Spt16 and Pob3 antibodies and manufacturers’ web sites for commercial antibodies). Rabbit antibodies were coupled to Dynabeads coated with Protein G (Thermo Fisher Scientific, 10004D) and mouse antibodies were coupled to Dynabeads coated with Pan Mouse IgG antibodies (Thermo Fisher Scientific, 11042). 50 μL of the appropriate Dynabeads pre-coupled with the indicated antibody were added to the chromatin sample and incubated overnight at 4°C. Beads were washed twice with Lysis buffer, twice with Lysis buffer 500 (Lysis buffer + 360 mM NaCl), twice with Wash buffer (10 mM Tris-HCl pH 8.0, 250 mM LiCl, 0.5% NP40, 0.5% Na-deoxycholate, 1 mM EDTA) and once with TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Input and immunoprecipitated chromatin were eluted and reverse-crosslinked with 50 μL TE/SDS (TE + 1% SDS) by incubating overnight at 65°C. Samples were treated with RNase A (345 μL TE, 3 μL 10 mg/mL RNAse A (Sigma-Aldrich, R6513), 2 μL 20 mg/mL Glycogen (Roche, 10901393001)) at 37°C for 2 hr and subsequently subjected to Proteinase K (15 μL 10% SDS, 7.5 μL 20 mg/mL Proteinase K (Thermo Fisher Scientific, 25530049)) digestion at 37°C for 2 hr. Samples were extracted twice with phenol/chloroform/isoamyl alcohol (25:24:1), followed by precipitation with 200 mM NaCl and 70% ethanol. Precipitated DNA was resuspended in 50 μL of TE before being used in ChIP-chip or ChIP-qPCR experiments.
Drosophila ChIP experiments were performed in three biological replicates. 8×106 S2R+ cells (expressing, or not, SSRP1–6Flag; treated with either dsGFP or dsChd1) were seeded in 100 mm plates and grown for 4 days. Cells were crosslinked in 1X PBS containing 1% formaldehyde at room temperature for 20 min and quenched with 125 mM glycine for 5 min. Crosslinked cells were harvested and washed twice with cold 1X PBS before snap-freezing. Cell pellets were resuspended in 130 μL S2-RIPA lysis buffer. Sonication was performed using Covaris E220 with the following parameters to obtain chromatin fragments between 100–500 bp: PIP = 140 W, DF = 20%, CPB = 200, duration = 150 s. Soluble fragmented chromatin was recovered by centrifugation. 12.5 μL of the chromatin sample was taken per IP and 1% was saved as Input sample. The following amounts of antibody per IP were used: anti-Rpb1 (BioLegend 664912, 5 μL), anti-Flag (Sigma-Aldrich F3165, 5 μg) and normal mouse IgG (Millipore 12–371, 2.5 μg). Antibodies were coupled to Dynabeads coated with Pan Mouse IgG antibodies. Antibody-coated beads were washed twice with S2-RIPA lysis buffer. 50 μL of washed antibody-coated beads were added to the chromatin sample and incubated overnight at 4°C. Beads were washed twice with Wash buffer A (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.1% Triton X-100), twice with Wash buffer B (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 5.2% sucrose, 1% Triton X-100, 0.2% SDS), twice with Wash buffer C (5 mM Tris-HCl pH 8.0, 25 mM HEPES, 250 mM NaCl, 0.5 mM EDTA, 0.5% Triton X-100, 0.05% Na-deoxycholate), twice with Wash buffer D (10 mM Tris pH 8.0, 250 mM LiCl, 10 mM EDTA, 0.5% IGEPAL CA-630, 0.5% Nadeoxycholate) then twice with TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Input and immunoprecipitated chromatin were eluted and reverse-crosslinked with 50 μL TE/SDS (TE + 1% SDS) by incubating overnight at 65°C. Samples were treated with RNase A (345 μL TE, 3 μL 10 mg/mL RNAse A, 2 μL 20 mg/mL Glycogen) at 37°C for 2 hr and subsequently subjected to Proteinase K (15 μL 10% SDS, 7.5 μL 20 mg/mL Proteinase K) digestion at 37°C for 2 hr. Samples were extracted twice with phenol/chloroform/isoamyl alcohol (25:24:1), followed by precipitation with 200 mM NaCl and 70% ethanol. Precipitated DNA was resuspended in 50 μL of TE before being used in qPCR experiments.
ChIP-qPCR
ChIP DNA and DNA from 1% of the Input were subjected to qPCR using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25741) on a ViiA 7 Real-Time PCR System (Thermo Fisher Scientific) (see Table S2 for oligonucleotide sequences). In Figure S6D, relative Chd1 expression was calculated by normalizing results against the geometric mean of reference genes RpL15 and RpL32 (Vandesompele et al., 2002). In Figures 2C, S6E and S6F, occupancy was reported as the % of Input using the following formula: 100*2^(CtInput-6.644-CtIP). Note that subtracting 6.644 to the CtInput is to adjust for the fact that 1% of Input was used. In Figure 6E, the data are represented as “fraction of t0”. To achieve this, the % of Input values were divided by the average % of Input of the t0 samples.
ChIP-chip
The ChIP and Input samples (see above) were blunted as follows. 40 μL of ChIP DNA or about 400 ng of Input DNA were added to 70 μL of blunting mix (11 μL 10X NEBuffer 2 (NEB, B7002S), 0.5 μL 10 mg/mL BSA, 0.5 μL 20 mM dNTPs and 0.2 μL (0.6 units) of T4 DNA polymerase (NEB, M0203L)) and incubated at 12˚C for 20 min. Then, the blunted DNA was extracted with phenol/chloroform/isoamyl alcohol (25:24:1) in the presence of 280 mM of NaOAc pH 5.2 and 10 μg of Glycogen, precipitated with ethanol and resuspended in 25 μL ddH2O. Blunted DNA was ligated to 100 pmol of annealed linkers (see Table S2) with 2.5 units of T4 DNA ligase (Thermo Fisher Scientific, 15224041) in the supplied buffer, in a final volume of 50 μL, overnight at 16˚C. Samples were precipitated with 6 μL of 3 M NaOAc pH 5.2 and 130 μL ethanol, and resuspended in 25 μL ddH2O. 15 μL of labeling mix (4 μL 10X ThermoPol Reaction buffer (NEB, B9004S), 2 μL of 5 mM aa-dUTP/dNTP mix (5 mM each dATP, dGTP and dCTP, 3 mM dTTP and 2 mM 5-(3-aminoallyl)-dUTP (Thermo Fisher Scientific, AM8439)) and 1.25 μL of 40 μM oligo 1) were added to each sample. Samples were placed in a thermocycler and when the temperature reached 55˚C, 10 μL of enzyme mix (1 μL 10X ThermoPol Reaction buffer, 1 μL (5 units) Taq DNA polymerase (Thermo Fisher Scientific, 18038240) and 0.01 μL (0.025 units) Pfu DNA polymerase (Thermo Fisher Scientific, EP0502)) were added and the following program was run. 1) 72˚C, 5 min; 2) 95˚C, 2 min; 3) 32 cycles: 95˚C, 30sec; 55˚C, 30sec; 72˚C, 1 min; 4) 72˚C, 4 min; 5) 4˚C, forever. PCR products were purified using QIAquick PCR purification kit (QIAGEN, 28106) with few modifications: PE buffer was replaced by Phosphate wash buffer (5 mM KPO4 pH 8.5 and 80% ethanol) and EB buffer was replaced by Phosphate elution buffer (4 mM KPO4 pH 8.5). Eluates were dried by SpeedVac and pellets were resuspended in 4.5 μL of freshly prepared 0.1 M Na2CO3 pH 9.0 buffer. 4.5 μL of Cy5 Mono-Reactive NHS Ester Fluorescent Dye (ChIP samples) or Cy3 Mono-Reactive NHS Ester Fluorescent Dye (control samples Input DNA or notag ChIP) (GE Healthcare, PA25001 and PA23001) were added. Samples were incubated 1 hr in the dark at room temperature. 35 μL of 0.1 M NaOAc pH 5.2 were added and samples were purified using QIAquick PCR Purification kit according to the manufacturers’ instructions. ChIP and control samples were combined and dried by SpeedVac. Combined samples were resuspended in 110 μL of fresh hybridization mix (5 μL 10 mg/mL Salmon Sperm DNA (Thermo Fisher Scientific, 15632011), 5 μL 8 mg/mL Yeast tRNA (Thermo Fisher Scientific, 15401011) and 100 μL DIG Easy Hyb (Roche, 11603558001)) and incubated 5 min at 95˚C followed by 5 min at 45˚C. Samples were hybridized to Agilent microarrays in SureHyb enabled chambers (Agilent, G2534A) with rotation at 42˚C for at least 18 hr. Microarrays were washed on an orbital shaker for 5 min at room temperature with Wash I buffer (6X SSPE, 0.005% N-Lauroylsarcosine), followed by a second wash with pre-warmed (31˚C) Wash II buffer (0.06X SSPE) for 5 min at room temperature. Microarrays were scanned using an InnoScan900 (Innopsys) at 2 μm resolution. The microarrays used, custom designed by Agilent Technologies, were described before (Jeronimo and Robert, 2014) and contain a total of about 180,000 Tm-adjusted 60-mer probes covering the entire yeast genome with virtually no gaps between probes.
