

Abstract
Background:
House dust mite (HDM)-challenged Apoe−/− mice display enhanced airway hyperreactivity and mucous cell metaplasia.
Objective:
To characterize the pathways that induce APOE expression by asthmatic macrophages and identify how APOE regulates IL-1β secretion.
Methods:
Macrophages were isolated from asthmatic bronchoalveolar lavage fluid (BALF) and derived from THP-1 cells and human monocytes.
Results:
HDM-derived cysteine and serine proteases induced APOE secretion from asthmatic BALF macrophages via protease-activated receptor 2. APOE at concentrations of < 2.5 nM, which are similar to levels found in epithelial lining fluid (ELF) from healthy adults, did not induce IL-1β release from asthmatic BALF macrophages. In contrast, APOE at concentrations ≥ 25 nM induced NLRP3 and pro-IL-1β expression by asthmatic BALF macrophages, as well as the caspase-1-mediated generation of mature IL-1β that was secreted from cells. HDM acted synergistically with APOE to both prime and activate the NLRP3 inflammasome. In a murine model of neutrophilic airway inflammation induced by HDM and poly(I:C), APOE reached a concentration of 32 nM in ELF with associated increases in BALF IL-1β. APOE-dependent NLRP3 inflammasome activation in macrophages was primarily mediated by a potassium efflux-dependent mechanism.
Conclusion:
APOE can function as an endogenous, concentration-dependent pulmonary danger signal that primes and activates the NLPR3 inflammasome in asthmatic BALF macrophages to secrete IL-1β. This may represent a mechanism by which APOE amplifies pulmonary inflammatory responses when concentrations in the lung are increased above normal levels, which may occur during viral exacerbations of house dust mite-induced asthma characterized by neutrophilic airway inflammation.
Keywords: Asthma, apolipoprotein E, house dust mite, interleukin-1β, macrophages, NLRP3 inflammasome
Capsule summary:
Apolipoprotein E is a concentration-dependent danger signal in the lung that can prime and activate the NLRP3 inflammasome in asthmatic macrophages to secrete IL-1β. This may promote and amplify inflammation when pulmonary APOE concentrations are increased above normal levels.
Introduction
Apolipoprotein E (APOE) is a 34-kDa protein that is primarily synthesized by the liver and plays an important role in lipid homeostasis by facilitating the clearance of triglyceride- and cholesterol-containing lipoprotein particles, such as chylomicron remnants and very low-density lipoproteins (VLDL), from plasma into cells via receptor-mediated endocytosis1, 2. APOE-containing high-density lipoprotein (HDL) particles can also facilitate reverse cholesterol transport out of macrophages, which has a protective role in atherosclerosis3, 4. However, the biological effects of APOE are not limited to vascular biology, as APOE is also synthesized in the central nervous system with the APOE ε4 allele representing the major genetic risk factor for the development of Alzheimer’s disease1. Furthermore, APOE can modulate innate and adaptive immune responses, as well as host defense against a variety of pathogens, including malaria, bacteria, mycobacteria, and viruses1, 5.
APOE is also expressed by multiple cell types in the lung, including alveolar macrophages, type I and type II alveolar epithelial cells, and pulmonary artery smooth muscle cells5–8. Studies using murine model systems have shown that APOE modulates normal lung development. For example, Apoe−/− mice have impaired alveologenesis with increased airway resistance and accelerated loss of lung recoil during aging, as well as elevated levels of desaturated phosphatidylcholine, a major surfactant lipid9–11. APOE also modulates the pathogenesis of allergic airways disease, as house dust mite (HDM)-challenged Apoe−/− and Ldlr−/− mice display a phenotype of enhanced airway hyperreactivity and mucous cell metaplasia, without increases in airway inflammation, which is consistent with a protective role in asthma8. Neutrophilic lung inflammation, oxidative stress, acute lung injury, and cigarette smoke-induced emphysema are also increased in Apoe−/− mice, which provides evidence for an anti-inflammatory role for APOE in the lung5, 12–15. Similarly, APOE has been shown to have a beneficial effect in the setting of systemic endotoxemia, as Apoe−/− mice display a phenotype of enhanced lipopolysaccharide (LPS)-mediated inflammation and reduced host defense against bacterial infections that may in part reflect direct binding and neutralization of LPS by APOE5, 16–18. Experiments utilizing murine macrophages have similarly demonstrated that APOE has anti-inflammatory functions. For example, murine macrophages from Apoe−/− mice have increased NF-κB signaling and inflammatory responses to LPS stimulation that were mediated by decreased levels of microRNA-146a, which is a key negative regulator of NF-κB signaling19. APOE converted murine RAW264.7 macrophages from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype, with decreased production of iNOS (inducible nitric oxide synthase), IL-12, and CCL320. Similarly, APOE suppressed the TLR3- and TLR4-mediated activation of pro-inflammatory c-Jun N-terminal kinase signaling pathways in murine RAW264.7 macrophages21.
Several studies, however, have shown that APOE can also augment inflammation. For example, administration of APOE3 to rats with polymicrobial sepsis increased mortality due to the augmented presentation of endogenous lipid antigens which activated natural killer T cells, increased Th1 cytokines, and enhanced liver injury22, 23. Similarly, treatment of murine peritoneal macrophages with human APOE activated p38 MAPK, c-Jun terminal kinase, and NF-κB signaling via a MyD88-dependent pathway24. APOE also induced IL-6 production by murine peritoneal macrophages via a CD14- and TLR2-dependent pathway25. Taken collectively, these studies suggest that APOE can also mediate context-dependent pro-inflammatory effects.
Here, we sought to characterize the pathways that induce APOE expression by asthmatic macrophages and to identify how APOE regulates IL-1β secretion by these cells. First, we show that the serine and cysteine protease components of HDM, which is a major perennial aeroallergen that causes allergic airway inflammation, induce APOE production by bronchoalveolar lavage fluid (BALF) macrophages isolated from asthmatic subjects via a protease-activated receptor 2 (PAR2)-dependent pathway26, 27. Second, we identify that APOE can independently, as well as synergistically with HDM, increase the expression of NLRP3 and pro-IL-1β by asthmatic BALF macrophages. APOE also activates caspase-1 to generate a mature IL-1β that is secreted from macrophages via a potassium efflux-dependent mechanism. In addition, we used a murine model of neutrophilic airway inflammation induced by HDM and the double-stranded RNA surrogate, poly(I:C), which mimics a concurrent viral-induced asthma exacerbation, to show that APOE can reach levels in lung epithelial lining fluid (ELF) that are sufficiently high to activate the NLRP3 inflammasome in asthmatic BALF macrophages, as well as increase BALF IL-1β. Thus, we propose that when concentrations of APOE are increased in the lung above normal levels, it may function as an endogenous, concentration-dependent danger signal that both primes and activates the NLRP3 inflammasome in asthmatic BALF macrophages leading to IL-1β secretion. This may represent a mechanism by which increased concentrations of APOE can promote and amplify inflammatory responses in the lung, which may be relevant during viral exacerbations of house dust mite-induced asthma characterized by neutrophilic airway inflammation.
Methods
Reagents:
House dust mite extract (# B84, Dermatophagoides pteronyssinus, Greer Laboratories, Inc., Lenoir, NC) was depleted of endotoxin using Pierce™ High Capacity Endotoxin Removal Spin Columns (# 88274, ThermoFisher Scientific, Waltham, MA) to generate a final concentration of 0.096 endotoxin units (EU) per microgram of HDM protein. Recombinant human IFN-γ(# 570204), TNF-α(# 570104), IL-13 (# 571104), IL-17A (# 570504), and M-CSF (# 574806) were from BioLegend (San Diego, CA). Poly (I:C) (# tlrl-pic) and polymyxin B (# tlrl-pmb) were from InvivoGen (San Diego, CA). E64 (# 5208), AEBSF (# 5175), protease activated receptor-1 (PAR-1) antagonist, RWJ 56110 (# 2614), and PAR-2 antagonist, FSLLRY-NH2 (# 4751), were from Tocris (Minneapolis, MN). Recombinant human APOE3 (# 350-02) and recombinant human IL-10 (# 200-10) were from PeproTech (Rocky Hill, NJ). The recombinant human APOE3 contained 0.021 EU/μg of protein. Dexamethasone (# D4902), LPS from Escherichia coli O111:B4 (# L4391), MitoTEMPO (# SML0737), CA-074 methyl ester (# C5857), A-438079 hydrochloride hydrate (# A9736), E-64d (# E8640), potassium chloride (KCL) (# 60142), methyl-β-cyclodextrin (# C4555), α-cyclodextrin (# C4680) and cholesterol-loaded, water soluble, methyl-β-cyclodextrin (# C4951) were from Millipore Sigma (St. Louis MO). Z-YVAD-FMK (# 218746) and MCC950 (# 5.38120.0001) were from Calbiochem (Burlington, MA).
Research Subjects.
Asthmatic subjects (n = 17) provided informed consent and participated in protocol 99-H-0076, which was approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute. Demographics for the asthmatic subjects are presented in Repository eTable 1. Asthma was confirmed in all subjects by either a positive response to an inhaled β2-agonist or a positive methacholine bronchoprovocation challenge test. Allergy status was determined by skin testing, which was performed either at NHLBI or by their local physician, or a history of severe allergy. Allergy skin testing at NHLBI was performed using a Multi-Test II applicator, which was a generous gift from Lincoln Diagnostics (Decatur, IL). Histamine was utilized as a positive control and glycerin was a negative control (Holister-Stier, Spokane, WA). Allergy was defined by the presence of skin test reactivity to at least one of six common aeroallergens; house dust mite (Dermatophagoides farinae), cockroaches (Periplaneta americana and Battella germanica), cat hair, Aspergillus fumigatus, Ragweed (giant and short) and grasses (Kentucky Bluegrass, Orchard Redtop, Timothy Sweet Vernal grass, Meadow Fescue, and Perennial Ryegrass).
Isolation and Culture of Bronchoalveolar Lavage Fluid (BALF) Macrophages.
Bronchoalveolar lavage was performed in each of three sub-segmental bronchi using 90 cc of normal saline that had been pre-warmed to 37° C. BALF was gently aspirated by hand suction and cells were transported to the laboratory on ice. BALF was filtered through gauze sponge (USP, type VII Gauze, 12 Ply) to remove debris and cells were pelleted by centrifugation at 275 × g for 10 min. After lysis of red blood cells with ACK buffer (#118-156-101; Quality Biological, Gaithersburg, MD), BALF cells were pelleted by centrifugation for 5 min at 275 × g and resuspended in RPMI-1640 media containing 10% fetal bovine serum (FBS) at a concentration of 2 × 105 macrophages per well for 96-well plate experiments or 1 × 106 macrophages per well were for 6-well plate experiments. Macrophages were adhered to plastic tissue culture plates for 2 hours and non-adherent cells were removed by washes with RPMI-1640 for 96-well plate experiments or RPMI-1640 plus 1% FBS for 6-well plate experiments, which yielded a purity of > 95% macrophages. Experiments were performed over 24 hours.
THP-1 macrophages.
The THP-1 human monocytic leukemia cell line (ATCC® TIB-202™) was purchased from American Type Culture Collection (Manassas, VA) and maintained in RPMI-1640 supplemented with 10% heat-inactivated FBS, HEPES (25 mM), penicillin-streptomycin-glutamine, and β-mercaptoethanol (0.05 mM) at a density of 3–8 × 105 cells/mL. THP-1 cells were differentiated into macrophages by seeding 5 × 105 cells per well in 24-well tissue culture plates, or 1 × 106 cells per well in 6-well tissue culture plates, with phorbol 12-myristate 13-acetate (PMA) (10 ng/mL, # P8139; Sigma-Aldrich) for 48 hours28. Cells were rested in media without PMA for 24 hours and stimulated with APOE for 4 hours in media that contained 1% FBS.
Human monocyte-derived macrophages.
Elutriated human monocytes from anonymous donors were provided under an IRB-approved protocol by the NIH Clinical Center Department of Transfusion Medicine. Cells were centrifuged at 275 × g for 5 minutes, supernatant was removed and cells were resuspended at a concentration of 50 × 106 cells/mL in 90% FBS and 10% dimethyl sulfoxide29. Cell suspensions of 1 mL each were placed into cryovials and frozen for future use. Upon thawing, monocytes were cultured in complete RPMI-1640 medium supplemented with 10% FBS, penicillin-streptomycin-glutamine, and recombinant human M-CSF (50 ng/mL), which was changed every 48 hours until the monocytes differentiated into macrophages after 7 days. To dislodge adherent cells, flasks were rinsed with pre-warmed Dulbecco’s phosphate-buffered saline without Ca2+ and Mg2+ × 3 and pre-warmed 0.25% trypsin-ethylenediaminetetraacetic acid solution (# 25200 056; ThermoFisher Scientific) was added for 15 minutes. Following trypsin neutralization by addition of RPMI-1640 containing 10% FBS, cells were centrifuged at 275 × g for 5 min and cells were seeded in 24-well dishes at a density of 2 × 105 cells per well for 24 hours in complete media supplemented with recombinant human M-CSF. Experiments were performed for either 7 or 24 hours in media containing 1% FBS.
Murine Airway Inflammation Models.
