

SUMMARY
The aim of this study was to retrospectively assess the value of whole genome sequencing (WGS) compared to conventional typing methods in the investigation and control of an outbreak of Shigella sonnei in the Orthodox Jewish (OJ) community in the UK. The genome sequence analysis showed that the strains implicated in the outbreak formed three phylogenetically distinct clusters. One cluster represented cases associated with recent exposure to a single strain, whereas the other two clusters represented related but distinct strains of S. sonnei circulating in the OJ community across the UK. The WGS data challenged the conclusions drawn during the initial outbreak investigation and allowed cases of dysentery to be implicated or ruled out of the outbreak that were previously misclassified. This study showed that the resolution achieved using WGS would have clearly defined the outbreak, thus facilitating the promotion of infection control measures within local schools and the dissemination of a stronger public health message to the community.
Key words: Bacterial typing, gastrointestinal infections, molecular epidemiology, public health, Shigella
INTRODUCTION
Shigella sonnei, the leading cause of bacterial dysentery in developed countries, emerged as a widespread disease in England and Wales in the late 1930s [1, 2]. In 1992, laboratory reports of S. sonnei infections to the Public Health Laboratory Service's Communicable Disease Surveillance Centre peaked at 17237 cases [2]. Since that time, the number of reported cases has declined, and between 2005 and 2011 an average of 800 isolates per year were submitted to the Gastrointestinal Bacteria Reference Unit (GBRU), Health Protection Agency Colindale, for confirmation of bacterial identification and S. sonnei-specific phage typing. In the UK S. sonnei outbreaks are associated with primary schools and nurseries with the usual mode of transmission being person-to-person spread, via the faecal–oral route of infection [3, 4]. The symptoms of S. sonnei range from mild to severe diarrhoea, which may contain mucus and/or blood in the faeces, and may include fever and abdominal pain.
Phage typing has been used routinely at GBRU to subtype S. sonnei from cases in England and Wales since 1994. The most common phage type (PT) between 2004 and 2011 was PT6, representing over 70% of isolates submitted to GBRU, the remaining isolates being largely assigned to PT2. The dominance of PT6 has resulted in phage typing having a limited use in most outbreak investigations in recent times. Consequently, since 2010, multi-locus variable number tandem repeat (VNTR) analysis (MLVA) has been the typing method of choice [5]. This technique shows a higher level of discrimination of S. sonnei compared to phage typing, and has been used to confirm the relatedness of a number of epidemiologically linked domestic cases, and in UK residents returning from abroad (GBRU in-house data).
In June 2011, the North East and North Central London Health Protection Unit (NENCL HPU) received notifications of four apparently unrelated cases of S. sonnei, two of which had become symptomatic after attending Jewish festivities in Manchester. Further investigations confirmed that all cases were members of the Orthodox Jewish (OJ) population residing within the same North London borough, and revealed that all had contacts within their individual family clusters (defined as members of the same household and/or extended family) that were currently or previously showing symptoms consistent with S. sonnei diarrhoea. An investigation was instigated to assess the extent of the outbreak, to try and identify links between cases, and to formulate appropriate control measures. A working case definition (see Materials and methods section) was implemented, and enhanced surveillance was introduced for all S. sonnei infections in areas containing OJ communities in North London.
Public health actions included exclusion advice for all symptomatic children, letters sent to all parents of any child attending a Jewish school within the borough (and receiving schools located just outside of the borough) to inform them of the potential outbreak and provide infection control advice, and infection control posters displayed in community centres and synagogues. A review of infection control polices was conducted by public health professionals at NENCL HPU, and education in infection control procedures was provided to the staff of the schools and nurseries affected by this outbreak [3, 4].
Outbreak investigation and control was assisted at the time by the use of MLVA of confirmed isolates of S. sonnei to help determine if the cases could be linked to a single outbreak strain or could be explained by a general increase in the background levels of infection previously seen the OJ community. The aim of this study was (i) to compare the differences in the conclusions reached using MLVA and genome sequencing methods, and (ii) to explore how the sequencing analysis may have affected the epidemiological investigation, had it been available. The broader impact of whole genome sequencing (WGS) for informing public health protection actions is discussed.
