

Abstract
Children with Langerhans cell histiocytosis (LCH) develop granulomatous lesions with characteristic clonal CD207+ dendritic cells that can arise as single lesions or life-threatening disseminated disease. Despite the wide range of clinical presentations, LCH lesions are histologically indistinguishable based on severity of disease, and uncertain classification as an immune versus neoplastic disorder has historically challenged development of optimal clinical strategies for patients with LCH. Recently, activating somatic mutations in MAPK pathway genes, most notably BRAFV600E, have been discovered in almost all cases of LCH. Further, the stage of myeloid differentiation in which the mutation arises defines the extent of disease and risk of developing LCH-associated neurodegeneration. MAPK activation in LCH precursor cells drives myeloid differentiation, inhibits migration, and inhibits apoptosis, resulting in accumulation of resilient pathologic DCs that recruit and activate T cells. Recurrent somatic mutations in MAPK pathway genes have also been identified in related histiocytic disorders: juvenile xanthogranuloma, Erdheim-Chester disease, and Rosai-Dorfman disease. New insights into pathogenesis support reclassification of these conditions as a myeloid neoplastic disorders. Continued research will uncover opportunities to identify novel targets and inform personalized therapeutic strategies based on cell of origin, somatic mutation, inherited risk factors and residual disease.
Keywords: Langerhans cell histiocytosis, juvenile xanthogranuloma, Erdheim-Chester disease, Rosai-Dorfman disease, histiocytic disorder
INTRODUCTION
Histiocytic disorders comprise a heterogeneous group of hematologic and immunologic conditions historically classified based on histologic similarities to cells of the mononuclear phagocyte system.1 However, with the rapidly increasing understanding of mechanisms of pathogenesis and ontogeny, a revised classification is proposed that includes cellular origins, tissue distribution and molecular lesions along with histologic features.2 (Tables 1 & 2). Langerhans cell histiocytosis (LCH), the most common histiocytic disorder in children, is the focus of this article. We will also briefly describe features related disorders: juvenile xanthogranuloma (JXG), Erdheim–Chester disease (ECD), and sinus histiocytosis with massive lymphadenopathy (SHML), also called Rosai-Dorfman disease (RDD).
Table 1.
Historical Classification of Histiocytic Disorders
| Dendritic cell related |
| Langerhans cell histiocytosis |
| Juvenile xanthogranuloma/Erdheim-Chester disease |
| Macrophage related |
| Hemophagocytic syndromes |
| Primary hemophagocytic lymphohistiocytosis |
| Secondary hemophagocytic syndromes |
| Rosai-Dorfman disease |
| Malignant diseases |
| Monocyte-related leukemias |
| Extramedullary monocytic tumor (myeloid sarcoma) |
| Macrophage-related histiocytic sarcoma |
| Dendritic cell malignancy (malignant histiocytosis) |
Adapted from 1
Table 2.
Proposed Revised Classification of Histiocytocytic Disorders
| L Group | LCH Intermediate-cell histiocytosis (ICH) Erdheim-Chester disease (ECD) Mixed LCH/ECD |
| C Group | Cutaneous non-LCH -Xanthomatous granuloma (XG) family: includes JXG -Non-XG family: includes cutaneous RDD Cutaneous non-LCH with major systemic component -XG family: xanthoma disseminatum -Non-XG: multicentric reticulohistiocytosis |
| R Group | Familial RDD Sporadic RDD -Classic RDD -Extranodal RDD -RDD with neoplasia or immune disease -Unclassified |
| M Group | Primary malignant histiocytosis Secondary malignant histiocytosis |
| H Group | Primary HLH Secondary HLH (non-Mendelian) HLH of unknown/uncertain origin |
LCH, Langerhans cell histiocytosis; ECD, Erdheim-Chester disease; JXG, juvenile xanthogranuloma; RDD, Rosai-Dorfman disease; HLH, hemophagocytic lymphohistiocytosis. Adapted from2
LANGERHANS CELL HISTIOCYTOSIS
Pathophysiology
LCH has captured the attention of physicians and scientists for more than 100 years. Clinical cases initially recognized in the early 1900s in children with unusual constellations of bone and pituitary lesions (Hand-Schüller-Christian disease), aggressive disseminated disease (Letterer-Siwe disease), or isolated or multifocal bone lesions (eosinophilic granuloma). Pathologists in the 1950s noted histologic similarity of biopsies from patients with these conditions and proposed a unifying hypothesis that these clinically distinct syndromes represent a common pathological entity, “Histiocytosis X”. Subsequently, Nezelof and colleagues identified Birbeck granules pathologic histiocytes of LCH lesions (Figure 1), a feature at that time was thought to be shared only with epidermal Langerhans cells.
Figure 1. Histologic Features of LCH.
