

Abstract
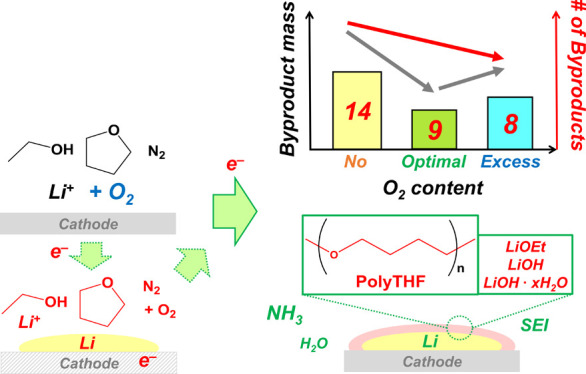
Although oxygen added to nonaqueous lithium-mediated electrochemical ammonia synthesis (LiMEAS) enhances Faradaic efficiency, its effect on chemical stability and byproducts requires understanding. Therefore, standardized high-resolution gas chromatography–mass spectrometry and nuclear magnetic resonance were employed. Different volatile degradation products have been qualitatively analyzed and quantified in tetrahydrofuran electrolyte by adding some oxygen to LiMEAS. Electrodeposited lithium and reduction/oxidation of the solvent on the electrodes produced organic byproducts to different extents, depending on the oxygen concentration, and resulted in less decomposition products after LiMEAS with oxygen. The main organic component in solid-electrolyte interphase was polytetrahydrofuran, which disappeared by adding an excess of oxygen (3 mol %) to LiMEAS. The total number of byproducts detected was 14, 9, and 8 with oxygen concentrations of 0, 0.8, and 3 mol %, respectively. The Faradaic efficiency and chemical stability of the LiMEAS have been greatly improved with addition of optimal 0.8 mol % oxygen at 20 bar total pressure.
Thermodynamically driven by an electric potential at ambient temperature and pressure, lithium-mediated electrochemical ammonia synthesis (LiMEAS) can potentially compete with the traditional thermochemical Haber–Bosch process1−3 with its high temperatures (400 °C) and pressures (150–200 bar). This method of ammonia (NH3) production is responsible for the emission of a large amount of CO2 (around 1%) associated with steam reforming emissions and is characterized by high capital costs related to the high-pressure and high-temperature requirements of the Haber–Bosch process.3 On the other hand, the nitrogen reduction reaction (N2RR) between lithium (Li) and nitrogen (N2) is driven by the affinity and chemical reactivity of Li toward N2 through the formation of lithium nitride, Li3N, or other N-containing Li materials at ambient temperature and pressure.4 LiMEAS can be even fully powered by renewable energies; it thus offers CO2-free operation, and because of its milder process conditions, it can be decentralized in smaller and less capital-intensive plants.
Even though it seems simple and is effective, LiMEAS is a chemically complicated process performed in an electrochemical cell by application of the potentials close to Li deposition. The LiMEAS mechanism is not fully understood to date, although the literature has some speculative models.5 For an assembly of a typical LiMEAS cell, a noble metal anode, e.g., platinum (Pt), and a transition metal cathode which does not interact and alloy with Li, e.g., molybdenum (Mo) or copper (Cu), are employed.6−8 The desired product, i.e., NH3, is synthesized when the electrodes are submerged in the nonaqueous organic electrolyte, which consists of a conducting salt, e.g., lithium perchlorate (LiClO4);9 an organic solvent, e.g., tetrahydrofuran (THF);6,10 and a potential proton source, such as alcohol or hydrogen (H2).5−7 The charge is usually passed at high overall cell potentials (>5 V). Our recent study described the stability of LiMEAS with the emphasis on the evolution of the electrolyte degradation products and showed that prolonged constant current reaction may lead to significant changes in the electrolyte composition and SEI, presumably deteriorating the efficiency of the whole LiMEAS.11 The decomposition reactions which have been identified are hydrolysis, ring-opening, and oxidation products of the electrolyte.11 There are many reports regarding the aging behavior of lithium ion batteries (LIB) cells with various approaches, including gas chromatography–mass spectrometry (GCMS).12−14 Our recent work on stability of LiMEAS monitored using GC with high-resolution MS for chemical and structural elucidation is a pioneering approach toward stability of LiMEAS in different organic electrolytes.11,14
Taking into consideration that Li metal is very reactive chemically, one would assume that its instability and reactivity with traces of moisture (H2O); O2; and organic solvent components, such as ethanol (EtOH) and THF, should deteriorate LiMEAS.15 However, this is not the case; it has been reported recently by our group that small amounts of O2 boost the LiMEAS.16 It is very important to mention that the electrochemical reduction of the electrolyte on the negative electrode of LIB results in the formation of the solid electrolyte interphase (SEI),12 which must be considered in LiMEAS as well. Both inorganic and organic components of this SEI are required for long-term performance if it is permeable for Li ions, thus protecting the highly reactive cathode against unwanted reductive side reactions.17−19 The SEI layer on the working electrode (WE) in LiMEAS would be different from LIBs affecting the nature and amount of byproducts, which dissolve in the electrolyte and stay as part of the SEI.20−23 The nature of the SEI is distinct to prevent further electrolyte decomposition, improving the synthesis process, and would depend on the conditions and various aspects and parameters of LiMEAS.