MATERIALS AND METHODS
Epidemiological investigation
The case definition used in this epidemiological investigation was diarrhoea (⩾3 unformed stools in a 24-h period), with or without blood in the stool and fever or abdominal cramps occurring after 1 May 2011 and membership of the Jewish community. A confirmed case was defined as a symptomatic patient, in whom S. sonnei infection had been diagnosed by a local laboratory. A probable case was defined as an illness which met the clinical definition, had an epidemiological link to a confirmed case but no isolate provided.
MLVA and phage typing
Twenty-three of the 27 laboratory-confirmed isolates of S. sonnei were submitted to GBRU and typed by MLVA. The standard MLVA protocol for S. sonnei targets eight MLVA loci for amplification in 20 μl reactions in two quadruplex PCR assays [5]. The resultant amplified products were sized on an ABI 3730 Genetic Analyzer with 600 LIZ (Applied Biosystems, USA) as the size standard, and data were analysed with Peakscanner software (Applied Biosystems). Fragment sizes were imported into Bionumerics software (Belgium) via a script that calculated the tandem repeat numbers for each locus. All 23 isolates were phage-typed retrospectively using the method described by Frost et al. [6].
WGS
Twenty-four isolates were selected for WGS, including 22/23 isolates submitted to GBRU that met the case definition (one isolate did not survive the archiving process) and an additional two isolates from members of the OJ communities in Manchester and North East England (included in the dataset because a number of the London cases reported recent travel to these areas). Tagged genomic library preparation and DNA sequencing with multiplexing was performed using Illumina MiSeq for cases designated B, F, G, H, I, J, K, and Q (see Table 1) or HiSeq 2000 (Illumnia, UK) (the remaining 16 samples) platforms, as described previously [7, 8].
Table 1.
The cases involved in the outbreak investigation
| Case | MLVA profile | VNTR cluster | Phage type | SNP cluster |
|---|---|---|---|---|
| A | 8-17-4-5-2-3-3-3 | MC | 6 | PC1 |
| B | 8-17-4-5-2-3-3-3 | MC | 6 | PC1 |
| C | 8-17-4-5-2-3-3-3 | MC | 6 | PC1 |
| D | 8-17-4-5-2-3-3-3 | MC | 6 | PC1 |
| E | 10-17-4-5-2-3-3-3 | MC | 6 | PC1 |
| F | 10-17-4-5-2-3-3-3 | MC | 6 | PC1 |
| G | 10-17-4-5-2-3-3-3 | MC | 6 | PC1 |
| H1 | 10-7-5-5-2-3-3-3 | MC | 6 | PC1 |
| I1 | 10-7-5-5-2-3-3-3 | MC | 6 | PC1 |
| J | 10-7-5-5-2-3-3-3 | MC | 6 | PC1 |
| K | 8-17-5-5-2-3-3-3 | MC | 6 | PC1 |
| L | 8-17-5-5-2-3-3-3 | MC | 6 | PC1 |
| M2 | 8-17-5-5-2-3-3-3 | MC | 6 | PC1 |
| N3 | 8-17-5-5-2-3-3-3 | MC | 6 | PC1 |
| O3 | 8-17-5-5-2-3-3-3 | MC | 6 | PC1 |
| P2 | 8-17-5-5-2-3-3-3 | MC | 6 | PC1 |
| Q | 7-17-5-5-2-3-3-3 | MC | 6 | PC1 |
| R | 9-16-4-5-2-3-3-3 | SC2 | 6 | PC1 |
| S4 | 12-14-7-5-2-3-3-3 | SC3 | 7 | PC2 |
| T4 | 12-14-7-5-2-3-3-3 | SC3 | 7 | PC2 |
| U | 12-14-7-5-2-3-3-3 | SC3 | 7 | n.a. |
| V | 10-17-7-5-2-3-3-3 | MC | 7 | PC2 |
| W | 8-12-0-6-2-3-3-3 | SC1 | P | PC3 |
MLVA, Multi-locus variable number tandem repeat analysis; VNTR, variable number tandem repeat; SNP, single nucleotide polymorphism; MC, Main cluster; SC, sporadic cluster; PC, phylogentic cluster, n.a., sequencing data was not available.