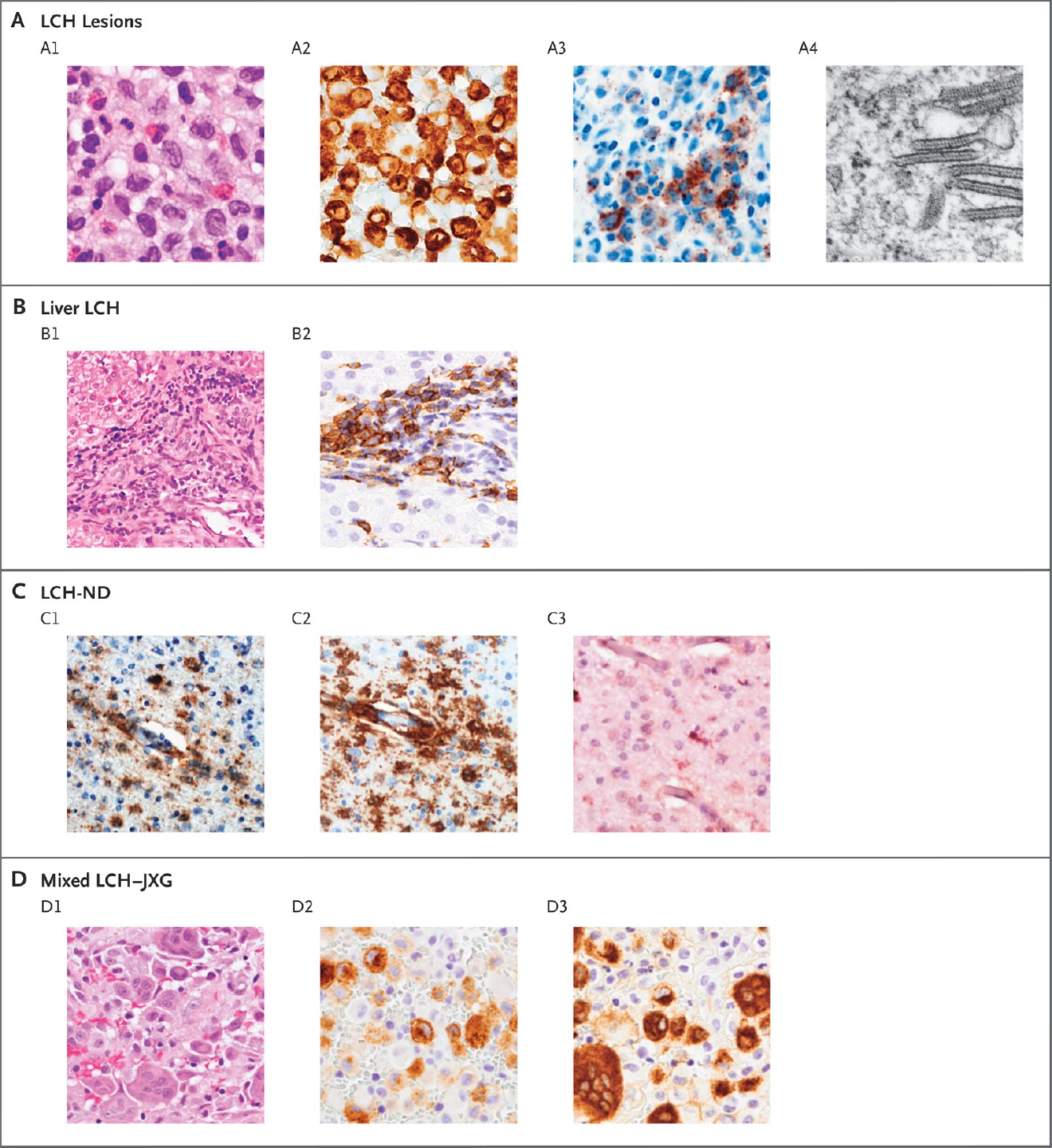
Panel A shows typical LCH lesions with large cells, pale cytoplasm, and reniform nuclei on hematoxylin and eosin staining (A1); CD207-positive immunostaining (A2); VE1-positive immunostaining for BRAF V600E protein (A3); and Birbeck granules visualized with electron microscopy (A4). Panel B shows liver involvement, which is frequently characterized by periportal infiltration by histiocytes (B1) and variable CD207-positive staining (B2). Panel C shows biopsy specimens from a patient with severe LCH-associated neurodegeneration (LCH-ND), characterized by perivascular VE1-positive staining (C1), CD163-positive staining (C2), and a P2RY12 infiltrate with occasional P2RY12-positive, tissue-resident microglia (C3). Panel D shows histiocytic lesions that are characteristic of both LCH and juvenile xanthogranuloma (JXG), with heterogeneous histologic features on hematoxylin and eosin staining (D1), including distinct cell populations that are CD207-positive (D2) and CD68-positive (D3).
(From NEJM, Langerhans Cell Histiocytosis, 379:856–8683; Copyright ©2018 Massachusetts Medical Society. Reprinted with permission.)
LCH lesions are granulomatous lesions consisting of pathologic “Langerhans cells” (LCs), lymphocytes (primarily T-cells), eosinophils, and macrophages. Like physiologic epidermal LCs (eLC), LCH lesion LCs express CD1a and CD207 (langerin) surface markers (Figure 1, Table 3). Common features among LCH and epidermal LC supported hypotheses of LCH as a reactive immune disorder, neoplastic disorder, or some combination of both (Reviewed in 3). In the 1990s, studies of X-inactivation hinted at the clonal nature of LCH lesion LCs. In 2010, Rollins and colleagues made a breakthrough discovery of recurrent somatic BRAFV600E mutations in over 50% of LCH lesions.4 Subsequently, alternative BRAF mutations (indels and fusions) and mutations in MAP2K1 (encoding MEK1) were also been described. Mutually exclusive somatic activating mutations in MAPK pathway genes have now been identified in approximately 85% LCH lesions.5 (Figure 2)
Table 3.
