

Abstract
One application area of computational methods in drug discovery is the automated design of small molecules. Despite the large number of publications describing methods and their application in both retrospective and prospective studies, there is a lack of agreement on terminology and key attributes to distinguish these various systems. We introduce Automated Chemical Design (ACD) Levels to clearly define the level of autonomy along the axes of ideation and decision making. To fully illustrate this framework, we provide literature exemplars and place some notable methods and applications into the levels. The ACD framework provides a common language for describing automated small molecule design systems and enables medicinal chemists to better understand and evaluate such systems.
Introduction
A great deal of attention is currently directed toward computational (especially Artificial Intelligence) methods in drug discovery and development.1−3 Many publications,4−7 conferences,8−10 and company meetings are devoted to describing, understanding, and evaluating the latest methods and results. As discussed in this Perspective, some groups have assembled these methods into complete systems that choose or design molecules with varying levels of machine intelligence and autonomy. Despite this great interest, there is little consistency or shared understanding on how to describe computational drug discovery systems such that relevant similarities and differences can be clearly understood and examined.
In this Perspective, we propose definitions to describe systems for one application area of computational methods in drug discovery: the automated design of small molecules, which we will refer to as Automated Chemical Design (ACD). We focus on small molecules (as opposed to other modalities such as protein therapeutics or antibodies) as it enables us to sharpen the discussion to the particular challenges for small molecules and draw relevant distinctions between systems. Specifically, the problem of chemical design is to locate, in the vast space11 of potential small molecules, a molecule with an acceptable set of properties to be a drug. These properties would naturally include engagement with the desired target, selectivity compared to undesired targets, favorable pharmacokinetic profile, and synthetic accessibility. We assume this process must be done iteratively through gradually improving molecules that satisfy some but not all of the desired properties.
We wish to emphasize the focus on design. The framework and definitions here intentionally do not address the automation of synthesis or testing of compounds. Automated design systems can just as easily send their choices to a team of chemists and biologists for execution as they can send instructions to automated laboratories. While we believe the automated execution of many lab experiments is an important direction in the field, it is not the focus of this framework. Similarly, although automated retrosynthetic planning is an important enabling technology for some systems, it is not fundamental to any of the definitions here.
Before describing the framework for categorizing automated chemical design systems, it is important to consider what value this automation could add. Automated design can potentially reduce iteration cycle time, require fewer compounds and iterations to produce a candidate, and scale to more programs. These potential advantages have been discussed elsewhere.12−14
Further, we wish to emphasize an under-discussed benefit of automation. Across many fields, the most reliable way to improve at a task is to experiment and get feedback. The process of becoming an expert medicinal chemist takes years of experience observing the effects of various decisions. Especially in medicinal chemistry, the feedback is noisy and it is difficult to properly attribute the causes of success and failure. For example, it is difficult or impossible to run a clean side by side experiment to compare different chemical exploration strategies, since chemists cannot simply “forget” everything they have learned and start again with a different strategy. However, machines are perfectly capable of being reset to the same initial state. This means that once a system is sufficiently automated, you can run clean, meaningful, and relatively easy to interpret experiments on the ideation and decision making process.15
Experiments also help focus effort on components of the process that will lead to the most meaningful improvements. For instance, it is common for machine learning researchers to build new generative models or better property prediction models with relatively abstract and reductionist objectives such as improving performance on a benchmark data set. If these research programs were pursued in the broader context of a complete design-make-test system, it would be easier to assess the real-world value of modeling improvements. This ability to experiment and get accurate feedback will translate to continual improvement in the system itself. Failures of the system to make progress on a target can be examined and understood at a level of detail that is nearly impossible for less automated systems. This naturally implies that organizations that are willing to invest in these experiments will have a powerful tool for driving continual improvement.
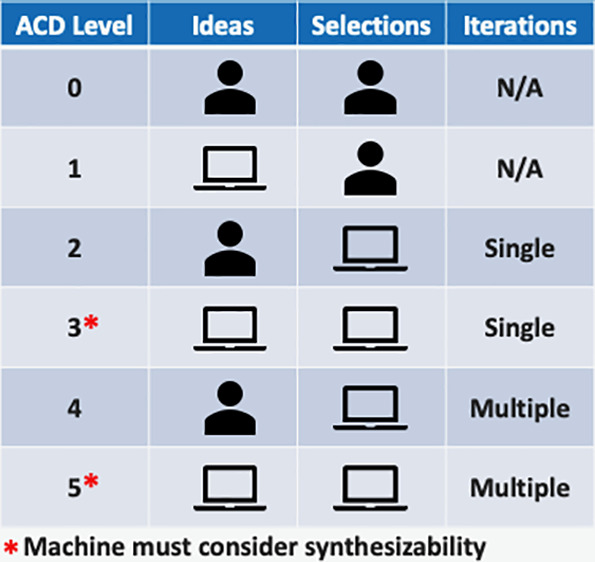
We break autonomy, as it relates to ACD, into six levels arranged on two axes as shown in Figure 1. The axes represent two distinct dimensions for defining automated design. While other authors have discussed key questions around levels of automation,13,16 the ACD framework is more complete with better defined distinctions between levels. We first precisely define the axes and levels and illustrate the levels with published systems from the literature. These examples are not intended to be a complete review; we focus on the most compelling examples that illustrate the technical accomplishments and challenges of each level. In our descriptions, we will be careful to distinguish when complete vs partial systems have been assembled using the definitions in Table 1. We will then discuss the challenges of moving to more effective and more automated design systems and review the key points of partnership between humans and machines even at the most automated levels.
Figure 1.

Summary graphic for the two dimensions of design automation and the named ACD levels defined in this Perspective. The x-axis represents where the final decision making power lies and the y-axis represents the source of molecular ideas.
Table 1. Definitions of Words Used to Describe Previous Literature as It Relates to the ACD Framework.
| ACD system | A complete system (both human and machine) that has been used to create and test molecules in the real world. |
| components | Algorithms and models that address some of the problems a complete ACD system must address (such as molecular property prediction or decision making under uncertainty). |
| potential ACD system | An assembly of components that would form an ACD system except that it has only been tested in virtual or retrospective ways. |
| approaching an ACD Level X system | A system which almost matches the description of an ACD system at level X, but adds a crucial point of human decision making. |
ACD Level 0
Description
In an ACD Level 0 system, the molecules are explicitly designed and selected by a chemist. This approach, which has been effectively applied for more than 100 years, is still the pervasive model in industrial and academic drug discovery research.
As represented on the x-axis in Figure 1, the decision making for what molecules to make next is solely in the hands of the chemist (“Chemist decides”). The chemist may be looking at machine computed or predicted properties to enable better or more efficient decision making, but the final decision making power resides with the chemist.
Good prediction of molecular properties is a powerful tool for design. When asked how he had so many good ideas, Linus Pauling famously said “Well, you just have lots of ideas and throw away the bad ones.″17 Predictive models enable you to “throw away the bad ones.” These predictive models can take a variety of forms. One can employ filtering rules that specify the types of molecules that should be selected or ignored. For instance, Lipinski’s Rule of 518 has been used to identify molecules that reside within “drug-like” space. The PAINS (pan-assay interference compounds) filters19 have been used to identify molecules that may interfere with some assays and provide a misleading readout. A wide array of additional computational techniques can also be applied to the prioritization task. Molecules can be scored based on the output of molecular similarity calculations,20 molecular shape,21 complementarity to a protein binding site,22 or many other factors. When optimizing a congeneric series, methods such as Free Energy Perturbation (FEP)23 can be used to estimate binding affinity and rank order molecules. Over the past decade, machine learning models have become an essential adjunct to computational and medicinal chemists.4 When used to predict a molecule’s properties or biological activity, a machine learning model identifies patterns of molecular features which can be related to the value being predicted. These values, often referred to as labels, can be either categorical, for example, “soluble” or “insoluble” or real-valued. In early machine learning work, molecules were typically represented by molecular fingerprint vectors24 where each position in the vector corresponded to the presence or absence of specific chemical substructures. More recently, several groups have developed methods that use neural networks to learn a molecular representation.25 These learned representations can be roughly divided into two categories: string representations and graph representations. String representations take their inspiration from work in language translation and produce molecular representations based on string encodings of molecules such as SMILES (Simplified Molecular Input Line Entry Specification).26 In graph-based machine learning, a molecule is represented as a graph with nodes corresponding to atoms and edges corresponding to bonds. A wide array of methods continues to be developed for passing information among nodes and edges and generating the final vector representation of the molecule.
These predictive models can help chemists make decisions at ACD Level 0, but they are especially critical as systems move to higher levels of autonomy.
As represented on the y-axis of Figure 1, at ACD Level 0, the ideas for molecules come from the chemist (“Chemist defines recipe”). The meaning of “recipe” will be more fully covered in ACD Level 1, but briefly, the chemist precisely defines all the molecules to be considered. The simplest and perhaps most common approach is for a chemist to sketch a specific set of molecules. For a slightly more complex process, a chemist can enumerate a set of molecules. This enumeration can be driven by one or more chemical reactions and building blocks; the use of precedented reactions and reagents allows a chemist to have some confidence that the generated molecules can be synthesized. There are, of course, numerous other approaches to enumerating chemical structures. One can apply R-group substitutions to attach functional groups at specific positions on a structure or utilize techniques such as positional analog scanning27 to generate sets of closely related molecules. Several alternate approaches28,29 analyze databases of bioactive molecules to identify common medicinal chemistry transformations. These transformations can then be programmatically applied to chemical structures to generate plausible analogs.
Finer-grained distinctions could be made about how much assistance the machine gives the chemist at ACD Level 0. For example, a system where a machine ranks a million possibilities and the chemist reviews the top 100 has more machine assistance than one where the chemist reviews every idea. However, to keep a clean and understandable definition, we put all systems where the chemist has the final say at this lowest degree of decision making autonomy.
