

Abstract
While most cases of pancreatitis in dogs are thought to be idiopathic, potential risk factors are identified. In this article we provide a state‐of‐the‐art overview of suspected risk factors for pancreatitis in dogs, allowing for improved awareness and detection of potential dog‐specific risk factors, which might guide the development of disease prevention strategies. Additionally, we review important advances in our understanding of the pathophysiology of pancreatitis and potential areas for therapeutic manipulation based thereof. The outcome of pathophysiologic mechanisms and the development of clinical disease is dependent on the balance between stressors and protective mechanisms, which can be evaluated using the critical threshold theory.
Keywords: colocalization, critical threshold theory, ER stress, impaired autophagy, mitochondrial dysfunction, oxidative stress, pathologic calcium signaling
Abbreviations
- AP
acute pancreatitis
- cPLI
canine pancreatic lipase immunoreactivity
- cTLI
canine trypsin like immunoreactivity
- DGGR
1,2‐O‐dilauryl‐rac‐glycero glutaric acid‐(6′‐methylresorudin) ester
- DM
diabetes mellitus
- ER
endoplasmic reticulum
- HAC
hyperadrenocorticism
- ICAM‐1
immunoglobulin‐like cell adhesion molecule 1
- IL
interleukin
- KBr
potassium bromide
- LAMP‐1
lysosomal membrane protein 1
- LAMP‐2
lysosomal membrane protein 2
- LFA‐1
leukocyte function antigen‐1
- MPTP
mitochondrial permeability transition pores
- NEFA's
nonesterified fatty acids
- NF‐κB
nuclear factor‐kappa beta
- OR
odds ratio
- PB
phenobarbital
- PSTI
pancreatic secretory trypsin inhibitor
- ROS
reactive oxygen species
- SPINK1
serine protease inhibitor Kazal type 1
- STAT3
signal transducer and activator of transcription 3
- TNF‐α
tumor necrosis factor alpha
- UPR
unfolded protein response
1. INTRODUCTION
The etiology and pathogenesis of spontaneous pancreatitis are poorly understood in both humans and dogs and data is largely extrapolated from experimental models and observations in clinical disease. Experimental models provide mechanistic insights; however, spontaneous disease is likely far more complex and dependent on multiple genetic and environmental factors, some of which might be unknown. Additionally, animal models provide species‐specific or etiology‐specific data, which might not be directly relevant to spontaneous disease. Clinical observations are also not without limitations, and the complexity of spontaneous disease often limits determination of causation. Furthermore, discrepancies in diagnostic standards and evolution of these criteria over time likely further complicate interpretation of observational data in companion animal species. In this review we will utilize a combination of data types to review the latest understanding of the etiology, risk factors, and pathogenesis of pancreatitis in dogs and their relation to clinical disease.
2. ETIOLOGY AND POTENTIAL RISK FACTORS FOR PANCREATITIS
The etiology of pancreatitis is much better understood in humans than in dogs. A systematic review and meta‐analysis revealed that gallstones were the most frequent cause of acute pancreatitis in humans, followed by alcoholic pancreatitis (with geographical differences). 1 The third most common cause is idiopathic acute pancreatitis (AP), which is suspected to be the most common cause in dogs. 1 It is important to note that research into the etiology of pancreatitis in dogs is limited. The term cryptogenic AP, might also, more accurately reflect the level of diagnostic investigation commonly performed into specific etiologies in dogs. In other words, pancreatitis in dogs might be classified as idiopathic, not because there is no underlying cause, but because the potential underlying causes have not been sufficiently studied.
Many factors have been identified as potential risks for AP in dogs. These factors are often established based on a temporal association with the onset of clinical signs, and it is important to consider that these risk factors might not represent a causative relationship. These risk factors can be broken down into: dietary factors, lipid disorders, drug and toxins, endocrinopathies, hereditary/breed predispositions, and miscellaneous causes (see Table 1).
TABLE 1.
Suggested risk factors for pancreatitis in dogs
| Category | Potential risk factor |
|---|---|
| Dietary factors | High fat diet 2 , 3 , 4 |
| Ingestion of unusual food items 5 , 6 | |
| Ingestion of table scraps 5 | |
| Ingestion of trash 5 | |
| Drugs/toxins | L‐asparaginase 7 , 8 |
| Phenobarbital and potassium bromide 9 , 10 , 11 , a | |
| Azathioprine 12 , 13 , 14 | |
| Potentiated sulfonamides 15 | |
| Organophosphates 12 , 16 | |
| Corticosteroids, 12 , 17 , b | |
| Furosemide 12 | |
| Atovaquone/proguanil (Malarone) 18 | |
| N‐methyl‐glucamine (Meglumine), 19 , b | |
| Clomipramine 20 | |
| Zinc 21 | |
| Endocrinopathies | Hyperadrenocorticism 12 , 22 , 23 , a |
| Hypothyroidism 12 , a | |
| Diabetes mellitus, 12 , 24 , 25 , a , c | |
| Hereditary/breed predispositions | SPINK 1 mutation, 26 , 27 , b |
| Acute: Terrier breeds, miniature poodles, dachshunds, cocker spaniel, Alaskan malamute, laika, miniature schnauzer 12 , 28 , 29 , 30 , 31 | |
| Chronic: Cavalier King Charles spaniel, collies, boxers 32 | |
| Lipid disorders | Hypertriglyceridemia 28 , 29 , 33 |
| Miscellaneous | Babesiosis 34 , 35 , 36 |
| Canine monocytic ehrlichiosis 37 | |
| Schistosomiasis (Heterobilharzia americana) 38 , 39 , 40 | |
| Honeybee envenomation 41 | |
| Organic acidemias 42 | |
| Immunoglobulin G4‐related disease 43 , 44 | |
| Increasing age 12 | |
| Obesity/overweight status 5 , 12 | |
| Neutered status 5 , 12 | |
| Previous surgery 5 | |
| Hepatitis/cholangitis 45 |
Note: Potential risk factors for AP in dogs. Many of these factors are implied by a temporal association alone and causation has not been established for many of these factors. Additionally, various definitions and indicators of AP were utilized in the referenced studies and clinical signs of AP were not always noted, thus some of these risk factors might represent risk factors for subclinical pancreatic injury rather than primary clinical AP. The relationship between the proposed risk factors and pancreatitis is often challenging to determine clinically and for some risk factors the direction of causation cannot be determined.
aMay be due to secondary lipid abnormalities.
Contradictory evidence exists.
Reverse direction of causation has been suggested.
2.1. Dietary factors
High fat diets induce or worsen the severity of pancreatitis in dogs leading to the belief that high‐fat diets are a predisposing factor to AP. 2 , 46 Unfortunately consensus criteria regarding what constitutes a high fat diet is lacking. However, a low‐fat diet is considered to contain less than 20% fat on a metabolizable energy basis. 47 Animal models document associations with high protein and fat diets in the development of AP. 48 , 49 , 50 , 51 Further potential evidence for the role of high‐fat diets in the development of AP comes from the results of a study evaluating ketogenic diets in the management of idiopathic epilepsy. 3 In that study 3/9 dogs fed a ketogenic diet (57% fat, 5.8% NFE, 25% crude protein; as dry matter) developed pancreatitis whereas only 2/31 dogs fed the control diet (16% crude fat, 54% NFE, 25% crude protein; as dry matter) developed pancreatitis. 3 This study is published in abstract form and information needed to calculate the fat content on an energy basis is unavailable. Additionally, the method of pancreatitis diagnosis is not reported. A more recent study prospectively evaluated the effect of dietary composition on serum concentrations of canine pancreatic lipase immunoreactivity (cPLI). 52 In that study there was no difference in serum cPLI concentration between a maintenance dog food (4.01 g of fat/100 kcal, 16% crude fat) and a low fat dog food (1.55 g of fat/100 kcal, 5% crude fat), but it is important to note that the dogs were all healthy dogs and that none of the diets are considered high in fat. 52 While the study investigated the effect of dietary fat content on serum cPLI concentration, this study cannot be extended to infer the effects of high fat diets on the development of pancreatitis. In another study 2/50 dogs receiving a struvite dissolution diet (5.7 g/100 kcal, 26.3% crude fat) were noted to develop pancreatitis. 4 A control group was not used as part of the study design and the basis for a diagnosis of pancreatitis is not reported. Given the current literature definitive conclusions on the significance of high levels of dietary fat in the development of pancreatitis cannot be made. Controlled clinical trials are required to evaluate this potential relationship. Further studies are also required to determine the influence of the type of fat on AP in dogs, given differences in endoplasmic reticulum stress noted in in vitro studies. 53 The role that diet plays in the development of AP is also complicated by its association with other predisposing factors including obesity and hypertriglyceridemia. Two retrospective studies have identified potential relationships between being overweight and development of AP. 5 , 12 In a retrospective study 39/101 dogs diagnosed with AP were overweight and being overweight is associated with a 1.3× increased risk of AP. 5 , 12 Unusual food items (odds ratio [OR]: 4.3), table scraps (OR: 2.2) and access to the trash (OR: 13.2) increases the risk of pancreatitis in dogs. 5 , 6 It should be noted that this study is an association study and thus the level of evidence for a causative relationship should be considered weak. Additional factors other than dietary fat alone might modulate the disease risk toward pancreatitis and severe disease in a given dog. Potential factors might include the level of intrapancreatic and visceral fat present and its composition. 54 , 55 This phenomenon is observed in people with alcoholic pancreatitis. 56 While a certain daily consumption of alcohol is a significant risk factor for developing acute pancreatitis, certainly only the minority of human patients with such alcohol consumption develop pancreatitis. 56 Phosphate status (eg, hypophosphatemia) is a risk modifying factor in alcohol induced pancreatitis in people. 57 Unfortunately, these risk modifying factors are poorly understood in dogs fed a high fat diet.
2.2. Lipid disorders
Hypertriglyceridemia is commonly investigated as a cause of AP given the results of ex vivo and in vivo studies. Triglycerides are hydrolyzed by pancreatic lipase, thus high levels of triglycerides might result in excessive production of free fatty acids which are toxic to pancreatic acinar cells. 58 Addition of triglycerides to a perfused canine pancreas results in structural changes including edema, hemorrhage, and weight gain suggestive of pancreatic injury. 59 Clinical studies evaluating the correlation between hypertriglyceridemia and AP have been focused on the miniature schnauzer. Miniature schnauzers with a history of pancreatitis are more likely to have hypertriglyceridemia than those without a history of pancreatitis (77% vs 33%) and there is an association between hypertriglyceridemia and elevated serum cPLI concentrations. 28 , 29 Additionally, severe hypertriglyceridemia (≥862 mg/dL) is associated with a 4.5× increased risk of an elevated cPLI concentration. 29 While the relationship between hypertriglyceridemia and AP has been described as bidirectional, there is a low prevalence of hypertriglyceridemia (18%) in dogs with AP. 33 This prevalence is lower than in earlier studies and likely reflects the study design, which controlled for causes of secondary pancreatitis in addition to medication and dietary factors that could affect serum triglyceride concentrations. Although the prevalence of hypertriglyceridemia was significantly different between the AP (18%) and the control (7.5%) group in that study, the magnitude of hypertriglyceridemia was low (median 67 mg/dL vs 54 mg/dL). 33 The results of these studies suggest that severe hypertriglyceridemia is unlikely to occur secondary to AP alone, and if severe hypertriglyceridemia is documented in a dog, it likely reflects a predisposing factor to AP and could be considered a therapeutic target. Of note, many dogs with hypertriglyceridemia do not develop pancreatitis, and risk modifying factors are unfortunately poorly understood.
2.3. Drugs/toxins
Several drugs and toxins are implicated in the development of AP. In human patients several different classification systems have been developed for drug associated pancreatitis. Initial systems were based on 3‐tiers with the relative weighting being determined by the number of cases and data from drug re‐exposure (if performed). 60 , 61 More‐recently, the Badlov classification system, utilizing a 4‐class system, was introduced. 62 Class I drugs are classified as those with at least 1 case report documenting recurrence of AP with re‐exposure to the drug. 62 Class II drugs are classified as those with a consistent latency (ie, time from initiation of treatment to development of disease) in 75% or more of reported cases. 62 Class III drugs are classified as drugs with 2 or more case reports, but no consistent latency period or rechallenge reports. 62 Class IV drugs are similar to Class III drugs. Except they have only 1 documented case report. 62 It is the authors' opinion that further detailed reports of cases of potential drug‐associated pancreatitis are needed before the development of a veterinary classification scheme.
L‐asparaginase is associated with pancreatitis in up to 16.2% of children being treated for acute lymphocytic leukemia or undifferentiated leukemia. 63 L‐asparaginase depletes the body of L‐asparagine, thus reducing protein synthesis, which also affects the pancreas and might predispose to AP. 64 Eleven of 52 dogs evaluated at various stages of a CHOP protocol for canine large‐cell lymphoma developed clinical signs compatible with pancreatitis; however, the serum cPLI concentrations were not consistently increased, suggesting that the gastrointestinal signs that occurred were likely due to gastrointestinal toxicoses secondary to chemotherapy rather than drug‐induced AP. 7 In the same study, 14% of dogs that received concurrent L‐asparaginase and vincristine had serum cPLI concentrations ≥400 mg/dL; however no clinical signs of pancreatitis were noted. 7 There was no statistically significant difference in serum cPLI concentrations before and after vincristine therapy, suggesting that L‐asparaginase was the cause of any subclinical increase of cPLI. 7 A further case study reported suspected L‐asparaginase induced pancreatitis 2 days after therapy. In that case report the diagnosis of AP was made based on the presence of consistent clinical signs in conjunction with an increased serum cPLI concentration, although abdominal ultrasonography was normal. 8 Further studies are required to determine the incidence and clinical significance of AP in dogs treated with L‐asparaginase.