Histologic Features of Histiocytic Disorders
| LCH | ECD/JXG | RDD | |
|---|---|---|---|
| HLA-DR | ++ | − | + |
| CD1a | ++ | − | − |
| CD14 | +/− | ++ | ++ |
| CD68 | +/− | ++ | ++ |
| CD163 | − | + | ++ |
| CD 207 (Langerin) | +++ | − | − |
| Factor XIIIa | − | ++ | − |
| Fascin | − | ++ | + |
| Birbeck granules | + | − | − |
| Hemophagocytosis | +/− | − | − |
| Emperiopolesis | − | − | + |
LCH, Langerhans cell histiocytosis; ECD, Erdheim Chester disease; JXG, juvenile xanthogranuloma; HLH, hemophagocytic lymphohistiocytosis; RDD, Rosai-Dorfman disease; LC, epidermal Langerhans cell
Adapted from 22
Figure 2. MAPK Pathway Mutations in Histiocytic Disorders.
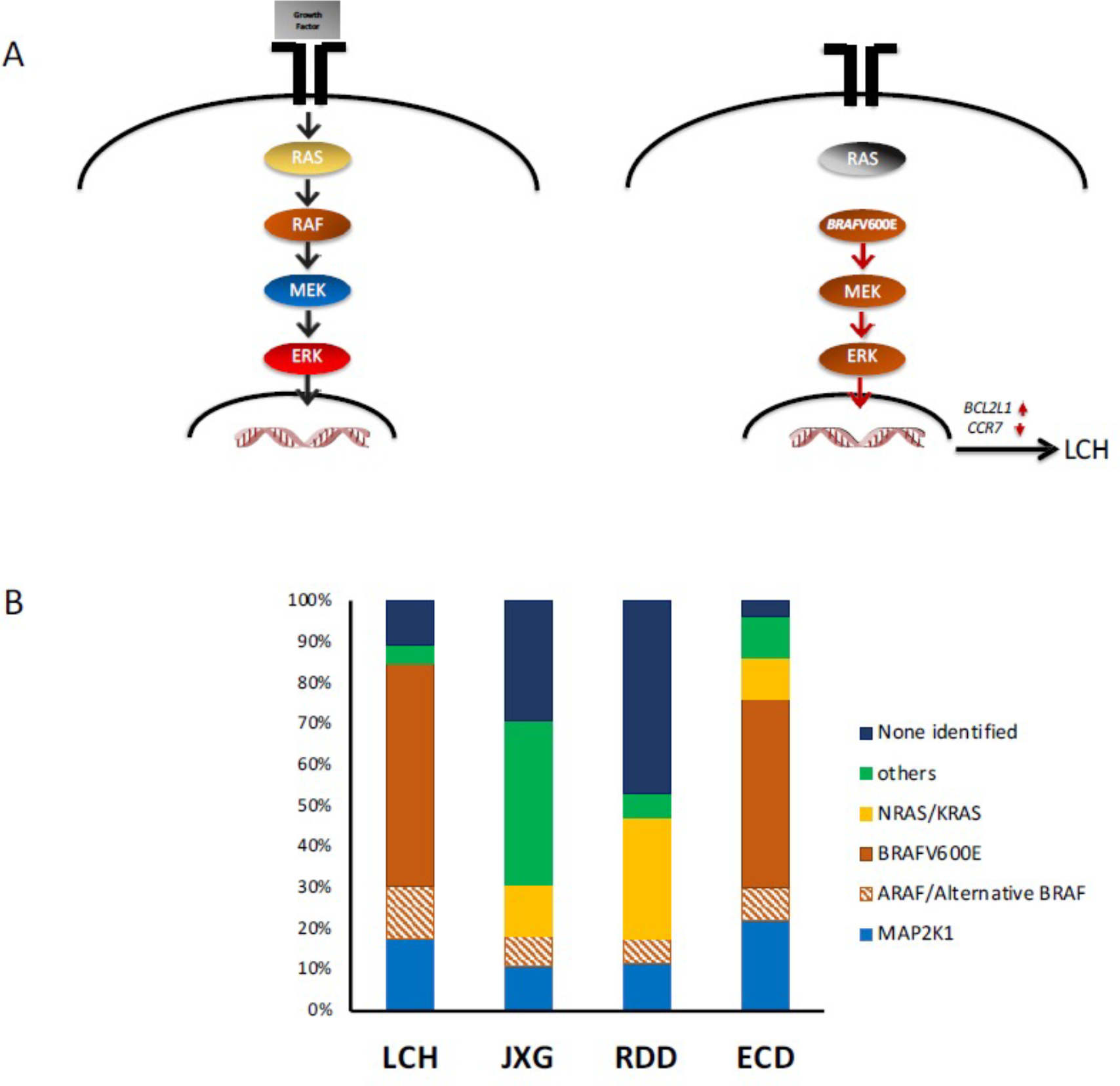
A. (left) Schematic of MAPK pathway. Under physiologic conditions, the growth factor (gray box) engages the tyrosine kinase receptor that transduces the signal to the nucleus. (right) Activating mutations (such as BRAF-V600E) drive constitutive ERK activation. In the case of LCH, this drives the expression of anti-apoptosis BCL2L1 (BCL-xL) and inhibits CCR7.
