

Abstract
Identifying the genetic basis of adaptation is a central goal of evolutionary biology. However, identifying genes and mutations affecting fitness remains challenging because a large number of traits and variants can influence fitness. Selected phenotypes can also be difficult to know a priori, complicating top–down genetic approaches for trait mapping that involve crosses or genome-wide association studies. In such cases, experimental genetic approaches, where one maps fitness directly and attempts to infer the traits involved afterwards, can be valuable. Here, we re-analyse data from a transplant experiment involving Timema stick insects, where five physically clustered single-nucleotide polymorphisms associated with cryptic body coloration were shown to interact to affect survival. Our analysis covers a larger genomic region than past work and revealed a locus previously not identified as associated with survival. This locus resides near a gene, Punch (Pu), involved in pteridine pigments production, implying that it could be associated with an unmeasured coloration trait. However, by combining previous and newly obtained phenotypic data, we show that this trait is not eye or body coloration. We discuss the implications of our results for the discovery of traits, genes and mutations associated with fitness in other systems, as well as for supergene evolution.
This article is part of the theme issue ‘Genetic basis of adaptation and speciation: from loci to causative mutations’.
Keywords: epistasis, survival, genetic mapping, unmeasured traits, inversion, supergene
1. Background
The identification of adaptive mutations is a long-standing goal of evolutionary biology. This goal is important because such mutations represent the ultimate source for evolutionary change and affect the dynamics of evolution. In this regard, theory predicts that the rate and dynamics of adaptation are affected by properties of selected mutations, particularly their effect sizes, and pleiotropic and epistatic effects [1–4]. Specifically, in the absence of gene flow, mutations fixed by natural selection as populations adapt to constant selection pressures through time are expected to have exponentially smaller effect [3], and to display intermediate level of pleiotropy and epistasis [4]. The fixation of mutations with high levels of pleiotropy or epistasis might constrain adaptation and prevent a population from reaching its fitness optimum. Recent work has also explored how gene flow affects these predictions [5–7]. A characterization of many selected mutations is thus necessary to test the expectations of theory and constitutes an important step towards predicting evolutionary outcomes in nature [8].
With recent advances in sequencing technologies, genes associated with selected traits have been identified in many systems, with causal mutations even being identified in some systems. Examples include genes and mutations affecting coat colour in the deer mouse Peromyscus maniculatus (Agouti; ΔSer mutation) [8], defensive body armour and pelvic apparatus in the three-spined stickleback Gasterosteus aculeatus (Eda and Pitx1, respectively; recurrent deletion of the Pel-501 bp enhancer in Pitx1) [9,10] and flowering time in the mouse-ear cress Arabidopsis thaliana (Frigida; multiple mutations) [11,12]. Despite these discoveries, identifying genes and mutations underlying adaptation remains challenging in most systems.
A common approach to identify such genes and mutations (for simplicity we refer to these as ‘genes’ hereafter, with the understanding that identification of causal mutations is desirable) begins with the identification of a selected trait, followed by dissection of its genetic basis, usually through crosses or genome-wide association mapping (GWA hereafter). We refer to this methodology as the top–down genetic approach. Verifying the causal effects of genes on selected traits can then be accomplished through functional genetics (e.g. using CRISPR-Cas 9 or other molecular manipulative tools) [13]. While useful, application of top–down genetic approaches to many systems is challenging because the traits associated with fitness variation are not known or are difficult to detect. For example, in the fruit fly Drosophila melanogaster and the mosquito Anopheles gambiae, adaptation to environmental clines can involve behavioural, physiological and phenological traits that cannot be directly observed and that require time-consuming or specific methodologies to measure [14–16]. Moreover, because adaptation is expected to involve multiple traits, identifying all the traits associated with fitness is still challenging even in systems where a subset of traits is known to be associated with fitness variation [17]. To circumvent the problems associated with a top–down strategy, another approach, the bottom–up genetic approach, begins by using genome scans of natural populations to detect associations between genes and environmental variables [18,19]. Following this initial step, the traits associated with these genes can then be identified through analysis of the molecular function of these genes, or through functional genetics by knocking out these genes and looking at resulting phenotypic changes [20].
Another approach, mixing elements of the top–down and bottom–up approaches, consists in starting with a manipulative field experiment. In this case, rather than surveying different populations to detect genetically diverged regions and genes in the genome, individuals from one environment are transplanted to another environment to identify loci displaying statistically significant changes between initial source and surviving transplant samples. Here, the initial analysis aims to identify associations between genes and the inclusive phenotype of survival or fitness, rather than correlations with particular environmental variables. After this initial analysis, it is then possible to identify the traits under selection through the molecular function associated with these genes and/or functional genomics (as described in the above paragraph). One advantage of such an experimental approach is that it may be less susceptible to spurious associations than genome scans, in particular, when the natural populations being surveyed are geographically or demographically structured [18]. This approach is, however, more time-consuming to implement than genome scans and only informs about adaptation happening in a particular population, over a limited time scale. Examples of such experiments have now been carried out in the deer mouse P. maniculatus, the three-spined stickleback G. aculeatus, Rhagoletis flies, Timema stick insects and A. thaliana [8,21–24].
One consideration potentially complicating the analysis of transplant experiments is that, depending on the selection regime experienced by the individuals, genes may often interact with each other to affect fitness (i.e. epistasis for fitness), even if they have additive effects on selected traits (figure 1a) [25]. Indeed, for nonlinear selection regimes (e.g. stabilizing selection and disruptive selection), or selection acting on trait combinations (e.g. correlational selection), epistasis for fitness is expected. This is because under such selection regimes, the fitness effects of a mutation that additively increases a trait value (e.g. body length) will depend on whether the mutation occurs in a genetic background where it moves the phenotype closer to or further from a fitness peak (figure 1a). In other words, the same mutation can have different (sometimes opposite) effects on fitness depending on the genetic background it resides in (figure 1a). But detecting epistasis is computationally challenging. For example, testing for interactions across all possible pairwise combinations for 1 million single-nucleotide polymorphisms (SNPs hereafter) requires assessing a total of 499 999.5 million interactions (). One method developed to overcome this problem, implemented in the software LT-MAPIT [26,27], does not focus on identifying significant interactions between pairs of SNPs but rather quantifies interaction effects between a given SNP and all other SNPs included in the analysis (termed marginal epistasis). LT-MAPIT thus provides a single test of marginal epistasis per SNP and drastically reduces the computational burden associated with epistasis analyses [26,27].
Figure 1.

(a) Disruptive selection on a trait with an additive genetic basis generates epistasis for fitness. The trait is controlled by four genes, each gene having two alleles (denoted here as plus and minus signs). For simplicity, we represented the situation for a haploid organism, but the same property would emerge for diploid organisms. The pluses and minuses represent the relative effects of alleles on the trait (i.e. plus alleles increase the trait value and minus alleles decrease it). The fitness effect of a particular allele depends on the alleles present at other genes (i.e. epistasis for fitness). For example, a plus allele is detrimental when accompanied by three minus alleles because it will move the mean phenotype towards a larger trait value, away from the extreme and associated with lower fitness. By contrast, the same plus allele will be favoured when accompanied by at least two more plus alleles. Figure modified and redrawn from [25]. (b) An inversion located on linkage group 8 is associated with cryptic body colouration in Timema cristinae (i.e. the Mel-Stripe locus depicted here). Body colouration loci are deleted in T. cristinae green haplotypes (indel locus). The deletion is located in the vicinity of a breakpoint of the inversion. The existence of other loci associated with divergently selected traits within Mel-Stripe, but away from the indel locus, is yet to be tested and is the core objective of the current study. Mel-Stripe is estimated to have a length of at least 13.4 megabase pairs, the indel locus approximately 1.4 megabase pairs. (Online version in colour.)