Female Balb/c mice (6 to 7 weeks of age) were purchased from Envigo (East Millstone, NJ) and experiments were performed using protocols approved by the Animal Care and Use Committee of the National Heart, Lung, and Blood Institute. Mice received intranasal administration of HDM, poly(I:C) (Sigma-Aldrich, St. Louis, MO), and/or APOE and bronchoalveolar lavage was performed. Urea was quantified in BALF and serum using the Urea Assay Kit (Sigma-Aldrich, St. Louis, MO) and the volume of ELF was determined as previously described30. APOE was quantified using the Mouse apoE ELISAPRO Kit (Mabtech, Cincinnati, OH) and IL-1β was quantified using the Mouse IL-1β DuoSet ELISA (R&D Systems, Minneapolis, MN).
APOE and IL-1β assays.
Cell culture media was collected and cleared of debris by centrifugation at 3000 × g for 5 minutes prior to analysis. The amount of human apolipoprotein E secreted into the media was quantified using a human APOE ELISA development kit (HRP) (# 3712-1H-20; Mabtech, Cincinnati, OH). Human IL-1β (# DY201) and human TNF-α (# DY210) secretion were quantified using DuoSet ELISA kits from R & D Systems (Minneapolis, MN). The limits of detection for apolipoprotein E, IL-1β and TNF-α were 0.1 ng/mL, 3.91 pg/mL, and 15.6 pg/mL, respectively.
Western blotting.
Cells were lysed with Mammalian Protein Extraction Reagent (# 78501; ThermoFisher Scientific) and centrifuged at 14,000 × g for 10 min to remove cellular debris. Western blots were performed on cell lysates or supernatants using NuPAGE™ 4–12% Bis-Tris gels (# NP0335BOX; ThermoFisher Scientific) and proteins were transferred to Invitrolon™ PVDF membranes (#LC2005; ThermoFisher Scientific). PVDF membranes were reacted with antibodies directed against ASC (# sc-514414; Santa Cruz Biotechnology, Dallas, TX) and NLRC4 (# 659702; BioLegend), as well as IL-1β (# 12703S), cleaved IL-1β (# 83186S), NLRP3 (# 13158S), Caspase-1 (# 2225S), Caspase-5 (# 46680), and Caspase-8 (# 9746), all from Cell Signaling, Danvers, MA. To control for equivalency of protein loading of cellular proteins, membranes were stripped with OneMinute Western Blot Stripping Buffer (#GM6001; GM Biosciences, Frederick, MD) and reacted with an antibody directed against β-actin (# A5441; Sigma-Aldrich). Ponceau S staining was used to verify the equivalent loading of proteins in culture media. HRP-conjugated secondary antibodies included anti-rabbit (# 7074S) and anti-mouse secondary antibodies (# 7076S), both from Cell Signaling. Chemiluminescence was detected using SuperSignal West Pico Chemiluminescent Substrate (# 34080; ThermoFisher Scientific) and images were acquired using an LAS4000 imaging system (GE Healthcare Life Sciences, Pittsburgh, PA).
Statistics.
Data analysis was performed using Graph Pad Prism version 7.0b. Data are presented as mean ± SEM. A P value < 0.05 was considered significant.
Results
House Dust Mite-derived Serine and Cysteine Proteases Induce Apolipoprotein E Secretion from Asthmatic BALF Macrophages via Protease-activated Receptor 2
To characterize the spectrum of inflammatory mediators that induce APOE production by asthmatic macrophages, cells isolated from BALF were treated ex vivo with type 1 (interferon-γ and TNF-α), type 2 (IL-13), and type 17 (IL-17A) cytokines, as well as ligands for toll-like receptor 3 (poly I:C) and toll-like receptor 4 (LPS). In addition, cells were treated with HDM, dexamethasone, and HDM plus dexamethasone. As shown in Figure 1A, only HDM induced APOE secretion by asthmatic macrophages, which was not inhibited by dexamethasone, whereas type 1, 2, or 17 cytokines, as well as ligands for TLR3 or TLR4 did not induce APOE release. Since protease activity is an important contributor to the allergenicity of HDM, we next assessed whether inhibitors of cysteine and serine proteases attenuate HDM-induced APOE secretion from asthmatic macrophages26. As shown in Figure 1B, both the cysteine protease inhibitor, E64, and the serine protease inhibitor, AEBSF, significantly reduced HDM-induced APOE secretion. Next, we assessed whether the ability of HDM-derived serine and cysteine proteases to induce APOE secretion by asthmatic macrophages was dependent upon protease-activated receptors. As shown in Figure 1C, HDM-induced APOE secretion was attenuated by an antagonist of protease-activated receptor 2 (PAR-2), but not by an antagonist of protease-activated receptor 1 (PAR-1). Taken collectively, these results are consistent with the conclusion that HDM-derived serine and cysteine proteases activate PAR-2 to induce APOE secretion by BALF macrophages isolated from asthmatic subjects.
Figure 1. House dust mite (HDM) induces apolipoprotein E (APOE) secretion from asthmatic bronchoalveolar lavage fluid (BALF) macrophages.
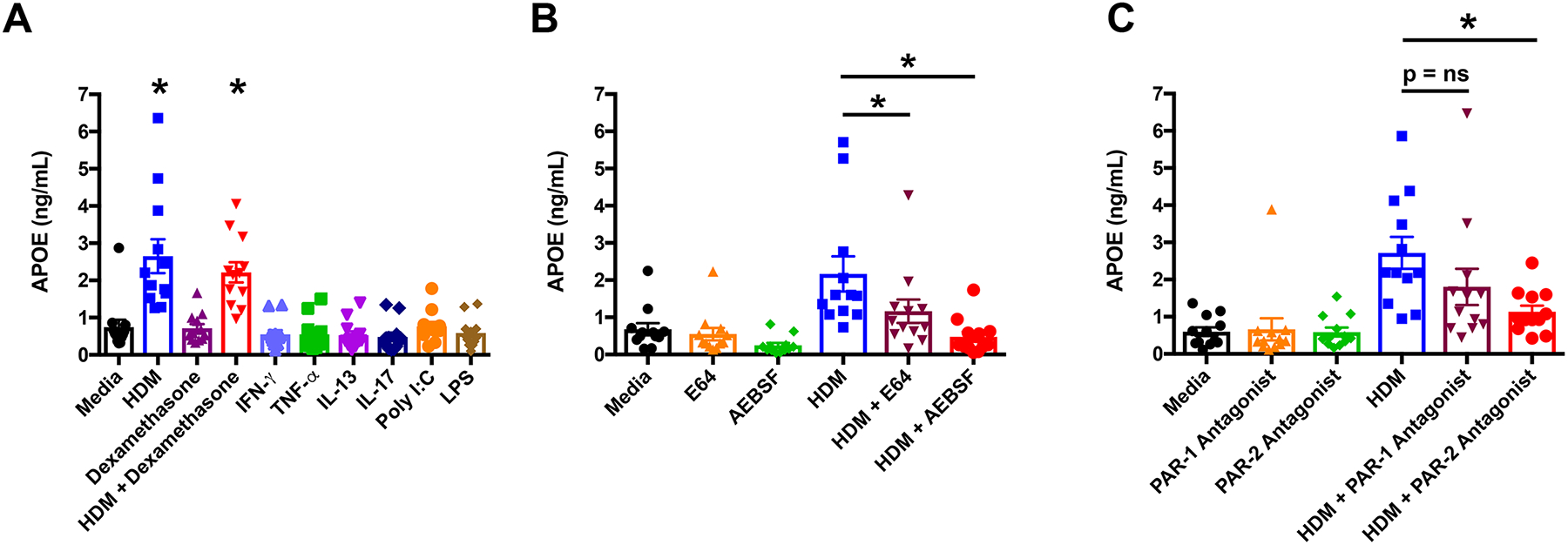
A. BALF macrophages from asthmatic subjects were treated for 24 hours with HDM (10 μg/mL), dexamethasone (0.1 μM), HDM + dexamethasone, IFN-γ (10 ng/mL), TNF-α (10 ng/mL), IL-13 (10 ng/mL), IL-17 (10 ng/mL), poly I:C (25 μg/mL), or LPS (10 ng/mL) (n = 12 subjects, P < 0.0001 versus media). B. BALF macrophages were treated with HDM alone or in combination with E64 (10 μM) or AEBSF (150 μM) (n = 12 subjects, P < 0.05 versus HDM). C. BALF macrophages were treated with HDM alone or in combination with a PAR-1 antagonist (10 μM) or PAR-2 antagonist (10 μM) (n = 12 subjects, P < 0.01 versus HDM). A one-way ANOVA with Dunnett’s multiple comparisons test was utilized for all experiments.
APOE Primes the NLRP3 Inflammasome and Induces the Secretion of Mature IL-1β from Asthmatic BALF Macrophages
One mechanism by which HDM can induce inflammation is via the activation of the NLRP3 inflammasome with resultant generation of the pro-inflammatory cytokine, IL-1β31, 32. Consistent with this, we show that asthmatic BALF macrophages treated ex vivo with HDM secreted increased amounts of IL-1β as compared to cells that were treated with media alone (Figure 2A). HDM also induced the secretion of TNF-α by asthmatic BALF macrophages (Repository eFigure 1), which is consistent with the conclusion that HDM skews asthmatic BALF macrophages towards a M1 phenotype33. In contrast, CCL17, which is a product of M2 macrophages, could not be detected in the media of HDM-treated cells. Since HDM can induce the secretion of both APOE and IL-1β from asthmatic BALF macrophages, we next considered whether APOE can modify IL-1β secretion from these cells. To address this question, asthmatic BALF macrophages were treated with HDM alone or in combination with 500 nM APOE, which is similar to the concentration of APOE present in serum34. As shown in Figure 2B, APOE alone as well as APOE in combination with HDM (HDM + APOE), induced significant increases in IL-1β secretion from asthmatic BALF macrophages. Furthermore, asthmatic BALF macrophages that were exposed to HDM + APOE secreted the largest amount of IL-1β, suggesting that HDM acted synergistically with APOE. As the recombinant human APOE preparation contained approximately 2.1 pg of lipopolysaccharide (LPS) per μg of protein, experiments were performed to exclude the possibility that APOE-mediated IL-1β secretion from asthmatic BALF macrophages reflected LPS contamination. As shown in Figure 2B, asthmatic BALF macrophages treated with the equivalent amount of LPS present in recombinant APOE did not secrete IL-1β, which supports the conclusion that the ability of APOE to induce IL-1β secretion by asthmatic BALF macrophages was not a consequence of LPS contamination.
Figure 2. APOE is Signal 1 and Signal 2 for NLRP3 inflammasome activation and IL-1β secretion by asthmatic BALF macrophages.
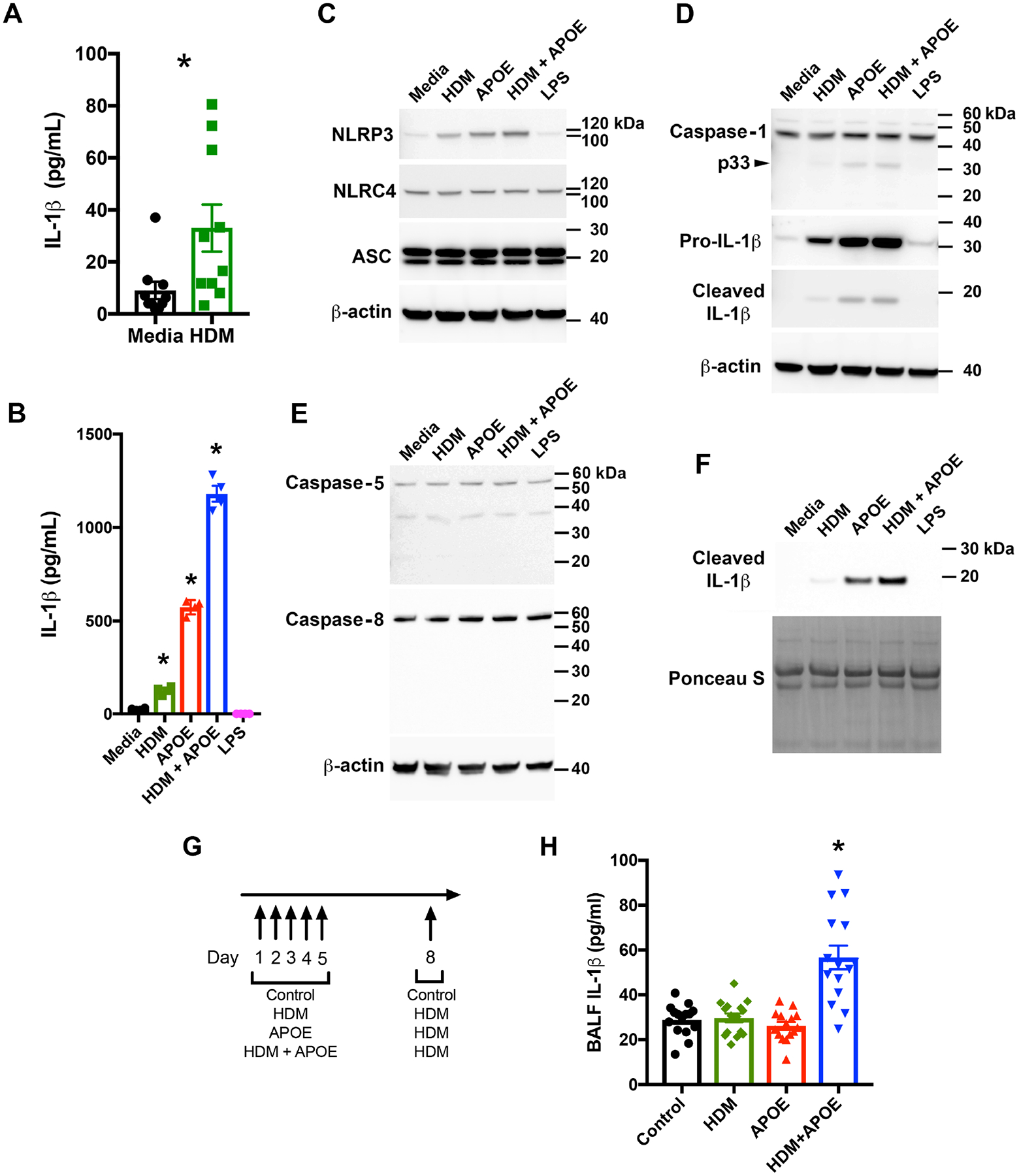
A. Macrophages were cultured with or without HDM (10 μg/mL) for 24 hours. (n = 10 asthmatics, P = 0.002, Wilcoxon matched-pairs signed rank test). B. Macrophages were cultured with HDM (10 μg/mL), APOE (500 nM), HDM + APOE, or LPS (38 pg/mL), for 24 hours. (n = 4, P < 0.05 vs. media, Sidak’s multiple comparisons test). This result is representative of 6 asthmatics. C, D and E. Western blots of macrophage lysates from the subject in panel B. F. Western blots of media from the subject in panel B. Western blots shown are representative of 3 independent experiments using macrophages from different asthmatics. G. Mice received saline (control), HDM (50 μg), APOE (12 μg), or HDM + APOE, daily for 5 days. On Day 8, the HDM, APOE and HDM + APOE groups received HDM (100 μg) and bronchoalveolar lavage was performed 4 hours later. H. IL-1β in BALF from mice challenged with saline (control), HDM, APOE, or HDM + APOE (n = 15 mice per group, * P = 0.0001 vs. control (saline), Dunnett’s multiple comparisons test).
