

Abstract
The toxic potential of H2O2 is limited, even if intracellular concentrations of H2O2 under conditions of oxidative stress increase to the micromolar concentration range. Its toxicity is mostly restricted to the oxidation of highly reactive thiol groups, some of which are functionally very important. Subsequently, the HO· radical is generated spontaneously from H2O2 in the Fenton reaction. The HO· radical is extremely toxic and destroys any biological structure. Due to the high reactivity, its action is limited to a locally restricted site of its generation. On the other hand, H2O2 with its stability and long half-life can reach virtually any site and distribute its toxic effect all over the cell. Thereby HO·, in spite of its ultra-short half-life (10–9 s), can execute its extraordinary toxic action at any target of the cell. In this oxidative stress scenario, H2O2 is the pro-radical, that spreads the toxic action of the HO· radical. It is the longevity of the H2O2 molecule allowing it to distribute its toxic action from the site of origin all over the cell and may even mediate intercellular communication. Thus, H2O2 acts as a spreader by transporting it to sites where the extremely short-lived toxic HO· radical can arise in the presence of “free iron”. H2O2 and HO· act in concert due to their different complementary chemical properties. They are dependent upon each other while executing the toxic effects in oxidative stress under diabetic metabolic conditions in particular in the highly vulnerable pancreatic beta cell, which in contrast to many other cell types is so badly protected against oxidative stress due to its extremely low H2O2 inactivating enzyme capacity.
Keywords: Oxidative stress, Hydrogen peroxide, Hydroxyl radical, Pancreatic beta cell
Introduction
In the process of oxidative phosphorylation, energy is harvested as ATP via a process of reduction of a dioxygen molecule to two water molecules. ROS (reactive oxygen species) are products of incomplete reduction of molecular O2 (oxygen). H2O2 (hydrogen peroxide) as the 2-electron reduction product of O2 is not fully reduced and more reactive than dioxygen. At variance, the O2·− (superoxide radical anion) and the HO· (hydroxyl radical) are 1-electron reduction products of O2 and of H2O2, respectively [for details, see Lenzen (2017)]:
Although O2 has the same oxidation state in HO· and H2O2, the standard potential of the HO· radicals is exceedingly higher:
This is so because of the stoichiometry and the fact that not the standard potentials, but the free energies are additive (Luther’s rule) (Scholz 2012):
Of the partially reduced O2 products, only H2O2 is kinetically sufficiently stable, a small electroneutral molecule in its non-dissociated form (pKa, 11.75), and endowed with chemical properties, which allow this reactive species to travel long distances and cross biological membranes due to which it can reach reaction sites far away from the site of its generation. These are physicochemical features which put H2O2 into a crucial role, when large amounts of H2O2 (in the micromolar concentration range) are generated under conditions of oxidative stress (Halliwell 2006; Halliwell and Gutteridge 2015; Sies 1986, 2014; Winterbourn 2008; Winterbourn 2013).
H2O2 is the intermediate reduction product of O2, generated in the peroxisomes and the ER (Lenzen 2017), while it is the reduction product of O2·− in mitochondria and the cytosol, where H2O2 is generated in an initial step by the SOD isoenzymes MnSOD and Cu/ZnSOD or spontaneously (Lenzen 2008; Lortz et al. 2005; Lushchak 2014; Winterbourn 2020). Thus, at all subcellular sites, H2O2 is ultimately one of the ROS of crucial importance as a mediator of toxicity under conditions of oxidative stress (Jones and Sies 2015; Lenzen 2017; Lushchak 2014).
We will present here a new interpretation of the role of H2O2 in the mediation of oxidative stress-induced dysfunction and death in the very vulnerable (Lenzen 2008 #34) and badly protected pancreatic beta cell (Lenzen 2017), when compared to many other cell types (Lenzen et al. 1996; Tiedge et al. 1997) in the development of a diabetic metabolic state. We explain that H2O2 is not only a reactive species with toxic potential locally at the site of H2O2 generation but also spreads because of its physicochemical properties the toxicity throughout the whole cell, thereby spreading the toxicity of the HO· radical ultimately generated in the Fenton reaction from H2O2.
