

Abstract
The immune system plays a vital role in maintaining the delicate balance between immune recognition and tumor development. Regardless, it is not uncommon that cancerous cells can intelligently acquire abilities to bypass the antitumor immune responses, thus allowing continuous tumor growth and development. Immune evasion has emerged as a significant factor contributing to the progression and immune resistance of pancreatic cancer. Compared with other cancers, pancreatic cancer has a tumor microenvironment that can resist most treatment modalities, including emerging immunotherapy. Sadly, the use of immunotherapy has yet to bring significant clinical breakthrough among pancreatic cancer patients, suggesting that pancreatic cancer has successfully evaded immunomodulation. In this review, we summarize the impact of genetic alteration and epigenetic modification (especially histone deacetylases, HDAC) on immune evasion in pancreatic cancer. HDAC overexpression significantly suppresses tumor suppressor genes, contributing to tumor growth and progression. We review the evidence on HDAC inhibitors in tumor eradication, improving T cells activation, restoring tumor immunogenicity, and modulating programmed death 1 interaction. We provide our perspective in targeting HDAC as a strategy to reverse immune evasion in pancreatic cancer.
Keywords: Histone acetylation, Histone deacetylases inhibitors, Immune evasion, Pancreatic cancers, Pancreatic ductal adenocarcinoma
Core Tip: There are several broad reviews covering histone deacetylases (HDAC) in cancer but none on its role in modulating immune evasion in pancreatic cancer. This is the first review to discuss the role of HDAC in the context of immune-evading pancreatic cancer. We also summarize the evidence of HDAC inhibitors in targeting immune-evading pancreatic cancer. This mini review also covers our perspective in the strategies to target overexpression of HDAC in pancreatic cancer.
INTRODUCTION
Pancreatic cancer is the seventh most common cause of cancer-related deaths worldwide[1]. Pancreatic ductal adenocarcinoma (PDAC) is the most ubiquitous type of pancreatic cancer and remains incurable for 95% of patients. Pancreatic cancer is characterized by a devastating prognosis, with the lowest overall 5-year survival rate among all cancers[2]. The average survival time for pancreatic cancer is less than six months if left untreated[3]. Pancreatic cancer is thus estimated to be the second leading cause of death in the United States by 2030[3].
The dismal prognosis of PDAC is partly contributed by the lack of early clinical symptoms and poor sensitivity of PDAC diagnostic tests. Moreover, the treatment modalities available to date are generally ineffective for management of PDAC. Currently, the mainstay treatment for PDAC is surgery but only approximately 10% of PDAC patients qualify for surgical resection upon diagnosis[4]. Among the patients who undergo surgical resection, only < 25% could survive for more than five years. Besides, chemotherapeutic strategies have been exhausted in PDAC treatment, in which the use of chemotherapy has been limited by its well-established low efficacy, high toxicity and drastic decline in quality of life[5]. As a result, the search for an effective treatment regimen for PDAC remains a significant challenge.
Recently, immunotherapy has been hailed as a breakthrough in the realm of cancer therapy, which warrants further exploration for PDAC treatment. The use of immunotherapy in PDAC, however, is largely limited by its immune evasion barrier[6]. Thus, understanding the diverse mechanisms underlying the immune evasion in pancreatic cancer may empower the search for methods to tackle and prevent the bypass of immune surveillance[6,7].
GENETIC ALTERATION IN PDAC POTENTIATES ITS IMMUNE EVASION CAPABILITY
PDAC is an exocrine pancreatic cancer derived from pancreatic ductal cells. The progression to PDAC is characterized by its transition from normal pancreatic ductal cells to pancreatic intraepithelial neoplasia (PanIN) or its precursor lesions[8]. PanIN may be differentiated into three grades based on its histological or architectural changes[8]. PanIN1A and PanIN1B are characterized by the low-grade dysplasia while PanIN2 is characterized by the loss of polarity, nuclear crowding, enlargement of cell, and its typical papillary development. PanIN3 represents mature lesions with drastic nuclear aberrations, luminal necrosis, and show epithelial cell budding into the ductal lumen[8]. High-grade PanIN is almost uniquely found in invasive PDAC[8]. These precursor lesions develop into invasive PDAC following the accumulation of genetic mutations (Figure 1). Pertinent genetic alterations include KRAS oncogene mutation, the initiating genetic event in PDAC, followed by the loss of function in essential tumor suppressor genes such as CDKN2a, TP53 and SMAD4 (Figure 1).
Figure 1.

Association of common genetic alterations and pancreatic ductal adenocarcinoma pathogenesis. The progression from the early to invasive stage of pancreatic ductal adenocarcinoma (PDAC) is supported by different genes alteration at different stages. KRAS mutation transforms the normal pancreatic ductal cells to pancreatic intraepithelial neoplasia (PanIN). The PanIN1A, PanIN1B and PanIN2 are low grade PanIN. Additional mutation such as CDKN2A is required to develop PanIN2. As the disease deteriorates, additional genes mutation such as TP53, SMAD4 and BRCA2 are involved to develop high grade PanIN3 and eventually invasive PDAC. PDAC: Pancreatic ductal adenocarcinoma. The figure in this review paper is created by using BioRender.com by the authors.
KRAS oncogene mutation is found in nearly all PDACs[9]. High expression of the mutated KRAS is also associated with poor prognosis[9]. The KRAS GTPase switches between being bound to guanosine diphosphate, GDP (inactive state) and being bound to guanosine triphosphate, GTP (active state)[10]. In cancer development, missense mutations cause RAS to be persistently bound to GTP allowing an unlimited cellular proliferation. The alteration of CDKN2a results in unimpeded G1/S transformation and unrestrained cell division, facilitating tumor to evade host immunomodulation[11]. As expected, PDAC patients with CDKN2a inactivation are associated with poor overall survival and prognosis[11].
The metastasized tumors also exhibit several genetic alterations, namely TP53, BRCA2 and SMAD4, indicating that these tumors have successfully evaded host immunosurveillance by leveraging on these genetic alterations[12-14]. TP53 tumor suppressor gene on chromosome 17p is often mutated in cancer[15]. Up to 75% of PDACs present with loss or mutation of TP53 expression[15]. The loss of TP53 gene expression decreases cell cycle arrest and apoptosis, allowing for damaged DNA to replicate and aggregate genetic alterations. Accumulation of TP53 mutants significantly increases the incidence of cancer metastasis[12]. Similarly, BRCA2 is essential in restoring damages to double-stranded DNA. BRCA2 mutations can cause alterations of TP53 tumor suppressor genes, leading to pancreatic tumorigenesis[13]. Loss of SMAD4 (an effector of the transforming growth factor β signaling pathway) promotes pancreatic cancer progression and increases rate of metastasis[14]. SMAD4 inactivation in PDAC is also associated with poor prognosis and short overall survival[14].
Nevertheless, the genetic alterations in PDAC are still unable to explain the complex immune evasion in PDAC, indicating that other more pertinent underlying factors may be the driving force for immune-evading PDAC.