Several studies suggest a relationship between anticonvulsant therapy and AP. Between 10.3% and 37.0% of dogs with idiopathic epilepsy being treated with potassium bromide (KBr) have biochemical variables and a clinical history consistent with pancreatitis. 9 This study was performed prior to the commercial development of cPLI assays. A group of researchers measured cPLI concentrations in 337 serum samples that had been submitted for measurement of serum phenobarbital (PB) levels, KBr levels, or a combination of both in dogs. 10 In that study, there was an increased risk of an elevated cPLI concentration in dogs being treated with KBr, PB, or a combination of PB and KBr. 10 Given the study design, no information was available regarding clinical signs or diagnostic imaging findings, potentially limiting its application. An additional study documented that 6.8% of dogs receiving PB or KBr have an elevated cPLI concentration. 11 One potential confounding factor is that long term treatment of dogs with PB with or without KBr is associated with the development of hypertriglyceridemia in 33% of dogs, which might be an independent risk factor for pancreatitis. 65 The combined results of these studies suggest that a clinically relevant proportion of dogs receiving PB, KBr, or a combination of both might develop increases in serum cPLI concentrations.
Azathioprine is implicated in the development of AP based on data in humans in addition to ex vivo models and isolated reports in dogs. Exposure of an ex vivo perfused canine pancreas to azathioprine results in marked effects on pancreatic function, as determined by secretory volume, bicarbonate and trypsin output. 66 Acute pancreatitis has been documented in 2 case reports of dogs receiving azathioprine in conjunction with prednisone for various immune mediated disorders. 13 , 14 Acute pancreatitis was suspected to be related to the azathioprine and not the prednisone in both reports due to improvement in clinical signs after discontinuation of the drug; however, repeat exposure was not performed and as such causation is difficult to prove. These reports were published before the development of commercial cPLI assays. Azathioprine was also noted in the history of 2 dogs, but in conjunction with corticosteroids, in a retrospective study evaluating risk factors for AP in dogs. 12 While additional studies are needed to fully determine the risk profile of azathioprine with regard to AP, the authors suggest that dogs that develop gastrointestinal upset or abdominal discomfort while receiving azathioprine, should be evaluated for pancreatitis.
Knowledge regarding the role of exogenous steroids in the etiology of AP is perpetually evolving. Early studies documented an increase in serum lipase activity after steroid administration; however, postmortem examination rarely revealed evidence of pancreatitis, leading to the suggestion that serum lipase activity was an unreliable marker of AP in dogs receiving corticosteroids. 67 , 68 Histopathology might however miss focal areas of inflammation and this limitation should be considered. In another study 18/101 dogs received corticosteroids within 7 days of diagnosis of AP and in a study of dogs with “pancreatic abscesses” (peri‐pancreatic fluid accumulations) 9/36 received corticosteroids prior to admission. 12 , 69 Given these seemingly contradictory results several additional studies have been performed utilizing cPLI assays. Six young female dogs with X‐linked hereditary nephritis receiving 2.2 mg/kg/d prednisone PO for 4 weeks had no significant change in serum cPLI concentration. 70 Another study evaluated the effects of 4 mg/kg prednisolone daily for 2‐3 weeks in 6 healthy beagle dogs. This study revealed an increase in cPLI concentration after prednisolone therapy; however, no ultrasonographic signs of pancreatitis were noted with the exception of 1 day when a single dog was assessed to have a hypoechoic pancreas. 71 Additionally, laparoscopic pancreatic biopsies noted no histologic evidence of pancreatitis. 71 The same caveat as above exists regarding the sensitivity of histopathology in the diagnosis of AP. The same research group evaluated the effects of 2.2 mg/kg prednisolone daily in dogs with various immune mediated diseases. In this study, 5/10 dogs showed serum cPLI concentrations ≥400 mg/dL; however, no clinical signs of pancreatitis were noted. 17 The potential contribution of the primary disease process to the cPLI concentration is unclear. A recent abstract also documented no effect of chronic administration of supraphysiologic doses of corticosteroids on cPLI concentration in 15 dogs. 72 Administration of corticosteroids prior to referral also had no effect on serum cPLI activities in a recent study of dogs with intervertebral disc disease. 73 The effect of exogenous corticosteroids on 1,2‐O‐dilauryl‐rac‐glycero glutaric acid‐(6′‐methylresorudin) ester (DGGR) lipase assays has also been evaluated. 74 In 17 dogs being treated with corticosteroids for several reasons DGGR lipase activity increased following initiation of treatment and reduced during drug tapering. 74 The magnitude of increase in DGGR lipase activity was however low, potentially reducing the clinical significance of this finding. 74 The combined results of the above studies suggest that exogenous corticosteroids are unlikely to result in clinically important AP in dogs, although markers of subclinical pancreatic injury/inflammation might be noted in some cases, and might be more importantly influenced by concurrent disease. Additionally, corticosteroids are being increasingly utilized at anti‐inflammatory doses in the management of AP in dogs. In 1 study a 1 mg/kg dose of prednisolone was shown to reduce C‐reactive protein concentrations, shorten time to clinical improvement, and decrease mortality in spontaneous AP in dogs. 75 A recent review, evaluating data from multiple species, suggested that corticosteroids might have a positive effect on outcome in the treatment of acute or acute on chronic pancreatitis in dogs. 76
Several early studies documented a potential relationship between organophosphates and the development of AP. Sublethal doses of organophosphate anticholinesterase ex vivo results in AP in dogs as determined by histopathology. 16 Additionally in vitro acetylcholinesterase inhibitors result in marked increases in pancreatic amylase output. 77 Five dogs were also noted to have a recent exposure to organophosphate insecticides prior to the development of AP in a large retrospective study. 12 Organophosphates might therefore be a risk factor for the development of pancreatitis in dogs, but more studies are needed to confirm this suspicion.
Potentiated sulfonamides, furosemide, clomipramine, atovaquone/proguanil (Malarone), and n‐methyl‐glucamine (Meglumine) are associated with the development of AP in isolated dogs. 12 , 15 , 18 , 19 , 20 Another prospective study reported that none of 20 dogs treated with meglumine and allopurinol developed increased cPLI concentrations or clinical signs consistent with pancreatitis. 78 Isolated reports of toxins such as zinc causing AP have been reported. 21 Various antibiotics, chemotherapeutics, and endocrine therapies were also noted within 7 days of presentation, in a large retrospective study of AP; however, case details were sparse thus limiting the clinical utility of these data. 12 Given the infrequent nature of these reports many drug reactions are suspected to be idiosyncratic. Nonetheless a thorough drug and toxin history should be taken in cases of suspected AP.
2.4. Endocrinopathies
Numerous endocrinopathies have been reported as risk factors for AP, including hyperadrenocorticism (HAC), hypothyroidism, and diabetes mellitus (DM). 12 , 22 , 23 , 24 , 25 , 79 , 80 A retrospective study documented a preexisting or subsequent diagnosis of HAC in 12/101 dogs with AP. 12 It is important to note that this study did not include a control group. Although large prospective studies have not been performed to evaluate this relationship, recent studies suggest a potential relationship between HAC and higher cPLI concentrations and DGGR lipase activities in the absence of clinical pancreatitis. 22 , 23 Given that most studies evaluating exogenous steroids document only minor elevations in pancreatic lipase we suspect that increased pancreatic lipase concentrations in dogs with HAC reflect subclinical pancreatic injury/inflammation that has yet to reach a threshold, required to cause clinical pancreatitis (see Figure 1A). Additional studies are needed to evaluate whether there is a relationship between AP and HAC in dogs and the direction of the relationship. Hypothyroidism was documented in 4/101 dogs diagnosed with AP in a retrospective study, and it has been hypothesized that secondary hyperlipidemia might be responsible for this relationship. 12 A previous diagnosis of hypothyroidism was also noted in a retrospective study of dogs with extrahepatic bile duct obstruction secondary to AP. 80
FIGURE 1.
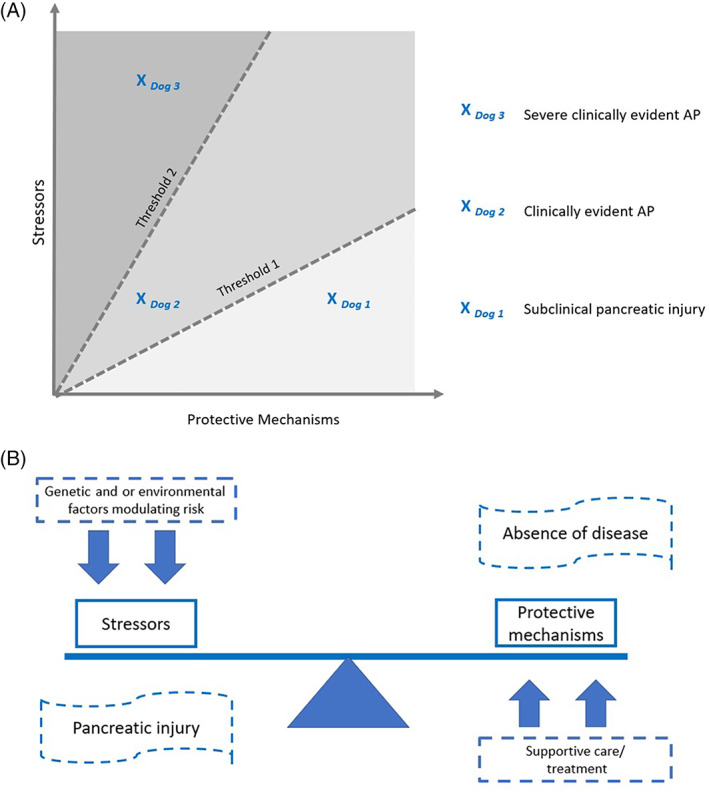
(A) Thresholds in the development of pancreatitis. A graphical representation of the critical threshold theory in which the balance between stressors and protective mechanisms determines whether clinical disease is present (threshold 1) and if so, its severity (threshold 2). Dog 1 and dog 2 have been exposed to the same stressor; however, dog 1 has greater protective mechanisms and as such, the disease burden falls below threshold 1 and the dog develops subclinical pancreatic injury. In contrast, dog 2's disease burden is above threshold 1 and the dog develops clinically evident AP. Similarly, dogs 2 and 3 have the same level of protective mechanisms, but the stressors are greater in dog 3. The disease burden for dog 3 is therefore above threshold 2 and the dog develops clinically severe AP. As depicted in (B) this balance is also likely influenced by factors such as supportive care, genetic and environmental factors, and amplification systems. While thresholds 1 and 2 are represented as dashed lines in the figure, we suspect that the thresholds are more fluid and vary between individuals. This figure was adapted with permission from Barreto et al. Gut. 2021;70:194‐203. (B) A balance between protective mechanisms, stressors, and modulating factors determines the risk of pancreatitis in each individual dog. A schematic representation of the fine balance between stressors and protective mechanisms that determine whether pancreatic injury and inflammation occurs in an individual dog. Note that genetic and or environmental factors on one side and supportive care on the other can modulate the risk toward or away from pancreatic injury, respectively
Many studies have identified pancreatitis concurrently with DM in dogs and this might represent shared risk factors for disease or a causative relationship between the 2 diseases. 12 , 25 , 30 , 81 Pancreatitis has also been shown to increase the hazard risk of death in dogs with DM. 81 Proposed mechanisms by which pancreatitis could theoretically lead to DM include: islet cell damage, glucose toxicity, initiation of autoimmune injury, inflammation induced insulin resistance, and the action of cytokines and adipokines. 24 One study indicated that 5/18 dogs with diabetes mellitus had histopathologic features consistent with advanced chronic pancreatitis, which was the presumed cause of DM in those dogs. 82 Additionally, in a study utilizing glucagon stimulation tests in 6 dogs with presumed chronic pancreatitis, 5/6 had reduced ß cell response and glucose intolerance. 83 Proposed mechanisms in which DM could in theory lead to pancreatitis include the development of auto‐antibodies or anti‐insulin antibodies. 24 Although the relationship might be bidirectional, many clinicians suspect that the predominant direction of the relationship is pancreatitis leading to DM. 24 Further discussion on the relationship between pancreatitis and DM is available. 24
2.5. Breed associated risk and potential hereditary pancreatitis
Certain breeds are reported to be at greater risk for the development of AP. These predispositions likely reflect a combination of direct genetic predispositions, a predisposition to a secondary factor such as hypertriglyceridemia, or other disease modifying factors. The most consistently reported at‐risk breeds for acute pancreatitis include the miniature schnauzer, miniature poodle, Yorkshire terrier, and other terrier breeds. 5 , 12 , 84 Other predisposed breeds include dachshunds, poodles, cocker spaniels, Alaskan malamutes, and laika. 30 Two potential explanations are suggested for the high incidence of AP in miniature schnauzers. One potential explanation is that AP occurs secondary to hypertriglyceridemia, which occurs commonly in this breed. 28 , 29 A second potential explanation is that AP might be heritable in the breed due to mutations in the serine protease inhibitor Kazal type 1 gene (SPINK1). The product of the SPINK1 gene, pancreatic secretory trypsin inhibitor (PSTI), is suspected to play a protective role in the prevention of premature zymogen activation. 85 A prospective study evaluated the presence of SPINK1 gene variants in 39 Miniature Schnauzers with pancreatitis, 25 healthy Miniature Schnauzers, and 23 healthy dogs of other breeds. 26 In that study 3 variants were identified, including 2 missense mutations in the second exon (N20K and N25T) in addition to a poly T insertion in the 3rd intron (IVS3 + 26‐27ins[T]33‐39,15_61dup11) and these variants were shown to correlate with the development of pancreatitis in this breed. 26 However, a subsequent study detected a high prevalence of SPINK1 variants in Miniature Schnauzers with and without pancreatitis and a relationship between the SPINK1 variant and clinical pancreatitis could not be established. 27 In this study however, diagnosis of pancreatitis was based solely on a semiquantitative point of care assay in approximately 30% of dogs, which might have affected the results of the study. 86 Additional potential explanations for the lack of an association between the SPINK1 variant and clinical disease is that the mutation might not be sufficient in isolation to cause pancreatitis and might require concurrent environmental or other genetic factors. Additional studies are therefore required to determine the potential role of SPINK1 variants and other gene mutations in AP in dogs. 87 Cavalier King Charles spaniels, collies and boxers are predisposed to chronic pancreatitis (CP). 32
2.6. Autoimmune pancreatitis IgG4‐related disease
The English Cocker spaniel is suggested to have a distinct form of CP characterized by ductal destruction, interlobar fibrosis, and T lymphocyte infiltrations on histopathology. 88 Immunohistochemistry has also shown a predominance of IgG4 positive plasma cells in multiple tissues, including the pancreas, suggesting the presence of a multiorgan immune mediated disease similar to IgG4‐related disease in human patients. 89 Many of these dogs have concurrent keratoconjunctivitis sicca, xerostomia, proteinuria, and other immune‐mediated disorders. 90 The diagnostic criteria for IgG4‐related disease(s) in dogs needs further clarification and consensus before a definitive diagnosis can be confirmed. 43 , 91 Also serum IgG4 concentrations have been noted to be high in other dogs with pancreatitis. 44 However, strict diagnostic criteria are required to prevent misclassification of other inflammatory or neoplastic diseases as IgG4‐related disease. 43 It is hoped that ongoing study will further help to characterize this form of CP in dogs.