Exemplars
One example of an efficient ACD Level 0 system is described in a 2016 paper by Gomez-Bombarelli et al.30 In this paper, the authors describe a collaborative system for the design of materials for organic light-emitting diodes (OLEDs). Their process begins with a set of 1.6 million molecules generated through a library enumeration process. These molecules were then prioritized using an ML model, and molecules with the best scores from the model were subjected to quantum chemical calculations. Following the calculation steps, a team of chemists used a web portal to select molecules based on human assessments of calculated values, novelty, and synthesizability. A consensus approach was then used to select molecules that would be processed in a subsequent round of selections. After several cycles of this design process, a number of novel materials for OLEDs were obtained. While this system incorporates a number of automated processes, the design of the initial library, as well as the selection of molecules at each iteration, was driven by human chemists.
ACD Level 1
Description
ACD Level 1 systems move up the scale of ideation automation, represented by the y-axis in Figure 1, to “Machine generates ideas.” We make one primary distinction to separate human centered and machine centered design with the concept of a “recipe”. At ACD Level 0, “Chemist defines recipe” means the chemist specifies precisely how to construct molecules from existing chemical matter and what molecular ingredients are available. We consider this degree of autonomy to use fixed, enumerated lists of ingredients, such as a set of building blocks available from a vendor. As the length of the list of ingredients grows, the chemist may not review every member of this list and therefore could be surprised at some of the generated molecules. However, this surprise comes from the list of ingredients and not how they were used; the creativity and design idea came solely from the chemist. For clarity, we consider any fully enumerated list where the chemist has designed each molecule individually to be a very simple form of a “recipe”.
Another key issue for a chemist-defined recipe is that the chemist is responsible for ensuring the synthesizability of the molecules. While no human or machine can perfectly predict whether a given synthesis will be successful (and chemists will not agree on some molecules), we expect the molecular ideas to generally be amenable to a skilled chemist producing a successful synthetic approach.
The transition to ACD Level 1 means that the machine no longer has simple rote directions. With rote directions, when given an output from the generation process, it will be obvious how that molecule resulted from those directions. For machine generated ideas, the process and reason for a particular output molecule will no longer be obvious. While there is some gray area between these, most ideation processes we have seen fall clearly to one side or the other. The hallmark of this transition is that the produced molecules can surprise the chemist, and not just because the chemist did not review the entire list of possible ingredients.
We note that within the scope of an ACD Level 1 system, the chemist may provide variable amounts of guidance or constraints. A typical constrained generation would be allowing the machine to only modify one vector off of a fixed core. These constraints can provide natural ways for the chemist to guide the machine’s efforts in much the way a medicinal chemistry team may decide to focus on one aspect of a molecule. Generating less constrained molecules is a technical challenge for generation as well as potentially making the decision making process harder.
Even though the machine is generating ideas, at ACD Level 1, the ultimate selection of molecules to be synthesized is performed by a human chemist. A key job for the chemist is to decide which of the machine’s ideas are synthesizable. Better machine ideation algorithms will produce a higher rate of synthesizable molecules, but this assessment by the chemist is fundamentally tied to the evaluation for fitness.
Exemplars
Over the last five years, we have seen a renaissance in the development of de novo design methods. Advances from fields such as image analysis, language translation, and reinforcement learning have been adapted to molecule generation. These methods have been coupled with predictive models and will likely become key components of ACD approaches. While there have been dozens, perhaps hundreds of recent publications describing generative models for chemistry,5,31,32 there has been a paucity of prospective studies that resulted in the synthesis and testing of molecules.
The earliest work we know of using these recent style of generative models is a potential ACD Level 1 system by Gomez-Bombarelli.33 In this paper, the authors used a type of neural network known as an autoencoder to encode molecules into a continuous vector representation known as a latent space. Points in this latent space can subsequently be decoded to generate new molecules represented as molecular graphs or text strings. By selecting latent space coordinates near the representation of a molecule in the latent space, one can generate representations of similar molecules. The traversal of latent space can be coupled with predictive models to generate the structures of molecules predicted to be optimal by the model. While the paper by Gomez-Bombarelli demonstrated the ability of latent space traversal to optimize the scores for computed functions, the lack of experimental testing leads this to be categorized as a potential ACD Level 1 system.
One complete ACD Level 1 system was described in a 2018 paper by Merk et al.34 The authors use a type of neural network called recurrent neural networks (RNNs). When applied to text, an RNN begins by analyzing a large corpus of documents and collecting distributions of words that tend to follow other words. Given these distributions, an RNN can, among other things, suggest that the phrase “for lunch” would follow the phrase “would you like to meet”. In a similar fashion, the authors use an RNN to analyze a set of molecules represented by Simplified Molecular Input Line Entry System (SMILES) strings and identify groups of characters, representing atoms, that tend to occur together. They train on a set of more than 500 000 bioactive molecules from the ChEMBL database. The RNN was subsequently tuned on a task-specific set of 25 fatty acid mimetics with reported activity against RXR and PPAR. A set of computational models was then used to rank 1000 structures generated by the RNN and 49 molecules were selected by visual inspection to assess synthesizability and novelty, resulting in the selection of five compounds. In subsequent testing in a reporter gene assay, four of the five compounds demonstrated agonist activity against at least one RXR or PPAR subtype.
A potential ACD Level 1 system with a similar technical approach was published by Popova et al.35 The authors add an additional Reinforcement Learning (RL) step during the generation. RL is a branch of machine learning that focuses on how to identify optimal actions in a framework where there is a series of actions that change the state of the world and the value of an action might not be known for many more steps. The generation of a molecule is considered a series of actions of adding characters to a SMILES string. The RL step learns to generate SMILES strings that a separately trained predictive model rates highly.
ACD Level 2
Description
The move to ACD Level 2 is a move up the decision making automation axis (“Machine decides, single iteration”) while the human still determines the chemical space to explore on the ideation axis (“Chemist defines recipe”). The machine makes the final decisions on which molecules to make next, but this is only done a single time without the opportunity for multiple feedback cycles. This is common for hit finding systems or a single round of expansion around known hits. Typically, many molecules would be identified and tested in parallel. The transition to an ACD Level 2 system represents a dramatic departure from the typical role of computation in drug discovery, from human-driven selection to computer-driven selection. Many of the implicit assumptions and biases in an ACD Level 1 system become explicit in an ACD Level 2 system, requiring a much more systematic definition of the search space and objectives in order to match ACD Level 0 or 1 performance. For example, chemist-driven selections in an ACD Level 1 system may implicitly consider multiple properties and trade-offs between them, like balancing potency and logP; in an ACD Level 2 system, these objectives must be defined explicitly and have associated models for predicting and combining them. In lower level systems, the chemist is relied on to fix poor choices by the machine and we are left wondering whether outcomes from the system can be attributed to the machine or to the skilled chemist.15 Moving to this degree of decision making autonomy allows for understanding the value of the machine’s decision making.
ACD Level 2 systems will typically have to consider multiple desired properties, typically termed multiparameter optimization (MPO), which integrates multiple scores into a decision making process. This MPO score can be a simple weighted sum of different scores or can employ more sophisticated functional forms to adjust the weights of specific parameters.36−40
It is important to restate that while ACD Level 2 requires that the decision making be automated, the execution of synthesis and assays does not have to be automated. Further, some of the designed molecules may fail synthesis. Even if humans are evaluating or attempting synthesis, the key point is that they are making decisions about synthesizability only, not about the value of a molecule to the overall goals.
Exemplars
A recent study by Sadybekov et al.41 on gigascale virtual docking is a prototypical example of an ACD Level 2 system. The authors designed a computer program that can search the approximately 11 billion molecules originating from the 129 reactions in the Enamine REAL Space. The algorithm exploits the combinatorial nature of the molecular libraries to examine only a small portion of the input space and identify high scoring molecules. Top-ranked molecules were clustered and standard cheminformatic filtering applied to identify compounds for synthesis. The program was prospectively tested on two protein systems (a cannabinoid receptor and kinase ROCK1) and successfully found submicromolar hits. This is a prototypical example of a ACD Level 2 system as the chemical search space was precisely defined by reaction schemes and reagents, while the final compounds for synthesis were selected algorithmically.
Stokes et al.42 trained a machine learning model on 2335 molecules with experimental data to predict E. coli growth inhibition and used it to screen 6111 molecules from the Drug Repurposing Hub. The top 99 unique molecules were tested experimentally, and 51 molecules showed antibacterial activity (vs only two from the bottom 63 predictions). These new experimental data were added to the previous data and a new model was trained and used to screen the WuXi antituberculosis library; however, none of the 300 molecules tested (200 top-scoring and 100 bottom-scoring) showed antibacterial activity. A third model was trained using the accumulated data and used to screen a subset of the ZINC15 database of commercially available molecules. From a set of more than 107 M molecules, 23 were selected with high prediction scores and low similarity to known antibiotics; eight of these 23 molecules showed growth inhibition against at least one of three species tested. The experiments in this paper are good examples of ACD Level 2 systems because each used a defined chemical space and only relied on algorithmic predictions or measurements for compound selection.
Konze et al.43 used FEP (Free Energy Perturbation) simulations to construct a potential ACD Level 2 system based on enumeration constrained by a synthetic route (PathFinder). The first step of the process uses template-based retrosynthesis to identify potential synthetic routes to a known starting compound. After the preferred route is chosen, candidate molecules are generated with reaction-based enumeration that applies the selected route to a set of building blocks. The enumerated library is then filtered based on property criteria and docking to reduce the candidate set to a size appropriate for more expensive FEP calculations. Since the library is still likely to be too large to run every molecule through a full FEP simulation, the authors used short FEP simulations to train a machine learning model for predicting potency: a random set of molecules are chosen for the initial training set, and the model is used to select the next batch of compounds for evaluation; this was repeated four times, after which the model was used to select compounds for full FEP simulations. This system tackles the synthesizability challenge by limiting compound generation to a single synthetic route. It is a potential ACD Level 2 system because the evaluation of selected compounds was purely in silico rather than experimental.
McCloskey et al.44 described a system approaching ACD Level 2. The authors trained machine learning models to predict protein binding using experimental data from DNA-encoded library (DEL) selections. These models were then used to search two enumerated sets of molecules: the MCule vendor catalog and an internal database based on building block chemistry. Top-scoring selected compounds were further pruned by sphere exclusion clustering and “automated or automatable filters” including restricted chemist review for reactivity and assay interference. The system was applied to three protein systems sEH (a hydrolase), ERα (a nuclear receptor), and c-KIT (a kinase), and successfully identified potent (IC50 < 10 nM) molecules for all three systems. This system has many of the components of an ACD Level 2 system, but due to the limited manual chemist review of selected compounds this system is classified as approaching ACD Level 2.