B. Stacked bar graphs represent percentages of MAPK pathway mutations in each histiocytic histologic subtype.
As discussed above, shared histology between epidermal LC and LCH lesion histiocytes prompted updated branding from “Histiocytosis X” to “Langerhans cell histiocytosis”. However, gene expression studies comparing LCH lesion CD207+ cells to eLC revealed LCH cells to be relatively less differentiated. Subsequently, high-sensitivity BRAFV600E PCR assays identified the mutation in hematopoietic stem cells from bone marrow aspirate and myeloid precursors from peripheral blood of patients with disseminated LCH. Notably, BRAFV600E was not identified in peripheral blood mononuclear cells from patients with single BRAFV600E+ lesions. Enforced expression of BRAFV600E in langerin+ cells in mice resulted in formation of some limited LCH-like lesions with minimal impact on overall health, but enforced expression in CD11c+ myeloid cells drove rapid formation of severe lesions in bone marrow, lung, liver and spleen resembling high risk LCH. Pathologic MAPK activation in LCH lesion cells results in up-regulation of anti-apoptotic program (Bcl-xL) and down-regulation of CCR7, which renders the cells trapped in lesions, unable to migrate to draining lymph nodes.6
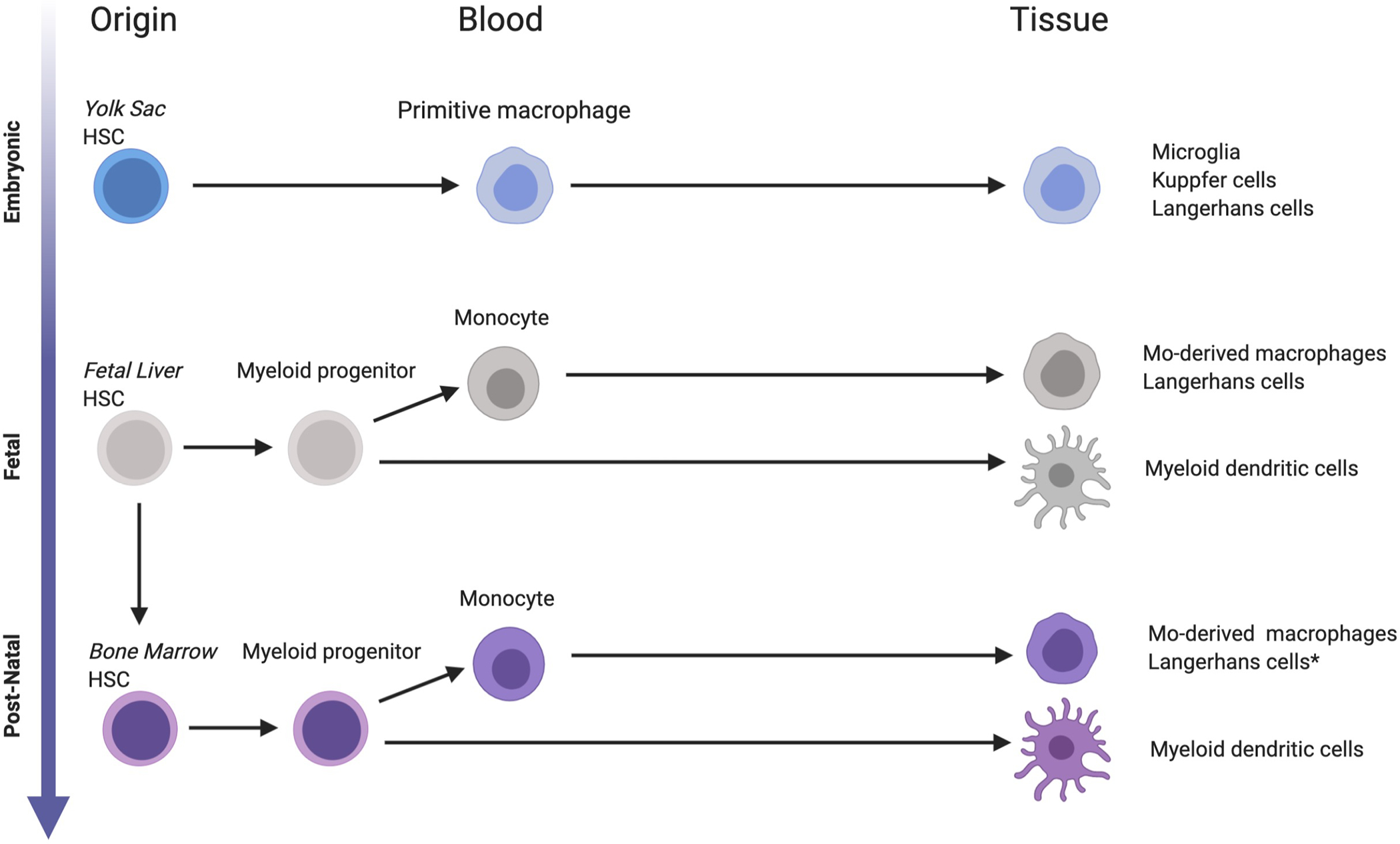
Addressing the decades-long debate of LCH pathogenesis arising from immune dysregulation versus transformation of eLC, findings over the past decade reframe LCH as a myeloid neoplastic disorder arising from myeloid precursors.3 Together, observations in LCH patients and mouse experiments support a model of ‘Misguided Myeloid Differentiation’ where state of differentiation of myeloid precursor in which activating MAPK mutation arises determines the extent and severity of disease (Figure 3).7
Figure 3. Ontogeny of tissue “histiocytes”.