ChIP-exo
ChIP-exo experiments were performed in two biological replicates essentially as described previously (Rossi et al., 2018) with some modifications. After following our standard ChIP protocol using a crosslinked 50 mL cell culture at an OD600~0.8 per IP and the appropriate antibody-coupled Dynabeads (see above), beads were washed twice with Lysis buffer, twice with Lysis buffer 500, twice with Wash buffer and once with 10mM Tris-HCl pH 8.0. Beads were then briefly centrifuged to remove all liquid before being used in the ChIP-exo library as follows. For A-tailing of the immunoprecipitated DNA, beads were resuspended in 50 μL of 1X NEBuffer 2 (NEB, B7002S) containing 0.1 mM dATP and 15 units of Klenow Fragment (3’→5’ exo-) (NEB, M0212M) and incubated at 37˚C for 30 min. After washing the beads with 10 mM Tris-HCl pH 8.0, dA-tailed DNA was ligated to the First adapter by resuspending the beads in 45 μL of 1X Rapid ligation buffer (Enzymatics, B1010) containing 375 nM of First ligation adapter (synthesized by IDT; see Table S2 for a list of adapters with their bar codes), 10 units of T4 polynucleotide kinase (NEB, M0201S) and 1,200 units of Rapid T4 DNA ligase (Enzymatics, L6030-HC-L) and incubating at room temperature for 60 min. After washing the beads with 10 mM Tris-HCl pH 8.0, the ligated DNA was subjected to a Fill-in reaction by resuspending the beads in 40 μL of 1X phi29 buffer (NEB) containing 0.2 mg/mL BSA (NEB), 180 μM dNTPs and 10 units of phi29 DNA polymerase (NEB, M0269S) and incubating at 30˚C for 20 min. After washing the beads with 10 mM Tris-HCl pH 8.0, DNA was subjected to λ exonuclease digestion by incubating the beads with 50 μL of λ exonuclease reaction buffer (NEB) containing 0.1% Triton X-100, 5% DMSO and 10 units of λ exonuclease (NEB, M0262S) at 37˚C for 30 min. To elute and reverse-crosslink DNA, beads were resuspended in 40 uL of reverse crosslink mix containing 25 mM Tris-HCl pH 7.5, 200 mM NaCl, 2 mM EDTA, 0.5% SDS and 30 μg of Proteinase K (Thermo Scientific) and incubated at 65˚C for 16 hr. Eluted DNA was then subjected to a 1.8X cleanup using KAPA Pure Beads (Roche, 07983280001) according to the manufacturers’ instructions before proceeding with Second adapter ligation as follows. Beads were resuspended in 40 μL of 1X T4 DNA ligase reaction buffer (NEB, B0202S) containing 375 nM of Second ligation adapter (synthesized by IDT; see Table S2) and 1,200 units of Rapid T4 DNA ligase (Enzymatics, L6030-HC-L), and incubated at room temperature for 60 min. The ligated DNA was then subjected to a 1.8X cleanup with KAPA Pure Beads and eluted in 15 μL ddH2O. Libraries were PCR-amplified in 40 μL reactions with 15–18 cycles using KOD Hot Start DNA polymerase (Millipore, 71086–3) and 500 nM of each primer P1.3 and P2.1 (synthesized by IDT; see Table S2). At this step, 10 μL from each sample was further amplified for six more cycles to visualize the quality of the library by electrophoresis on a 2% TAE-agarose gel stained with ethidium bromide. Once the library validated, DNA was subjected to a 1X cleanup with KAPA Pure Beads, followed by a double size selection (0.6X-1X) leading to DNA fragments in the 150–450 bp range. Libraries were qualified on Agilent 2100 Bioanalyzer using High Sensitivity DNA kit (Agilent Technologies, 5067–4626) and quantified by qPCR using NEBNext Library Quant kit for Illumina (NEB, E7630). Equal molarity of each library were pooled and subjected to sequencing on an Illumina HiSeq 4000 platform at the McGill University and Génome Québec Innovation Centre to generate 50 bp paired-end reads.
MNase-ChIP-seq
MNase-ChIP experiments were performed in two biological replicates, as previously described (Rando, 2010) with minor modifications. Briefly, yeast cultures were grown in 500 mL of YPD to an OD600 of 0.7–0.9 before crosslinking with 1% formaldehyde (Fisher Scientific) at room temperature for 15 min and quenched with 125 mM glycine at room temperature for 5 min. Crosslinked cells were collected by centrifugation and washed twice with ice-cold ddH2O. Yeast cell wall digestion was then performed by resuspending cell pellets in 39 mL Buffer Z (1 M sorbitol and 50 mM Tris-HCl pH 7.4) containing 10 mM β-mercaptoethanol and 10 mg Zymolyase 100T (US Biological, Z1004) and incubating at 30˚C with agitation (20–40 min). When digestion was completed, the resulting spheroplasts were pelleted by centrifugation at 3,000 g for 5 min at 4°C and gently resuspended in 2.5 mL of NP buffer (1 M sorbitol, 50 mM NaCl, 10 mM Tris-HCl pH 7.4, 5 mM MgCl2, 1 mM CaCl2, 500 μM spermidine (Sigma-Aldrich, 85558), 1 mM β-mercaptoethanol and 0.075% NP-40) before being exposed to MNase (Worthington, LS004798) digestion titration to generate the appropriate size of chromatin fragments as follow. 700 μl of spheroplasts were incubated with 4.5, 9, 18, and 36 units of MNase (resuspended in 10 mM Tris-HCl pH 7.4 and 30% glycerol) at 37˚C for 20 min and the reaction was stopped by adding 10 mM EDTA. To ensure IP conditions, 200 μL of adjusting buffer (50 mM HEPES-KOH pH7.5, 140 mM NaCl, 1% Triton X100, and 0.1% Na-deoxycholate) were added to the digested chromatin sample. To determine the appropriate condition of MNase digestion, 45 μL (5%) of each MNase digested sample was taken and incubated with 5 μL of 10X TE/SDS at 65˚C for 16 hr to reverse crosslinking. Samples were then treated with RNase A (345 μL TE, 3 μL 10 mg/mL RNAse A, 2 μL 20 mg/mL Glycogen) at 37°C for 2 hr and subsequently subjected to Proteinase K (15 μL 10% SDS, 7.5 μL 20 mg/mL Proteinase K) digestion at 37°C for 2 hr. Samples were twice phenol/chloroform/isoamyl alcohol (25:24:1) extracted followed by precipitation with 200 mM NaCl and 100 % ethanol. Precipitated DNA was resuspended in 30 μL of TE, treated with 1μL of 10 mg/mL RNAse A at 37°C for 1 hr before being analyzed by electrophoresis on a 1.8% TAE-agarose gel stained with ethidium bromide. The digested samples were also qualified on Agilent 2100 Bioanalyzer using High Sensitivity DNA Kit (Agilent Technologies). The condition giving approximately 65% mono-, 25% di- and 10% tri-nucleosomes was subjected to IP as follows. 45μL (5%) were saved as an Input sample and the remaining digested chromatin was incubated overnight with the appropriate antibody-coupled Dynabeads as per our standard ChIP protocol (see above). Beads were then washed and samples reverse crosslinked, treated with RNase A, and subsequently subjected to Proteinase K before being twice phenol/chloroform/isoamyl alcohol extracted followed by precipitation with NaCl and ethanol. Precipitated DNA was resuspended in 50 μL of TE before being used in sequencing libraries.
Before proceeding with MNase-ChIP-seq libraries, ChIP and Input samples were subjected to a 2X cleanup using KAPA Pure Beads (Roche) according to the manufacturers’ instructions. Samples were eluted with 50 μL of Elution buffer (10 mM Tris pH 8.0) and quantified/qualified on an Agilent 2100 Bioanalyzer instrument using the High Sensitivity DNA kit. 1 ng of Input DNA and 40 μL of ChIP DNA were used for library preparation as follows. The ends of DNA were repaired by incubating in 70 μL of 1X NEBuffer 2 containing 0.6 units of T4 DNA polymerase (NEB, M0203L), 2 units of T4 polynucleotide kinase (NEB, M0201S), 0.09 nM dNTPs and 0.045 μg/μL of BSA at 12˚C for 30 min. Repaired DNA was then subjected to a 2X cleanup using KAPA Pure Beads before dA-tailing as follows. Beads containing the repaired DNA were resuspended in 50 μL of 1X NEBuffer 2 containing 0.1 mM dATP and 25 units of Klenow Fragment (3’→5’ exo-) (NEB, M0212M), and incubated at 37˚C for 30 min. After a 2X cleanup with KAPA Pure Beads, dA-tailed DNA was ligated to index adapters (Roche, SeqCap Adapter kit A (07141530001) and SeqCap Adapter kit B (07141548001) or Illumina Truseq DNA UD Indexes (20023784)) as follow. The beads were resuspended in 45 μL of 1X Ligase buffer containing 8 nM of adapter and 2.5 units of T4 DNA ligase and incubated at room temperature for 60 min. The ligated DNA was then subjected to a 1X cleanup with KAPA Pure Beads. Libraries were PCR-amplified with 10–12 cycles using KOD Hot Start DNA polymerase (Millipore, 71086–3) and cleaned up using 1X KAPA Pure Beads. Libraries were qualified on Agilent 2100 Bioanalyzer using High Sensitivity DNA kit and quantified by qPCR using NEBNext Library Quant kit for Illumina (NEB, E7630). Equal molarity of each library was pooled and subjected to sequencing on Illumina HiSeq 4000 and NovaSeq 6000 platforms at the McGill University and Génome Québec Innovation Centre to generate 50 bp paired-end reads.
