

Summary
Musk is a precious raw material and ingredient in Chinese traditional medicine. The production of unqualified musk has become a puzzling problem in forest musk deer (FMD) breeding. However, what the essential differences between so‐called unqualified musk and mature qualified musk have not yet been elucidated. In this study, 12 musk samples were collected and separated into two groups according to their external properties. One group is white or black cream‐like secretion with sour or unpleasant odour (MM); the other group is brown or blackish brown solid secretion with pleasant fragrance (DM). Next‐generation sequencing and gas chromatography‐mass spectrometry were used to explore the essential differences between the DM and MM groups in microbial and chemical compositions. The results indicate that the DM group has more heterogenous microbial structure but simpler relationships among microbial communities. LEfSe analysis showed that 14 taxa at the genus level could be used to distinguish the DM and MM groups and Bacillus, Paracoccus, tenoteophomonas, Mycobacterium and Leuconostoc were more abundant in the DM group (P < 0.05). In addition, six compounds were identified to specifically distinguish the DM and MM groups under the OPLS‐DA model. PICRUSt analysis revealed that metabolic pathways such as carbohydrate metabolism, nucleotide metabolism, energy metabolism, transport and catabolism were enriched in the DM group. All these findings of differences in microbiota and chemical compositions would provide potential clues for MM quality improvement and new evidence for the scientific establishment of a quality evaluation standard for musk.
Next‐generation sequencing and gas chromatography‐mass spectrometry were used to explore the essential differences between different quality of musk in microbial and chemical compositions. The results indicate that 14 taxa at the genus level and six compounds were identified to specifically distinguish DM and MM groups under the LEfSe analysis and OPLS‐DA model. All these findings of differences in microbiota and chemical compositions would provide potential clues for MM quality improvement and new evidence for the scientific establishment of a quality evaluation standard for musk.

Introduction
Natural musk, a rare and precious raw material and ingredient in traditional Chinese medicine, was first recorded in Shen Nong's Herbal Classic and has a medicinal history of nearly 2000 years in China (Zhou et al., 2010). Secreted from the musk gland in adult male Moschus berezovskii, Moschus sifanicus and Moschus moschiferus, natural musk is reported to have cardiac, anti‐inflammatory and anti‐tumour properties as well as bidirectionally regulated effects on central nervous system and the ability to prevent ischaemic cardiac disorder (Hu et al., 2009; Tian et al., 2011; Fan et al., 2017; Ali et al., 2018) and has been collected by Chinese Pharmacopoeia. Natural musk has been widely used in lots of drugs. Statistical analysis showed that some drugs including bezoar angong pill, pien tze huang and liushen pill have an output value of more than $1.43 billion per year (Wang et al., 2006). Not surprisingly, the high medicinal and economic value has made natural musk scarce resources. The acquisition of musk caused the population of forest musk deer (FMD) to suffer a drastic decline (Hawkins, 1950; Yang et al., 2003; Meng et al., 2006; Cai et al., 2017), and FMD is becoming endangered animals. To protect FMD populations and provide a sustainable musk supply, FMD was listed as a category I key species under the Wild Animal Protection Law in China in 2003 (Wang and Harris, 2015), and musk deer have been artificially bred and farmed in China since the 1950s. To date, FMD has been bred in Shaanxi, Sichuan, Anhui and Gansu Provinces (Ren, 2003; Wang et al., 2006; Xiang et al., 2011). During the process of breeding, musk is collected only once a year. However, among the musk collections, the so‐called unqualified musk, which is white or black in colour, contains higher water content or smells sour and unpleasant, appeared randomly and was regarded as unsuitable for further use. To avoid the so‐called unqualified musk, further maturation is needed and the musk was often collected in the second or third year. Thus, the so‐called unqualified musk not only resulted in severe economic losses and wasted labour but also greatly injured the confidence of the FMD breeder.
Considering the so‐called unqualified musk is not suitable and qualified ingredient for further use in medicine, it has become a serious problem for FMD breeders. As so far, the standard for musk qualification is only through evaluating the concentration of muscone, which should over 2.0%, according to the Chinese Pharmacopoeia (2015). However, whether this evaluation standard can be effectively used to distinguish the so‐called unqualified musk and mature qualified musk is unknown. Are there essential differences between them in chemical characterization and microbial community structures? All these issues have not yet been elucidated in related studies. It is the important basis for understanding the composition of musk as well as its ecological and pharmacological effects.
In this study, 12 musk samples were collected and separated into two groups. One group, named MM, is mushy, white or black or blackish brown in colour, cream‐like in texture, has a higher water content and smells sour or unpleasant; the other group, named DM, is dry, brown or blackish brown in colour, powdery or granular or bar‐like in texture and smells pleasant. The two groups were separated mainly focus on the colour, water content and odour. The differences between the DM and MM groups in microbial community structures were detected by high‐throughput sequencing, and the chemical components were analysed by GC/MS. Principal component analysis (PCA), redundant analysis (RDA) and orthogonal partial least‐squares‐discriminant analysis (OPLS‐DA) were used to cluster the chemical components and microbial community structure for analysis. The core microorganism and main chemical components that could be used to distinguish the DM and MM groups were concluded. These results could provide good guidance for comprehensive assessment of the quality of musk, as well as provide novel evidence for the scientific establishment of a quality evaluation standard for musk.
Results
Appearance characteristics of 12 collected musk samples
During the process of harvesting musk, the so‐called unqualified musk was always found in the FMD breeding centre. In order to know whether there exist essential differences between mature qualified musk and so‐called unqualified musk and whether the essential differences were highly related to certain core microorganism, 12 samples were collected and analysed. The detailed sample information was described in Table S1. Twelve collected musk samples were separated into two groups according to their appearance and odour (Fig. 1). The MM group, is white or black in colour, cream‐like in texture, has a higher water content and smells sour or unpleasant; the DM group is brown or blackish brown in colour, powdery or granular or bar‐like in texture, dry and smell pleasant.
Fig. 1.

Appearance characterization of MM and DM samples.
