

Abstract
Purpose
Inherited retinal diseases are a group of clinically and genetically heterogeneous disorders with approximately 270 genes involved. IMPG2 is associated with adult-onset vitelliform macular dystrophy. Here, we investigated two unrelated patients with vitelliform macular dystrophy to identify the underlying genetic cause.
Methods
Whole-exome sequencing identified a putative causal complex allele consisting of c.3023-15T>A and c.3023G>A (p.(Gly1008Asp)) in IMPG2 in both individuals. To assess its effect, in vitro splice assays in HEK293T and further characterization in patient-derived photoreceptor precursor cells (PPCs) were conducted.
Results
The results of the midigene splice assays in HEK293T showed that the complex allele causes a variety of splicing defects ranging from a small deletion to (multiple-)exon skipping. This finding was further validated using patient-derived PPCs that showed a significant increase of out-of-frame transcripts lacking one or multiple exons compared to control-derived PPCs. Overall, control PPCs consistently showed low levels of aberrantly spliced IMPG2 transcripts that were highly elevated in patient-derived PPCs. These differences were even more obvious upon inhibition of nonsense-mediated decay with cycloheximide.
Conclusions
We report a heterozygous complex allele in IMPG2 causative for adult-onset vitelliform macular dystrophy in two unrelated individuals with mild visual loss and bilateral vitelliform lesions. The predicted causal missense mutation c.3023G>A, located in the consensus splice acceptor site, enhances the splicing effect of the upstream variant c.3023-15T>A, leading to the generation of aberrant transcripts that decrease the full-length IMPG2 levels. These results suggest a haploinsufficiency mechanism of action and highlight the complementarity of using different models to functionally assesses splicing defects.
Keywords: adult-onset vitelliform macular dystrophy, IMPG2, complex allele, non-canonical splice site variant, in vitro splice assay; photoreceptor precursor cells; aberrant splicing
Inherited retinal diseases (IRDs) are a group of rare genetic disorders that can lead to severe visual loss or blindness. These IRDs display a broad range of clinical and genetic heterogeneity1,2 with more than 270 associated genes (https://sph.uth.edu/retnet/home.htm). Adult-onset vitelliform macular dystrophy (VMD) (OMIM#616152) is one of the most prevalent forms of inherited macular dystrophy,3 occurring 1 in 7400 to 8200 in Olmsted County, Minnesota, USA; but the worldwide prevalence remains unknown.4 Patients with adult-onset VMD present with gradual vision loss and/or metamorphopsia between the ages of 30 and 50 years, or remain asymptomatic and do not manifest significant visual impairment owing to incomplete penetrance.3,5,6
The first identified genes associated with VMDs were BEST1 and PRPH2, mainly causing autosomal-dominant juvenile VMD (OMIM#153700)7–9 and autosomal-dominant adult-onset VMD, respectively. The phenotype–genotype association between VMDs and IMPG110–12 and IMPG211–13 have been described more recently. Pathogenic variants in IMPG2 are responsible for autosomal recessive retinitis pigmentosa (RP)14 and autosomal-dominant VMDs that display incomplete penetrance and variable expression.3,12 The clinical symptoms of VMD caused by variants in IMPG2 are generally less severe compared with variants in IMPG1.12 Mutations in IMPG1 and IMPG2 together underlie 8% of the VMDs in which a pathogenic variant has not been identified in BEST1 and PRPH2.11
IMPG1 and IMPG2 encode two structurally similar glycoproteins synthesized by photoreceptors and are major components of the interphotoreceptor matrix. The interphotoreceptor matrix is a retina-specific extracellular matrix that surrounds the photoreceptor inner and outer segments and plays an essential role in the interaction of RPE and photoreceptors, amongst others, the exchange of retinoids and nutrients of the visual cycle, photoreceptor differentiation and maintenance, and the alignment of the photoreceptor cells.15–18 In the absence of IMPG2, IMPG1 aggregates between the photoreceptor outer segment and RPE. This accumulation of material inhibits a normal interaction between the RPE and photoreceptor outer segment that is vital for normal functioning retina. In vivo studies in fish19 showed that cones seem to be more sensitive to alteration of the interphotoreceptor matrix than rods, which may explain the location of vitelliform lesions in the macula.12
To date, there are 82 pathogenic and likely pathogenic variants that have been reported for IMPG2 in the LOVD database (www.lovd.nl/IMPG2), 12 of which affect a splice site. Furthermore, 346 variants are recorded for IMPG2 in ClinVar, of which 42 variants are currently classified as pathogenic (http://www.ncbi.nlm.nih.gov/clinvar/IMPG2). With the advancement of genetic therapies for retinal diseases, identifying the molecular mechanism of disease is crucial to design potential therapeutic approaches, such as gene augmentation using viral vectors or antisense oligonucleotide-based splicing modulation.20,21
Nowadays, the importance of non-coding regions of the genome has become more evident and a large number of splice site variants have been identified for IRD-associated genes.14,22–25 In this study, we report the pathogenicity of the first heterozygous complex allele, NC_000003.12(NM_016247.3):c.[3023-15T>A;3023G>A], found in IMPG2 in two unrelated families with adult-onset VMD.
Methods
This study was conducted following the tenets of the Declaration of Helsinki. Approval was obtained from the local ethics committee of Radboud University Medical Center in Nijmegen, the Netherlands, and written informed consent was collected from all individuals described in this work.
Clinical Examination
Two unrelated individuals with adult-onset VMD were clinically examined at the Radboud University Medical Center in Nijmegen, the Netherlands (Fig. 1). The medical histories were assessed, and the individuals underwent ophthalmic examinations, including best-corrected visual acuity, slit lamp examination, and fundoscopy. Multimodal imaging included color fundus photographs captured using a Topcon TRC50IX retinal camera (Topcon Corporation, Tokyo, Japan), and spectral-domain optical coherence tomography, and fundus autofluorescence using Spectralis (Heidelberg Engineering, Heidelberg, Germany). An electro-oculogram (EOG) was performed using Espion (Diagnosys LLC, Lowell, MA) according to the standard of the International Society of Clinical Electrophysiology of Vision.26 An Arden ratio of 1.85 or greater was considered normal.
Figure 1.