Western blots were next performed to characterize further the role of APOE in priming and activating the NLRP3 inflammasome in asthmatic BALF macrophages. As shown in Figures 2C and 2D, HDM alone, APOE alone, and HDM + APOE, induced the expression of both NLRP3 and pro-IL-1β in asthmatic BALF macrophage cell lysates, whereas the expression of NLRC4 was not modified and NLRP1 was not detected. HDM + APOE, as well as APOE alone, also induced the processing of the full-length, 46-kDa (p46) pro-caspase-1 to generate the active, 33-kDa (p33) caspase-1 species that cleaves pro-IL-1β to its mature 17-kDa form (Figure 2D)35. In contrast, neither HDM + APOE nor APOE alone, induced the processing of pro-caspase-5 or pro-caspase-8 (Figure 2E). HDM + APOE, as well as APOE alone, induced the secretion the mature 17-kDa IL-1β isoform into the culture medium (Figure 2F), whereas HDM alone minimally induced the secretion of mature IL-1β (Figure 2F) and also minimally induced the cleavage of pro-caspase-1 in BALF macrophage cell lysates (Figure 2D). Pro-caspase-1 cleavage products could not be detected in the culture medium of asthmatic BALF macrophages by Western blotting. Treatment of asthmatic BALF macrophages with the amount of LPS present in recombinant APOE did not induce the expression of NLRP3 or pro-IL-1β nor did it induce the cleavage of caspase-1 or pro-IL-1β, which again shows that the effects of APOE were not mediated by the quantity of LPS contaminating the APOE reagent. Taken collectively, these data are consistent with the conclusion that both HDM and APOE can prime asthmatic BALF macrophages by increasing the expression of NLRP3 and pro-IL-1β. APOE, and to a lesser extent HDM, also induce the autoproteolytic activation of caspase-1 and the processing of pro-IL-1β to a 17-kDa mature IL-1β that is secreted to the extracellular space. Furthermore, APOE functions synergistically with HDM to prime and activate the NLRP3 inflammasome, which further enhances the secretion of mature IL-1β from asthmatic BALF macrophages.
We then used a murine model system to confirm that HDM acts synergistically with APOE to induce increases in BALF IL-1β in vivo. As shown in Figure 2G, Balb/c mice received intranasal administration of either saline, HDM, APOE, or HDM plus APOE (HDM+APOE) followed by a subsequent HDM challenge. Only mice treated with HDM+APOE showed significant increases in BALF IL-1β, whereas BALF IL-1β in mice that received HDM alone or APOE alone were not different from saline treated mice, which served as a control (Figure 2H). This shows that HDM can act synergistically with APOE in vivo to induce IL-1β production in the mouse lung.
APOE Induces Concentration-dependent Priming and Activation of the NLRP3 Inflammasome in Asthmatic BALF Macrophages
Having shown that concentrations of APOE similar to those found in human serum can both prime and activate the NLRP3 inflammasome with resultant IL-1β secretion by asthmatic BALF macrophages, we investigated whether lower APOE concentrations mediate a similar effect. In particular, we assessed whether APOE at concentrations similar to those present in lung ELF from normal healthy adults, which ranges from 0.3 to 2.4 nM, can prime and activate the NLRP3 inflammasome36. APOE at concentrations of 0.25 nM and 2.5 nM did not increase the expression of NLRP3 (Figure 3A) nor did it activate the cleavage of pro-IL-1β to a mature form (Figure 3B) that is secreted (Figure 3C) from asthmatic BALF macrophages. APOE at a concentration of 2.5 nM increased the expression of pro-IL-1β whereas 0.25 nM APOE did not (Figure 3B). In contrast, higher APOE concentrations (25 nM to 500 nM) both increased the expression of NLRP3 (Figure 3A) and pro-IL-1β (Figure 3B), as well as induced the cleavage of pro-IL-1β to its 17-kDa mature form (Figure 3B) that is secreted from asthmatic BALF macrophages to the medium (Figure 3C). This finding is consistent with the conclusion that concentrations of APOE similar to those found in normal ELF from healthy individuals are not sufficient to either prime the NLRP3 inflammasome nor induce the secretion of mature IL-1β from asthmatic BALF macrophages. In contrast, concentrations of APOE that exceed levels found in normal ELF in healthy individuals both activated the NLRP3 inflammasome and induced IL-1β secretion from asthmatic BALF macrophages.
Figure 3. APOE induces concentration-dependent IL-1β secretion by asthmatic BALF macrophages.
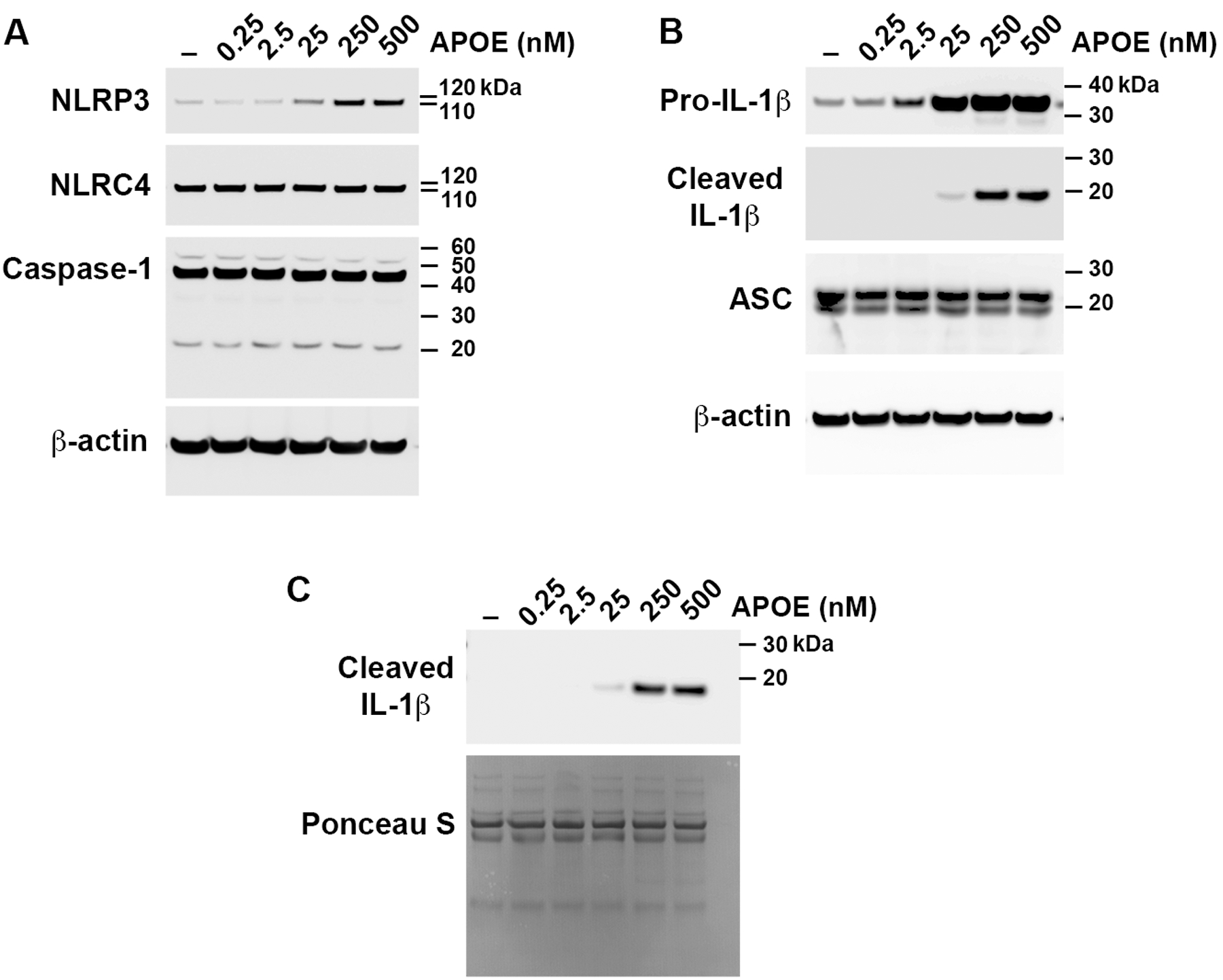
Asthmatic BALF macrophages were cultured with increasing concentrations of APOE (0.25 nM to 500 nM) for 24 hours and Western blots were performed on cell lysates (Panels A and B) and cell culture media (Panel C). The Western blot shown is representative of 3 independent experiments using BALF macrophages from different asthmatic subjects.
Additional experiments assessed whether APOE concentrations in the lung can reach levels ≥ 25 nM, which can activate the NLRP3 inflammasome and induce IL-1β secretion by ex vivo cultures of asthmatic BALF macrophages. Since IL-1β contributes to the pathogenesis of neutrophilic airway inflammation in asthma, we used a murine model of neutrophilic airway inflammation induced by HDM with subsequent administration of the double-stranded RNA surrogate, poly(I:C), to mimic a concurrent viral-induced asthma exacerbation (Figure 4A)37, 38. Of note, poly(I:C) alone does not induce IL-1β secretion in the mouse lung37, 39, 40. As shown in Figure 4B, HDM+poly(I:C) induced a neutrophilic-predominant inflammatory phenotype that was associated with significant increases in BALF IL-1β (Figure 4C). Furthermore, the concentration of APOE in ELF from mice challenged with HDM+poly(I:C) was 32 ± 5 nM (Figure 4D), which is a concentration that activated the NLRP3 inflammasome in ex vivo cultures of asthmatic BALF macrophages. This shows that in the setting of viral exacerbations complicating HDM-induced neutrophilic airway inflammation, APOE concentrations in the murine lung are increased to the range that activated the NLRP3 inflammasome in ex vivo cultures of asthmatic BALF macrophages.
Figure 4. Murine viral exacerbation model of HDM-induced neutrophilic airway inflammation.