The central role of hydrogen peroxide as a pro-radical in the mitochondria and the peroxisomes
H2O2 can traverse lipid membranes (Lushchak 2011), in particular through some members of the aquaporin family, the so-called peroxiporins by facilitated diffusion (Bienert and Chaumont 2014; Laporte et al. 2020; Lenzen 2017), thereby entering the cytosolic compartment irrespective of the subcellular site of its generation, may it be the mitochondria or the peroxisomes. The number of fast reacting high affinity sites, namely the thiol (sulfhydryl, SH) groups of the antioxidative enzymes of the glutathione peroxidase (GPx) and peroxiredoxin (Prx) enzyme families is small due to the low expression levels in the mitochondria and the peroxisomes of the pancreatic beta cells (Lenzen 2017). Therefore, only little of the high amounts of H2O2 generated in these organelles during cytokine toxicity in the T1DM scenario (Lenzen 2017) in the mitochondria and glucolipotoxicity in the T2DM scenario (Gehrmann et al. 2010; Lenzen 2017) in the peroxisomes are quickly inactivated at the sites of H2O2 generation. These are optimal prerequisites for longevity and long-distance travel of H2O2. This allows a distribution of H2O2 over the entire organelle of origin and beyond in the cytosolic space across the surrounding lipid membranes through facilitated diffusion via peroxiporins. Under these conditions, H2O2 can travel as long and as far until it meets free iron (II) or copper (I) ions or other suitable electron donors (Lenzen 2017). Instead of an interaction of H2O2 with a protective high affinity thiol, it can hit such a free ion, which acts as a catalyst for the formation of the most highly toxic hydroxyl radical (HO·) (Lenzen 2017). The high toxicity of the HO· radical has thermodynamic and kinetic reasons. Its very high standard oxidative potential provides the thermodynamic driving force for many oxidations, and its small radius and uncharged state provides a great mobility, thereby allowing high rates of chemical reactions.
Relation between the subcellular organelles in the generation of hydrogen peroxide and the hydroxyl radical in the pancreatic beta cell
The volume of the beta cell covered by the mitochondria and the peroxisomes is very low, 4% and less than 1% of the cytosolic ground substance, respectively (Dean 1973; Lenzen and Panten 1983). The cytosolic ground substance comprises more than 50% of the beta cell volume (Dean 1973). The consequence of the very low beta cell mitochondrial volume and the even smaller peroxisomal volume is that any crossing of H2O2 into the surrounding cytoplasmic space results in a decrease of the H2O2 concentration in these organelles through the dilution in a larger cellular space. This can result in a lower H2O2 toxicity in the organelle of origin. Vice versa, it is not surprising that a knockout of a peroxiporin (e.g., aquaporin-8) can result in an increased H2O2 toxicity in the mitochondria through an increased steady-state H2O2 level after exposure of the insulin-producing cells to proinflammatory cytokines (Lortz et al. 2005). On the other hand, this widespread distribution over the whole cytosolic space may allow H2O2 to reach many different intracellular sites. However, the ultimate reason for the toxicity of H2O2 and the pronounced vulnerability of the pancreatic beta cell is the low level expression of H2O2-eliminating enzymes (Lenzen 2008, 2017, 2021). This explains the long persistence of the H2O2 molecule in the beta cell and maximizes the chance to be converted to the highly toxic HO· at sites all over the beta cell, in particular when H2O2 meets weakly complexed iron (II) ions (Halliwell and Gutteridge 2015; Lenzen 2017; Winterbourn 2013). It has been speculated that HO· radicals may follow a diffusion mechanism, which has a similarity with the famous Grotthuss mechanism (Osakai 2012) of H+ and HO− diffusion (so-called proton tunneling). Of course, in the case of the HO· radicals a hydrogen atom abstraction should occur instead of a proton abstraction (in fact, only bonds are moving). Very recently, it has been shown that HO· radicals do not diffuse via a hydrogen atom abstraction (Vassilev et al. 2005). Rather the diffusion rate of the HO· radical (like that of H+ and HO− ions) is typical for its physical size. An exceptionally high diffusion rate would also contribute to a higher reaction rate with whatever targets. Thus, the high reactivity of the HO· radical, which also extends to the noble metals (Au, Pt, Pd, Ag) (Nowicka et al. 2010a, b, 2011), has to be solely attributed to its radical nature.