In the current study we demonstrate that small amounts of oxygen (O2) present together with N2 gas enhance not only the FE but also the stability of the electrolyte in LiMEAS. For analyzing and identifying mainly organic degradation products or byproducts in both the electrolyte and SEI, high-resolution GCMS and nuclear magnetic resonance (NMR) were employed. We compared the outcome and stability of LiMEAS with different O2 concentrations in nonaqueous LiMEAS both qualitatively and quantitatively. It has been shown that 0.8 mol % of O2 is an optimal amount for promotion of LiMEAS toward NH3 with improved stability of the nonaqueous organic electrolyte based on THF.8,16
Typical electrochemical results obtained for LiMEAS in an autoclave (see Figure S1) at elevated total pressure (20 bar) and ambient temperature with Mo WE of 1.8 cm2 area are shown in Figure 1. A total amount of 50 C charge was passed in 116 min with current density of −4 mA cm–2. The regular LiMEAS in THF without O2 (Figure 1a) shows an unstable voltage profile where the WE potential drops significantly after 10 min. In contrast, the electrolyte with optimal O2 content of 0.8 mol % demonstrates a more stable WE potential (Figure 1b). In addition, the most stable WE potential is seen with excess (3.0 mol %) of O2 (Figure 1c). The more oxygen in the system, the greater the electrochemical stability. This can be attributed to the complete reaction of Li with O2 forming oxide materials and suppressed accessibility for N2 to reach the metallic Li for N2RR as the Faradaic efficiency (FE) of LiMEAS drops as well. Moreover, the WE potential with excess O2 is the most stable. This could be attributed to the most stable SEI and permeability of Li ions in the SEI. The presence of O2 minimizes the decomposition products as supported by the GCMS/NMR data. Consequently, most of the further reduction occurs either as hydrogen evolution reaction (HER) or electrolyte decomposition. Thus, the LiMEAS run with 0.8 mol % O2 in the gas feed shows intermediate WE stability that is markedly better than that observed in the absence of O2. The electrochemical behavior observed for the experiments of this work is in line with results we reported previously.8,10,16,24 One difference can be seen in the linear sweep voltammmetry (LSV), shown in Figure S2, where a clear feature at −1.5 V vs Pt pseudoreference (Ptpseudo) can be seen with the presence of O2, which represents oxygen reduction reaction according to the literature.19 For all of the cases in Figure 1, the counter electrode (CE) potential is around 2 V vs Ptpseudo. The noises and some instability of CE were most likely due to mechanical contact issues. We assume that the main reason for such a high CE potential is electrolyte (THF and EtOH) oxidation and formation of the products reported elsewhere.11
Figure 1.
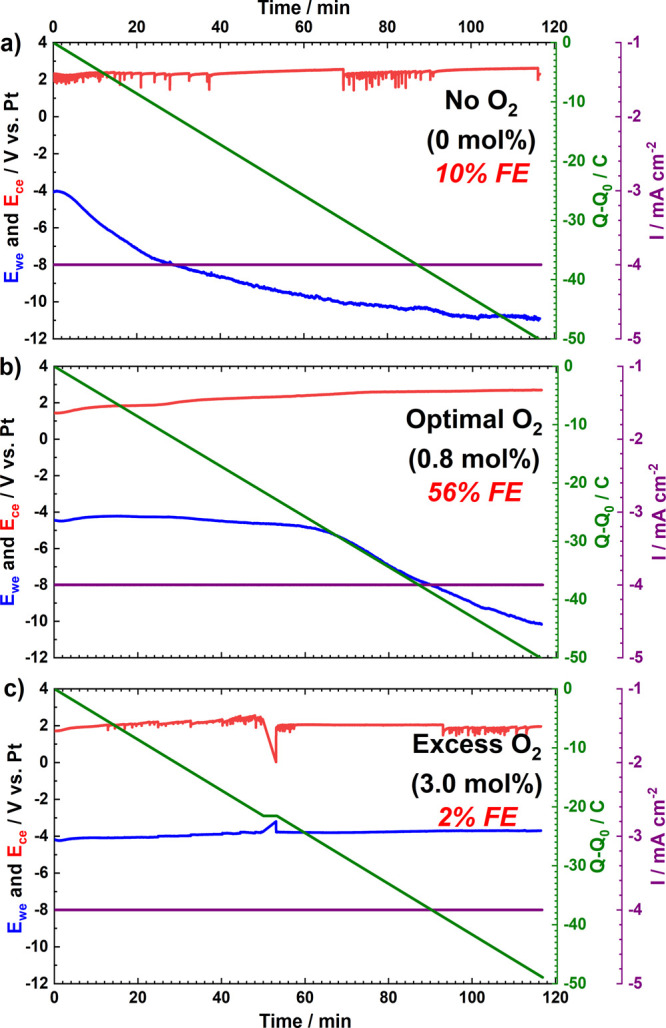
Chronopotentiometry (CP) results of LiMEAS with the presence of (a) 0, (b) optimal 0.8, and (c) excess 3 mol % of O2 added. All the experiments were performed at 20 bar (N2 ± O2) and at −4 mA cm–2 with 50 C of charge passed.
The FE of LiMEAS in THF electrolytes was evaluated using an indophenol method described in more detail in the Supporting Information.25,26 The LiMEAS resulted in FE of 10%, 56%, and 2% (all <1% error within 3 different measurements) with 0, 0.8, and 3.0 mol % of O2, respectively. The advantages of adding a small amount of O2 to the system over typical non-O2 reaction can be clearly seen. Therefore, the optimal amount of O2 enhances the FE and electrochemical stability of the system (Figure 1).