2.7. Miscellaneous risk factors
Many miscellaneous risk factors have been proposed for AP in dogs, including weight/neuter status, infectious diseases (eg, Babesia spp., Heterobilharzia americana, Leishmania infantum), hepatitis, honeybee envenomation, snake envenomation, hypercalcemia and organic acidemias. 5 , 12 , 41 , 42 , 45 Although dogs of any age can be affected by AP, most dogs are middle‐aged to older. 12 , 30 , 31 , 92 Overweight animals and those who are neutered are also reported to be predisposed to AP. 5 , 12 , 84 , 93 It is however, important to note that many of these factors are influenced by additional factors, such as diet, lifestyle, and exercise level. 94 A retrospective study revealed clinicopathologic evidence of AP in 16 dogs and histological evidence of AP in 4 dogs diagnosed with a Babesia rossi infection. 34 An additional study documented that 28% of dogs with a B. rossi infection had an increased serum cPLI concentration. 36 Other babesia species (eg, B. gibsoni) have been implicated in the development of AP, albeit at a lower prevalence. 35 Potential mechanisms include hypotension, secondary immune‐mediated hemolytic anemia, hemoconcentration, and alterations in lipid metabolism. 34 Canine monocytic ehrlichiosis has also been reported to be a predisposing factor to AP in dogs, with 20% of cases in a recent study having subclinical elevations in cPLI. 37 Necropsy studies of canine schistosomiasis (ie, Heterobilharzia americana infection) also document a high prevalence of parasitic infiltration of the pancreas. 38 Gross lesions consistent with pancreatitis were also noted during exploratory laparotomy in a dog subsequently diagnosed with schistosomiasis. 39 Several studies also noted elevated pancreatic lipase concentrations in systemically ill dogs such as those with cardiac disease, gastric‐dilatation and volvulus, intervertebral disc disease, parvovirus, and foreign bodies. 73 , 95 , 96 , 97 , 98 , 99 In the authors' opinion elevations in pancreatic lipase concentrations in such dogs likely reflect a secondary pancreatic injury that might not be clinically relevant, although this can be difficult to definitively determine. But, in some diseases (eg, chronic enteropathies) increased serum cPLI concentrations have been associated with a worse outcome. 100
Previous surgery, other than neutering, is associated with an increase in risk of AP (OR: 21.1), likely due to hypotension, tissue manipulation, or a combination of both. 5 A high prevalence of AP has also been noted in a recent retrospective study of dogs and cats with cholecystitis, although the direction of the relationship is unknown. 45 Therefore, many factors in a dogs history should be considered when evaluating a dog with suspected AP.
3. PATHOGENESIS OF ACUTE PANCREATITIS
Over 100 years ago it was proposed that pancreatitis was caused by autodigestion of the pancreas from premature activation of digestive zymogens. 101 Since that time several experimental and clinical studies have documented a key role of intra‐acinar trypsinogen activation. 102 , 103 , 104 , 105 , 106 , 107 , 108 Despite the long‐standing concept of premature trypsinogen activation, the initiating cellular mechanisms and the potential role of alternate or complementary pathways have long been the source of ongoing research. While discussing these concepts we will relate the results of experimental data to implications for clinical practice by highlighting potential therapeutic targets. Development of such therapeutics will be critical in improving the outcome of this disease for which clinicians have traditionally been restricted to providing only supportive and symptomatic care.
In this section we will also review studies that suggest the presence of trypsinogen independent pathways of acinar cell injury and pancreatic and systemic inflammation. We will then discuss several proposed mechanisms that might initiate both trypsin‐dependent and independent cellular damage. It is important to consider that the described pathogenic mechanisms do not always result in clinical disease and that the critical threshold theory (Figure 1A) is useful to help describe the relationship between pathologic stressors and clinical disease expression and severity (Figure 1B).
In 2011, a study utilizing trypsinogen isoform‐7 gene knockout mice and wild type mice to determine the role of trypsinogen in experimentally‐induced pancreatitis (cerulein model) was published. 109 Trypsinogen activation led to in vitro acinar cell death during early pancreatitis, which was responsible for ~50% of pancreatic injury. 109 Interestingly, however, the knockout mice demonstrated similar levels of local and systemic inflammation when compared to the wild‐type cohort. 109 The results of this experimental study suggested that the inflammatory response and approximately 50% of acinar cell damage during pancreatitis was independent of premature trypsinogen activation. 109 This prompted reconsideration of the traditional trypsin‐centric pathogenesis and allowed for consideration of potential alternate or complementary pathways. One potential caveat to this is that mice have various trypsinogen isoforms, and the clinical significance of each isoform might vary. A further study documented that intra‐acinar activation of trypsinogen was able to induce AP but not CP, suggesting that different pathogenic mechanisms might exist between acute and chronic disease. 108 Although trypsin‐independent mechanisms for pancreatitis have been suggested, they likely act in parallel with trypsin‐dependent mechanisms, therefore therapeutic manipulation of trypsin and other proteases might still be of benefit. Reduced trypsinogen activation peptide concentrations, reduced pancreatic necrosis, and reduced mortality is seen in rats treated with the protease inhibitor nafamostat. 110 Multiple studies have also investigated protease inhibitors in humans, with varied, but most often disappointing, results. 111 , 112 , 113 Concern also exists regarding the vasoconstrictive properties of these drugs and the potential perpetuation of pancreatic necrosis. 114 Additionally, proteases other than trypsin, might play protective roles in the pathophysiology of pancreatitis. Chymotrypsin is a protease that regulates trypsin activity and is noted in early pancreatic injury. 109 Chymotrypsinogen knockout mice have been shown to have an increased severity of induced pancreatitis, when compared to a wild type cohort, thus protease inhibitors used in pancreatitis should ideally target trypsin, but spare chymotrypsin. 115 Somatostatin analogues have the potential of inhibiting exocrine secretions of the pancreas, including trypsinogen, thus these drugs have also been considered in disease management. Unfortunately, the somatostatin analogue, octreotide, is shown to have no benefit in an experimentally induced (bile injection model)pancreatitis in dogs. 116 Indeed, it is suspected that zymogen granule release is inhibited early during the pathogenesis of pancreatitis, a process that would be expected to be even further propagated by somatostatin or its therapeutic analogues. 117 Given the results of the above studies, and the presence of trypsin‐independent mechanisms of pancreatic injury and inflammation, it is likely that detailed preclinical study would be needed before protease inhibitors or somatostatin analogues are considered for clinical use in dogs.
A major alternative or complementary pathway to the trypsinogen activation pathway of AP includes activation of nuclear factor‐kappa beta (NF‐κB), which has been documented in pancreatitis for over 20 years. 118 Intra‐acinar NF‐κB activation occurs alongside, and independent of, trypsinogen activation. 109 , 119 , 120 The critical role of NF‐κB in the inflammatory response is evidenced by experimental studies in knockout mice, where absence of NF‐κB activation results in reduced pancreatic inflammation relative to wild type mice. 121 , 122 , 123 The level of NF‐κB activation correlates with the severity of pancreatitis in mice. 124 Nuclear factor‐κB is also involved in the systemic inflammatory response to pancreatitis. 125 , 126 While these studies lay strong evidence for a proinflammatory action of NF‐κB in pancreatitis, some data suggests that NF‐κB might play a multifaceted role in AP, and might be protective in some circumstances. 127 , 128 , 129 These discrepancies can also be explained with different approaches in the preclinical models (overactivation vs genetic deletion). Additionally, currently used inhibitors are not specific for the NF‐κB pathway. Furthermore, inhibition of single subunits of the pathway might be compensated for by other components of the NF‐κB family of proteins, such as RelB or c‐Rel. Given the results of these studies targeted inhibition of the proinflammatory properties of NF‐κB could be of therapeutic benefit but requires further study. Other key components of the inflammatory response include tumor necrosis factor alpha (TNF‐α), interleukin (IL)‐1, IL‐1ß, IL‐2, IL‐6, IL‐8, and IL‐18. 93 , 130 , 131 , 132 , 133 , 134 Association studies correlating cytokines and chemokines with AP severity have been performed to identify surrogate biomarkers as predictors for severe AP. In humans serum levels of IL‐6 and the IL‐6 dependent acute phase protein C‐reactive protein are the most reliable predictors of severe AP. 135 , 136
Interleukin 6 has both anti‐inflammatory and proinflammatory properties depending on the signaling type. Classic signaling requires the presence of an IL‐6 receptor on a limited number of target cells and mediates its anti‐inflammatory properties. 137 Transsignaling however involves the interaction of IL‐6 with a soluble IL‐6 receptor (sIL‐6R) which then mediates gp130 activation, and IL‐6's proinflammatory actions. 137 IL‐6 transsignaling has effects on many cell types and can further increase the expression of signal transducer and activator of transcription 3 (STAT3) in pancreatic acinar cells. 138 Increased STAT3 expression further promotes the inflammatory cascade via release of cytokines and chemokines. 138 Cytokine secretion in macrophages is dependent on nuclear translocation of RelA. 138 Thus IL‐6 has also been shown to have a direct impact on the course of disease (Figure 2). Cytokines and chemokines in the circulation induce further secretion of acute‐phase proteins in the liver, activate complement factors, and activate bradykinin‐kinin systems, thus increasing capillary permeability and in turn leading to hypovolemia and edema.
FIGURE 2.
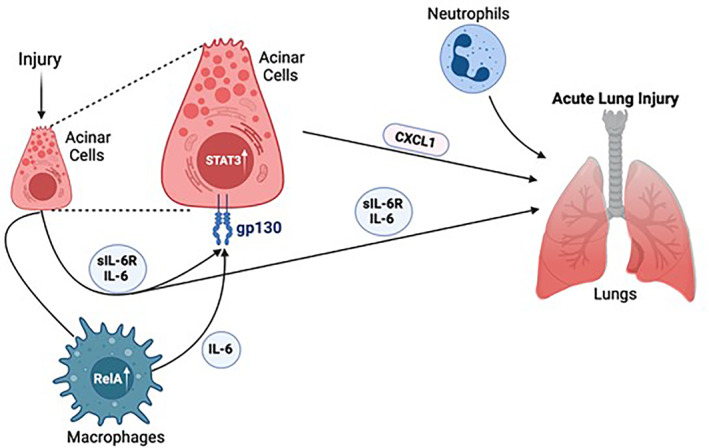
Systemic inflammatory response syndrome in AP. IL‐6 transsignaling involves the interaction between IL‐6 and a soluble IL‐6 receptor which increases gp130‐dependent STAT3 activation. Nuclear translocation of STAT3 results in release of cytokines such as CXCL1 which leads to acute lung injury. Cytokine secretion in macrophages depend on nuclear translocation of RelA
Neutrophils and macrophages also play a key role in both the local as well as systemic inflammatory response in acute pancreatitis. 139 , 140 Neutrophil invasion into the pancreas is suspected to occur due to the release of CXC ligand 2 (CXCL2) and other chemoattractants in early AP. 141 , 142 Experimental depletion of neutrophils results in reduced pancreatic injury and increased survival, highlighting the critical nature of neutrophils in disease pathogenesis. 143 , 144 Neutrophils also play a role in the development of systemic complications of AP including acute lung injury. 145 , 146 Neutrophil extravasation into the tissues is dependent on rolling, activation, adhesion, and migration of neutrophils from the capillaries. Leukocyte function antigen‐1 (LFA‐1), which binds to immunoglobulin‐like cell adhesion molecule 1 (ICAM‐1) during vascular adhesion, plays an important role in the recruitment of neutrophils to the pancreas and other tissues, CXCL2 formation, and subsequent tissue injury (Figure 3). 147 This pathway is amenable to therapeutic intervention and LFA‐1 inhibitors such as fuzapladib sodium hydrate prevent extravasation of neutrophils into the tissue and have been approved for the treatment of pancreatitis in dogs in Japan. Data from a proof‐of‐concept study utilizing an experimental model of pancreatitis in dogs was recently presented at the ACVIM Forum. 148 This data suggests that fuzapladib improved survival rate in this experimental model, in a dose‐dependent fashion. A subsequent clinical trial in Japan revealed improved clinical scores, and a faster resolution of serum C‐reactive protein increases in dogs treated with fuzapladib. 148 A multicenter clinical trial in spontaneous pancreatitis in dogs is ongoing in the United States.
FIGURE 3.