Household clusters are indicated by superscript numbers.
Illumina reads were mapped to the reference S. sonnei strain SS046 (EMBL ID: CP000038) [9] using Bowtie [10]. The sequence alignment map output from Bowtie was sorted and indexed to produce a binary alignment map (BAM). Samtools mpileup [11] was used to create a variant call format (VCF) file from each of the BAMs, which was further parsed to extract only single nucleotide polymorphism (SNP) positions which were of high quality in all genomes. High quality was defined as SNPs having an overall VCF SNP quality score of >50, a genotype quality score >30 for each strain and either homozygous wild-type or variant type in a diploid model. All discriminatory SNPs were manually validated visually using the BAM viewer Tablet [12]. Pseudosequences of polymorphic positions were created, and approximate maximum-likelihood trees were created using Fasttree [13] under the Jukes–Cantor model of nucleotide evolution.
RESULTS
Epidemiological investigation
Following the presumptive outbreak notification in June 2010 a questionnaire was developed based on that used in a previous S. sonnei outbreak within the OJ community in Hackney, North London in 2008 [14], and administered to a member of each family cluster linked to the S. sonnei outbreak. Information was obtained on demographics, geographical location, food consumption, occupation, recent travel history, and synagogue and school attendance of all members within each cluster.
A ‘look-back’ exercise was performed to assist in determining whether there had been a rise in the number of cases above background levels. All public health epidemiological surveillance records of confirmed cases of S. sonnei were examined to identify members of the OJ community, going back to December 2009. In addition, surveillance records from 2007–2009 were examined, and estimates for numbers of confirmed cases from the OJ community were made for those years, based on names of patients.
As a result of this investigation gastrointestinal illness was reported in 86 people, of whom 82 met the case definition for possible, probable or confirmed outbreak case of S. sonnei, across 18 family clusters and six further individuals. Of these, 27 cases were laboratory confirmed at the local laboratory. Figure 1 displays the epidemic curve of the presumptive outbreak.
Fig. 1.

Epidemic curve showing onset week of all cases of S. Sonnei meeting the case definitions as described previously (see Materials and methods section). OJ, Orthodox Jewish.
Eighty-nine percent (73/82) of cases had known contact with children aged <5 years prior to onset of symptoms. Forty-five (55%) of the cases attended a school or playgroup, most were aged <5 years (39%, 32/82) and six were teachers or teaching assistants associated with these childcare facilities. A large number of cases were also seen in those aged 25–34 years (22%, 18/82); i.e. parents of children aged <5 years. The first reported case within each family (presumed to be the index case) was aged ⩽10 years in 79% (19/24) of household clusters. Of these index cases, nine attended primary school, four attended nursery, three were part of informal playgroups and three did not attend any group (all of which were aged ⩽1 year).
Molecular typing by MLVA
MLVA was used to type 23 isolates from this presumptive outbreak revealing nine different profiles (Table 1). The MLVA profiles were used to construct a minimum spanning tree (MST) to determine the relationship between isolates from this study compared to other domestically acquired S. sonnei strains isolated from diarrhoeal cases in the UK in 2011 (Fig. 2).
Fig. 2.