Several images of the WE in the Ar-filled glovebox after LiMEAS for 2 h in THF are shown in Figure S3. Even though the experimental conditions were the same, the images are difficult to analyze. The WE with 0 and excess of 3 mol % O2 show only a thin deposit on the front side that is facing the CE. A slightly thicker deposit is observed for the optimum O2 content of 0.8 mol %, both on the front and back sides of the electrode. It should be noted that the formation of the dendrites was not observed on the surface of the electrodes, and the yellow color is due to image quality, not a yellow deposit. A slightly more homogeneous surface was obtained for the case of LiMEAS with optimal 0.8 mol % O2 concentration, especially in the peripheral shinny metallic part of the electrode. The effect of O2 on the FE and the WE potential was investigated in detail in the literature.16
Herein, we investigate the effect of O2 on the stability of the electrolyte with GCMS and NMR. For the GCMS analysis, all the samples were analyzed for 20 min each with repeated injections, and the temperature program used is shown in the Supporting Information (Figure S4). Overall GCMS results are shown in Figure 2 and Figures S5 and S6 in the Supporting Information as 3D chromatograms after 2 h of constant current LiMEAS in THF-based electrolytes with different O2 concentrations. The freshly obtained electrolytes were transferred to the GCMS after LiMEAS followed by immediate analysis of the electrolytes to limit reaction of the electrolyte with air and moisture. The experimental details regarding the GCMS experiments and analysis of the samples are described in brief in the Supporting Information and were reported by us in detail in ref (11). Longer GCMS recording time did not reveal any eluents from the samples, and all the byproducts of LiMEAS eluted in less than 15 min. It can be seen that the increase of the O2 content in the system reduces the number and the amount of the byproducts formed during LiMEAS. The thorough analysis of the organic volatile species in the electrolytes after LiMEAS resulted in 10, 5, and 5 compounds detected by GCMS for the 0, 0.8, and 3 mol % O2, respectively. The results are shown in Tables S1–S3 of the Supporting Information. As shown in Figure 2 and summarized in Figure 3, the total number of organic volatile species detectable with GCMS decreases with increasing O2 content; the sum of the total amounts (organic and nonorganic), however, increases because of the permanent increase of H2O, which is caused by the oxygen reduction reaction (ORR). On the other hand, the addition of excess O2 does not have an effect on the structure of the byproducts introducing some novel or different compounds, only the number and the amount of them. The chromatograms become cleaner from organic species with increasing O2 (Tables S1–S3). The quantity and mass of the decomposition products and their concentration measured in the electrolytes decrease when the optimal 0.8 mol % O2 is added. On the other hand, the excess of O2 induces much less decomposition products than that without O2. This suggests that denser or thicker surface films are formed on the basis of inorganic Li compounds on the surface of the electrodes because of reaction between Li and O2. Consequently, these inorganic Li materials are not involved in further decomposition reactions of the electrolyte.
Figure 2.
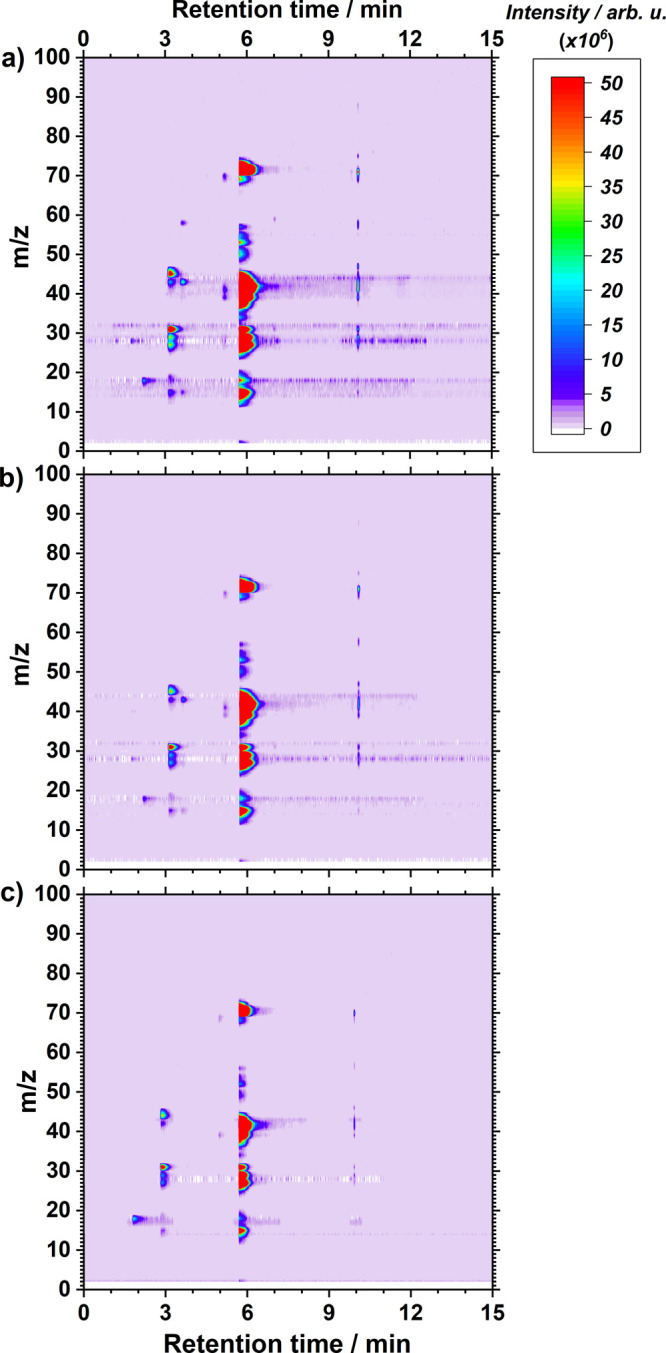
GCMS contour chromatograms visualizing electrolyte composition after LiMEAS with the presence of (a) 0, (b) 0.8, and (c) 3.0 mol % of O2 added to the system.