Multimodal imaging of two patients with adult-onset vitelliform macular dystrophy, caused by the IMPG2 complex allele c. [3023G>A;3023-15T>A]. (A–G) Imaging of the right (A–C) and left eye (D–G) of patient A:II-1 is shown. (A, D) Fundus photography shows bilateral presence of yellowish vitelliform lesions. (B, E) Fundus autofluorescence images show a central circular lesion with hypo- and hyperautofluorescent spots. (C, F) These correspond with the dome-shaped neuroepithelium detachment with residue vitelliform material and RPE atrophy. (E, G) In the left eye, a second vitelliform lesion was seen adjacent to the superior arcade. The ‘G’ in image E depicts the location of the lesion shown in image G. (H–O) Imaging of the right (H–J) and left eye (K–O) of patient B:II-1 is shown. (H, K) Fundus photography shows bilateral presence of yellowish vitelliform lesions. (I, L) Fundus autofluorescence images show a central circular lesion with hypo- and hyperautofluorescent spots. On spectral-domain optical coherence tomography vitelliform lesions were seen. The ‘N’ and ‘O’ in image L depict the location of the lesion shown in image N and O. (J) In the right eye, the lesion is in the vitelliform stage. The left eye shows neurosensory retinal detachment with partial resorption of the vitelliform material. The remaining vitelliform material has shifted inferiorly owing to gravity. (N, O) The superior and inferior sectional view of the vitelliform lesion are shown, respectively. Material has also migrated to the inner foveal layers.
Genetic Analysis
Genomic DNA was extracted from peripheral lymphocytes according to standard protocols.24 Whole exome sequencing was performed for both affected individuals at BGI‐Europe (Copenhagen, Denmark). In short, the exome was enriched by using Agilent's SureSelectXT Human all Exon V5 (Agilent Technologies, Santa Clara, CA) and next‐generation sequencing was performed by an Illumina HiSeq 4000 sequencer (Illumina, Inc., San Diego, CA) to a mean sequence depth of at least 75-fold. The Burrows‐Wheeler Aligner was applied to align reads to the human reference genome (hg19). Nucleotide and copy number variant calling were performed with the Genome Analysis Toolkit and CoNIFER 0.2.0, respectively.27 Finally, variants were annotated using a bespoke in‐house annotation strategy.24
Variant Prioritization
Only variants in visual impairment-associated genes were assessed using the local diagnostic gene panel version DG-2.11 (patient A:II-1) or DG-2.15 (patient B:II-1) containing 395 and 434 genes, respectively. Variant prioritization was carried out based on a minor allele frequency of less than 1% in the ExAC,28 less than 5% in dbSNP, less than 1% in an in-house database consisting of 15,576 mostly Caucasian individuals, and less than 2% in an in-house database consisting of 454 mostly Asian individuals.29 Subsequently, all coding and splice site variants (nucleotides –17 to –1 and +1 to +6) were manually classified according to a five-class system based on the American College of Medical Genetics–Association for Molecular Pathology classification system.30 Evaluating of prioritized variants is performed following the practice guidelines by Wallis et al.31 An analysis of the putative effects on splicing is described in the Supplemental Methods.
In Vitro Splice Assays
The functional analysis of the putative causal splice site variants, c.3023-15T>A and c.3023G>A in IMPG2 was carried out using midigene splice assays. Two different but overlapping constructs were designed to cover the IMPG2 gene according to a previously described protocol.32 The first wild-type (WT) and mutant constructs (midigene 14-15, MG14-15) consist of a 10.4-kb insert including exons 14 to 15 encompassing the 9.7-kb intron 14, and the second WT and mutant constructs (MG15-18) containing a 4.8-kb insert including exons 15 to 18. Both genomic regions of interest were amplified respectively, using primers that contain attB1 and attB2 tags at their 5ʹ ends to facilitate Gateway cloning (Table S1). Transfections were conducted in triplicate and PCR in duplicate to represent biological and technical replicates, respectively. Details on the midigenes are provided in the Supplementary Methods.
Induced Pluripotent Stem Cell (iPSC) Differentiation Into Photoreceptor Precursor Cells (PPCs)
The generation of the patient-derived iPSC line from blood samples was performed by the Stem Cell Technology Center of the Radboud University Medical Center as described in previous publications.33–37 One patient (patient B:II-1) harboring the complex allele c.[3023-15T>A;3023G>A] in IMPG2 and one control line were used to obtain PPCs as previously described33–35,38 (see Supplementary Methods). Two wells for each cell line were differentiated in each replicate. At day 29, one of the wells was treated with 100 µg/mL of cycloheximide solution (CHX, Sigma-Aldrich) (CHX+ condition), and the other well was kept non-treated (CHX– condition). The following day (Supplementary Fig. S3), cells were rinsed with PBS, scraped, and harvested for RNA analysis. All differentiation experiments were performed in duplicate.
Quantitative PCR and Reverse Transcriptase (RT)-PCR Studies in PPCs
RNA isolation of the iPSC (day 0) and PPC cells (day 30) was performed as described above. One microgram of total RNA was used for cDNA synthesis using SuperScript VILO Master Mix (ThermoFisher Scientific, Waltham, MA) according to the manufacturer's protocol. A quantitative PCR analysis was conducted with the GoTaq Real-Time Quantitative PCR Master kit (Promega, Madison, WI) and samples were processed in an Applied Biosystem QuantStudio 5 Digital system. The expression levels of eight PPC markers and one housekeeping gene (GUSB) were studied for assessing the differentiation level. The complete list of primers is provided in Table S2. Each sample was normalized against the expression of the housekeeping gene and compared with iPSC (day 0) using the 2−(ΔΔCt) method.39
For RT-PCR, 50 ng cDNA were used for the amplification of exons 14 to 15 or exons 14 to 18. The RT-PCR was performed under the same conditions listed previously. The analysis of the transcripts was conducted by Sanger sequencing. The percentage estimation of each transcript was calculated using both FIJI software and the TapeStation system, as described in the Supplementary Methods. Primers are listed in Table S3. In addition, RNA from different control or IRD-differentiated (non–IMPG2-associated) PPCs used in previous or current studies in the group was used for cDNA synthesis and RT-PCR analysis (PPC1-6) were used as extra controls to evaluate the levels of aberrant IMPG2 splicing.33–35
Statistical Studies
Data related to the different spliced transcripts are represented as the mean ± standard deviation and analyzed using GraphPad Prism 10 software (GraphPad, San Diego, CA). To assess differences between the control and patient PPCs lines with the CHX– treatment or with CHX+ treatment conditions were compared by one-way ANOVA test followed by Bonferroni correction. The same test was used to compare control CHX– and patient CHX– conditions with respect to the MG4-15 minigenes. A P value of 0.05 or less was considered statistically significant.