Lyu et al.22 performed experiments that directly compared their ACD Level 1 and ACD Level 2 systems. Both experiments began by docking 170 million molecules into AmpC β-lactamase and the D4 dopamine receptor before diverging into “person” and “machine” workflows. In the “person” approach, the 1000 top scoring molecules were visually inspected and 124 were selected to be synthesized and screened. In the “machine” approach, the top 114 molecules were selected purely based on the docking score. The hit rates from the two screens were similar at approximately 24%, but the “person” approach identified more potent molecules. As the field of automated chemical design progresses, it will be important to conduct additional objective comparisons of ACD Level 2 and lower level systems.
ACD Level 3
Description
An ACD Level 3 system is distinguished primarily by the definition of the search space. In an ACD Level 2 system, the chemical space is explicitly defined by a recipe, and the machine chooses compounds exclusively from that set of molecules. In an ACD Level 3 system, this constraint is relaxed to allow for automated non-recipe approaches to molecule generation that explore chemical space with fewer restrictions. Note that like ACD Level 2, an ACD Level 3 system still requires machine-driven selection; a system with machine-generated molecules that relies on human selection would be classified as ACD Level 1. The key technical challenge for moving to an ACD Level 3 system is assessing the synthetic feasibility of candidate molecules that are generated by the machine. In lower-level systems, the responsibility for determining what can be made falls to humans as they define the chemical space or select synthesizable compounds, but in an ACD Level 3 system, the machine is required to generate the chemical space and prioritize compounds without human intervention. The machine should produce mostly synthesizable compounds (see the “Synthesizability” section below for more discussion of this challenge).
Exemplars
Morris et al.45 described an ACD Level 3 system that combined property modeling and prediction of synthetic routes to identify compounds active against SARS-CoV-2 without any human intervention. The chemical space was defined by “chemically reasonable perturbations” and molecule fragmentation to generate building blocks for recombination; the final space contained nearly 9 M molecules. Molecules were ranked by a property prediction model, and the best-scoring compounds were fed into a synthesis planning model to prioritize compounds with likely synthetic routes containing ≤3 steps, and the top five molecules were synthesized (one with great difficulty) and tested experimentally; one of these five molecules showed micromolar biochemical activity and activity in live virus cell assays. The algorithmic estimation of synthetic feasibility played a critical role in compound selection, highlighting its importance for real-world applications.
A 2019 paper by Zhavoronkov et al.46 extended the work of Gomez-Bombarelli33 and demonstrated the application of latent space exploration to the design of inhibitors of DDR1, a tyrosine kinase that has been implicated in fibrosis. In this work, the authors defined a latent space based on a set of known molecules including DDR1 inhibitors to generate a set of 30 000 structures which were subsequently prioritized using a variety of computational models and filters. This prioritization process yielded a set of 40 molecules that were subsequently reviewed by human chemists to assess synthesizability. Note that this system still qualifies as ACD Level 3 because the human review was limited to synthesizability. Of the 40 selected molecules, 6 were synthesized and assayed for in vitro DDR1 activity. Subsequent pharmacokinetic assays also demonstrated that one molecule exhibited acceptable bioavailability in a mouse model.
A recent paper from Novartis described the use of a generative model to identify antimalarial compounds.47 The model was trained using 21 065 compounds with experimental data, and three compounds were used as seeds for the generative model to produce a pool of 282 candidate molecules. Each molecule was scored with an activity prediction model, and the top four molecules were selected for synthesis. Of these four compounds, only two were made, and both were active in antimalarial activity assays in the single- to double-digit nanomolar range. It is not clear how the number of top-scoring compounds to select was determined; in an ACD Level 3 system, it is expected that this would either be predetermined or algorithmically decided.
Besnard et al.48 iteratively optimized the structure of donepezil, a D4 dopamine receptor inverse agonist, for activity against the D2 receptor and blood–brain barrier penetration. The chemical space was defined by repeated rounds of enumeration using transformations mined from the literature—each round considered novelty, rule-of-five properties, and synthetic accessibility to reduce the pool for the next round—resulting in thousands of candidate structures. In the first experiment, eight compounds were selected by the authors for synthesis based on predicted polypharmacological profiles, and all showed D2 affinity between 156 nM and 1.7 μM. Subsequent experiments focused on reducing activity against α1-adrenoceptors and increasing D4 potency and selectivity, identifying a highly selective benzolactam series that was not present in the training data. Additionally, the authors identified a morpholino chemotype with D4 affinities in the double-digit nanomolar to single-digit micromolar range. This system approaches ACD Level 3 due to its machine-driven chemical space exploration, but the system as described falls into ACD Level 1 since the final compounds were chosen by humans for reasons beyond synthesizability. Additionally, the use of a synthetic accessibility score is likely insufficient to produce generally synthesizable molecules that are a feature of a ACD Level 3 system.
Bos et al.49 described an ACD Level 1 system that approaches ACD Level 3 that combines matched molecular pair transformation, reaction-based enumeration (see Konze et al.43), recursive trimming, and R-group decoration to create large pools of candidate molecules (>100M) for identifying DAO inhibitors. Each pool was reduced with property and complexity filters, and compounds unsuitable for structure-based evaluation with docking or FEP were discarded. The CNS MPO score,37 predicted potency, and lipophilic ligand efficiency (LLE) metric were used for ranking, and several compounds showed <1 μM experimental activity. This system only approaches ACD Level 3 because the final compounds were prioritized by chemists.
ACD Level 4
Description
An ACD Level 4 system moves to the highest degree on the decision making axis (“Machine decides, multiple iterations”). Multiple rounds of decision making explicitly forces the machine to consider exploration-exploitation trade-offs rather than just myopically focus on the next best molecule. These iterations refer to synthesis and testing in the real world (not just in silico rounds of search through chemical space) and must be performed without human intervention in the decision making process. If a human chemist intervenes to redefine the problem or space of molecules to consider after every round, we consider that to be a repeated application of a single iteration autonomous system and not true multiple-iteration automation.
Systems at this level typically use a class of machine learning methods known as active learning. In contrast to standard learning methods (as illustrated in Figure 2) where a model is trained and then directly utilized to find potent compounds, active learning models are refined in multiple rounds of training.50−53 Initially a model is trained and then this model is used to select candidate molecules from a source pool. The selected molecules are then assayed and in conjunction with the original data set used to retrain the model in preparation for the next round of selection.
Figure 2.

Graphical representation of the difference between standard machine learning and active machine learning.
Active learning systems are composed of two main components: an evaluation function that predicts a score and its associated uncertainty, and an acquisition function or selection policy that uses these values to select the next set of molecules. Within the machine learning community, the main goal of active learning is to quickly refine the predictive power of the underlying model. However, within the drug design community, the goal of active learning is twofold: to both quickly identify optimal compounds (“exploitation”) as well as to refine the model and hence disambiguate the underlying structure activity landscape (“exploration”). The primary differences in active learning methods relate to the acquisition function and how uncertainty estimates are utilized. Popular methods include upper-confidence bound,54 expected-improvement,55 and Thompson sampling.56 Drug discovery projects typically select a batch of compounds to synthesize and test due to the long latency in that process. Most active learning algorithms are designed to select single compounds and effective strategies for optimal selection of a batch of compounds are an ongoing research challenge.
Exemplars
The earliest ACD Level 4 system we are aware of is by Weber et al.57 They described the use of a genetic algorithm to identify thrombin inhibitors by repeated selection from a pool of 160 000 possible Ugi reaction products. After starting with an initial set of 20 randomly selected compounds, subsequent rounds selected batches of 20 compounds each for synthesis and testing, and the top 20 compounds from the full library were used as parents for the next round of selection. The authors ran the experiment for 20 rounds (a total of 400 compounds) and identified submicromolar thrombin inhibitors (the initial random set was in the hundreds of micromolar range). A least two of the compounds shared structural features with previously known inhibitors, emphasizing the role of human chemists in defining the chemical space.
Another example of an ACD Level 4 system was built by the team at Cyclofluidic.58 In 2013, they published a report detailing the discovery of BCR-Abl inhibitors using a closed-loop flow chemistry system that prioritized compounds over many rounds of active learning.59 The chemical space was defined by three templates chosen by their potential for hinge-binding and occupation of the DFG-out conformation of the target. Each template contained two vectors; four substituents were used for R1, while 27 were chosen for R2 by structure-based assessment of their likely interactions with the target. In total, 270 compounds were available for flow synthesis followed by inline assays with a cycle time of 1–2 h. Compounds were selected by machine learning models with both exploration and exploitation strategies based on predicted potency and reactant frequency. The first experiment ran 29 rounds of exploration and identified several inhibitors (the Abl1 model used for compound selection was updated after each round), including a 60 nM inhibitor of Abl1. The second experiment focused on improving potency and ran for 20 rounds, and the third experiment alternated between exploration and exploitation for an additional 41 rounds. Overall, 90 rounds of selection resulted in 64 successfully synthesized/assayed compounds and the identification of single-digit nanomolar inhibitors of Abl1 and Abl2. Although this system meets the criteria for ACD Level 4, the limited size of the explored chemical space highlights the fact that reaching a certain level of autonomy does not guarantee any specific level of utility relative to lower-level systems.
The “robot scientist” Eve, described by Williams et al.60 is another example of an ACD Level 4 system. The authors developed assays for DHFR inhibition and gathered single-concentration data for a library containing about 14k compounds. A subset of this data was chosen as a training set for a QSAR model, which was then used to select a batch of 96 compounds for dose–response assays. For subsequent rounds of selection (three in total), the newly acquired data were added to the training set and the model was retrained. The authors also performed computational simulations using the original single-point data to investigate the behavior of the system over many more rounds of selection. The paper does not provide a description of how the training set was chosen; this is likely to have a profound effect on the success of the active learning and the apparent effectiveness of different compound selection strategies.