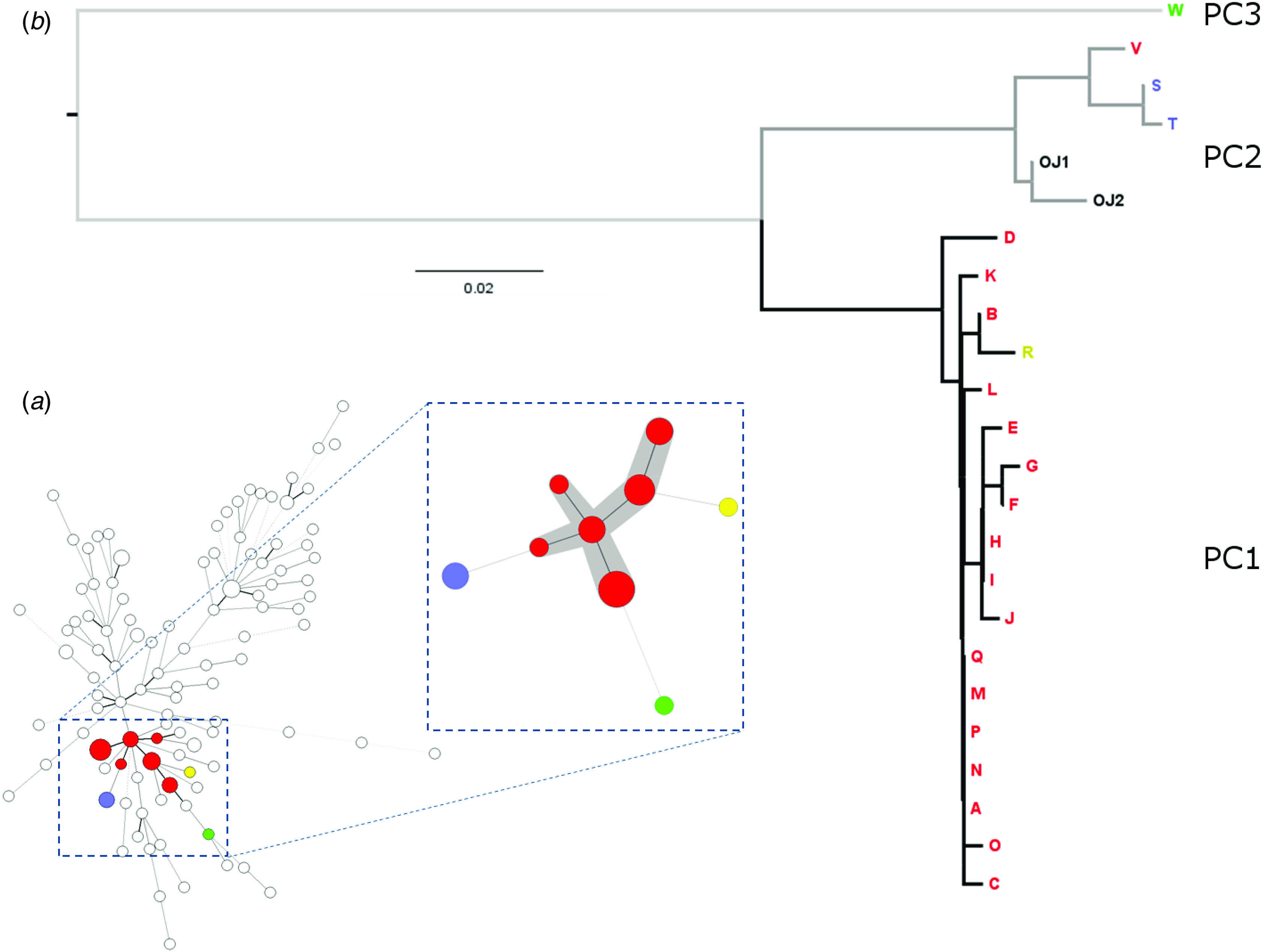
(a) Minimum spanning tree illustrating the population structure of domestically acquired S. sonnei in the UK in 2011 based on multi-locus variable number tandem repeat analysis (MLVA) profiles with the cluster of strains associated with the Orthodox Jewish (OJ) community highlighted. The numbers of strains in each cluster are represented by the size of the circles. Clusters highlighted grey differ by single locus variants. MLVA cluster key: red (18 cases) = main cluster (MC); blue (two cases) = sporadic cluster 3, (SC3); green (one case) = sporadic cluster 1 (SC1); yellow (one case) = sporadic cluster 2 (SC2) (see Table 1). (b) Maximum-likelihood tree of OJ community split into three phylogentic clusters (PCs). Cases that were included in the outbreak investigation are labelled A–W, strains OJ1 and OJ2 represent OJ cases not included in the outbreak investigation.