Tissue macrophages and dendritic cells seed tissues at various stages of development. Microglia, Küpffer cells and Langerhans cells arise from yolk sac-derived progenitors, followed by fetal liver, then adult hematopoiesis. *Langerhans cells may be generated from adult HSC monocyte-derived LCs following inflammation or tissue damage.
Epidemiology
The incidence of LCH is estimated to be approximately 5–10 cases per million children per year and 1–2 cases per million adults per year with male to female ratio 1.2:1. Registry studies report an increased incidence in Hispanic populations and rare occurrence of LCH in children with African ancestry.8 Notably, a GWAS trio study that identified an increased risk of LCH in patients with a germline SMAD6 variant, which is enriched in Hispanic populations.9
Clinical presentation and diagnostic work-up
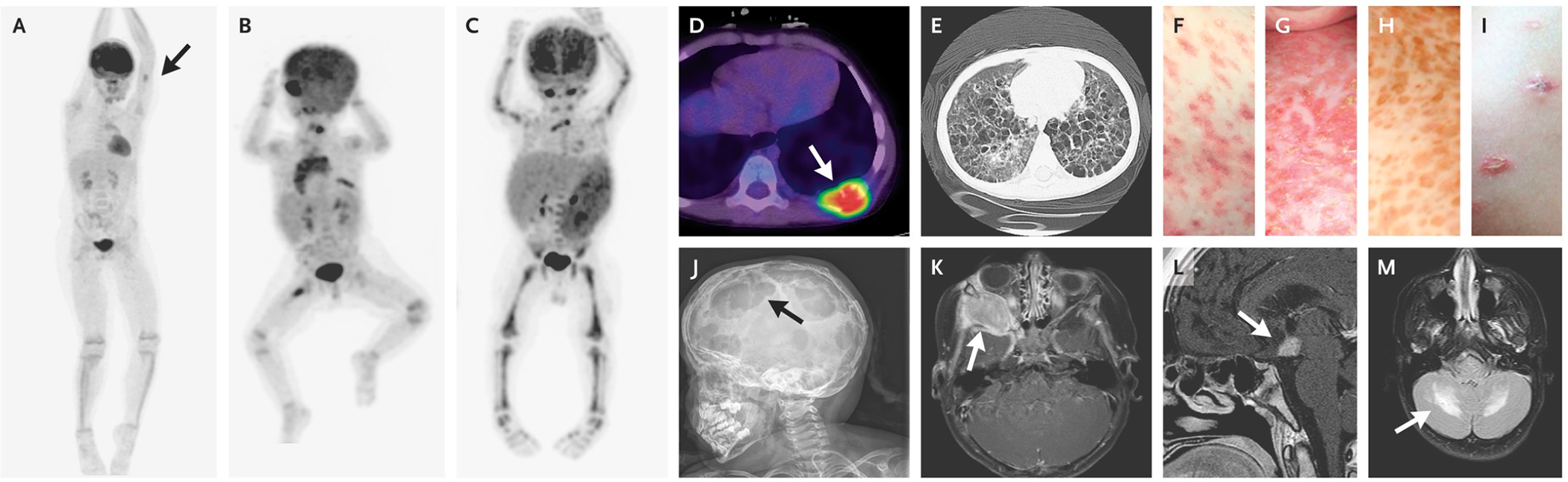
LCH has a wide range of clinical manifestations that can be difficult to recognize due to overlap with more common conditions (Figure 4). However, once LCH is considered, diagnosis is fairly straightforward with biopsy. A step-wise approach to diagnosis and staging is outlined in Table 4. Characteristic presentations include lytic bone lesions (~80% of cases), rash (~20–40% of cases), soft tissue swelling (often in proximity to bony lesions), external ear drainage, lymph node or thymic enlargement, and gum hypertrophy with premature eruption of teeth. More severe systemic involvement reflected by cytopenias, heptasplenomegaly and/or impaired liver function portends a higher risk of morbidity and mortality. Based on findings from Histiocyte Society trials, LCH is clinically divided into “high risk” (liver, spleen and/or bone marrow involvement) and “low risk” (lesions anywhere else), reflecting relative risk of death.10 The significance of ‘risk sites’ in adults remains uncertain.11 Historical nomenclature (e.g. Letterer-Siwe) has been replaced with a more generalized attribution of “LCH” along with description of extent of disease (e.g. low risk single system; low-risk multisystem; high risk multisystem).
Figure 4. Clinical Presentations of LCH.
Positron-emission tomographic (PET) images show a single bone lesion involving the humerus (Panel A, arrow); low-risk lesions involving the orbit, lymph nodes, bone (multifocal lesion), and thymus (Panel B); and high-risk lesions involving the liver, spleen, and bone marrow (Panel C). Other classic presentations include a lytic bone lesion (Panel D, arrow), cystic lung lesions (Panel E), and various skin lesions (Panels F through I). Examples of LCH lesions involving the skull and brain include multifocal skull lesions (Panel J, arrow), an orbital lesion (Panel K, arrow), a pituitary lesion (Panel L, arrow), and LCH-associated neurodegeneration (Panel M, arrow).