MM samples are in the top row and DM samples are in the bottom row.
Differences in microbial diversity between the DM and MM groups
Characterization of the 16S rRNA sequence of musk samples
The microbial diversity of the DM and MM groups was analysed by high‐throughput amplicon sequencing. A dataset consisting of 904 391 high‐quality, classifiable 16S rRNA gene sequences were generated, 57 045–82 520 valid sequences (Mean length = 426 ± 2 bp) were obtained from each sample and gene sequences with an average of 73 541 ± 9467 and 77 921 ± 4944 per sample in the DM and MM groups were obtained. Statistical results of the sequencing data are summarized in Table S2. The raw sequences of this study have been deposited in the Sequence Read Archive (accession number PRJNA545203). The sequences were assigned to 265 operational taxonomic unit (OTU) at the similarity threshold of 97% and categorized into seven phyla, nine classes, 15 orders, 34 families and 42 genera.
Diversity analysis of microbiota in the DM and MM groups
Rarefaction curves were constructed for each individual sample of the DM and MM groups showing the number of observed OTUs (Fig. 2A). The curve becomes asymptotic as the OTU number is saturated, and each sample added an increasingly smaller number of new OTUs, which indicated adequate coverage for the sample being tested. Remarkably dissimilar shapes of the OTU rarefaction curves were observed when comparing samples of the DM and MM groups. The samples of the MM group displayed uniform rarefaction curves, whereas the variation in the shape of the rarefaction curves from the samples of the DM group was much higher (Fig. 2A). The high variability of OTU richness of the DM group, as depicted by the rarefaction curves, could possibly be caused by the more complicated chemical components. The more heterogenous microbial structure is more easily adapted to the environment of musk gland scent pod. While in the MM group, certain chemical components might give rise to increasing kinds of microorganism, which made the microbial structure more homogeneous. The alpha diversity measures also showed that Shannon and Simpson indexes were significantly different between the DM and MM groups (P < 0.05), reflecting that richness and evenness were significantly different between them (Fig. 2B). These analyses verified that microbes in the DM and MM groups were really significantly different at the OTU level.
Fig. 2.
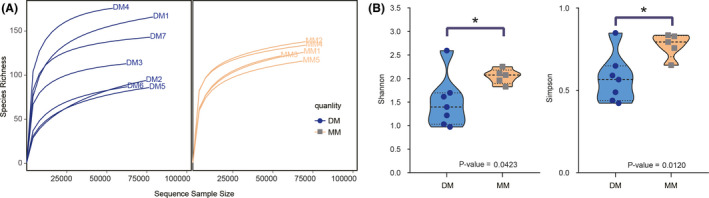
Rarefaction curves and variations in alpha diversity of the microbiota between the DM and MM groups.
(A) Rarefaction curves of DM and MM samples; (B) Comparisons of Shannon and Simpson diversity indices between DM and MM samples by Mann–Whitney test.
To further analyse the differences in microbiota composition between the DM and MM groups, a Bray‐Curtis dissimilarity matrix was calculated on normalized and square‐root transformed read abundance data. Overall similarities in bacterial community structures among samples were displayed using principal coordinate analysis (PCoA). PCoA analyses revealed that the musk microbiotas of the DM group were distinct from those of the MM group. Two principal components explained 58.8% of the total variation, with the first principal component having a greater power of separation (Fig. 3). To statistically support the visual clustering of the bacterial communities in the PCoA analyses, PERMANOVA test was used. These suggested that the microbial communities in these two groups were significantly dissimilar from each other.
Fig. 3.
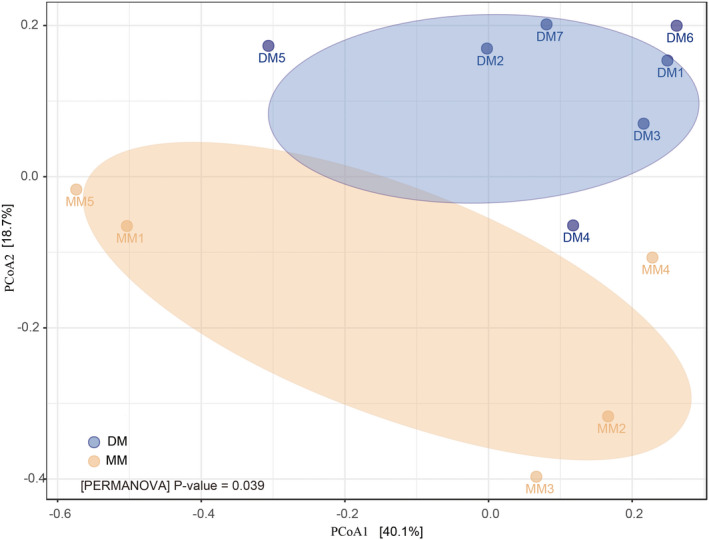
Principal coordinate (PCoA) analysis of the community structure using UniFrac distances. Blue and yellow circles represent the musk microbiota from the DM and MM groups, respectively. Distances between circles on the ordination plot reflect relative dissimilarities in community structures.
Analysis of microbiota differences between the DM and MM groups
The bacterial phyla and genera in relative abundance in the DM and MM groups are shown in Fig. 4A and B. In all, more than 99% of the sequences in all samples belong to four bacterial phyla (namely, Actinobacteria, Firmicutes, Proteobacteria and Bacteroidetes). Among them, Actinobacteria and Firmicutes were present in higher proportions in the DM than in the MM group (66.98% ± 0.29 versus 37.90% ± 0.18 and 29.52% ± 0.03 versus 11.20% ± 0.05, respectively). In contrast, Proteobacteria and Bacteroidetes were more abundant in the MM than in the DM group (49.35% ± 0.16 versus 3.29% ± 0.06, P < 0.01 and 1.33% ± 0.01 versus 0.12% ± 0.002, P < 0.05, respectively). At the genus level, the main dominant genera in the MM and DM groups were different (Fig. 4B). In the MM group, the main dominant genera are Corynebacterium, Pseudomonas and Proteus; in the DM group, the main dominant genera are Corynebacterium, Staphylococcus and Trueperella. Between the DM and MM groups, Corynebacterium (31.10% ± 0.21 versus 63.98% ± 0.28, P = 0.026), Clostridium_XlVa (P = 0.045), Streptophyta (P = 0.033) and Faecalibacterium (P = 0.005) were significantly different in relative abundance.