With the renewed interest in automated molecular design, it becomes easy to overlook historical research in this field. However, automated molecular design in the pharmaceutical industry has been an active area of research for over 5 decades. In the early 1970s, Darvas61 described a potential ACD Level 4 design system based upon a simplex optimization method coupled with a two-dimensional descriptor space. Starting from three compounds with known activity, a simplex is constructed in the descriptor and activity space and the direction of increase of biological activity calculated. A new compound is selected from the source pool that is located along this direction. The activity of the selected compound is obtained, and the process repeats using the newly obtained data point. The paper demonstrated statistical superiority of the method to identify optimal compounds as compared to random selection. Although this is a small study by modern standards, it embodies the essential components of an ACD Level 4 system: iterative design and algorithmic selection of molecules.
Another potential ACD Level 4 system is the information theory based iterative design strategy introduced by Bradley et al.,62 which was used for lead identification and optimization of CDK-2 ligands. To mimic a typical discovery program, this study used two different source pools of molecules: an early stage pool for initial model building and a late-stage pool for compound optimization. The early stage pool consisted of 13 359 diverse chemical structures containing 207 actives while the late-stage pool contained approximately 4500 compounds with 161 actives spread across 22 different scaffolds. Using the early stage pool, an initial binary molecular descriptor space was defined using approximately 4 million 3D pharmacophores. Subsequently, each molecule was encoded into a binary vector denoting the presence or absence of a particular pharmacophore in any of its molecular conformations. This space was subsequently pruned by retaining only those pharmacophores which were populated to a sufficient degree by molecules in the early screening pool. This resulted in retaining approximately 1.8 million pharmacophores. Using an information theory-based selection algorithm, a small set of molecules were selected from the late-stage pool, their activities exposed, and then used to refine the pharmacophore space by retaining only pharmacophores overrepresented in the newly discovered active compounds. This process was repeated four times, ultimately pruning the space to 82K pharmacophores. In the final step, compounds in the late-stage pool enriched in the final pharmacophores were predicted as “active” compounds. The procedure discovered 11 of 14 active scaffolds in the late-stage pool and outperformed other profiled methods. This study is an excellent embodiment of a potential ACD Level 4 system containing both iterative design and algorithmic selection of molecules.
A further example of a potential ACD Level 4 system is the retrospective study conducted by Warmuth et al.51 which utilized support vector machines (SVM) coupled with an active learning strategy. The methodology was evaluated on two different ligand binding systems: Thrombin and CDK2. The Thrombin data set contained approximately 2000 compounds with 190 actives while the CDK2 data set consisted of 17 500 compounds and 383 active molecules. The experimental setup was designed to be analogous to an iterative drug discovery project, in that an initial classifier is constructed using available data and subsequently refined as more information becomes available. For this study, the initial classifier was constructed using 5% of the available data. This classifier, coupled with a selection policy, is then used to select a new batch of compounds, and retrained using the exposed labels of the newly selected batch. As is definitional for an ACD Level 4 system, compounds were selected iteratively and algorithmically. Notably, in an ACD Level 4 system the algorithmic selection policy can be automatically tuned to the stage of the project. One policy can be utilized at the beginning of a project when it is important to quickly identify active chemical matter, while at later stages of the project, when understanding the structure activity landscape becomes important, a different policy can be used.
An example of an approaching ACD Level 4 system is the methodology utilized by Fujiwara et al.,63 who applied a “query-by-committee” active learning strategy to explore SAR landscapes and identify potent compounds. This study used a well-known approach termed “bagging” to create a collection of machine learning models, where each model is trained on a unique random sample of the currently available data. A batch of molecules for testing is then selected by identifying a set of molecules where there is maximal disagreement among the created group of machine learning models. The activities of these selected molecules are obtained, and the process iterates. The method was retrospectively tested on three systems, and outperformed conventional hit selection strategies in each case. The method was prospectively tested on biogenic amine receptors by using the algorithm to select 50 compounds from a source pool of approximately 50 000 molecules. From the machine-selected molecules, 10 compounds were manually selected, ordered, and assayed with four compounds exhibiting inhibitory potency of more than 50%. The additional manual selection step means that this prospective application only approaches ACD Level 4; as described, the system would be ACD Level 0.
A further example of a potential ACD Level 4 system is research by Ahmadi et al.64 In this system, a machine learning algorithm known as Gaussian process regression is used as the base algorithm while the acquisition function employs an expected improvement methodology. The method was retrospectively tested on 12 data sets and, compared to baseline methods, required significantly fewer iterations to identify the most potent compounds. However, the utility of this study to inform prospective drug discovery projects is limited; during the active learning update cycle, the current study selected only one compound per round, which is in stark contrast to typical drug discovery efforts where larger batches of compounds are selected for synthesis and assays in each cycle.
ACD Level 5 and Future Challenges
Description
ACD Level 5 is the most autonomous on both the decision making and ideation axes. We know of no examples of complete ACD Level 5 systems. The closest potential ACD Level 5 system we are aware of is Green et al.65 which describes a fully automated platform (BRADSHAW) developed for internal use at GSK that supports molecule generation, property prediction, multiobjective optimization, and compound selection. The platform is configured by composing multiple “Tasks” that incorporate best-practice implementations of steps like MMP (Matched Molecular Pairs) transforms,66,67 filtering by similarity or SMARTS (SMILES Arbitrary Target Specification),68 and assigning multiobjective scores based on linear desirability functions for each property. The system anticipates active learning applications by allowing compound selections to include both exploitation of known desirable chemical matter as well as exploration into new chemical space by targeting areas where the property models are uncertain. Additionally, the system considers the challenge of starting a program with very little data by including methods for selecting an initial set of compounds. The use of the platform in the paper is purely retrospective.
Future Challenges
The creation of an ACD Level 5 system that is truly effective at drug design represents both a shared goal of the community and significant technical challenge. One challenge is the further improvement and evaluation of algorithms and models for generating molecular ideas. While generation algorithms have sometimes been shown to produce synthesizable molecules that improve desired properties, the robustness, completeness, and effectiveness of the existing algorithms remains to be better understood and improved. Several benchmarks have been introduced (GuacaMol69 and MOSES70) to standardize measurements of the novelty, diversity, and properties of molecules produced by molecular generators, though questions remain about the quality of the metrics used.71 The Molecular Turing Test72 is a well done example of how to evaluate whether a molecular generator produces molecules similar to those that would be selected by chemists. These evaluations should continue and drive algorithm development.
Another significant challenge is that we do not yet understand the effectiveness of current optimization algorithms for the peculiarities of chemical design. Chemical space is discrete and thought to be quite “rough”, meaning that nearby molecules can have vastly different properties.73 Improvements in optimization algorithms for this complex space and/or improvements in the notion of “nearby” molecules which make the optimization problem simpler are ongoing challenges. For example, some molecules that seem very different when examined as 2D molecular graphs may adopt very similar electrostatic shapes in 3D. This change from a 2D to a 3D view can result in very different performance of models and optimization algorithms.
Especially as the space of molecules to be explored is made more global, machines will have to grapple with estimating the long-term value of exploring a region of chemical space and not just the value of a particular molecule. In other words, choosing one chemical series over another and not just one molecule over another. This is fundamentally a much more difficult estimation problem; even great medicinal chemistry teams struggle to estimate whether they will succeed with a given series. While absolute estimates (for example, “we need to make between 100 and 200 more compounds to reach a development candidate”) would be beneficial, especially for managing an entire portfolio, relative comparisons (for example, “this series is more likely to be successful than that one”) may be sufficient for the decision making needed at higher ACD levels.
In addition to the levels defined above, there are a few issues, common to multiple levels, that merit additional discussion. Proper handling of model uncertainty and addressing the synthesizability of designed molecules are two larger challenges we discuss further in the following sections.
Uncertainty
Automated chemical design requires an exploration of the underlying structure–activity landscape so that regions of chemical space yielding productive molecules can be quickly identified. While various selection policies are employed by different algorithms to identify the next set of molecules to synthesize and test, what unifies these approaches is an estimate of predictive uncertainty. For example, if a candidate molecule is identified to be inactive with high certainty, then it would make little sense to acquire and test that molecule. Conversely, if a molecule is predicted to be moderately active, but with a sufficiently high uncertainty that it could be extremely active, then that molecule should perhaps be tested as it provides information pertinent to disambiguating the SAR landscape. Unfortunately, there exist few computational procedures for estimating uncertainty that provide theoretical guarantees of performance. This lack of well-founded uncertainty estimates is a potential source of frustration for automated chemical design as it hinders accurate decision making.
Well-calibrated uncertainty estimates that enable accurate decision making possess two fundamental properties: coverage and discriminability. Coverage ensures that the true value of a predicted point falls within the uncertainty estimate for the predictive value of that point. Discriminability relates to the confidence of the prediction and is directly related to the width of the confidence interval, with smaller widths implying higher confidence. For an uncertainty estimate to be useful, it must contain both of these properties. The true value for a prediction should ideally lie within the uncertainty estimate, and the uncertainty estimate should be meaningfully tight; excessively large uncertainty estimates provide little value.
Gaussian Process Regression74 is the most common traditional machine learning technique with well founded uncertainty estimates and is frequently used in active learning problems such as those encountered in ACD systems. For other machine learning models, numerous other techniques of varying computational expense and quality have been developed (for example, Monte Carlo dropout,75 deep kernel learning,76 epistemic neural networks,77 and Jackknife+78). Especially for neural network models, better techniques for estimating uncertainty is an active area of research.79
Synthesizability
A typical lead optimization program goes through many design, make, test cycles before a development candidate is nominated. It is imperative that these cycles be made as short as possible to increase the probability of success as shorter cycle times will translate into more cycles over a fixed time horizon, amplifying the benefits of an active learning approach. The synthesis of organic compounds during the “make” step is the most time-consuming and has the most variability in the cycle.80 While automatic synthesis planning and execution are exciting areas of research, neither is required for an ACD system; however, they can be used to drive down cycle times to accelerate ACD systems.