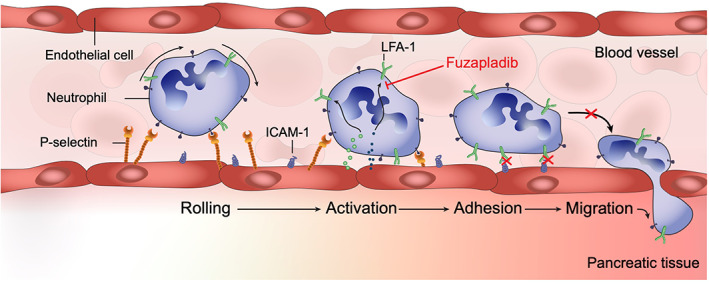
Neutrophil extravasation into the pancreas and therapeutic manipulation with fuzapladib sodium hydrate. Fuzadib sodium hydrate inhibits leukocyte function antigen‐1, which prevents neutrophils from extravasating from capillaries into the surrounding tissue, which inhibits the systemic inflammatory response syndrome (SIRS). ICAM‐1, immunoglobulin‐like cell adhesion molecule 1; LFA‐1, leukocyte function antigen‐1
Multiple mechanisms have been proposed to explain trypsinogen and NF‐κB activation during the early stages of pancreatitis. These mechanisms include deranged calcium signaling, colocalization of zymogens and lysosomes, impaired autophagy, endoplasmic reticulum stress and maladaptive unfolded protein response, mitochondrial dysfunction, and oxidative stress. For simplicity and given the focus of this review, we will discuss each of these mechanisms sequentially; however, there is likely a complex interaction between these mechanisms in spontaneous disease.
Deranged calcium signaling is suspected to play a role in the pathogenesis of AP. Under physiologic conditions a stimulus results in a transient spike in calcium in the apical region of the acinar cell. 149 However, during pancreatitis there is a global and persistent increase in cytosolic calcium. 150 , 151 Calcium overload then causes the premature activation of trypsinogen, in addition to the development of vacuoles, and acinar cell damage (Figure 4). 152 , 153 Pathological calcium overload is associated with release of calcium from the endoplasmic reticulum (ER) and acidic calcium stores, which acts as a positive feedback mechanism. 154 , 155 A variety of receptors are involved in pathologic calcium signaling. 155 , 156 , 157 Calcium overload causes mitochondrial permeability transition pores (MPTP) to open, leading to a loss of mitochondrial membrane potential and mitochondrial dysfunction. 158 , 159 Mitochondrial dysfunction is a key acinar event in early pancreatitis. 160 , 161 Loss of mitochondrial function leads to ATP depletion. Removal of cytosolic calcium is ATP‐dependent, therefore mitochondrial dysfunction might further the global and persistent increase in cytosolic calcium, thus forming a self‐perpetuating increase in cytosolic calcium, which is characteristic of pathological calcium signaling during pancreatitis. 151 , 160 , 161 , 162 Pathologic calcium signaling ultimately results in downstream activation of calcineurin, trypsinogen, and NF‐κB. 163 , 164 Calcineurin knockout mice have been shown to have reduced zymogen activation and acinar cell injury in response to an experimental model of pancreatitis. 165 Additionally, genetic mutations in calcium channels such as transient receptor potential vanilloid subfamily member 6 (TRPV6) have been shown to be associated with nonalcoholic chronic pancreatitis (CP) in human patients. 166 Given these findings, manipulation of calcineurin has garnered considerable attention as a potential therapeutic target for the treatment of pancreatitis in humans. Pharmacologic inhibition of calcineurin via cyclosporine A and tacrolimus has protective effects in a rodent model of pancreatitis. 163 , 165 The protective effects of calcineurin inhibition appear to be dependent on the source of its expression. 167 Studies are ongoing to investigate cyclosporine therapy in both canine and feline CP (personal communication JMS). Given the immunosuppressive properties of cyclosporine it might have multiple beneficial effects in pancreatitis, particularly in those with suspected immune‐mediated etiology. 168 Research into drugs manipulating calcium signaling and mitochondrial dysfunction are ongoing in experimental models and might offer future promise in the management of pancreatitis. 169 , 170 , 171
FIGURE 4.
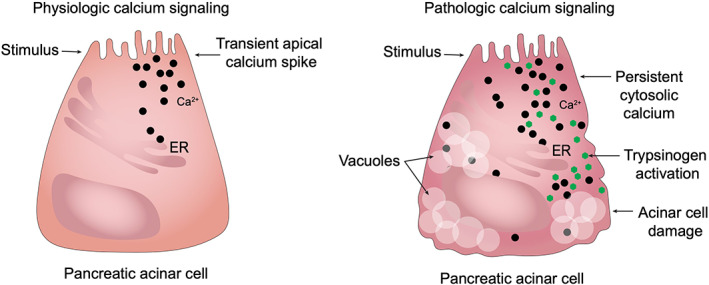
Persistent cytosolic calcium accumulation in AP. Under physiologic conditions there is a transient spike in apical calcium concentration, which results in release of digestive zymogens from the apical border of the pancreatic acinar cell. During AP there is a global and persistent increase in cytosolic calcium concentration, which causes calcium overload, premature trypsinogen activation and subsequent acinar cell damage. Deranged calcium signaling is associated with mitochondrial dysfunction
Under physiologic conditions, digestive enzymes are packaged as inactive enzymes, called zymogens, and are stored in granules. Lysosomal enzymes are packaged into lysosomes, thus preventing the interaction of zymogens and lysosomal enzymes until zymogens enter the duodenum. During pancreatitis, lysosomes redistribute and colocalizes with zymogen granules in the subcellular compartment, resulting in the formation of large vacuoles. 172 , 173 This colocalization allows the lysosomal enzyme cathepsin B to prematurely activate trypsinogen (Figure 5). 174 Maximal activation of trypsinogen by cathepsin B is reported to occur at an acidic pH, but might also occur at a higher pH. 174 , 175 This hypothesis was further evaluated in a cathepsin B gene knockout mouse cohort. 176 In these mice, trypsin activity and pancreatic necrosis in response to cerulein stimulation is markedly reduced relative to the wild‐type cohort, suggesting that cathepsin B plays a key role in the induction of pancreatitis. 176 Absence of the cathepsin B gene did not however completely prevent the onset of pancreatitis. 176 Similar results have been noted in several studies using cathepsin B inhibitors. 177 , 178 While colocalization is a popular theory and is well supported by the literature, there are some studies that suggest that we do not fully understand this process. For example, colocalization of cathepsin B with secretory vesicles is seen in regulated secretion from the exocrine pancreas and redistribution of lysosomal enzymes into the zymogen‐rich subcellular compartment fails to result in pancreatitis in some studies. 175 , 179 Thus, it is suspected that cathepsin B redistribution is involved in pancreatitis, but alone, it might not be sufficient to induce disease and might be influenced by other factors, such as cathepsin D. 180 Cathepsin B gene mutations have also been associated with some forms of pancreatitis in humans, but the role of cathepsin B has yet to be studied in dogs. 181 , 182 Further studies are needed to understand apparent discrepancies in the results of prior studies, and to determine whether these pathways could act as therapeutic targets.
FIGURE 5.
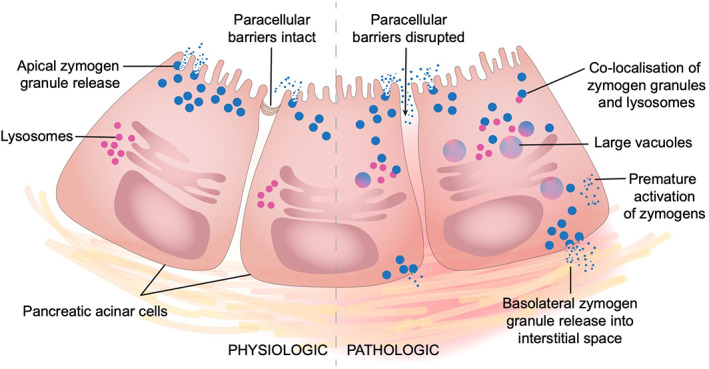
Colocalization theory. Under physiologic conditions, digestive zymogens and lysosomes do not interact with each other within pancreatic acinar cells. During AP an apical block results in redistribution of lysosomes, which then colocalize with digestive zymogens. Colocalization allows the lysosomal enzyme cathepsin‐B to activate the digestive zymogens within the pancreatic acinar cell, resulting in cell damage. Basolateral release of granules might occur leading to damage to the interstitial space
Autophagy is cytoprotective and involves the catabolic removal of various obsolete cellular components preventing cell damage and promoting survival in response to insults. 183 Basal autophagy has been shown to play an important role in the homeostasis of pancreatic acinar cells. 184 Genetic disruption of Atg5 induced pancreatic degradation also supports the role of autophagy in exocrine pancreatic homeostasis. 185 During pancreatitis impaired autophagy occurs as a result of lysosomal dysfunction. 162 Colocalization of organelles is autophagic in nature. 186 Lysosomal dysfunction results in degradation of lysosomal membrane proteins 1 (LAMP‐1) and 2 (LAMP‐2). Lysosomal membrane proteins play an important role in lysosomal stability. The role of LAMP‐2 in pancreatitis is demonstrated in knockout mice, which were shown to have impaired autophagy and developed pancreatitis. 187 Ultimately these processes result in acinar cell vacuolation, trypsinogen activation, inflammation and necrosis. 162 Pharmacological manipulation of impaired autophagy was studied in mice. 188 Inhibition of a transcriptional regulator of autophagy alleviated pancreatic acinar cell injury in experimental pancreatitis. 188 , 189 Further studies are needed to determine whether pharmacological manipulation of autophagy offers promise in the management of AP. Impaired autophagy might also be interrelated with other pathomechanisms of AP including mitochondrial dysfunction and endoplasmic reticulum (ER) stress. 190
The exocrine pancreas is rich in ER, which plays a key role in protein synthesis. Endoplasmic reticulum stress is a key acinar event during early pancreatitis and is associated with accumulation of unfolded proteins in the ER. 191 , 192 , 193 Under physiologic conditions, ER stress is countered by an unfolded protein response (UPR), thus preventing cell injury. 194 This physiologic response involves autophagic processes. 194 During excess and prolonged ER stress, the UPR becomes maladaptive and upregulated NF‐κB triggers proinflammatory genes (Figure 6). 122 , 124 , 194 Thus, dysregulated UPR results in the initiation of acinar cell injury and apoptosis. 194 Interestingly, drugs such as HMG‐coA reductase inhibitors, have been shown to act on the UPR and reduce ER stress. 195 Observational studies also document a lower incidence of pancreatitis in humans treated with HMG‐coA reductase inhibitors (OR: 0.29). 196 Furthermore preexisting statin therapy is associated with a reduced severity of pancreatitis in humans. 197 To the authors' knowledge no studies have been performed investigating the use of HMG‐coA reductase inhibitors in dogs with pancreatitis.
FIGURE 6.
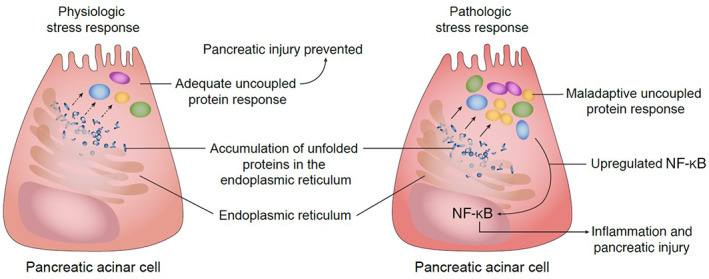
Endoplasmic reticulum stress and the unfolded protein response. Under physiologic conditions endoplasmic reticulum stress is countered by the unfolded protein response, which prevents cellular injury. However, in AP excessive and prolonged ER stress results in a maladaptive UPR and subsequent upregulation of NF‐κB, resulting in acinar cell injury
Oxidative stress has long been implicated in the pathogenesis of pancreatitis and free‐radical concentrations have been correlated with the degree of inflammatory response and disease severity. 198 , 199 , 200 , 201 Antioxidants have been shown to reduce pancreatic damage in experimental models; but this contrasts with the results of clinical trials in spontaneous disease. 202 , 203 , 204 A decisive study might help to explain the lack of efficacy of antioxidants in some clinical trials. 205 Booth et al. showed that reactive oxygen species (ROS) promoted apoptosis of pancreatic acinar cells, and inhibition of ROS resulted in pathologic necrosis, suggesting an unexpected protective role of ROS within the pancreatic acinar cells during pancreatitis. 205 Contrasting findings were noted regarding oxidative stress in neutrophils, suggesting a dual role of ROS in pancreatic injury. 205 , 206 This dual role likely complicates manipulation of oxidative stress pathways. Given these results, further studies are needed to investigate the role of oxidative stress in dogs with pancreatitis before therapeutic manipulation is considered. Studies are being performed at the primary author's institution to evaluate reactive metabolite concentrations, plasma antioxidant potential, and urinary F2‐isoprostane concentrations in dogs with spontaneous AP.
An additional factor implicated in determining the clinical course of AP is the role of nonesterified fatty acids (NEFA's). 54 , 55 Lipolysis of visceral fat by pancreatic lipase results in the formation of NEFA's, which results in systemic inflammation and organ failure (Figure 7). 54 Nonesterified fatty acids have been shown to alter the severity of AP, independent of the necroinflammatory response. 207 Free fatty acids have also been shown to induce hypocalcemia. 208 The mechanism of NEFA‐induced organ failure is suspected to involve inhibition of mitochondrial complex I and V and release of intracellular calcium. 209 The systemic effects of NEFAs have been evidenced by induction of mitochondrial damage in the kidney. 209 , 210 Therapeutic manipulation of these pathways has been considered in the management of AP. Calcium supplementation and Ringer's lactate to counter the hypocalcemia induced by NEFA's reduces C‐reactive protein concentrations and the systemic inflammatory response during early pancreatitis. 211 Additionally, inhibition of lipases is considered a therapeutic target, with the aim of reducing the generation of NEFA's. Orlistat, a lipase inhibitor, reduces organ failure and mortality in experimental AP. 207 Other case reports have suggested that orlistat might induce pancreatitis in some people and thus more studies are needed to investigate the effects of lipase inhibitors on disease outcome before they can be considered for clinical use. 212
FIGURE 7.