HISTONE DEACETYLASES IN CANCER
Epigenetic abnormalities are also crucial in carcinogenesis and the pathophysiology of cancer. Histone modification is one of the essential epigenetic processes involved in tumorigenesis and progression[16]. Histone acetylation is strictly controlled by a balance between histone acetyltransferase and histone deacetylases (HDAC) with opposing enzymatic activities. Histone acetylation is associated with an increased transcription level, while deacetylation is correlated with its repression. Histone deacetylation increases the ionic interactions between positively charged histones and negatively charged DNA, this limits the access to transcription machinery and represses gene transcription[17]. HDACs also remove acetyl groups and repress the transcription of essential genes such as tumor suppressor genes[18]. HDACs can also regulate the transcription of tumor suppressor gene via the formation of corepressor complexes or direct interaction with the transcription factors[19]. Notably, HDACs may also deacetylate nonhistone proteins, resulting in dysregulation of cellular homeostasis including cell-cycle progression and apoptosis[18,19]. There are 18 potential HDACs grouped into four classes, based on their homology to yeast proteins. Class I (HDAC 1–3 and 8), Class II (HDAC 4–7, 9 and 10) and Class IV (HDAC 11) HDACs are zinc dependent while Class III HDACs are nicotinamide adenine dinucleotide (Figure 2). Class III HDACs are also referred to as sirtuins (SIRT 1–7) (Figure 2)[20].
Figure 2.
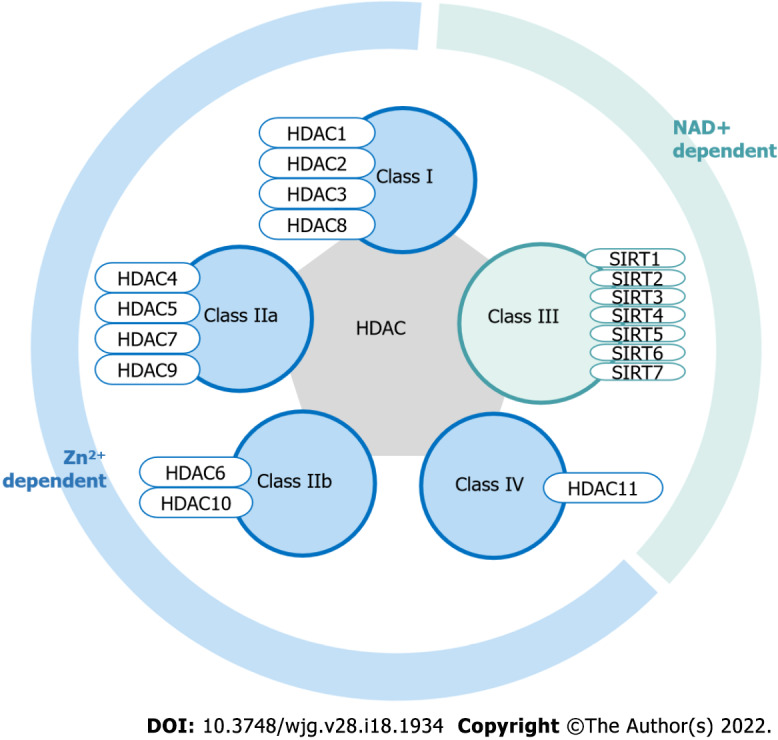
Histone deacetylases classification. Histone deacetylases (HDAC) can be divided into two major classes, namely Zn-dependent and NAD dependent. Class I (HDAC 1–3 and 8), Class IIa (HDAC 4, 5, 7 and 9), Class IIb (HDAC 6 and 10) and Class IV (HDAC 11) are Zn-dependent HDAC. Class III (SIRT1 to 7) is NAD+ dependent HDAC. HDAC: Histone deacetylases. The figure in this review paper is created by using BioRender.com by the authors.
The dysregulation of post-translational histone modification, especially histone acetylation, leads to gene transcription dysregulation. Overexpression of HDAC results in significant suppression of tumor suppressor genes, contributing to tumor growth and progression[17,20]. In PDAC, more than half of PDACs were stained positive for HDAC 1. The high expression of HDAC 1 has been correlated with a poorer distant metastasis-free survival[21]. Separately, another study also showed that overexpression of HDAC 1 was linked to a lower overall survival[22]. Treatment with HDAC 1 inhibitors decreased the invasion and metastasis ability of PDAC[21]. In addition, HDAC 2[23], HDAC 7 [24] and HDAC 8[24] overexpression is commonly found in PDAC.
Recently, HDAC has been highlighted for its contribution towards immune evasion. For example, HDAC 3 transcriptionally regulates programmed death ligand 1 (PD-L1) expression[25]. It is reported that higher expression of HDAC 3 is positively correlated with increased PD-L1 expression[25]. Such phenomena suppress immune cells that carry PD-L1 receptors, thus disputing immunosurveillance[25]. HDAC overexpression is also a common observation among other immune-evading solid tumors[17]. In particular, the high expression HDAC 1 is found in gastric and prostate cancers; while HDAC 2 overexpression is associated with gastric, cervical and breast cancers[17]. HDAC 1–3 are highly expressed in renal cell cancer and Hodgkin’s lymphoma[17]. Overexpression of HDACs is linked to a significant decline in overall survival and prognosis[20]. It is believed that mutation or loss of HDAC expression is correlated with increased oncogene expression[17]. For instance, Rb tumor suppressor gene needs the concomitant action of HDAC to suppress transcription of other essential oncogenes. Loss of HDAC reduces the protective effect of Rb tumor suppressor gene[26]. Due to the roles and targetability of HDACs, targeting HDAC has garnered much attention as an effective anticancer therapeutic strategy.
HDAC INHIBITORS AS CANCER THERAPY
HDAC inhibitors bind directly to the active sites on HDAC enzymes, and inhibit the deacetylation effect of HDAC. Each HDAC inhibitor contains a cap, connecting unit, linker and a zinc-binding group that chelates the cation in the target HDAC[27]. HDAC inhibitors can be classified based on their specificity towards HDACs, namely the pan-HDAC inhibitors, Class I or Class II specific inhibitors. HDAC inhibitors can change the acetylation status of both nonhistone proteins and chromatin, causing a viable gene expression alteration, induction of apoptosis and cell cycle arrest[27]. HDAC inhibitors can target not only the tumor cell itself, but also the tumor microenvironment and immune milieu, making the use of HDAC inhibitor a promising strategy to eradicate immune evaded PDAC[27].
The overexpression of HDACs in cancer allows cancer cells to have increased sensitivity to HDAC inhibitors, leading to the induction of growth arrest, differentiation inhibition and eventual tumor cell death without compromising the nontumor cells[27]. Given PDAC’s therapeutic resistance to conventional therapy, it is not surprising that the use of HDAC inhibitor as an alternative treatment option has been studied. Ivaltinostat (CG200745), a pan-HDAC inhibitor, demonstrates inhibitory effects on PDAC tumor growth by upregulating proapoptotic proteins BAX and p21[28]. Treatment of PDAC cells with belinostat (PXD101), a Class I and II HDAC inhibitor, also induces cell cycle arrest and tumor regression[29]. A Phase I clinical study using escalating doses of dacinostat (LAQ824), another pan-HDAC inhibitor, showed that the drug was well tolerated by PDAC patients[30]. A significant accumulation of histone acetylation was reported among the patients treated with dacinostat[30]. However, most patients in this trial discontinued dacinostat treatment due to disease progression[30], indicating an unresolved limitation of prescribing HDAC inhibitor as monotherapy for pancreatic cancer.