During this investigation, and using this methodology, S. sonnei MLVA single locus variants (SLVs) that could be epidemiologically linked in space and time were assumed to be part of the same outbreak cluster. During the early phase of the outbreak, the MLVA profiles belonging to the first set of isolates typed, differed by more than one SLV and so, based on our criterion appeared to be unrelated. However, as the outbreak progressed, additional strains linked to form a central cluster consisting of six of the profiles, representing isolates from 18 individual cases, shown as six red circles highlighted in grey in Figure 2.
These data showed that there was a subset of strains circulating at this time in the community that were both epidemiologically and genetically more closely related. However, it was unclear as to whether the criterion of linking only SLVs in order to define outbreaks caused by a single evolving strain was too tight or too loose a definition when attempting to link cases. Furthermore, the precise nature of the genetic relationship of isolates within this subset (i.e. whether the cluster represented one strain or multiple strains) remained uncertain. What was certain was that these confounders and a lack of resolution hampered the epidemiological investigation and made it difficult to provide a clear public health message to the community.
Retrospective WGS analysis
To attempt to understand how robust the interpretations made using the MLVA data had been during this investigation, we retrospectively sequenced 24 isolates, including 22 that met the case definition as well as an additional two isolates from members of the OJ communities in Manchester and North East England isolated in the same year (see Table 1).
From these WGS, SNPs were called against the S. sonnei reference strain SS046, concatenated for each strain, and used to construct a maximum-likelihood phylogenetic tree based on all 395 variable sites (see Materials and methods section). Figure 2 showed that the strains matching the case definition formed three distinct phylogenetic clusters (PC1–3). PC1 contained 18 isolates which differed by a maximum of seven SNPs and PC2 contained five isolates which differed by a maximum of 13 SNPs. PC3 contained one isolate. There was a minimum of 26 SNPs between cases in PC1 and PC2; 92 between PC1 and PC3; and 94 between PC2 and PC3. Two of the five isolates comprising PC2 belonged to the UK OJ community but did not match the case definition with respect to dates of onset of disease or location and were therefore not epidemiologically implicated in the outbreak. Sequenced strains belonging to cases in the same family cluster are highlighted in Table 1.
The conclusion from the phylogenetic analysis was that the apparent increase in the incidence of S. sonnei dysentery could be largely attributed to a single phylogenetically distinct S. sonnei lineage: PC1 was composed of 18/24 of the strains that had been epidemiologically linked to the OJ community. Whereas PC2 (5/24 strains) and PC3 (1/24 strains) represent related but distinct background strains of S. sonnei that were found in cases within the OJ community in the UK but were not linked to the June 2011 outbreak. This investigation therefore revealed at least three distinct S. sonnei lineages were causing disease in the OJ community in regions across the UK during this time.
Comparison of MLVA and genome sequencing
Several discrepancies were observed between the WGS relationships shown from the phylogeny compared to the MLVA typing data. MLVA typing illustrated the existence of an SLV cluster containing six profiles and three outlier profiles. The WGS SNP-based analysis showed that the MLVA outlier case R was a member of PC1, and one of the SLV MLVA profiles (case V) was an epidemiologically unrelated lineage not closely related to the outbreak strain (Fig. 2).
Additional retrospective epidemiological analyses using genome sequencing
The combined MLVA and WGS data were used to reappraise the epidemiological data collected at the time of the investigation. The majority of S. sonnei cases lived within close proximity of each other and centred around a highly populated OJ area within North London, served by a number of local schools. No association was identified between a particular MLVA profile or WGS phylogenetic clusters and attendance at any given school, consistent with human-to-human transmission across the community.