Thus, H2O2 plays the role of a pro-radical in the weakly protected beta cell (Lenzen 2008). That means that virtually any protein, carbohydrate, lipid or nucleic acid is a potential target for the toxic action of the highly toxic HO· radical with its extremely high reactivity (Lenzen 2017; Lushchak 2014). This explains the universal unspecific toxicity of the HO· radical towards any chemical structure at the site of its generation against which no protection whatsoever is possible. A protection is possible only by elimination of H2O2, which, however, is limited in the pancreatic beta cell due to the low level of expression of H2O2-eliminating enzymes (Lenzen 2017).
The role of the superoxide radical anion
When the primary reactive oxygen species generated is not H2O2 but the O2·− radical anion, O2·− is efficiently transformed into H2O2 through SOD isoenzymes (Lortz et al. 2005). This is in particular true for the highly expressed MnSOD in the beta cell mitochondria (Lortz et al. 2005) so that also when O2·− is initially generated, it is instantaneously available as H2O2 owing to the action of SOD (Mehmeti et al. 2011). Thus SOD acts as a pro-oxidant enzyme (Lortz et al. 2005; Lushchak et al. 2005), whenever the conversion of H2O2 into the HO· radical is fostered in a cell type like the beta cell with its low abundance of H2O2-eliminating enzymes (Lenzen 2017). The hydrophilic O2·− as a precursor of the hydrophilic H2O2 is a pro-oxidant, which as an anion is restricted in its mobility to the compartment of generation. In contrast to the negatively charged O2·−, the mobility of the electroneutral H2O2 molecule is virtually not restricted and thus reaches more sites than O2·− for mediating toxicity.
The special situation in the pancreatic beta cell ER
The ER in secretory cells such as the pancreatic beta cells is a subcellular compartment in which a very oxidized state prevails. The strongly oxidative milieu in the ER is the result of high amounts of H2O2 generated as well as of the low GSH/GSSG ratio (Appenzeller-Herzog 2011). This is achieved in an oxidation step mediated by PDI oxidoreductases (protein disulfide isomerases) [for details, see Lenzen (2017)].The ER acts as a H2O2 store, keeping a greater amount of the generated H2O2 in the ER due to a limited efflux of H2O2 into the cytosolic space. Insulin biosynthesis is a process which takes place constantly in the pancreatic beta cells. Its rate is high in particular at increased blood glucose concentrations in the postprandial state. Further accelerated is the increased rate of insulin biosynthesis in the prediabetic phase during the development of the T2DM state due to the prevailing insulin resistance under these conditions. This goes along with an increased rate of insulin misfolding, to which other islet cell hormones in other islet cell types are not prone, thus further accelerating ER stress (Lenzen 2017). In the prediabetic state, the insulin resistance induced hyperinsulinemia can be compensated by increased rates of insulin biosynthesis until the compensatory mechanisms ultimately collapse along with a gradually decreasing insulin synthesizing capacity. This results in continuously decreasing plasma insulin levels in the circulation due to progressive development of beta cell dysfunction along with an open state of ER stress.