Co-immunoprecipitation experiments
Co-IP experiments between Spt16 and Chd1 in WT, chd1-K407R and chd1ΔN cells were performed in four biological replicates. Yeast strains were grown in 50 mL of YPD medium to an OD600 of 0.7–0.9, collected by centrifugation, washed in cold ddH2O, and resuspended in 700 μL of IP buffer (50 mM HEPES-KOH pH 7.5, 100 mM NaCl, 0.5 mM EDTA, 0.05% NP-40, 20% glycerol, 1 mM DTT and (1 mM PMSF, 1 mM Benzamidine, 10 μg/mL Aprotinin, 1 μg/mL Leupeptin, 1 μg/mL Pepstatin)). About the same amount of OD600 units was used for all samples. Cells were lysed by bead beating for 5 min and the soluble extract was recovered by centrifugation (15 min at 4°C at 18,000 × g). Cleared lysates were then treated or not with 500 units of Benzonase (Sigma-Aldrich, E1014) for 1 hr at 4°C. About 3–3.5 mg of protein (600 μl) was taken per IP and 6 μL (1%) were saved as Input sample. Benzonase-treated and untreated protein extracts were incubated for 60 min at 4°C with 50 μL of pre-washed magnetic protein G Dynabeads (Thermo Fisher Scientific) coupled to 15 μL of anti-HA F7 mouse monoclonal antibody (Santa Cruz Biotechnology). Samples were then washed three times for 5 min and twice shortly with room temperature IP buffer and resuspended in Laemmli buffer. Samples were incubated at 95oC for 5 min, briefly centrifuged and analyzed by SDS-PAGE and western blot using anti-Spt16 rabbit polyclonal (a gift from T. Formosa, 1:1,000 dilution) and anti-HA mouse monoclonal (Santa Cruz Biotechnology, 1:1,000 dilution) antibodies. Membranes were incubated with donkey anti-rabbit IRDye 800CW and donkey anti-mouse IRDye 680RD antibodies (LI-COR Biosciences, 926–32213 and 926–68072) according to the manufacturers’ instructions and scanned on the Odyssey infrared imaging system (LI-COR Biosciences).
Live cell, single-molecule imaging using wide-field microscopy
To perform single-molecule imaging of FACT in living yeast, HaloTag was fused to C-terminus of the Spt16 subunit of FACT in a strain background harboring pdr5Δ for efficient JF ligand labeling. SMT experiments were performed essentially as described (Ranjan et al., 2020). Yeast cultures growing in suspension were labelled with 10 nM (fast-tracking) or 1 nM (slow-tracking and time-lapse) JF552 dye for 2–3 hr at 30°C. 10 ms-frame rate (fast-tracking) movies were recorded with continuous 555 nm laser irradiation at ~1 kW/cm2. For slow tracking of H2B and Spt16, we used lower excitation power (~50 W/ cm2) and imaged a 256×256 pixel field for ~3–5 min at 250 ms/frame. For time-lapse imaging, we modified the slow-tracking regime to alternate 250 ms excitation and 250 ms or 500 ms dark time and imaged each field of view for ~5 min at 500 ms or 750 ms/frame, respectively.
We used the software DiaTrack v3.04/3.05 (Vallotton and Olivier, 2013) for single-molecule localization and tracking. For fast-tracking data, localization was carried out under High Precision mode (HWHM = 1 pixel) and customized “remove blur/dim” settings to minimize false detections. Then, tracking was performed with 6-pixel (642 nm) maximum allowance between consecutive localizations. Trajectories found outside of the nucleus were filtered out by masking the nuclear regions based on the maximum intensity Z-projection of each movie.
Slow-tracking data were similarly localized, and for tracking, we applied a 2-pixel maximum allowance between consecutive localizations based on H2B displacement observed under the same imaging regime (Nguyen et al., 2020).
QUANTIFICATION AND STATISTICAL ANALYSIS
ChIP-chip Data analysis
The ChIP-chip data were normalized using the Limma Loess method, and replicates were combined as described previously (Ren et al., 2000). The data were subjected to one round of smoothing using a Gaussian sliding window with a standard deviation of 100 bp to generate data points in 10 bp intervals (Guillemette et al., 2005).
ChIP-exo Data analysis
Paired-end reads from ChIP-exo data were aligned to the yeast S. cerevisiae genome (sacCer3 from UCSC Genome Browser) using BWA version 0.7.17 (Li and Durbin, 2009). Unmapped reads, reads not mapped properly in pairs, and duplicates were removed from the analysis using SAMtools version 1.8 (Li et al., 2009). For each paired-end read, only the first read was kept for further analysis. The aligned files were converted to BED using BEDTools version 2.28.0 (Quinlan and Hall, 2010). BED files containing reads from replicates were further analyzed individually in addition to being merged. The reads were moved 6 bp towards the 3’ end to correct for the offset of exonuclease stops and the site of crosslinking using a custom java script called bed-tools-j version 2.1 available at https://github.com/francoisrobertlab/bedtools-j with the parameter “moveannotations -d 6 -r -dn”. Three genome coverage files were computed using the 5’ position of the moved reads with BEDTools. Coverage files were generated for reads mapping the positive strand, the negative strand, and the merged strands. The genome coverage files were converted to the bigWig format using the utilities from UCSC Genome Browser (Casper et al., 2018). The analysis was partially automated using a custom script called seqtools version 1.0 available at https://github.com/francoisrobertlab/seqtools.
MNase-ChIP-seq Data analysis
Paired-end reads from MNase-ChIP-seq data where aligned on the S. cerevisiae genome version sacCer3 using Bowtie2 version 2.3.4.3 (Langmead and Salzberg, 2012) with the parameter “-X 1000” to include longer fragments. Unmapped and low-quality fragments were removed using SAMtools version 1.9 (Li et al., 2009) with the following parameter “view -f 2 -F 2048”. Duplicates were removed using SAMtools with the following parameters “fixmate -m” and “markdup -r”. The aligned files were converted to BED with BEDTools version 2.27.1 (Quinlan and Hall, 2010) using seqtools with the command “seqtools bam2bed” resulting in BED files containing the DNA fragments. The BED files of replicates were merged using seqtools with command “seqtools merge”. Genome coverage files were generated using seqtools with commands “mnasetools prepgenomecov” and “seqtools genomecov” that require BEDTools version 2.27.1 and KentUtils (downloaded on July 16th 2018). The genome coverage files were converted to the bigWig format using the utilities from UCSC Genome Browser (Casper et al., 2018). The configuration files used with seqtools for the MNase-ChIP-seq data analysis are available on GitHub at https://github.com/francoisrobertlab/mnasechip-cj2020.
Heatmaps and metagene analyses
The heatmap from Figure 1A and the metagenes from Figures 1A, 1C, 1D, 1E, 2A, 2B, 2E, 7C, S1A, S1C and S2 were generated using VAP (Brunelle et al., 2015; Coulombe et al., 2014). Data were aligned on the TSS (Pelechano et al., 2013) of the +1 nucleosome coordinates (Chereji et al., 2018) as indicated. The heatmap from Figure 1A shows all genes with defined +1 nucleosome coordinates, sorted by decreasing RNAPII occupancy. Most metagenes show signal over “transcribed genes”. These genes, defined as those with an average RNAPII ChIP-chip log2 ratio over the ORF >1, are independently defined in each experiment to account for differences in expression in different genetic backgrounds and growth conditions. Consequently, the number of “transcribed genes” slightly varies between figures. The number of genes included in each metagene is indicated in figure legends (see also Table S3). In Figures 1C, and S1A, the “High”, “Medium” and “Low” transcription groups are defined as those with an average RNAPII ChIP-chip log2 ratio over the ORF >2, between 1 and 2 and <1, respectively.
Heatmaps of coverage over genes versus fragment size
To generate Figures 4B, 4C, S4B, S4D and S4F, fragments from the MNase-ChIP-seq data were grouped in bins of 10 bp using seqtools with command “seqtools split -- binLength 10 --binMinLength 50 -- binMaxLength 500” and genome coverage files were generated using seqtools with commands “mnasetools prepgenomecov” and “seqtools genomecov” that require BEDTools version 2.27.1 (Quinlan and Hall, 2010) and KentUtils (downloaded on July 16th 2018). The heatmap matrices were then generated using seqtools with command “seqtools vap” that requires VAP (Coulombe et al., 2014) (Note: split and genome coverage must be executed prior to “seqtools vap”). Heatmaps were then generated using Java TreeView Version 1.1.6r4 (Saldanha, 2004).
Two-dimensional occupancy (2DO) plots
In Figures 5 and S5, Plot2DO (Beati and Chereji, 2020) revision number 87fadb4acd23214f83e5440b0ccb02c623fa62d9 with parameters “-t dyads -r Plus1 -l 100 -L 400 -m 0.02” was used to generate 2DO heatmaps from the MNase-ChIP-seq data. In some analyzes, the fragments not overlapping the top 10% of genes were removed using seqtools with command “seqtools intersect- a Top10percentTxbdGenes.txt”, where “Top10percentTxbdGenes.txt” contains the top 10% most transcribed genes and their coordinates.