Fig. 4.
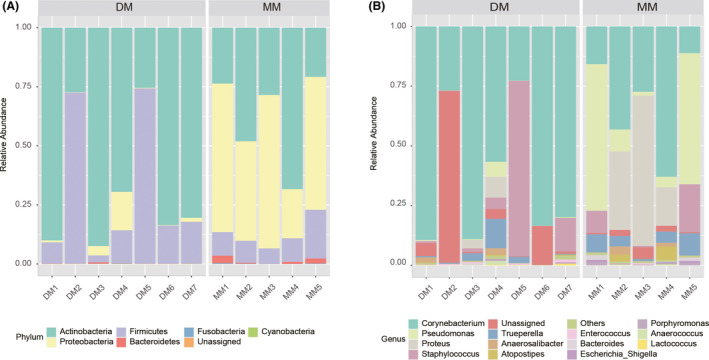
Composition variation of the DM and MM microbiota analysed employing 16S rRNA as biomarkers. Bars represent the relative abundance of bacterial taxa at the phylum and genus levels.
(A) Relative abundance of phyla; (B) relative abundance of genera; unassigned: sequences which could not be classified.
The hierarchically clustered heatmap based on the bacterial composition at the phylum level revealed that the bacterial communities in the DM and MM groups could be clustered together as different patterns. In Fig. 5A, each branch on the tree represents one musk microbiota, each row represents a dominant bacterial phylum. The values in the heatmap represent the square root‐transformed relative percentage of each bacterial phyla. A redder colour indicates larger numbers and more plenty microbiota, while a bluer colour represents smaller numbers. The core microbiomes in the DM and MM groups were different (Fig. 5B).
Fig. 5.
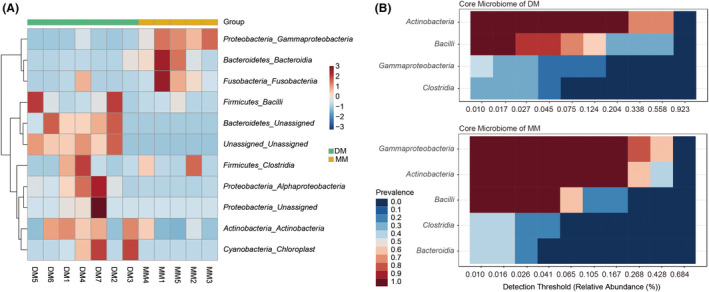
Heatmap analysis of the bacterial distribution between the DM and MM groups.
(A) Heatmap was based on hierarchical clustering (Bray‐Curtis distance metric and complete clustering method) and shows the bacterial distribution among 12 individual samples; (B) heatmap shows the abundance of the core microbiome in the DM and MM groups, respectively.
Linear discriminant analysis (LDA) and linear discriminant analysis effect size (LEfSe) determinations further indicated that at the genus level, 14 taxa can be used to distinguish the DM and MM groups (Fig. 6A). Bacillus, Paracoccus, tenoteophomonas, Mycobacterium and Leuconostoc were more abundant in the DM group (P < 0.05), while Pseudomonas, Porphyromonas, Arsenophonus, Cetobacterium, Succinivibrio, Roseburia, Prevotella, Ruminococcus and Streptococcus were in higher lever in the MM group (P < 0.05) (Fig. 6B and C).
Fig. 6.
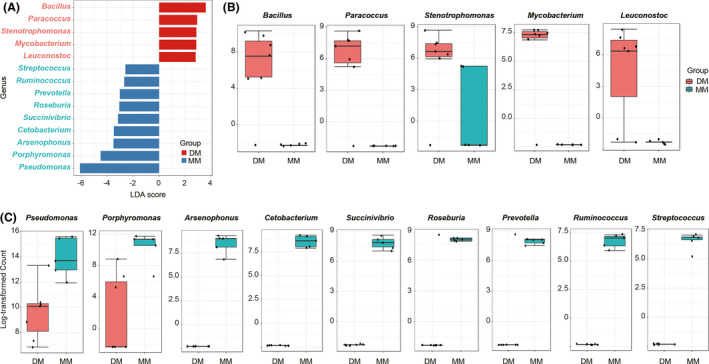
LEfSe analysis of bacterial composition between the DM and MM groups.
(A) LDA score plot of microbial taxa with significant group differences; (B) genus with a significant higher lever in the DM group; (C) genus with a significant higher lever in the MM group.
Network‐based analysis
The networks were considered to offer insight into microbial interactions (Jiao et al., 2016). Figure 7 depicts a complex network‐based analysis of the DM and MM microbiome obtained using the Cytoscape program. In the DM group, the microbial communities seemed more stable, and the interactions among the microbes were simpler. While in the MM group, the interactions between microbial communities were more complicated. Every node performs different functions in modules, and edge between every two nodes shows different relationships (Newman, 2006). Positive relationships may result from commensalism or mutualism, while negative relationships may be ascribed to competition or predation (Hu et al., 2019). In Fig. 7, the networks for the MM group had 42 nodes and 102 edges, while the network for the DM group comprised 17 nodes and 23 edges. In the MM group, the relationships between the edges were 53.92% positive and 46.08% negative, and 33.33% reached significance at the level of P < 0.01. In contrast, the positive relationships in DM group accounted for 100% of the total relationships and 39.13% reached significance at the level of P < 0.01. These results indicated that in the DM group, the relationship among the microbial community was simpler, which might alleviate their competition, strengthen their associations and be more beneficial for the microbes to participate in metabolites. This analysis further elucidates that the microbial communities in the DM and MM groups may play different roles in the transformation of the chemical composition of musk.
Fig. 7.