In combination, HDAC inhibitors show a better effect with other chemotherapeutic agents. The combination of two Class I HDAC inhibitors, romidepsin and ricolinostat (ACY-1215), showed potent synergy with gemcitabine in a panel of PDAC cell lines[31]. Combination of entinostat (Class I HDAC inhibitor), vorinostat (Class I and II HDAC inhibitor) and cyclooxygenase (COX)-2 inhibitors showed complete stalling of PDAC cell growth[32]. In addition, the use of vorinostat and trichostatin A (Class I and II HDAC inhibitor), showed induction of apoptosis in caspase-independent pathways, even for antineoplastic drug-resistant PDAC cell lines[24]. Combination of trichostatin A and proteasome inhibitor PS-341 downregulated antiapoptotic factors and synergistically induced apoptosis[33]. In a separate study, trichostatin A together with silibinin demonstrated a synergistic growth inhibitory effect on PDAC cells by inducing G2/M cell cycle arrest and apoptosis[34].
Meanwhile, the combination of ivaltinostat, gemcitabine and erlotinib significantly reduced PDAC tumor size up to 50%[28]. Ivaltinostat enhanced gemcitabine sensitivity in gemcitabine-resistant pancreatic cancer cells[28]. The use of valproic acid (Class I HDAC inhibitor) confers a synergistic effect with gemcitabine on PDAC cells, lending support to the postulation that targeting HDAC may be a promising strategy to overcome therapeutic resistance and circumvent immune evasion strategies[35].
HDAC inhibitors demonstrated anti-angiogenic effects by regulating angiogenic related transcriptional factors such as von Hippel-Lindau (VHL), hypoxia inducible factor-1a (HIF-1a) and vascular endothelial growth factor (VEGF)[36]. Rosengren RJ team found novel HDAC inhibitors, Jazz90 and Jazz167, which had a superior potency and anti-angiogenic effects than conventional HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) in prostate cancer cell lines[37]. Independently, Vyas A team synthesised novel SAHA analogues with greater anti-angiogenic effects using the chick chorioallantonic membrane assay[38]. Several clinical trials are ongoing to evaluate the clinical benefits of HDAC inhibitors in targeting angiogenesis in cancer, however, to the best of our knowledge, there is no solid evidence on its benefits among pancreatic cancer patients. More studies are needed to investigate the potential of HDAC inhibitors as anti-angiogenesis agents in PDAC.
In view of their promising combinatory therapeutic efficacy, there has been growing interest in the use of HDAC inhibitors with other therapeutic agents for pancreatic cancer patients (Table 1). However, the use of HDAC inhibitors has been studied in the treatment of lymphoma and other nonpancreatic tumors[39]. For example, HDAC inhibitor has been approved by the FDA for various cancer treatments. Romidepsin[40] and vorinostat[41] have been approved for refractory cutaneous T cell lymphoma while panobinostat[42] (Class I, II and IV HDAC inhibitor) has been approved for multiple myeloma. Likewise, belinostat[42] and tucidinostat[39] (Class I, II, and III inhibitor) have been approved for the treatment of peripheral T cell lymphoma.
Table 1.
Investigational histone deacetylases inhibitors in pancreatic cancer patients
|
HDAC1 specificity
|
Intervention
|
Clinical trial phase
|
Study start date
|
Status
|
Clinical trial reference code
|
| Pan-HDAC | Vorinostat + Marizomib | I | March 2008 | Completed | NCT00667082 |
| Pan-HDAC | Vorinostat + Capecitabine + Radiation Therapy | I | October 2009 | Completed | NCT00983268 |
| Pan-HDAC | Vorinostat + Gemcitabine + Sorafenib + Radiation Therapy | I | January 2015 | Active | NCT02349867 |
| Pan-HDAC | Vorinostat + Radiation Therapy | I and II | March 2009 | Terminated | NCT00831493 |
| Pan-HDAC | Vorinostat + 5-fluorouracil + Radiation Therapy | I and II | August 2009 | Terminated | NCT00948688 |
| Pan-HDAC | Panobinostat + Bortezomib | II | September 2010 | Terminated | NCT01056601 |
| Class I | Entinostat | I | March 2001 | Completed | NCT00020579 |
| Class I | Entinostat + Nivolumab | II | November 2017 | Completed | NCT03250273 |
| Class I | Entinostat + ZEN003694 | I and II | March 2022 | Not yet recruiting | NCT05053971 |
| Class I | Entinostat + Molibresib | I | September 2020 | Withdrawn | NCT03925428 |
| Class I | Entinostat + FOLFOX2 | I | January 2021 | Withdrawn | NCT03760614 |
| Class I | Tacedinaline + Gemcitabine | II | October 1999 | Completed | NCT00004861 |
HDAC: Histone deacetylases.
FOLFOX regimen consists of folinic acid, 5-fluorouracil, and oxaliplatin.
HDAC INHIBITORS REVERSE IMMUNE EVASION IN CANCER
HDAC inhibitors enhance immune cell activation
The PDAC tumor microenvironment is composed of regulatory T cells, tumor-associated macrophages (TAMs) and myeloid-deprived suppressive cells (MDSCs) that inhibit ability of cytotoxic T cells (CTLs) in tumor recognition and clearance[6,43]. Cells in the tumor microenvironment can also produce immunosuppressive cytokines including interleukin (IL)-1, IL-6, IL-10, and tumor necrosis factor-α (TNF-α) to cause T cell anergy or tolerance, resulting in immune evasion[44]. Other important cells in the tumor microenvironment include the fibrotic matrix, pancreatic stellate cells and cancer associated fibroblasts (CAFs); all of which can adhere to infiltrating T lymphocytes and prevent their entry into cancer cells, resulting in T cell anergy[44]. In particular, the activated CAFs create a dense stroma that dominates in PDAC, mediating tumor growth and survival by the production of extracellular matrix proteins, growth factors and cytokines[6,45]. CAFs represent the majority type of cells in PDAC stroma and have been acknowledged to be one of the key contributing factors to the immune evasion of cancer cells[45]. CAFs can limit access of infiltrating immune cells to cancer cells through the release of dense collagen networks, resulting in a physical hurdle that disrupts T cell dispersal and inhibition of T cell migration in areas of increased collagen deposition, such as that of PDAC tumors[45]. CAFs have been demonstrated to upregulate immune checkpoints on Cluster of Differentiation (CD)4+ T cells and CTLs, thus resulting in reduced immune function of T cells[45]. Within the immunosuppressive tumor microenvironment, PDACs can disrupt the immunogenic effects of CTLs, by which the CTLs that presented in PDACs may be poorly cytotoxic and nonfunctional[46]. Multiplex staining has demonstrated that proximity of T cells to PDACs is correlated with patient prognosis and survival[47]. As a result, exclusion of T cells from the tumor microenvironment correlates with the tumor initiation and progression.