Identification of coordinates of +2 nucleosomes
Coordinates of +1 nucleosomes were from (Chereji et al., 2018) while coordinates of +2 nucleosomes were determined from the chemical cleavage data (Chereji et al., 2018) as follow. BigWig dyads from (Chereji et al., 2018) were downloaded and merged using seqtools with command “seqtools mergebw”. Then the position of the +2 dyad was computed using seqtools with command “chectools dyadposition-g 13059_2018_1398_MOESM2_ESM.txt -s merge.bw”, where “13059_2018_1398_MOESM2_ESM.txt” is the file from (Chereji et al., 2018) containing the +1 nucleosomes coordinates. The seqtools script assesses the position of the +2 nucleosome by finding the position of the maximum signal from the merged bigWig file at a distance between 141 and 191 bp from the position of the +1 nucleosome for each gene.
Distributions of MNase-ChIP-seq fragments relative to nucleosome dyads
In Figures 7B and S7, the distribution of the center of MNase-ChIP-seq DNA fragments of different size, relative to the dyad of the +1 or +2 nucleosomes were generated as follow. The fragments from MNase-ChIP-seq data were grouped in bins of 10 bp using seqtools with commands “seqtools slowsplit -binMinLength 63 --binMaxLength 73”, “seqtools slowsplit --binMinLength 85 --binMaxLength 95”, “seqtools slowsplit --binMinLength 98 --binMaxLength 108” and, “seqtools slowsplit --binMinLength 120 --binMaxLength 130”. The genome coverage was computed from the resulting files using seqtools with command “mnasetools prepgenomecov” and then “seqtools genomecov --scale 1.0” which requires BEDTools 2.27.1 (Quinlan and Hall, 2010) and KentUtils (downloaded on July 16th 2018). The dyad coverage was computed from the genome coverage files using seqtools with command “mnasetools dyadcov -g first_dyad.txt --smoothing 20”, where “first_dyad.txt” contains the position of the +1 nucleosome from (Chereji et al., 2018). The dyad coverage for the second dyad used “mnasetools dyadcov -g second_dyad.txt --smoothing 20”, where “second_dyad.txt” contains the position of the +2 nucleosome from “chectools dyadposition” execution. Gaussian fits were computed using seqtools with command “mnasetools fitgaussian--svg --amin 0”, while double Gaussian fits were computed using seqtools with command “mnasetools fitdoublegaussian --gaussian --svg --amin1 0 --amin2 0”. The fitting was computed using the lmfit Python library version 1.0.0 available at https://lmfit.github.io/lmfit-py/ using the GaussianModel and ConstantModel.
Analysis for 10 ms-frame rate movies (fast-tracking):
Using Sojourner R package (https://rdrr.io/github/sheng-liu/sojourner/), diffusion coefficient values were calculated for all “masked” trajectories ≥ 5 frames, based on the slope of their MSD plots (using 2–5 Δt time lags). Trajectories with R2 < 0.8 were filtered out. Diffusion coefficient is calculated as (where d = 2):
Spot-On kinetic modeling (Hansen et al., 2018) was applied to all “masked” trajectories ≥ 3 frames. The following parameters were applied for Jump Length Distribution: Bin width (μm): 0.01, Number of time-points: 6, Jumps to consider: 4, Use entire trajectories No, Max jump (μm): 2. Additionally, the following parameters were applied for 2-state Model Fitting: Dbound (μm2/s): 0.0005–0.1, Dfree (μm2/s): 0.15–25, Fbound: 0–1, Localization error (μm): Fit from data (0.01–0.1), dZ (μm): 0.6, Use Z correction, Model Fit: CDF, Iterations: 3. For Pol II-depleted mutant dataset showing minimal chromatin-binding frequency, the Dbound value was constrained to the output Dbound values of DMSO-treated control datasets: [0.065–0.069].
Analysis for continuous and time-lapse 250 ms-frame rate movies (slow-tracking):
Tracking results from DiaTrack were processed in Sojourner to determine the apparent dwell times (temporal lengths of trajectories). The cumulative frequency distribution (1-CDF) of dwell times was fitted with the double-exponential decay function, where fsb and ksb represent the frequency and apparent dissociation rate constant of stable binding, respectively, and ktb represents the apparent dissociation rate constant of transient binding (Mazza et al., 2012):
We used the bootstrap method (Shrout and Bolger, 2002) to generate 100 resampled datasets and calculate 95% confidence interval. We then computed 1-CDF and performed fitting for each dataset. The mean values for fsb, ksb, and ktb were taken, with their standard deviations providing an assessment of fit quality.
We adapted the approach described by Hansen et. al. (Hansen et al., 2017) to correct the apparent dissociation rates. First, we imaged H2B under the same regime, with the assumption that its decay kinetics measures limits imposed by photobleaching and chromatin motions. The 1-CDF for H2B was fitted as described, and its ksb was used for correction as follows:
The overall fraction of molecules stably bound (Ceq), was calculated using the Fbound and fsb values as follow:
Calculating search kinetics
The search time τsearch is the average time a single molecule takes to sample Ntrials chromatin sites, alternating between binding each site (with average dwell time τtb) and diffusing in the nucleoplasm (with average duration τfree), before reaching any one of its specific targets. We calculated the τsearch for FACT in WT and chd1Δ cells according to the following equation (Chen et al., 2014; Lionnet and Wu, 2021; Loffreda et al., 2017; Tatavosian et al., 2018),
To obtain τsearch, we used FB and results obtained from fast- and slow-tracking experiments, respectively, to derive Ntrials and τfree as outlined in detail in (Nguyen et al., 2020) (Figure S3A). As FACT dwell times in WT cells required time-lapse imaging to resolve, we took τsb and τtb values for both WT and chd1Δ as means±SD from 250 ms and 500 ms TL results. However, because the stable fraction is highly sensitive to the frame rate of the imaging regime (longer frame rates increase motion blurring of transient events, leading to overcounting of stable events), we used fsb values (means±SD from two biological replicates) from the continuous slow-tracking regime (250 ms frame rate) instead of TL regimes (500 ms-750 ms frame rates).
Mathematical modeling
Models were implemented in Matlab 2019b and used a fixed time-step stochastic algorithm in which it is assumed that only one reaction can occur in each time step. At each time step, a random number was drawn from a uniform distribution (Matlab rand() function, Mersenne Twister random number generator). This random number was then compared against a list of the cumulative summed probabilities per unit time of all possible reactions as described in the pseudo-code below (with description in roman font and pseudo xcode in italics). Table 1 contains parameter values and their abbreviations as used in the pseudo-code. The simulation keeps track of the number of FACT complexes and the numbers that are in the extending, retracting, fully extended, fully retracted and stalled states, FACTNo, ExtNo, RetNo, FullyExtNo, FullyRetNo and StalledNo, respectively.
Table 1:
Parameter values, expressed as probabilities per unit time, and their abbreviations used in the main model for the WT and chd1Δ cases (which differ only in the extension rate or stalling during extension), see Figure 6.
| Parameter | Abbr. | WT (Fig. 6B) | chd1Δ slow extension (Fig. 6C) | chd1Δ stalled extension (Fig. 6D) |
|---|---|---|---|---|
| Binding | BR | 0.00001 | 0.00001 | 0.00001 |
| Extension | ER | 0.03 | 0.0075 | 0.03 |
| Extension to Retraction | ERS | 0.003 | 0.003 | 0.003 |
| Retraction | RR | 0.03 | 0.03 | 0.03 |
| Retraction to Extension | RES | 0.003 | 0.003 | 0.003 |
| Unbinding | UBR | 0.000005 | 0.00001 | 0.00001 |
| Stalling | SR | 0 | 0 | 0.00001 |
| Relaxation Time Steps | TSR | 106 | 106 | 106 |
| Measurement Time Steps | TSM | 106 | 106 | 106 |
| Measurement Interval | MI | 10 | 10 | 10 |
| Realisations | R | 100000 | 100000 | 100000 |
Generate random number:
Diceroll = rand;
Compare random number against possible reactions:
if rand < (BR)
Bind a FACT complex at the +1 Nucleosome location (if it is unoccupied)
else if rand < (BR+ExtNo*ER)
Select a random FACT complex from those that are in the extending state and, if possible, extend by one base pair at the TSS-distal side – if the complex abuts with a preceding complex, do not extend; if the complexes leading edge reaches the end of the gene, flag as fully extended
else if rand < (BR+ExtNo*ER+RetNo*RR)
Select a random FACT complex from those that are in the retracting state and, if possible, retract by one base pair at the TSS-proximal side
else if rand < (BR+ExtNo*ER+RetNo*RR+FullyExtNo*ERS)
Select a random FACT complex from those that are in the fully extended state and transition to the retracting state
else if rand < (BR+ExtNo*ER+RetNo*RR+FullyExtNo*ERS+FullyRetNo*RES)
Select a random FACT complex from those that are in the fully retracted state and transition to the extending state – if the FACT complex has reached the end of the gene, unbind
else if rand < (BR+ExtNo*ER+RetNo*RR+FullyExtNo*ERS+FullyRetNo*RES+FactNo*UBR)
Select a random FACT complex and unbind
else if rand <
(BR+ExtNo*ER+RetNo*RR+FullyExtNo*ERS+FullyRetNo*RES+FactNo*UBR+ExtNo*SR)
Select an extending FACT complex and transition to a stalled state
else
Leave the system unchanged
end
The system was simulated on a synthetic gene of length 1,510 bp. Nucleosome locations were defined as regions of 150 bp, with the first at the beginning of the gene and spacing regions of 20 bp between each nucleosome location. The minimum extent of a FACT complex was taken to be 150 bp and the maximum to be 320 bp (the extent of two nucleosomes and a spacing region).