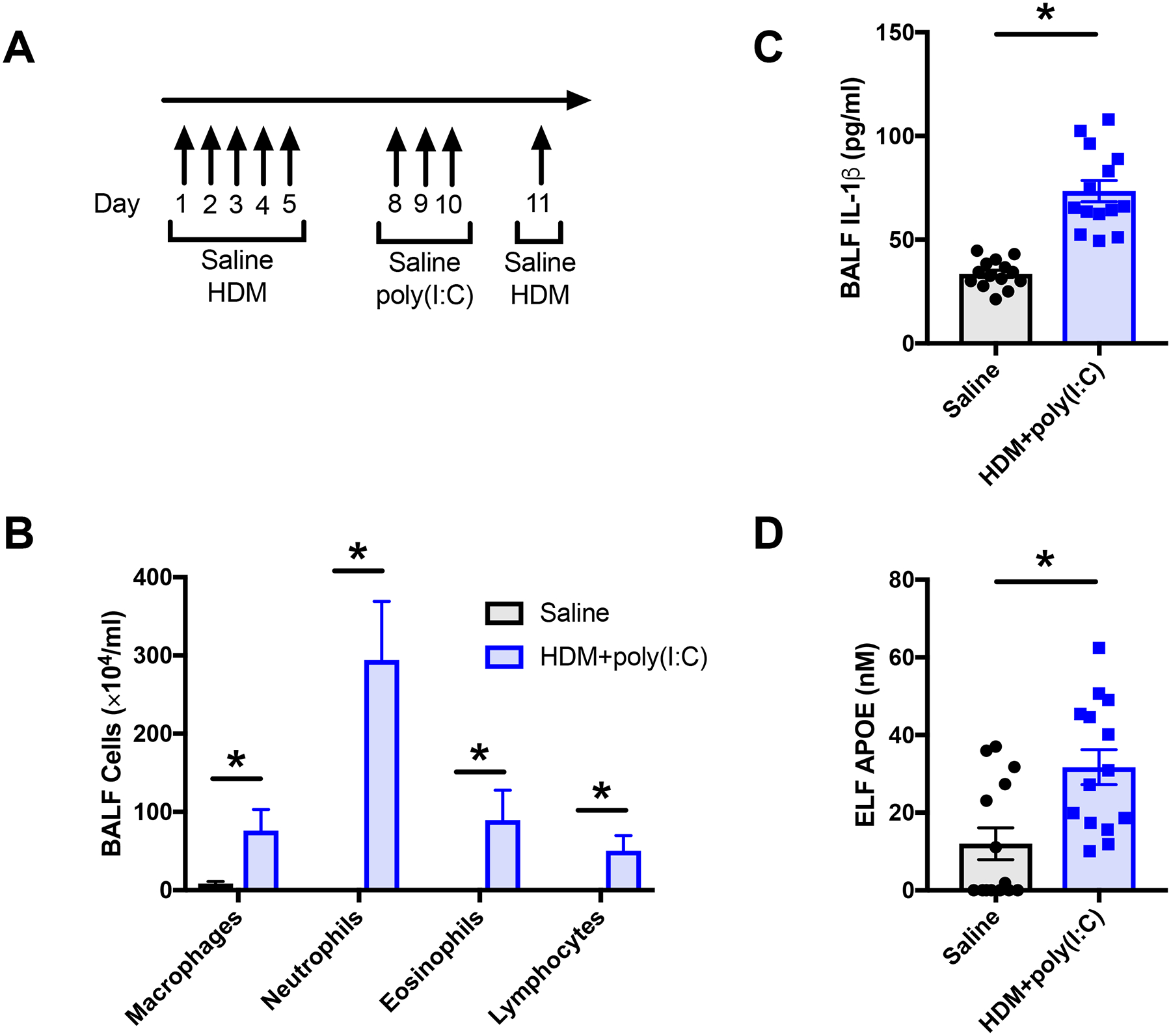
A. Mice were sensitized by intranasal administration of saline (control) or HDM (50 μg) daily for 5 days (n = 14 mice per group). To mimic a viral exacerbation, the HDM group mice received intranasal administration of the dsRNA surrogate, poly(I:C) (150 μg) daily, on days 8 to 10. On day 11, the HDM group was challenged by intranasal administration of HDM (100 μg) and lungs were harvested for bronchoalveolar lavage 4 hours later. B. The amount of IL-1β in BALF was quantified by ELISA (*P < 0.0001, unpaired t test). C. BALF cell counts (*P < 0.0001, Mann-Whitney test). D. The amount of APOE in lung epithelial lining fluid (ELF) was quantified by ELISA (*P = 0.0046, Mann-Whitney test).
APOE Induces NLRP3 Inflammasome Activation and IL-1β Secretion by THP-1 Macrophages
To confirm and elucidate further the mechanisms by which APOE mediates IL-1β secretion in macrophages, we next performed experiments utilizing THP-1 macrophages that had been differentiated in the presence of phorbol ester (i.e., PMA), which also serves as a priming signal that induces the expression of both NLRP3 and pro-IL-1β41, 42. As shown in Figure 5A and 5B, both APOE alone (50 nM), and in combination with HDM (HDM + APOE), increased the expression of NLRP3 and pro-IL-1β above basal levels. Furthermore, both APOE alone and HDM + APOE induced the cleavage of pro-caspase-1 to generate the p33 and p20 fragments in cell lysates (Figure 5B), as well as the secretion of the catalytically inactive p20 fragment into the medium (Figure 5C)35. Similarly, both APOE alone and HDM + APOE induced the cleavage of pro-IL-1β in cell lysates to generate a mature, 17-kDa IL-1β isoform that could be detected both in cell lysates and medium (Figure 5B and 5C). Activation of the NLRP3 inflammasome in THP-1 macrophages by APOE alone or HDM + APOE appeared to induce a more robust cleavage of pro-IL-1β and subsequent generation of a mature, 17-kDa IL-1β isoform than was seen in asthmatic BALF macrophages, which likely reflects differences between primary macrophages and THP-1 cells that have been differentiated into macrophages with PMA. In contrast, the amount of LPS present in the APOE reagent did not induce the expression of pro-IL-1β, increase the expression of NLRP3, or stimulate the cleavage of caspase-1 or pro-IL-1β. These experiments show that APOE activates the NLRP3 inflammasome in THP-1 macrophages, which induces the autoproteolytic cleavage of caspase-1 and the maturation of pro-IL-1β.
Figure 5. APOE induces the expression of pro-IL-1β and NLRP3, as well as the secretion of cleaved IL-1β, by THP-1 macrophages.
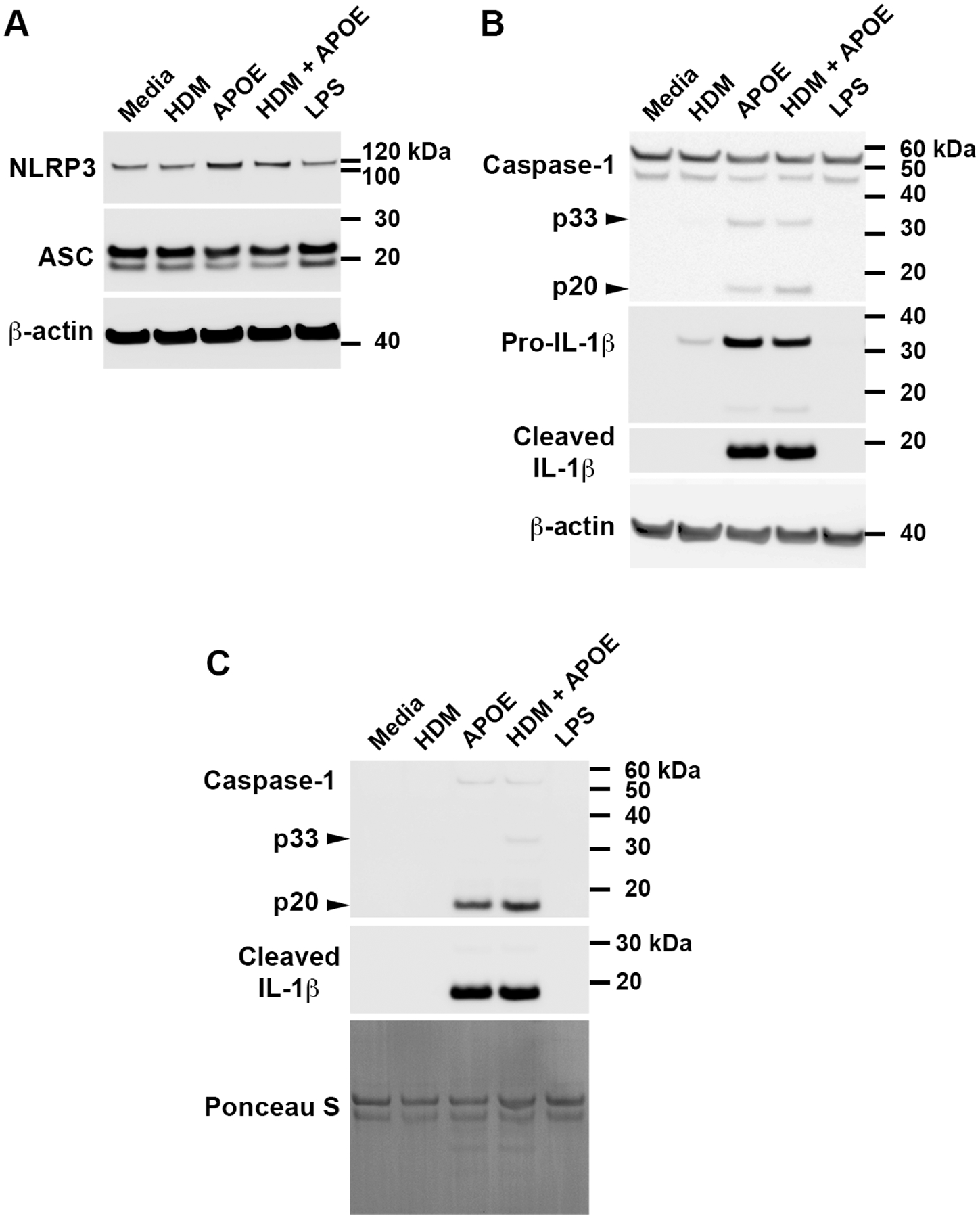
THP-1 macrophages were cultured with HDM alone (10 μg/mL), APOE alone (50 nM), HDM + APOE, or LPS alone (38 pg/mL), for 4 hours. Western blots were performed on cell lysates (Panels A and B) and cell culture media (Panel C). The Western blot shown is representative of 4 independent experiments that showed similar results.
The ability of APOE to induce IL-1β secretion from THP-1 macrophages was confirmed by ELISA. Consistent with the Western blot results, APOE induced IL-1β secretion from THP-1 macrophages in a concentration-dependent fashion, with 50 nM APOE inducing maximal effects (Figure 6A). Based on this result, a concentration of 50 nM APOE was utilized for subsequent THP-1 macrophage experiments. Consistent with the results from the Western blot experiments, APOE alone induced a similar amount of IL-1β secretion as when cells were treated with HDM + APOE (Figure 6B), which may reflect that these cells are already primed by PMA. Since HDM alone did not induce significant increases in IL-1β secretion after 24 hours, additional experiments were performed to assess whether longer periods of HDM exposure might be required for stimulation of IL-1β secretion by monocyte-derived macrophages. As shown in Repository eFigure 2, HDM stimulation for 48 or 72 hours also did not induce significant increases in IL-1β secretion by human monocyte-derived macrophages. Thus, in contrast to asthmatic BALF macrophages, THP-1 monocyte-derived macrophages and human monocyte-derived macrophages did not secrete IL-1β in response to HDM stimulation. To exclude non-specific effects related to stimulation of cells with recombinant APOE, we next assessed whether another recombinant protein, human IL-10, could induce IL-1β secretion from THP-1 macrophages. As shown in Figure 5C, IL-10 (54 nM) did not induce IL-1β secretion from THP-1 macrophages, which is consistent with the conclusion that the ability of APOE to induce IL-1β secretion was not a non-specific effect of recombinant proteins. To confirm further that the ability of APOE to induce IL-1β secretion was not a consequence of LPS contamination, we showed that the amount of LPS present in the APOE reagent did not induce IL-1β secretion from THP-1 macrophages (Figure 6D). Furthermore, we showed that the ability of APOE to induce IL-1β secretion was not attenuated when polymyxin B was added to APOE in order to bind contaminating LPS (Figure 6E). Since IL-1β has been implicated in the pathogenesis of steroid-resistant asthma, we also assessed whether the ability of APOE to induce IL-1β secretion from THP-1 macrophages could be inhibited by dexamethasone43. As shown in Figure 6F, although dexamethasone significantly attenuated APOE-mediated IL-1β secretion, levels remained increased as compared to controls. This shows that corticosteroids do not completely suppress APOE-mediated IL-1β secretion by THP-1 macrophages.
Figure 6. APOE-mediated IL-1β Secretion by THP-1 macrophages is partially suppressed by corticosteroids.
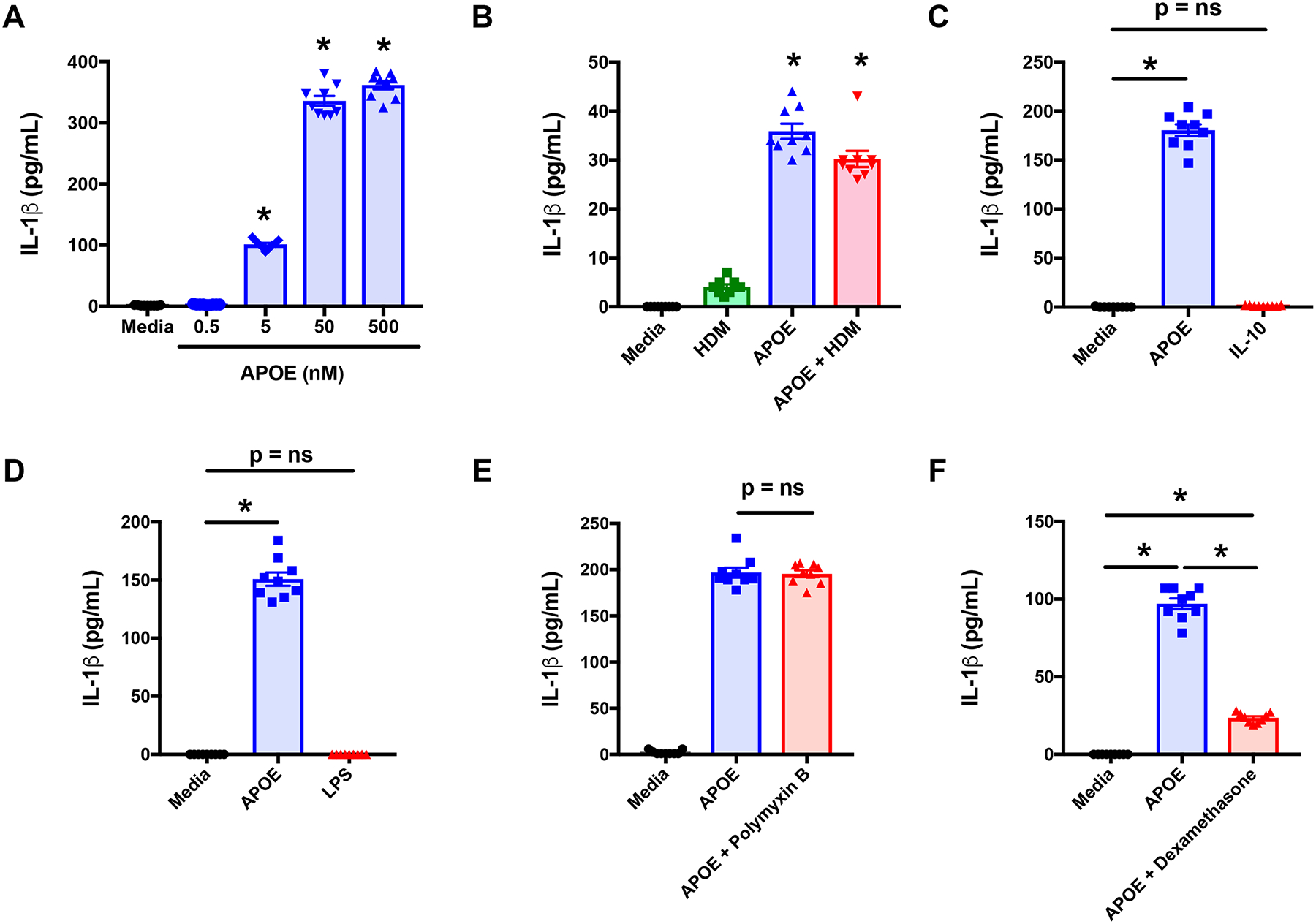
A. THP-1 macrophages were treated with APOE (0.5 – 500 nM). (n = 9, * p < 0.004 versus APOE (0 nM)). B. THP-1 macrophages were treated with HDM (10 μg/mL), APOE (50 nM) or APOE + HDM (n = 9, * p < 0.0066 versus media). C. THP-1 macrophages were treated with APOE (50 nM) or IL-10 (54 nM) (n = 9, * p = 0.0001 versus media). D. THP-1 macrophages were treated with APOE (50 nM) or LPS (38 pg/mL). (n = 9, * p < 0.001 vs. media) E. THP-1 macrophages were treated with APOE (50 nM) or APOE plus Polymyxin B (25 μg/mL) (n = 9, p = ns, APOE vs. APOE + Polymyxin B). F. THP-1 macrophages were treated with APOE (50 nM) or APOE plus Dexamethasone (0.1 μM) (n = 9, * p < 0.0001). Pooled data from 3 independent experiments using a 4 hour treatment are shown. Panels A and C were analyzed using Dunnett’s multiple comparisons test and Panels B, D through F were analyzed using Sidak’s multiple comparisons test.