HDAC inhibitors can modulate immune evasion by modulating immune cell functions[48]. HDAC inhibitors can also increase the expression of major histocompatibility complex (MHC) and its costimulatory molecules leading to T-cell activation[48]. For example, AR42 (a pan-HDAC inhibitor) enhances its adaptive immunity through improving the functions and capabilities of CTLs and natural killer (NK) cells in murine melanoma[49]. Meanwhile, trichostatin A suppresses CD4 T cells from undergoing apoptosis, leading to enhanced antitumor effect[50]. Entinostat increases CTL cytotoxic function and T cell signatures in ovarian tumors[51]. Entinostat also reverses CTL–T regulatory (Treg) cell ratios in the ovarian tumor microenvironment, thus facilitating CTL accumulation at the tumor site[51]. Therefore, HDAC inhibitors have a pivotal role in sustaining T-cell-mediated antitumor immunity. Further studies are required to understand their underlying mechanisms in reversing T cell resistance.
HDAC inhibitors enhance tumor immunogenicity
The intrinsic resistance of PDAC tumors to immune eradication is mainly due to its nonimmunogenic characteristic[6,7]. Tumors can further evade CTL-induced tumor lysis via immunoediting, or changes in immunogenicity of cancer cells[52]. Host immune system can alter the expression profile of tumors, in turn enabling them to evade immune detection[53]. The development of PDAC has been described to reflect the three Es of cancer immunoediting, which are elimination, equilibrium and escape[6,54]. The elimination phase occurs during cancer immunosurveillance, when immune effector cells are enlisted to the cancerous tissue to eliminate PDAC cancer cells. Immune effector cells, including CTLs can eliminate most of the vulnerable tumor cells, leaving the resistant tumor clones behind. These resistant tumor clones then expand and remain undetected by the immune system. During the equilibrium phase, a unique equilibrium between antitumor and protumor immune cells is sustained until tumor escape mechanisms are established. During the escape phase, an immunosuppressive microenvironment is formed with the presence of TAMs and MDSCs, creating an effective barrier against the effector immune cells such as CTLs[6,54]. Taken together, such immunoediting has allowed PDAC to bypass immune detection, whereby PDAC continues to grow, progress and metastasize.
HDAC inhibitors may reverse immune evasion in tumors by enhancing tumorigenicity. Trichostatin A induced suppression of tumor growth by improving the immunogenicity of the metastatic tumor cells in a murine study[21,55]. Trichostatin A also increased the MHC Class I expression that translated into enhanced susceptibility to being killed by cytotoxic T cells[55]. Entinostat altered the tumor microenvironment by increasing the MHC Class II expression and its transactivator[56]. Entinostat also re-expressed the natural killer cell receptor and ligand, leading to a decrease in the immunosuppressive effects by host immune cells[57]. Another study using vorinostat and entinostat revealed that breast and prostate carcinoma cells became more sensitive to T-cell-mediated lysis after treatment with HDAC inhibitors. Treatment with vorinostat increased CTL sensitivity, leading to tumor lysis, demonstrating the enhancement of antigen-specific CTL-mediated killing by HDAC inhibitors[58]. Entinostat can sensitize immune checkpoint inhibitors by ablating MDSC-mediated immunosuppressive effects in PDAC tumor-bearing mice[59]. Such exciting findings have rendered the researchers to launch their human clinical trial to investigate the combination of entinostat with nivolumab (a checkpoint inhibitor) in unresectable PDAC patients (Clinical Trial NCT03250273)[60]. Other studies showed that epigenetic modulators[61,62] (including HDAC) could improve tumor immunogenicity and could be a promising translational intervention in cancer.
Given the high frequency of KRAS mutations in pancreatic cancer patients, mRNA vaccine that targets a specific mutant has gained attraction[63]. In theory, mRNA vaccine encodes KRAS mutant-specific antigens into the host’s cytoplasm, leading to the eradication of tumor cells with KRAS mutation by host’s immune system[63]. mRNA-5671/V941 is being investigated as monotherapy or in combination with immunotherapy for pancreatic cancer patients with KRAS mutation (NCT03948763). However, it remains uncertain whether cancer vaccine could be given along with HDAC inhibitors since HDAC inhibitors may also enhance tumor immunogenicity. Barouch DH group demonstrated that the combination of romidepsin, I-BET151 and cancer vaccine enhanced the CTL cell response in a mouse model, indicating the possible synergism between HDAC inhibitor and cancer vaccine[64]. Thus, further studies are warranted to explore the efficacy of cancer vaccine as a monotherapy or in combination with other therapeutic agents.
HDAC inhibitors counteract PD-L1 and PD-1 interaction
Tumor-infiltrating lymphocytes including CTLs produce a high level of programmed death (PD)-1 while PDAC cells counteract by overproducing the specific ligand of PD-1, which is PD-L1. The interaction between PD-1 and PD-L1 results in T-cell depletion[6,65]. Studies have shown that PD-1 and PD-L1 interaction impedes T-cell growth and that tumor-cell-borne PD-L1, and thus induce apoptosis of tumor-specific T-cell clones[66]. Such transformation turns CTLs into a dysfunctional state of exhaustion that characterized by the loss of CTL proliferation ability as well as the loss of CTL cytotoxic functions[67,68]. As a result, CTLs will be downregulated, and tumor cells can then escape the cytotoxic killing by CTLs.
PD-L1 is overexpressed in PDAC and the higher level of PD-L1 has been linked to a poorer prognosis for PDAC patients[52,67]. In addition, a higher HDAC 3 expression has been linked to the increased PD-L1 expression on PDAC cells[25]. HDAC 3 modulates PD-L1 expression via the signal transducer and activator of transcription 3 pathway[22]. HDAC 3 inhibitor (RGFP966) reduces PD-L1 mRNA and protein expression levels, thus enhancing immunosurveillance and aiding the reversal of immune evasion[25].
Another study reported that combination of CG-745 (HDAC Class I and IIb inhibitor) and anti-PD-1 antibody showed synergistic tumor eradication in two syngeneic cancer mouse models[69]. Further mechanistic studies indicated CG-745 increased T-cell activation and macrophage M1 polarization, helping the anti-PD-1 anticancer effect[69]. In addition, HDAC inhibition with entinostat improved the antitumor effect of PD-1 blockade in two syngeneic cancer mouse models[70]. The combination of entinostat and PD-1 inhibitor reduced the tumor burden and improved its survival[70]. Additional analyses indicated that entinostat upregulated PD-L1 in tumors, blocked the immunosuppressive function of MDSCs, and reduced COX-2, inducible nitric oxide synthase (iNOS) and arginase-1 mRNA expression[70]. Romidepsin enhanced the PD-1 blockade in a murine tumor model, leading to tumor rejection[71]. Individual treatment with romidepsin or PD-1 blocker did not result in significant tumor suppression[71]. Further analyses indicated that romidepsin increased PD-L1 Levels in the tumor[71]. Romidepsin and PD-1 blocker synergized by unleashing the interferon-dependent response in T-cell recruitment to the upregulated PD-L1 tumor cells[71]. Unlike entinostat[70], romidepsin-treated tumor did not alter the MDSC population[71], indicating that HDAC inhibitors have varied impacts on PD-1 and PD-L1 interaction, which can be independent of MDSC modulation. Further study is warranted to explore how HDAC inhibitor affects PD-1 and PD-L1 interaction. Such findings support the notion that HDAC inhibitors can modulate PD-1 and PD-L1 interaction in tumors apart from their canonical role in inhibiting HDAC.