From this retrospective investigation we were able to rule out one case of dysentery from the outbreak (case V, Fig. 2) where the isolate belonged to PC2, while a family resident in an adjoining house presented with S. sonnei found to belong to PC1. This was explained by the case recently visiting family and friends in the North of England and so was likely to have acquired the infection during their travels and not locally.
Conversely, we ruled in case H who had travelled to Australia to attend a Jewish ceremony and became unwell while there. Also attending this ceremony in Australia were friends and family from the UK, some of whom were reportedly unwell showing similar symptoms immediately prior to travelling. The sequencing data shows that this isolate differs by only 1–2 SNPs from four other isolates within PC1. The ‘onset of symptoms date’ of these four cases preceded that of case H and so combined with the WGS and epidemiological data suggests that this isolate was acquired by the individual while in Australia, most likely from another symptomatic UK resident originating from the same community in North London. Transmission of S. sonnei by travel and intercommunity person-to-person contact in OJ communities has been previously described [15, 16].
DISCUSSION
MLVA is a useful tool for the epidemiological analysis of monomorphic bacterial species, such as S. sonnei, that have evolved over different timescales [17]. However, during the investigations described in this study, MLVA was unable to elucidate whether the cases were associated with recent exposure to a single strain or with a number of different strains circulating within a relatively closed community. Genome sequencing has already contributed to the phylogenetic analysis of the global dissemination of S. sonnei [18]. We aimed to use the same approach locally, and in a much shorter time-frame, to clarify the relationship between strains circulating within the OJ community in North London during June and July 2011.
Within the set of isolates analysed, the sequencing data suggested a close relationship (0–7 SNPs) between the strains in PC1. Epidemiological data on date of onset indicated that for the majority of families, there was an index case (generally a young child) with staggered onset in the rest of the family. The risks associated with attendance at schools and/or nurseries and subsequent household transmission is well documented [16, 19]. The conclusion from the SNP analysis was that cases in PC1 had been recently exposed to a single evolving lineage. By contrast, PC2 and PC3 represented strains of S. sonnei circulating within the OJ community at a background level.
Superimposing the information gained from the sequencing data onto that from the MLVA challenges some of the conclusions drawn in the initial outbreak investigation. The most fundamental difference is that we now conclude that the majority of cases of S. sonnei identified in the outbreak investigation came from an outbreak of one evolving strain suggesting a localized source, rather than concurrently from a number of different but closely related strains with different epidemiological explanations. This clarification would have had a positive impact on the outbreak investigation with respect to (i) reducing the time spent determining that an outbreak was occurring, (ii) promoting infection control policies across the schools and (iii) developing effective strategies for communicating with this community.
A large focus of the investigation was on ensuring that an outbreak was occurring, rather than the enhanced surveillance picking up increased background levels of the disease circulating in the community, which while still important require different intervention strategies. The sequencing data would have clearly identified the outbreak and the ‘look-back’ exercise that attempted to estimate levels within the community from previous years would not have been necessary.
Given that S. sonnei outbreaks often centre round primary schools and nurseries, each school was investigated as a potential source of this outbreak. The variation observed in the profiles resulting from the MLVA hampered this investigation as it overestimated the diversity of the strains circulating within this outbreak producing a complex, transmission pattern that took time to dissect.
WGS-based epidemiological analysis facilitated an estimation of true genetic distance between strains of S. sonnei. The sequencing data facilitated the most precise case definition, and clarified the inclusion or exclusion of cases, especially those cases from outside the main location of the outbreak. The sequencing data also provided good evidence that a common strain was being transmitted largely between children and that the schools/nurseries in the area were the conduit for these transmission events, rather than the source. This clarification may have facilitated a more strategic approach to public health actions across the schools within this community, and a rapid implementation of exclusion and infection control policies.