Network analysis of microbes in the DM group (A) and in the MM group (B). Nodes indicate taxonomic affiliations at the OTU level. Red lines indicate positive correlations; blue lines indicate negative correlations. The solid edge represents the extremely significant correlative relationship (P < 0.01); the dashed edge represents the significant correlative relationship (P < 0.05).
Musk chemical composition of the DM and MM groups
PCA analysis
Musk in the DM and MM groups were separated by diethyl ether solution treatment and detected by GC‐MS. The abundances of the main chemical constituents of the musk samples of the DM and MM groups were determined (Fig. S1 and Table S4). To screen for key metabolites with statistical importance to distinguish the DM and MM groups, a PCA model was established. PCA analysis shows the overall distribution trend among all samples of the DM and MM groups, and possible discrete points can be identified (Table S5 and Fig. S2). Under the positive mode (EI+), PC1 and PC2 show segregation between the samples in the two groups (Fig. S2), which indicates that metabolic profiling differed between the MM and DM groups.
Orthogonal partial least‐squares‐discriminant (OPLS‐DA) analysis
The OPLS‐DA analysis demonstrated that there were significant differences between the DM and MM groups (Fig. S3). As shown in Fig. S3, samples in the DM and MM groups were separated into two clusters (the left cluster is the MM group and the right cluster is the DM group), which indicated that the chemical components of the two groups were remarkably different. The parameters of the OPLS‐DA model (R2Y, Q 2) obtained by sevenfold cross‐validation are shown in Table S6. The value Q 2 > 0.5 confirmed that the OPLS‐DA model (R2Y, Q 2) was stable and reliable. To identify these differential markers, the overall differences between the two datasets were further analysed. Every point on the curves represents one chemical compound (Fig. S4). The closer to the middle a point is, the less different is the chemical compound in the two groups. The chemical compounds beside the upper and lower parts were significantly different components. The compounds in the lower left and upper right sections were more abundant in the MM group and the DM group, respectively. In total, the data points in both sections of the curves represent highly reliable compounds in the sample.
According to OPLS‐DA model, the VIP value was used to measure the expression pattern of each metabolite in terms of effect intensity and explanatory ability to identify biologically relevant significantly differential metabolites. In this study, differentially expressed metabolites were selected preliminarily according to the parameter VIP > 1. Univariate analysis was then performed to verify whether the difference was significant. The metabolites satisfying VIP > 1 and P < 0.05 were considered significantly differentially expressed metabolites. Metabolites satisfying VIP > 1 and 0.05 < P < 0.1 were differentially expressed metabolites. These metabolites can provide a basis for comparing the DM and MM groups. To further identify different compounds, different points were marked with matched peak numbers in the TIC results and listed with retention time and specific charge. The differentially expressed metabolites are shown in Table 1. In total, six compounds were identified as differentially expressed metabolites between the MM and DM groups.
Table 1.
Differential expressed metabolites in the DM and MM groups.
| Number | Chemical name | Molecular formula | CAS | Retention time (R.T.) |
|---|---|---|---|---|
| 1 | Heneicosane | C21H44 | 629‐94‐7 | 32.11 |
| 2 | 1,2‐Benzenedicarboxylic acid | C8H6O4 | 88‐99‐3 | 29.43 |
| 3 | Normuscone | C15H28O | 502‐72‐7 | 25.86 |
| 4 | Butylated hydroxytoluene | C15H24O | 128‐37‐0 | 17.79 |
| 5 | Cholesterol | C27H46O | 57‐88‐5 | 48.80 |
| 6 | Cholesta‐3,5‐diene | C27H44 | 747‐90‐0 | 45.06 |
RDA analysis of the relationship of the microbiota and chemical components between the MM and DM groups
RDA analysis reflected the relationship of the different compounds and microbiota with the MM and DM samples. As shown in Fig. 8, chemicals, such as muscone, androstenedione, benzoic acid and butylated hydroxytoluene, and microbes in Acitinobacteria and Firmicutes were more strongly correlated with samples in the DM group. While chemicals, such as cholesta‐3,5‐diene, eicosane, heptacosane, heneicosane and cholesterol, and microbes in Proteobacteria were more strongly correlated with samples in the MM group. The relationship between the DM and MM groups in microbiota and chemical components were consistent with the results obtained using the OPLS‐DA model and microbial diversity analysis.
Fig. 8.
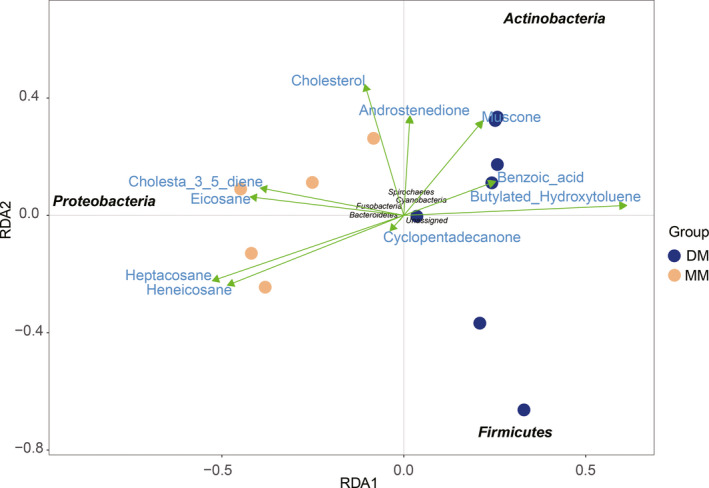
RDA analysis of the relationship of the microbiota and chemical components between the DM and MM groups. The length of the line between the arrow and the origin represents the degree of correlation between the compound and community distribution and species distribution. The longer the line, the greater the correlation.