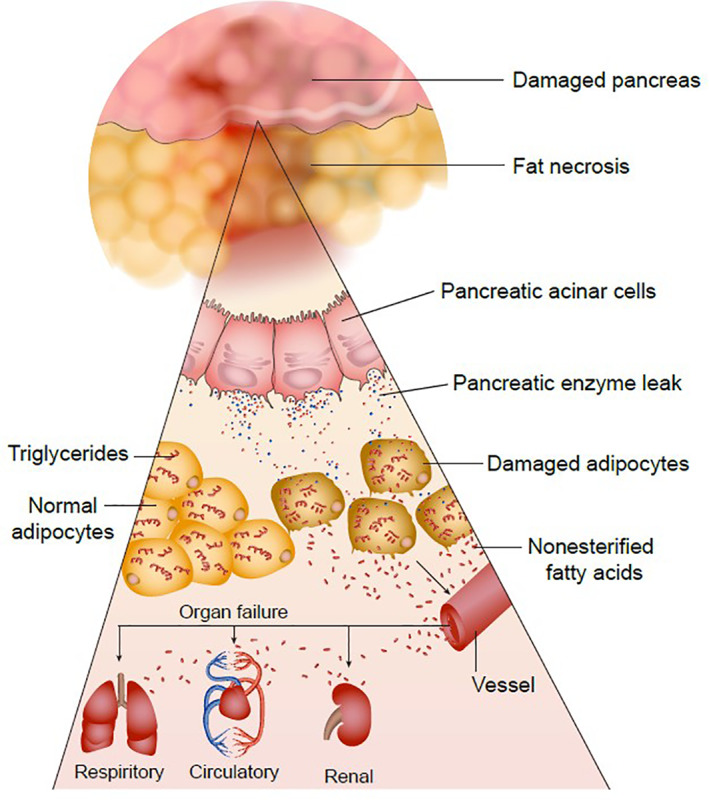
Pancreatic enzyme leakage and the role of fatty acids in the pathogenesis of AP. Leakage of pancreatic enzymes, particularly classical pancreatic lipase, into the intrapancreatic and peri‐pancreatic fat results in the generation of nonesterified fatty acids. These nonesterified fatty acids lead to systemic inflammation and organ failure; thus, the level of visceral adipose tissue might influence the clinical course of AP Source: This figure was adapted with permission from de Oliveira et al. J Clin Invest. 2020;130(4):1931‐1947
Having discussed potential initiating mechanisms of pancreatitis, it is important to note that, the development of pancreatitis and the disease severity is dependent on a balance between stressors and protective mechanisms, with several genetic and environmental factors modifying the individuals risk profile (Figure 1B). 139 Protective mechanisms of the pancreas include: synthesis and storage of digestive enzymes as inactive zymogens, physical separation of lysosomal enzymes from zymogens, pancreatic secretory trypsin inhibitor, suboptimal pH for autoactivation in zymogen granules, unidirectional flow in pancreatic ducts, and circulating antiproteases in the vascular space. 93 Acinar cell injury results in the release of inflammatory mediators, which are then amplified by pancreatic stellate cells resulting in a necroinflammatory amplification loop that furthers acinar cell injury as well as a systemic inflammatory response. 139 A critical threshold of acinar cell injury is likely required prior to the development of clinical disease (Figures 1A,B and 8). 139 This critical threshold theory is important when interpreting abnormalities in pancreatic lipase or imaging findings suggestive of pancreatitis, in the absence of clinical disease. This theory would suggest that while various pathophysiologic mechanisms might be at play in each dog, clinical disease only develops if a certain clinical threshold is reached, and severe pancreatitis occurs only when another threshold is reached. This theory might provide a useful model for pancreatitis in dogs. It is important to note that these thresholds are likely not true thresholds, but rather theoretical ones and that the actual thresholds might vary widely between dogs.
FIGURE 8.
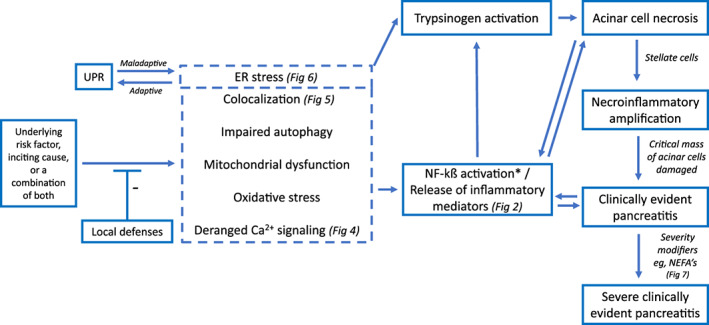
Proposed pathogenesis of pancreatitis. This figure outlines the key pathophysiologic mechanisms proposed in the development of clinically evident acute pancreatitis. UPR, unfolded protein response; *, context specific proinflammatory role Source: This figure was adapted with permission from Barreto et al. Gut. 2021;70:194‐203
4. CONCLUSIONS
While most cases of pancreatitis are thought to be idiopathic, our knowledge regarding the etiology of pancreatitis in dogs is more limited than it is for humans and a thorough diagnostic work‐up evaluating potential risk factors is not always performed. Thus, the term cryptogenic AP might be more appropriate in most cases. A thorough medical, surgical, drug, and dietary history is indicated in all dogs with suspected pancreatitis, and the results of which might be of particular importance in dogs with recurrent episodes of pancreatitis. If a temporal association is noted between a drug and pancreatitis the relationship might be causative or associative. Where it is deemed likely that a drug or other risk factor is associated with pancreatitis, objective markers such as cPLI might be useful to identify reduction in pancreatic inflammation with risk factor elimination. However, repeat exposure would be required to prove causation. Many systemic disorders have been identified as potential risk factors for pancreatitis in dogs, and where biochemical markers or imaging are suggestive of pancreatic inflammation in the absence of clinical signs, this might reflect secondary acinar cell damage below the critical threshold required for clinical disease.
In recent years, important insights have been gained into the pathophysiologic mechanisms of pancreatitis, many of which appear to have complex interactions and situation specific expressions. The use of animal models, and more specifically knockout mice, has been critical to determine these interactions and elucidate potential explanations for apparently contradictory studies. The mechanistic insights from these studies offer promise in the identification of therapeutic targets, but species specific or etiology specific mechanisms might mean that the results of experimental studies might not be directly relevant to dogs with spontaneous disease.
CONFLICT OF INTEREST DECLARATION
Dr Steiner and Dr Lim are associated with the Gastrointestinal laboratory at Texas A&M University, which offers Spec cPL testing on a fee‐for‐service basis. Dr Steiner acts as a paid consultant and Speaker for Idexx Laboratories, which offers Spec cPL testing on a fee‐for‐service basis. Dr Steiner is also a paid consultant of ISK, who manufactures fuzapladib. No other authors have a conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Authors declare no IACUC or other approval was needed.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
ACKNOWLEDGMENT
No funding was received for this study.
Cridge H, Lim SY, Algül H, Steiner JM. New insights into the etiology, risk factors, and pathogenesis of pancreatitis in dogs: Potential impacts on clinical practice. J Vet Intern Med. 2022;36(3):847‐864. doi: 10.1111/jvim.16437
[Correction added on 28 May 2022, after first online publication: The phrase “uncoupled protein response” has been replaced with “unfolded protein response” on pages 1 (abbreviations), 10, 11 (Figure 6), and 12 (Figure 8).]
REFERENCES
- 1. Zilio MB, Eyff TF, Azeredo‐Da‐Silva ALF, et al. A systematic review and meta‐analysis of the aetiology of acute pancreatitis. HPB (Oxford). 2019;21(3):259‐267. [DOI] [PubMed] [Google Scholar]
- 2. Lindsay S, Enenman C, Chaikoff I. Pancreatitis accompanying hepatic disease in dogs fed a high fat, low protein diet. Medicine (Baltimore). 1948;45:635. [Google Scholar]
- 3. Patterson E, Munana K, Kirk C, et al. Results of a ketogenic food trial for dogs with idiopathic epilepsy. J Vet Intern Med. 2005;19:421. [Google Scholar]
- 4. Wingert AM, Murray OA, Lulich JP, Hoelmer AM, Merkel LK, Furrow E. Efficacy of medical dissolution for suspected struvite cystoliths in dogs. J Vet Intern Med. 2021;35(5):2287‐2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lem KY, Fosgate GT, Norby B, Steiner JM. Associations between dietary factors and pancreatitis in dogs. J Am Vet Med Assoc. 2008;233(9):1425‐1431. [DOI] [PubMed] [Google Scholar]
- 6. Shukla A. Acute pancreatitis attributed to dietary indiscretion in a female mixed breed canine. Can Vet J. 2010;51(2):201‐203. [PMC free article] [PubMed] [Google Scholar]
- 7. Wright Z, Steiner J, Suchodolski J, Rogers K, Barton C, Brown M. A pilot study evaluating changes in pancreatic lipase immunoreactivity concentrations in canines treated with L‐asparaginase (ASNase), vincristine, or both for lymphoma. Can J Vet Res. 2009;73(2):103‐110. [PMC free article] [PubMed] [Google Scholar]
- 8. Schleis SE, Rizzo SA, Phillips JC, et al. Asparaginase‐associated pancreatitis in a dog. Can Vet J. 2011;52:1009‐1012. [PMC free article] [PubMed] [Google Scholar]
- 9. Gaskill CL, Cribb AE. Pancreatitis associated with potassium bromide/phenobarbital combination therapy in epileptic dogs. Can Vet J. 2000;41(7):555‐558. [PMC free article] [PubMed] [Google Scholar]
- 10. Steiner JM, Xenoulis PG, Anderson J, et al. Serum pancreatic lipase immunoreactivity concentrations in dogs treated with potassium bromide and/or phenobarbital. Vet Ther. 2008;9(1):37‐44. [PubMed] [Google Scholar]
- 11. Albarracín V, Teles M, Meléndez‐Lazo A, Rodón J, Pastor J. Canine pancreas‐specific lipase and c‐reactive protein in dogs treated with anticonvulsants (phenobarbital and potassium bromide). Top Companion Anim Med. 2015;30(2):57‐61. [DOI] [PubMed] [Google Scholar]
- 12. Cook A, Breitschwerdt E, Levine J, et al. Risk factors associated with acute pancreatitis in dogs: 101 cases (1985‐1990). J Am Vet Med Assoc. 1993;203(5):673‐679. [PubMed] [Google Scholar]
- 13. Houston DM, Taylor JA. Acute pancreatitis and bone marrow suppression in a dog given azathioprine. Can Vet J. 1991;32(8):496‐497. [PMC free article] [PubMed] [Google Scholar]
- 14. Moriello K, Bowen D, Meyer D. Acute pancreatitis in two dogs given azathioprine and prednisone. J Am Vet Med Assoc. 1987;191(6):695‐696. [PubMed] [Google Scholar]
- 15. Trepanier LA. Idiosyncratic toxicity associated with potentiated sulfonamides in the dog. J Vet Pharmacol Ther. 2004;27(3):129‐138. [DOI] [PubMed] [Google Scholar]
- 16. Liu S, Oguchi Y, Borner JW, Runge W, Dressel TD, Goodale RL. Increased canine pancreatic acinar cell damage after organophosphate and acetylcholine or cholecystokinin. Pancreas. 1990;5(2):177‐182. [DOI] [PubMed] [Google Scholar]
- 17. Ohta H, Morita T, Yokoyama N, et al. Serial measurement of pancreatic lipase immunoreactivity concentration in dogs with immune‐mediated disease treated with prednisolone. J Small Anim Pract. 2017;58(6):342‐347. [DOI] [PubMed] [Google Scholar]
- 18. Choi H, Ko H, Shin I, Kim H. Malarone® induced pancreatitis and alopecia in a dog: a case report. BMC Vet Res. 2019;15(1):9‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aste G, Di Tommaso M, Steiner JM, et al. Pancreatitis associated with N‐methyl‐glucamine therapy in a dog with leishmaniasis. Vet Res Commun. 2005;29(S2):269‐272. [DOI] [PubMed] [Google Scholar]
- 20. Kook PH, Kranjc A, Dennler M, Glaus TM. Pancreatitis associated with clomipramine administration in a dog. J Small Anim Pract. 2009;50(2):95‐98. [DOI] [PubMed] [Google Scholar]
- 21. Mikszewski J, Saunders H, Hess R. Zinc‐associated acute pancreatitis in a dog. J Small Anim Pract. 2003;44:177‐180. [DOI] [PubMed] [Google Scholar]
- 22. Mawby DI, Whittemore JC, Fecteau KA. Canine pancreatic‐specific lipase concentrations in clinically healthy dogs and dogs with naturally occurring hyperadrenocorticism. J Vet Intern Med. 2014;28(4):1244‐1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Linari G, Dondi F, Segatore S, et al. Evaluation of 1,2‐O‐dilauryl‐rac‐glycero glutaric acid‐(6′ ‐ methylresorufin) ester (DGGR) lipase assay in dogs with naturally occurring hypercortisolism. J Vet Intern Med. 2020;34(1):359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davison LJ. Diabetes mellitus and pancreatitis—cause or effect? J Small Anim Pract. 2015;56(1):50‐59. [DOI] [PubMed] [Google Scholar]
- 25. Kim H, Kang JH, Heo TY, et al. Evaluation of hypertriglyceridemia as a mediator between endocrine diseases and pancreatitis in dogs. J Am Anim Hosp Assoc. 2019;55(2):92‐100. [DOI] [PubMed] [Google Scholar]
- 26. Bishop MA, Xenoulis PG, Levinski MD, Suchodolski JS, Steiner JM. Identification of variants of the SPINK1 gene and their association with pancreatitis in Miniature Schnauzers. Am J Vet Res. 2010;71(5):527‐533. [DOI] [PubMed] [Google Scholar]
- 27. Furrow E, Armstrong PJ, Patterson EE. High prevalence of the c.74A>C SPINK1 variant in Miniature and Standard Schnauzers. J Vet Intern Med. 2012;26(6):1295‐1299. [DOI] [PubMed] [Google Scholar]
- 28. Xenoulis PG, Levinski MD, Suchodolski JS, Steiner JM. Serum triglyceride concentrations in Miniature Schnauzers with and without a history of probable pancreatitis. J Vet Intern Med. 2011;25(1):20‐25. [DOI] [PubMed] [Google Scholar]
- 29. Xenoulis PG, Suchodolski JS, Ruaux CG. Association between serum triglyceride and canine pancreatic lipase immunoreactivity concentrations in Miniature Schnauzers. J Am Anim Hosp Assoc. 2010;46(4):229‐234. [DOI] [PubMed] [Google Scholar]
- 30. Pápa K, Máthé Á, Abonyi‐Tóth Z, et al. Occurrence, clinical features and outcome of canine pancreatitis (80 cases). Acta Vet Hung. 2011;59(1):37‐52. [DOI] [PubMed] [Google Scholar]
- 31. Hess R, Kass P, Shofer F, Van Winkle T, Washabau RJ. Evaluation of risk factors for fatal acute pancreatitis in dogs. J Am Vet Med Assoc. 1999;214(1):46‐51. [PubMed] [Google Scholar]