Moving to an ACD Level 3 or ACD Level 5 system requires the machine to consider the synthesizability of the molecules it designs. While chemist review for synthesizability can still happen at these levels, it is required that only a small fraction of the molecules be filtered (that is, the machine does most of the work). If the chemist has to filter a large fraction of the molecules, it becomes difficult for them to do this in a repeatable, unbiased way, mitigating much of the expected value of the ACD system.81
Predictions of synthesizability are related to, but distinct from, prediction of routes. Several methods of predicting synthesizability are structure based82−84 which provide a score but no suggested route or reaction conditions. A synthetic pathway approach assesses synthesizability using a computer-aided synthesis planning (CASP) program.85,86 These programs are more interpretable and can provide suggested reaction conditions to aid in synthesis execution. Synthetic pathway approaches are not prevalent in existing ACD systems because their runtimes are currently impractical for large chemical spaces.
De novo molecular design methods have a high risk of proposing molecules that may be difficult or even impossible to synthesize.87 Recent methods that combine de novo design and synthesis planning,88 or that bias generation87 by a synthesizability heuristic, are likely to produce more actionable molecules.
Several methods have been proposed to generate synthesizable chemical spaces including SAVI89 and SynthI.90 These methods maintain a set of expert encoded reaction transforms and a set of available building blocks to assemble molecules and optionally score them based on likelihood of synthetic success. As an example, SynthI has methods for transforming building blocks into synthons with their reaction centers annotated to indicate the type of reaction center. These synthons can then be combined using SMIRKS (a reaction transform language) transforms.91
The downside of these template-based approaches is the need for experts to maintain the reaction rules and the difficulty of staying up to date with advances in chemistry. It is estimated that over 10 000 template-based rules must be encoded to match the knowledge of a skilled chemist.85 However, given the relatively few reactions92 regularly in use in pharmaceutical companies and the diversity available from common reactions,93 this drawback may be academic in practice.
Newer methods have recently been proposed to train deep learning models to predict reaction outcomes.94 These methods operate by encoding sets of reactants and products from the chemical literature to learn reaction pathways. When presented with a new molecule, the programs can propose synthetic routes. These methods, while promising, suffer from a lack of widely available curated data sets—especially negative results. While internal electronic lab notebooks (ELNs) provide a compelling source of training data, reactions and their outcomes are often not captured in a consistent fashion. In order to address this shortcoming, internal ELNs should be outfitted with strict business rules such that over time these databases can become a rich source of training data. Hopefully, public efforts such as the Open Reaction Database95 can fill this need over time for the broader community.
Ranking of synthetic difficulty will also be an important area of research.84 Leveraging synthetic risk in the active learning process can provide an important component of exploitation vs exploration by explicitly tying synthetic difficulty to expected reward. It is important to not think of synthesizability as a binary condition but a measure of difficulty. It will be necessary to accurately estimate the effort and risk in synthesizing a given molecule so the system can make optimal choices between easier but less informative molecules and more informative but more challenging molecules. The field of optimal design96 can be leveraged to maximize the reward over variable duration cost functions.
Ordering of ACD Levels
Figure 3 summarizes the ACD systems discussed above. Note that as listed in Table 1, we intentionally do not call something an ACD system unless it has been used to create and test molecules in the real world.
Figure 3.

The most notable ACD systems discussed in this paper are grouped into the levels defined in Figure 1. Systems described as approaching a particular ACD Level in the text are categorized by their actual ACD Level, and no potential ACD systems are listed.
We chose to assign linear levels from these two axes of automation with the decision making axis as primary. While there are significant technical challenges on both axes, we believe moving to more automated decision making is a more holistic challenge. For automated decision making to be effective, all relevant criteria have to be handled by the machine to avoid the machine going toward dead-end molecules. This change in the control of decisions represents a level of trust being placed in the system. Automated decision making also enables clean experimentation on the system (as discussed in ACD Level 2) and is therefore key to producing continually improving systems.
Note that systems do not have to advance step by step on this single linear scale. For example, a system may be gradually improved from an ACD Level 0 system to a ACD Level 2 system without ever being an ACD Level 1 system.
Human–Machine Partnership
The ACD levels above describe increasing levels of autonomy and decision making vested into the machine. But even at the highest levels, a human-machine partnership is core to a successful system (see the work of Jensen et al.16 for additional discussion of this topic). The decisions that require the most context and integration of disparate types of information are best done by human experts and are done so even in an ACD Level 5 system.
First and foremost, humans define the goals for a molecule. This requires partial understanding of the relevant biological processes (from clinical and preclinical data), the clinical setting for the treatment that would be acceptable, the useful level of disease modification, and the relationship of this potential treatment to alternatives.
Second, all of our assays for molecules are merely proxies for the true goal of effective treatment in humans. Even the best assays can produce misleading results. This can be for fundamental reasons (for example, rats are biologically different from humans) or technical reasons specific to an assay. For instance, a redox cycling compound97 may produce a false positive readout with some assay formats. In many cases, an ACD system would mistakenly classify such a compound and choose to further optimize it. Noticing and debugging these issues often requires a deep understanding of the underlying physics and chemistry of the assay. Different assays notionally measuring the same physical phenomenon can produce inconsistent results and a key job of the human experts is to define the assay cascade that provides the most convincing evidence for translation to human disease. A careful assay cascade design will help a machine avoid chasing assay artifacts, but we also expect that more effective ACD systems will need to learn or be told how to avoid some of these artifacts in the first place. However, given the lack of publicly available data and multiple paths to assay interference, this may be a challenging task.
Third, the notion of novelty that underlies chemical patents can often be ambiguous and difficult for a human, let alone a computer, to comprehend. While it may be possible to extract specific chemical examples from patents and automatically compare these with molecules generated by an algorithm, the interpretation of Markush claims is an artform that has yet to be mastered by a machine. At present, humans are responsible for assessing the novelty of computer generated molecules. Given the complexity of this evaluation, the lack of standards for reporting patent claims, and the obfuscation present in patent filings, the assessment of novelty may be the province of human chemists for the foreseeable future.
Fourth, humans will continue to provide oversight of the overall process at several levels. Automated systems can get stuck in dead ends and need to be reset to make progress. New exogenous information may arrive (such as competitive developments or new biological insights) that redefine the goals. The overall decision of whether to continue the program given the observed progress is fundamentally a human decision involving the context of the organization and the human impact of the unmet medical need.
The job of the medicinal chemist has continually evolved as technology has come into common use. Tools such as structure searching systems, that medicinal chemists take for granted today have only been in use for the last 30 years. The property calculations that every chemist uses did not become commonplace until the early 2000s. We expect the increasing use of ACD systems will continue this trend of changing a chemist’s allocation of time on the wide variety of activities needed to successfully produce a drug.98
Conclusions
One goal with the ACD framework we propose is to enable better probing and understanding of the function and value of automated design systems. While it may be tempting to use the ACD levels directly as a value judgment such as “An ACD Level 3 system is better than an ACD Level 2 system”, this is not necessarily the case. Just because a system operates with a certain level of autonomy does not imply that that the system is either applicable to many problems or well suited for its intended purpose. For example, one can imagine an ACD Level 5 system that can only operate on unbranched hydrocarbon chains. While it may autonomously find the best unbranched hydrocarbon chain for a purpose, there are not many problems for which that is useful. Similarly, an ACD Level 2 system may simply make poor choices, such that a human-driven ACD Level 0 system can discover an acceptable molecule faster. The evaluation of the usefulness of the system is separate from establishing its level of autonomy.
However, the ACD levels do inspire questions that a user or customer of an ACD system should ask to more deeply understand the value of the system, similar to those proposed by others.16 We list a number of such questions in Table 2. Not every question applies to systems at every level, but these should lead to a more rigorous conversation between creators and potential users of the systems.
Table 2. Important Questions to Help Teams Evaluate the Quality of ACD Systems (As Opposed to Categorizing the System into the Appropriate Level).
| How specific is the system to this particular problem vs generally applicable to many problems? |
| Is the system efficient (in terms of time, number of compounds, or money spent) in finding an acceptable molecule? |
| Does the system outperform simple approaches like random selection from the chemical space? |
| How many cycles did the system run autonomously? |
| How complex were the goals given to the machine (for example, optimizing for potency vs balancing multiple competing objectives)? |
| How much choice and guidance does the human exercise? |
| How big was the chemical space of molecules given to the machine? |
| Did the system explore only local modifications to known molecules or larger changes? |
| Did the machine generate a similar space of ideas as medicinal chemists would generate? |
| Were the machine’s ideas generally synthesizable? |
On a related note, there are many useful applications of computation and AI methods in drug discovery that do not move a system to higher ACD levels. Systems to provide biological insight from literature or images can help humans pick the right targets. Analysis of clinical data can help identify the right patients for a treatment. Better AI or physics-based predictions in an ACD Level 0 system can significantly improve decision making. The definition of the levels in this perspective is capturing just one of the many useful trajectories of computation in drug discovery.
The current great interest in further developing autonomous computational approaches to molecular design will create many systems that can be classified into the ACD levels. We hope that the nomenclature we have introduced here will facilitate discussion, understanding, and focused research throughout the community.
Acknowledgments
We would like to acknowledge our colleagues at Relay Therapeutics, Mark Murcko and Connor Coley, for helpful discussions.
Glossary
Abbreviations Used
- ACD
Automated Chemical Design
- CASP
Computer-Aided Synthesis Planning
- DEL
DNA-Encoded Library
- ELN
Electronic Lab Notebook
- FEP
Free Energy Perturbation
- LLE
Lipophilic Ligand Efficiency
- MMP
Matched Molecular Pairs
- MPO
Multi-Parameter Optimization
- OLED
Organic Light Emitting Diode
- PAINS
Pan-Assay Interference Compounds
- RL
Reinforcement Learning
- RNN
Recurrent Neural Network
- SMARTS
SMILES Arbitrary Target Specification
- SMILES
Simplified Molecular Input Line Entry System
Biographies
Brian Goldman got his B.A. in Biochemistry and Ph.D. in Computational Chemistry from the University of California at Santa Cruz. He spent 20 years at Vertex Pharmaceuticals developing and applying a wide range of computational methods to drug discovery programs. His research is focused on designing and applying machine learning methods to drug discovery.