Having demonstrated that APOE increases the expression of NLRP3 in THP-1 macrophages, experiments were performed to define the role of the NLRP3 inflammasome and caspase-1 in mediating IL-1β secretion from these cells. As shown in Figure 7A, MCC950, which is a highly active and specific NLRP3 inhibitor, significantly attenuated APOE-induced IL-1β secretion from THP-1 macrophages44. We next confirmed that APOE-mediated NLRP3 inflammasome activation induced caspase-1 to cleave pro-IL-1β into its mature form, which is secreted to the extracellular space. As shown in Figure 7B, the caspase-1 inhibitor, Z-YVAD-FMK, significantly attenuated APOE-mediated IL-1β secretion from THP-1 macrophages41, 45, 46. We then demonstrated that IL-1β secretion from asthmatic BALF macrophages was similarly mediated by NLRP3 and caspase-1. As shown in Figure 7C, both the NLRP3 inhibitor, MCC950, and the caspase-1 inhibitor, Z-YVAD-FMK, significantly inhibited the secretion of the 17-kDa cleaved IL-1β isoform into the media from asthmatic BALF macrophages that had been stimulated with APOE. Taken collectively, these results show that APOE acts via the NLRP3 inflammasome and caspase-1 to induce the cleavage and secretion of mature IL-1β from both THP-1 macrophages and asthmatic BALF macrophages.
Figure 7. APOE-mediated IL-1β secretion by THP-1 and asthmatic macrophages is mediated by activation of the NLRP3 inflammasome and caspase-1.
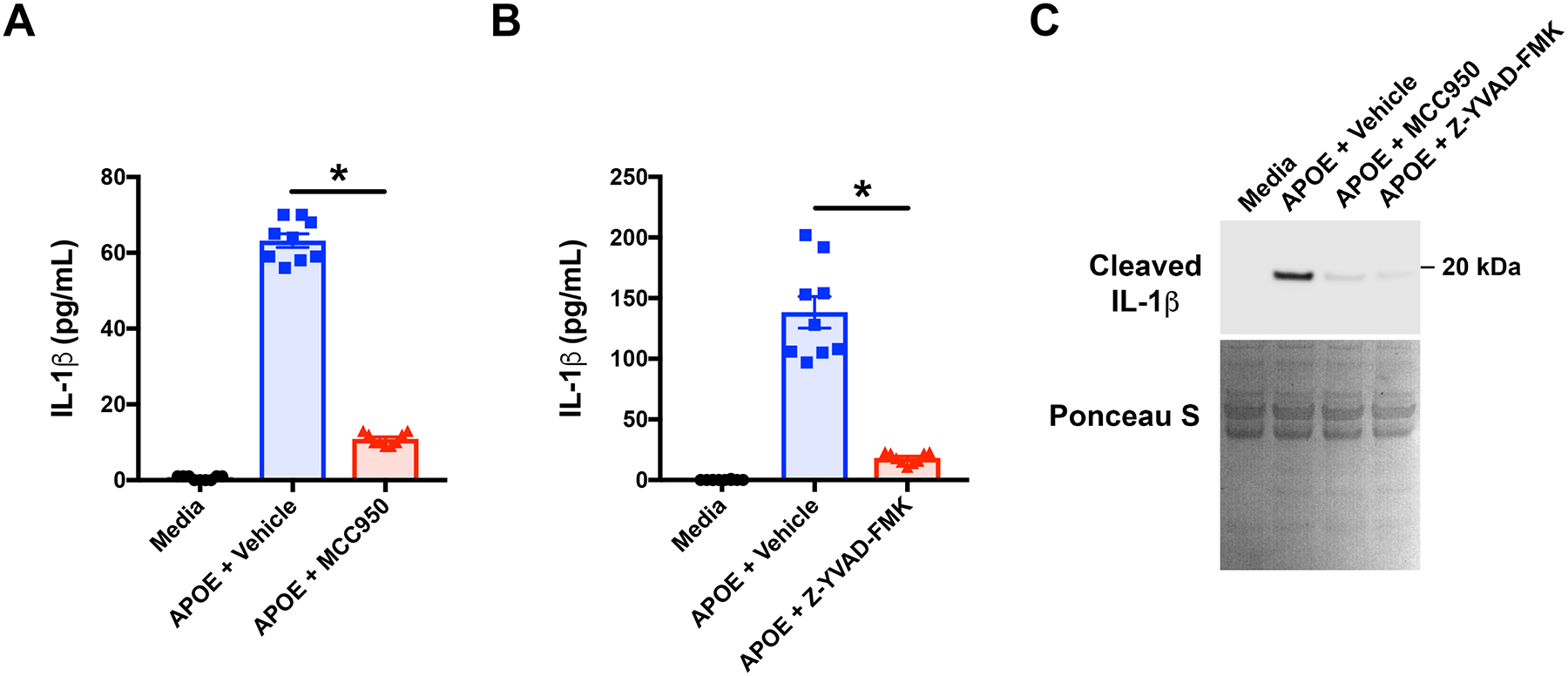
A. THP-1 macrophages were treated with APOE plus the NLRP3 inhibitor, MCC950 (1 μM), or vehicle (DMSO), for 4 hours and the quantity of IL-1β secreted into the media was quantified by ELISA. (n = 9, * p < 0.0001 APOE + MCC950 vs. APOE + Vehicle, Sidak’s multiple comparisons test). B. THP-1 macrophages were treated with APOE plus the selective caspase-1 inhibitor, Z-YVAD-FMK (10 μM), or Vehicle (DMSO) for 4 hours and the quantity of IL-1β secreted into the media was quantified by ELISA. (n = 9, * p < 0.0001 APOE + Z-YVAD-FMK vs. APOE + vehicle, Sidak’s multiple comparisons test). C. Asthmatic BALF macrophages were cultured with media, as well as APOE plus MCC950 (1 μM), Z-YVAD-FMK (10 μM), or vehicle (DMSO) for 24 hours. Proteins secreted into the media were separated by SDS-PAGE and reacted with antibodies that detected cleaved, mature, 17-kDa IL-1β. Total proteins were stained with Ponceau S to show equivalency of protein loading in each well. The Western blot shown is representative of 4 independent experiments using BALF macrophages from different asthmatic subjects.
APOE Activates the NLRP3 Inflammasome in Macrophages via a Potassium Efflux-dependent Mechanism
We next sought to define the mechanism by which APOE activates the NLRP3 inflammasome in macrophages. Mitochondrial dysfunction with associated production of reactive oxygen species (ROS) has been implicated in NLRP3 activation, although its exact role remains unclear47, 48. Treatment of THP-1 macrophages with the mitochondria-targeted anti-oxidant, MitoTEMPO, which is a specific scavenger of mitochondrial superoxide, did not attenuate, but instead increased APOE-mediated IL-1β secretion (Figure 8A). Next, we considered whether APOE might efflux cholesterol from macrophages, thereby causing a reduction in cellular cholesterol levels that activates the NLRP3 inflammasome3, 4, 49, 50. As shown in Figure 8B, acute depletion of cellular cholesterol using methyl-β-cyclodextrin (MβCD) induced IL-1β secretion from THP-1 macrophages, whereas its inactive analog, αCD, did not, which is consistent with activation of the NLRP3 inflammasome by cholesterol depletion. However, incubation of THP-1 macrophages with cholesterol-loaded MβCD to maintain cellular cholesterol levels51, did not prevent, but instead increased, APOE-mediated increases in IL-1β secretion (Figure 8C). Lysosomal damage with the release of cathepsins, represents the mechanism by which endogenous crystals, such as cholesterol, urate and amyloid-β, as well as exogenous crystals, such as silica, urate and aluminum salts, activate the NLRP3 inflammasome41, 45–47, 52. Furthermore, it has been demonstrated that multiple cathepsins mediate this effect47, 53. Therefore, we next considered whether APOE might facilitate cholesterol transport into macrophages as a mechanism to activate the NLRP3 inflammasome. However, as shown in Figure 8D, cathepsin inhibitors, E64d and CA-074me, only caused small reductions in APOE-mediated IL-1β secretion by THP-1 macrophages41, 45, 46, 52–54. Next, we assessed whether the APOE-mediated activation of the NLRP3 inflammasome involves ATP, which functions as a danger signal to bind the purinergic receptor P2X7 (P2RX7) and activate the NLRP3 inflammasome55. Treatment with the P2RX7 antagonist, A438079, also caused only a small reduction in APOE-mediated IL-1β secretion from THP-1 macrophages (Figure 8E)56.
Figure 8. Potassium efflux, P2RX7 and lysosomal cathepsins mediate APOE-induced IL-1β secretion by THP-1 macrophages.
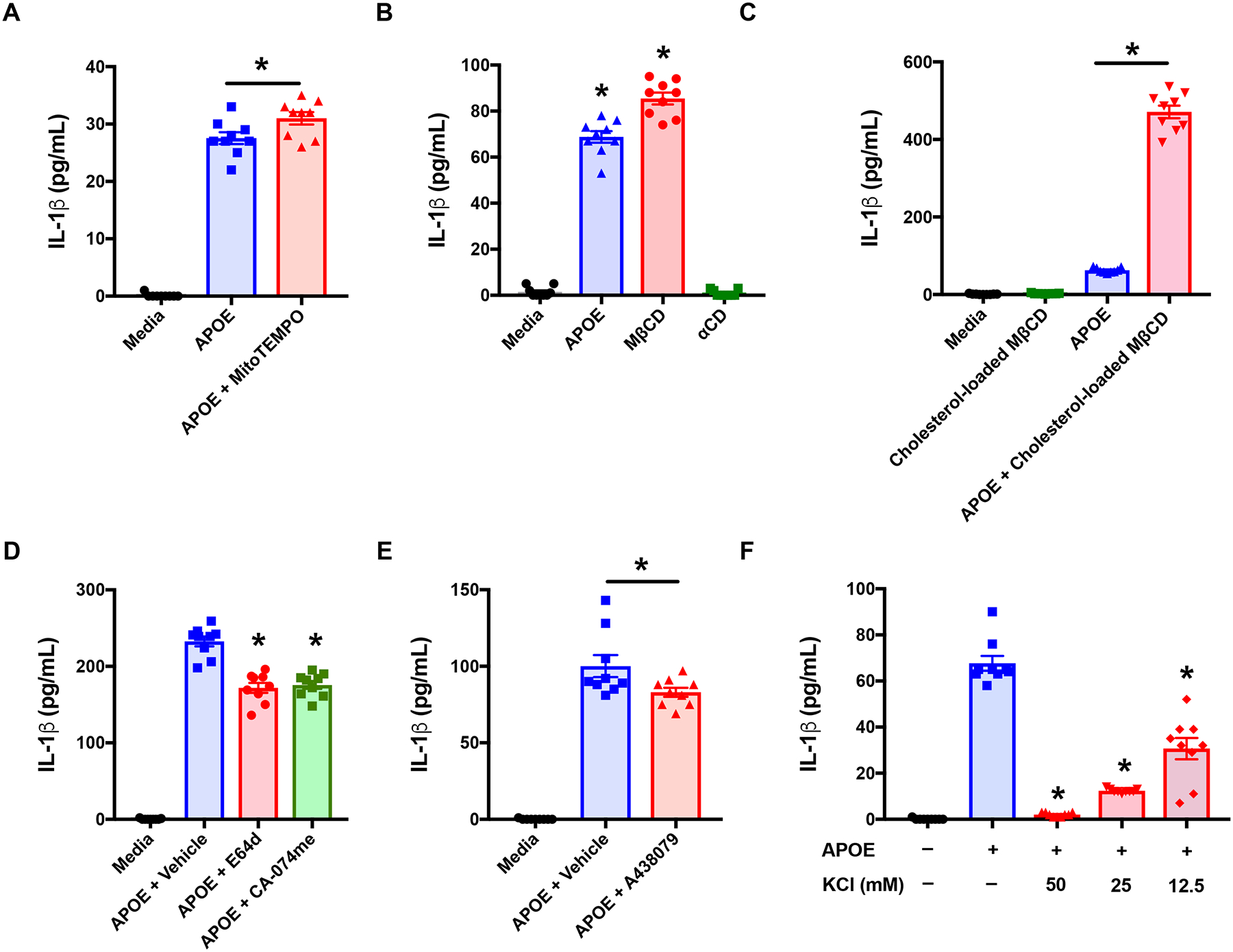
A. Cells were treated with APOE (50 nM) +/− MitoTEMPO (500 μM) for 4 hours. (n = 9, * p < 0.01, APOE vs. APOE + MitoTEMPO). B. Cells were treated with APOE (50 nM), methyl-β-cyclodextrin (MβCD, 5 mM) or α-cyclodextrin (αCD, 5 mM), for 2 hours. (n = 9, * p < 0.0001, versus media). C. Cells were treated with cholesterol-loaded MβCD (100 μg/mL), APOE (50 nM), or APOE + cholesterol-loaded MβCD for 2 hours. (n = 9, * p < 0.0001 APOE vs. APOE + cholesterol-loaded MβCD). D. Cells were treated with APOE (50 nM) plus vehicle (DMSO) or APOE plus E64d (10 μM) or CA-074me (10 μM) for 4 hours. (n = 9, * p < 0.0001 versus APOE + Vehicle). E. Cells were treated with APOE (50 nM) plus A438079 (25 μM) or vehicle (DMSO) for 4 hours. (n = 9, * p < 0.02 versus APOE + Vehicle). F. Cells were treated with APOE (50 nM) in media containing KCL (0 to 50 mM) for 2 hours. (n = 9, * p < 0.0001, versus APOE). Sidak’s multiple comparisons test was used for Panels A and C and Dunnett’s multiple comparisons test was used for Panels B and F.