Results
Clinical Description
Two unrelated individuals of Dutch ancestry were referred to our center and were examined (Table). Patient A:II-1, a 50-year-old man, presented with complaints of slowly worsening mild metamorphopsia and mild visual loss symptoms, starting from the age of 40. He was in good general health and had no other ocular conditions or complaints. His best-corrected visual acuity was at the first visit 0.9 and 0.7 (Snellen decimals) for the left and right eyes, respectively. Amsler chart assessment revealed mild central metamorphopsia in the left eye. The EOG showed a normal Arden ratio (3.02 in the right eye, 3.04 in the left eye). Patient B:II-1, a 52-year-old man, presented with slow progressive visual loss for years and difficulty with driving at night. He was also in good general health and had no other ocular complaints. Based on the patient's history, the left eye was presumed to be amblyopic. Family history was negative. The best-corrected visual acuity was 0.9 and 0.6 (Snellen decimals) for the right and left eyes, respectively. The EOG showed a normal Arden ratio (2.2 in the right eye, 2.3 in the left eye).
Table.
Clinical Characteristics of Two Unrelated Patients With the Novel Complex IMPG2 Allele c.[3023G>A;3023-15T>A]
| Visual Acuity | Refraction Error | EOG (Arden Ratio) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Gender | Age (y) | Initial Symptom | RE | LE | RE | LE | Lens Status | Ophthalmoscopy Results | Amsler Chart Assessment | RE | LE |
| A:II-1 | M | 50 | Metamorphopsia | 0.9 | 0.7 | S+0.50 C-0.25 × 65 | S+0.50 C-0.25 × 65 | Clear | Vital and well-demarcated optic disc. Vitelliform lesion in the macula, no drusen. No abnormalities in the periphery. | RE: no metamorphopsia. LE: mild central metamorphosia | 3.02 | 3.04 |
| B:II-1 | M | 52 | Visual loss, difficult driving at night | 0.9 | 0.6 | S+0.75 C-1.00 × 5 | S+1.50 C-2.5 × 175 | Clear | Vital and well-demarcated optic disc. Vitelliform lesion in the macula, no drusen. No abnormalities in the periphery. | No metamorphopsia | 2.2 | 2.3 |
LE, left eye; M, male; EOG, electro-oculogram; RE, right eye.
Ophthalmoscopy revealed the bilateral presence of yellowish foveal lesions (Fig. 1). On spectral-domain optical coherence tomography, the lesions presented as dome-shaped central elevations of the neuroepithelium with disruption of the ellipsoid zone and RPE atrophy in both patients. In patient B:II-1, the dome was filled with a hyper-reflective material. In patient A:II-1, the vitelliform material was mostly absorbed, leaving an empty cavity. In both patients, the size of the subfoveal lesions increased slightly during a follow-up of 2 years. Fundus autofluorescence imaging demonstrated central circular lesions with hyperautofluorescent spots corresponding with the vitelliform material, and hypoautofluorescence in areas where the material was resorbed.
Molecular Genetic Findings
After variant prioritization and analysis of the coding and splice site regions, we identified two heterozygous variants in cis in IMPG2 (NG_028284.1) in both probands, c.3023G>A (p.(Gly1008Asp)), and c.3023-15T>A (p.(?)), which where verified with Sanger sequencing to be present in cis in the parents as well (Fig. 2). Variant c.3023G>A had a total allele frequency of 0.000004003 in gnomAD (1/249,796 alleles; allele frequency of 0.000008915 in the European non-Finnish population, i.e. 1/ 112,174 alleles) and a PhyloP score of 7.44 (range, −14.1_6.4; predicted pathogenic ≥2.7),40 a CADD_PHRED score of 34 (range, 1_99; predicted pathogenic ≥15),41 and a Grantham score of 94 (range, 215; predicted pathogenic ≥80).42 Additionally, the in silico missense prediction tools SIFT and MutationTaster considered this variant as “Deleterious” and “Disease-causing,” respectively. Because the variant was located in the noncanonical SAS, its effect on splicing was also evaluated using splice site prediction algorithms embedded in Alamut version 2.10. In the presence of this variant, all five splice prediction scores showed a reduction for the strength of the SAS of exon 15 (Supplementary Fig. S1A, Supplementary Table S4). Moreover, the SpliceAI algorithm predicted a loss of the SAS and SDS of exon 15 with delta scores of 0.25 and 0.03, respectively (Table S5).
Figure 2.
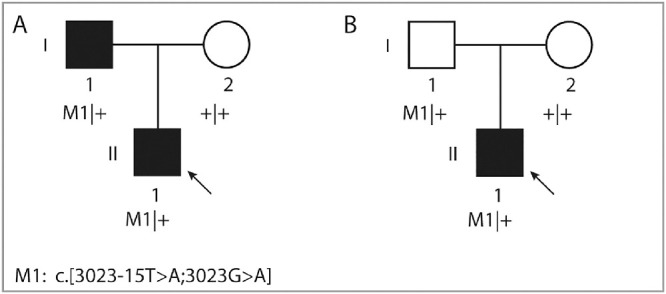
Pedigrees of two unrelated affected individuals and segregation analysis of the complex IMPG2 allele. The arrow indicates the proband in each family. Individual A:I-1, who carries the complex allele, has no vision impairment but does show abnormalities in imaging. Individual B:I-1 carries the same allele, but is asymptomatic owing to incomplete penetrance.