In the present study, we use a manipulative field experiment to identify genes associated with survival in Timema stick insects. Specifically, we re-analyse survival data from a previous mark-release-and-recapture transplant experiment in Timema chumash stick insects [23], employing LT-MAPIT to determine if we may have missed loci contributing to fitness due to the focus of previous work on a single narrow genetic region controlling body cryptic coloration [23]. Timema stick insects are a genus of wingless herbivorous insects that rely on cryptic body coloration to escape visual predators such as birds and lizards [28,29]. In many Timema species, individuals exist with green or grey/brown (i.e. melanistic) body coloration, making them, respectively, more camouflaged on the leaves or stems of their host-plants [28,–30]. In the transplant experiment, marked T. chumash of a single population were moved from Mountain Mahogany (Cercocarpus) to a combination of two host-plant species present in the same geographical area (Adenostoma and Ceanothus). These two host-plant species generated correlational selection on cryptic body coloration, favouring very green or very brown individuals and selecting against intermediate body coloration. Before release, a single leg was dissected from all the experimental individuals in order to genotype them at markers across their genome. In past work, five SNPs in close proximity to each other (in an approx. 1 megabase pair genomic region; referred as the indel locus hereafter; see below for details) on linkage group eight (LG8 hereafter) were found to be associated with cryptic body coloration and to interact with each other to explain survival in the transplant environment. Whether additional loci outside of the indel locus affect survival was not tested and is thus our focus here.
There are a priori reasons to suspect that such loci may exist outside of the indel locus. In several species of the genus (i.e. T. californicum, T. cristinae, T. landelsensis, T. petita and T. poppensis), the genomic region harbouring the five aforementioned body coloration SNPs is deleted in green haplotypes but present in the brown haplotype [30]. Interestingly, this deletion is associated with an approximately 10.5 megabase pair inversion (referred as the Mel-Stripe locus hereafter) in T. cristinae [23,30], raising the possibility that genes controlling variation of undetected selected traits reside within the Mel-Stripe locus but away from the indel locus (figure 1b). If so, then this finding would inform different non-exclusive hypotheses for why inversions are selected for [31]. Indeed, inversions can either be selected for because of the advantage of a breakpoint mutation [32,33], or because they strongly reduce recombination between alleles at different genes they contain, thus helping maintain favoured allelic combinations [31,33,34]. These mechanisms could also act in conjunction, as might occur in T. cristinae [31].
To accomplish our goal, we first performed a ‘traditional’ GWA mapping analysis (i.e. not accounting for epistatic effects) on survival using SNPs within the Mel-Stripe locus, which yielded limited evidence for genetic associations with survival. We next tested for epistasis for survival within the Mel-Stripe locus using LT-MAPIT and identified two loci associated with survival. One of these loci was previously known, is located within the indel locus and contains the gene Scarlet (st) which we previously hypothesized to be associated with cryptic body coloration in Timema [30]. The other previously unidentified locus is away from the indel locus and contains two interesting genes, Chitinase 5 (Cht5) and Punch (Pu). Chitinases are associated with cold or heat stress tolerance in several insect species [35,36] suggesting that this locus could be associated with heat tolerance in Timema. However, the most intriguing candidate, Punch, controls the first step of pteridine pigments production and is associated with eye and body coloration in many insect species [37–40]. This led us to hypothesize that this locus could be primarily associated with eye colour variation in T. chumash. We therefore collected new data on eye coloration from photographs taken of the individuals used in the transplant experiment and then performed ‘traditional’ GWA mapping for this trait. Eye coloration mapped to the indel locus and, as we show below, eye coloration and cryptic body coloration are strongly genetically correlated in T. chumash. No association was detected, however, between eye or body coloration and the region containing and surrounding the gene Punch. Our combined results therefore appear to refute the hypothesis that our measured coloration traits were the target of selection associated with the SNP residing near Punch in the transplant experiment.
Nevertheless, our results indicate that at least one selected locus, whether Punch, Chitinase 5 or some other gene, likely resides within Mel-Stripe, away from the indel locus. We discuss the general implications of this finding and how methods such as those employed here could facilitate the detection of traits, genes and ultimately causal variants associated with fitness in the wild in other organisms.
2. Methods
(a) . Transplant experiment with Timema chumash
Full details concerning the transplant experiment with T. chumash are described in a previous publication [23]. We provide a brief overview of the relevant information for the current study here. Over 700 insects were collected from a single natural population (Angeles National Forest, CA, HF5 34° 15.584′ N, 118° 6.254′ W), on the host-plant Mountain Mahogany (Cercocarpus sp.), from which we selected 437 healthy adults for use in the transplant experiment. We gave all selected individuals a unique id number, photographed them, gave them an individual mark on the ventral side using Sharpie pens (i.e. dots of different colour combinations) and released them back into the area from which they were collected in one of two host-plants treatments (i.e. different host-plant species dominating the vegetation in this population; details below). Before release, we took a leg (i.e. tissue sample) from each transplanted individual for DNA sequencing purposes. In the first treatment, we released 219 individuals onto isolated vegetation patches composed of intertwined plant individuals, one of each of two plant species (Ceanothus sp. and Adenostoma sp.; referred as AC treatment hereafter). In the second treatment, we released 218 individuals onto an isolated Mountain Mahogany host-plant (Cercocarpus sp.; referred as MM treatment hereafter). We recaptured surviving individuals approximately 72 h after release. Past studies with similar experimental design have shown that dispersal of Timema across bare ground is essentially non-existent such that recapture is a good proxy for survival [41,42].
For all analyses except for GWA of body and eye coloration, we only used data from the AC treatment, as this is the only treatment where past work found evidence for correlational selection on cryptic body coloration [23]; thus epistasis for fitness is only strongly expected in the AC treatment. However, eye and body coloration can be measured independently from treatment. Thus, for GWA on eye and body coloration, we used data both from the AC and MM treatments.
We here re-analyse published genomic data from the transplanted individuals. These data are published and were generated in the aforementioned past study [23] using a standard genotyping-by-sequencing approach with two restriction enzymes (i.e. ddRAD) [43]. Details concerning filtering, read alignment and variant calling are described in past work [23]. For the current study, we generated new data on eye coloration and conducted novel analyses of the genetic basis of this trait.
(b) . Association mapping for survival within the Mel-Stripe locus using GEMMA
We first quantified associations between genotypes at bi-allelic SNPs (we also used only bi-allelic SNPs for all subsequent analyses) within the Mel-Stripe locus and survival using a mapping approach that does not explicitly consider epistasis, implemented in the software GEMMA [44,45]. For these analyses, we excluded SNPs with a minor allele frequency less than 0.01 and fit a probit Bayesian sparse linear mixed model. We set five MCMC chains with the following parameters: a burnin of 1 million iterations, a run of 3 million iterations and a record every hundred iterations. Following past work, we calculated posterior probabilities for all model estimates from the combined output of the five MCMC chains [23,28,30].
(c) . Estimating marginal epistasis for survival within the Mel-Stripe locus using LT-MAPIT
We tested for SNPs that exhibit epistatic effects on survival (i.e. interact with other SNPs) using the software LT-MAPIT [26,27]. Briefly, this method detects SNPs with non-zero marginal epistatic effects defined as the combined pairwise interaction effects between a given focal SNP and all other SNPs included in the same analysis [26]. LT-MAPIT was originally designed for case-control studies and is therefore an appropriate method to use to analyse our binary survival data. We set the disease prevalence parameter in LT-MAPIT as the survival empirically observed in the transplant experiment (51 recaptured individuals/219 released individuals = 23.28%). We conducted additional analyses that consider individual pairs of SNPs using different methods, as described below.
(d) . Finding Drosophila melanogaster homologs for genes in the vicinity of LT-MAPIT outlier single-nucleotide polymorphism 2
We attempted to inform which potential traits might be associated with LT-MAPIT outlier SNP 2 by selecting all predicted genes located within 200 kilobase pairs from this outlier SNP and looking for their homologs (if any) in the D. melanogaster genome. We then searched for described phenotypic effects of these homologs in D. melanogaster and other insects, which allowed us to hypothesize what trait(s) might be associated with these genes in Timema. Specifically, we identified D. melanogaster homologs for our predicted genes with the blastn function on the NCBI website (https://blast.ncbi.nlm.nih.gov/Blast.cgi%23) [46] using only the coding sequence of our predicted genes as a query and restricting our search to D. melanogaster sequences only (taxid:7227). We obtained the coding sequence of our predicted genes of interest from our 1.3c2 T. cristinae reference genome and annotation [30,47] using the gestfasta function from the bedtools software (bedtools v. 2.28.0; see https://bedtools.readthedocs.io/en/latest/). If we successfully identified a homolog in D. melanogaster for our predicted genes of interest, we then looked for molecular function and phenotypic effects of the D. melanogaster homolog genes in flybase (https://flybase.org/) [38] and searched in the literature for phenotypic effects of these homologs in other insects.