Figure 3.
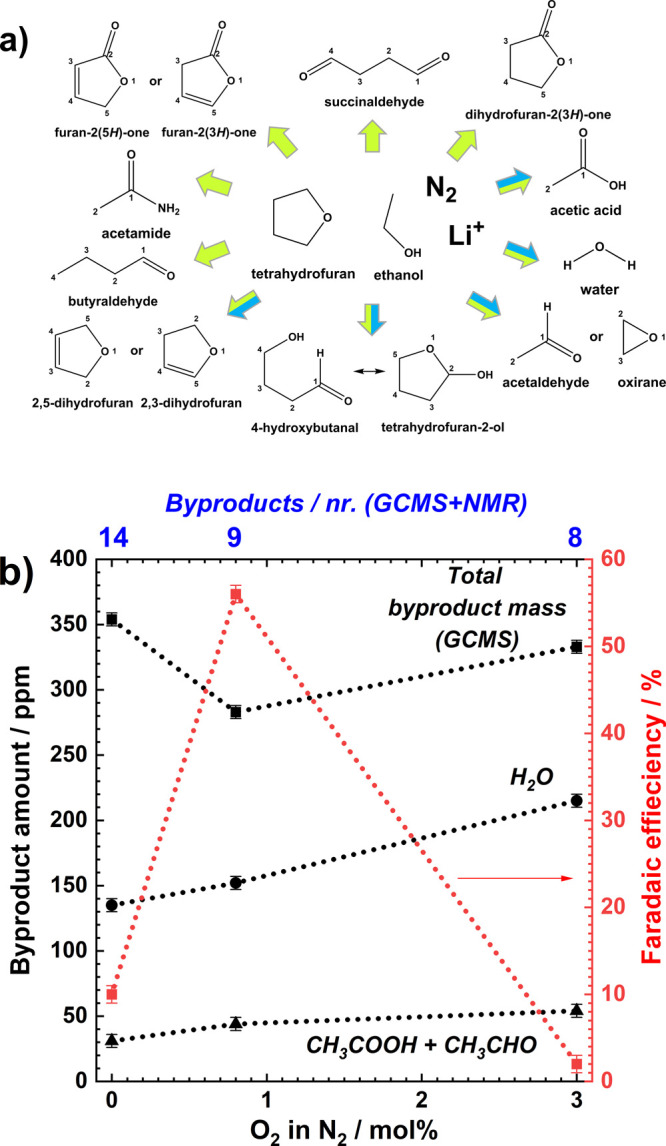
GCMS results. (a) The spectrum of volatile byproducts detected by GCMS in THF-based electrolyte after LiMEAS without (green) and with (blue) addition of O2; (b) total number (blue scale), concentration of byproducts, H2O, acetic acid/acetaldehyde (CH3COOH/CH3CHO) (black), and Faradaic efficiency (red) vs O2 content. The error bars represent three separate measurements of the same experiment.
The decomposition extent of the THF-based electrolyte after LiMEAS with addition of O2 to the system is exhibited in Figure 3. The LiMEAS without O2 leads to all the compounds shown by green arrows (10). However, the addition of O2 to LiMEAS reduces the number of the byproducts formed during the electrolysis represented by blue arrows (5), and the mass of organic decomposition products decreases as shown in Figure 3a,b. The detected compounds were characterized and their amounts were evaluated as summarized in Tables S1–S3. It can be seen that O2 added to the LiMEAS electrochemical system significantly improves FE for ammonia and stability of the system. We hypothesize that this is due to the changes in the formation and stabilization of the SEI layer which inhibit further decomposition of the electrolyte compared to LiMEAS with only N2 present, which gives relatively better stability of the electrolyte and better protection of the electrode. The second argument is that some O2 present suppresses the HER reaction from EtOH by employing it in oxidation reactions toward acetaldehyde and acetic acid on the transition metal–transition metal oxide surface, as has been reported previously in the literature.27,28 We have observed that EtOH at less than 1 vol %, as used in this study, increases FE but decreases stability of the system; that is, more dendrites are formed and more flakes are seen in the electrolyte (not reported in this study). The LiMEAS demonstrates enhanced performance and stability with the addition of the optimal amount of O2.
As can be seen in Figure 3b, the total amount of the organic decomposition products detected by GCMS shown in Figure 3a is decreases from 0 to 0.8 mol % and increases from 0.8 to 3 mol % of O2, respectively. In total, the amount of H2O and EtOH oxidation products, i.e., acetic acid and acetaldehyde, increases with addition of O2.