Variant c.3023-15T>A has an allele frequency of 0.000004016 in gnomAD (1/248,992 alleles; allele frequency of 0.000008967 in European non-Finnish population). Based on the aforementioned splice site prediction algorithms, not only did the c.3023-15T>A weaken the nearby SAS, it also favored the use of an alternative SAS at position c.3023-14 (Supplementary Fig. S1B, Table S4), which is also predicted by SpliceAI with a delta score of 0.37. Furthermore, the SpliceAI algorithm predicted a loss of the SAS and SDS of exon 15 with delta scores of 0.47 and 0.12, respectively (Table S5). Finally, the Alamut prediction scores for the combination of both variants in cis also indicated a decrease in SAS scores in exon 15 together with the creation of a new cryptic SDS at the position c.3023-14 (Fig. 3A). The segregation analysis confirmed that in both families, the mutant alleles in probands were inherited from one of the parents in a heterozygous state. No putative pathogenic variants were detected in BEST1 and PRPH2 in both probands.
Figure 3.

HEK293T cell splice assay results for variants c.3023-15T>A, c.3023G>A, and c.[3023-15T>A;3023G>A] in IMPG2. (A) Schematic representation of the IMPG2 gene and enlargement of the intron 14–exon 15 boundary sequence in the WT and c.[3023-15T>A;3023G>A] variant. The SpliceSiteFinder‐like (SSFL, range 0–100) and Human Splicing Finder (HSF, range 0–100) scores for the splice acceptor site are indicated below each sequence. The green triangles indicate the position of the splice acceptor site in WT and mutant sequences. The two red “a” nucleotides highlight the c.3023-15T>A and c.3023G>A variants in the mutant sequence. The red numbers represent the altered scores for the mutant compared with the WT sequence. (B, C) Schematic representation of the mutant pCI‐NEO‐RHO vector, containing exon 14–15 (B) and exon 15–18 (C) of IMPG2 flanked by RHO exon 3 and 5, which was used to transfect HEK293T cells with WT and mutant constructs, respectively. The gel images of RT-PCR products in MG14-15 (B) and MG15-18 (C) are shown. The MG14-15 WT and c.3023A constructs reveal the 391-nt WT and p.(Gly1008Asp) fragments. Similarly, in the MG15-18 WT and c.3023A constructs, the 778-nt fragments corresponding to WT and p.(Gly1008Asp) fragments were observed. Three additional fragments were observed in MG15-18_WT and MG15-18_3023A constructs. The larger one corresponds with exon 16 skipping and the middle one corresponds with exons 16/17 skipping, and the smaller fragment corresponds with exons 16/17/18 skipping. In RT-PCR products resulting from the transfection of constructs carrying c.3023-15A and c.3023-15A;3023A in MG14-15 and MG15-18, one main additional fragment was observed which corresponds to a 343-nt (B) or 730-nt (C) fragment representing a 48-nt deletion of exon 15. In addition, two fragments were only detected in the MG15-18 constructs (C) carrying the c.3023-15A and c.3023-15A;3023A corresponding with a 541-nt and 330-nt transcript. These fragments harbor the 48-nt deletion of exon 15 together with the exon 16 skipping and exons 16/17 skipping, respectively. For the MG15-18_c.3023-15A construct, the 589-nt fragment was also observed that corresponds with exon 16 skipping. RT‐PCR analysis of RHO exon 5 was performed as a control for efficient transfection. The right panel indicates the quantification of each fragment in all eight midigenes obtained by TapeStation system. The asterisks indicate the heteroduplex fragments. HSF, Human Splicing Finder; Mut, mutant; nt, nucleotide; SSFL, SpliceSiteFinder‐like.
To assess the pathogenicity of the single variants c.3023G>A or c.3023-15T>A, and of the combined variants c.[3023G>A;3023-15T>A] in IMPG2, midigene splice assays were carried out using WT and mutant midigenes. Because the genomic context of a variant plays a significant role in the splicing process, we aimed to design constructs containing at least exons 14, 15, and 16.32 However, the genomic region encompassing these three exons and flanking intronic sequences (12.8 kb) exceeded the Gateway-cloning capacity (<12 kb).32 Therefore, we used two different midigene constructs, one containing exons 14 and 15 (MG14–15) and another one containing exons 15 to 18 (MG15–18). After RNA isolation and RT-PCR analysis of the MG14-15_WT, we detected the predicted 391-nt fragment containing IMPG2 WT mRNA. Despite the predicted splicing effect in Alamut and SpliceAI algorithms for c.3023G>A, MG14-15_3023A did not show a splicing defect in HEK293T cells. Transfection of MG14-15_3023-15A and MG14-15_3023-15A;3023A revealed an additional mRNA fragment of 343 nt. This mutant fragment corresponded with an in-frame 48-nt deletion of IMPG2 exon 15 (p.(Gly1008_Ser1023del)), which starts in a SAS located at position c.3071 (Fig. 3A). The WT and mutant mRNA fragments in the MG14-15_3023-15A were calculated to constitute 90.8% and 9.2% of the RNA products, respectively, whereas the WT and mutant fragments of MG14-15_3023-15A;3023A were present in 18.3% and 81.7% of the RNA products, respectively (Fig. 3B).
For the MG15-18_WT construct, an IMPG2 WT mRNA fragment of 778 nt was detected, which represented 52.9% of the total RNA product. In addition, we observed a 589-nt fragment (30.1%) and a 378-nt fragment (15.8%) corresponding with exon 16 skipping (p.(Arg1078_Ser1141del)) and exons 16 and 17 skipping (p.(Cys1079Lysfs*4)) of IMPG2 mRNA, respectively. Furthermore, a 169-nt fragment (1.3%) corresponding with the skipping of exons 15 to 17 was detected resulting in an in-frame deletion (p.(Leu935_Glu1211del)). MG15-18_3023A showed a similar RNA pattern as the MG15-18_WT vector, i.e., 50.2% WT, 28.6% exon 16 skipping, and 20.7% exons 16 and 17 skipping products (Fig. 3C, Supplementary Fig. S4)
MG15-18_3023-15A transcript analysis showed that the WT fragment accounted for 57.3% of the total transcripts. Additionally, there was a 730-nt fragment (9.7%; Supplementary Fig. S2) corresponding with a 48-nt deletion of exon 15, a 589-nt fragment (9.1%) corresponding with exon 16 skipping, and a 169-nt fragment (16.1%) corresponding with the skipping from exons 15 to 17. Besides these, we observed two minor residual 541-nt fragment (1.4%) and 330-nt (3.2%) fragments corresponding with the skipping of the 48-nt shorter exon 15 and the exon 16 or showed the skipping of exons 16 and 17, respectively. The MG15-18_3023-15A;3023A construct resulted in the same splicing events in which 11.5% corresponded with the WT fragment, 21.8% with the deletion of 48-nt of exon 15 transcript, and 49.6% showed the skipping of exons 15, 16, and 17; the remaining transcripts together constituted 17.1% of all products (Fig. 3C, Supplementary Fig. S4).