Steven Kearnes got his B.S. in Biochemistry from Brigham Young University and his Ph.D. in Structural Biology from Stanford University. His research is focused on applying deep learning to problems in drug discovery, including graph-convolutional neural networks and DNA-encoded libraries.
Trevor Kramer got his B.A.from Hampshire College in Computer Science. He spent 20 years at Vertex Pharmaceuticals in Research Informatics building decision support systems for chemistry. His research is focused on designing synthetically accessible chemical spaces.
Patrick Riley got his B.S, M.S., and Ph.D. from Carnegie Mellon University in Computer Science with a focus on Artificial Intelligence. He spent 16 years at Google, first working on web search and data analytics and then coleading a research group applying computational methods to a variety of natural science problems, including chemistry problems. He is now the SVP, AI at Relay Therapeutics.
W. Patrick Walters got his B.S. in Chemistry from The University of California Santa Barbara and his Ph.D. in Organic Chemistry from the University of Arizona, where he studied the application of Artificial Intelligence in conformational analysis. He spent 20 years at Vertex Pharmaceuticals where he was the Global Head of Modeling and Informatics. He is currently the Chief Data Officer at Relay Therapeutics.
Author Contributions
All authors contributed to the conceptual framework and to writing the manuscript.
The authors declare the following competing financial interest(s): All authors are employees of Relay Therapeutics, a for-profit pharmaceutical company.
References
- Bender A.; Cortes-Ciriano I. Artificial Intelligence in Drug Discovery: What Is Realistic, What Are Illusions? Part 2: A Discussion of Chemical and Biological Data. Drug Discovery Today 2021, 26, 1040. 10.1016/j.drudis.2020.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider P.; Walters W. P.; Plowright A. T.; Sieroka N.; Listgarten J.; Goodnow R. A.; Fisher J.; Jansen J. M.; Duca J. S.; Rush T. S.; Zentgraf M.; Hill J. E.; Krutoholow E.; Kohler M.; Blaney J.; Funatsu K.; Luebkemann C.; Schneider G. Rethinking Drug Design in the Artificial Intelligence Era. Nat. Rev. Drug Discovery 2020, 19, 353. 10.1038/s41573-019-0050-3. [DOI] [PubMed] [Google Scholar]
- Walters W. P.; Barzilay R. Critical Assessment of AI in Drug Discovery. Expert Opin. Drug Discovery 2021, 16, 937–947. 10.1080/17460441.2021.1915982. [DOI] [PubMed] [Google Scholar]
- Vamathevan J.; Clark D.; Czodrowski P.; Dunham I.; Ferran E.; Lee G.; Li B.; Madabhushi A.; Shah P.; Spitzer M.; Zhao S. Applications of Machine Learning in Drug Discovery and Development. Nat. Rev. Drug Discovery 2019, 18, 463. 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers J.; Fabian B.; Brown N. De Novo Molecular Design and Generative Models. Drug Discovery Today 2021, 26, 2707. 10.1016/j.drudis.2021.05.019. [DOI] [PubMed] [Google Scholar]
- Jiménez-Luna J.; Grisoni F.; Schneider G. Drug Discovery with Explainable Artificial Intelligence. Nature Machine Intelligence 2020, 2 (10), 573–584. 10.1038/s42256-020-00236-4. [DOI] [Google Scholar]
- Miljković F.; Rodríguez-Pérez R.; Bajorath J. Impact of Artificial Intelligence on Compound Discovery, Design, and Synthesis. ACS Omega 2021, 6 (49), 33293–33299. 10.1021/acsomega.1c05512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIT AI Powered Drug Discovery and Manufacturing Conference 2020https://www.aidm.mit.edu/ (accessed 2022–04–05).
- SLAS 2021 AI-Powered Drug Discovery Symposium https://www.slas.org/events-calendar/slas-2021-ai-powered-drug-discovery-symposium/ (accessed 2022. –04–05).
- Machine Learning for Molecules Workshop @ NeurIPS 2020 https://ml4molecules.github.io/ (accessed 2022. –04–05).
- Walters W. P. Virtual Chemical Libraries. J. Med. Chem. 2019, 62 (3), 1116–1124. 10.1021/acs.jmedchem.8b01048. [DOI] [PubMed] [Google Scholar]
- Schneider G. Automating Drug Discovery. Nat. Rev. Drug Discovery 2018, 17 (2), 97–113. 10.1038/nrd.2017.232. [DOI] [PubMed] [Google Scholar]
- Green C. P.; Engkvist O.; Pairaudeau G. The Convergence of Artificial Intelligence and Chemistry for Improved Drug Discovery. Future Med. Chem. 2018, 10 (22), 2573–2576. 10.4155/fmc-2018-0161. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Eyke N. S.; Jensen K. F. Autonomous Discovery in the Chemical Sciences Part II: Outlook. Angew. Chem., Int. Ed. Engl. 2020, 59, 23414. 10.1002/anie.201909989. [DOI] [PubMed] [Google Scholar]
- Kearnes S. Pursuing a Prospective Perspective. TRECHEM 2021, 3 (2), 77–79. 10.1016/j.trechm.2020.10.012. [DOI] [Google Scholar]
- Jensen K. F.; Coley C. W.; Eyke N. S. Autonomous Discovery in the Chemical Sciences Part I: Progress. Angew. Chem., Int. Ed. Engl. 2020, 59, 22858. 10.1002/anie.201909987. [DOI] [PubMed] [Google Scholar]
- Richter R.Linus Pauling, Crusading Scientist; Richter Productions, 1977. [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev. 1997, 23 (1–3), 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53 (7), 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Maggiora G.; Vogt M.; Stumpfe D.; Bajorath J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2014, 57 (8), 3186–3204. 10.1021/jm401411z. [DOI] [PubMed] [Google Scholar]
- Nicholls A.; McGaughey G. B.; Sheridan R. P.; Good A. C.; Warren G.; Mathieu M.; Muchmore S. W.; Brown S. P.; Grant J. A.; Haigh J. A.; Nevins N.; Jain A. N.; Kelley B. Molecular Shape and Medicinal Chemistry: A Perspective. J. Med. Chem. 2010, 53 (10), 3862–3886. 10.1021/jm900818s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu J.; Wang S.; Balius T. E.; Singh I.; Levit A.; Moroz Y. S.; O’Meara M. J.; Che T.; Algaa E.; Tolmachova K.; Tolmachev A. A.; Shoichet B. K.; Roth B. L.; Irwin J. J. Ultra-Large Library Docking for Discovering New Chemotypes. Nature 2019, 566 (7743), 224–229. 10.1038/s41586-019-0917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Wu Y.; Deng Y.; Kim B.; Pierce L.; Krilov G.; Lupyan D.; Robinson S.; Dahlgren M. K.; Greenwood J.; Romero D. L.; Masse C.; Knight J. L.; Steinbrecher T.; Beuming T.; Damm W.; Harder E.; Sherman W.; Brewer M.; Wester R.; Murcko M.; Frye L.; Farid R.; Lin T.; Mobley D. L.; Jorgensen W. L.; Berne B. J.; Friesner R. A.; Abel R. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137 (7), 2695–2703. 10.1021/ja512751q. [DOI] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50 (5), 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Chuang K. V.; Gunsalus L. M.; Keiser M. J. Learning Molecular Representations for Medicinal Chemistry. J. Med. Chem. 2020, 63 (16), 8705–8722. 10.1021/acs.jmedchem.0c00385. [DOI] [PubMed] [Google Scholar]
- Weininger D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28 (1), 31–36. 10.1021/ci00057a005. [DOI] [Google Scholar]
- Pennington L. D.; Aquila B. M.; Choi Y.; Valiulin R. A.; Muegge I. Positional Analogue Scanning: An Effective Strategy for Multiparameter Optimization in Drug Design. J. Med. Chem. 2020, 63 (17), 8956–8976. 10.1021/acs.jmedchem.9b02092. [DOI] [PubMed] [Google Scholar]
- Polishchuk P. CReM: Chemically Reasonable Mutations Framework for Structure Generation. J. Cheminform. 2020, 12 (1), 28. 10.1186/s13321-020-00431-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awale M.; Hert J.; Guasch L.; Riniker S.; Kramer C. The Playbooks of Medicinal Chemistry Design Moves. J. Chem. Inf. Model. 2021, 61 (2), 729–742. 10.1021/acs.jcim.0c01143. [DOI] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Aguilera-Iparraguirre J.; Hirzel T. D.; Duvenaud D.; Maclaurin D.; Blood-Forsythe M. A.; Chae H. S.; Einzinger M.; Ha D.-G.; Wu T.; Markopoulos G.; Jeon S.; Kang H.; Miyazaki H.; Numata M.; Kim S.; Huang W.; Hong S. I.; Baldo M.; Adams R. P.; Aspuru-Guzik A. Design of Efficient Molecular Organic Light-Emitting Diodes by a High-Throughput Virtual Screening and Experimental Approach. Nat. Mater. 2016, 15 (10), 1120–1127. 10.1038/nmat4717. [DOI] [PubMed] [Google Scholar]
- Tong X.; Liu X.; Tan X.; Li X.; Jiang J.; Xiong Z.; Xu T.; Jiang H.; Qiao N.; Zheng M. Generative Models for De Novo Drug Design. J. Med. Chem. 2021, 64 (19), 14011–14027. 10.1021/acs.jmedchem.1c00927. [DOI] [PubMed] [Google Scholar]