(e) . Eye coloration measurements from photographs
Following past work where we measured body coloration from photographs of insects used in the transplant experiment [23], we corrected raw photographs (i.e. .NEF format) taken during the experiment for temperature (set at 6150 K) in the software RawTherapee (v. 5.8; https://www.rawtherapee.com/) and exported them as JPEG images. We scored eye coloration from these JPEG images using ImageJ [48] (v. 1.52r; https://imagej.nih.gov/ij/) circling the right eye (when not possible, we measured the left eye) with the polygon tools and using the Colour Histogram add-on (electronic supplementary material, figure S1). Following past work, we measured the RGB colour channels (red, green and blue) and processed them following [49] to obtain RG and GB estimates (the ratio of red over green and the ratio of green over blue, respectively) [23,28,30]. As for body coloration, we therefore studied two eye coloration traits: the RG and GB estimates we described above.
(f) . Genome-wide association mapping for eye and body coloration
We conducted GWA on the new eye coloration traits using GEMMA [44,45] and also on body coloration traits to allow eventual estimation of the genetic correlation between eye and body coloration (details below). For this, we fit a Bayesian sparse linear mixed model using the same parameters described above for survival.
(g) . Estimating the number of unlinked genetic variants (i.e. quantitative trait nucleotide) for eye coloration within the indel locus
We followed past work to obtain the total number of genetic variants affecting eye coloration within the indel locus using our GEMMA models [30]. Briefly, GEMMA outputs a posterior inclusion probability (PIP) value for each SNP, which corresponds to the proportion of recorded MCMC steps in which the SNP was found to have a measurable effect on the phenotype. PIP values are therefore bounded between 0 (the SNP was never found to have a measurable effect on the phenotype) and 1 (the SNP was found to always have a measurable effect on the phenotype), and represent a measure of the weight of evidence for trait–genotype association. One can therefore estimate the number of causal variants affecting each trait in a genomic region by summing the PIPs for all SNPs in that region. For example, for a polygenic trait with recombination among loci, the one or few SNPs that best tag each causal variant are expected to consistently be associated with the trait across MCMC steps (i.e. exhibit high PIP values). Thus, PIPs across such SNPs sum to an estimate of the number of total causal variants. We emphasize that this approach for estimating the number of quantitative trait nucleotides (QTN), by virtue of relying on linkage disequilibrium and the mechanics of the GEMMA model, works even if the casual variants themselves are not unambiguously identified.
These analyses revealed that some SNPs within the indel locus are associated with both eye coloration traits (RG and GB), due to pleiotropy or close genetic proximity (i.e. high linkage disequilibrium), potentially inflating our estimate of variant number. We therefore corrected our estimate for the number of total causal variants affecting eye coloration within the indel locus with the following method. For each SNP within the indel locus, we summed its PIP values for both RG and GB. If this summed value was above one, we set it to one. We then summed these values over all SNPs within the indel locus. Our corrected estimate is certainly an underestimate of the true number of variants within the indel locus; the real number of unlinked variants will be somewhere in between the corrected estimate and the uncorrected estimate. Nonetheless, by following these approaches, we were able to estimate reasonable bounds on the range of number of QTN.
(h) . Genetic correlation between eye and body coloration
We estimated the genetic correlation between eye and body coloration using polygenic scores estimated with GEMMA's ‘predict’ option [44,45]. Specifically, for each trait, we masked the phenotype of a quarter of the sampled individuals (i.e. 109 individuals) and ran a Bayesian sparse linear mixed model GWA mapping with one MCMC chain for the remaining individuals (the same parameters were used as for survival described above). We repeated this process four times for each trait, allowing us to get predicted phenotypic values (i.e. polygenic scores) for each individual. The correlation between predicted phenotypic values for eye and body coloration (i.e. the genetic correlation) was estimated using Pearson's correlation coefficient.
(i) . Comparing the genetic bases of eye and body coloration
To test if eye and body coloration might be controlled by similar genetic regions, we compared the lists of the most highly associated SNPs for each trait, between eye and body coloration traits (RG and GB). Specifically, because GEMMA analyses indicated that most traits were controlled by approximately 10 SNPs, we selected the 10 most-associated SNPs for each trait and looked for intersections between these lists (i.e. SNPs present in both lists).
To test if the observed frequency of sharing/overlap of the most-associated SNPs between eye and body coloration traits could arise by chance, we generated a null distribution of overlap expected under random sampling. Specifically, we sampled 10 items from an ensemble with a number of elements (i.e. cardinality) similar to the total number of input SNPs in our GEMMA analysis. We repeated this operation to obtain a second sample and recorded the number of items picked in both samples. We repeated these two operations a million times to obtain the expected distribution of shared elements in two samples under random sampling. We compared the observed number of shared SNPs to this null distribution to obtain a p-value.
(j) . Quantifying our ability to predict survival based on LT-MAPIT outlier single-nucleotide polymorphisms
We tested whether allowing for epistatic interactions between LT-MAPIT outlier SNPs and other SNPs within Mel-Stripe improved our ability to predict survival. Indeed, one can potentially detect loci whose epistatic effects on survival are so small that they might not help in improving our ability to predict survival from genomic data. This does not necessarily mean that these loci are falsely associated, but that the variation in survival they explain might just not be enough to improve our predictive power. To determine this, we fit binomial generalized linear models with Bayesian model averaging. Ten-fold cross-validation was used to assess predictive performance for the full model (with epistasis; model 1—see below) and reduced model (without epistasis; model 2—see below) while averaging predictions of survival over sub-models including different subsets of covariates. We fit these models using the bic.glm function in the R BMA package (BMA v. 3.18.15) [50]. We assigned all covariates prior inclusion probabilities of 0.5 (i.e. equally likely to be in or left out of the model). For cross-validation, each observation was left out of one of the 10 training sets. Specifically, we tested the following full (with epistasis; model 1) and reduced (without epistasis; model 2) models:
and
where β0 = a constant, β1–6 = coefficients for different terms in the model, outlier1i = genotype estimate at the LT-MAPIT outlier SNP 1 (near the st gene) for individual i, outlier2i = genotype estimate at the LT-MAPIT outlier SNP 2 (near the Punch gene) for individual i, PCA1i = value on the first axis from a PCA realized on all SNPs within the Mel-Stripe locus excluding the LT-MAPIT outlier SNPs for individual i.
(k) . Estimation of recombination within the Mel-Stripe locus in Timema cristinae
To further inform the implications of our results on supergene evolution, we specifically analysed the characteristic of the Mel-Stripe inversion in T. cristinae. Specifically we assessed if recombination suppression varied in the Mel-Stripe inversion region in T. cristinae. Indeed, because of double recombination events, recombination suppression between the two inversion haplotypes might be lower in the centre of the inversion than near breakpoints [51,52]. This could have implications for recombination suppression between the putatively selected locus that we located within the inversion (Punch) and the previously identified adaptive breakpoint deletion we estimated in T. cristinae. We therefore estimated linkage disequilibrium (i.e. as a proxy of recombination) in the genomic region surrounding and including the Mel-Stripe inversion in T. cristinae. Specifically, we reanalysed genotyping-by-sequencing data (GBS; double-digestion-restriction-site-associated-DNA libraries) from 602 insects collected in 2013 from a single polymorphic population of T. cristinae (population code FHA; GPS coordinates: 34.52, −119.8) [28,30]. Briefly, we extracted DNA from legs and estimated genotypes at thousands of markers across the genome using a standard genotyping-by-sequencing approach with two restriction enzymes (i.e. ddRAD) [43]. Details concerning filtering, read alignment and variant calling are described in past work [30]. The dataset included 175 918 SNPs with 8 149 SNPs within the two LG8 scaffolds containing Mel-Stripe (702.1 and 128) or the scaffolds directly adjacent to these (2963 and 1845), which we focus on here (this focal region covers approximately 51 megabases, including the approximately 10 megabase Mel-Stripe locus). We first estimated allele frequencies for the SNPs in this dataset using an expectation-maximization algorithm that accounts for uncertainty in genotypes caused by sequence error and finite sequence coverage [53]. This was done with estpEM (v. 0.1) with a tolerance threshold of 0.001 and 40 maximum iterations ([54,55]; DRYAD https://doi.org/10.5061/dryad.nq67q). We then obtained empirical Bayesian estimates of genotypes as gij = L(gij = 0) (1 − pi)2 + L(gij = 1) 2 pi (1 − pi) + L(gij = 2) p2, where gij is the genotype estimate (number of non-reference alleles) for SNP i and individual j, L(·) is the genotype likelihood from samtools/bcftools (as computed in [30]), and pi is the non-reference allele frequency from estpEM. Lastly, we computed linkage disequilibrium for all pairs of SNPs in 100 kilobase windows along the four genome scaffolds considered here, which included all of the Mel-Stripe locus. Linkage disequilibrium (LD hereafter) was measured as the squared genotypic correlation for pairs of SNPs. We used the mean estimate of pairwise LD within each window as our summary of LD for that window.