(From NEJM, Langerhans Cell Histiocytosis, 379:856–8683; Copyright ©2018 Massachusetts Medical Society. Reprinted with permission.)
TABLE 4.
Clinical Evaluations for Newly Diagnosed LCH
| Initial Evaluation |
| • History, Physical examination • Laboratory Studies ○ Complete blood count ○ Serum chemistry ○ Liver function test ○ Sedimentation rate (ESR) ○ Lactate dehydrogenase (LDH) ○ Serum ferritin ○ Immunoglobulin profile ○ PT/INR, aPTT (with evidence of liver dysfunction) • Imaging ○ Skeletal survey with 2 views of chest and 4 views of the skull (if PET/CT not obtained). |
| Confirmation of LCH and extent of disease evaluation |
| • PET/CT • Diagnostic Biopsy-excisional biopsy is preferred. Curettage of bone lesions is optimal and complete excision is not required. The presence of abnormal clusters of CD1a+/CD207+ histiocytes are diagnostic (Note: normal skin and lymph node biopsies may include scattered physiologic CD207+ Langerhans cells). CD163, fascin, and factor XII help identify mixed histiocytic lesions (such as JXG/LCH, ECD/LCH) |
| Additional studies(based on lab and/or clinical features) |
| • All patients < 2 years of age, any patients with cytopenias, liver and spleen involvement ○ Bilateral bone marrow aspirate and biopsy • Any skull lesions (based on physical exam or lytic lesions on skull x-rays) -CT Skull/maxillofacial scans • CNS-risk lesions ○ MRI brain with and without contrast for patients with CNS-Risk lesions ○ For auditory canal or temporal bone involvement, also perform a hearing evaluation ○ For clinical suspicion of DI, pituitary dysfunction, or thickened pituitary stalk on MRI brain- urine specific gravity, urine and serum osmolality/water deprivation test. (Note: If there is an isolated DI and thickened pituitary stalk without any other features suggestive of LCH, perform diagnostic LP for cytology and AFP/B-HCG to rule out germ cell tumor). ○ Other endocrine evaluation as indicated ○ Baseline neurocognitive evaluation for patients with DI or evidence of LCH-ND • Spinal cord or vertebral involvement-MRI spine with and without contrast • Pulmonary involvement-CT chest (chest x-rays and PET/CT may miss small pulmonary nodules, cysts, or thymic involvement). • Elevated transaminases, elevated direct bilirubin or decreased albumin-Abdominal US or MRI • History of malabsorption or hypoalbuminemia- Lower GI endoscopy |
Beyond risk attribution, some characteristic sites merit discussion. Skin lesions in infants may represent isolated skin disease with potential to spontaneously resolve or a component of more extensive systemic disease requiring chemotherapy. Gastrointestinal tract involvement is rare but can present with severe diarrhea, hematochezia, malabsorption, and hypoproteinemia. Isolated pulmonary involvement is more common in young adults with a history of chronic smoking in the third or fourth decades of life but is occasionally seen in children and adolescents. Pulmonary involvement may lead to a severe, chronic debilitating course and often presents with spontaneous pneumothorax. Central diabetes insipidus (DI) affects approximately 25% of patients, most commonly seen in children with systemic disease and the orbit and skull involvement. Most cases of DI present with initial systemic disease, but can also present as isolated pituitary disease, or arise as a site of relapse. While posterior pituitary involvement is more common, other endocrine manifestations associated with LCH may include growth hormone deficiency, adrenal insufficiency, hyperprolactinemia, or hypogonadism caused by hypothalamic infiltration of the anterior pituitary gland.12
LCH-associated neurodegeneration (LCH-ND) is one of the most severe complications of LCH. LCH-ND may develop with the onset of LCH or several years after the patient has completed therapy and is presumed to be in remission. The presence of “CNS-risk” bone lesions (orbit, mastoid, maxilla, temporal, sphenoid, zygomatic, clivus) or pituitary lesions at the time of initial diagnosis is thought to increase the risk of developing LCH-ND. While the true risks of “CNS-risk” require additional investigation, current practice is generally to treat isolated CNS risk lesions with systemic chemotherapy. BRAFV600E mutation is also associated with increased risk of LCH-ND. Patients with LCH-ND typically present with prolonged decline in cognitive abilities, worsening school performance, and/or development of cerebellar symptoms. Characteristic magnetic resonance imaging (MRI) findings include T2 hyper-intense diffuse or polymorphic lesions involving the white matter of the cerebellum, pons, basal ganglia, and less often the cerebral hemispheres.13 (Figure 4). The etiology of LCH-ND remained uncertain for years, with limited biopsy studies demonstrating lymphocytic infiltration and activated microglia interpreted as a paraneoplastic or autoimmune phenomenon. More recent studies demonstrate microglia-like mononuclear cells at sites of neurodegeneration with BRAFV600E, supporting clonal origin with systemic LCH lesions.14 (Figure 1)
Therapy
Local LCH
Treatment options depend on the site and extent of the disease. Isolated skin lesions sometimes resolve spontaneously, with topical steroids, or with oral immune suppression (e.g. methotrexate or hydroxyurea). Single bone lesions in readily accessible and non-CNS-risks sites may be treated with curettage and/or steroid injection. Notably, unlike other pediatric “cancer”, LCH does not require complete excision with “clean” margins – in fact, extensive bone resection can impair bone remodeling.