Potassium efflux is recognized as a common trigger that activates the NLRP3 inflammasome in response to diverse stimuli, such as ATP, nigericin, and particulate matter, however, potassium efflux-independent mechanisms of NLRP3 activation have also been identified57, 58. To assess the role of potassium efflux in APOE-mediated NLRP3 activation in macrophages, extracellular potassium was added to the culture medium of THP-1 cells to prevent a reduction in intracellular potassium concentrations57. As shown in Figure 8F, extracellular potassium, at concentrations ranging from 12.5 mM to 50 mM, significantly inhibited APOE-mediated IL-1β secretion from THP-1 macrophages. Furthermore, the ability of 50 mM KCl to inhibit IL-1β secretion was almost complete. Taken collectively, these results suggest that potassium-efflux is the primary mechanism by which APOE induces NLRP3 inflammasome activation and IL-1β secretion from THP-1 macrophages.
To confirm the role of potassium efflux in APOE-mediated NLRP3 activation in macrophages, additional experiments were performed using human monocyte-derived macrophages (HMDMs). As shown in Figure 9A and 9B, neither the cathepsin inhibitors, E64d and CA-074me, nor the P2RX7 antagonist, A438079, inhibited APOE-mediated IL-1β secretion from HMDMs. In contrast, extracellular potassium, at concentrations ranging from 6.25 mM to 25 mM, significantly attenuated APOE-mediated IL-1β secretion from HMDMs (Figure 9C). Taken collectively, these results are consistent with the conclusion that APOE mediates NLRP3 activation and IL-1β secretion from macrophages by a potassium efflux-dependent mechanism.
Figure 9. APOE-mediated IL-1β Secretion by Human Monocyte-derived Macrophages is Primarily Mediated via a Potassium Efflux-dependent Mechanism.
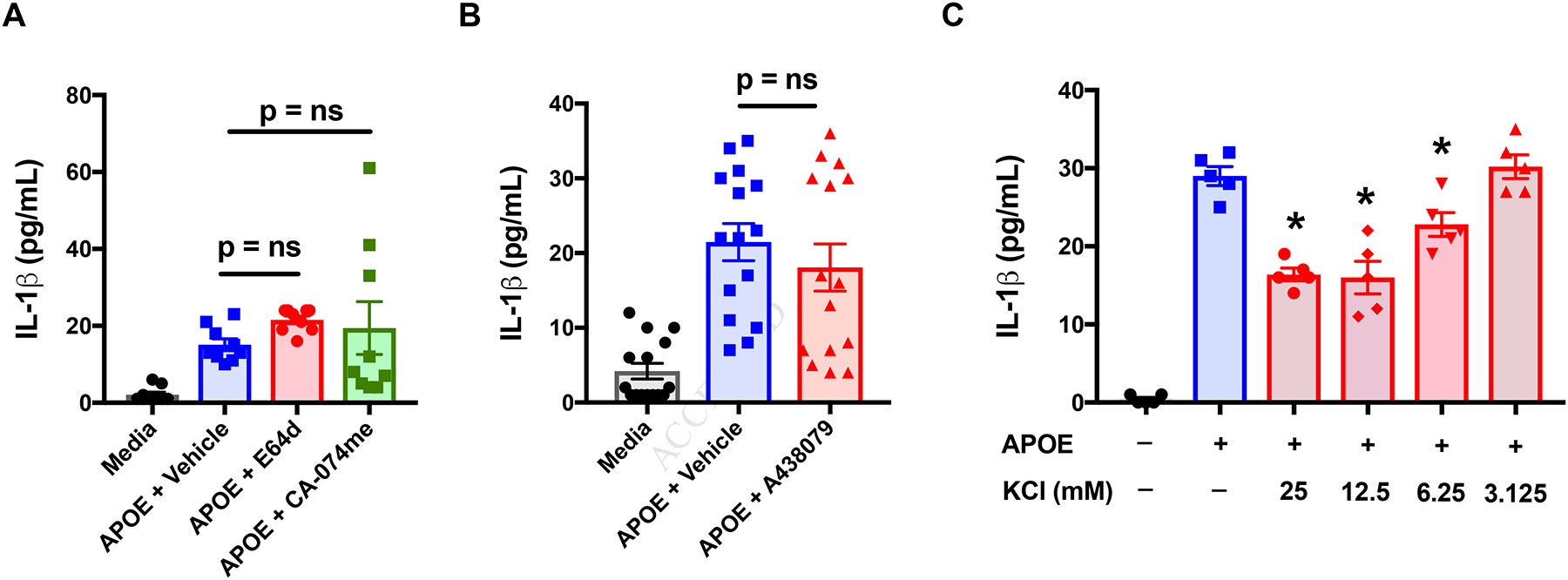
A. Human monocyte-derived macrophages (MDMs) were treated with APOE (50 nM) plus vehicle (DMSO) or APOE plus either E64-d (10 μM) or CA-074me (10 μM) for 24 hours (n = 15, p = ns, versus APOE + Vehicle, Dunnett’s multiple comparisons test). B. Human MDMs were treated with APOE (50 nM) plus either A438079 (25 μM) or vehicle (DMSO) for 24 hours. (n = 15, p = ns, versus APOE + Vehicle, Dunnett’s multiple comparisons test). The data shown in Panels A and B are pooled results from 5 donors. C. Human MDMs were treated with APOE (500 nM) in cell culture media that had been supplemented with KCL (0 to 25 mM) for 7 hours. (n = 5, * p < 0.016, versus APOE, Dunnett’s multiple comparisons test). This result is representative of experiments performed using human MDMs from 3 donors.
Discussion
Although APOE is expressed by pulmonary macrophages and modulates HDM-induced airway hyperreactivity and mucous cell metaplasia in mice, its role in regulating airway inflammatory responses in asthmatic patients is not well defined8. The first goal of this study was to identify the inflammatory mediators that induce APOE production by asthmatic airway macrophages. We found that HDM, but not other pro-inflammatory mediators, such as type 1, 2, and 17 cytokines, or TLR3 and TLR4 ligands, were capable of inducing APOE secretion by asthmatic BALF macrophages. Furthermore, HDM-mediated increases in APOE secretion from asthmatic BALF macrophages were not prevented by dexamethasone. This is consistent with our prior finding in mice, which showed that sensitization and challenge with HDM increased Apoe mRNA levels in the lung that was resistant to inhibition by corticosteroids8. Collectively, these results suggest that APOE derived from asthmatic airway macrophages may play a role in modulating airway inflammatory responses to HDM. HDM is a complex allergen that is comprised of both mites and fecal pellets26, 59. Important components of HDM allergen are serine and cysteine proteases that signal via protease-activated receptors, such as PAR-2, to amplify allergic airway inflammation. Furthermore, multiple HDM allergens can activate PAR-2, while PAR-2-deficient mice have reduced HDM-induced allergic airway inflammation60. Consistent with this, we found that HDM-induced APOE secretion by asthmatic BALF macrophages involved HDM-derived serine and cysteine proteases, as well as PAR-2.
Not only did HDM induce APOE secretion by asthmatic BALF macrophages, we also found that it induced IL-1β secretion. This is consistent with prior reports which showed that HDM can both increase pro-IL-1β expression in human monocyte-derived macrophages and activate the NLRP3 inflammasome in keratinocytes via a cysteine protease-dependent pathway31, 32. As HDM induced the secretion of both APOE and IL-1β from asthmatic BALF macrophages, we hypothesized that APOE might also modulate NLRP3 inflammasome activation and IL-1β secretion by these cells. Consequently, the second goal of this study was to identify how APOE regulates NLRP3 inflammasome activation and IL-1β secretion from asthmatic airway macrophages. IL-1β is a prototypical pro-inflammatory cytokine that plays a central role in mediating innate immune responses in the lung in response to infection or noxious stimuli, such as allergens, cigarette smoke, or particulate matter43, 61–63. Both the NLRP3 inflammasome and IL-1β have also been linked to neutrophilic airway inflammation and increased disease severity in severe, steroid-resistant asthma64–68. Consistent with this, neutrophilic airway inflammation, disease severity, and steroid resistance in asthmatics correlate with sputum levels of NLRP3 and IL-1β66.
The pathway generating mature IL-1β is complex and tightly regulated. IL-1β is initially synthesized as a 31-kDa precursor that is processed by proteolytic cleavage to generate a mature 17-kDa protein, which binds to and signals via the IL-1 receptor61. Caspase-1 is the key protease that processes pro-IL-1β to its mature form, however, non-canonical, caspase-1-independent pathways have also been identified that involve caspase-5 or caspase-842, 69. Similar to IL-1β, caspase-1 is initially generated as an inactive precursor that is activated by auto-proteolytic processing induced by formation of a high-molecular weight multimeric protein complex, termed the inflammasome35, 61–63, 70. Five canonical inflammasomes have been identified, which include the sensor proteins, NLRP1 (nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing protein 1), NRLP3, and NLRC4 (NLR family caspase recruitment domain (CARD)-containing 4), as well as pyrin and AIM2 (absent in melanoma 2)61, 63, 70, 71. The best characterized of these is the NLRP3 inflammasome.
Two signals are typically required for activation of the NLRP3 inflammasome. Signal 1 typically involves Toll-like receptor signaling, by a PAMP (pathogen-associated molecular pattern), such as LPS, which then induces the NF-κB-mediated synthesis of NLRP3 and pro-IL-1β, thereby priming the inflammasome61. Signal 2 is typically a PAMP or a host-derived DAMP (danger-associated molecular pattern) of either endogenous origin, such as cholesterol crystals, uric acid crystals, adenosine tri-phosphate (ATP), and amyloid-β, or exogenous origin, such as adjuvant aluminum hydroxide, smoke, particulate matter and allergens45, 46, 52, 63. Upon sensing signal 2, NLRP3 nucleates the adaptor molecule, ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain), which then recruits caspase-1, followed by autocatalytic cleavage and activation of caspase-1 to generate its active p33 subunit that cleaves pro-IL-1β and generates a mature IL-1β that is secreted from cells35. Here, we show that APOE functions as both signal 1, which induces expression of NLRP3 and pro-IL-1β to prime the inflammasome, as well as signal 2, which activates caspase-1 within the NLRP3 to cleave pro-IL-1β and generate its mature 17-kDa form that is secreted from asthmatic BALF macrophages. Furthermore, APOE acts synergistically with HDM to enhance the expression of NLRP3 and pro-IL-1β by asthmatic BALF macrophages, as well as to increase the amount of IL-1β that is secreted from these cells. Similarly, APOE acted synergistically with HDM in vivo to induce IL-1β release into BALF.