- Vanhaelen Q.; Lin Y.-C.; Zhavoronkov A. The Advent of Generative Chemistry. ACS Med. Chem. Lett. 2020, 11 (8), 1496–1505. 10.1021/acsmedchemlett.0c00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernández-Lobato J. M.; Sánchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent Sci. 2018, 4 (2), 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merk D.; Friedrich L.; Grisoni F.; Schneider G. De NovoDesign of Bioactive Small Molecules by Artificial Intelligence. Mol. Inform. 2018, 37 (1–2), 1700153–1700154. 10.1002/minf.201700153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova M.; Isayev O.; Tropsha A. Deep Reinforcement Learning for de Novo Drug Design. Sci. Adv. 2018, 4 (7), eaap7885 10.1126/sciadv.aap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Ye K.; van Vlijmen H. W. T.; Emmerich M. T. M.; Ijzerman A. P.; van Westen G. J. P. DrugEx v2: De Novo Design of Drug Molecules by Pareto-Based Multi-Objective Reinforcement Learning in Polypharmacology. J. Cheminform. 2021, 13 (1), 85. 10.1186/s13321-021-00561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach to Enable Alignment of Druglike Properties. ACS Chem. Neurosci. 2010, 1 (6), 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin H. Probabilistic Approach to Generating MPOs and Its Application as a Scoring Function for CNS Drugs. ACS Med. Chem. Lett. 2016, 7 (1), 89–93. 10.1021/acsmedchemlett.5b00390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Honeycutt J. D.; Metz J. T. Evolving Molecules Using Multi-Objective Optimization: Applying to ADME/Tox. Drug Discovery Today 2010, 15 (11–12), 451–460. 10.1016/j.drudis.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Ahmed A.; Saeed F.; Salim N.; Abdo A. Condorcet and Borda Count Fusion Method for Ligand-Based Virtual Screening. J. Cheminform. 2014, 6 (1), 19. 10.1186/1758-2946-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadybekov A. A.; Sadybekov A. V.; Liu Y.; Iliopoulos-Tsoutsouvas C.; Huang X.-P.; Pickett J.; Houser B.; Patel N.; Tran N. K.; Tong F.; Zvonok N.; Jain M. K.; Savych O.; Radchenko D. S.; Nikas S. P.; Petasis N. A.; Moroz Y. S.; Roth B. L.; Makriyannis A.; Katritch V. Synthon-Based Ligand Discovery in Virtual Libraries of over 11 Billion Compounds. Nature 2022, 601 (7893), 452–459. 10.1038/s41586-021-04220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes J. M.; Yang K.; Swanson K.; Jin W.; Cubillos-Ruiz A.; Donghia N. M.; MacNair C. R.; French S.; Carfrae L. A.; Bloom-Ackermann Z.; Tran V. M.; Chiappino-Pepe A.; Badran A. H.; Andrews I. W.; Chory E. J.; Church G. M.; Brown E. D.; Jaakkola T. S.; Barzilay R.; Collins J. J. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180 (4), 688–702. 10.1016/j.cell.2020.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze K. D.; Bos P. H.; Dahlgren M. K.; Leswing K.; Tubert-Brohman I.; Bortolato A.; Robbason B.; Abel R.; Bhat S. Reaction-Based Enumeration, Active Learning, and Free Energy Calculations To Rapidly Explore Synthetically Tractable Chemical Space and Optimize Potency of Cyclin-Dependent Kinase 2 Inhibitors. J. Chem. Inf. Model. 2019, 59 (9), 3782–3793. 10.1021/acs.jcim.9b00367. [DOI] [PubMed] [Google Scholar]
- McCloskey K.; Sigel E. A.; Kearnes S.; Xue L.; Tian X.; Moccia D.; Gikunju D.; Bazzaz S.; Chan B.; Clark M. A.; Cuozzo J. W.; Guié M.-A.; Guilinger J. P.; Huguet C.; Hupp C. D.; Keefe A. D.; Mulhern C. J.; Zhang Y.; Riley P. Machine Learning on DNA-Encoded Libraries: A New Paradigm for Hit Finding. J. Med. Chem. 2020, 63 (16), 8857–8866. 10.1021/acs.jmedchem.0c00452. [DOI] [PubMed] [Google Scholar]
- Morris A.; McCorkindale W.; Consortium T. C. M.; Drayman N.; Chodera J. D.; Tay S.; London N.; Lee A. A. Discovery of SARS-CoV-2 Main Protease Inhibitors Using a Synthesis-Directed de Novo Design Model. Chem. Commun. 2021, 57 (48), 5909–5912. 10.1039/D1CC00050K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhavoronkov A.; Ivanenkov Y. A.; Aliper A.; Veselov M. S.; Aladinskiy V. A.; Aladinskaya A. V.; Terentiev V. A.; Polykovskiy D. A.; Kuznetsov M. D.; Asadulaev A.; Volkov Y.; Zholus A.; Shayakhmetov R. R.; Zhebrak A.; Minaeva L. I.; Zagribelnyy B. A.; Lee L. H.; Soll R.; Madge D.; Xing L.; Guo T.; Aspuru-Guzik A. Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol. 2019, 37 (9), 1038–1040. 10.1038/s41587-019-0224-x. [DOI] [PubMed] [Google Scholar]
- Godinez W. J.; Ma E. J.; Chao A. T.; Pei L.; Skewes-Cox P.; Canham S. M.; Jenkins J. L.; Young J. M.; Martin E. J.; Guiguemde W. A. Design of Potent Antimalarials with Generative Chemistry. Nature Machine Intelligence 2022, 4 (2), 180–186. 10.1038/s42256-022-00448-w. [DOI] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X.-P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R. C.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated Design of Ligands to Polypharmacological Profiles. Nature 2012, 492 (7428), 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos P. H.; Houang E. M.; Ranalli F.; Leffler A. E.; Boyles N. A.; Eyrich V. A.; Luria Y.; Katz D.; Tang H.; Abel R.; Bhat S.. AutoDesigner, a De Novo Design Algorithm for Rapidly Exploring Large Chemical Space for Lead Optimization: Application to the Design and Synthesis of D-Amino Acid Oxidase Inhibitors. J. Chem. Inf. Model. 2022. 10.1021/acs.jcim.2c00072. [DOI] [PubMed] [Google Scholar]
- Reker D.; Schneider G. Active-Learning Strategies in Computer-Assisted Drug Discovery. Drug Discovery Today 2015, 20 (4), 458–465. 10.1016/j.drudis.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Warmuth M. K.; Liao J.; Rätsch G.; Mathieson M.; Putta S.; Lemmen C. Active Learning with Support Vector Machines in the Drug Discovery Process. J. Chem. Inf. Comput. Sci. 2003, 43 (2), 667–673. 10.1021/ci025620t. [DOI] [PubMed] [Google Scholar]
- Reker D.; Schneider P.; Schneider G.; Brown J. B. Active Learning for Computational Chemogenomics. Future Med. Chem. 2017, 9 (4), 381–402. 10.4155/fmc-2016-0197. [DOI] [PubMed] [Google Scholar]
- Ding X.; Cui R.; Yu J.; Liu T.; Zhu T.; Wang D.; Chang J.; Fan Z.; Liu X.; Chen K.; Jiang H.; Li X.; Luo X.; Zheng M. Active Learning for Drug Design: A Case Study on the Plasma Exposure of Orally Administered Drugs. J. Med. Chem. 2021, 64 (22), 16838–16853. 10.1021/acs.jmedchem.1c01683. [DOI] [PubMed] [Google Scholar]
- Auer P.; Cesa-Bianchi N.; Fischer P. Finite-Time Analysis of the Multiarmed Bandit Problem. Mach. Learn. 2002, 47 (2), 235–256. 10.1023/A:1013689704352. [DOI] [Google Scholar]
- Jones D. R.; Schonlau M.; Welch W. J. Efficient Global Optimization of Expensive Black-Box Functions. J. Global Optimiz. 1998, 13 (4), 455–492. 10.1023/A:1008306431147. [DOI] [Google Scholar]
- Russo D. J.; Van Roy B.; Kazerouni A.; Osband I.; Wen Z. A Tutorial on Thompson Sampling. Found. Trends Mach. Learn. 2018, 11 (1), 1–96. 10.1561/2200000070. [DOI] [Google Scholar]
- Weber L.; Wallbaum S.; Broger C.; Gubernator K. Optimization of the Biological Activity of Combinatorial Compound Libraries by a Genetic Algorithm. Angew. Chem., Int. Ed. Engl. 1995, 34 (20), 2280–2282. 10.1002/anie.199522801. [DOI] [Google Scholar]
- Parry D. M. Closing the Loop: Developing an Integrated Design, Make, and Test Platform for Discovery. ACS Med. Chem. Lett. 2019, 10 (6), 848–856. 10.1021/acsmedchemlett.9b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai B.; Dixon K.; Farrant E.; Feng Q.; Gibson K. R.; van Hoorn W. P.; Mills J.; Morgan T.; Parry D. M.; Ramjee M. K.; Selway C. N.; Tarver G. J.; Whitlock G.; Wright A. G. Rapid Discovery of a Novel Series of Abl Kinase Inhibitors by Application of an Integrated Microfluidic Synthesis and Screening Platform. J. Med. Chem. 2013, 56 (7), 3033–3047. 10.1021/jm400099d. [DOI] [PubMed] [Google Scholar]
- Williams K.; Bilsland E.; Sparkes A.; Aubrey W.; Young M.; Soldatova L. N.; De Grave K.; Ramon J.; de Clare M.; Sirawaraporn W.; Oliver S. G.; King R. D. Cheaper Faster Drug Development Validated by the Repositioning of Drugs against Neglected Tropical Diseases. J. R. Soc. Interface 2015, 12 (104), 20141289. 10.1098/rsif.2014.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvas F. Application of the Sequential Simplex Method. J. Med. Chem. 1974, 17 (8), 799–804. 10.1021/jm00254a004. [DOI] [PubMed] [Google Scholar]
- Bradley E. K.; Miller J. L.; Saiah E.; Grootenhuis P. D. J. Informative Library Design as an Efficient Strategy to Identify and Optimize Leads: Application to Cyclin-Dependent Kinase 2 Antagonists. J. Med. Chem. 2003, 46 (20), 4360–4364. 10.1021/jm020472j. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y.; Yamashita Y.; Osoda T.; Asogawa M.; Fukushima C.; Asao M.; Shimadzu H.; Nakao K.; Shimizu R. Virtual Screening System for Finding Structurally Diverse Hits by Active Learning. J. Chem. Inf. Model. 2008, 48 (4), 930–940. 10.1021/ci700085q. [DOI] [PubMed] [Google Scholar]
- Ahmadi M.; Vogt M.; Iyer P.; Bajorath J.; Fröhlich H. Predicting Potent Compounds via Model-Based Global Optimization. J. Chem. Inf. Model. 2013, 53 (3), 553–559. 10.1021/ci3004682. [DOI] [PubMed] [Google Scholar]
- Green D. V. S.; Pickett S.; Luscombe C.; Senger S.; Marcus D.; Meslamani J.; Brett D.; Powell A.; Masson J. BRADSHAW: A System for Automated Molecular Design. J. Comput. Aided Mol. Des. 2020, 34 (7), 747–765. 10.1007/s10822-019-00234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain J.; Rea C. Computationally Efficient Algorithm to Identify Matched Molecular Pairs (MMPs) in Large Data Sets. J. Chem. Inf. Model. 2010, 50 (3), 339–348. 10.1021/ci900450m. [DOI] [PubMed] [Google Scholar]
- Griffen E.; Leach A. G.; Robb G. R.; Warner D. J. Matched Molecular Pairs as a Medicinal Chemistry Tool. J. Med. Chem. 2011, 54 (22), 7739–7750. 10.1021/jm200452d. [DOI] [PubMed] [Google Scholar]
- Daylight theory: SMARTS—A language for describing molecular patterns https://www.daylight.com/dayhtml/doc/theory/theory.smarts.html (accessed 2022-04–01).