3. Results
(a) . Association mapping for survival within the Mel-Stripe locus, without epistasis
We first tested for associations between SNPs within Mel-Stripe and survival (figure 2), using a multi-SNP approach that does not account for epistasis. As expected, because of the selective regime imposed by the transplant experiment for cryptic body coloration in the AC treatment (i.e. disruptive/correlational selection for cryptic body coloration) [23], this approach explained little variation in survival (2% of variance explained; 0–23% as 95% equal-tail probability intervals). All SNPs exhibited appreciable PIP, but we did not detect individual SNPs with exceptionally high PIPs (figure 2). This pattern is most likely an artefact of the MCMC approach when using a relatively small number of SNPs and when strong associations do not exist for any SNP with the trait studied. In other words, SNPs are largely redundant (i.e. they each explain little variation) and have a high prior probability to be randomly picked by the MCMC chain over 3 million iterations, leading to somewhat inflated PIPs for all SNPs examined [44,45].
Figure 2.

Association mapping of survival within the Mel-Stripe locus when epistasis is not explicitly considered. We conducted this analysis with the software GEMMA [44,45]. The shaded grey rectangle represents the position of the indel locus within Mel-Stripe. The space between two ticks on the x-axis represents 1 megabase pair. PIP: posterior inclusion probability. The high PIP values of all SNPs (approx. 0.2) are an artefact of the MCMC method and the small number of SNPs tested, all SNPs within Mel-Stripe having some weak association with survival (see main text for further explanation).
(b) . Epistasis for survival within the Mel-Stripe locus
We next tested for evidence of epistasis between SNPs within Mel-Stripe associated with survival using LT-MAPIT [26,27], a method that tests for epistasis between a particular focal SNP and the remaining input SNPs (here all other SNPs within the Mel-Stripe locus; figure 3). This method quantifies interaction effects between the focal SNP and a variable summarizing the remaining genetic variation within Mel-Stripe (i.e. marginal epistasis), in a fashion similar to a principal component axis. From this analysis, we identified five SNPs with nominally significant marginal epistasis (p-value ≤ 0.05), two of which were clear outliers with particularly strong evidence for epistasis (figure 3). One of these outlier SNPs (outlier 1, hereafter) is located within the indel locus, while the other is located within the Mel-Stripe locus but away from the indel locus (outlier 2 hereafter; figure 3).
Figure 3.
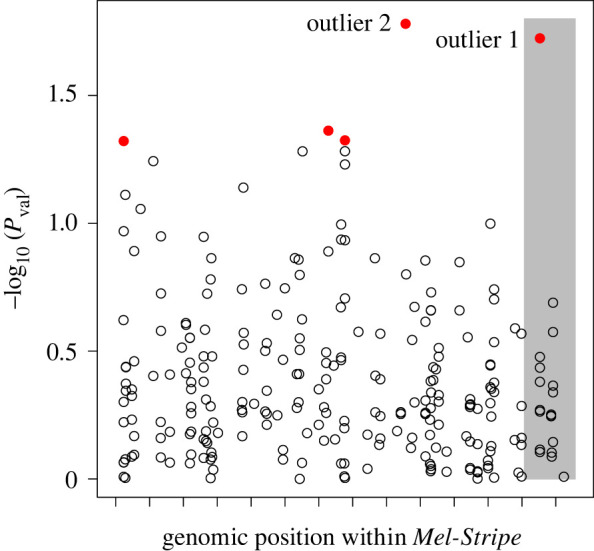
Association mapping of survival within the Mel-Stripe locus, where marginal epistasis is explicitly considered. We conducted this analysis with LT-MAPIT [26,27]. The shaded grey rectangle represents the position of the indel locus within Mel-Stripe. The space between two ticks on the x-axis represents 1 megabase pair. Red dots (solid grey in print) correspond to SNPs having non-zero marginal epistasis for survival with p-value ≤ 0.05. Pval: p value for marginal epistasis test. (Online version in colour.)
(c) . Potential function of the two LT-MAPIT outlier single-nucleotide polymorphisms
We next examined the predicted genes in physical proximity (i.e. located within 200 kilobase pairs) of the two LT-MAPIT outlier SNPs in order to identify candidate genes and traits potentially associated with these two loci (table 1; electronic supplementary material, tables S1 and S2).
Table 1.
Homologs in Drosophila melanogaster for predicted genes located less than 200 kb way from LT-MAPIT outlier SNP 2. Only genes with identified molecular functions were considered here. Tcri: gene number in T. cristinae reference genome 1.3c2; D.mel: homolog gene in Drosophila melanogaster; molecular function in D.mel: molecular function of this gene in D. melanogaster; effects in D.mel: observed phenotypic effects of this gene in D. melanogaster; effects in other insects: observed phenotypic effects of this gene or genes in the same family in other insects species.
| Tcri | D.mel | molecular function in D.mel | effects in D.mel | effects in other insects |
|---|---|---|---|---|
| g6060 | Chitinase 5 (Cht5) | encodes an enzyme involved in the formation of chitin-based extracellular matrix at barrier tissues [38] | lethality [38] | cold/heat tolerance [35,36] |
| g6064 | Punch (Pu) | isoform B is required for eye pigment production, isoform C may be required for normal embryonic development and segment pattern formation [38] | abnormal eye coloration, lethality, sterility [38] | eye and body coloration [40] |
| g6057 | — | — | — | — |
| g6058 | — | — | — | — |
| g6068 | Kramer (Kmr) | predicted to enable phosphatidylinositol biphosphate binding activity, involved in regulation of establishment of planar polarity [38] | lethality, abnormal planar polarity [38] | — |
Outlier SNP 1 is located within the indel locus and situated approximately 56 kilobase pairs from the st gene (predicted gene g6239), coding the protein scarlet. The st gene is known to affect different aspects of coloration in several insect species [56–58] and was one of the prime candidate genes for cryptic body coloration identified in past Timema work [23,30].
Outlier SNP 2 is not within the indel locus, being approximately 3.7 megabase pairs away from it. There are several predicted genes within this region and they exhibit various molecular functions. This includes a chitinase II, a GTP cyclohydrase I enzyme, a TORC2 component and the target of rapamycin complex 2 (table 1; electronic supplementary material, table S2). The predicted gene coding for a chitinase II (g6060) is an intriguing candidate. This gene is located approximately 19 kilobase pairs away from LT-MAPIT outlier 2, is homologous to the Chitinase 5 (Cht5) gene in D. melanogaster and codes for an enzyme involved in the formation of chitin-based extracellular matrix at barrier tissues [38]. Interestingly, enzymes of the same family have been associated with cold or heat tolerance in several insect species [35,36], leading us to hypothesize that LT-MAPIT outlier 2 could be associated with heat tolerance in Timema. Further experiments are yet needed to test this hypothesis.