The system was simulated for a relaxation period of 106 time steps, following this, the simulation was continued for an additional 106 steps during which a measurement of protected fragment sizes and positions was taken every 10 time steps. Where presented in figures, the fragment sizes and positions are displayed as block averages – each displayed pixel is the mean of the 20-by-20 region around it in the original measurement. This was to increase visibility as the simulations lack noise in the positioning and size of nucleosomes and FACT complexes.
In addition to the core mechanisms described above, the possibility for new FACT complexes to bind with uniform probability to any unoccupied nucleosome location in the gene was tested.
Supplementary Material
Movie S1. Animation of the “inchworm-like” model of FACT recruitment spreading for the WT case. The left panel shows a representation of the regions of DNA protected from MNase digestion by FACT complexes for 100 realisations of a simulation across a synthetic gene. Bars represent FACT complexes, the x-axis is location in the gene and each track on the y-axis represents an individual realisation (tracks are separated by blank spaces). Bars are colour-coded according to the state of the FACT complex: bright green for extending, dark green for fully extended, bright blue for retracting and dark blue for fully retracted. The right panel shows the density of fragments, of given sizes (y-axis) and locations (x-axis), protected from MNase digestion by FACT complexes as they are sampled from the simulation. Parameters of the simulation are as for the WT case as in the main text (Figure 6B and Table 1) except that the number of time steps shown is 4×105, there are 100 realisations and there is no relaxation period prior to measurements for the protected fragments being taken. Related to Figure 6.
Movie S2. Animation of the “inchworm-like” model of FACT recruitment spreading for the model 1 chd1Δ case (same as WT except for a reduced extension rate and increase unbinding of the FACT complex). The left panel shows a representation of the regions of DNA protected from MNase digestion by FACT complexes for 100 realisations of a simulation across a synthetic gene. Bars represent FACT complexes, the x-axis is location in the gene and each track on the y-axis represents an individual realisation (tracks are separated by blank spaces). Bars are colour-coded according to the state of the FACT complex: bright green for extending, dark green for fully extended, bright blue for retracting and dark blue for fully retracted. The right panel shows the density of fragments, of given sizes (y-axis) and locations (x-axis), protected from MNase digestion by FACT complexes as they are sampled from the simulation. Parameters of the simulation are as for the chd1Δ case as in the main text (Figure 6C and Table 1) except that the number of time steps shown is 4×105, there are 100 realisations and there is no relaxation period prior to measurements for the protected fragments being taken. Related to Figure 6.
Movie S3. Animation of the “inchworm-like” model of FACT recruitment spreading for the model 2 chd1Δ case (same as WT except for increase unbinding and the possibility of the FACT complex stalling while in the extending phase). The left panel shows a representation of the regions of DNA protected from MNase digestion by FACT complexes for 100 realisations of a simulation across a synthetic gene. Bars represent FACT complexes, the x-axis is location in the gene and each track on the y-axis represents an individual realisation (tracks are separated by blank spaces). Bars are colour-coded according to the state of the FACT complex: bright green for extending, dark green for fully extended, bright blue for retracting, dark blue for fully retracted and red for stalled. The right panel shows the density of fragments, of given sizes (y-axis) and locations (x-axis), protected from MNase digestion by FACT complexes as they are sampled from the simulation. Parameters of the simulation are as for the chd1Δ case as in the main text (Figure 6D and Table 1) except that the number of time steps shown is 4×105, there are 100 realisations and there is no relaxation period prior to measurements for the protected fragments being taken. Related to Figure 6.
Table S3. List of genes used in different figures in this study. Related to STAR Methods.
Table 2:
Parameter values, expressed as probabilities per unit time used for the modelling data shown in Figure S6.
| Parameter | Fig. S6A left | Fig. S6A middle | Fig. S6A right | Fig. S6B left | Fig. S6B middle | Fig. S6B right | Fig. S6C top | Fig. S6C bottom | Fig. S6D middle | Fig. S6D right |
|---|---|---|---|---|---|---|---|---|---|---|
| BR | 0.00001 | 0.00001 | 0.00009 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 |
| ER | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.0075 | 0.03 | 0.0075 | 0.03 |
| ERS | 0.03 | 0.03 | 0.03 | 0.003 | 0.003 | 0.0003 | 0.003 | 0.003 | 0.003 | 0.003 |
| RR | 0.03 | 0.03 | 0.03 | 0.0075 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 |
| RES | 0.03 | 0.003 | 0.03 | 0.003 | 0.003 | 0.003 | 0.003 | 0.003 | 0.003 | 0.003 |
| UBR | 0 | 0 | 0 | 0.000005 | 0.00001 | 0.000005 | 0.000005 | 0.000005 | 0.00001 | 0.00001 |
| SR | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.00001 | 0 | 0.00001 |
| TSR | 106 | 106 | 106 | 106 | 106 | 106 | 106 | 106 | 106 | 106 |
| TSM | 106 | 106 | 106 | 106 | 106 | 106 | 106 | 106 | 106 | 106 |
| MI | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| R | 10000 | 10000 | 10000 | 10000 | 10000 | 10000 | 100000 | 100000 | 100000 | 100000 |
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Spt16 | T. Formosa | N/A |
| Rabbit polyclonal anti-Pob3 | T. Formosa | N/A |
| Mouse monoclonal anti-RNA Polymerase II Rpb3 (1Y26) | Neoclone; BioLegend | Cat# 665003, RRID:AB_2564529; Cat# 665004, RRID:AB_2565221 |
| Mouse monoclonal anti-Flag (M2) | Sigma-Aldrich | Cat# F3165, RRID:AB_259529 |
| Mouse monoclonal anti-HA (F7) | Santa Cruz Biotechnology | Cat# sc-7392, RRID:AB_627809 |
| Rabbit IgG | Millipore | Cat# 12-370, RRID:AB_145841 |
| Rabbit polyclonal anti-Chd1 | A. Lusser | N/A |
| Mouse monoclonal anti-α-tubulin | Sigma-Aldrich | Cat# T5168, RRID:AB_477579 |
| Mouse monoclonal anti-RNA Polymerase II Rpb1 (8WG16) | BioLegend | Cat# 664912, RRID:AB_2650945 |
| IRDye800CW Donkey anti-rabbit | LI-COR Biosciences | Cat# 926-32213 RRID:AB_621848 |
| IRDye680RD Donkey anti-mouse | LI-COR Biosciences | Cat# 926-68072 RRID:AB_10953628 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1-Naphthyl PP1 (1NAPP1) | Tocris Bioscience | 3063 |
| Rapamycin | Bio Basic | R706203 |
| DRB | Sigma-Aldrich | D1916 |
| RQ1 DNase I | Promega | M610A |
| FuGENE HD | Promega | E2311 |
| 37% Formaldehyde | Fisher Scientific | BP531-500 |
| PMSF | Roche | 10837091001 |
| Benzamidine | Sigma-Aldrich | B-6506 |
| Aprotinin | Roche | 10981532001 |
| Leupeptin | Roche | 11017101001 |
| Pepstatin | Roche | 11359053001 |
| Acid washed glass beads (400-600um) | Sigma-Aldrich | G8772-1KG |
| Dynabeads Protein G | Thermo Fisher Scientific | 10004D |
| Dynabeads M-450 pre-coated with pan anti-mouse IgG | Thermo Fisher Scientific | 11042 |
| RNaseA | Sigma-Aldrich | R6513- |
| Glycogen (20 mg/mL) | Roche | 10901393001 |
| Proteinase K (20 mg/mL) | Thermo Fisher Scientific | 25530049 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | A25741 |
| NEBuffer 2 | New England Biolabs | B7002S |
| T4 DNA polymerase | New England Biolabs | M0203L |
| T4 DNA ligase | Thermo Fisher Scientific | 15224041 |
| ThermoPol Reaction buffer | New England Biolabs | B9004S |
| 5-(3-aminoallyl)-dUTP (aa-dUTP) | Thermo Fisher Scientific | AM8439 |
| Taq DNA polymerase | Thermo Fisher Scientific | 18038240 |
| Pfu DNA polymerase | Thermo Fisher Scientific | EP0502 |
| Cy3 Mono-Reactive NHS Ester Fluorescent Dye | GE Healthcare | PA23001 |
| Cy5 Mono-Reactive NHS Ester Fluorescent Dye | GE Healthcare | PA25001 |
| Salmon Sperm DNA | Thermo Fisher Scientific | 15632011 |
| Yeast tRNA | Thermo Fisher Scientific | 15401011 |
| DIG Easy Hyb | Roche | 11603558001 |
| Klenow Fragment (3’→5’ exo-) | New England Biolabs | M0212M |
| T4 polynucleotide kinase | New England Biolabs | M0201S |
| Rapid ligation buffer | Enzymatics | B1010 |
| Rapid T4 DNA ligase | Enzymatics | L6030-HC-L |
| phi29 DNA polymerase | New England Biolabs | M0269S |
| λ exonuclease | New England Biolabs | M0262S |
| KAPA Pure Beads | Roche | 07983280001 |
| T4 DNA ligase reaction buffer | New England Biolabs | B0202S |
| KOD Hot Start DNA polymerase | Millipore | 71086-3 |
| Zymolyase 100T | US Biological | Z1004 |
| Spermidine | Sigma-Aldrich | 85558 |
| MNase (Micrococcal nuclease) | Worthington | LS004798 |
| Index adapters | Roche | (SeqCAp Adapter kit A, (07141530001) and SeqCAp Adapter kit B, (07141548001)) |
| Truseq DNA UD Indexes | Illumina | 20023784 |
| Critical Commercial Assays | ||
| QIAquick PCR Purification Kit | QIAGEN | 28106 |
| RNeasy Mini kit | QIAGEN | 74134 |
| SuperScript IV Reverse Transcriptase kit | Thermo Fisher Scientific | 18090050 |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 |
| NEBNext Library Quant Kit | New England Biolabs | E7630 |
| Deposited Data | ||
| Raw and processed microarray and sequencing data | This study | GEO: GSE155144 |
| Single-molecule tracking data | This study | Mendeley Data : DOI: 10.17632/rnf4nc3g6y.1 |
| RNAPII in kin28as and CTD mutants ChIP-chip processed data | (Jeronimo and Robert, 2014) | GEO: GSE55402 |
| H4 ChIP-chip processed data | (Jeronimo et al., 2015) | GEO: GSE62880 |
| H2B ChIP-chip processed data | (Jeronimo et al., 2019) | GEO: GSE113270 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: S2R+ cells | Laboratory of Eric Lécuyer | N/A |