CONCLUSION
PDAC is often synonymous with a “death sentence”. The high mortality and poor outcome of PDAC are mainly due to PDAC being refractory to most forms of contemporary therapeutic strategies. Given that the hallmark of PDAC is a highly dense stroma and immense microenvironment, immunotherapy stands out as a promising novel approach to PDAC treatment. Chen and Mellman proposed that cancer immunity is a series of ongoing cyclical events (Figure 3)[72]. Disruption in the major events in the cancer–immunity cycle leads to immune evasion in cancer[6].
Figure 3.

Role of histone deacetylases inhibitors in targeting cancer immune evasion. Cancer–immunity cycle is a continuously cyclical process to amplify the immune response leading to cancer eradication. The cancer–immunity cycle has seven steps: Step 1: Dying cancer cells release neoantigen (Step 2). These neoantigens are captured by antigen-presenting cells and present the antigens on the major histocompatibility complex to T cells (Step 3), allowing the T cells to be primed and activated. Once T cells are activated (Step 4), T cells are transported to the tumor site and (Step 5) infiltrate the tumor. Once inside the tumor (Step 6), T cells recognize the tumor cells, and (Step 7) kill the tumor cells. Histone deacetylases inhibitors will support Steps 2–7 of the cancer–immunity cycle. Such effects can be synergized with other therapeutic agents. CAR: Chimeric antigen receptor; CTLA4: Cytotoxic T-lymphocyte associated antigen 4; PD-1: Programmed death protein 1; PD-L1: Programmed death ligand 1; VEGF: Vascular endothelial growth factor; HDAC: Histone deacetylases. The figure in this review paper is created by using BioRender.com by the authors.
HDAC inhibitors have grained traction in medical research as a promising approach to counteracting immune evasion strategies to strengthen Steps 2–7 of the cancer–immunity cycle (Figure 3). HDAC overexpression as a contributing factor to immune evasion and subsequent carcinogenesis have been increasingly recognized. The use of HDAC inhibitors to eliminate cancerous cells (Step 7 of the cancer–immunity cycle, Figure 3) is also gaining traction. Further research is warranted to investigate the effectiveness of HDAC inhibitors in cancer patients. HDAC inhibitor use may well be the key to a long-awaited treatment regimen for cancer patients. The direction for HDAC inhibitors in cancer treatment seems to lean towards combination therapy (Figure 3), with chemotherapy, radiotherapy or immunotherapy. HDAC inhibitors have been widely used with checkpoint inhibitor antibodies in preclinical models. Such combinations have demonstrated successful enhancement of antitumor efficacy and an increase in immune-cell activation.
Studies on HDAC inhibition to circumvent immune evasion are still limited. Current use of HDAC inhibitor to treat PDAC has had promising results, but studies are often still in the early preclinical stage, with much still unknown about the effect of HDAC inhibitors in PDAC patients. There remain much hope and scope in investigating HDAC inhibitors in clinical trials, which will help shed light on the effectiveness of HDAC inhibitors as an adjunct therapy in PDAC.
The ability of HDAC inhibitors to inhibit histone deacetylation may also have its limitations. HDAC inhibitors have poor physiochemical features and unfavorable pharmacokinetics[73]. HDAC inhibitors may also nonspecifically block angiogenesis, which may disrupt drug delivery through blood vessels to solid tumors[73]. In addition, the anti-inflammatory properties of HDAC inhibitors have been postulated to induce apoptosis among immune cells[73]. Current evidence supports the understanding that HDAC inhibitors may counter immune-cell suppression and apoptosis by enhancing anti-PD1 blockade effects[25]. Moreover, there must be an intact immune function in the host as a prerequisite for HDAC inhibitors to modulate antitumor immune response[6]. Moreover, HDAC inhibitors have yet to utilize the current advanced drug delivery systems that offer site-specific drug delivery[74] with enhanced tumor targeting and reduced toxicity[73]. We believe that, by targeting HDAC in the immune evaded pancreatic cancer, the greatest therapeutic outcome will emerge.
Footnotes
Conflict-of-interest statement: There is no conflict of interest with any authors contributed to this manuscript.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Corresponding Author's Membership in Professional Societies: International Pharmaceutical Federation (FIP), No. 36982; European Association Cancer Research (EACR), No. EACR27345; Royal Society of Chemistry, No. 538435; Malaysian Pharmaceutical Society, No. 8141.
Peer-review started: January 7, 2022
First decision: March 9, 2022
Article in press: April 4, 2022
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: China
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Balcerczyk A, Poland; Isaji S, Japan S-Editor: Fan JR L-Editor: A P-Editor: Fan JR
Contributor Information
Wynne Sim, School of Medicine, International Medical University, Kuala Lumpur 57000, Malaysia.
Wei-Meng Lim, School of Pharmacy, International Medical University, Kuala Lumpur 57000, Malaysia; Center for Cancer and Stem Cell Research, Institute for Research, Development, and Innovation, International Medical University, Kuala Lumpur 57000, Malaysia.
Ling-Wei Hii, School of Pharmacy, International Medical University, Kuala Lumpur 57000, Malaysia; Center for Cancer and Stem Cell Research, Institute for Research, Development, and Innovation, International Medical University, Kuala Lumpur 57000, Malaysia.
Chee-Onn Leong, Center for Cancer and Stem Cell Research, Institute for Research, Development, and Innovation, International Medical University, Kuala Lumpur 57000, Malaysia; AGTC Genomics, Kuala Lumpur 57000, Malaysia.