However, the most intriguing candidate gene (predicted gene g6064) is located approximately 118 kilobase pairs away from outlier 2 and is homologous to the Punch (Pu) gene in D. melanogaster (table 1). This gene codes for a GTP cyclohydrase I enzyme, which is involved in the first step of the production of pteridine pigments in D. melanogaster and other insects, and is associated with eye and body coloration in multiple insect species [37–40,59]. This led us to hypothesize that this SNP could also be associated with T. chumash eye coloration, a trait that we observed to be quite variable but which is previously unstudied in this species (electronic supplementary material, figure S2). We test this latter hypothesis in the following section.
(d) . Genetic basis of eye coloration in Timema chumash
To test our eye coloration hypothesis, we measured eye coloration in all experimental individuals from photographs and conducted a GWA analysis for this trait. Here, because we did not have a strong a priori expectation concerning the genetic architecture of eye coloration, we did not restrict our analysis to the Mel-Stripe locus but instead, tested for associations across the entire genome.
Our models revealed that eye coloration is controlled by a modest number of SNPs (RG: 6 SNPs with detectable effects, range 2 to 19 for 95% equal-tail probability interval, ETPI hereafter; GB: 6 SNPs with detectable effects, range 3 to 16 for 95% ETPI). Genetic variation for loci with measurable phenotypic effects explained a substantial amount of phenotypic variation in our models (obtained by multiplying the proportion of variance in phenotypes explained (PVE) and proportion of genetic variance explained by the sparse effects terms (PGE) hyper-parameters; RG: 51%, range 31% to 76% for 95% ETPI; GB: 49%, range 32% to 68% for 95% ETPI). Our results indicate that SNPs associated with eye coloration are located on different chromosomes (electronic supplementary material, figures S3 and S4); however, SNPs within the indel locus showed the highest associations with eye coloration traits (figure 4). We estimated that the indel locus contained a maximum of four QTN for eye coloration traits (two QTN each, for RG and GB, but these QTN overlapped between coloration traits; the true number of independent QTN is thus likely somewhere between two and four). However, the region surrounding Punch did not display an association with eye coloration traits suggesting that, contrary to our hypothesis, LT-MAPIT outlier SNP 2 is not associated with eye coloration.
Figure 4.

Association mapping of eye coloration within the Mel-Stripe locus, when epistasis is not explicitly considered. (a) RG trait and (b) GB trait. We conducted these analyses with GEMMA [44,45]. The graphics display the results from the Mel-Stripe locus only, but because these traits are being mapped for the first time the overall analysis included genome-wide data. The shaded grey rectangles represent the position of the indel locus within Mel-Stripe. The space between adjacent ticks on the x-axis represents 1 megabase pair. PIP: Posterior inclusion probability. Because of the MCMC approach, GEMMA controls for linkage disequilibrium in the results. Only SNPs explaining trait variance will be consistently retained across MCMC steps in the model.
(e) . Test for shared genetic basis of body and eye coloration
Given that body and eye colorations are at least in part controlled by the indel locus, we tested whether eye and body coloration share similar genetic bases. Indeed, this is expected given that we found here that body and eye coloration are strongly phenotypically correlated (Pearson's correlation coefficients on phenotypic values: RG = 0.87, p-value < 2.2 × 10−16; GB = 0.77, p-value < 2.2 × 10−16; figure 5). Moreover, explicit estimation of the genetic correlation between eye and body coloration traits revealed strong genetic correlations (Pearson's correlation coefficients on polygenic scores: RG = 0.92, p-value < 2.2 × 10−16, GB = 0.88, p-value < 2.2 × 10−16; figure 5).
Figure 5.

Phenotypic and genetic correlations between eye and body coloration traits (RG and GB). (a,b) Phenotypic correlations. (c,d) Genetic correlations, here the trait values where obtained from predictions from genotype in the software GEMMA [44,45]. The black lines represent the fit of a linear model.
Our results indicate that some SNPs were found to be most associated with both eye and body coloration, and that this number of shared SNPs is greater than what can be expected by chance (eye and body RG: five shared SNPs, p-value < 1 × 10−6; eye and body GB: four shared SNPs, p-value < 1 × 10−6). This suggests that genes near these SNPs have pleiotropic effects on both body and eye coloration, or that multiple genes independently controlling body and eye coloration are in close physical proximity.
(f) . Predicting survival based on LT-MAPIT outlier single-nucleotide polymorphisms
Finally, we asked whether allowing for epistatic interactions between LT-MAPIT outlier SNPs and the rest of the genetic variation within Mel-stripe improved our ability to predict survival relative to a model without epistasis.
When fit with all of the observations, we found that the sub-models predicting the best survival for the full (with epistasis) and reduced (without epistasis) models were those that included only an intercept term. These intercept-only sub-models had posterior probabilities of 0.593 and 0.781 for the full and reduced models, respectively. Moreover, posterior probabilities that individual covariates (additive or epistatic effects) affected survival were approximately 10% or less (table 2). Using all of the data for model fitting and prediction, correlations between survival and predicted survival were slightly higher for the model with epistasis (r = 0.125, 95% CI = −0.007–0.254, p = 0.064) than for the model without epistasis (r = 0.092, 95% CI = −0.041–0.222, p = 0.175). However, in both cases, a correlation of 0 could not be strictly rejected.
Table 2.
Summary of parameter estimates from Bayesian model averaging of sub-models with (full model) or without (reduced model) epistasis that were used to predict survival. The covariates included in each model are listed, and the posterior probability that each associated regression parameter is non-zero (Prob ! = 0 column) is given along with the model-averaged point estimate (posterior mean; estimate (s.d.) column) and posterior standard deviation for each coefficient (in parentheses in the estimate (s.d.) column); outlier 1, genotype probability at the LT-MAPIT outlier SNP 1 (near the st gene); outlier 2, genotype probability at the LT-MAPIT outlier SNP 2 (near the Punch gene); PCA1, individual value on the first axis from a PCA realized on all SNPs within the Mel-Stripe locus excluding the LT-MAPIT outlier SNPs; outlier 1 * outlier 2, the interaction between the LT-MAPIT outlier SNPs; outlier 1 * PCA1, the interaction between the outlier 1 SNP and the first axis from a PCA realized on all SNPs within the Mel-Stripe locus excluding both LT-MAPIT outliers SNPs; outlier 2 * PCA1, the interaction between the outlier 2 SNP and the first axis from a PCA realized on all SNPs within the Mel-Stripe locus excluding both LT-MAPIT outlier SNPs.
| covariate | full model (with epistasis) |
reduced model (without epistasis) |
||
|---|---|---|---|---|
| Prob ! = 0 | estimate (s.d.) | Prob ! = 0 | estimate (s.d.) | |
| PCA1 | 0.051 | 0.006 (0.048) | 0.067 | 0.008 (0.055) |
| outlier 1 | 0.075 | 0.012 (0.055) | 0.099 | 0.015 (0.063) |
| outlier 2 | 0.040 | −0.001 (0.032) | 0.053 | −0.001 (0.037) |
| outlier 1 * outlier 2 | 0.070 | 0.018 (0.092) | ||
| outlier 1 * PCA1 | 0.052 | 0.017 (0.121) | ||
| outlier 2 * PCA1 | 0.118 | −0.037 (0.125) | ||
Moreover, when using predictions from cross-validation, an approach that specifically measures predictive performance and avoids over-fitting, we failed to predict survival. Indeed, we observed negative correlations between predicted and observed survival (full model, r = −0.179, 95% CI = −0.305 to −0.048, p = 0.0078; reduced model, r = −0.216, 95% CI = −0.339 to −0.086, p = 0.0013). It therefore appears that we have poor ability to actually predict survival with or without epistatic terms in our model, although we were able to map a portion of its genetic basis. This result is perhaps unsurprising for a complex and integrative trait like survival, but forms a major point of our discussion below.