- 32. Watson PJ, Roulois AJA, Scase T, Johnston PEJ, Thompson H, Herrtage ME. Prevalence and breed distribution of chronic pancreatitis at post‐mortem examination in first‐opinion dogs. J Small Anim Pract. 2007;48(11):609‐618. [DOI] [PubMed] [Google Scholar]
- 33. Xenoulis PG, Cammarata PJ, Walzem RL, Suchodolski JS, Steiner JM. Serum triglyceride and cholesterol concentrations and lipoprotein profiles in dogs with naturally occurring pancreatitis and healthy control dogs. J Vet Intern Med. 2020;34(2):644‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Möhr AJ, Lobetti RG, Van Der Lugt JJ. Acute pancreatitis: a newly recognised potential complication of canine babesiosis. J S Afr Vet Assoc. 2000;71(4):232‐239. [DOI] [PubMed] [Google Scholar]
- 35. Masuda M, Otsuka‐Yamasaki Y, Shiranaga N, et al. Retrospective study on intercurrent pancreatitis with babesia gibsoni infection in dogs. J Vet Med Sci. 2019;81(11):1558‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Köster LS, Steiner JM, Suchodolski JS, Schoeman JP. Serum canine pancreatic‐specific lipase concentrations in dogs with naturally occurring Babesia rossi infection. J S Afr Vet Assoc. 2015;86(1):4‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mylonakis ME, Xenoulis PG, Theodorou K, et al. Serum canine pancreatic lipase immunoreactivity in experimentally induced and naturally occurring canine monocytic ehrlichiosis (Ehrlichia canis). Vet Microbiol. 2014;169(3–4):198‐202. [DOI] [PubMed] [Google Scholar]
- 38. Rodriguez JY, Lewis BC, Snowden KF. Distribution and characterization of Heterobilharzia americana in dogs in Texas. Vet Parasitol. 2014;203(1–2):35‐42. [DOI] [PubMed] [Google Scholar]
- 39. Rodriguez JY, Camp JW, Lenz SD, Kazacos KR, Snowden KF. Identification of Heterobilharzia americana infection in a dog residing in Indiana with no history of travel. J Am Vet Med Assoc. 2016;248(7):827‐830. [DOI] [PubMed] [Google Scholar]
- 40. Corapi WV, Ajithdoss DK, Snowden KF, Spaulding KA. Multi‐organ involvement of Heterobilharzia americana infection in a dog presented for systemic mineralization. J Vet Diagn Investig. 2011;23(4):826‐831. [DOI] [PubMed] [Google Scholar]
- 41. Groover J, Schaer M, Londoño L. Suspected acute pancreatitis in a dog following honeybee envenomation. Can Vet J. 2020;61(4):411‐414. [PMC free article] [PubMed] [Google Scholar]
- 42. McCallum K, Watson P. Hereditary selective cobalamin malabsorption and concurrent pancreatitis in a young Border Collie. Vet Rec Case Rep. 2018;6:1‐6. [Google Scholar]
- 43. Watson P, Coddou F, Blacklaws B, Bazelle J, Day M, Constantino‐Casas F. Letter to the editor regarding immunoglobulin G4‐related disease in a dog. J Vet Intern Med. 2020;34(2):542‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moon MY, Kim J, Kim HJ. Evaluation of serum immunoglobulin G4 concentrations in canine pancreatitis. Korean J Vet Res. 2021;61(1):1‐7. [Google Scholar]
- 45. Peters LM, Glanemann B, Garden OA, Szladovits B. Cytologic findings of 140 bile samples from dogs and cats and associated clinical pathologic data. J Vet Intern Med. 2016;30(1):123‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haig T. Experimental pancreatitis intensified by a high fat diet. Surg Gynecol Obs. 1970;131(5):914‐918. [PubMed] [Google Scholar]
- 47. Kathrani A. Dietary and nutritional approaches to the management of chronic enteropathy in dogs and cats. Vet Clin North Am Small Anim Pract. 2021;51(1):123‐136. [DOI] [PubMed] [Google Scholar]
- 48. Maki T, Kakizaki G, Sato T, et al. Effect of diet on experimental pancreatitis in rat. Tohoku J Exp Med. 1967;92:301‐309. [DOI] [PubMed] [Google Scholar]
- 49. Ramo O. Antecedent long term ethanol consumption in combination with different diets alters the severity of experimental acute pancreatitis in rats. Gut. 1987;28:64‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ramo O, Apaja‐Sarkkinen M, Jalovaara P. Experimental acute pancreatitis in rats receiving different diets and ethanol. Correlation between histological findings and mortality. Res Exp Med. 1987;187(1):33‐41. [DOI] [PubMed] [Google Scholar]
- 51. Czakó L, Szabolcs A, Vajda Á, et al. Hyperlipidemia induced by a cholesterol‐rich diet aggravates necrotizing pancreatitis in rats. Eur J Pharmacol. 2007;572:74‐81. [DOI] [PubMed] [Google Scholar]
- 52. James FE, Mansfield CS, Steiner JM, Williams DA, Robertson ID. Pancreatic response in healthy dogs fed diets of various fat compositions. Am J Vet Res. 2009;70:614‐618. [DOI] [PubMed] [Google Scholar]
- 53. Danino H, Ben‐Dror K, Birk R. Exocrine pancreas ER stress is differentially induced by different fatty acids. Exp Cell Res. 2015;339(2):397‐406. [DOI] [PubMed] [Google Scholar]
- 54. de Oliveira C, Khatua B, Noel P, et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J Clin Invest. 2020;130(4):1931‐1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singh VP. The role of fatty acids in the pathogenesis of pancreatitis. In: ACVIM Forum (On Demand) 2021Proceedings: 428–430.
- 56. Irving HM, Samokhvalov AV, Rehm J. Alcohol as a risk factor for pancreatitis. A systematic review and meta‐analysis. J Pancreas. 2009;10(4):387‐392. [PMC free article] [PubMed] [Google Scholar]
- 57. Farooq A, Richman CM, Swain SM, Shahid RA, Vigna SR, Liddle RA. The role of phosphate in alcohol‐induced experimental pancreatitis. Gastroenterology. 2021;161(3):982‐995.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Havel R. Pathogenesis, differentiation and management of hypertriglyceridemia. Adv Intern Med. 1969;15:117‐154. [PubMed] [Google Scholar]
- 59. Saharia P, Margolis S, Zuidema G, Cameron JL. Acute pancreatitis with hyperlipemia: studies with an isolated perfused canine pancreas. Surgery. 1977;82(1):60‐67. [PubMed] [Google Scholar]
- 60. Mallory A, Kern F. Drug‐induced pancreatitis: a critical review. Gastroenterology. 1980;78(4):813‐820. [PubMed] [Google Scholar]
- 61. Trivedi C, Pitchumoni C. Drug‐induced pancreatitis. J Clin Gastroenterol. 2005;39(8):709‐716. [DOI] [PubMed] [Google Scholar]
- 62. Badalov N, Baradarian R, Iswara K, Li J, Steinberg W, Tenner S. Drug‐induced acute pancreatitis: an evidence‐based review. Clin Gastroenterol Hepatol. 2007;5(6):648‐661. [DOI] [PubMed] [Google Scholar]
- 63. Weetman R, Baehner R. Latent onset of clinical pancreatitis in children receiving L‐asparaginase therapy. Cancer. 1974;34:780‐785. [DOI] [PubMed] [Google Scholar]
- 64. Chabner B, Friedmann A. Asparaginase. In: Chabner B, Longlo D, eds. Cancer Chemotherapy and Biotherapy. 4th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2006:476‐482. [Google Scholar]
- 65. Kluger EK, Malik R, Ilkin WJ, Snow D, Sullivan DR, Govendir M. Serum triglyceride concentration in dogs with epilepsy treated with phenobarbital or with phenobarbital and bromide. J Am Vet Med Assoc. 2008;233(8):1270‐1277. [DOI] [PubMed] [Google Scholar]
- 66. Broe P, Cameron J. Azathioprine and acute pancreatitis: studies with an isolated perfused canine pancreas. J Surg Res. 1983;34(2):159‐163. [DOI] [PubMed] [Google Scholar]
- 67. Parent J. Effects of dexamethasone on pancreatic tissue and on serum amylase and lipase activity in dogs. J Am Vet Med Assoc. 1982;180(7):743‐746. [PubMed] [Google Scholar]
- 68. Fittschen C, Bellamy JEC. Prednisone treatment alters the serum amylase and lipase activities in normal dogs without causing pancreatitis. Can J Comp Med. 1984;48(2):136‐140. [PMC free article] [PubMed] [Google Scholar]
- 69. Anderson JR, Cornell KK, Parnell NK, Salisbury SK. Pancreatic abscess in 36 dogs: A retrospective analysis of prognostic indicators. J Am Anim Hosp Assoc. 2008;44(4):171‐179. 10.5326/0440171 [DOI] [PubMed] [Google Scholar]
- 70. Steiner JM, Teague SR, Lees GE, Willard MD, Williams DA, Ruaux CG. Stability of canine pancreatic lipase immunoreactivity concentration in serum samples and effects of long‐term administration of prednisone to dogs on serum canine pancreatic lipase immunoreactivity concentrations. Am J Vet Res. 2009;70(8):1001‐1005. [DOI] [PubMed] [Google Scholar]
- 71. Ohta H, Kojima K, Yokoyama N, et al. Effects of immunosuppressive prednisolone therapy on pancreatic tissue and concentration of canine pancreatic lipase immunoreactivity in healthy dogs. Can J Vet Res. 2018;82(4):278‐286. [PMC free article] [PubMed] [Google Scholar]
- 72. Cocker S, Richter K, Steiner J. Serum pancreatic lipase immunoreactivity concentrations after chronic administration of supraphysiologic doses of glucocorticoids to dogs. J Vet Intern Med. 2016;30:1453. [DOI] [PubMed] [Google Scholar]
- 73. Schueler RO, White G, Schueler RL, Steiner JM, Wassef A. Canine pancreatic lipase immunoreactivity concentrations associated with intervertebral disc disease in 84 dogs. J Small Anim Pract. 2018;59(5):305‐310. [DOI] [PubMed] [Google Scholar]
- 74. Mendoza B, Dias MJ, Hernandez J, et al. Effect of prednisolone therapy on serum levels of 1,2‐O‐dilauryl‐rac‐glycero glutaric acid‐(6′‐methylresorufin) ester lipase in dogs. J Vet Intern Med. 2020;43(6):2330‐2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Okanishi H, Nagata T, Nakane S, Watari T. Comparison of initial treatment with and without corticosteroids for suspected acute pancreatitis in dogs. J Small Anim Pract. 2019;60(5):298‐304. [DOI] [PubMed] [Google Scholar]
- 76. Bjørnkjær‐Nielson K, Bjørnvad C. Corticosteroid treatment for acute/acute‐on‐chronic experimental and naturally occurring pancreatitis in several species: a scoping review to inform possible use in dogs. Acta Vet Scand. 2021;63(28):1‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Oguchi Y, Dressel TD, Borner JW. Inhibition of acetyl‐ and butyrlcholinesterase and amylsase release from canine pancreas. Pancreas. 1989;4:423‐428. [DOI] [PubMed] [Google Scholar]
- 78. Xenoulis PG, Saridomichelakis MN, Chatzis MK, et al. Prospective evaluation of serum pancreatic lipase immunoreactivity and troponin I concentrations in Leishmania infantum‐infected dogs treated with meglumine antimonate. Vet Parasitol. 2014;203(3–4):326‐330. [DOI] [PubMed] [Google Scholar]
- 79. Pöppl AG, de Carvalho GLC, Vivian IF, Corbellini LG, González FHD. Canine diabetes mellitus risk factors: a matched case‐control study. Res Vet Sci. 2017;114:469‐473. [DOI] [PubMed] [Google Scholar]
- 80. Palermo SM, Brown DC, Mehler SJ, Rondeau MP. Clinical and prognostic findings in dogs with suspected extrahepatic biliary obstruction and pancreatitis. J Am Anim Hosp Assoc. 2020;56(5):270‐279. [Google Scholar]
- 81. Mattin M, O'Neill D, Church D, et al. An epidemiological study of diabetes mellitus in dogs attending first opinion practice in the UK. Vet Rec. 2014;174(14):349. [DOI] [PubMed] [Google Scholar]
- 82. Alejandro R, Feldman E, Shienvold F, et al. Advances in canine diabetes mellitus research: etiopathology and results of islet transplantation. J Am Vet Med Assoc. 1988;193(9):1050‐1055. [PubMed] [Google Scholar]
- 83. Watson P, Herrtage M. Use of glucagon stimulation tests to assess beta‐cell function in dogs with chronic pancreatitis. J Nutr. 2004;134(8):2081S‐2083S. [DOI] [PubMed] [Google Scholar]
- 84. Hess R, Saunders M, Van Winkle T, et al. Clinical, clinicopathologic, radiographic, and ultrasonographic abnormalities in dogs with fatal acute pancreatitis: 70 cases (1986‐1995). J Am Vet Med Assoc. 1998;213(5):665‐670. [PubMed] [Google Scholar]
- 85. Rinderknecht H. Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig Dis Sci. 1986;31:314‐321. [DOI] [PubMed] [Google Scholar]
- 86. Bishop M, Xenoulis PG, Steiner JM. Letter to the editor. J Vet Intern Med. 2013;27:427‐428. [DOI] [PubMed] [Google Scholar]
- 87. Watson P. Letter to the editor. J Vet Intern Med. 2013;27:1291. [DOI] [PubMed] [Google Scholar]
- 88. Watson P, Roulois A, Scase T, et al. Characterization of chronic pancreatitis in English Cocker Spaniels. J Vet Intern Med. 2011;25:797‐804. [DOI] [PubMed] [Google Scholar]
- 89. Coddou M, Constantino‐Casas F, Blacklaws B, et al. Identification of IgG4‐related disease in the English Cocker Spaniel and dogs of other breeds [27th ECVIM‐CA Congress Abstract]. J Vet Intern Med. 2018;32:538. [Google Scholar]
- 90. Coddou M, Constantino‐Casas F, Blacklaws B, et al. Clinical features of English Cocker Spaniels with chronic pancreatitis mimic human IgG4‐related disease [27th ECVIM Congress]. J Vet Intern Med. 2018;32:580. [Google Scholar]