Discussion
The critical roles of microbiota in maintenance of mammals’ health by providing benefits to their host, such as breakdown of indigestible food, supply of energy for epithelial cells and a barrier against invasive pathogens, have emerged recently (Xue et al., 2011). However, it is still largely unknown how about the distribution of microbiota in musk. Li reported different musk microbiota profiles between mated and unmated FMD males (Li et al., 2016). Actinobacteria and Firmicutes presented in higher proportions in the musk microbiotas of unmated FMD (26.63% versus 5.5%, 32.32% versus 5.45%, respectively), while Proteobacteria were more abundant in the musk microbiota of mated FMD (29.44% versus 11.88%). Considering the puzzling problem of the existence of so‐called unqualified musk, we wonder whether microbial community can be used to distinguish so‐called unqualified musk (MM) from mature qualified musk (DM). We found that more than 99% of the sequences in all DM and MM samples belonged to four bacterial phyla. Specifically, Actinobacteria and Firmicutes were present in higher proportions in the DM group, while Proteobacteria and Bacteroidetes were more abundant in the MM group.
Accumulating evidence has demonstrated that microbiota is indispensable in generation of various odorous metabolites (Carthey et al., 2018; Hang et al., 2019). Thus, there is a possibility that microbiota contribute to the unique and intense perfume of musk by regulating odorous metabolites production and may be a candidate for musk quality control. It was reported that the better the quality was, the higher was the proportion of Actinobacteria and Firmicutes, and the lower was the proportion of Proteobacteria (Li et al., 2016). Li reported previously that the microbiota composition of musk samples in three different mature states varied significantly (P < 0.05) (Li et al., 2018), especially focused on Firmicutes, Proteobacteria, Actinobacteria and Bacteroidetes, as we reported here. All these data collectively demonstrated that these four bacterial phyla are indispensable in musk evaluation.
Muscone, an organic compound secreted by various animals from the stink glands (Ward and Dorp, 1981; Shirasu et al., 2014), is the primary contributor to the fascinating scent of musk and has been used for years to musk evaluation (Chinese Pharmacopoeia, 2015). However, Jiang reported previously that it is not sufficient to judge the quality of musk simply by muscone concentration and comprehensive assessment of chemical fingerprint chromatography and various chemical components including polypeptides, macrocyclic ketones, sterides, pyridine, fatty acids and esters may be essential and useful (Jiang et al., 2018). Wang also verified that using muscone concentration alone to judge the quality of musk is not comprehensive. They suggested evaluating the quality of musk by combined indexes in which muscone, androstanols and cholesteryl substance should all over 2.0% (Wang, 2011). When we detected muscone concentrations in the DM and MM groups, we found that the concentration of muscone in 80% of the so‐called unqualified musk samples were lower than 1.42% with the lowest only about 0.033%. However, there still existed a so‐called unqualified musk sample, in which the muscone concentration was as high as 6.06% and a mature qualified musk sample in which the muscone concentration was as low as 0.29%. Thus, it is not reliable to evaluate the quality of musk only through the concentration of muscone, according to the Chinese Pharmacopoeia (2015).
To effectively distinguish the DM and MM groups in terms of chemical aspect, metabolite analysis was performed and a total of six compounds were identified between them. These compounds include heneicosane, 1,2‐benzenedicarboxylic acid, normuscone, butylated hydroxytoluene, cholesterol and cholesta‐3,5‐diene. Li also found previously that the better quality musk produced by the unmated FMD had higher proportions of cholesterol, normuscone, benzene acetic acid, 3‐ethyl‐3‐hydroxy‐5ɑ‐androstan‐17‐one, cholestan‐3‐ol, cholest‐7‐en‐3β‐ol and 4ɑ‐methyl‐5ɑ‐cholest‐8(14)‐en‐3β‐ol (Li et al., 2016). This suggested that higher concentrations of androstanols and cholesteryl should be a beneficial attachment for better quality musk identification besides higher concentration of muscone.
Traditional Chinese medicine functions based on the coordination of multiple kinds of chemical components (Sun et al., 2008; Liao et al., 2014). It is rational that musk components and microbiota interact with each other in the musk. On one hand, musk components and their changes in the musk gland may modify the microbiota composition (Li et al., 2016). On the other hand, existence and succession of the bacterial community is regarded as one of the important factors causing changes in the musk components. Thus, it was hypothesized that there is a complex relationship among musk components, microbial succession and its related metabolic components. The maturation process of aloes is an example of the complex relationship between microbial community succession and metabolites (Li, 2017). This study investigated the quality of traditional Chinese medicine musk in terms of both chemical components and microbiota. It is the first study to define and characterize the so‐called unqualified musk in terms of appearance, microbial community and chemical components. The results showed that both the chemical and microbial components and proportions were greatly different in the DM and MM groups. So, using the concentration of muscone as the only standard to evaluate the quality of musk could not totally reflect the quality of musk in accordance with the Chinese Pharmacopoeia and the overall view of TCM clinical drug use. Whether muscone can be used as the sole standard to evaluate the quality of natural musk and whether artificial synthesized muscone could completely replace the function of natural musk are questions that still need to be discussed. In addition, whether microorganisms play a role in the maturation of musk needs further study. For example, the functions and metabolic pathways of microorganisms in musk should be better analysed by means of metagenomics to establish a more systematic and comprehensive indicator system for the evaluation of musk quality.
Experimental procedures
Musk sample collection
Musk samples analysed in this study were collected from adult male FMD (M. berezovskii) maintained in Zhenping forest musk deer breeding centre (Ankang, Shaanxi, China). The region is at an altitude of 950–1000 m (30.21°–31.70°N, 109.18°–109.63°E) and belongs to north subtropical humid climate, with an annual average temperature of 12.1°C and annual average rainfall of 1015 mm. The collection work started in the middle of September, when the musk components were considered to be completely mature and does not affect mating in November. Before collecting musk, each of the musk deer was placed in its own cage, and the area surrounding the musk gland scent pods and experimental tools were sterilized with alcohol. All samples of the musk were obtained directly from the musk gland scent pods with a special spoon (Zhang et al., 2009), which would not hurt the FMD. The collected fresh musk samples were placed in sterile cryogenic vials and immediately stored in dry ice for transporting to the lab, where they are temporarily frozen at −80°C until further analyses. All experimental protocols were approved by the Institutional Animal Protection Committee of Shaanxi Academy of Sciences (No. 2017001). All methods were carried out in accordance with International Guiding Principles for Biomedical Research Involving Animals issued by CIOMS (Organization, 1986).