| S. cerevisiae: Strain background W303-1A | Laboratory of Rodney Rothstein | ATCC:208352 |
| S. cerevisiae: Strain background BY4741 | Laboratory of Jef D. Boeke | ATCC-201388 |
| S. cerevisiae: Strain background FY4 | Laboratory of Fred Winston | N/A |
| See Table S1 for all yeast strains | N/A | N/A |
| Oligonucleotides | ||
| See Table S2 for all oligonucleotides | N/A | N/A |
| Recombinant DNA | ||
| pMPY-3xHA | (Schneider et al., 1995) | N/A |
| pML104 | (Laughery et al., 2015) | N/A |
| pML107 | (Laughery et al., 2015) | N/A |
| pKF296 | (Kunzelmann et al., 2016) | N/A |
| pRB17 | (Bottcher et al., 2014) | N/A |
| pRB14 | (Bottcher et al., 2014) | N/A |
| Software and Algorithms | ||
| DiaTrack (version 3.04) | (Vallotton and Olivier, 2013) | http://www.diatrack.org/product.html |
| Versatile Aggregate Profiler (version 1.1.0) | (Brunelle et al., 2015; Coulombe et al., 2014) | http://lab-jacques.recherche.usherbrooke.ca/vap/ |
| UCSC Genome Browser | (Casper et al., 2018) | http://genome.ucsc.edu/ |
| BWA (version 0.7.17) | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/ |
| SAMtools (version 1.8) | (Li et al., 2009) | http://samtools.sourceforge.net |
| BEDTools (versions 2.27.1 and 2.28.0) | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/ |
| Bowtie 2 (version 2.3.4.3) | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Java TreeView (Version 1.1.6r4) | (Saldanha, 2004) | https://sourceforge.net/projects/jtreeview/ |
| Plot2DO | (Beati and Chereji, 2020) | https://github.com/rchereji/plot2DO |
| Sojourner R package | (Liu et al., 2021) | https://rdrr.io/github/sheng-liu/sojourner/ |
| MATLAB | MathWorks | https://www.mathworks.com/ |
Highlights.
High-resolution mapping and single-molecule tracking of FACT in yeast
FACT binds partially unwrapped nucleosomes in transcribed genes, not RNAPII
FACT distribution along genes requires Chd1
Processive mechanism for FACT function
ACKNOWLEDGMENTS
We are grateful to N. Francis for critical reading of the manuscript and the Robert lab members for comments. We also thank A. Bataille for advice about ChIP-exo library preparation and analysis; M. Sarsenova and O. Rocheleau-Leclair for help generating some strains; and T. Formosa, D. Stillman, R.A. Young, S. Hahn, S. Buratowski, F. Winston, S. Churchman, A. Lusser, and K. Förstemann for sharing reagents. This work was funded by grants from the Canadian Institutes of Health Research (CIHR, MOP162334) and Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN-2018–04519) to F.R. and from the National Institute of Health (GM132290–01) to C.W. This research was also enabled in part by support provided by « Calcul Québec » (http://www.calculquebec.ca) and Compute Canada (www.computecanada.ca). A.A. was supported by a Biotechnology and Biological Sciences Research Council (BBSRC) grant to J.M. (BB/S009035/1), J.M.K. by a Korean Foundation for Advanced Studies Fellowship, and E.L. by an Alexander Graham Bell Canada Doctoral Scholarship from NSERC. F.R. holds a Research Chair from the « Fonds de Recherche Québec –Santé » (FRQS).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adelman K, Wei W, Ardehali MB, Werner J, Zhu B, Reinberg D, and Lis JT (2006). Drosophila Paf1 modulates chromatin structure at actively transcribed genes. Mol Cell Biol 26, 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataille AR, Jeronimo C, Jacques PE, Laramee L, Fortin ME, Forest A, Bergeron M, Hanes SD, and Robert F. (2012). A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol Cell 45, 158–170. [DOI] [PubMed] [Google Scholar]
- Beati P, and Chereji RV (2020). Creating 2D Occupancy Plots Using plot2DO. Methods Mol Biol 2117, 93–108. [DOI] [PubMed] [Google Scholar]
- Berson A, Sartoris A, Nativio R, Van Deerlin V, Toledo JB, Porta S, Liu S, Chung CY, Garcia BA, Lee VM, et al. (2017). TDP-43 Promotes Neurodegeneration by Impairing Chromatin Remodeling. Curr Biol 27, 3579–3590 e3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch JL, Tan BC, Panov KI, Panova TB, Andersen JS, Owen-Hughes TA, Russell J, Lee SC, and Zomerdijk JC (2009). FACT facilitates chromatin transcription by RNA polymerases I and III. EMBO J 28, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher R, Hollmann M, Merk K, Nitschko V, Obermaier C, Philippou-Massier J, Wieland I, Gaul U, and Forstemann K. (2014). Efficient chromosomal gene modification with CRISPR/cas9 and PCRbased homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res 42, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle M, Coulombe C, Poitras C, Robert MA, Markovits AN, Robert F, and Jacques PE (2015). Aggregate and Heatmap Representations of Genome-Wide Localization Data Using VAP, a Versatile Aggregate Profiler. Methods Mol Biol 1334, 273–298. [DOI] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. (2005). Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592. [DOI] [PubMed] [Google Scholar]
- Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, Carmo-Fonseca M, and de Almeida SF (2013). Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res 41, 2881–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper J, Zweig AS, Villarreal C, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Karolchik D, et al. (2018). The UCSC Genome Browser database: 2018 update. Nucleic Acids Res 46, D762–D769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Zhang Z., Li L., Chen BC., Revyakin A., Hajj B., Legant W., Dahan M., Lionnet T., Betzig E., et al. (2014). Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156, 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereji RV, Ramachandran S, Bryson TD, and Henikoff S. (2018). Precise genome-wide mapping of single nucleosomes and linkers in vivo. Genome Biol 19, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, and Winston F. (2008). Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol 6, e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, and Weissman JS (2011). Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin P, Jeronimo C, Poitras C, and Robert F. (2019). RNA Polymerase II CTD Tyrosine 1 Is Required for Efficient Termination by the Nrd1-Nab3-Sen1 Pathway. Mol Cell 73, 655–669 e657. [DOI] [PubMed] [Google Scholar]
- Coulombe C, Poitras C, Nordell-Markovits A, Brunelle M, Lavoie MA, Robert F, and Jacques PE (2014). VAP: a versatile aggregate profiler for efficient genome-wide data representation and discovery. Nucleic Acids Res 42, W485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crickard JB, Lee J, Lee TH, and Reese JC (2017). The elongation factor Spt4/5 regulates RNA polymerase II transcription through the nucleosome. Nucleic Acids Res 45, 6362–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin S, Laramee L, Jacques PE, Forest A, Bergeron M, and Robert F. (2010). DSIF and RNA polymerase II CTD phosphorylation coordinate the recruitment of Rpd3S to actively transcribed genes. PLoS Genet 6, e1001173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehara H, Kujirai T, Fujino Y, Shirouzu M, Kurumizaka H, and Sekine SI (2019). Structural insight into nucleosome transcription by RNA polymerase II with elongation factors. Science 363, 744–747. [DOI] [PubMed] [Google Scholar]
- Farnung L, Ochmann M, Engeholm M, and Cramer P. (2021). Structural basis of nucleosome transcription mediated by Chd1 and FACT. Nat Struct Mol Biol 28, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnung L, Vos SM, and Cramer P. (2018). Structure of transcribing RNA polymerase II-nucleosome complex. Nat Commun 9, 5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnung L, Vos SM, Wigge C, and Cramer P. (2017). Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature 550, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming AB, Kao CF, Hillyer C, Pikaart M, and Osley MA (2008). H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol Cell 31, 57–66. [DOI] [PubMed] [Google Scholar]
- Foltman M, Evrin C, De Piccoli G, Jones RC, Edmondson RD, Katou Y, Nakato R, Shirahige K, and Labib K. (2013). Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep 3, 892–904. [DOI] [PubMed] [Google Scholar]
- Formosa T. (2013). The role of FACT in making and breaking nucleosomes. Biochim Biophys Acta 1819, 247–255. [PubMed] [Google Scholar]
- Formosa T, Eriksson P, Wittmeyer J, Ginn J, Yu Y, and Stillman DJ (2001). Spt16-Pob3 and the HMG protein Nhp6 combine to form the nucleosome-binding factor SPN. EMBO J 20, 3506–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa T, and Winston F. (2020). The role of FACT in managing chromatin: disruption, assembly, or repair? Nucleic Acids Res 48, 11929–11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs G, Voichek Y, Rabani M, Benjamin S, Gilad S, Amit I, and Oren M. (2015). Simultaneous measurement of genome-wide transcription elongation speeds and rates of RNA polymerase II transition into active elongation with 4sUDRB-seq. Nat Protoc 10, 605–618. [DOI] [PubMed] [Google Scholar]