4. Discussion
We used a manipulative field experiment and survival data to attempt to identify candidate genes associated with fitness in Timema stick insects. In particular, by mapping survival in a transplant experiment with T. chumash and explicitly taking epistasis into account, we detected a genomic region in the Mel-Stripe locus on LG8 not previously known to be associated with survival in Timema. We collected new eye coloration data to try to determine the nature of the phenotype controlled by this region, but showed it was not this particular phenotype. Specifically, although the functional annotation of a gene, Punch, in proximity to the newly discovered SNP associated with survival suggested a possible association with eye coloration, we found no evidence for this in a subsequent GWA analysis of the trait. Thus, the phenotype encoded by this region might still be related to coloration, but an aspect that we did not measure in the study. Alternatively, the presence of the Chitinase 5 gene in this region suggests that survival could have been affected by heat tolerance, a factor known to affect adaptation in multiple insect species [35,36], including Timema [47]. However, we suspect it is unlikely that heat tolerance contributed strongly to mortality during the period of a few days at the same locality where the experimental animals were collected. This raises the interesting possibility that Chitinase 5 has other functions in Timema, perhaps even related to coloration. Our results illustrate the challenges of detecting selected traits, even using approaches such as those employed here. We note that one way to potentially identify the selected trait(s) associated with this region (and in general) could involve genetic manipulations of these two candidate genes using functional tools such as CRISPR-Cas9 and RNAi and looking for any resulting phenotypic changes in transformed individuals [60].
Another aspect of our results is that despite finding evidence that selection is likely acting on a previously unknown locus in the Mel-Stripe locus and one locus in the indel locus in T. chumash, the inclusion of these two LT-MAPIT outlier SNPs, even when including their epistatic effects with other genes across these regions, did not have notable consequences for increasing the predicted survival of insects in the transplant experiment. In this regard, it is important to appreciate that the deterministic component of survival generally represents the sum total of many traits encoded by many genes, often of relatively small effect size, collectively affecting fitness. Thus, while a particular variant may show a significant association with fitness, this does not mean that the mutation will necessarily make a substantial contribution to predicting whether an individual possessing the mutation will survive, given the many other loci and phenotypes that are likely involved. Moreover, this issue of prediction is likely exacerbated by the contribution of random elements to survival (e.g. genetic drift), which can act jointly with selection and on the same genetic regions.
Our results therefore highlight the utility of manipulative experiments for identifying potential genes under selection, but also the challenges that can remain in verifying the specific loci, mutations and phenotypes involved. Nevertheless, we propose that the manipulative approach employed here could be useful for the study of adaptation in many organisms. Indeed, a similar manipulative approach was used in the three-spine stickleback G. aculeatus where marine fishes were transplanted into four experimental freshwater ponds and phenotypic and genetic evolution were tracked for the two subsequent generations [17,21]. This experiment confirmed that reduced defensive body armour is selected for in the freshwater environment, along with its underlying gene (Eda) [17,21]. Interestingly, this experiment also confirmed that defensive body armour is likely not the sole trait controlled by the Eda gene [17], which also appears to influence at least four other selected traits including lateral plate count, neuromast number, neuromast pattern and, to some extent, body shape [61]. All of these traits are genetically correlated due to either pleiotropy, close physical linkage, or their combination within the Eda gene region [61]. The analytical methods employed in the three-spine stickleback studies are well suited for traits experiencing directional selection. The analytical methods we employed in this study are well tailored for traits experiencing nonlinear selection (e.g. stabilizing or disruptive selection), or selection acting in concert on multiple traits (e.g. correlational selection) and therefore constitute a useful addition for the identification of adaptive genes and mutations in natural populations. These methods will be especially useful for the study of balanced polymorphisms: that is, polymorphisms maintained within natural populations because of selective processes [62,63]. This excludes neutral polymorphisms, or transient polymorphisms where one form is in the process of replacing another within the population [63].
Our results also provide insight into the evolution of genetic architecture. Specifically, we here provide the first evidence in Timema for a selective locus residing within the Mel-Stripe locus but away from the indel locus (figure 1b). This finding sheds new light on the chromosomal inversion associated with colour morphs in T. cristinae [30,64], which spans the Mel-Stripe locus (and based on patterns of LD appears to suppress recombination fairly evenly throughout this locus; electronic supplementary material, figure S5), and even more generally, on regions of suppressed recombination on LG8 that extend beyond the indel locus (these regions of suppressed recombination appear widespread in Timema, although direct evidence for inversions in other Timema species awaits further data [30]). In T. cristinae, the selective advantage of the Mel-Stripe inversion may involve the combination of the deletion at one breakpoint affecting body coloration [30], and another locus (potentially Punch) within the inversion. If true, then two possible scenarios could account for the evolution of this inversion in T. cristinae. In the first scenario, the inversion might have initially been selected because of the adaptive breakpoint mutation, with genetic variation at the second locus evolving afterwards. Such a ‘breakpoint first’ scenario is conceptually similar to models describing the accumulation of genetic incompatibilities in inversions after their formation proposed by Navarro and Barton in a model of parapatric divergence [65]. In the second scenario, the inversion may have simultaneously trapped pre-existing genetic variation within the Mel-Stripe locus with a newly generated adaptive breakpoint mutation, which shares some conceptual similarities with the local adaptation scenario for the spread of inversions proposed by Kirkpatrick & Barton [66], and modified by Feder and colleagues to allow for allopatry and secondary contact [67]. These scenarios expand upon the conditions under which inversions may contribute to adaptation, the most well-known being the ability for inversions to spread because of their effects on suppressing recombination and maintaining favourable allelic combinations (i.e. keeping such combinations intact [66]). Distinguishing between the two scenarios noted above in Timema will now be important to evaluate the contribution and order of evolution of mutations and genome rearrangement in adaptation. Future work in T. cristinae should allow such characterization, specifically by independently dating the inversion and the adaptive genetic variation it contains [31].
In conclusion, our study highlights that manipulative experiments can be useful to identify adaptive genes and mutations, especially when traits associated with fitness variation are not known. The methods we employed, because they explicitly consider epistasis, are particularly suited for the study of nonlinear forms of selection (e.g. balanced polymorphisms), which may be widespread in nature [63]. Our results also highlight several challenges associated with elucidating the genetic basis of adaptation and integrative traits like fitness. With the creative use of modern sequencing technologies, analytical advances, natural history information and experiments, we believe the field is poised to continue to tackle these challenges.
Acknowledgements
We are thankful to Pr. Todd Oakley and Emily Lau for providing us with 95% alcohol to conduct this experiment.
Contributor Information
Romain Villoutreix, Email: romain.villoutreix@gmail.com.
Patrik Nosil, Email: patrik.nosil@cefe.cnrs.fr.
Data accessibility
All data and scripts are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.dbrv15f2w [68] and Zenodo: https://doi.org/10.5281/zenodo.5884987.
The data are provided in the electronic supplementary material [69]. We conducted analyses, summarized the results and generated graphics with custom perl and R scripts [70] (perl version 5.16.3; R version 3.6.0 or 4.0.2).
Authors' contributions
R.V.: conceptualization, data curation, formal analysis, investigation, methodology, project administration, visualization, writing—original draft and writing—review and editing; C.F.de C.: data curation, writing—original draft and writing---review and editing; Z.G.: conceptualization, formal analysis, investigation, methodology, project administration, writing—original draft and writing—review and editing; T.L.P.: data curation, project administration, writing—original draft and writing—review and editing; J.L.F.: writing—original draft and writing—review and editing; P.N.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, writing—original draft and writing—review and editing.
All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Conflict of interest declaration
We declare we have no competing interests.
Funding
This study is part of a project that has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant agreement no. 770826 EE-Dynamics). R.V. and P.N. were supported by the aforementioned ERC grant (Grant agreement No. 770826 EE-Dynamics). C.F.de C. was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (2020/07556-8; Dimensions US-BIOTA-Sao Paulo 18/03428-5). J.L.F. was supported by grants from the National Science Foundation and the United States Department of Agriculture. Z.G. was supported by the US NSF (grant no. DEB 1638768). This work greatly benefited from the use of the University of Utah CHPC cluster.