The musk samples were separated into two groups by appearance characteristics, which were regarded as different quality. One group, named DM, is dry, brown or blackish brown in colour, powdery or granular or bar‐like in texture and smells pleasant; the other group, named MM, is mushy or has a higher water content, white or black in colour, cream‐like in texture and smells sour or unpleasant. Twelve musk samples were selected for the study, contained five MM and seven DM samples. The detailed collecting information of musk samples are recorded in Table S1.
Microbial community analysis of musk
Extraction of musk genomic DNA, PCR amplification and sequencing
Total bacterial DNA was extracted from each 0.25 g musk sample using an MO BIO Ultra Clean Fecal DNA Kit (Carlsbad, CA, USA) according to manufacturer’s instruction. The quality and concentration of the extracted DNA were measured using a NanoDrop spectrophotometer (ND‐1000; NanoDrop Technologies, Wilmington, DE, USA). The V3‐V4 variable regions of the 16S rRNA gene in musk were amplified by PCR using specific primers (338F: 5′‐ACTCCTACGGGAGGCAGCA‐3′; 806R: 5′‐GGACTACHVGGGTWTCTAAT‐3′) with 8 bp barcodes.
PCR amplifications were conducted using Bio‐Rad S1000 (Bio‐Rad Laboratory, CA, USA) with the following conditions: 94°C for 5 min (initialization), followed by 30 cycles of 94°C for 30 s (denaturation), 52°C for 30 s (annealing), and 72°C for 30 s (extension), followed by 10 min of final elongation at 72°C. Negative controls (no template) were run to test reagent contamination. The PCR products were purified with an EZNA Gel Extraction Kit (Omega, Norcross, GA, USA) and then mixed proportionally according to the concentration measured by NanoDrop ND‐1000 spectrophotometer (NanoDrop Technologies). Sequencing libraries were generated using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (New England Biolabs, Beverly, MA, USA) and was sequenced on an Illumina HiSeq2500 platform.
Characterization of the similarities of the OTUs of musk
Raw sequence reads were filtered using Usearch 10 (Edgar, 2010). Sequences were grouped into OTUs using Unoise3 against the SILVA bacterial database (Christian et al., 2012) defined as ≥ 97% as a threshold (Edgar, 2018; Rob et al., 2018). RDP classifier software (version 2.10) was used to classify the sequences according to the taxonomy proposed by Garrity et al. (2007) maintained at the Ribosomal Database Project with a confidence threshold of 80% (http://rdp.cme.msu.edu/). The representative sequences of OTUs with their relative abundance were used to calculate richness, Shannon, Simpson and Chao1 diversity indices by the command alpha_div in Usearch 10. Two‐way ANOVA and Student’s t‐test were used for significance testing of alpha diversity between groups (Schloss et al., 2009).
Beta diversity was estimated by computing unweighted UniFrac distances between samples by vegan package in R (version 3.6.1). PCoA was performed to get principal coordinates and visualize from complex, multidimensional data. PCoA analysis was displayed by QIIME2 and ggplot2 package in R software (Bolyen et al., 2019). The PERMANOVA test was used for the significance testing of beta diversity differences between groups. LDA effect size LEfSe (Segata et al., 2011) was used to determine the specific microbiota between the two groups. A size‐effect threshold was 3.5 on the logarithmic. LDA score was used for identifying bacterial taxa. Differences in phylum and genus relative abundances are presented as means ± SD. Student’s t‐test was used to compare the data between groups for significance testing. Network analysis was predicted using the Cytoscape interactive platform (version 3.6.1). Phylogenetic molecular ecological networks (pMENs) were calculated based on the microbial interactions in musk analysed with the R package ‘igraph’ via the random matrix theory (r ≤ 0.8, P ≤ 0.01).
Metabolic profiling of musk
Primary reagents
HPLC‐grade ether was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Distilled water (18.2 MΩ) was prepared using a Milli‐Q water purification system (Millipore, Billerica, MA, USA).
Sample preparation for metabolic profiling
After microbial analysis, the remaining fresh musk was dried by exposure to phosphorus pentoxide (P2O5) and was weighed every 30 min until its weight is unchanged. Then, 100 μl hexacosane was added to each 0.1 g of musk as the internal standard, and the sample was dissolved in 25 ml of ether (purity >99.5%) followed by extracted with ultrasonication for 2 h at 4°C. Finally, the dissolved samples were concentrated on a rotary evaporator, appropriately diluted in 1 ml ether and passed through a 0.45 μm PVDF membrane filter (2005). The liquid is used for chemical composition analysis.
Gas chromatography
Samples were analysed using a gas chromatograph‐mass spectrometer equipped with a Class 5000 data processing system (GC/MS system; Agilent model 7890‐5975, Santa Clara, CA, USA). The capillary column was a fused‐silica HP‐5MS (30 m × 0.25 mm i.d.) with a 0.25‐mm thick film of 5% phenylmethylsilicone. The temperature programme was started at 60°C with an initial hold of 5 min and was then increased at a rate of 10°C·min‐1 to 150°C with a hold of 5 min and finally increased at a rate of 5°C·min‐1 to 280°C with a final hold of 5 min. The temperatures of the injection port and transfer line were both 290°C. The flow rate of the helium carrier was 1.2 ml·min‐1. A 1 μl aliquot of the sample was injected into the carrier solution; then, the stream was introduced to the ion source of MS.
Mass spectrometer
The MS parameters in positive ion mode (EI+ MODE) were as follows: EM voltage, 1255 V; repulsion pole, 32.09 and emission current, 34.8 A. Mass spectra were obtained by standard electron impact ionization scanning from m/z 45 to m/z 550 at a rate of 0.25 s/cycle, and the linear velocity was 38.5 m/s. All data acquisition and processing were carried out with mstop software (Agilent, Santa Clara, CA, USA).