Insulin comprises the major portion of the proteins synthesized in the pancreatic beta cell. For each proinsulin molecule folded in the ER three molecules of H2O2 are generated (Lenzen 2017) giving rise to a concentration of H2O2 higher than in many other cell types. This high level of H2O2 is also due to the fact that the ER membrane is rather impermeable for H2O2 (Konno et al. 2015). This is advantageous, since a significant proportion of H2O2 generated in the ER during proinsulin folding is consumed again for PDI re-oxidation (Hudson et al. 2015; Lenzen 2017). At the same time, high H2O2 levels in the ER are a risk factor with potential for beta cell dysfunction, as documented by the fact that an increase of the low expression level of the antioxidative enzyme Prdx4 in rodent beta cells through overexpression goes along with enhanced glucose-induced insulin secretion due to increased proinsulin mRNA transcription and insulin content (Mehmeti et al. 2012, 2014). An increased expression of the peroxiredoxin Prdx4 (Mehmeti et al. 2014, 2012) and the glutathione peroxidases GPx7 and GPx8 (Mehmeti et al. 2017) resident in the ER of many cell types can also improve the antioxidative capacity in the ER of rodent beta cells, though GPx7 and GPx8 are not expressed constitutively in their ER (Mehmeti et al. 2017). At variance from Prdx4, however, expression of these glutathione peroxidases in the ER has no positive effect on proinsulin folding (Lenzen 2017; Mehmeti et al. 2017).
Such a reinforcement of the antioxidative and functional capacity of the ER is thus an option to improve the resistance of the pancreatic beta cell against the challenges of diabetic metabolic stress. Exactly, this is the option which has been exercised by the ER in the human beta cell. This is likely the best possible choice of the human beta cell to protect the ER thereby allowing the human beta cell to remain viable over the long lifespan of the human being. Nevertheless, a Westernized lifestyle that is often associated with insufficient exercise and overweight remains a constant challenge also for a well-protected ER. Even the best protected proinsulin-folding machinery in the human ER can be overwhelmed through a lifestyle, which overcharges the functional capacity of the beta cell.
Though the rough ER compartment is relatively large, comprising one seventh of the beta cell volume (Dean 1973), the H2O2 generation during protein oxidative folding and its consumption during PDI re-oxidation takes place around the proinsulin molecule. The ER as a whole is occupied by the proinsulin-folding apparatus and rather oxidized through limited H2O2 transition into the surrounding cytosol. Due to this, there is no need for a long-distance travel of H2O2 within the ER.
ER stress under type 1 and type 2 diabetic conditions: a comparison
When glucose tolerance starts to deteriorate towards the end of the prediabetic phase along with the start of a shrinking of the beta cell mass in the pancreas the pressure on the remaining beta cells through metabolic stress increases steadily. This poses an increased functional demand on the ER in each remaining beta cell, both in the quickly decreasing beta cell mass under T1DM conditions as well as in the gradually decreasing beta cell mass with its continuously increasing dysfunction under T2DM conditions (Lenzen 2017). In particular in the type 2 scenario, this ER stress can induce a process lasting many years or even decades, comprising both an initial phase of compensatory hyperinsulinemia due to insulin resistance and a subsequent phase of a slowly developing hypoinsulinemia due to deterioration of the insulin biosynthetic capacity in the beta cell ER and the gradual decease of the beta cell volume (Lenzen 2017).
This is a scenario which overstresses the remaining beta cells, which are still capable of providing insulin in the diabetic metabolic state and thus gives rise also to more misfolded proinsulin (Lenzen 2017). Therefore, the induction of a beta cell rest through provision of insulin by exogenous administration or by a blockade of insulin release by administration of a KATP channel opener such as diazoxide (Lenzen 2017) is a feasible measure. The desired result of this resting state of the insulin biosynthesis apparatus can be ideally a cessation of proinsulin folding and thus a reduction of H2O2 generation, thereby reducing the ER stress.