Chun-Wai Mai, State Key Laboratory of Oncogenes and Related Genes, Renji-Med X Clinical Stem Cell Research Center, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China; Department of Pharmaceutical Chemistry, Faculty of Pharmaceutical Sciences, UCSI University, Kuala Lumpur 56000, Malaysia. mai.chunwai@outlook.com.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 3.Doleh Y, Lal LS, Blauer-Petersen C, Antico G, Pishvaian M. Treatment patterns and outcomes in pancreatic cancer: Retrospective claims analysis. Cancer Med. 2020;9:3463–3476. doi: 10.1002/cam4.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strobel O, Neoptolemos J, Jäger D, Büchler MW. Optimizing the outcomes of pancreatic cancer surgery. Nat Rev Clin Oncol. 2019;16:11–26. doi: 10.1038/s41571-018-0112-1. [DOI] [PubMed] [Google Scholar]
- 5.Chin V, Nagrial A, Sjoquist K, O'Connor CA, Chantrill L, Biankin AV, Scholten RJ, Yip D. Chemotherapy and radiotherapy for advanced pancreatic cancer. Cochrane Database Syst Rev. 2018;3:CD011044. doi: 10.1002/14651858.CD011044.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gan LL, Hii LW, Wong SF, Leong CO, Mai CW. Molecular Mechanisms and Potential Therapeutic Reversal of Pancreatic Cancer-Induced Immune Evasion. Cancers (Basel) 2020;12 doi: 10.3390/cancers12071872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Looi CK, Chung FF, Leong CO, Wong SF, Rosli R, Mai CW. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J Exp Clin Cancer Res. 2019;38:162. doi: 10.1186/s13046-019-1153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyazaki T, Ohishi Y, Miyasaka Y, Oda Y, Aishima S, Ozono K, Abe A, Nagai E, Nakamura M. Molecular Characteristics of Pancreatic Ductal Adenocarcinomas with High-Grade Pancreatic Intraepithelial Neoplasia (PanIN) Are Different from Those without High-Grade PanIN. Pathobiology. 2017;84:192–201. doi: 10.1159/000455194. [DOI] [PubMed] [Google Scholar]
- 9.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17:153–168. doi: 10.1038/s41575-019-0245-4. [DOI] [PubMed] [Google Scholar]
- 10.Pantsar T. The current understanding of KRAS protein structure and dynamics. Comput Struct Biotechnol J. 2020;18:189–198. doi: 10.1016/j.csbj.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo W, Yang G, Qiu J, Luan J, Zhang Y, You L, Feng M, Zhao F, Liu Y, Cao Z, Zheng L, Zhang T, Zhao Y. Novel discoveries targeting gemcitabine-based chemoresistance and new therapies in pancreatic cancer: How far are we from the destination? Cancer Med. 2019;8:6403–6413. doi: 10.1002/cam4.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voutsadakis IA. Mutations of p53 associated with pancreatic cancer and therapeutic implications. Ann Hepatobiliary Pancreat Surg. 2021;25:315–327. doi: 10.14701/ahbps.2021.25.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vietri MT, D'Elia G, Caliendo G, Albanese L, Signoriello G, Napoli C, Molinari AM. Pancreatic Cancer with Mutation in BRCA1/2, MLH1, and APC Genes: Phenotype Correlation and Detection of a Novel Germline BRCA2 Mutation. Genes (Basel) 2022;13 doi: 10.3390/genes13020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed S, Bradshaw AD, Gera S, Dewan MZ, Xu R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J Clin Med. 2017;6 doi: 10.3390/jcm6010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan AA, Liu X, Yan X, Tahir M, Ali S, Huang H. An overview of genetic mutations and epigenetic signatures in the course of pancreatic cancer progression. Cancer Metastasis Rev. 2021;40:245–272. doi: 10.1007/s10555-020-09952-0. [DOI] [PubMed] [Google Scholar]
- 16.Yang G, Yuan Y, Yuan H, Wang J, Yun H, Geng Y, Zhao M, Li L, Weng Y, Liu Z, Feng J, Bu Y, Liu L, Wang B, Zhang X. Histone acetyltransferase 1 is a succinyltransferase for histones and non-histones and promotes tumorigenesis. EMBO Rep. 2021;22:e50967. doi: 10.15252/embr.202050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Zhao J. Targeted Cancer Therapy Based on Acetylation and Deacetylation of Key Proteins Involved in Double-Strand Break Repair. Cancer Manag Res. 2022;14:259–271. doi: 10.2147/CMAR.S346052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JH, Bollschweiler D, Schäfer T, Huber R. Structural basis for the regulation of nucleosome recognition and HDAC activity by histone deacetylase assemblies. Sci Adv. 2021;7 doi: 10.1126/sciadv.abd4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Wang F, Chen X, Wang J, Zhao Y, Li Y, He B. Zinc-dependent Deacetylase (HDAC) Inhibitors with Different Zinc Binding Groups. Curr Top Med Chem. 2019;19:223–241. doi: 10.2174/1568026619666190122144949. [DOI] [PubMed] [Google Scholar]
- 20.Milazzo G, Mercatelli D, Di Muzio G, Triboli L, De Rosa P, Perini G, Giorgi FM. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes (Basel) 2020;11 doi: 10.3390/genes11050556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shinke G, Yamada D, Eguchi H, Iwagami Y, Asaoka T, Noda T, Wada H, Kawamoto K, Gotoh K, Kobayashi S, Takeda Y, Tanemura M, Mori M, Doki Y. Role of histone deacetylase 1 in distant metastasis of pancreatic ductal cancer. Cancer Sci. 2018;109:2520–2531. doi: 10.1111/cas.13700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang R, Zhang X, Sophia S, Min Z, Liu X. Clinicopathological features and prediction values of HDAC1, HDAC2, HDAC3, and HDAC11 in classical Hodgkin lymphoma. Anticancer Drugs. 2018;29:364–370. doi: 10.1097/CAD.0000000000000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krauß L, Urban BC, Hastreiter S, Schneider C, Wenzel P, Hassan Z, Wirth M, Lankes K, Terrasi A, Klement C, Cernilogar FM, Öllinger R, de Andrade Krätzig N, Engleitner T, Schmid RM, Steiger K, Rad R, Krämer OH, Reichert M, Schotta G, Saur D, Schneider G. HDAC2 Facilitates Pancreatic Cancer Metastasis. Cancer Res. 2022;82:695–707. doi: 10.1158/0008-5472.CAN-20-3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai MH, Xu XG, Yan SL, Sun Z, Ying Y, Wang BK, Tu YX. Depletion of HDAC1, 7 and 8 by Histone Deacetylase Inhibition Confers Elimination of Pancreatic Cancer Stem Cells in Combination with Gemcitabine. Sci Rep. 2018;8:1621. doi: 10.1038/s41598-018-20004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu G, He N, Cai C, Cai F, Fan P, Zheng Z, Jin X. HDAC3 modulates cancer immunity via increasing PD-L1 expression in pancreatic cancer. Pancreatology. 2019;19:383–389. doi: 10.1016/j.pan.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Y, Jin X, Ma J, Ding D, Huang Z, Sheng H, Yan Y, Pan Y, Wei T, Wang L, Wu H, Huang H. HDAC5 Loss Impairs RB Repression of Pro-Oncogenic Genes and Confers CDK4/6 Inhibitor Resistance in Cancer. Cancer Res. 2021;81:1486–1499. doi: 10.1158/0008-5472.CAN-20-2828. [DOI] [PubMed] [Google Scholar]