Statistical analysis
The raw MS data were converted to MzXML files using ProteoWizard MS Convert and processed using XCMS for feature detection, retention time correction and alignment. The metabolites were identified by accurate mass (<25 ppm) and MS data, which were matched with NIST2.0 and our standards database.
In the extracted ion features, only the variables having nonzero measurement values in more than 1/3 of the total samples were kept. SIMCA‐P 14.1 (Umetrics, Umea, Sweden) was used for PCA, partial least‐squares‐discriminant analysis (PLS‐DA) and orthogonal partial least‐squares‐discriminant analysis (OPLS‐DA) after Pareto‐scaling. Single‐dimensional statistical analysis included Student’s t test and fold change. The volcano plot was obtained by R software (Wang et al., 2019). Differences were considered statistically significant when P < 0.05.
Conflict of interests
The authors declare that they have no competing interests.
Supporting information
Fig. S1. Two examples of GC‐MS chromatograms from the musk diethyl ether extraction of MM (A) and DM (B), (C) blank TIC and (D) QC sample TIC.
Fig. S2. PCA score plot of the DM and MM groups in positive ion mode.
Fig. S3. Score scatter‐plot of DM and MM samples in positive ion mode.
Fig. S4. The replacement test chart based on the positive mode of DM and MM samples.
Table S1. Musk sampled record of forest musk deer.
Table S2. Sequencing information of the 12 musk samples.
Table S3. Phylum and genus‐level assignments of operational taxomomic units (OTUs) in the DM and MM groups.
Table S4. Chemical constituents of forest musk deer preputial gland secretion.
Table S5. PCA model parameters.
Table S6. OPLS‐DA model parameters.
Acknowledgements
This research was funded by Science and Technology Research Project of Shaanxi Province Academy of Sciences (2018nk‐01, 2018k‐01) and the National Key Research and Development Program of China (No. 2018YFD1001000). The authors would like to express their gratitude towards the technical director of the FMD breeding centre for helping with gaining access to the musk sampled in this project.
Microbial Biotechnology (2022) 15(6), 1783–1794
Funding information
This research was funded by Science and Technology Research Project of Shaanxi Province Academy of Sciences (2018nk‐01, 2018k‐01) and the National Key Research and Development Program of China (No. 2018YFD1001000).
Contributor Information
Yan Wang, Email: wxxyu@163.com.
Yi Wan, Email: wanyi6565@163.com.
Data Availability Statement
The data sets generated and analysed during the current study are available in this published article (and its supplementary information files), and the NCBI sequence reads archive (SRA) under accession number PRJNA545203.
References
- Ali, G. , Shaker, S. , and Naeim, A.N. (2018) The role of musk in relieving the neurodegenerative changes induced after exposure to chronic stress. Am J Alzheimer’s Dis Other Demen 33: 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen, E. , Rideout, J.R. , Dillon, M.R. , Bokulich, N.A. , Abnet, C.C. , Al‐Ghalith, G.A. , et al. (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37: 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y. , Sun, J. , Yang, Y. , Wang, J. , Fu, W. , Zhu, P. , et al. (2017) Studies on body condition scoring and influencing factors in captive forest musk deer. Acta Ecol Sin 37: 1617–1622. [Google Scholar]
- Carthey, A.J.R. , Gillings, M.R. , and Blumstein, D.T. (2018) The extended genotype: microbially mediated olfactory communication. Trends Ecol Evol 33: 885–894. [DOI] [PubMed] [Google Scholar]
- Christian, Q. , Elmar, P. , Pelin, Y. , Jan, G. , Timmy, S. , Pablo, Y. , et al. (2012) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: 590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2018) Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 14: 2371–2375. [DOI] [PubMed] [Google Scholar]
- Fan, D. , Qi, J. , and Zhang, M. (2017) Insights of Chinese medicine on ventricular remodeling: multiple‐targets, individualized‐treatment. Chinese J Integr Med 23: 643–647. [DOI] [PubMed] [Google Scholar]
- Garrity, G. , Lilburn, T. , Cole, J. , Harrison, S.H. , Euzéby, J. , and Tindall, B.J. (2007) Taxonomic outline of the bacteria and archaea, Release 7. 7 March 6, 1‐5.
- Hang, J. , Zhao, G. , Zeng, D. , Feng, X. , Zhang, C. , Liang, Z. , and Lei, M. (2019) Microbial diversity of musk across its maturation process in forest musk deer. China J Chin Materia Med 44: 4448–4453. [DOI] [PubMed] [Google Scholar]
- Hawkins, T.H. (1950) Musk and the musk deer. Nature 166: 262–362. [DOI] [PubMed] [Google Scholar]
- Hu, S. , Tian, D. , Chen, G. , Liu, J. , Ke, Y. , and Bian, K. (2009) Protective effect of heart protecting Musk Pill on heart in spontaneously hypertensive rats. LISHIZHEN Med Materia Medica Res 20: 2458–2461. [Google Scholar]
- Hu, T. , Wang, X. , Zhen, L. , Gu, J. , Zhang, K. , Wang, Q. , et al. (2019) Effects of inoculating with lignocellulose‐degrading consortium on cellulose‐degrading genes and fungal community during co‐composting of spent mushroom substrate with swine manure. Bioresour Technol 291: 121876. [DOI] [PubMed] [Google Scholar]
- Jiang, Q. , Luo, Y. , Tan, T. , Yang, M. , and Liao, Z. (2018) Comparison of volatile chemical compositions of moschus from different species and producing areas by GC‐MS and chemometric analysis. Chinese J Exp Tradit Med Formulae 24: 49–55. [Google Scholar]
- Jiao, S. , Liu, Z. , Lin, Y. , Yang, J. , Chen, W. , and Wei, G. (2016) Bacterial communities in oil contaminated soils: biogeography and co‐occurrence patterns. Soil Biol Biochem 98: 64–73. [Google Scholar]
- Li, D. , Chen, B. , Zhang, L. , Gaur, U. , Ma, T. , Jie, H. , et al. (2016) The musk chemical composition and microbiota of Chinese forest musk deer males. Sci Rep 6: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. (2017) Application of GC‐MS and LC‐MS for screening and identification of different markers in agarwood'. Guangzhou: Guangzhou University of Chinese Medicine. [Google Scholar]
- Li, Y. , Zhang, T. , Qi, L. , Yang, S. , Xu, S. , Cha, M. , et al. (2018) Microbiota changes in the musk gland of male forest musk deer during musk maturation. Front Microbiol 9: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, B. , Song, J. , Xie, C. , Han, J. , and Chen, S.L. (2014) Study on traceability system of genuine medicinal materials. China J Chin Materia Med 39: 3881–3888. [PubMed] [Google Scholar]
- Meng, X. , Zhou, C. , Hu, J. , Li, C. , Meng, Z. , Feng, J. , et al. (2006) Musk deer farming in China. Anim Sci 82: 1–6. [Google Scholar]
- Newman, M.E. (2006) Modularity and community structure in networks. Proc Natl Acad Sci USA 103: 8577–8582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organization, P.A.H. (1986) International guiding principles for biomedical research involving animals issued by CIOMS. Vet Q 8: 350–352. [DOI] [PubMed] [Google Scholar]
- Chinese Pharmacopoeia . (2015) The Pharmacopoeia committee of the People's Republic of China. Vol.I, 384‐385.