To validate the splicing defects observed in midigenes in the full RNA context and eventually design a therapeutic strategy to modulate splicing, we generated iPSCs from patient B:II-1. Interestingly, the quantification of the splicing analysis revealed that the 48-nt in-frame deletion was not as prominent in patient-derived PPCs as it was observed in the midigene MG14-15 system (Fig. 4A). This finding, together with the results from transcript analysis on MG15-18 midigene constructs, prompted us to study a larger genetic context from exons 14 to 18 to elucidate whether other splicing events were taking place. The amplification of this region showed clear and consistent low levels of full-length IMPG2 transcript. In contrast, several spliced transcripts were observed, among which the joint skipping of exon 15 and exon 16 was the most prominent (Fig. 4B). The deletion of these two exons is out of frame and, therefore, it is expected that the transcript will be degraded by nonsense-mediated decay. Indeed, we could confirm that this spliced transcript was detected at higher levels upon CHX treatment (a 3× increase). In addition, this splicing event was detected at low levels in control PPCs (9.2% and 4.3% in the CHX– and CHX+ conditions, respectively). In-frame skipping of exon 16 was detected in both control and patient PPCs, whereas the 48-nt in-frame deletion and the skipping of exons 15, 16, and 17 together was mainly observed in patient PPCs and seem to be absent in the control PPC (Figs. 4B, 4C).
Figure 4.

IMPG2 transcript analysis in PPCs. (A) Analysis of transcripts amplified from exon 14 to exon 15 between control- (CON) and patient (PAT)-derived PPCs and two MG constructs: WT and c.[3023-15T>A;3023G>A] (MUT). The left panel shows a representative RT-PCR gel picture. The bar chart on the right represents the semiquantification of the percentage of each transcript as mean ± standard deviation. The PPC lines were evaluated with cycloheximide treatment (CHX+) and without cycloheximide treatment (CHX–). The experiment was performed in duplicate. For the statistical analysis, CON CHX– was compared with MG14-15 WT and PAT CHX– was compared with MG14-15 MUT using a one-way ANOVA test. Statistical significance is indicated as **** P < 0.0001 using one-way ANOVA test. (B) Analysis of transcripts amplified from exon 14 to exon 18 between CON and PAT PPCs. The left panel shows a representative RT-PCR gel. The right upper panel represents the semiquantification of the percentage of each identified transcript as mean ± standard deviation. The right lower panel represents the percentage of full-length transcript (WT) and the sum of all spliced transcripts. The PPC lines were evaluated with cycloheximide treatment (CHX+) and without cycloheximide treatment (CHX–). The experiment was performed in duplicate. Statistical analysis was performed by comparing CON and PAT CHX-untreated conditions and CON and PAT CHX-treated conditions. Statistical significance is indicated as ** P < 0.01 using one-way ANOVA test. For the final RNA and protein annotation, only the fragments with >10% of total RNA were included. (C) Summary of the different transcripts detected in (A) and (B). The number of each transcript is indicated on the left side of the referred the band. (+) indicates weak expression and (++) indicates high expression. bp, base pair.
The PPC characterization showed that both cell lines were differentiated toward photoreceptor precursor fate. Based on the expression levels of several markers, the patient PPCs were more differentiated compared with controls (Supplementary Fig. S5). To validate that the splicing events were not explained by a differential stage of differentiation, we analyzed the same region using the RNA of six other PPCs derived from control and non–IMPG2-associated IRD individuals from local previous and ongoing studies.33–35 In all cell lines, we were able to detect skipping of exon 16, as shown previously. In contrast, the skipping of exons 15 and 16 as well as the skipping of exons 15, 16, and 17 were mainly observed in patient PPCs, with a minor presence in the other six lines and the control; the 48-nt in-frame deletion was exclusively found in the patient PPCs (Supplementary Fig. S6).
Discussion
In this study, we described two unrelated individuals with adult-onset VMD owing to a novel complex allele in the IMPG2 gene, that is, the heterozygous c.3023G>A (p.(Gly1008Asp)) variant in cis with c.3023-15T>A. Only in the presence of the c.3023G>A variant, the 3023-15T>A variant causes a strong splicing defect observed in midigene splice assay. The combination of both variants showed multiple splicing events in patient-derived PPCs.
Phenotype
The affected individuals (A:II-1 and B:II-1) presented with symptoms of mild visual loss and in patient A:II-1 also mild metamorphopsia, in the fourth decade with normal EOG Arden ratios. Imaging revealed the bilateral presence of foveal exudative neuroepithelium detachments, similar to vitelliform lesions seen in the classic Best disease caused by BEST1 pathogenic variants. Classic BEST1-associated vitelliform lesions are, however, generally larger and present usually in the juvenile age,43 while cases carrying pathogenic IMPG2 variants present with onset after the fourth decade.11–13 The EOG is notably normal as opposed to abnormal or subnormal Arden ratio found in patients with Best disease. Our clinical findings are in line with previous reports of the VMD phenotype caused by IMPG2 variants.11–13 Persons carrying IMPG2 variants can remain asymptomatic,5,6,44 as it was the case for the father (A:I-1) of patient A:II-1, who carried the same complex allele and had the same phenotype on imaging, although reported to be asymptomatic (Supplementary Fig. S7). The father (B:I-1) of patient B:II-1 carried the pathogenic allele, but was clinically unaffected. This finding could suggest two possible scenarios: the presentation of IMPG2-associated autosomal-dominant inheritance with incomplete penetrance or a haploinsufficiency inheritance.