Our NMR study after LiMEAS involves two main types of samples: (i) solid residual on the WE and (ii) solid residual from dried electrolyte. For the former, the WE was dried in the glovebox atmosphere overnight and then washed with two different deuterated solvents, i.e., CDCl3 and D2O; for the latter, the used electrolyte was first dried at 60 °C to evaporate THF and EtOH, then the solid residual was dissolved in deuterated solvent. Figure 4a presents NMR spectra of the dry residue on the working electrode after LiMEAS washed with CDCl3. As expected, no obvious signal of THF and EtOH is visible as they were removed by drying the sample prior to NMR analysis. This seems to be sufficient for removing most of the volatile solvent because there are no obvious signals of the electrolyte components. On the other hand, the main organic component of the solid film on the surface of the WE is the THF polymerization product, polytetrahydrofuran (polyTHF), according to the chemical shifts and multiplicity of the peaks.29,30 This polymer is the main organic and polymeric constituent of the SEI in LiMEAS together with inorganic species reported previously,8,10,24 which is formed on the surface of the WE and responsible for mass transport in and out of the electrode surface. The characteristic signals at 3.7–3.8 ppm belong to the protons of the methylene groups attached to the carbon close to the heteroatom, i.e., oxygen in −OCH2CH2CH2CH2O– chains.30 The chemical shifts at 1.8–2.0 and 1.7 ppm belong to different protons attached to the carbons between other carbon atoms (−OCH2CH2CH2CH2O−) of polyTHF. The higher the chemical shift, the closer the proton is attached to the heteronucleus, i.e., oxygen in the chain. The latter polymer has been reported and characterized in the literature as a substance produced in acid-treated polymerization of THF;31 however, it has not been reported in any electrochemical systems or as an SEI component to date. PolyTHF forms on the surface of WE with or without O2 added to the system. However, with excess O2 (3 mol %), the surface of the electrode is most likely completely dominated by hydrated lithium oxide materials (LixOyHz) because of increased reaction of electroplated Li and O2, as can be seen in Figure S7 of the Supporting Information. For other types of protons in the materials, no aromatic or unsaturated protons have been detected in the samples, as there are no peaks at 6–8 ppm and higher chemical shifts, indicating that polyTHF is the main organic matrix of the SEI. Only some additional peaks can be seen in Figure 4a, which are very difficult to attribute to some structure of the molecules, and we speculate that according to their chemical shifts, they belong to lithium ethoxide (LiOCH2CH3 or LiOEt). The 1H NMR spectrum of the WE after LiMEAS with excess (3.0 mol %) O2 is relatively clean with respect to protons detected in organic molecules including polyTHF, indicating less organic decomposition products. Overall, the NMR results lead to the conclusion that excess O2 reacts with electroplated Li and stops its activity toward electrolyte decomposition on the WE but also shut down the ammonia synthesis.
Figure 4.
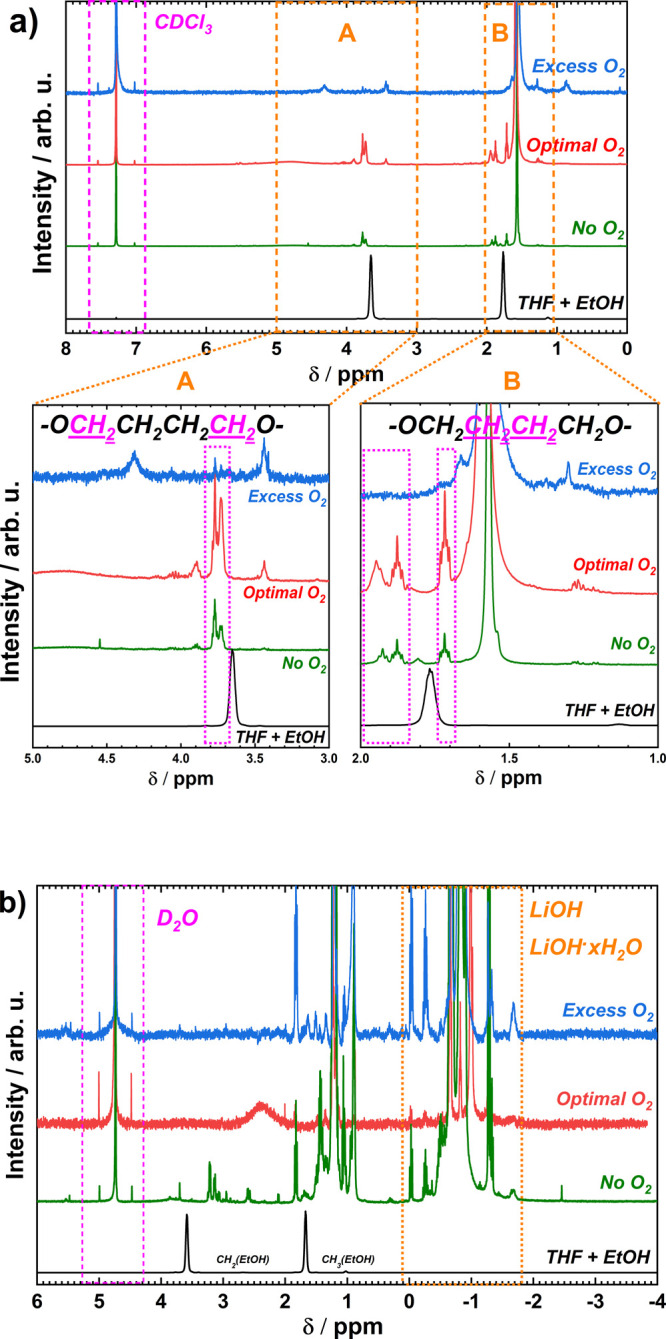
1H NMR results of the surface species on the working electrode (WE) washed with CDCl3 after LiMEAS with different O2 content in the feed gas. A and B show magnified parts of the spectra for specific aliphatic protons of the polyTHF. (b) The 1H NMR results of the surface species on the WE washed with D2O after LiMEAS with different O2 content in the feed gas. A and B represent selected regions in the spectrum.