Conclusions
The pancreatic beta cell is characterized by a number of unique features (Lenzen 2021). One of these is the extreme vulnerability to ROS toxicity due to its weak antioxidative defense equipment compared to many other better protected cell types (Lenzen 2008, 2017; Lenzen et al. 1996; Tiedge et al. 1997). This is particularly true in states of diabetic metabolic stress. A crucial element of the beta cell demise in the developing diabetic state is the extreme toxicity of the HO· radical. All biological structures in the beta cell are potential targets for this toxic action. In contrast, H2O2 causes only limited cellular dysfunction. However, due to its longevity and capability to reach all sites in the cell, H2O2 acts as a spreader for the toxicity of the HO· radical reaching virtually all sites of the cell, where H2O2 in the presence of “free iron” can generate the HO· radical (Fig. 1). This allows HO· to execute its extremely toxic action leading to pancreatic beta cell dysfunction and ultimately to beta cell death in the vulnerable beta cell with its low enzymatic capacity for H2O2 inactivation as firmly documented (Grankvist et al. 1981; Lenzen 2008, 2017; Lenzen et al. 1996; Tiedge et al. 1997).
Fig. 1.
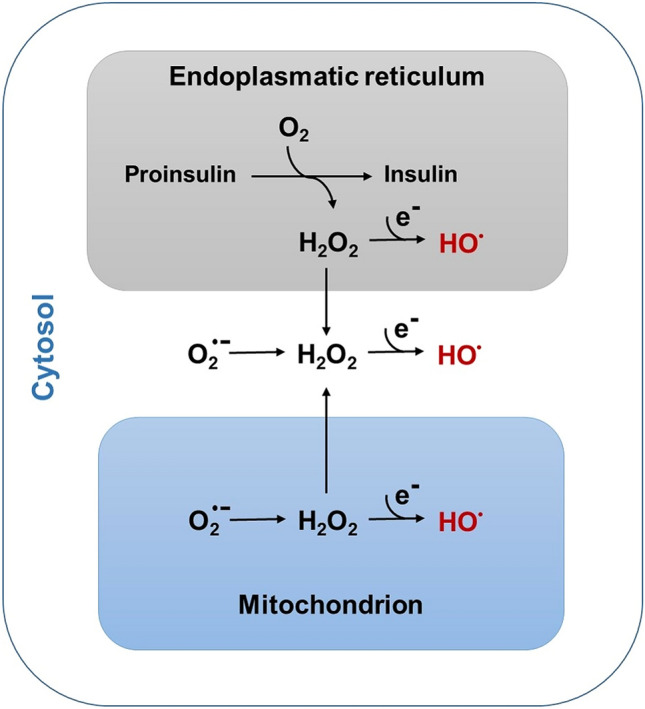
Formation of HO· radicals as part of the homeostasis of reactive oxygen species in pancreatic beta cells. This figure depicts the different pathways of hydrogen (H2O2) generation in the different subcellular organelles, with H2O2 either originating from the superoxide radical in the cytosol and in the mitochondria or directly during proinsulin-folding in the endoplasmic reticulum (ER). The figure provides no quantitative information on the amounts of H2O2 formation in the different subcellular compartments
Acknowledgements
This article is dedicated to the memory of my old friend, the distinguished toxicologist and free radical scientist Dr. Rex Munday (1942-2017), Ruakura Agricultural Research Centre, Hamilton, New Zealand.
Abbreviations
- GPx
Glutathione peroxidase
- H2O2
Hydrogen peroxide
- O2·−
Superoxide radical
- HO·
Hydroxyl radical
- Prx
Peroxiredoxin
- SOD
Superoxide dismutase
Author contributions
SL had the idea for the concept of this review article and all three authors collected data from the literature, interpreted and integrated them into the new concept and draw the conclusions with their perspectives. The review originated during the cooperation within the Graduiertenkolleg 1947 in Greifswald, funded by the German Research Council.
Funding
Open Access funding enabled and organized by Projekt DEAL. No funding has been received to assist with the preparation of this review article.