Pathogenic variants in IMPG2 are also associated with RP (OMIM#268000), a retinal disorder that is distinct from VMD. However, it still has to be elucidated why certain variants cause VMD and others RP. To date, no clear association between the severity of a pathogenic variant and the described phenotype has been reported. In a recent study, the authors described a pathogenic variant in IMPG1, which led to different phenotypes, even within families.19 Such findings emphasize the role of modifiers for IMPG1 and IMPG2-associated diseases that remain to be identified.
Genotype
We reported the complex allele, c.[3023-15T>A;3023G>A] identified in IMPG2, r.[3023_3422del,3023g>a,3023_3633del]; p.[Gly1008Valfs*17,Gly1008Asp,Asp1009Asnfs*14] wherein the variant c.3023G>A enhances the splicing defect of c.3023-15T>A, responsible for adult-onset VMD in two unrelated individuals.
Both individual variants have been observed in one individual in gnomAD. However, it could not be determined yet if this is the same individual; co-occurrence can only be determined for coding variants in gnomAD. The c.3023G>A variant has been reported previously by Brandl et al.12 The variant was classified as likely pathogenic owing to a damaging PhyloP, CADD-PHRED, and Grantham score. The presence of the c.3023-15T>A variant, and thus the complex allele, was validated upon request for this patient (personal communication (10 March 2021) Prof. H. Stöhr and Prof U. Kellner). This confirmation emphasizes the importance of complex allele identification, which may be overlooked when only one of two variants in cis are reported as the cause of the disease while the combination of both variants is pathogenic.
Complex alleles may harbor several variants in cis, where these may or may not collectively contribute to a pathogenic effect. Complex alleles have frequently been reported in the ABCA4 gene, causing Stargardt disease45–48 and were reported in the PRPH2 gene in a case of RP with macular dystrophy.49 The identification of complex alleles is important because they have diagnostic, prognostic, and therapeutic consequences. Additional mutations in cis might be missed when mutational scanning protocols stop when two mutations are found in trans in the autosomal-recessive conditions. The identification of complex alleles can genetically explain unsolved cases who carry variants with unknown significance.
Initially, we hypothesized that the c.3023-15T>A or the c.[3023-15T>A;3023G>A] variants would lead to a 13-nt exon elongation owing to the cryptic SAS at position c.3023-14 as suggested by the Alamut and SpliceAI prediction algorithms. After transcript analysis of cells transfected with MG14-15 and MG15-18 carrying the c.3023-15T>A or the c.[3023-15T>A;3023G>A] variants, a 48-nt deletion of the 5ʹ part of exon 15 was observed. This in-frame deletion of the first 16 amino acids of exon 15 from glycine 1008 to serine 1023 would affect the epithelium growth factor (EGF)-like-2 domain in IMPG2 protein. However, the analysis of the splicing effect using a larger construct (MG15-18) as well as the analyses using patient-derived PPCs confirmed that this splicing event was only present in a minority of the transcripts. In contrast, two other out-of-frame spliced transcripts were confirmed in patient-derived PPCs, which were detected at low levels in the WT PPCs. The skipping of exons 15 and 16, and the skipping of exons 15, 16, and 17, produce out-of-frame transcripts that are susceptible to nonsense-mediated decay. Nevertheless, if a truncated protein would be synthesized the skipping of these exons would disrupt either an EGF-like-2 domain or the transmembrane domain.12 The EGF-like-2 domain is involved in the structural stability of the IMPG2 protein through intra-chain disulfide bonding.50 Our data show that the transcript lacking exons 15 and 16 is degraded by nonsense-mediated decay; its presence was observed three times more in the CHX–treated patient PPCs compared with the untreated PPCs. In addition, the skipping of exon 16, which leads to an in-frame transcript from arginine 1078 to serine 1141, was detected in both control and patient PPCs. The deletion of this exon could lead to partial disruption of the EGF-like-2 domain. Furthermore, it also affects two potential hyaluronic acid binding sites that allow for the interaction of heparin and chondroitin sulfates, which participate in retinal development and function.51,52 However, the significance and possible function of this transcript remain to be resolved.
In this work, we studied a putative effect on splicing caused by the complex allele c.[3023-15T>A;3023G>A] in the IMPG2 gene by employing midigenes. This artificial system has a size restriction for the insert. This limitation hampered the cloning of all relevant exons and introns into one construct, leading to the loss of the relevant genetic context for both the MG14-15 as well as the MG15-18 construct. As a consequence, the MG14-15 constructs only enabled the detection of the deletion of 48-nt of exon 15 as a main splice event. The MG15-18 constructs, which allowed us to study a larger genetic context, showed similar splice events as those observed in patient-derived PPCs. However, these MG15-18 constructs also detected two transcripts that likely represent artifacts from the midigene system. These 541-nt and 330-nt transcripts showed the 48-nt shorter exon 15 and exon 16 skipping, as well as the skipping of exon 16 and 17, respectively, which remained undetected in the patient-derived PPCs. In addition, the most prominent effect in patient-derived PPCs (the skipping of exons 15 and 16) was not detected in the midigene system owing to the aforementioned limited cloning capacity (Fig. 3C; Fig. 4 and Supplementary Fig. S6). In addition, the overexpression of the plasmid and the high abundance of different transcripts leads to heteroduplexes or unspecific transcripts (Fig. 3B and C). Moreover, midigenes were transfected in non-retinal cells (HEK293T), which offer great transfection efficiencies, although they lack the retinal molecular environment. When using PPCs, our results clearly determined that the main effect of the complex allele is the increase of skipping of exons 15 and 16. This transcript showed a 15-fold increase in patient-derived PPCs compared with control PPCs and six other PPCs in the presence of CHX (Supplementary Fig. S4C).