- 91. Moore AR, Colopy LJ, Shiu KB, Snyder LA, Avery AC, Rout ED. Response to letter to editor regarding immunoglobulin G4‐related disease in a dog. J Vet Intern Med. 2020;34(2):544‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Xenoulis PG. Diagnosis of pancreatitis in dogs and cats. J Small Anim Pract. 2015;56(1):13‐26. [DOI] [PubMed] [Google Scholar]
- 93. Mansfield C. Pathophysiology of acute pancreatitis: potential application from experimental models and human medicine to dogs. J Vet Intern Med. 2012;26:875‐887. [DOI] [PubMed] [Google Scholar]
- 94. Walton S. Diagnosing acute pancreatitis in dogs. Todays Vet Pract. 2020;Jan/Feb:47‐54. [Google Scholar]
- 95. Kalli IV, Adamama‐Moraitou KK, Patsika MN, et al. Prevalence of increased canine pancreas‐specific lipase concentrations in young dogs with parvovirus enteritis. Vet Clin Pathol. 2017;46(1):111‐119. [DOI] [PubMed] [Google Scholar]
- 96. Haworth MD, Hosgood G, Swindells KL, Mansfield CS. Diagnostic accuracy of the SNAP and Spec canine pancreatic lipase tests for pancreatitis in dogs presenting with clinical signs of acute abdominal disease. J Vet Emerg Crit Care. 2014;24(2):135‐143. [DOI] [PubMed] [Google Scholar]
- 97. Spinella G, Dondi F, Grassato L, et al. Prognostic value of canine pancreatic lipase immunoreactivity and lipase activity in dogs with gastric dilatation‐volvulus. PLoS One. 2018;13(9):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cochran L, Hill S, Suchodolski S. Serum pancreatic lipase immunoreactivity in dogs with gastric foreign bodies [2016 ACVIM Forum Abstract]. J Vet Intern Med. 2016;30:1453. [Google Scholar]
- 99. Park JS, Park JH, Seo KW, Song KH. Correlation between NT‐proBNP and lipase levels according to the severity of chronic mitral valve disease in dogs. J Vet Sci. 2019;20(4):e43. doi: 10.4142/jvs.2019.20.e43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kathrani A, Steiner JM, Suchodolski J, et al. Elevated canine pancreatic lipase immunoreactivity concentration in dogs with inflammatory bowel disease is associated with a negative outcome. J Small Anim Pract. 2009;50(3):126‐132. [DOI] [PubMed] [Google Scholar]
- 101. Chiari H. About the digestion of the human pancreas (in German). ZeitschriftfurHeilkunde. 1896;17:69‐96. [Google Scholar]
- 102. Saluja AK, Bhagat L, Lee HS, Bhatia M, Frossard JL, Steer ML. Secretagogue‐induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 1999;276(4):G835‐G842. [DOI] [PubMed] [Google Scholar]
- 103. Saluja A, Saluja M, Villa A, et al. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J Clin Invest. 1989;84(4):1260‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Willemer S, Klöppel G, Kern HF, et al. Immunocytochemical and morphometric analysis of acinar zymogen granules in human acute pancreatitis. Virchows Arch A Pathol Anat Histopathol. 1989;415(2):115‐124. [DOI] [PubMed] [Google Scholar]
- 105. Hofbauer B, Saluja AK, Lerch MM, et al. Intra‐acinar cell activation of trypsinogen during caerulein‐induced pancreatitis in rats. Am J Physiol Gastrointest Liver Physiol. 1998;275(2):G352‐G362. [DOI] [PubMed] [Google Scholar]
- 106. Mithöfer K, Fernández‐Del Castillo C, Rattner D, et al. Subcellular kinetics of early trypsinogen activation in acute rodent pancreatitis. Am J Physiol Gastrointest Liver Physiol. 1998;274(1):G71‐G79. [DOI] [PubMed] [Google Scholar]
- 107. Perides G, Laukkarinen JM, Vassileva G, Steer ML. Biliary acute pancreatitis in mice is mediated by the G‐protein‐coupled cell surface bile acid receptor Gpbar1. Gastroenterology. 2010;138(2):715‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gaiser S, Daniluk J, Liu Y, et al. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut. 2011;60(10):1379‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Dawra R, Sah RP, Dudeja V, et al. Intra‐acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology. 2011;141(6):2210‐2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mikami Y, Takeda K, Matsuda K, et al. Rat experimental model of continuous regional arterial infusion of protease inhibitor and its effects on severe acute pancreatitis. Pancreas. 2005;30(3):248‐253. [DOI] [PubMed] [Google Scholar]
- 111. Uhr W, Buchler M, Malfertheiner P, et al. A randomised, double blind, multicentre trial of octreotide in moderate to severe acute pancreatitis. Gut. 1999;45(1):97‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zhang J, Niu L, Yang Y, Hao Y, Feng J. Systematic review and meta‐analysis of the efficacy of trypsin inhibitors in patients with severe pancreatitis. Ann Palliat Med. 2021;10(6):6577‐6587. [DOI] [PubMed] [Google Scholar]
- 113. Lagoo JY, D'Souza MC, Kartha A, et al. Role of Ulinastatin, a trypsin inhibitor, in severe acute pancreatitis in critical care setting: a retrospective analysis. J Crit Care. 2018;45:27‐32. [DOI] [PubMed] [Google Scholar]
- 114. Klar E, Rattner D, Compton C, et al. Adverse effec of therapeutic vasoconstrictors on experimental acute pancreatitis. Ann Surg. 1991;214(2):168‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jancsó Z, Hegyi E, Sahin‐Tóth M. Chymotrypsin reduces the severity of secretagogue‐induced pancreatitis in mice. Gastroenterology. 2018;155(4):1017‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ko T, Bertram M, Prinz R, et al. Effect of somatostatin analogue and cholecystokinin receptor antagonist on bile‐induced acute canine pancreatitis. Am Surg. 1992;58(4):213‐219. [PubMed] [Google Scholar]
- 117. Gorelick F, Thrower E. The acinar cell and early pancreatitis responses. Clin Gastroenterol Hepatol. 2009;7:S10‐S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Gukovsky I, Gukovskaya AS, Blinman TA, Zaninovic V, Pandol SJ. Early NF‐κB activation is associated with hormone‐induced pancreatitis. Am J Physiol Gastrointest Liver Physiol. 1998;275(6):G1402‐G1414. [DOI] [PubMed] [Google Scholar]
- 119. Hietaranta AJ, Saluja AK, Bhagat L, Singh VP, Song AM, Steer ML. Relationship between NF‐κB and trypsinogen activation in rat pancreas after supramaximal caerulein stimulation. Biochem Biophys Res Commun. 2001;280(1):388‐395. [DOI] [PubMed] [Google Scholar]
- 120. Ji B, Gaiser S, Chen X, Ernst SA, Logsdon CD. Intracellular trypsin induces pancreatic acinar cell death but not NF‐κB activation. J Biol Chem. 2009;284(26):17488‐17498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Rakonczay Z, Hegyi P, Takács T, et al. The role of NF‐κB activation in the pathogenesis of acute pancreatitis. Gut. 2008;57(2):259‐267. [DOI] [PubMed] [Google Scholar]
- 122. Baumann B, Wagner M, Aleksic T, et al. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest. 2007;117(6):1502‐1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Altavilla D, Famulari C, Passaniti M, et al. Attenuated cerulein‐induced pancreatitis in nuclear factor‐κ B‐deficient mice. Lab Invest. 2003;83(12):1723‐1732. [DOI] [PubMed] [Google Scholar]
- 124. Huang H, Liu Y, Daniluk J, et al. Activation of nuclear factor‐kB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology. 2013;144:202‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD. NF‐κB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology. 2002;122(2):448‐457. [DOI] [PubMed] [Google Scholar]
- 126. Song L, Wörmann S, Ai J, et al. BCL3 reduces the sterile inflammatory response in pancreatic and biliary tissues. Gastroenterology. 2016;150:499‐512. [DOI] [PubMed] [Google Scholar]
- 127. Gukovsky I, Gukvoskaya A. Nuclear factor‐kB in pancreatitis: jack of all trades, but which one is more important? Gastroenterology. 2013;14:26‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Neuhöfer P, Liang S, Einwächter H, et al. Deletion of IkB alpha activates RelA to reduce acute pancreatitis in mice through up‐regulation of Spi2A. Gastroenterology. 2013;144:192‐201. [DOI] [PubMed] [Google Scholar]
- 129. Algül H, Paxian S, Schmid RM, et al. Pancreas‐specific RelA/p65 truncation increases susceptibility of acini to inflammation‐associated cell death following cerulein pancreatitis. J Clin Invest. 2007;117(6):1490‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ruaux CG, Pennington HL, Worrall S, Atwell RB. Tumor necrosis factor‐α at presentation in 60 cases of spontaneous canine acute pancreatitis. Vet Immunol Immunopathol. 1999;72(3–4):369‐376. [DOI] [PubMed] [Google Scholar]
- 131. Paek J, Kang JH, Kim HS, Lee I, Seo KW, Yang MP. Serum adipokine concentrations in dogs with acute pancreatitis. J Vet Intern Med. 2014;28(6):1760‐1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kuzi S, Mazaki‐Tovi M, Suchodolski JS, et al. Protease inhibitors, inflammatory markers, and their association with outcome in dogs with naturally occurring acute pancreatitis. J Vet Intern Med. 2020;34(5):1801‐1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Choi S, Kim Y, Kang MS, et al. Serum concentration of inflammatory cytokines in dogs with suspected acute pancreatitis. Vet Sci. 2021;8(3):51. doi: 10.3390/vetsci8030051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lee PJ, Papachristou GI. New insights into acute pancreatitis. Nat Rev Gastroenterol Hepatol. 2019;16(8):479‐496. [DOI] [PubMed] [Google Scholar]
- 135. Pooran N, Indaram A, Singh P, Bank S. Cytokines (IL‐6, IL‐8, TNF): early and reliable predictors of severe acute pancreatitis. J Clin Gastroenterol. 2003;37(3):263‐266. [DOI] [PubMed] [Google Scholar]
- 136. Stirling AD, Moran NR, Kelly ME, Ridgway PF, Conlon KC. The predictive value of C‐reactive protein (CRP) in acute pancreatitis—is interval change in CRP an additional indicator of severity? HPB (Oxford). 2017;19(10):874‐880. [DOI] [PubMed] [Google Scholar]
- 137. Scheller J, Chalaris A, Schmidt‐Arras D, Rose‐John S. The pro‐ and anti‐inflammatory properties of the cytokine interleukin‐6. Biochim Biophys Acta Mol Cell Res. 2011;1813(5):878‐888. [DOI] [PubMed] [Google Scholar]
- 138. Zhang H, Neuhöfer P, Song L, et al. IL‐6 trans‐signaling promotes pancreatitis‐associated lung injury and lethality. J Clin Invest. 2013;123(3):1019‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Barreto SG, Habtezion A, Gukovskaya A, et al. Critical thresholds: key to unlocking the door to the prevention and specific treatments for acute pancreatitis. Gut. 2021;70:194‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wan J, Ren Y, Yang X, Li X, Xia L, Lu N. The role of neutrophils and neutrophil extracellular traps in acute pancreatitis. Front Cell Dev Biol. 2021;8:1‐8. doi: 10.3389/fcell.2020.565758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Newman S, Steiner J, Woosley K, Barton L, Ruaux C, Williams D. Localization of pancreatic inflammation and necrosis in dogs. J Vet Intern Med. 2004;18(4):488‐493. [DOI] [PubMed] [Google Scholar]
- 142. Pastor CM, Rubbia‐Brandt L, Hadengue A, Jordan M, Morel P, Frossard JL. Role of macrophage inflammatory peptide‐2 in cerulein‐induced acute pancreatitis and pancreatitis‐associated lung injury. Lab Invest. 2003;83(4):471‐478. [DOI] [PubMed] [Google Scholar]
- 143. Chen G, Xu F, Li J, Lu S. Depletion of neutrophils protects against L‐arginine‐induced acute pancreatitis in mice. Cell Physiol Biochem. 2015;35(6):2111‐2120. [DOI] [PubMed] [Google Scholar]
- 144. Kyriakides C, Jasleen J, Wang Y, Moore FD Jr, Ashley SW, Hechtman HB. Neutrophils, not complement, mediate the mortality of experimental hemorrhagic pancreatitis. Pancreas. 2001;22(1):40‐46. [DOI] [PubMed] [Google Scholar]
- 145. Willemer S, Fedderson C, Karges W, et al. Lung injury in acute experimental pancreatitis in rats. I. Morphological studies. Int J Pancreatol. 1991;8:305‐321. [DOI] [PubMed] [Google Scholar]
- 146. Guice K, Oldham K, Caty M, et al. Neutrophil‐dependent oxygen‐radical mediated lung injury associated with acute pancreatitis. Ann Surg. 1989;210:740‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Awla D, Abdulla A, Zhang S, et al. Lymphocyte function antigen‐1 regulates neutrophil recruitment and tissue damage in acute pancreatitis. Br J Pharmacol. 2011;163(2):413‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Steiner J. How pancreatitis research in animals and animal models guides clinical practice. In: ACVIM Forum 2021 (On Demand); 2021.