Availability of data and material
Data for this review article originate from the publications in the reference list.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
Not applicable.
Consent for publication
All the authors agreed to the publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Appenzeller-Herzog C. Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. J Cell Sci. 2011;124(6):847–855. doi: 10.1242/jcs.080895. [DOI] [PubMed] [Google Scholar]
- Bienert GP, Chaumont F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochem Biophys Acta. 2014;1840(5):1596–1604. doi: 10.1016/j.bbagen.2013.09.017. [DOI] [PubMed] [Google Scholar]
- Dean PM. Ultrastructural morphometry of the pancreatic β-cell. Diabetologia. 1973;9(2):115–119. doi: 10.1007/BF01230690. [DOI] [PubMed] [Google Scholar]
- Gehrmann W, Elsner M, Lenzen S. Role of metabolically generated reactive oxygen species for lipotoxicity in pancreatic beta-cells. Diabetes Obes Metab. 2010;12(Suppl 2):149–158. doi: 10.1111/j.1463-1326.2010.01265.x. [DOI] [PubMed] [Google Scholar]
- Grankvist K, Marklund SL, Täljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J. 1981;199(2):393–398. doi: 10.1042/bj1990393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006;141(2):312–22. doi: 10.1104/pp.106.077073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge MC. Free radicals in biology and medicine. 5. Oxford: Oxford University Press; 2015. [Google Scholar]
- Hudson DA, Gannon SA, Thorpe C. Oxidative protein folding: from thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic Biol Med. 2015;80:171–182. doi: 10.1016/j.freeradbiomed.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP, Sies H. The redox code. Antioxid Redox Signal. 2015;23(9):734–746. doi: 10.1089/ars.2015.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T, Pinho Melo E, Lopes C, et al. ERO1-independent production of H2O2 within the endoplasmic reticulum fuels Prdx4-mediated oxidative protein folding. J Cell Biol. 2015;211(2):253–259. doi: 10.1083/jcb.201506123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte A, Lortz S, Schaal C, Lenzen S, Elsner M. Hydrogen peroxide permeability of cellular membranes in insulin-producing cells. Biochim Biophys Acta. 2020;1862(2):183096. doi: 10.1016/j.bbamem.2019.183096. [DOI] [PubMed] [Google Scholar]
- Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans. 2008;36(Pt 3):343–347. doi: 10.1042/BST0360343. [DOI] [PubMed] [Google Scholar]
- Lenzen S. Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic beta-cells. Biochim Biophys Acta. 2017;1861(8):1929–1942. doi: 10.1016/j.bbagen.2017.05.013. [DOI] [PubMed] [Google Scholar]
- Lenzen S (2021) The pancreatic beta cell: an intricate relation between anatomical structure, the signalling mechanism of glucose-induced insulin secretion, the low antioxidative defence, the high vulnerability and sensitivity to diabetic stress. Chemtexts 7, Article Number 13. 10.1007/s40828-021-00140-3
- Lenzen S, Panten U. Characterization of succinate dehydrogenase and alpha-glycerophosphate dehydrogenase in pancreatic islets. Biochem Med. 1983;30(3):349–356. doi: 10.1016/0006-2944(83)90027-3. [DOI] [PubMed] [Google Scholar]
- Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20(3):463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- Lortz S, Gurgul-Convey E, Lenzen S, Tiedge M. Importance of mitochondrial superoxide dismutase expression in insulin-producing cells for the toxicity of reactive oxygen species and proinflammatory cytokines. Diabetologia. 2005;48(8):1541–1548. doi: 10.1007/s00125-005-1822-3. [DOI] [PubMed] [Google Scholar]