We show that the mechanism by which APOE serves as signal 2 to activate the NLRP3 inflammasome in asthmatic airway macrophages is primarily dependent upon potassium efflux from cells. A decrease in cytosolic potassium levels due to efflux from cells has been recognized to represent a common pathway that is necessary and sufficient to activate the NLRP3 inflammasome in response to a variety of stimuli, such as ATP, aluminum hydroxide, silica, uric acid, and calcium pyrophosphate crystals57, 72. Furthermore, NEK7, which is a member of the NIMA-related kinase family, has been found to function as a NLRP3-binding protein that acts downstream of K+ efflux and induces NLRP3 inflammasome assembly and activation73. However, potassium efflux-independent activation of NLRP3 inflammasome and IL-1β secretion has been found in bone marrow-derived dendritic cells stimulated with the small nucleoside analog, imiquimod, which is a ligand for Toll-like receptor 758. Therefore, we assessed whether APOE induced NLRP3 activation and IL-1β secretion from macrophages by a potassium-dependent or potassium-independent mechanism. Consistent with the central role of potassium efflux in activating NLRP3 in response to a variety of stimuli, we show that APOE-mediated IL-1β secretion from macrophages was significantly inhibited when potassium chloride was added to the culture medium to prevent APOE-mediated reductions in intracellular potassium levels72.
Spatial heterogeneity exists regarding the concentration of APOE present in different anatomic compartments. In the circulation, APOE is primarily complexed with lipoproteins and is present at concentrations of 12 to 32.5 μg/mL (352 to 955 nM) in asthmatic serum34, 74. APOE also can be found at lower levels under physiological conditions74. For example, the concentration of APOE in ELF from healthy adults ranges from 10 to 80 ng/mL (0.3 to 2.4 nM), with a median concentration of 60 ng/mL (1.8 nM)36. Here, we show that concentrations of APOE similar to levels present in ELF from healthy individuals neither activated the NLRP3 inflammasome nor induced the secretion of mature IL-1β from asthmatic BALF macrophages. In contrast, APOE at concentrations ≥ 25 nM, induced the cleavage of pro-IL-1β to its mature 17-kDa form that was secreted from cells. These findings suggest that APOE acts as a concentration-dependent danger signal for asthmatic BALF macrophages that induces NLRP3 activation and IL-1β secretion when APOE concentrations are pathologically increased above basal levels. Using a viral exacerbation model of HDM-mediated murine airways disease characterized by neutrophilic airway inflammation and increased IL-1β production37, we showed that APOE concentrations in ELF reach a concentration of 32 nM, which is sufficiently high to activate the NLRP3 inflammasome in ex vivo cultures of asthmatic BALF macrophages. Furthermore, the disruption of microvascular integrity with leakage of APOE from plasma into the lung may represent an additional mechanism that further increases local concentrations. Consistent with this, increased microvascular permeability due to the contraction of post-capillary venular endothelial cells and the exudation of plasma proteins is a characteristic feature of allergic asthmatic inflammation75–79. HDM proteases induce and enhance vascular permeability, which may in part be mediated by the activation of the kinin-kallikrein system to generate bradykinin80, 81. Similarly, experimental murine allergic asthma caused by ovalbumin induces the production of hypoxia inducible factor‒α and insulin-like growth factor-I, which act through vascular endothelial growth factor to induce vascular permeability and plasma exudation into the lung78.
Our lab previously reported that APOE has a protective effect in HDM-induced asthma based upon experiments which showed that airway hyperreactivity and mucous cell metaplasia were increased in HDM-challenged Apoe−/− mice, whereas eosinophil-predominant airway inflammation was not modified8. Here, we sought to assess whether APOE might modify NLRP3 inflammasome activation and IL-1β secretion by asthmatic BALF macrophages. Based upon our prior result using a murine model of HDM-induced airways disease, we anticipated that APOE might suppress HDM-induced IL-1β secretion8. Instead, we found that APOE activated the NLRP3 inflammasome in asthmatic BALF macrophages and induced IL-1β secretion when concentrations exceeded levels that had previously been found in ELF from healthy individuals36. Furthermore, using a murine viral exacerbation model of HDM-induced neutrophilic airway inflammation, we show that APOE concentrations in mouse ELF are sufficiently high to activate the NLRP3 inflammasome in ex vivo cultures of asthmatic BALF macrophages. Taken collectively, we propose that APOE can reach concentrations in the lung that may activate the NLRP3 inflammasome and induce IL-1β secretion. This may be relevant for allergic asthmatics with HDM-induced disease who develop asthma exacerbations caused by double-stranded RNA viruses, such as influenza, rhinovirus, or respiratory syncytial virus, as IL-1β has been shown to mediate neutrophilic airway inflammation in this setting37, 38. In addition, our contrasting results from murine models induced by HDM in Apoe-null mice on a C57BL/6 background8, versus HDM+poly(I:C) in wild type mice on a Balb/c background, suggest that APOE has context-dependent effects in the setting of neutrophilic as compared to eosinophilic airway inflammation, as well as when APOE levels are increased as compared to when APOE is absent from the lung.
In conclusion, we have identified that HDM induces APOE secretion from asthmatic BALF macrophages by a mechanism that involves activation of PAR-2 signaling by HDM-derived serine and cysteine proteases. Furthermore, we for the first time identify a role for APOE as an endogenous, concentration-dependent danger signal that activates the NLRP3 inflammasome and induces IL-1β secretion by asthmatic BALF macrophages via a mechanism that involves potassium efflux from cells. Concentrations of APOE that are normally present in the lungs of healthy individuals were not sufficient to activate the NLRP3 inflammasome nor induce IL-1β secretion. In contrast, APOE concentrations that exceeded those normally present in the lung induced the expression of both NLRP3 and pro-IL-1β, as well as activated caspase-1 to generate mature IL-1β that is secreted from cells. Thus, we propose that pathologic increases in local APOE concentrations in the lung may serve to promote and amplify pulmonary inflammatory responses to HDM via the generation of macrophage-derived IL-1β. This may contribute to the pathogenesis of neutrophilic airway inflammation when a viral respiratory infection complicates house dust mite-induced asthma (Figure 10).
Figure 10. Proposed model of APOE-mediated NLRP3 inflammasome activation and IL-1β secretion by asthmatic macrophages.
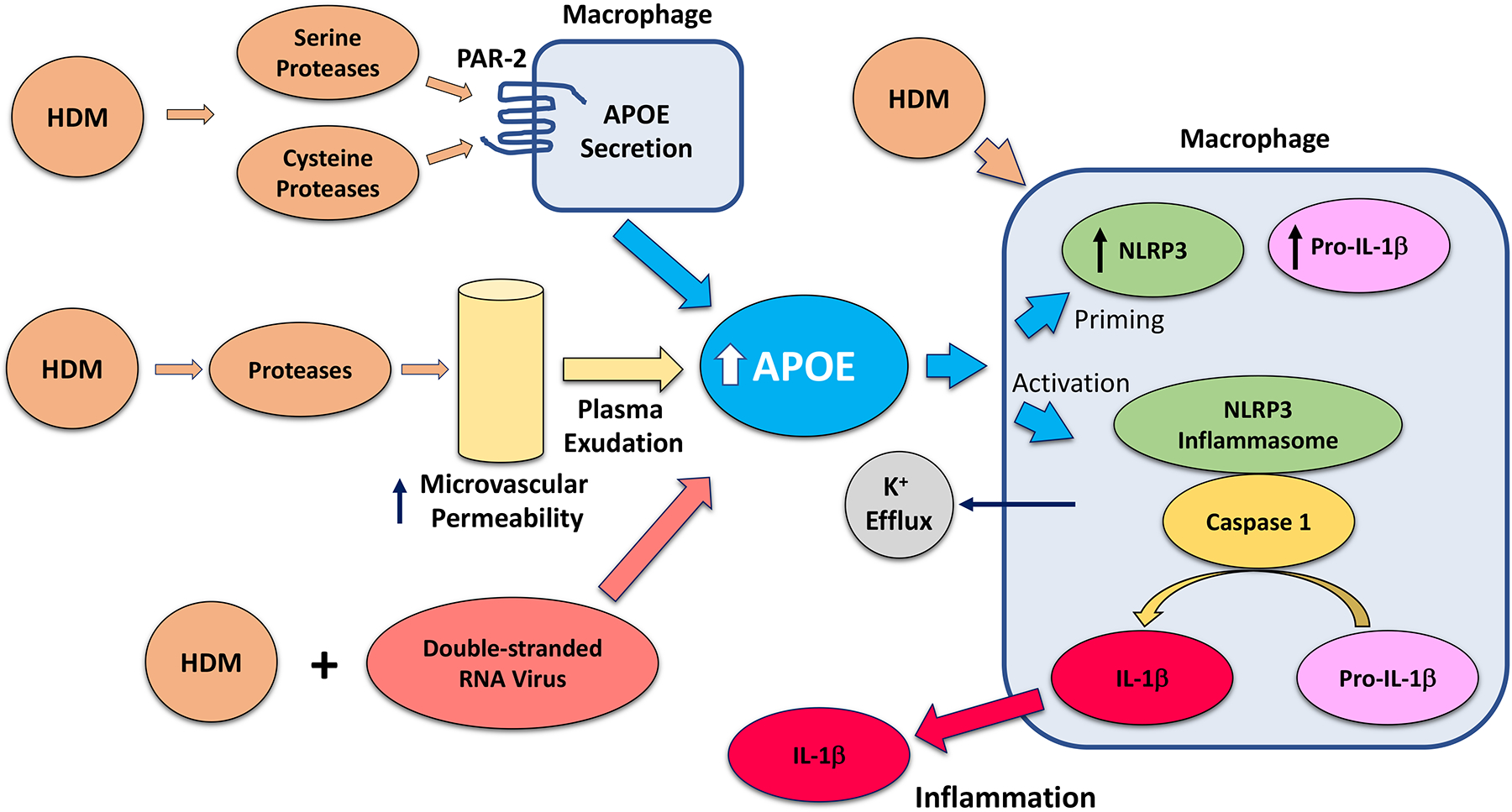
APOE in the asthmatic lung can be increased via several mechanisms; (i) serine and cysteine proteases within house dust mite (HDM) induce APOE secretion by asthmatic macrophages, (ii) HDM-derived proteases induce microvascular permeability and exudation of plasma that contains APOE into the airway80, 81, and (iii) viral exacerbations of HDM-mediated airways disease in mice induce increases in epithelial lining fluid (ELF) APOE concentrations that augment IL-1β production and neutrophilic airway inflammation. APOE, at concentrations that exceed those normally found in ELF from healthy individuals, induces the expression of NLRP3 and pro-IL-1β, which primes the inflammasome in macrophages. APOE also activates the NLRP3 inflammasome in macrophages by a potassium efflux-dependent mechanism, which results in the caspase-1-mediated generation of a mature 17-kDa IL-1β that is secreted. HDM can also act synergistically with APOE to both prime and activate the NLRP3 inflammasome and induce IL-1β secretion from asthmatic macrophages. Collectively, this may represent a mechanism how pathologic increases in local APOE concentrations in the lung can serve as an endogenous danger signal that amplifies neutrophilic airway inflammation in the setting of viral exacerbations of house dust mite-induced asthma.
Supplementary Material
Key Messages:
House dust mite-derived cysteine and serine proteases induce the secretion of apolipoprotein E from asthmatic macrophages via protease-activated receptor 2.
Apolipoprotein E, at concentrations that exceed those normally found in lung epithelial lining fluid (ELF) from healthy individuals, both primes and activates the NLRP3 inflammasome in asthmatic macrophages to induce IL-1β secretion. Results from murine studies showed that apolipoprotein E can reach levels in ELF that are sufficiently high to activate the macrophage NLRP3 inflammasome and increase bronchoalveolar lavage fluid IL-1β in the setting of viral exacerbations of house dust mite-induced asthma characterized by neutrophilic airway inflammation. This may allow increases in lung apolipoprotein E to act as an endogenous danger signal that amplifies pulmonary inflammatory responses.
Acknowledgements
This research was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, NIH, Bethesda, Maryland.
Abbreviations used:
- AIM
absent in melanoma
- APOE
apolipoprotein E
- ASC
apoptosis-associated speck-like protein containing a caspase activation and recruitment domain
- BALF
bronchoalveolar lavage fluid
- DAMP
danger-associated molecular pattern
- ELF
epithelial lining fluid
- HDL
high-density lipoproteins
- HDM
house dust mite
- iNOS
inducible nitric oxide synthase
- IL
interleukin
- LPS
lipopolysaccharide
- NLRC
NLR family caspase recruitment domain (CARD)-containing
- NLRP
nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing protein
- PAMP
pathogen-associated molecular pattern
- PAR
protease-activated receptor
- PMA
phorbol 12-myristate 13-acetate
- TLR
Toll-like receptor
- TNF-α
tumor necrosis factor-α
- VLDL
very low-density lipoproteins
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure Statement: The authors do not have any conflicts of interest to disclose.