- Gebhardt JC, Suter DM, Roy R, Zhao ZW, Chapman AR, Basu S, Maniatis T, and Xie XS (2013). Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat Methods 10, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind CK, Qiu H, Ginsburg DS, Ruan C, Hofmeyer K, Hu C, Swaminathan V, Workman JL, Li B, and Hinnebusch AG (2010). Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol Cell 39, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette B, Bataille AR, Gevry N, Adam M, Blanchette M, Robert F, and Gaudreau L. (2005). Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol 3, e384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurova K, Chang HW, Valieva ME, Sandlesh P, and Studitsky VM (2018). Structure and function of the histone chaperone FACT - Resolving FACTual issues. Biochim Biophys Acta Gene Regul Mech 1861, 892–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Li Q., McCullough L., Kettelkamp C., Formosa T., and Zhang Z. (2010). Ubiquitylation of FACT by the cullin-E3 ligase Rtt101 connects FACT to DNA replication. Genes Dev 24, 1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Pustova I, Cattoglio C, Tjian R, and Darzacq X. (2017). CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife 6, e25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Woringer M, Grimm JB, Lavis LD, Tjian R, and Darzacq X. (2018). Robust modelbased analysis of single-particle tracking experiments with Spot-On. eLife 7, e33125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, and Laemmli UK (2008). The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31, 925–932. [DOI] [PubMed] [Google Scholar]
- Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, and Henikoff S. (2011). Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci U S A 108, 18318–18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho B, Baryshnikova A, and Brown GW (2018). Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst 6, 192–205 e193. [DOI] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al. (2004). A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, Poitras C, and Robert F. (2019). Histone Recycling by FACT and Spt6 during Transcription Prevents the Scrambling of Histone Modifications. Cell Rep 28, 1206–1218 e1208. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, and Robert F. (2014). Kin28 regulates the transient association of Mediator with core promoters. Nat Struct Mol Biol 21, 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, Watanabe S, Kaplan CD, Peterson CL, and Robert F. (2015). The Histone Chaperones FACT and Spt6 Restrict H2A.Z from Intragenic Locations. Mol Cell 58, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, and Pugh BF (2009). A compiled and systematic reference map of nucleosome positions across the Saccharomyces cerevisiae genome. Genome Biol 10, R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Howe L, Tafrov ST, Grant PA, Sternglanz R, and Workman JL (2000). The something about silencing protein, Sas3, is the catalytic subunit of NuA3, a yTAF(II)30-containing HAT complex that interacts with the Spt16 subunit of the yeast CP (Cdc68/Pob3)-FACT complex. Genes Dev 14, 1196–1208. [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Visanpattanasin P, Jou V, Liu S, Tang X, Zheng Q, Li KY, Snedeker J, Lavis LD, Lionnet T, et al. (2021). Single-molecule imaging of chromatin remodelers reveals role of ATPase in promoting fast kinetics of target search and dissociation from chromatin. bioRxiv, 2021.2004.2021.440742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. (2006). Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Ahn SH, Zhong G, Kobor MS, Cagney G, Emili A, Shilatifard A, Buratowski S, and Greenblatt JF (2002). RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol 22, 6979–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, Richards DP, Beattie BK, Emili A, Boone C, et al. (2003). Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol 23, 4207–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ., Peng WT., Cagney G., Robinson MD., Haw R., Zhong G., Guo X., Zhang X., Canadien V., Richards DP., et al. (2004). High-definition macromolecular composition of yeast RNA-processing complexes. Mol Cell 13, 225–239. [DOI] [PubMed] [Google Scholar]
- Kujirai T, Ehara H, Fujino Y, Shirouzu M, Sekine SI, and Kurumizaka H. (2018). Structural basis of the nucleosome transition during RNA polymerase II passage. Science 362, 595–598. [DOI] [PubMed] [Google Scholar]
- Kunzelmann S, Bottcher R, Schmidts I, and Forstemann K. (2016). A Comprehensive Toolbox for Genome Editing in Cultured Drosophila melanogaster Cells. G3 (Bethesda) 6, 1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SH, Florens L, Swanson SK, Washburn MP, Abmayr SM, and Workman JL (2010). Heterochromatin protein 1 (HP1) connects the FACT histone chaperone complex to the phosphorylated CTD of RNA polymerase II. Genes Dev 24, 2133–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WKM, and Pugh BF (2017). Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol 18, 548–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughery MF, Hunter T, Brown A, Hoopes J, Ostbye T, Shumaker T, and Wyrick JJ (2015). New vectors for simple and streamlined CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. Yeast 32, 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom DL, Squazzo SL, Muster N, Burckin TA, Wachter KC, Emigh CA, McCleery JA, Yates JR 3rd, and Hartzog GA (2003). Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol 23, 1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionnet T, and Wu C. (2021). Single-molecule tracking of transcription protein dynamics in living cells: seeing is believing, but what are we seeing? Curr Opin Genet Dev 67, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yoo S, Tang X, Sung Y, and Wu C. (2021). Sojourner: Statistical analysis of single molecule trajectories. [Google Scholar]
- Liu Y, Kung C, Fishburn J, Ansari AZ, Shokat KM, and Hahn S. (2004). Two cyclin-dependent kinases promote RNA polymerase II transcription and formation of the scaffold complex. Mol Cell Biol 24, 1721–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou K, Zhang N, Wei H, Tan YZ, Zhang Z, Carragher B, Potter CS, D’Arcy S, and Luger K. (2020). FACT caught in the act of manipulating the nucleosome. Nature 577, 426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffreda A, Jacchetti E, Antunes S, Rainone P, Daniele T, Morisaki T, Bianchi ME, Tacchetti C, and Mazza D. (2017). Live-cell p53 single-molecule binding is modulated by C-terminal acetylation and correlates with transcriptional activity. Nat Commun 8, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- Malik I, Qiu C, Snavely T, and Kaplan CD (2017). Wide-ranging and unexpected consequences of altered Pol II catalytic activity in vivo. Nucleic Acids Res 45, 4431–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BJE, Chruscicki AT, and Howe LJ (2018). Transcription Promotes the Interaction of the FAcilitates Chromatin Transactions (FACT) Complex with Nucleosomes in Saccharomyces cerevisiae. Genetics 210, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PB, and Struhl K. (2003). The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Mol Cell Biol 23, 8323–8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PB, and Struhl K. (2005). Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell 17, 831–840. [DOI] [PubMed] [Google Scholar]
- Mazza D, Stasevich TJ, Karpova TS, and McNally JG (2012). Monitoring dynamic binding of chromatin proteins in vivo by fluorescence correlation spectroscopy and temporal image correlation spectroscopy. Methods Mol Biol 833, 177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough L, Connell Z, Petersen C, and Formosa T. (2015). The Abundant Histone Chaperones Spt6 and FACT Collaborate to Assemble, Inspect, and Maintain Chromatin Structure in Saccharomyces cerevisiae. Genetics 201, 1031–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LL, Connell Z, Xin H, Studitsky VM, Feofanov AV, Valieva ME, and Formosa T. (2018). Functional roles of the DNA-binding HMGB domain in the histone chaperone FACT in nucleosome reorganization. J Biol Chem 293, 6121–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murawska M., Schauer T., Matsuda A., Wilson MD., Pysik T., Wojcik F., Muir TW., Hiraoka Y., Straub T., and Ladurner AG. (2020). The Chaperone FACT and Histone H2B Ubiquitination Maintain S. pombe Genome Architecture through Genic and Subtelomeric Functions. Mol Cell 77, 501–513 e507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesher E, Safina A, Aljahdali I, Portwood S, Wang ES, Koman I, Wang J, and Gurova KV (2018). Role of Chromatin Damage and Chromatin Trapping of FACT in Mediating the Anticancer Cytotoxicity of DNA-Binding Small-Molecule Drugs. Cancer Res 78, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VQ, Ranjan A, Liu S, Tang X, Ling YH, Wisniewski J, Mizuguchi G, Li KY, Jou V, Zheng Q, et al. (2020). Spatio-Temporal Coordination of Transcription Preinitiation Complex Assembly in Live Cells. bioRxiv, 2020.2012.2030.424853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet M, Scafe C, Sexton J, and Young R. (1987). Eucaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol Cell Biol 7, 1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak R, Singh P, Ananthakrishnan S, Adamczyk S, Schimmel O, and Govind CK (2018). Acetylation-Dependent Recruitment of the FACT Complex and Its Role in Regulating Pol II Occupancy Genome-Wide in Saccharomyces cerevisiae. Genetics 209, 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Chavez S, and Perez-Ortin JE (2010). A complete set of nascent transcription rates for yeast genes. PLoS One 5, e15442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, and Steinmetz LM (2013). Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Ahmad K, and Henikoff S. (2017). Transcription and Remodeling Produce Asymmetrically Unwrapped Nucleosomal Intermediates. Mol Cell 68, 1038–1053 e1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ (2010). Genome-wide mapping of nucleosomes in yeast. Methods Enzymol 470, 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan A, Nguyen VQ, Liu S, Wisniewski J, Kim JM, Tang X, Mizuguchi G, Elalaoui E, Nickels TJ, Jou V, et al. (2020). Live-cell single particle imaging reveals the role of RNA polymerase II in histone H2A.Z eviction. eLife 9, e55667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. (2000). Genome-wide location and function of DNA binding proteins. Science 290, 2306–2309. [DOI] [PubMed] [Google Scholar]