References
- 1.Fisher RA. 1930. The genetical theory of natural selection. Oxford, UK: Clarendon. [Google Scholar]
- 2.Gillespie JH. 1984. Molecular evolution over the mutational landscape. Evolution 38, 1116-1129. ( 10.2307/2408444) [DOI] [PubMed] [Google Scholar]
- 3.Orr HA. 2005. Theories of adaptation: what they do and don't say. Genetica 123, 3-13. ( 10.1007/s10709-004-2702-3) [DOI] [PubMed] [Google Scholar]
- 4.Østman B, Hintze A, Adami C. 2012. Impact of epistasis and pleiotropy on evolutionary adaptation. Proc. R. Soc. B 279, 247-256. ( 10.1098/rspb.2011.0870) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeaman S. 2013. Genomic rearrangements and the evolution of clusters of locally adaptive loci. Proc. Natl Acad. Sci. USA 110, E1743-E1751. ( 10.1073/pnas.1219381110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeaman S. 2015. Local adaptation by alleles of small effect. Am. Nat. 186, S74-S89. ( 10.1086/682405). [DOI] [PubMed] [Google Scholar]
- 7.Yeaman S, Whitlock MC. 2011. The genetic architecture of adaptation under migration–selection balance. Evolution 65, 1897-1911. ( 10.1111/j.1558-5646.2011.01269.x) [DOI] [PubMed] [Google Scholar]
- 8.Barrett RDH, et al. 2019. Linking a mutation to survival in wild mice. Science 363, 499-504. ( 10.1126/science.aav3824) [DOI] [PubMed] [Google Scholar]
- 9.Chan YF, et al. 2010. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327, 302-305. ( 10.1126/science.1182213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schluter D, Marchinko KB, Arnegard ME, Zhang H, Brady SD, Jones FC, Bell MA, Kingsley DM. 2021. Fitness maps to a large-effect locus in introduced stickleback populations. Proc. Natl Acad. Sci. USA 118, e1914889118. ( 10.1073/pnas.1914889118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Méndez-Vigo B, Picó FX, Ramiro M, Martínez-Zapater JM, Alonso-Blanco C. 2011. Altitudinal and climatic adaptation is mediated by flowering traits and FRI, FLC, and PHYC genes in arabidopsis. Plant Physiol. 157, 1942-1955. ( 10.1104/pp.111.183426) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, Jiménez-Gómez JM. 2020. Functional analysis of FRIGIDA using naturally occurring variation in Arabidopsis thaliana. Plant J. 103, 154-165. ( 10.1111/tpj.14716) [DOI] [PubMed] [Google Scholar]
- 13.Spielmann M, et al. 2016. Exome sequencing and CRISPR/Cas genome editing identify mutations of ZAK as a cause of limb defects in humans and mice. Genome Res. 26, 183-191. ( 10.1101/gr.199430.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adrion JR, Hahn MW, Cooper BS. 2015. Revisiting classic clines in Drosophila melanogaster in the age of genomics. Trends Genet. 31, 434-444. ( 10.1016/j.tig.2015.05.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng CD, Tan JC, Hahn MW, Besansky NJ. 2018. Systems genetic analysis of inversion polymorphisms in the malaria mosquito Anopheles gambiae. Proc. Natl Acad. Sci. USA 115, E7005-E7014. ( 10.1073/pnas.1806760115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng CD, White BJ, Kamdem C, Mockaitis K, Costantini C, Hahn MW, Besansky NJ. 2012. Ecological genomics of Anopheles gambiae along a latitudinal cline: a population-resequencing approach. Genetics 190, 1417-1432. ( 10.1534/genetics.111.137794) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rennison DJ, Heilbron K, Barrett RDH, Schluter D. 2015. Discriminating selection on lateral plate phenotype and its underlying gene, Ectodysplasin, in threespine stickleback. Am. Nat. 185, 150-156. ( 10.1086/679280) [DOI] [PubMed] [Google Scholar]
- 18.Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R. 2015. A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 24, 4348-4370. ( 10.1111/mec.13322) [DOI] [PubMed] [Google Scholar]
- 19.Song Z, et al. 2016. Genome scans for divergent selection in natural populations of the widespread hardwood species Eucalyptus grandis (Myrtaceae) using microsatellites. Sci. Rep. 6, 34941. ( 10.1038/srep34941) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffiths AJ, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM. 2000. An introduction to genetic analysis, 7th edn. New York, NY: W. H. Freeman. [Google Scholar]
- 21.Barrett RDH, Rogers SM, Schluter D. 2008. Natural selection on a major armor gene in threespine stickleback. Science 322, 255-257. ( 10.1126/science.1159978) [DOI] [PubMed] [Google Scholar]
- 22.Michel AP, Sim S, Powell THQ, Taylor MS, Nosil P, Feder JL. 2010. Widespread genomic divergence during sympatric speciation. Proc. Natl Acad. Sci. USA 107, 9724-9729. ( 10.1073/pnas.1000939107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nosil P, Villoutreix R, de Carvalho CF, Feder JL, Parchman TL, Gompert Z. 2020. Ecology shapes epistasis in a genotype–phenotype–fitness map for stick insect colour. Nat. Ecol. Evol. 4, 1673-1684. ( 10.1038/s41559-020-01305-y) [DOI] [PubMed] [Google Scholar]
- 24.Wilczek AM, Cooper MD, Korves TM, Schmitt J. 2014. Lagging adaptation to warming climate in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA 111, 7906-7913. ( 10.1073/pnas.1406314111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitlock MC, Phillips PC, Moore FBG, Tonsor SJ. 1995. Multiple fitness peaks and epistasis. Annu. Rev. Ecol. Syst. 26, 601-629. ( 10.1146/annurev.es.26.110195.003125) [DOI] [Google Scholar]
- 26.Crawford L, Zeng P, Mukherjee S, Zhou X. 2017. Detecting epistasis with the marginal epistasis test in genetic mapping studies of quantitative traits. PLoS Genet. 13, e1006869. ( 10.1371/journal.pgen.1006869) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crawford L, Zhou X. 2018. Genome-wide marginal epistatic association mapping in case-control studies. bioRxiv. ( 10.1101/374983) [DOI]
- 28.Comeault AA, et al. 2015. Selection on a genetic polymorphism counteracts ecological speciation in a stick insect. Curr. Biol. 25, 1975-1981. ( 10.1016/j.cub.2015.05.058) [DOI] [PubMed] [Google Scholar]
- 29.Sandoval CP. 1994. Differential visual predation on morphs of Timema cristinae (Phasmatodeae:Timemidae) and its consequences for host range. Biol. J. Linn. Soc. 52, 341-356. ( 10.1111/j.1095-8312.1994.tb00996.x) [DOI] [Google Scholar]
- 30.Villoutreix R, et al. 2020. Large-scale mutation in the evolution of a gene complex for cryptic coloration. Science 369, 460-466. ( 10.1126/science.aaz4351) [DOI] [PubMed] [Google Scholar]
- 31.Villoutreix R, Ayala D, Joron M, Gompert Z, Feder JL, Nosil P. 2021. Inversion breakpoints and the evolution of supergenes. Mol. Ecol. 30, 2738-2755. ( 10.1111/mec.15907) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobzhansky TG. 1947. Adaptive changes induced by natural selection in wild populations of Drosophila. Evolution 1, 1-16. ( 10.2307/2405399) [DOI] [Google Scholar]
- 33.Kirkpatrick M. 2010. How and why chromosome inversions evolve. PLoS Biol. 8, e1000501. ( 10.1371/journal.pbio.1000501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darlington CD, Mather K. 1949. The elements of genetics. London, UK: George Allen & Unwin Ltd. [Google Scholar]
- 35.Gu X, Li Z, Su Y, Zhao Y, Liu L. 2019. Imaginal disc growth factor 4 regulates development and temperature adaptation in Bactrocera dorsalis. Sci. Rep. 9, 931. ( 10.1038/s41598-018-37414-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu XY, Li J, Liu X, Li X, Ma J. 2014. Characterization and expression analysis of six chitinase genes from the desert beetle Microdera punctipennis in response to low temperature. Cryo Lett. 35, 438-448. [PubMed] [Google Scholar]