- Ren, Z. (2003) Musk Deer Farming and Musk Production. Beijing: Jindun Press. [Google Scholar]
- Rob, K. , Alison, V. C., T. B. , Alexander, A. , Chris, C. , and Justine, D. , et al. (2018) Best practices for analysing microbiomes. Microbiome 16, 410‐422. [DOI] [PubMed] [Google Scholar]
- Schloss, P.D. , Westcott, S.L. , Ryabin, T. , Hall, J.R. , Hartmann, M. , Hollister, E.B. , et al. (2009) Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata, N. , Izard, J. , Waldron, L. , Gevers, D. , Miropolsky, L. , Garrett, W.S. , and Huttenhower, C. (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasu, M. , Yoshikawa, K. , Takai, Y. , Nakashima, A. , Takeuchi, H. , Sakano, H. , and Touhara, K. (2014) Olfactory receptor and neural pathway responsible for highly selective sensing of musk odors. Neuron 81: 165–178. [DOI] [PubMed] [Google Scholar]
- Sun, G. , Song, W. , Yang, S. , and Wang, Z. (2008) Research method for capillary electrophoresis fingerprints of traditional Chinese medicines. Central South Pharmacy 6: 6. [Google Scholar]
- Tian, D. , Ling, S. , Chen, G. , Li, Y. , Liu, J. , Ferid, M. , and Bian, K. (2011) Hypertensive nephropathy treatment by heart‐protecting musk pill: a study of anti‐inflammatory therapy for target organ damage of hypertension. Int J Gen Med 4: 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F. , Chen, L. , Chen, H. , Chen, S. , and Liu, Y. (2019) Analysis of flavonoid metabolites in citrus peels (Citrus reticulata “Dahongpao”) using UPLC‐ESI‐MS/MS. Molecules 24: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. (2011) Study on the Quality Control of Moschus. Shanghai: East China University of Science and Technology. [Google Scholar]
- Wang, H. , Liu, W. , Zhong, L. , and Wang, W. (2006) Development prospect and artificial cultivation situation of Musk deer. J Shaanxi Normal Univ (Nature Science edition) 34: 203–206. [Google Scholar]
- Wang, Y. & Harris, R. (2015) Moschus berezovskii. The IUCN Red List of Threatened Species. 2015: e.T13894A103431781. 10.2305/IUCN.UK.20154.RLTS.T13894A61976926.en [DOI]
- Ward, J.P. , van Dorp, D.A. (1981) The animal musks and a comment of their biogenesis. Experientia 37: 917–922. [Google Scholar]
- Xiang, L. , Lu, Q. , and Meng, X. (2011) Preliminary studies on the behavioral assessment of the domestication degree of captive alpine musk deer. Pakistan J. Zool 43: 751–757. [Google Scholar]
- Xue, X. , Feng, T. , Yao, S. , Wolf, K.J. , Liu, C.‐G. , Liu, X. , et al. (2011) Microbiota downregulates dendritic cell expression of miR‐10a, which targets IL‐12/IL‐23p40. J Immunol 187: 5879–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q. , Meng, X. , Xia, L. , and Feng, Z. (2003) Conservation status and causes of decline of musk deer (Moschus spp.) in China. Biol Cons 109: 333–342. [Google Scholar]
- Zhang, H. , Wang, M. , Li, J. , Shao, J. , and Ma, Y. (2009) Living extraction of musk from Moschus berezovskii in captivity. Chinese J Wildlife 30: 175–176. [Google Scholar]
- Zhou, J. , Jin, C. , Luo, Y. , Wu, Y. , Li, J. , Luo, Y. , and He, X. (2010) Identification of musk by fourier transform infrared spectroscopy. Guangpuxue Yu Guangpu Fenxi 30: 2368–2371. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Two examples of GC‐MS chromatograms from the musk diethyl ether extraction of MM (A) and DM (B), (C) blank TIC and (D) QC sample TIC.
Fig. S2. PCA score plot of the DM and MM groups in positive ion mode.
Fig. S3. Score scatter‐plot of DM and MM samples in positive ion mode.
Fig. S4. The replacement test chart based on the positive mode of DM and MM samples.
Table S1. Musk sampled record of forest musk deer.
Table S2. Sequencing information of the 12 musk samples.
Table S3. Phylum and genus‐level assignments of operational taxomomic units (OTUs) in the DM and MM groups.
Table S4. Chemical constituents of forest musk deer preputial gland secretion.
Table S5. PCA model parameters.
Table S6. OPLS‐DA model parameters.
Data Availability Statement
The data sets generated and analysed during the current study are available in this published article (and its supplementary information files), and the NCBI sequence reads archive (SRA) under accession number PRJNA545203.