- 149. Raraty M, Ward J, Erdemli G, et al. Calcium‐dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci U S A. 2000;97(24):13126‐13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Petersen OH, Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol. 2008;70:273‐299. [DOI] [PubMed] [Google Scholar]
- 151. Sah RP, Garg P, Saluja AK. Pathogenic mechanisms of acute pancreatitis. Curr Opin Gastroenterol. 2012;28(5):507‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Krüger B, Albrecht E, Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Pathol. 2000;157(1):43‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Voronina S, Sherwood M, Barrow S, Dolman N, Conant A, Tepikin A. Downstream from calcium signalling: mitochondria, vacuoles, and pancreatic acinar cell damage. Acta Physiol. 2009;195(1):161‐169. [DOI] [PubMed] [Google Scholar]
- 154. Li J, Zhou R, Zhang J, Li ZF. Calcium signaling of pancreatic acinar cells in the pathogenesis of pancreatitis. World J Gastroenterol. 2014;20(43):16146‐16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Petersen OH, Gerasimenko OV, Tepikin AV, Gerasimenko JV. Aberrant Ca2+ signalling through acidic calcium stores in pancreatic acinar cells. Cell Calcium. 2011;50(2):193‐199. [DOI] [PubMed] [Google Scholar]
- 156. Lewarchik CM, Orabi AI, Jin S, et al. The ryanodine receptor is expressed in human pancreatic acinar cells and contributes to acinar cell injury. Am J Physiol Gastrointest Liver Physiol. 2014;307(5):574‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kim MS, Hong JH, Li Q, et al. Deletion of TRPC3 in mice reduces store‐operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology. 2009;137(4):1509‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Mukherjee R, Mareninova OA, Odinokova IV, et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut. 2016;65(8):1333‐1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Maleth J, Hegyi P. Ca2+ toxicity and mitochondrial damage in acute pancreatitis: translational overview. Philos Trans R Soc B. 2016;371(1700):20150425. doi: 10.1098/rsbt.2015.0425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Gukovsky I, Pandol SJ, Gukovskaya AS. Organellar dysfunction in the pathogenesis of pancreatitis. Antioxid Redox Signal. 2011;15(10):2699‐2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mukherjee R, Criddle DN, Gukvoskaya A, et al. Mitochondrial injury in pancreatitis. Cell Calcium. 2008;44(1):14‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Gukovsky I, Pandol SJ, Mareninova OA, Shalbueva N, Jia W, Gukovskaya AS. Impaired autophagy and organellar dysfunction in pancreatitis. J Gastroenterol Hepatol. 2012;27(S2):27‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Shah AU, Sarwar A, Orabi AI, et al. Protease activation during in vivo pancreatitis is dependent on calcineurin activation. Am J Physiol Gastrointest Liver Physiol. 2009;297(5):967‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Muili KA, Jin S, Orabi AI, et al. Pancreatic acinar cell nuclear factor κb activation because of bile acid exposure is dependent on calcineurin. J Biol Chem. 2013;288(29):21065‐21073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Muili KA, Ahmad M, Orabi AI, et al. Pharmacological and genetic inhibition of calcineurin protects against carbachol‐induced pathological zymogen activation and acinar cell injury. Am J Physiol Gastrointest Liver Physiol. 2012;302(8):898‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Masamune A, Kotani H, Sörgel FL, et al. Variants that affect function of calcium channel TRPV6 are associated with early‐onset chronic pancreatitis. Gastroenterology. 2020;158(6):1626‐1641.e8. doi: 10.1053/j.gastro.2020.01.005 [DOI] [PubMed] [Google Scholar]
- 167. Wen L, Javed TA, Dobbs AK, et al. The protective effects of calcineurin on pancreatitis in mice depend on the cellular source. Gastroenterology. 2020;159(3):1036‐1050.e8. doi: 10.1053/j.gastro.2020.05.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Archer T, Boothe D, Langston V, et al. Oral cyclosporine treatment in dogs: a review of the literature. J Vet Intern Med. 2014;28:1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Gerasimenko JV, Gryshchenko O, Ferdek PE, et al. Ca2+ release‐activated Ca2+ channel blockage as a potential tool in antipancreatitis therapy. PNAS. 2013;110(32):13186‐13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wen L, Voronina S, Javed MA, et al. Inhibitors of ORAI1 prevent cytosolic calcium‐associated injury of human pancreatic acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology. 2015;149:481‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Javed MA, Wen L, Awais M, et al. TRO40303 ameliorates alcohol‐induced pancreatitis through reduction of fatty acid ethyl ester‐induced mitochondrial injury and necrotic cell death. Pancreas. 2018;47(1):18‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Saluja A, Hashimoto S, Saluja M, Powers RE, Meldolesi J, Steer ML. Subcellular redistribution of lysosomal enzymes during caerulein‐induced pancreatitis. Am J Physiol Gastrointest Liver Physiol. 1987;253(4):G508‐G516. [DOI] [PubMed] [Google Scholar]
- 173. Watanabe O, Baccino FM, Steer ML, Meldolesi J. Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: early morphological changes during development of experimental pancreatitis. Am J Physiol Gastrointest Liver Physiol. 1984;9(4):457‐467. [DOI] [PubMed] [Google Scholar]
- 174. Figarella C, Miszczuk‐Kamska B, Barrett A. Possible lysosomal activation of pancreatic zymogens. Activation of both human trypsinogens by cathepsin B and spontaneous acid. Activation of human trypsinogen 1. Biol Chem Hoppe Seyler. 1988;369:293‐298. [PubMed] [Google Scholar]
- 175. Lerch MM, Saluja AK, Dawra R, Saluja M, Steer ML. The effect of chloroquine administration on two experimental models of acute pancreatitis. Gastroenterology. 1993;104(6):1768‐1779. [DOI] [PubMed] [Google Scholar]
- 176. Halangk W, Lerch MM, Brandt‐Nedelev B, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106(6):773‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Van Acker GJD, Saluja AK, Bhagat L, et al. Cathepsin B inhibition prevents trypsinogen activation and reduces pancreatitis severity. Am J Physiol Gastrointest Liver Physiol. 2002;283(3):G794‐G800. [DOI] [PubMed] [Google Scholar]
- 178. Saluja AK, Donovan EA, Yamanaka K, Yamaguchi Y, Hofbauer B, Steer ML. Cerulein‐induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology. 1997;113(1):304‐310. [DOI] [PubMed] [Google Scholar]
- 179. Tooze J, Hollinshead M, Hensel G, Kern HF, Hoflack B. Regulated secretion of mature cathepsin B from rat exocrine pancreatic cells. Eur J Cell Biol. 1991;56(2):187‐200. [PubMed] [Google Scholar]
- 180. Aghdassi AA, John DS, Sendler M, et al. Cathepsin D regulates cathepsin b activation and disease severity predominantly in inflammatory cells during experimental pancreatitis. J Biol Chem. 2018;293(3):1018‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Mahurkar S, Idris MM, Reddy DN, et al. Association of cathepsin B gene polymorphisms with tropical calcific pancreatitis. Gut. 2006;55(9):1270‐1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Weiss F, Behn C, Simon P, et al. Cathepsin B gene polymorphism Val26 is not associated with idiopathic chronic pancreatitis in european patients. Gut. 2007;56(9):1322‐1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349‐364. [DOI] [PubMed] [Google Scholar]
- 184. Antonucci L, Fagman JB, Kim JY, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A. 2015;112(45):E6166‐E6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Diakopoulos KN, Lesina M, Wörmann S, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex‐ and nutrition‐dependent processes. Gastroenterology. 2015;148(3):626‐638.e17. [DOI] [PubMed] [Google Scholar]
- 186. Hashimoto D, Ohmuraya M, Hirota M, et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol. 2008;181(7):1065‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Mareninova OA, Sendler M, Malla SR, et al. Lysosome‐associated membrane proteins (LAMP) maintain pancreatic acinar cell homeostasis: LAMP‐2 deficient mice develop pancreatitis. Cell Mol Gastroenterol Hepatol. 2015;1(6):678‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Shen S, Li B, Dai J, et al. BRD4 inhibition protects against acute pancreatitis through restoring impaired autophagic flux. Front Pharmacol. 2020;11:618. doi: 10.3389/fphar.2020.00618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Sakamaki J, Wilkinson S, Hahn M, et al. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol Cell. 2017;66(4):517‐532.e9. doi: 10.1016/j.molcel.2017.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Biczo G, Vegh ET, Shalbueva N, et al. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology. 2018;154(3):689‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Kubisch CH, Sans MD, Arumugam T, Ernst SA, Williams JA, Logsdon CD. Early activation of endoplasmic reticulum stress is associated with arginine‐induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2006;291(2):238‐245. [DOI] [PubMed] [Google Scholar]
- 192. Sah RP, Garg SK, Dixit AK, Dudeja V, Dawra RK, Saluja AK. Endoplasmic reticulum stress is chronically activated in chronic pancreatitis. J Biol Chem. 2014;289(40):27551‐27561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Logsdon C, Ji B. The role of protein synthesis and digestive enzymes in acinar cell injury. Nat Rev Gastroenterol Hepatol. 2013;10:362‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Barrera K, Stanek A, Okochi K, et al. Acinar cell injury induced by inadequate unfolded protein response in acute pancreatitis. World J Gastrointest Pathophysiol. 2018;9(2):37‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Chen JC, Wu ML, Huang KC, Lin WW. HMG‐CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc Res. 2008;80(1):138‐150. [DOI] [PubMed] [Google Scholar]
- 196. Wu BU, Pandol SJ, Liu ILA. Simvastatin is associated with reduced risk of acute pancreatitis: findings from a regional integrated healthcare system. Gut. 2015;64(1):133‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Ruiz‐Rebollo ML, Muñoz‐Moreno MF, Mayo‐Iscar A, Udaondo‐Cascante MA, Nistal RB. Statin intake can decrease acute pancreatitis severit. Pancreatology. 2019;19(6):807‐812. [DOI] [PubMed] [Google Scholar]
- 198. Sanfey H, Bulkley G, Cameron J. The role of oxygen‐derived free radicals in the pathogenesis of acute pancreatitis. Ann Surg. 1984;200:405‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Verlaan M, Roelofs H, van Schaik A, et al. Assessment of oxidative stress in chronic pancreatitis patients. World J Gastroenterol. 2006;12(35):5705‐5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Park BK, Chung JB, Lee JH, et al. Role of oxygen free radicals in patients with acute pancreatitis. World J Gastroenterol. 2003;9(10):2266‐2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Que RS, Cao LP, Ding GP, Hu JA, Mao KJ, Wang GF. Correlation of nitric oxide and other free radicals with the severity of acute pancreatitis and complicated systemic inflammatory response syndrome. Pancreas. 2010;39(4):536‐540. [DOI] [PubMed] [Google Scholar]
- 202. Schoenberg MH, Buchler M, Gaspar M, et al. Oxygen free radicals in acute pancreatitis of the rat. Gut. 1990;31(10):1138‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Siriwardena AK, Mason JM, Balachandra S, et al. Randomised, double blind, placebo controlled trial of intravenous antioxidant (n‐acetylcysteine, selenium, vitamin C) therapy in severe acute pancreatitis. Gut. 2007;56(10):1439‐1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Monfared SSMS, Vahidi H, Abdolghaffari AH, Nikfar S, Abdollahi M. Antioxidant therapy in the management of acute, chronic and post‐ERCP pancreatitis: a systematic review. World J Gastroenterol. 2009;15(36):4481‐4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Booth DM, Murphy JA, Mukherjee R, et al. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology. 2011;140(7):2116‐2125. [DOI] [PubMed] [Google Scholar]
- 206. Sah RP, Saluja A. Molecular mechanisms of pancreatic injury. Curr Opin Gastroenterol. 2011;27(5):444‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Patel K, Trivedi RN, Durgampudi C, et al. Lipolysis of visceral adipocyte triglyceride by pancreatic lipases converts mild acute pancreatitis to severe pancreatitis independent of necrosis and inflammation. Am J Pathol. 2015;185(3):808‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Dettelbach D, Deftos L, Stewart A. Intraperitoneal free fatty acids induce severe hypocalcemia in rats: a model for the hypocalcemia of pancreatitis. J Bone Min Res. 1990;5(12):1249‐1255. [DOI] [PubMed] [Google Scholar]
- 209. Navina S, Acharya C, DeLany J, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Transl Med. 2011;3(107):107ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Khatua B, Trivedi RN, Noel P, et al. Carboxyl ester lipase may not mediate lipotoxic injury during severe acute pancreatitis. Am J Pathol. 2019;189(6):1226‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Khatua B, Yaron JR, El‐Kurdi B, et al. Ringer's lactate prevents early organ failure by providing extracellular calcium. J Clin Med. 2020;9(1):263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Ahmad F, Mahmud S. Acute pancreatitis following orlistat therapy: report of two cases. JOP. 2010;11(1):61‐63. [PubMed] [Google Scholar]