- 37.Francikowski J, et al. 2019. Characterisation of white and yellow eye colour mutant strains of house cricket, Acheta domesticus. PLoS ONE 14, e0216281. ( 10.1371/journal.pone.0216281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larkin A, et al. 2020. FlyBase: updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res. 49, D899-D907. ( 10.1093/nar/gkaa1026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu G, et al. 2021. Genome-wide identification and gene-editing of pigment transporter genes in the swallowtail butterfly Papilio xuthus. BMC Genomics 22, 120. ( 10.1186/s12864-021-07400-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vargas-Lowman A, et al. 2019. Cooption of the pteridine biosynthesis pathway underlies the diversification of embryonic colors in water striders. Proc. Natl Acad. Sci. USA 116, 19 046-19 054. ( 10.1073/pnas.1908316116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gompert Z, Comeault AA, Farkas TE, Feder JL, Parchman TL, Buerkle CA, Nosil P. 2014. Experimental evidence for ecological selection on genome variation in the wild. Ecol. Lett. 17, 369-379. ( 10.1111/ele.12238) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nosil P, Crespi BJ. 2006. Experimental evidence that predation promotes divergence in adaptive radiation. Proc. Natl Acad. Sci. USA 103, 9090-9095. ( 10.1073/pnas.0601575103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parchman TL, Gompert Z, Mudge J, Schilkey FD, Benkman CW, Buerkle CA. 2012. Genome-wide association genetics of an adaptive trait in lodgepole pine. Mol. Ecol. 21, 2991-3005. ( 10.1111/j.1365-294X.2012.05513.x) [DOI] [PubMed] [Google Scholar]
- 44.Zhou X, Carbonetto P, Stephens M. 2013. Polygenic modeling with Bayesian sparse linear mixed models. PLoS Genet. 9, e1003264. ( 10.1371/journal.pgen.1003264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou X, Stephens M. 2012. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821-824. ( 10.1038/ng.2310) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215, 403-410. ( 10.1016/S0022-2836(05)80360-2) [DOI] [PubMed] [Google Scholar]
- 47.Nosil P, Villoutreix R, de Carvalho CF, Farkas TE, Soria-Carrasco V, Feder JL, Crespi BJ, Gompert Z. 2018. Natural selection and the predictability of evolution in Timema stick insects. Science 359, 765-770. ( 10.1126/science.aap9125) [DOI] [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671-675. ( 10.1038/nmeth.2089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Endler JA. 2012. A framework for analysing colour pattern geometry: adjacent colours. Biol. J. Linn. Soc. 107, 233-253. ( 10.1111/j.1095-8312.2012.01937.x) [DOI] [Google Scholar]
- 50.Raftery AE. 1995. Bayesian model selection in social research. Sociol. Methodol. 25, 111-163. ( 10.2307/271063) [DOI] [Google Scholar]
- 51.Hoffmann AA, Rieseberg LH. 2008. Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Ann. Rev. Ecol. Evol. Syst. 39, 21-42. ( 10.1146/annurev.ecolsys.39.110707.173532) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stump AD, Pombi M, Goeddel L, Ribeiro JMC, Wilder JA, Torre AD, Besansky NJ. 2007. Genetic exchange in 2La inversion heterokaryotypes of Anopheles gambiae. Insect. Mol. Biol. 16, 703-709. ( 10.1111/j.1365-2583.2007.00764.x) [DOI] [PubMed] [Google Scholar]
- 53.Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987-2993. ( 10.1093/bioinformatics/btr509) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Riesch R, et al. 2017. Transitions between phases of genomic differentiation during stick-insect speciation. Nat. Ecol. Evol. 1, 0082. ( 10.1038/s41559-017-0082) [DOI] [PubMed] [Google Scholar]
- 55.Soria-Carrasco V, et al. 2014. Stick insect genomes reveal natural selection's role in parallel speciation. Science 344, 738-742. ( 10.1126/science.1252136) [DOI] [PubMed] [Google Scholar]
- 56.Chapman RF. 2012. The insects: structure and function, 5th edn. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 57.Tearle RG, Belote JM, McKeown M, Baker BS, Howells AJ. 1989. Cloning and characterization of the scarlet gene of Drosophila melanogaster. Genetics 122, 595-606. ( 10.1093/genetics/122.3.595) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao JT, Bennett CL, Stewart GJ, Frommer M, Raphael KA. 2003. The scarlet eye colour gene of the tephritid fruit fly: Bactrocera tryoni and the nature of two eye colour mutations. Insect. Mol. Biol. 12, 263-269. ( 10.1046/j.1365-2583.2003.00410.x) [DOI] [PubMed] [Google Scholar]
- 59.Okude G, Futahashi R. 2021. Pigmentation and color pattern diversity in Odonata. Curr. Opin. Genet. Dev. 69, 14-20. ( 10.1016/j.gde.2020.12.014) [DOI] [PubMed] [Google Scholar]
- 60.Concha C, et al. 2019. Interplay between developmental flexibility and determinism in the evolution of mimetic Heliconius wing patterns. Curr. Biol. 29, 3996-4009.e4. ( 10.1016/j.cub.2019.10.010) [DOI] [PubMed] [Google Scholar]
- 61.Archambeault SL, Bärtschi LR, Merminod AD, Peichel CL. 2020. Adaptation via pleiotropy and linkage: association mapping reveals a complex genetic architecture within the stickleback Eda locus. Evol. Lett. 4, 282-301. ( 10.1002/evl3.175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ford EB. 1965. Genetic polymorphism. London, UK: Studies Faber & Faber. [Google Scholar]
- 63.Ford EB. 1971. Ecological genomics, 3rd edn. London, UK: Chapman and Hall Ltd. [Google Scholar]
- 64.Lindtke D, Lucek K, Soria-Carrasco V, Villoutreix R, Farkas TE, Riesch R, Dennis SR, Gompert Z, Nosil P. 2017. Long-term balancing selection on chromosomal variants associated with crypsis in a stick insect. Mol. Ecol. 26, 6189-6205. ( 10.1111/mec.14280) [DOI] [PubMed] [Google Scholar]
- 65.Navarro A, Barton NH. 2003. Chromosomal speciation and molecular divergence–accelerated evolution in rearranged chromosomes. Science 300, 321-324. ( 10.1126/science.1080600) [DOI] [PubMed] [Google Scholar]
- 66.Kirkpatrick M, Barton N. 2006. Chromosome inversions, local adaptation and speciation. Genetics 173, 419-434. ( 10.1534/genetics.105.047985) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feder JL, Gejji R, Powell THQ, Nosil P. 2011. Adaptive chromosomal divergence driven by mixed geographic mode of evolution. Evolution 65, 2157-2170. ( 10.1111/j.1558-5646.2011.01321.x) [DOI] [PubMed] [Google Scholar]
- 68.Villoutreix R, de Carvalho CF, Gompert Z, Parchman TL, Feder JL, Nosil P. 2022. Testing for fitness epistasis in a transplant experiment identifies a candidate adaptive locus in Timema stick insects. Dryad Digital Repository. ( 10.5061/dryad.dbrv15f2w) [DOI] [PMC free article] [PubMed]
- 69.Villoutreix R, de Carvalho CF, Gompert Z, Parchman TL, Feder JL, Nosil P. 2022. Testing for fitness epistasis in a transplant experiment identifies a candidate adaptive locus in Timema stick insects. FigShare. [DOI] [PMC free article] [PubMed]
- 70.Team R C. 2020. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Villoutreix R, de Carvalho CF, Gompert Z, Parchman TL, Feder JL, Nosil P. 2022. Testing for fitness epistasis in a transplant experiment identifies a candidate adaptive locus in Timema stick insects. Dryad Digital Repository. ( 10.5061/dryad.dbrv15f2w) [DOI] [PMC free article] [PubMed]
- Villoutreix R, de Carvalho CF, Gompert Z, Parchman TL, Feder JL, Nosil P. 2022. Testing for fitness epistasis in a transplant experiment identifies a candidate adaptive locus in Timema stick insects. FigShare. [DOI] [PMC free article] [PubMed]
Data Availability Statement
All data and scripts are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.dbrv15f2w [68] and Zenodo: https://doi.org/10.5281/zenodo.5884987.
The data are provided in the electronic supplementary material [69]. We conducted analyses, summarized the results and generated graphics with custom perl and R scripts [70] (perl version 5.16.3; R version 3.6.0 or 4.0.2).