The amount of each transcript was assessed by quantifying each fragment in duplicate using independent experiments using both Fiji software and the TapeStation system. The first one was measured manually, and the latter was automatically determined by the TapeStation software. Both systems showed similar trends and the same conclusions would have been drawn (Supplementary Figs. S7 and S8). However, the TapeStation seemed to be more sensitive and accurate, in particular for smaller sized fragments, which could be overlooked by Fiji if they are faint on the electrophoresis gel owing to long runs to better separate the larger bands. In control and non–IMPG2-associated IRD PPCs, a consistent and robust 22.4% to 36.5% of spliced IMPG2 transcripts is observed. Interestingly, in the patient-derived PPCs, which are cells heterozygous for this complex allele, the spliced IMPG2 accounted for up to 83.7% of the total IMPG2 in the presence of CHX. Therefore, only a small percentage of WT transcript remains. Although the actual effects of the complex allele on the protein content are not defined by these experiments, our results support the hypothesis of a haploinsufficiency, in which the remaining levels of full-length IMPG2 transcripts may determine the severity of the disease, and thereby explain the incomplete penetrance observed in these families. In addition, semiquantitative analyses by, for example, Western blot, could confirm this hypothesis. Unfortunately, the lack of reliable antibodies for Western blot, together with the unavailability to material of other carriers, complicates these type of studies.
In conclusion, we identified a novel complex allele c.[3023G>A;3023-15T>A] in IMPG2 causing adult-onset VMD as observed in two unrelated families. We demonstrated the effect of c.3023G>A together with c.3023-15T>A on splicing both in the midigene system and patient-derived PPCs. The identification of complex alleles is essential owing to their diagnostic, prognostic, and therapeutic consequences. Furthermore, the use of patient-derived PPCs was crucial to assess the mechanism of disease, highlighting the importance of patient-derived material and the relevance of using complementary models to validate splicing defects. This point is particularly true in those cases in which it is not possible to clone the complete region of interest in a midigene.
Supplementary Material
Acknowledgments
The authors thank the affected individuals and their families for participating in this study and our funding bodies. We thank Rob W.J. Collin for the scientific discussions and support. We thank Saskia van der Velde-Visser for DNA sample preparation, Michael Kwint for his guidance using TapeStation, and the Stem Cell Technology Center for the generation of the iPSCs. The work of IV-D, CHZL, and ZF is funded by the Foundation Fighting Blindness USA Project Program Award, grant no. PPA‐0517–0717‐RAD (to FPMC, SR, AG, and CBH). The research was also supported by the Algemene Nederlandse Vereniging ter Voorkoming van Blindheid, Oogfonds, Landelijke Stichting voor Blinden en Slechtzienden, Rotterdamse Stichting Blindenbelangen, Stichting Blindenhulp, Stichting tot Verbetering van het Lot der Blinden, and Stichting Blinden-Penning (to SR and FPMC).
Disclosure: I. Vázquez-Domínguez, None; C.H.Z. Li, None; Z. Fadaie, None; L. Haer-Wigman, None; F.P.M. Cremers, None; A. Garanto, None; C.B. Hoyng, None; S. Roosing, None
References
- 1. Ellingford JM, Barton S, Bhaskar S, et al.. Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology. 2016; 123: 1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berger W, Kloeckener-Gruissem B, Neidhardt J.. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010; 29: 335–375. [DOI] [PubMed] [Google Scholar]
- 3. Chowers I, Tiosano L, Audo I, Grunin M, Boon CJ.. Adult-onset foveomacular vitelliform dystrophy: a fresh perspective. Prog Retin Eye Res. 2015; 47: 64–85. [DOI] [PubMed] [Google Scholar]
- 4. Dalvin LA, Pulido JS, Marmorstein AD.. Vitelliform dystrophies: prevalence in Olmsted County, Minnesota, United States. Ophthalmic Genet. 2017; 38: 143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gass JD. A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc. 1974; 72: 139–156. [PMC free article] [PubMed] [Google Scholar]
- 6. Agarwal A, Gass JDM, Gass JDM.. Gass' atlas of macular diseases. 5th ed. Edinburgh: Elsevier Saunders; 2012. [Google Scholar]
- 7. Boon CJ, Klevering BJ, Leroy BP, Hoyng CB, Keunen JE, den Hollander AI.. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009; 28: 187–205. [DOI] [PubMed] [Google Scholar]
- 8. Petrukhin K, Koisti MJ, Bakall B, et al.. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998; 19: 241–247. [DOI] [PubMed] [Google Scholar]
- 9. Travis GH, Christerson L, Danielson PE, et al.. The human retinal degeneration slow (RDS) gene: chromosome assignment and structure of the mRNA. Genomics. 1991; 10: 733–739. [DOI] [PubMed] [Google Scholar]
- 10. Manes G, Meunier I, Avila-Fernández A, et al.. Mutations in IMPG1 cause vitelliform macular dystrophies. Am J Human Genet. 2013; 93: 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meunier I, Manes G, Bocquet B, et al.. Frequency and clinical pattern of vitelliform macular dystrophy caused by mutations of interphotoreceptor matrix IMPG1 and IMPG2 genes. Ophthalmology. 2014; 121: 2406–2414. [DOI] [PubMed] [Google Scholar]
- 12. Brandl C, Schulz HL, Charbel Issa P, et al.. Mutations in the genes for interphotoreceptor matrix proteoglycans, IMPG1 and IMPG2, in patients with vitelliform macular lesions. Genes (Basel). 2017; 8: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shah SM, Schimmenti LA, Marmorstein AD, Bakri SJ. Adult-onset vitelliform macular dystrophy secondary to a novel IMPG2 gene variant. Retin Cases Brief Rep. 2021; 15: 356–358. [DOI] [PubMed] [Google Scholar]
- 14. Bandah-Rozenfeld D, Collin RW, Banin E, et al.. Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa. Am J Human Genet. 2010; 87: 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishikawa M, Sawada Y, Yoshitomi T.. Structure and function of the interphotoreceptor matrix surrounding retinal photoreceptor cells. Exp Eye Res. 2015; 133: 3–18. [DOI] [PubMed] [Google Scholar]
- 16. Hollyfield JG. Hyaluronan and the functional organization of the interphotoreceptor matrix. Invest Ophthalmol Vis Sci. 1999; 40: 2767–2769. [PubMed] [Google Scholar]
- 17. Enoch JM, Laties AM.. An analysis of retinal receptor orientation. II. Predictions for psychophysical tests. Invest Ophthalmol. 1971; 10: 959–970. [PubMed] [Google Scholar]