In a community that has previously not engaged fully on public health matters, it was imperative to have a strong public health message. The lack of clarity in the possible conclusions drawn from the MLVA prevented the broadcasting of specific risks associated with the outbreak allowing only for the rise in the number of cases to be publicized. Greater confidence that an outbreak was occurring would have facilitated a more pro-active approach with regard to the media helping to spread public health messages on infection control more effectively. These public health messages would have had more influence on the community if we had been able to confirm the situation as an outbreak. While the level of cooperation with health protection actions was generally good in this instance, a stronger public health message would enhance engagement with communities in the future. Although WGS is not currently routinely available in England for all gastrointestinal outbreak investigations, decreasing costs and the implementation of bench-top sequencers in many hospital and reference microbiology laboratories indicate that it will be more widely available in the near future. In conclusion, WGS and SNP analysis facilitated a more precise case definition, clarified the inclusion or exclusion of outbreak-related cases and provided a clear evidence base for decision-making on the appropriate public health actions.
ACKNOWLEDGEMENTS
We thank Vivienne do Nascimento, Yoshini Weersinghe and Laura Stock for their technical assistance with the MLVA and phage typing and David Harris for his efforts in whole genome sequencing. This work was supported by the Wellcome Trust (grant no. 098051).
DECLARATION OF INTEREST
None.
REFERENCES
- 1.Kotloff KL, et al. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bulletin of the World Health Organization 1999; 77: 651–666. [PMC free article] [PubMed] [Google Scholar]
- 2.Bentley CA, et al. Phage typing and drug resistance of Shigella sonnei isolated in England and Wales. Epidemiology and Infection 1996: 116: 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCann R, et al. An outbreak of Shigella sonnei dysentery among a religious community in Salford and Bury. Health Protection Agency North West Office, Health Protection Bulletin 2004, pp. 3–5. [Google Scholar]
- 4.Working Group of the former PHLS Advisory Committee on Gastrointestinal Infections. Preventing person-to-person spread following gastrointestinal infections: guidelines for public health physicians and environmental health officers, 2004. [PubMed]
- 5.Liang SY, et al. Multilocus variable-number tandem-repeat analysis for molecular typing of Shigella sonnei. Journal of Clinical Microbiology 2007; 45: 3574–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frost JA, et al. An outbreak of Shigella sonnei infection associated with consumption of iceberg lettuce. Emergering Infectious Diseases 1995; 1: 26–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris SR, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science 2010; 327: 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quail MA, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 2012; 13: 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang F, et al. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Research 2005; 33: 6445–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langmead B, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 2009; 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milne I, et al. Tablet – next generation sequence assembly visualization. Bioinformatics 2010; 26: 401–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price MN, et al. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 2010; 5: 9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Addiman S, et al. Is history repeating itself? Shigella sonnei PT P outbreak in the Orthodox Jewish community in Hackney, London. Health Protection Conference, 15–17 September 2008; Warwick, UK. Poster presentation.
- 15.Sobel J, et al. A prolonged outbreak of Shigella sonnei infections in traditionally observant Jewish communities in North America caused by a molecularly distinct bacterial subtype. Journal of Infectious Diseases 1998; 177: 1405–1409. [DOI] [PubMed] [Google Scholar]
- 16.De SK, et al. Outbreak of Shigella sonnei infections in the Orthodox Jewish community of Antwerp, Belgium, April to August 2008. European Surveillance 2011; 16: 19838. [PubMed] [Google Scholar]
- 17.Chiou CS, et al. Utility of multilocus variable-number tandem-repeat analysis as a molecular tool for phylogenetic analysis of Shigella sonnei. Journal of Clinical Microbiology 2009; 47: 1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holt KE, et al. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nature Genetics 2012; 44: 1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrett V, et al. A recurring outbreak of Shigella sonnei among traditionally observant Jewish children in New York City: the risks of daycare and household transmission. Epidemiology and Infection 2006; 134: 1231–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]