Front-line Therapy
Multiple lesions indicate potential for remote clonal progenitors. Patients with multiple lesions therefore typically require systemic chemotherapy. The current standard of care for initial therapy is vinblastine/prednisone for one year (with mercaptopurine added for high-risk LCH), based on the Histiocyte Society LCH-III trial. LCH-III study demonstrated higher rates of progression-free survival (PFS) in patients treated for one year vs. six months (5-year PFS 54% vs. 37%; p=0.03) and no benefit of adding methotrexate for patients with high-risk LCH.15 Notably, fewer than 50% of patients with high risk LCH were cured with vinblastine/prednisone. While vinblastine/prednisone may be the current standard, improved strategies are clearly needed. The Histiocyte Society is currently testing the impact of further treatment prolongation (2 versus 1 year with vinblastine/prednisone/(mercaptopurine) for frontline therapy (NCT02205762). Another phase 3 trial is currently randomizing 1 year of vinblastine/prednisone/mercaptopurine versus 1 year of cytarabine monotherapy for frontline LCH (NCT02670707).
Salvage Therapy
Nucleoside Analogs
Optimal approaches for patients who have relapsed or refractory LCH following front-line therapy have not been established. In 2 phase 2 studies, high dose cytarabine/cladribine was be effective, but associated with significant treatment-related morbidity and mortality. By comparison, lower-dose cladribine monotherapy induced high response rates, but only 3% of patients were cured after six months of therapy. Institutional series support the potential efficacy of intermediate-dose cytarabine or clofarabine monotherapy for relapsed and refractory LCH. While nucleoside analogs are promising, prospective trials are needed to determine optimal agent(s), dose and duration. Allogeneic hematopoietic cell may also be curative, but associated with >25% mortality. (Reviewed in 12)
Targeted Therapy
New concepts of LCH pathogenesis offer an opportunity to move treatment for LCH beyond empiricism to rational strategies. Early adult trials have reported extremely high response rates to MAPK pathway-directed targeted therapy with BRAFV600E or MEK inhibition for LCH and ECD.16;17 Retrospective series report similar findings for children with LCH. However, despite high response rates, MAPK pathway inhibition does not appear to be curative.18;19 Additionally, duration of therapy, durability of response, and potential for re-response after stopping the medication, and patterns of response/resistance are unknown. In LCH patients treated with MAPK pathway inhibitors, PBMCs with BRAF-V600E (presumed precursors) do not consistently clear from circulation, even in patients with complete clinical responses, and patient almost universally relapsed with cessation of therapy.18;19 Thus, there is an urgent need to improve therapeutic strategies (e.g., MAPK inhibitors with chemotherapy or anti-apoptotic agents or epigenetic modifiers) to safely cure patients with LCH and related disorders.
Adult LCH
The natural history of LCH in adults is understudied. Except for the predominance of lung disease, LCH appears to involve the same potential organ distribution as seen in children, though the incidence may be different. For example, pulmonary LCH usually occurs as a single-system disease in patients, 90% of the cases in adults who are heavy chronic smokers. Other differences include a higher incidence of oral and genital mucosa involvement in adults. Whole exome sequencing studies demonstrate higher somatic mutation burden in adult versus pediatric LCH (where median exome mutation is ~1). When LCH arises de novo in adults, it may reflect acquisition of mutation(s) through clonal hematopoiesis, reflected by mixed-phenotype myeloproliferative neoplastic disorders in some patients.
For adults with single LCH lesions, management strategies similar to the pediatric population include curettage (clean margins are not required) with or without intra-lesional corticosteroids for single bone lesions. However, there is no standard of care for the management of multisystem disease. Vinblastine/prednisone may have higher toxicity in adult patients, favoring alternatives such as cytarabine monotherapy. Other options include cladribine, clofarabine, hydroxyurea, methotrexate, 6MP and MAPK pathway inhibitors.11
Non-Langerhans Cell Histiocytic Disorders
Juvenile Xanthogranuloma (JXG) is a histiocytic disorder that shares many features with macrophage histology, and is histologically indistinguishable from ECD (Table 3). It most commonly affects infants and young children with a slight male predominance and presents as one or more “fleshy skin nodules.” However, in some patients, it may be systemic (<5% pediatric cases), involving multiple organs including deeper soft tissue, central nervous system, bone, lung, liver, spleen, pancreas, adrenal glands, intestines, kidneys, lymph nodes, bone marrow, orbit, and heart. The etiology and basis for prevalence in children are not well defined. An association between JXG and neurofibromatosis (types 1 and 2) and juvenile myelomonocytic leukemia and other molecular alterations such as CSF1R, KRAS, NRAS, and MAP2K1 (rarely BRAFV600E) implicates constitutive MAPK activation in pathogenesis.5 (Figure 2)
Erdheim-Chester Disease (ECD) typically arises in patients between the ages of 40 and 70, with a male predominance. Typical presentations include xanthelasma on the upper eyelids, skin rash, and bilateral lower limb bone pain. Consensus diagnostic criteria for ECD require the presence of (1) foamy CD68+/CD1a− histiocytes (Table 3), often with admixed inflammation and fibrosis, and (2) radiographic findings of bilateral and symmetric abnormalities in the diaphyseal and metaphyseal regions of the long bones of the legs. More severe manifestations can include cardiopulmonary insufficiency, renal failure due to retroperitoneal/perinephric infiltration, or CNS symptoms such as cerebellar signs, DI, or cognitive dysfunction. Like LCH, ECD is characterized by activating somatic MAPK pathway gene mutations (BRAFV600E being the most common in approximately 50% of patients) (Figure 2). ECD historically carried a dismal prognosis that has been significantly improved with MAPK inhibitors. In 2017 vemurafenib was approved for BRAFV600E+ ECD; and MEK inhibition has also been reported with high response rates. (Reviewed in 20)