- 18. Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005; 85: 845–881. [DOI] [PubMed] [Google Scholar]
- 19. Olivier G, Corton M, Intartaglia D, et al.. Pathogenic variants in IMPG1 cause autosomal dominant and autosomal recessive retinitis pigmentosa. J Med Genet. 2020; 58: 570–578. [DOI] [PubMed] [Google Scholar]
- 20. Havens MA, Duelli DM, Hastings ML.. Targeting RNA splicing for disease therapy. Wiley Interdiscip Rev RNA. 2013; 4: 247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hammond SM, Wood MJ.. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011; 27: 196–205. [DOI] [PubMed] [Google Scholar]
- 22. Cremers FP, van de Pol DJ, van Driel M, et al.. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet. 1998; 7: 355–362. [DOI] [PubMed] [Google Scholar]
- 23. Tatour Y, Tamaiev J, Shamaly S, et al.. A novel intronic mutation of PDE6B is a major cause of autosomal recessive retinitis pigmentosa among Caucasus Jews. Mol Vis. 2019; 25: 155–164. [PMC free article] [PubMed] [Google Scholar]
- 24. Verbakel SK, Fadaie Z, Klevering BJ, et al.. The identification of a RNA splice variant in TULP1 in two siblings with early-onset photoreceptor dystrophy. Mol Genet Genomic Med. 2019; 7: e660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yusuf IH, Shanks ME, Clouston P, MacLaren RE.. A splice-site variant in FLVCR1 produces retinitis pigmentosa without posterior column ataxia. Ophthalmic Genet. 2018; 39: 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Constable PA, Bach M, Frishman LJ, Jeffrey BG, Robson AG, International Society for Clinical Electrophysiology of V. ISCEV standard for clinical electro-oculography (2017 update). Doc Ophthalmol. 2017; 134: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krumm N, Sudmant PH, Ko A, et al.. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012; 22: 1525–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lek M, Karczewski KJ, Minikel EV, et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karczewski KJ, Francioli LC, Tiao G, et al.. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; 581: 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Richards S, Aziz N, Bale S, et al.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wallis Y, Payne S, McAnulty C, et al.. Practice guidelines for the evaluation of pathogenicity and the reporting of sequence variants in clinical molecular genetics. Association for Clinical Genetic Science and the Dutch Society of Clinical Genetic Laboratory Specialists. Association for Clinical Genetic Science (VKGL). 2013. [Google Scholar]
- 32. Sangermano R, Khan M, Cornelis SS, et al.. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018; 28: 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Albert S, Garanto A, Sangermano R, et al.. Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying Stargardt disease. Am J Hum Genet. 2018; 102: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garanto A, Duijkers L, Tomkiewicz TZ, Collin RWJ.. Antisense oligonucleotide screening to optimize the rescue of the splicing defect caused by the recurrent deep-intronic ABCA4 variant c.4539+2001G>A in Stargardt disease. Genes (Basel). 2019; 10: 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khan M, Arno G, Fakin A, et al.. Detailed phenotyping and therapeutic strategies for intronic ABCA4 variants in Stargardt disease. Mol Ther Nucleic Acids. 2020; 21: 412–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koolen L, Gagliardi G, ten Brink SCA, et al.. Generation and characterization of human induced pluripotent stem cells (iPSCs) from three individuals without age-related macular degeneration. Stem Cell Res. 2022; 60: 102670. [DOI] [PubMed] [Google Scholar]
- 37. Koolen L, Gagliardi G, ten Brink SCA, et al.. Generation and characterization of human induced pluripotent stem cells (iPSCs) from three patients with age-related macular degeneration carrying rare variants in the CFH gene. Stem Cell Res. 2022; 60: 102669. [DOI] [PubMed] [Google Scholar]
- 38. Flamier A, Barabino A, Bernier G.. Differentiation of human embryonic stem cells into cone photoreceptors. Bio-Protocol. 2016; 6: e1870. [DOI] [PubMed] [Google Scholar]
- 39. Livak KJ, Schmittgen TD.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 40. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A.. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010; 20: 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014; 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974; 185: 862. [DOI] [PubMed] [Google Scholar]
- 43. Boon CJ, Klevering BJ, den Hollander AI, et al.. Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Arch Ophthalmol. 2007; 125: 1100–1106. [DOI] [PubMed] [Google Scholar]
- 44. Deutman AF, Hoyng CB, van lith-Verhoeven JJC.. Macular dystrophies. In Ryan S, ed. Retina. 4th ed. New York: Elsevier; 2006. [Google Scholar]
- 45. Ścieżyńska A, Oziębło D, Ambroziak AM, et al.. Next-generation sequencing of ABCA4: high frequency of complex alleles and novel mutations in patients with retinal dystrophies from Central Europe. Exp Eye Res. 2016; 145: 93–99. [DOI] [PubMed] [Google Scholar]
- 46. Runhart EH, Khan M, Cornelis SS, et al.. Association of sex with frequent and mild ABCA4 alleles in Stargardt disease. JAMA Ophthalmol. 2020; 138: 1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cremers FPM, Lee W, Collin RWJ, Allikmets R.. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog Retin Eye Res. 2020; 79: 100861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salles MV, Motta FL, Dias da Silva E, et al.. Novel complex ABCA4 alleles in Brazilian patients with Stargardt disease: genotype-phenotype correlation. Invest Ophthalmol Vis Sci. 2017; 58: 5723–5730. [DOI] [PubMed] [Google Scholar]
- 49. Bareil C, Hamel C, Arnaud B, Demaille J, Claustres M.. A complex allele (1064delTC and IVS2 + 22ins7) in the peripherin/RDS gene in retinitis pigmentosa with macular dystrophy. Ophthalmic Genet. 1997; 18: 129–138. [DOI] [PubMed] [Google Scholar]
- 50. Handford PA, Mayhew M, Baron M, Winship PR, Campbell ID, Brownlee GG.. Key residues involved in calcium-binding motifs in EGF-like domains. Nature. 1991; 351: 164–167. [DOI] [PubMed] [Google Scholar]
- 51. Chen Q, Cai S, Shadrach KG, Prestwich GD, Hollyfield JG.. Spacrcan binding to hyaluronan and other glycosaminoglycans. Molecular and biochemical studies. J Biol Chem. 2004; 279: 23142–23150. [DOI] [PubMed] [Google Scholar]
- 52. Salido EM, Ramamurthy V.. Proteoglycan IMPG2 shapes the interphotoreceptor matrix and modulates vision. J Neurosci. 2020; 40: 4059–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.