- 27.He X, Hui Z, Xu L, Bai R, Gao Y, Wang Z, Xie T, Ye XY. Medicinal chemistry updates of novel HDACs inhibitors (2020 to present) Eur J Med Chem. 2022;227:113946. doi: 10.1016/j.ejmech.2021.113946. [DOI] [PubMed] [Google Scholar]
- 28.Lee HS, Park SB, Kim SA, Kwon SK, Cha H, Lee DY, Ro S, Cho JM, Song SY. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci Rep. 2017;7:41615. doi: 10.1038/srep41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dovzhanskiy DI, Arnold SM, Hackert T, Oehme I, Witt O, Felix K, Giese N, Werner J. Experimental in vivo and in vitro treatment with a new histone deacetylase inhibitor belinostat inhibits the growth of pancreatic cancer. BMC Cancer. 2012;12:226. doi: 10.1186/1471-2407-12-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park S, Park JA, Jeon JH, Lee Y. Traditional and Novel Mechanisms of Heat Shock Protein 90 (HSP90) Inhibition in Cancer Chemotherapy Including HSP90 Cleavage. Biomol Ther (Seoul) 2019;27:423–434. doi: 10.4062/biomolther.2019.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laschanzky RS, Humphrey LE, Ma J, Smith LM, Enke TJ, Shukla SK, Dasgupta A, Singh PK, Howell GM, Brattain MG, Ly QP, Black AR, Black JD. Selective Inhibition of Histone Deacetylases 1/2/6 in Combination with Gemcitabine: A Promising Combination for Pancreatic Cancer Therapy. Cancers (Basel) 2019;11 doi: 10.3390/cancers11091327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peulen O, Gonzalez A, Peixoto P, Turtoi A, Mottet D, Delvenne P, Castronovo V. The anti-tumor effect of HDAC inhibition in a human pancreas cancer model is significantly improved by the simultaneous inhibition of cyclooxygenase 2. PLoS One. 2013;8:e75102. doi: 10.1371/journal.pone.0075102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai J, Demirjian A, Sui J, Marasco W, Callery MP. Histone deacetylase inhibitor trichostatin A and proteasome inhibitor PS-341 synergistically induce apoptosis in pancreatic cancer cells. Biochem Biophys Res Commun. 2006;348:1245–1253. doi: 10.1016/j.bbrc.2006.07.185. [DOI] [PubMed] [Google Scholar]
- 34.Feng W, Cai D, Zhang B, Lou G, Zou X. Combination of HDAC inhibitor TSA and silibinin induces cell cycle arrest and apoptosis by targeting survivin and cyclinB1/Cdk1 in pancreatic cancer cells. Biomed Pharmacother. 2015;74:257–264. doi: 10.1016/j.biopha.2015.08.017. [DOI] [PubMed] [Google Scholar]
- 35.Li H, Zhang Z, Gao C, Wu S, Duan Q, Wu H, Wang C, Shen Q, Yin T. Combination chemotherapy of valproic acid (VPA) and gemcitabine regulates STAT3/Bmi1 pathway to differentially potentiate the motility of pancreatic cancer cells. Cell Biosci. 2019;9:50. doi: 10.1186/s13578-019-0312-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karagiannis D, Rampias T. HDAC Inhibitors: Dissecting Mechanisms of Action to Counter Tumor Heterogeneity. Cancers (Basel) 2021;13 doi: 10.3390/cancers13143575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rana Z, Diermeier S, Walsh FP, Hanif M, Hartinger CG, Rosengren RJ. Anti-Proliferative, Anti-Angiogenic and Safety Profiles of Novel HDAC Inhibitors for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Pharmaceuticals (Basel) 2021;14 doi: 10.3390/ph14101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moku G, Vangala S, Yakati V, Gali CC, Saha S, Madamsetty VS, Vyas A. Novel Suberoylanilide Hydroxamic Acid Analogs Inhibit Angiogenesis and Induce Apoptosis in Breast Cancer Cells. Anticancer Agents Med Chem. 2022;22:914–925. doi: 10.2174/1871520621666210901102425. [DOI] [PubMed] [Google Scholar]
- 39.Bondarev AD, Attwood MM, Jonsson J, Chubarev VN, Tarasov VV, Schiöth HB. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br J Clin Pharmacol. 2021;87:4577–4597. doi: 10.1111/bcp.14889. [DOI] [PubMed] [Google Scholar]
- 40.VanderMolen KM, McCulloch W, Pearce CJ, Oberlies NH. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. J Antibiot (Tokyo) 2011;64:525–531. doi: 10.1038/ja.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 42.Fenichel MP. FDA approves new agent for multiple myeloma. J Natl Cancer Inst. 2015;107:djv165. doi: 10.1093/jnci/djv165. [DOI] [PubMed] [Google Scholar]
- 43.Liu X, Xu J, Zhang B, Liu J, Liang C, Meng Q, Hua J, Yu X, Shi S. The reciprocal regulation between host tissue and immune cells in pancreatic ductal adenocarcinoma: new insights and therapeutic implications. Mol Cancer. 2019;18:184. doi: 10.1186/s12943-019-1117-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas D, Radhakrishnan P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer. 2019;18:14. doi: 10.1186/s12943-018-0927-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15:366–381. doi: 10.1038/s41571-018-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol. 2019;234:8509–8521. doi: 10.1002/jcp.27782. [DOI] [PubMed] [Google Scholar]
- 47.Gorchs L, Fernández Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, Rangelova E, Kaipe H. Human Pancreatic Carcinoma-Associated Fibroblasts Promote Expression of Co-inhibitory Markers on CD4+ and CD8+ T-Cells. Front Immunol. 2019;10:847. doi: 10.3389/fimmu.2019.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banik D, Moufarrij S, Villagra A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20092241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Booth L, Roberts JL, Poklepovic A, Kirkwood J, Dent P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget. 2017;8:83155–83170. doi: 10.18632/oncotarget.17950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao K, Wang G, Li W, Zhang L, Wang R, Huang Y, Du L, Jiang J, Wu C, He X, Roberts AI, Li F, Rabson AB, Wang Y, Shi Y. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene. 2015;34:5960–5970. doi: 10.1038/onc.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCaw TR, Goel N, Brooke DJ, Katre AA, Londoño AI, Smith HJ, Randall TD, Arend RC. Class I histone deacetylase inhibition promotes CD8 T cell activation in ovarian cancer. Cancer Med. 2021;10:709–717. doi: 10.1002/cam4.