The 1H NMR investigation of the chemical species on the WE soluble in H2O with different O2 content in the feed gas is shown in Figure 4b. For this purpose, D2O was used as a solvent, and the results were identical for all three O2 concentrations. The main components of materials formed on the surface of the WE are lithium hydroxide, LiOH, and lithium hydroxide hydrates (LiOH·xH2O). No lithium hydride (LiH) was detected ((−5) to (−20) ppm), as this would react with any traces of H2O to form LiOH. We have previously reported and described these materials as SEI components in LiMEAS using XRD and XPS,10 and they have also been reported as components of SEI in LIBs.32 Also, aliphatic protons belonging to most likely deuterated ethanol (DOEt) can be seen.
The 1H NMR spectra of the residue after LiMEAS dried at 60 °C from THF and EtOH and dissolved in CDCl3 are shown in Figure S7. The main components are a mixture of materials recorded and already identified by GCMS and the polyTHF described above and in Figure 4. Moreover, the aliphatic region of the NMR spectra seems to be similar without and with optimal 0.8 mol % of O2. On the other hand, the excess of O2 reduces the amount of signals with distinct chemical shifts, indicating that less organic species are formed as byproducts of LiMEAS, confirming the results discussed above and previously.11 The samples with no and optimal oxygen contain unsaturated carbons because of protons detected at 5–6.5 ppm (Figure S7). This supports the GCMS findings shown in Figure 3 when it comes to byproducts of the oxidation of the solvent containing double or triple carbon bonds (Figure S7). No signals at 7–8 ppm could be detected except CDCl3, which indicates no aromatic protons.
It is therefore important to mention that we cannot evaluate the whole extent of decomposition as the byproduct spectrum after LiMEAS because other inorganic substances, such as Li2CO3 and lithium chloride, LiCl, are not detectable by GCMS and 1H or 13C NMR. The latter methods, as with many in the field, have to be supplemented by, e.g., XRD or XPS. Thus, the “real” amount of byproducts would be different than assumed here because of nonmeasurable inorganic species. We have seen and reported several inorganic phases before by XRD and XPS,8,10,16,24 and they are reproducible.
In summary, the stabilizing effect of oxygen in LiMEAS was demonstrated in THF-based electrolyte by GCMS and NMR analysis. It was shown that adding O2 to the gas feed suppresses the electrolyte degradation and chemical side reactions, leading to a more efficient and stable LiMEAS process and enhancing the chemical stability of the system. A stable voltage profile for more than 2 h at a current density of −4 mA cm–2 and the least amount of the organic byproducts is achieved with excess O2, however at the expense of reduced FE. The total number of byproducts detected by GCMS and NMR decreased with increasing O2 and was 14, 9, and 8 with O2 concentration of 0, 0.8, and 3 mol %, respectively. The 0.8 mol % oxygen is optimal for very efficient and stable LiMEAS. The high performance of LiMEAS with optimal amount of O2 presented in this work was attributed to the formation of a stable and efficient SEI on the surface of the working electrode run in THF-based electrolyte and the subsequent suppression of hydrogen evolution reaction from the proton source, EtOH or THF, in LiMEAS. The use of O2 in the THF-based electrolyte is very promising as it makes it possible to obtain more efficient and stable LiMEAS, while simultaneously reducing the costs associated with purifying air to N2.
Acknowledgments
We gratefully acknowledge the funding by Villum Fonden, part of the Villum Center for the Science of Sustainable Fuels and Chemicals (V-SUSTAIN grant 9455), Innovationsfonden (Eammonia grant 9067-00010B), and the European Research Council (ERC) under the European 430 Union’s Horizon 2020 Research and Innovation Programme (grant agreement No 741860). We thank the floor managers and the whole SurfCat research group at DTU Physics for being together and helping with some equipment and experiments. The NMR measurements were performed at the NMR Center at DTU Chemistry. Therefore, we acknowledge Kasper Enemark-Rasmussen and Charlotte Held Gotfredsen for assistance with NMR.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.2c00768.