- Lushchak VI. Adaptive response to oxidative stress: Bacteria, fungi, plants and animals. Comp Biochem Phys C. 2011;153(2):175–190. doi: 10.1016/j.cbpc.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Lushchak VI. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem-Biol Interact. 2014;224:164–175. doi: 10.1016/j.cbi.2014.10.016. [DOI] [PubMed] [Google Scholar]
- Lushchak V, Semchyshyn H, Mandryk S, Lushchak E. Possible role of superoxide dismutases in the yeast Saccharomyces cerevisiae under respiratory conditions. Arch Biochem Biophys. 2005;441(1):35–40. doi: 10.1016/j.abb.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Mehmeti I, Gurgul-Convey E, Lenzen S, Lortz S. Induction of the intrinsic apoptosis pathway in insulin-secreting cells is dependent on oxidative damage of mitochondria but independent of caspase-12 activation. Biochem Biophys Acta. 2011;1813(10):1827–1835. doi: 10.1016/j.bbamcr.2011.06.022. [DOI] [PubMed] [Google Scholar]
- Mehmeti I, Lortz S, Lenzen S. The H2O2-sensitive HyPer protein targeted to the endoplasmic reticulum as a mirror of the oxidizing thiol-disulfide milieu. Free Radic Biol Med. 2012;53(7):1451–1458. doi: 10.1016/j.freeradbiomed.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Mehmeti I, Lortz S, Elsner M, Lenzen S. Peroxiredoxin 4 improves insulin biosynthesis and glucose-induced insulin secretion in insulin-secreting INS-1E cells. J Biol Chem. 2014;289(39):26904–26913. doi: 10.1074/jbc.M114.568329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmeti I, Lortz S, Avezov E, Jörns A, Lenzen S. ER-resident antioxidative GPx7 and GPx8 enzyme isoforms protect insulin-secreting INS-1E beta-cells against lipotoxicity by improving the ER antioxidative capacity. Free Radical Biol Med. 2017;112:121–130. doi: 10.1016/j.freeradbiomed.2017.07.021. [DOI] [PubMed] [Google Scholar]
- Nowicka AM, Hasse U, Hermes M, Scholz F. Hydroxyl radicals attack metallic gold. Angew Chem Int Edit. 2010;49(6):1061–1063. doi: 10.1002/anie.200906358. [DOI] [PubMed] [Google Scholar]
- Nowicka AM, Hasse U, Sievers G, et al. Selective knockout of gold active sites. Angew Chem Int Edit. 2010;49(17):3006–3009. doi: 10.1002/anie.201000485. [DOI] [PubMed] [Google Scholar]
- Nowicka AM, Hasse U, Donten M, Hermes M, Stojek ZJ, Scholz F. The treatment of Ag, Pd, Au and Pt electrodes with OHaEuro cent radicals reveals information on the nature of the electrocatalytic centers. J Solid State Electr. 2011;15(10):2141–2147. doi: 10.1007/s10008-011-1488-3. [DOI] [Google Scholar]
- Osakai T. Grotthuss mechanism. In: Bard AF, Inzelt G, Scholz F, editors. Electrochemical dictionary. 2. Berlin: Springer; 2012. p. 435. [Google Scholar]
- Scholz F. Luther's rule. In: Bard AF, Inzelt G, Scholz F, editors. Electrochemical dictionary. 2. Berlin: Springer; 2012. p. 514. [Google Scholar]
- Sies H. Biochemistry of oxidative stress. Angew Chem Int Ed Engl. 1986;25:1058–1071. doi: 10.1002/anie.198610581. [DOI] [Google Scholar]
- Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem. 2014;289(13):8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46(11):1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- Vassilev P, Louwerse MJ, Baerends EJ. Hydroxyl radical and hydroxide ion in liquid water: a comparative electron density functional theory study. J Phys Chem B. 2005;109(49):23605–23610. doi: 10.1021/jp044751p. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4(5):278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013;528:3–25. doi: 10.1016/B978-0-12-405881-1.00001-X. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC. Biological chemistry of superoxide radicals. Chemtexts. 2020;6(1):7. doi: 10.1007/s40828-019-0101-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data for this review article originate from the publications in the reference list.