Rosai-Dorfman Disease (RDD) or Sinus Histiocytosis with Massive Lymphadenopathy (SHML) likely represents a variety of conditions that share pathologic CD68+/CD1a- cells with emperipolesis (trafficking of viable lymphocytes through histiocytes) (Table 3). It can occur either as an isolated disorder or in conjunction with another autoimmune, malignant, or hereditary disease. Chronic, painless, massive cervical lymphadenopathy is the most common presentation. Other nodal areas and extranodal sites (such as skin, upper respiratory mucosa, ocular structures, bones, CNS) may also be involved. Treatment is variable and depends on the number of involved sites and can range from observation to systemic chemotherapy. MAPK mutations have been reported in RDD lesions, but less reliably than the other histiocytic disorders discussed above (Figure 2). (Reviewed in 21)
Conclusions
Clinical advances for patients with LCH and related disorders have historically been stalled by undefined mechanisms of pathogenesis. However, accelerated advances over the past decade have defined LCH as an inflammatory myeloid neoplastic disorder with extent of disease determined by the cell of origin in which activating MAPK somatic mutations arise. The challenge we now face is how to translate biological discovery into improved outcomes for children and adults with histiocytic disorders. Continued research on LCH, JXG, ECD and RDD will uncover opportunities to identify novel targets and inform personalized therapeutic strategies based on cell of origin, somatic mutation, and inherited risk factors.
Funding Information:
Dr. Allen receives support from the HistioCure Foundation, St. Baldrick’s Foundation (Innovation Grant, Consortium Grant), the Moves for Miles Childhood Cancer Foundation, and the Leukemia and Lymphoma Society (Translational Research Program).
Footnotes
Conflict of Interest: The authors have no conflicts to report with respect to this manuscript.
Data Availability:
Does not apply to this review article.
Reference List
- (1).Favara BE, Feller AC, Pauli M et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997;29:157–166. [DOI] [PubMed] [Google Scholar]
- (2).Emile JF, Abla O, Fraitag S et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Allen CE, Merad M, McClain KL. Langerhans-Cell Histiocytosis. N Engl J Med 2018;379:856–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Badalian-Very G, Vergilio JA, Degar BA et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116:1919–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Durham BH, Lopez RE, Picarsic J et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 2019;25:1839–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hogstad B, Berres ML, Chakraborty R et al. RAF/MEK/extracellular signal-related kinase pathway suppresses dendritic cell migration and traps dendritic cells in Langerhans cell histiocytosis lesions. J Exp Med 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Berres ML, Lim KP, Peters T et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2014;211:669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ribeiro KB, Degar B, Antoneli CB, Rollins B, Rodriguez-Galindo C. Ethnicity, race, and socioeconomic status influence incidence of Langerhans cell histiocytosis. Pediatr Blood Cancer 2015;62:982–987. [DOI] [PubMed] [Google Scholar]
- (9).Peckham-Gregory EC, Chakraborty R, Scheurer ME et al. A genome-wide association study of LCH identifies a variant in SMAD6 associated with susceptibility. Blood 2017;130:2229–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gadner H, Grois N, Potschger U et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood 2008;111:2556–2562. [DOI] [PubMed] [Google Scholar]
- (11).Girschikofsky M, Arico M, Castillo D et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis 2013;8:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood 2020;135:1319–1331. [DOI] [PubMed] [Google Scholar]
- (13).Yeh EA, Greenberg J, Abla O et al. Evaluation and treatment of Langerhans cell histiocytosis patients with central nervous system abnormalities: Current views and new vistas. Pediatr Blood Cancer 2017. [DOI] [PubMed] [Google Scholar]
- (14).McClain KL, Picarsic J, Chakraborty R et al. CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gadner H, Minkov M, Grois N et al. Therapy prolongation improves outcome in multi-system Langerhans cell histiocytosis. Blood 2013. [DOI] [PubMed] [Google Scholar]
- (16).Diamond EL, Subbiah V, Lockhart AC et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Haroche J, Cohen-Aubart F, Emile JF et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121:1495–1500. [DOI] [PubMed] [Google Scholar]
- (18).Donadieu J, Larabi IA, Tardieu M et al. Vemurafenib for Refractory Multisystem Langerhans Cell Histiocytosis in Children: An International Observational Study. J Clin Oncol 2019;JCO1900456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Eckstein OS, Visser J, Rodriguez-Galindo C, Allen CE. Clinical responses and persistent BRAFV600E+ blood cells in children with LCH treated with MAPK pathway inhibition. Blood 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Goyal G, Heaney ML, Collin M et al. Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood 2020;135:1929–1945. [DOI] [PubMed] [Google Scholar]
- (21).Abla O, Jacobsen E, Picarsic J et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood 2018;131:2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jaffe R The diagnostic histopathology of Langerhans cell histiocytosis. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Basic Science Clinical Features, and Therapy. Cambridge University Press; 2005;14–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Does not apply to this review article.