Experimental section; supplementary methods and calculations; Figures S1–S7 showing the setup and the electrodes after LiMEAS, LSV results, GCMS temperature program, and additional GCMS and NMR results; and Tables S1–S3 which summarize and quantify the byproducts with different O2 content added to LiMEAS system (PDF)
Transparent Peer Review report available (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chen J. G.; Crooks R. M.; Seefeldt L. C.; Bren K. L.; Bullock R. M.; Darensbourg M. Y.; Holland P. L.; Hoffman B.; Janik M. J.; Jones A. K.; Kanatzidis M. G.; King P.; Lancaster K. M.; Lymar S. V.; Pfromm P.; Schneider W. F.; Schrock R. R. Beyond fossil fuel-driven nitrogen transformations. Science (New York, N.Y.) 2018, 360, eaar6611. 10.1126/science.aar6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Wang T.; Gong J. Alternative Strategies Toward Sustainable Ammonia Synthesis. Transactions of Tianjin University 2020, 26, 67–91. 10.1007/s12209-020-00243-x. [DOI] [Google Scholar]
- Garagounis I.; Vourros A.; Stoukides D.; Dasopoulos D.; Stoukides M. Electrochemical Synthesis of Ammonia: Recent Efforts and Future Outlook. Membranes 2019, 9, 112. 10.3390/membranes9090112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane E. F.; Tompkins F. C. Nitridation of lithium. Trans. Faraday Soc. 1962, 58, 997–1007. 10.1039/tf9625800997. [DOI] [Google Scholar]
- Lazouski N.; Schiffer Z. J.; Williams K.; Manthiram K. Understanding Continuous Lithium-Mediated Electrochemical Nitrogen Reduction. Joule 2019, 3, 1127–1139. 10.1016/j.joule.2019.02.003. [DOI] [Google Scholar]
- Tsuneto A.; Kudo A.; Sakata T. Lithium-mediated electrochemical reduction of high pressure N2 to NH3. J. Electroanal. Chem. 1994, 367, 183–188. 10.1016/0022-0728(93)03025-K. [DOI] [Google Scholar]
- Tsuneto A.; Kudo A.; Sakata T. Efficient Electrochemical Reduction of N2 to NH3 Catalyzed by Lithium. Chem. Lett. 1993, 22, 851–854. 10.1246/cl.1993.851. [DOI] [Google Scholar]
- Li K.; Shapel S. G.; Hochfilzer D.; Pedersen J. B.; Krempl K.; Andersen S. Z.; Sažinas R.; Saccoccio M.; Li S.; Chakraborty D.; Kibsgaard J.; Vesborg P. C. K.; Nørskov J. K.; Chorkendorff I. Increasing Current Density of Li-Mediated Ammonia Synthesis with High Surface Area Copper Electrodes. ACS Energy Lett. 2022, 7, 36–41. 10.1021/acsenergylett.1c02104. [DOI] [Google Scholar]
- Marom R.; Haik O.; Aurbach D.; Halalay I. C. Revisiting LiClO4 as an Electrolyte for Rechargeable Lithium-Ion Batteries. J. Electrochem. Soc. 2010, 157, A972–A983. 10.1149/1.3447750. [DOI] [Google Scholar]
- Andersen S. Z.; Colic V.; Yang S.; Schwalbe J. A.; Nielander A. C.; McEnaney J. M.; Enemark-Rasmussen K.; Baker J. G.; Singh A. R.; Rohr B. A.; Statt M. J.; Blair S. J.; Mezzavilla S.; Kibsgaard J.; Vesborg P. C. K.; Cargnello M.; Bent S. F.; Jaramillo T. F.; Stephens I. E. L.; Norskov J. K.; Chorkendorff I. A rigorous electrochemical ammonia synthesis protocol with quantitative isotope measurements. Nature 2019, 570, 504–508. 10.1038/s41586-019-1260-x. [DOI] [PubMed] [Google Scholar]
- Sažinas R.; Andersen S. Z.; Li K.; Saccoccio M.; Krempl K.; Pedersen J. B.; Kibsgaard J.; Vesborg P. C. K.; Chakraborty D.; Chorkendorff I. Towards understanding of electrolyte degradation in lithium-mediated non-aqueous electrochemical ammonia synthesis with gas chromatography-mass spectrometry. RSC Adv. 2021, 11, 31487–31498. 10.1039/D1RA05963G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiskanen S. K.; Kim J.; Lucht B. L. Generation and Evolution of the Solid Electrolyte Interphase of Lithium-Ion Batteries. Joule 2019, 3, 2322–2333. 10.1016/j.joule.2019.08.018. [DOI] [Google Scholar]
- Liu D.; Shadike Z.; Lin R.; Qian K.; Li H.; Li K.; Wang S.; Yu Q.; Liu M.; Ganapathy S.; Qin X.; Yang Q. H.; Wagemaker M.; Kang F.; Yang X. Q.; Li B. Review of Recent Development of In Situ/Operando Characterization Techniques for Lithium Battery Research. Adv. Mater. 2019, 31, 1806620. 10.1002/adma.201806620. [DOI] [PubMed] [Google Scholar]
- Stenzel Y. P.; Horsthemke F.; Winter M.; Nowak S. Chromatographic Techniques in the Research Area of Lithium Ion Batteries: Current State-of-the-Art. Separations 2019, 6, 26. 10.3390/separations6020026. [DOI] [Google Scholar]
- Zhuang G. R.; Wang K.; Chen Y.; Ross P. N. Study of the reactions of Li with tetrahydrofuran and propylene carbonate by photoemission spectroscopy. J. Vacuum Sci. & Technol. A: Vacuum, Surfaces, and Films 1998, 16, 3041–3045. 10.1116/1.581475. [DOI] [Google Scholar]