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Knudsen ES, Vail P, Balaji U, Ngo H, Botros IW, Makarov V, Riaz N, Balachandran V, Leach S, Thompson DM, Chan TA, Witkiewicz AK. Stratification of Pancreatic Ductal Adenocarcinoma: Combinatorial Genetic, Stromal, and Immunologic Markers. Clin Cancer Res. 2017;23:4429–4440. doi: 10.1158/1078-0432.CCR-17-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, Wells DK, Cary CIO, Grbovic-Huezo O, Attiyeh M, Medina B, Zhang J, Loo J, Saglimbeni J, Abu-Akeel M, Zappasodi R, Riaz N, Smoragiewicz M, Kelley ZL, Basturk O Australian Pancreatic Cancer Genome Initiative; Garvan Institute of Medical Research; Prince of Wales Hospital; Royal North Shore Hospital; University of Glasgow; St Vincent’s Hospital; QIMR Berghofer Medical Research Institute; University of Melbourne, Centre for Cancer Research; University of Queensland, Institute for Molecular Bioscience; Bankstown Hospital; Liverpool Hospital; Royal Prince Alfred Hospital, Chris O’Brien Lifehouse; Westmead Hospital; Fremantle Hospital; St John of God Healthcare; Royal Adelaide Hospital; Flinders Medical Centre; Envoi Pathology; Princess Alexandria Hospital; Austin Hospital; Johns Hopkins Medical Institutes; ARC-Net Centre for Applied Research on Cancer, Gönen M, Levine AJ, Allen PJ, Fearon DT, Merad M, Gnjatic S, Iacobuzio-Donahue CA, Wolchok JD, DeMatteo RP, Chan TA, Greenbaum BD, Merghoub T, Leach SD. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512–516. doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Foucher ED, Ghigo C, Chouaib S, Galon J, Iovanna J, Olive D. Pancreatic Ductal Adenocarcinoma: A Strong Imbalance of Good and Bad Immunological Cops in the Tumor Microenvironment. Front Immunol. 2018;9:1044. doi: 10.3389/fimmu.2018.01044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Setiadi AF, Omilusik K, David MD, Seipp RP, Hartikainen J, Gopaul R, Choi KB, Jefferies WA. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008;68:9601–9607. doi: 10.1158/0008-5472.CAN-07-5270. [DOI] [PubMed] [Google Scholar]
- 56.McCaw TR, Li M, Starenki D, Liu M, Cooper SJ, Arend RC, Forero A, Buchsbaum DJ, Randall TD. Histone deacetylase inhibition promotes intratumoral CD8+ T-cell responses, sensitizing murine breast tumors to anti-PD1. Cancer Immunol Immunother. 2019;68:2081–2094. doi: 10.1007/s00262-019-02430-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ni L, Wang L, Yao C, Ni Z, Liu F, Gong C, Zhu X, Yan X, Watowich SS, Lee DA, Zhu S. The histone deacetylase inhibitor valproic acid inhibits NKG2D expression in natural killer cells through suppression of STAT3 and HDAC3. Sci Rep. 2017;7:45266. doi: 10.1038/srep45266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krug LM, Kindler HL, Calvert H, Manegold C, Tsao AS, Fennell D, Öhman R, Plummer R, Eberhardt WE, Fukuoka K, Gaafar RM, Lafitte JJ, Hillerdal G, Chu Q, Buikhuisen WA, Lubiniecki GM, Sun X, Smith M, Baas P. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 2015;16:447–456. doi: 10.1016/S1470-2045(15)70056-2. [DOI] [PubMed] [Google Scholar]
- 59.Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, Woolman S, Armstrong TD, Connolly RM, Azad NA, Jaffee EM, Roussos Torres ET. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol Res. 2018;6:1561–1577. doi: 10.1158/2326-6066.CIR-18-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baretti M, Durham JN, Walker R, Mitcheltree A-L, Christmas B, Cope L, Jaffee EM, Azad NS. Entinostat in combination with nivolumab for patients with advanced cholangiocarcinoma and pancreatic adenocarcinoma. J Clin Oncol . 2018;36(15_suppl):TPS4151–TPS. [Google Scholar]
- 61.Cao J, Yan Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer. 2020;6:580–592. doi: 10.1016/j.trecan.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villanueva L, Álvarez-Errico D, Esteller M. The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol. 2020;41:676–691. doi: 10.1016/j.it.2020.06.002. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, Ma JA, Zhang HX, Jiang YN, Luo WH. Cancer vaccines: Targeting KRAS-driven cancers. Expert Rev Vaccines. 2020;19:163–173. doi: 10.1080/14760584.2020.1733420. [DOI] [PubMed] [Google Scholar]
- 64.Badamchi-Zadeh A, Moynihan KD, Larocca RA, Aid M, Provine NM, Iampietro MJ, Kinnear E, Penaloza-MacMaster P, Abbink P, Blass E, Tregoning JS, Irvine DJ, Barouch DH. Combined HDAC and BET Inhibition Enhances Melanoma Vaccine Immunogenicity and Efficacy. J Immunol. 2018;201:2744–2752. doi: 10.4049/jimmunol.1800885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim S, Kwon D, Koh J, Nam SJ, Kim YA, Kim TM, Kim CW, Jeon YK. Clinicopathological features of programmed cell death-1 and programmed cell death-ligand-1 expression in the tumor cells and tumor microenvironment of angioimmunoblastic T cell lymphoma and peripheral T cell lymphoma not otherwise specified. Virchows Arch. 2020;477:131–142. doi: 10.1007/s00428-020-02790-z. [DOI] [PubMed] [Google Scholar]
- 66.Luo F, Cao J, Lu F, Zeng K, Ma W, Huang Y, Zhang L, Zhao H. Lymphocyte activating gene 3 protein expression in nasopharyngeal carcinoma is correlated with programmed cell death-1 and programmed cell death ligand-1, tumor-infiltrating lymphocytes. Cancer Cell Int. 2021;21:458. doi: 10.1186/s12935-021-02162-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang W, Zhang K, Zhang P, Zheng J, Min C, Li X. Research Progress of Pancreas-Related Microorganisms and Pancreatic Cancer. Front Oncol. 2020;10:604531. doi: 10.3389/fonc.2020.604531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu C, Talukder A, Savage NM, Singh N, Liu K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology. 2017;6:e1291106. doi: 10.1080/2162402X.2017.1291106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim YD, Park SM, Ha HC, Lee AR, Won H, Cha H, Cho S, Cho JM. HDAC Inhibitor, CG-745, Enhances the Anti-Cancer Effect of Anti-PD-1 Immune Checkpoint Inhibitor by Modulation of the Immune Microenvironment. J Cancer. 2020;11:4059–4072. doi: 10.7150/jca.44622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, Chintala S, Ordentlich P, Kao C, Elzey B, Gabrilovich D, Pili R. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin Cancer Res. 2017;23:5187–5201. doi: 10.1158/1078-0432.CCR-17-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, Massengill C, Noyes DR, Martinez GV, Afzal R, Chen Z, Ren X, Antonia SJ, Haura EB, Ruffell B, Beg AA. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res. 2016;22:4119–4132. doi: 10.1158/1078-0432.CCR-15-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 73.Hafez DA, Hassanin IA, Teleb M, Khattab SN, Elkhodairy KA, Elzoghby AO. Recent advances in nanomedicine-based delivery of histone deacetylase inhibitors for cancer therapy. Nanomedicine (Lond) 2021;16:2305–2325. doi: 10.2217/nnm-2021-0196. [DOI] [PubMed] [Google Scholar]
- 74.Maniam G, Mai CW, Zulkefeli M, Fu JY. Co-encapsulation of gemcitabine and tocotrienols in nanovesicles enhanced efficacy in pancreatic cancer. Nanomedicine (Lond) 2021;16:373–389. doi: 10.2217/nnm-2020-0374. [DOI] [PubMed] [Google Scholar]