References
- 1.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res 2009; 50 Suppl:S183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol 2009; 29:431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin CY, Duan H, Mazzone T. Apolipoprotein E-dependent cholesterol efflux from macrophages: kinetic study and divergent mechanisms for endogenous versus exogenous apolipoprotein E. J Lipid Res 1999; 40:1618–27. [PubMed] [Google Scholar]
- 4.Tall AR, Costet P, Wang N. Regulation and mechanisms of macrophage cholesterol efflux. J Clin Invest 2002; 110:899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao X, Gordon EM, Figueroa DM, Barochia AV, Levine SJ. The Emerging Roles of Apolipoprotein E and Apolipoprotein A-I in the Pathogenesis and Treatment of Lung Disease. Am J Respir Cell Mol Biol 2016; 55:159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin CT, Xu YF, Wu JY, Chan L. Immunoreactive apolipoprotein E is a widely distributed cellular protein. Immunohistochemical localization of apolipoprotein E in baboon tissues. J Clin Invest 1986; 78:947–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, et al. An antiproliferative BMP-2/PPARγ/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest 2008; 118:1846–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao X, Fredriksson K, Yu ZX, Xu X, Raghavachari N, Keeran KJ, et al. Apolipoprotein E negatively regulates house dust mite-induced asthma via a low-density lipoprotein receptor-mediated pathway. American Journal of Respiratory and Critical Care Medicine 2010; 182:1228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massaro D, Massaro GD. Apoetm1Unc mice have impaired alveologenesis, low lung function, and rapid loss of lung function. Am J Physiol Lung Cell Mol Physiol 2008; 294:L991–7. [DOI] [PubMed] [Google Scholar]
- 10.Massaro D, Massaro GD. Developmental alveologenesis: new roles for ApoE and LDL receptor. Pediatr Res 2011; 70:458–61. [DOI] [PubMed] [Google Scholar]
- 11.Ryan AJ, Medh JD, McCoy DM, Salome RG, Mallampalli RK. Maternal loading with very low-density lipoproteins stimulates fetal surfactant synthesis. Am J Physiol Lung Cell Mol Physiol 2002; 283:L310–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arunachalam G, Sundar IK, Hwang JW, Yao H, Rahman I. Emphysema is associated with increased inflammation in lungs of atherosclerosis-prone mice by cigarette smoke: implications in comorbidities of COPD. J Inflamm (Lond) 2010; 7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobsen NR, Moller P, Jensen KA, Vogel U, Ladefoged O, Loft S, et al. Lung inflammation and genotoxicity following pulmonary exposure to nanoparticles in ApoE−/− mice. Part Fibre Toxicol 2009; 6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venosa A, Malaviya R, Choi H, Gow AJ, Laskin JD, Laskin DL. Characterization of Distinct Macrophage Subpopulations During Nitrogen Mustard-Induced Injury and Fibrosis. Am J Respir Cell Mol Biol 2016; 54:436–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashita CM, Fessler MB, Vasanthamohan L, Lac J, Madenspacher J, McCaig L, et al. Apolipoprotein E-deficient mice are susceptible to the development of acute lung injury. Respiration 2014; 87:416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Bont N, Netea MG, Demacker PN, Verschueren I, Kullberg BJ, van Dijk KW, et al. Apolipoprotein E knock-out mice are highly susceptible to endotoxemia and Klebsiella pneumoniae infection. J Lipid Res 1999; 40:680–5. [PubMed] [Google Scholar]
- 17.Roselaar SE, Daugherty A. Apolipoprotein E-deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo. J Lipid Res 1998; 39:1740–3. [PubMed] [Google Scholar]
- 18.Van Oosten M, Rensen PC, Van Amersfoort ES, Van Eck M, Van Dam AM, Breve JJ, et al. Apolipoprotein E protects against bacterial lipopolysaccharide-induced lethality. A new therapeutic approach to treat gram-negative sepsis. J Biol Chem 2001; 276:8820–4. [DOI] [PubMed] [Google Scholar]
- 19.Li K, Ching D, Luk FS, Raffai RL. Apolipoprotein E enhances microRNA-146a in monocytes and macrophages to suppress nuclear factor-κB-driven inflammation and atherosclerosis. Circ Res 2015; 117:e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baitsch D, Bock HH, Engel T, Telgmann R, Muller-Tidow C, Varga G, et al. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology 2011; 31:1160–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y, Kodvawala A, Hui DY. Apolipoprotein E inhibits toll-like receptor (TLR)-3- and TLR4-mediated macrophage activation through distinct mechanisms. Biochem J 2010; 428:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chuang K, Elford EL, Tseng J, Leung B, Harris HW. An expanding role for apolipoprotein E in sepsis and inflammation. Am J Surg 2010; 200:391–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kattan OM, Kasravi FB, Elford EL, Schell MT, Harris HW. Apolipoprotein E-mediated immune regulation in sepsis. J Immunol 2008; 181:1399–408. [DOI] [PubMed] [Google Scholar]
- 24.Smoak KA, Aloor JJ, Madenspacher J, Merrick BA, Collins JB, Zhu X, et al. Myeloid differentiation primary response protein 88 couples reverse cholesterol transport to inflammation. Cell metabolism 2010; 11:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy O, Calippe B, Lavalette S, Hu SJ, Raoul W, Dominguez E, et al. Apolipoprotein E promotes subretinal mononuclear phagocyte survival and chronic inflammation in age-related macular degeneration. EMBO Mol Med 2015; 7:211–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gregory LG, Lloyd CM. Orchestrating house dust mite-associated allergy in the lung. Trends in Immunology 2011; 32:402–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calderon MA, Linneberg A, Kleine-Tebbe J, De Blay F, Hernandez Fernandez de Rojas D, Virchow JC, et al. Respiratory allergy caused by house dust mites: What do we really know? J Allergy Clin Immunol 2015; 136:38–48. [DOI] [PubMed] [Google Scholar]
- 28.Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res 2007; 56:45–50. [DOI] [PubMed] [Google Scholar]
- 29.Jin X, Kruth HS. Culture of Macrophage Colony-stimulating Factor Differentiated Human Monocyte-derived Macrophages. J Vis Exp 2016; Jun 30;(112). doi: 10.3791/54244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rennard SI, Basset G, Lecossier D, O’Donnell KM, Pinkston P, Martin PG, et al. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J Appl Physiol (1985) 1986; 60:532–8. [DOI] [PubMed] [Google Scholar]
- 31.Dai X, Sayama K, Tohyama M, Shirakata Y, Hanakawa Y, Tokumaru S, et al. Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes. J Allergy Clin Immunol 2011; 127:806–14 e1–4. [DOI] [PubMed] [Google Scholar]
- 32.Sundaram K, Mitra S, Gavrilin MA, Wewers MD. House Dust Mite Allergens and the Induction of Monocyte Interleukin 1beta Production That Triggers an IkappaBzeta-Dependent Granulocyte Macrophage Colony-Stimulating Factor Release from Human Lung Epithelial Cells. Am J Respir Cell Mol Biol 2015; 53:400–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barochia AV, Kaler M, Cuento RA, Gordon EM, Weir NA, Sampson M, et al. Serum Apolipoprotein A-I and Large High-density Lipoprotein Particles are Positively Correlated with FEV1 in Atopic Asthma. Am J Respir Crit Care Med 2015; 191:990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med 2018; 215: 827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mendivil CO, Koziel H, Brain JD. Metabolic hormones, apolipoproteins, adipokines, and cytokines in the alveolar lining fluid of healthy adults: compartmentalization and physiological correlates. PLoS One 2015; 10:e0123344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Alba J, Otal R, Calama E, Domenech A, Prats N, Gozzard N, et al. Double-stranded RNA evokes exacerbation in a mouse model of corticosteroid refractory asthma. Clin Sci (Lond) 2015; 129:973–87. [DOI] [PubMed] [Google Scholar]
- 38.Mahmutovic Persson I, Menzel M, Ramu S, Cerps S, Akbarshahi H, Uller L. IL-1β mediates lung neutrophilia and IL-33 expression in a mouse model of viral-induced asthma exacerbation. Respir Res 2018; 19:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stowell NC, Seideman J, Raymond HA, Smalley KA, Lamb RJ, Egenolf DD, et al. Long-term activation of TLR3 by poly(I:C) induces inflammation and impairs lung function in mice. Respir Res 2009; 10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Starkhammar M, Kumlien Georen S, Swedin L, Dahlen SE, Adner M, Cardell LO. Intranasal administration of poly(I:C) and LPS in BALB/c mice induces airway hyperresponsiveness and inflammation via different pathways. PLoS One 2012; 7:e32110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417–26. [DOI] [PubMed] [Google Scholar]
- 43.Hansbro PM, Kim RY, Starkey MR, Donovan C, Dua K, Mayall JR, et al. Mechanisms and treatments for severe, steroid-resistant allergic airway disease and asthma. Immunol Rev 2017; 278:41–62. [DOI] [PubMed] [Google Scholar]
- 44.Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015; 21:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol 2010; 11:897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 2008; 9:857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci 2016; 41:1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221–5. [DOI] [PubMed] [Google Scholar]
- 49.Mazzone T, Reardon C. Expression of heterologous human apolipoprotein E by J774 macrophages enhances cholesterol efflux to HDL3. J Lipid Res 1994; 35:1345–53. [PubMed] [Google Scholar]
- 50.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 2015; 15:104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson LE, Shridar M, Smith P, Murrell-Lagnado RD. Plasma membrane cholesterol as a regulator of human and rodent P2X7 receptor activation and sensitization. J Biol Chem 2014; 289:31983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010; 464:1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1beta Activation. J Immunol 2015; 195:1685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nomura J, So A, Tamura M, Busso N. Intracellular ATP Decrease Mediates NLRP3 Inflammasome Activation upon Nigericin and Crystal Stimulation. J Immunol 2015; 195:5718–24. [DOI] [PubMed] [Google Scholar]
- 55.Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017; 47:15–31. [DOI] [PubMed] [Google Scholar]
- 56.Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis 2015; 6:e1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013; 38:1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gross CJ, Mishra R, Schneider KS, Medard G, Wettmarshausen J, Dittlein DC, et al. K(+) Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016; 45:761–73. [DOI] [PubMed] [Google Scholar]
- 59.Randall TA, London RE, Fitzgerald MC, Mueller GA. Proteases of Dermatophagoides pteronyssinus. Int J Mol Sci 2017; 18 (6). pii: E1204. doi: 10.3390/ijms18061204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Boer JD, Van’t Veer C, Stroo I, van der Meer AJ, de Vos AF, van der Zee JS, et al. Protease-activated receptor-2 deficient mice have reduced house dust mite-evoked allergic lung inflammation. Innate Immun 2014; 20:618–25. [DOI] [PubMed] [Google Scholar]
- 61.Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015; 42:991–1004. [DOI] [PubMed] [Google Scholar]
- 62.Kim RY, Pinkerton JW, Gibson PG, Cooper MA, Horvat JC, Hansbro PM. Inflammasomes in COPD and neutrophilic asthma. Thorax 2015; 70:1199–201. [DOI] [PubMed] [Google Scholar]
- 63.Pinkerton JW, Kim RY, Robertson AAB, Hirota JA, Wood LG, Knight DA, et al. Inflammasomes in the lung. Mol Immunol 2017; 86:44–55. [DOI] [PubMed] [Google Scholar]
- 64.Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol 2011; 127:153–60, 60 e1–9. [DOI] [PubMed] [Google Scholar]
- 65.Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, et al. Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. The Journal of allergy and clinical immunology 2010; 125:1028–36 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim RY, Pinkerton JW, Essilfie AT, Robertson AAB, Baines KJ, Brown AC, et al. Role for NLRP3 Inflammasome-mediated, IL-1beta-Dependent Responses in Severe, Steroid-Resistant Asthma. Am J Respir Crit Care Med 2017; 196:283–97. [DOI] [PubMed] [Google Scholar]
- 67.Rossios C, Pavlidis S, Hoda U, Kuo CH, Wiegman C, Russell K, et al. Sputum transcriptomics reveal upregulation of IL-1 receptor family members in patients with severe asthma. J Allergy Clin Immunol 2018; 141:560–70. [DOI] [PubMed] [Google Scholar]
- 68.Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J 2014; 43:1067–76. [DOI] [PubMed] [Google Scholar]
- 69.Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun 2015; 6:8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016; 16:407–20. [DOI] [PubMed] [Google Scholar]
- 71.Sharma D, Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol 2016; 213:617–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 2007; 14:1583–9. [DOI] [PubMed] [Google Scholar]
- 73.He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016; 530:354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crouchet E, Lefevre M, Verrier ER, Oudot MA, Baumert TF, Schuster C. Extracellular lipid-free apolipoprotein E inhibits HCV replication and induces ABCG1-dependent cholesterol efflux. Gut 2017; 66:896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohrui T, Sekizawa K, Aikawa T, Yamauchi K, Sasaki H, Takishima T. Vascular permeability and airway narrowing during late asthmatic response in dogs treated with Metopirone. J Allergy Clin Immunol 1992; 89:933–43. [DOI] [PubMed] [Google Scholar]
- 76.Kanazawa H, Hirata K, Yoshikawa J. Involvement of vascular endothelial growth factor in exercise induced bronchoconstriction in asthmatic patients. Thorax 2002; 57:885–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kanazawa H, Asai K, Hirata K, Yoshikawa J. Vascular involvement in exercise-induced airway narrowing in patients with bronchial asthma. Chest 2002; 122:166–70. [DOI] [PubMed] [Google Scholar]
- 78.Kim SR, Lee KS, Lee KB, Lee YC. Recombinant IGFBP-3 inhibits allergic lung inflammation, VEGF production, and vascular leak in a mouse model of asthma. Allergy 2012; 67:869–77. [DOI] [PubMed] [Google Scholar]
- 79.Goldie RG, Pedersen KE. Mechanisms of increased airway microvascular permeability: role in airway inflammation and obstruction. Clin Exp Pharmacol Physiol 1995; 22:387–96. [DOI] [PubMed] [Google Scholar]
- 80.Maruo K, Akaike T, Matsumura Y, Kohmoto S, Inada Y, Ono T, et al. Triggering of the vascular permeability reaction by activation of the Hageman factor-prekallikrein system by house dust mite proteinase. Biochim Biophys Acta 1991; 1074:62–8. [DOI] [PubMed] [Google Scholar]
- 81.Stewart GA, Boyd SM, Bird CH, Krska KD, Kollinger MR, Thompson PJ. Immunobiology of the serine protease allergens from house dust mites. Am J Ind Med 1994; 25:105–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.