- Li K.; Andersen S. Z.; Statt M. J.; Saccoccio M.; Bukas V. J.; Krempl K.; Sažinas R.; Pedersen J. B.; Shadravan V.; Zhou Y.; Chakraborty D.; Kibsgaard J.; Vesborg P. C. K.; Nørskov J. K.; Chorkendorff I. Enhancement of lithium-mediated ammonia synthesis by addition of oxygen. Science 2021, 374, 1593–1597. 10.1126/science.abl4300. [DOI] [PubMed] [Google Scholar]
- Wang L.; Menakath A.; Han F.; Wang Y.; Zavalij P. Y.; Gaskell K. J.; Borodin O.; Iuga D.; Brown S. P.; Wang C.; Xu K.; Eichhorn B. W. Identifying the components of the solid-electrolyte interphase in Li-ion batteries. Nat. Chem. 2019, 11, 789–796. 10.1038/s41557-019-0304-z. [DOI] [PubMed] [Google Scholar]
- Aurbach D.; et al. Identification of Surface Films Formed on Lithium in Propylene Carbonate Solutions. J. Electrochem. Soc. 1987, 134, 1611–1620. 10.1149/1.2100722. [DOI] [Google Scholar]
- Aurbach D.; Daroux M.; Faguy P.; Yeager E. The electrochemistry of noble metal electrodes in aprotic organic solvents containing lithium salts. J. Electroanal. Chem. and Interface Electrochem. 1991, 297, 225–244. 10.1016/0022-0728(91)85370-5. [DOI] [Google Scholar]
- Schultz C.; Vedder S.; Streipert B.; Winter M.; Nowak S. Quantitative investigation of the decomposition of organic lithium ion battery electrolytes with LC-MS/MS. RSC Adv. 2017, 7, 27853–27862. 10.1039/C7RA03839A. [DOI] [Google Scholar]
- Kraft V.; Weber W.; Grützke M.; Winter M.; Nowak S. Study of decomposition products by gas chromatography-mass spectrometry and ion chromatography-electrospray ionization-mass spectrometry in thermally decomposed lithium hexafluorophosphate-based lithium ion battery electrolytes. RSC Adv. 2015, 5, 80150–80157. 10.1039/C5RA16679A. [DOI] [Google Scholar]
- Gourdin G.; Collins J.; Zheng D.; Foster M.; Qu D. Spectroscopic Compositional Analysis of Electrolyte during Initial SEI Layer Formation. J. Phys. Chem. C 2014, 118, 17383–17394. 10.1021/jp504181b. [DOI] [Google Scholar]
- Yang L.; Smith C.; Patrissi C.; Schumacher C. R.; Lucht B. L. Surface reactions and performance of non-aqueous electrolytes with lithium metal anodes. J. Pow. Sour. 2008, 185, 1359–1366. 10.1016/j.jpowsour.2008.09.037. [DOI] [Google Scholar]
- Andersen S. Z.; Statt M. J.; Bukas V. J.; Shapel S. G.; Pedersen J. B.; Krempl K.; Saccoccio M.; Chakraborty D.; Kibsgaard J.; Vesborg P. C. K.; Nørskov J.; Chorkendorff I. Increasing stability, efficiency, and fundamental understanding of lithium-mediated electrochemical nitrogen reduction. Energy & Environ. Sci. 2020, 13, 4291–4300. 10.1039/D0EE02246B. [DOI] [Google Scholar]
- Searle P. L. The berthelot or indophenol reaction and its use in the analytical chemistry of nitrogen. A review. Analyst 1984, 109, 549–568. 10.1039/an9840900549. [DOI] [Google Scholar]
- Verdouw H.; Van Echteld C. J. A.; Dekkers E. M. J. Ammonia determination based on indophenol formation with sodium salicylate. Water Res. 1978, 12, 399–402. 10.1016/0043-1354(78)90107-0. [DOI] [Google Scholar]
- Mallat T.; Baiker A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. 10.1021/cr0200116. [DOI] [PubMed] [Google Scholar]
- Redina E. A.; Greish A. A.; Mishin I. V.; Kapustin G. I.; Tkachenko O. P.; Kirichenko O. A.; Kustov L. M. Selective oxidation of ethanol to acetaldehyde over Au–Cu catalysts prepared by a redox method. Catal. Today 2015, 241, 246–254. 10.1016/j.cattod.2013.11.065. [DOI] [Google Scholar]
- Dey A. N.; Rudd E. J. Electroinitiated Polymerization of Tetrahydrofuran. J. Electrochem. Soc. 1974, 121, 1294–1298. 10.1149/1.2401671. [DOI] [Google Scholar]
- Pan C. W.; Xia K.; Parker S. A.; Tillman E. S. Identity of Low-Molecular-Weight Species Formed in End-To-End Cyclization Reactions Performed in THF. Polymers 2018, 10, 844. 10.3390/polym10080844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruckmayr G.; Dreyfuss P.; Dreyfuss M. P. Polyethers, Tetrahydrofuran and Oxetane Polymers. Kirk-Othmer Encyclopedia of Chem. Technol. 2000, 1–30. 10.1002/0471238961.2005201816182103.a01. [DOI] [Google Scholar]
- Liu T.; Liu Z.; Kim G.; Frith J. T.; Garcia-Araez N.; Grey C. P. Understanding LiOH Chemistry in a Ruthenium-Catalyzed Li-O2 Battery. Angew. Chem., Int. Ed. Engl. 2017, 56, 16057–16062. 10.1002/anie.201709886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.