- Rossi MJ, Lai WKM, and Pugh BF (2018). Simplified ChIP-exo assays. Nat Commun 9, 2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruone S, Rhoades AR, and Formosa T. (2003). Multiple Nhp6 molecules are required to recruit Spt16-Pob3 to form yFACT complexes and to reorganize nucleosomes. J Biol Chem 278, 45288–45295. [DOI] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview--extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Schneider BL, Seufert W, Steiner B, Yang QH, and Futcher AB (1995). Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast 11, 1265–1274. [DOI] [PubMed] [Google Scholar]
- Schwabish MA, and Struhl K. (2004). Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol Cell Biol 24, 10111–10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sdano MA, Fulcher JM, Palani S, Chandrasekharan MB, Parnell TJ, Whitby FG, Formosa T, and Hill CP (2017). A novel SH2 recognition mechanism recruits Spt6 to the doubly phosphorylated RNA polymerase II linker at sites of transcription. eLife 6, e28723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R, Kaja A, Ferdoush J, Lahudkar S, Barman P, and Bhaumik SR (2017). An mRNA Capping Enzyme Targets FACT to the Active Gene To Enhance the Engagement of RNA Polymerase II into Transcriptional Elongation. Mol Cell Biol 37, e00029–00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty A, Kallgren SP, Demel C, Maier KC, Spatt D, Alver BH, Cramer P, Park PJ, and Winston F. (2017). Spt5 Plays Vital Roles in the Control of Sense and Antisense Transcription Elongation. Mol Cell 66, 77–88 e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrout PE, and Bolger N. (2002). Mediation in experimental and nonexperimental studies: new procedures and recommendations. Psychol Methods 7, 422–445. [PubMed] [Google Scholar]
- Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, Hartzog GA, and Arndt KM (2003). Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J 22, 1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart SK., Mackintosh SG., Edmondson RD., Taverna SD., and Tackett AJ. (2009). Mapping the local protein interactome of the NuA3 histone acetyltransferase. Protein Sci 18, 1987–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, Rondon AG, Aguilera A, Struhl K, Reed R, et al. (2002). TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417, 304–308. [DOI] [PubMed] [Google Scholar]
- Takase Y, Takagi T, Komarnitsky PB, and Buratowski S. (2000). The essential interaction between yeast mRNA capping enzyme subunits is not required for triphosphatase function in vivo. Mol Cell Biol 20, 9307–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatavosian R, Duc HN, Huynh TN, Fang D, Schmitt B, Shi X, Deng Y, Phiel C, Yao T, Zhang Z, et al. (2018). Live-cell single-molecule dynamics of PcG proteins imposed by the DIPG H3.3K27M mutation. Nat Commun 9, 2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarz P, Santos-Rosa H, Robson SC, Sylvestersen KB, Nelson CJ, Nielsen ML, and Kouzarides T. (2014). Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature 505, 564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettey TT, Gao X, Shao W, Li H, Story BA, Chitsazan AD, Glaser RL, Goode ZH, Seidel CW, Conaway RC, et al. (2019). A Role for FACT in RNA Polymerase II Promoter-Proximal Pausing. Cell Rep 27, 3770–3779 e3777. [DOI] [PubMed] [Google Scholar]
- Teves SS, Weber CM, and Henikoff S. (2014). Transcribing through the nucleosome. Trends Biochem Sci 39, 577–586. [DOI] [PubMed] [Google Scholar]
- Tsunaka Y, Fujiwara Y, Oyama T, Hirose S, and Morikawa K. (2016). Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev 30, 673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallotton P, and Olivier S. (2013). Tri-track: free software for large-scale particle tracking. Microsc Microanal 19, 451–460. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, and Speleman F. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, and Workman JL (2015). Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16, 178–189. [DOI] [PubMed] [Google Scholar]
- Vinayachandran V, Reja R, Rossi MJ, Park B, Rieber L, Mittal C, Mahony S, and Pugh BF (2018). Widespread and precise reprogramming of yeast protein-genome interactions in response to heat shock. Genome Res 28, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Liu Y., Edwards G., Krzizike D., Scherman H., and Luger K. (2018). The histone chaperone FACT modulates nucleosome structure by tethering its components. Life Sci Alliance 1, e201800107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KH, Jin Y, and Struhl K. (2014). TFIIH phosphorylation of the Pol II CTD stimulates mediator dissociation from the preinitiation complex and promoter escape. Mol Cell 54, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Eriksson P, Bhoite LT, and Stillman DJ (2003). Regulation of TATA-binding protein binding by the SAGA complex and the Nhp6 high-mobility group protein. Mol Cell Biol 23, 1910–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Ayala AX, Chung I, Weigel AV, Ranjan A, Falco N, Grimm JB, Tkachuk AN, Wu C, Lippincott-Schwartz J, et al. (2019). Rational Design of Fluorogenic and Spontaneously Blinking Labels for Super-Resolution Imaging. ACS Cent Sci 5, 1602–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Animation of the “inchworm-like” model of FACT recruitment spreading for the WT case. The left panel shows a representation of the regions of DNA protected from MNase digestion by FACT complexes for 100 realisations of a simulation across a synthetic gene. Bars represent FACT complexes, the x-axis is location in the gene and each track on the y-axis represents an individual realisation (tracks are separated by blank spaces). Bars are colour-coded according to the state of the FACT complex: bright green for extending, dark green for fully extended, bright blue for retracting and dark blue for fully retracted. The right panel shows the density of fragments, of given sizes (y-axis) and locations (x-axis), protected from MNase digestion by FACT complexes as they are sampled from the simulation. Parameters of the simulation are as for the WT case as in the main text (Figure 6B and Table 1) except that the number of time steps shown is 4×105, there are 100 realisations and there is no relaxation period prior to measurements for the protected fragments being taken. Related to Figure 6.
Movie S2. Animation of the “inchworm-like” model of FACT recruitment spreading for the model 1 chd1Δ case (same as WT except for a reduced extension rate and increase unbinding of the FACT complex). The left panel shows a representation of the regions of DNA protected from MNase digestion by FACT complexes for 100 realisations of a simulation across a synthetic gene. Bars represent FACT complexes, the x-axis is location in the gene and each track on the y-axis represents an individual realisation (tracks are separated by blank spaces). Bars are colour-coded according to the state of the FACT complex: bright green for extending, dark green for fully extended, bright blue for retracting and dark blue for fully retracted. The right panel shows the density of fragments, of given sizes (y-axis) and locations (x-axis), protected from MNase digestion by FACT complexes as they are sampled from the simulation. Parameters of the simulation are as for the chd1Δ case as in the main text (Figure 6C and Table 1) except that the number of time steps shown is 4×105, there are 100 realisations and there is no relaxation period prior to measurements for the protected fragments being taken. Related to Figure 6.
Movie S3. Animation of the “inchworm-like” model of FACT recruitment spreading for the model 2 chd1Δ case (same as WT except for increase unbinding and the possibility of the FACT complex stalling while in the extending phase). The left panel shows a representation of the regions of DNA protected from MNase digestion by FACT complexes for 100 realisations of a simulation across a synthetic gene. Bars represent FACT complexes, the x-axis is location in the gene and each track on the y-axis represents an individual realisation (tracks are separated by blank spaces). Bars are colour-coded according to the state of the FACT complex: bright green for extending, dark green for fully extended, bright blue for retracting, dark blue for fully retracted and red for stalled. The right panel shows the density of fragments, of given sizes (y-axis) and locations (x-axis), protected from MNase digestion by FACT complexes as they are sampled from the simulation. Parameters of the simulation are as for the chd1Δ case as in the main text (Figure 6D and Table 1) except that the number of time steps shown is 4×105, there are 100 realisations and there is no relaxation period prior to measurements for the protected fragments being taken. Related to Figure 6.
Table S3. List of genes used in different figures in this study. Related to STAR Methods.
Data Availability Statement
The microarray (ChIP-chip) and DNA sequencing (ChIP-exo and MNase-ChIP-seq) data generated during this study are available at the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE155144.
The single-molecule tracking data generated during this study are available in Mendeley Data, DOI: 10.17632/rnf4nc3g6y.1 (https://data.mendeley.com/datasets/rnf4nc3g6y/1).
The in-house scripts used to generate some of the analyses during this study are available at https://github.com/francoisrobertlab.
The code related to the mathematical modeling of the MNase-ChIP-seq data is available at https://github.com/aangel-code/FACT-inchworm-model.