- Brown N.; Fiscato M.; Segler M. H. S.; Vaucher A. C. GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 2019, 59 (3), 1096–1108. 10.1021/acs.jcim.8b00839. [DOI] [PubMed] [Google Scholar]
- Polykovskiy D.; Zhebrak A.; Sanchez-Lengeling B.; Golovanov S.; Tatanov O.; Belyaev S.; Kurbanov R.; Artamonov A.; Aladinskiy V.; Veselov M.; Kadurin A.; Johansson S.; Chen H.; Nikolenko S.; Aspuru-Guzik A.; Zhavoronkov A.. Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol., 2020, 11 10.3389/fphar.2020.565644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz P.; Van Rompaey D.; Wegner J. K.; Hochreiter S.; Klambauer G. On Failure Modes in Molecule Generation and Optimization. Drug Discovery Today Technol. 2019, 32–33, 55–63. 10.1016/j.ddtec.2020.09.003. [DOI] [PubMed] [Google Scholar]
- Bush J. T.; Pogany P.; Pickett S. D.; Barker M.; Baxter A.; Campos S.; Cooper A. W. J.; Hirst D.; Inglis G.; Nadin A.; Patel V. K.; Poole D.; Pritchard J.; Washio Y.; White G.; Green D. V. S. A Turing Test for Molecular Generators. J. Med. Chem. 2020, 63 (20), 11964–11971. 10.1021/acs.jmedchem.0c01148. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Stumpfe D.; Bajorath J. Advancing the Activity Cliff Concept. F1000Res. 2013, 2, 199. 10.12688/f1000research.2-199.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. K. I.; Rasmussen C. E.. Gaussian Processes for Regression. In Advances in Neural Information Processing Systems 8; Touretzky S., D M. C. M., M E. H., Eds.; Max-Planck-Gesellschaft: Cambridge, MA, 1996; pp 514–520. [Google Scholar]
- Gal Y.; Ghahramani Z.. Dropout as a Bayesian Approximation: Representing Model Uncertainty in Deep Learning. In Proceedings of The 33rd International Conference on Machine Learning; Balcan M. F., Weinberger K. Q., Eds.; Proceedings of Machine Learning Research, PMLR: New York, NY, 20–22 Jun 2016; Vol. 48, pp 1050–1059.
- Wilson A. G.; Hu Z.; Salakhutdinov R.; Xing E. P.. Deep Kernel Learning, 2015. [Google Scholar]
- Osband I.; Wen Z.; Asghari M.; Ibrahimi M.; Lu X.; Van Roy B.. Epistemic Neural Networks, 2021. [Google Scholar]
- Barber R. F.; Candès E. J.; Ramdas A.; Tibshirani R. J. Predictive Inference with the Jackknife+. Ann. Stat. 2021, 49 (1), 486–507. 10.1214/20-AOS1965. [DOI] [Google Scholar]
- Abdar M.; Pourpanah F.; Hussain S.; Rezazadegan D.; Liu L.; Ghavamzadeh M.; Fieguth P.; Cao X.; Khosravi A.; Acharya U. R.; Makarenkov V.; Nahavandi S. A Review of Uncertainty Quantification in Deep Learning: Techniques, Applications and Challenges. Inf. Fusion 2021, 76, 243–297. 10.1016/j.inffus.2021.05.008. [DOI] [Google Scholar]
- Andersson S.; Armstrong A.; Björe A.; Bowker S.; Chapman S.; Davies R.; Donald C.; Egner B.; Elebring T.; Holmqvist S.; Inghardt T.; Johannesson P.; Johansson M.; Johnstone C.; Kemmitt P.; Kihlberg J.; Korsgren P.; Lemurell M.; Moore J.; Pettersson J. A.; Pointon H.; Pontén F.; Schofield P.; Selmi N.; Whittamore P. Making Medicinal Chemistry More Effective—application of Lean Sigma to Improve Processes, Speed and Quality. Drug Discovery Today 2009, 14 (11), 598–604. 10.1016/j.drudis.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Takaoka Y.; Endo Y.; Yamanobe S.; Kakinuma H.; Okubo T.; Shimazaki Y.; Ota T.; Sumiya S.; Yoshikawa K. Development of a Method for Evaluating Drug-Likeness and Ease of Synthesis Using a Data Set in Which Compounds Are Assigned Scores Based on Chemists’ Intuition. J. Chem. Inf. Comput. Sci. 2003, 43 (4), 1269–1275. 10.1021/ci034043l. [DOI] [PubMed] [Google Scholar]
- Thakkar A.; Chadimova V.; Bjerrum E. J.; Engkvist O.; Reymond J.-L. Retrosynthetic Accessibility Score (RAscore)--Rapid Machine Learned Synthesizability Classification from AI Driven Retrosynthetic Planning. Chem. Sci. 2021, 12 (9), 3339–3349. 10.1039/D0SC05401A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Rogers L.; Green W. H.; Jensen K. F. SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 2018, 58 (2), 252–261. 10.1021/acs.jcim.7b00622. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Schuffenhauer A. Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform. 2009, 1 (1), 8. 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymkuć S.; Gajewska E. P.; Klucznik T.; Molga K.; Dittwald P.; Startek M.; Bajczyk M.; Grzybowski B. A. Computer-Assisted Synthetic Planning: The End of the Beginning. Angew. Chem., Int. Ed. Engl. 2016, 55 (20), 5904–5937. 10.1002/anie.201506101. [DOI] [PubMed] [Google Scholar]
- Lin K.; Xu Y.; Pei J.; Lai L. Automatic Retrosynthetic Route Planning Using Template-Free Models. Chem. Sci. 2020, 11 (12), 3355–3364. 10.1039/C9SC03666K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W.; Coley C. W. The Synthesizability of Molecules Proposed by Generative Models. J. Chem. Inf. Model. 2020, 60 (12), 5714–5723. 10.1021/acs.jcim.0c00174. [DOI] [PubMed] [Google Scholar]
- Gao W.; Mercado R.; Coley C. W.. Amortized Tree Generation for Bottom-Up Synthesis Planning and Synthesizable Molecular Design, 2021, arXiv:2110.06389. [Google Scholar]
- Patel H.; Ihlenfeldt W.-D.; Judson P. N.; Moroz Y. S.; Pevzner Y.; Peach M. L.; Delannée V.; Tarasova N. I.; Nicklaus M. C. SAVI, in Silico Generation of Billions of Easily Synthesizable Compounds through Expert-System Type Rules. Sci. Data 2020, 7 (1), 384. 10.1038/s41597-020-00727-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabolotna Y.; Volochnyuk D. M.; Ryabukhin S. V.; Gavrylenko K.; Horvath D.; Klimchuk O.; Oksiuta O.; Marcou G.; Varnek A.. SynthI: A New Open-Source Tool for Synthon-Based Library Design. J. Chem. Inf. Model. 2021 10.1021/acs.jcim.1c00754. [DOI] [PubMed] [Google Scholar]
- Daylight theory: SMIRKS—A reaction transform language https://www.daylight.com/dayhtml/doc/theory/theory.smirks.html (accessed 2022. −04 −01).
- Wang Y.; Haight I.; Gupta R.; Vasudevan A. What Is in Our Kit? An Analysis of Building Blocks Used in Medicinal Chemistry Parallel Libraries. J. Med. Chem. 2021, 64 (23), 17115–17122. 10.1021/acs.jmedchem.1c01139. [DOI] [PubMed] [Google Scholar]
- Tomberg A.; Boström J. Can Easy Chemistry Produce Complex, Diverse, and Novel Molecules?. Drug Discovery Today 2020, 25 (12), 2174–2181. 10.1016/j.drudis.2020.09.027. [DOI] [PubMed] [Google Scholar]
- Schwaller P.; Laino T.; Gaudin T.; Bolgar P.; Hunter C. A.; Bekas C.; Lee A. A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent Sci. 2019, 5 (9), 1572–1583. 10.1021/acscentsci.9b00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearnes S. M.; Maser M. R.; Wleklinski M.; Kast A.; Doyle A. G.; Dreher S. D.; Hawkins J. M.; Jensen K. F.; Coley C. W. The Open Reaction Database. J. Am. Chem. Soc. 2021, 143 (45), 18820–18826. 10.1021/jacs.1c09820. [DOI] [PubMed] [Google Scholar]
- Powell W. B.; Ryzhov I. O.. Optimal Learning; John Wiley & Sons: Hoboken, NJ, 2012. [Google Scholar]
- Johnston P. A. Redox Cycling Compounds Generate H2O2 in HTS Buffers Containing Strong Reducing Reagents--Real Hits or Promiscuous Artifacts?. Curr. Opin. Chem. Biol. 2011, 15 (1), 174–182. 10.1016/j.cbpa.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffen E. J.; Dossetter A. G.; Leach A. G. Chemists: AI Is Here; Unite To Get the Benefits. J. Med. Chem. 2020, 63 (16), 8695–8704. 10.1021/acs.jmedchem.0c00163. [DOI] [PubMed] [Google Scholar]