

Abstract
Introduction
Immunotherapy (IO) has transformed the treatment paradigm for a wide variety of solid tumours. However, assessment of response can be challenging with conventional radiological imaging (eg, iRECIST), which do not precisely capture the unique response patterns of tumours treated with IO. Emerging data suggest that circulating tumour DNA (ctDNA) can aid in response assessment in patients with solid tumours receiving IO. The short half-life of ctDNA puts it in a unique position for early treatment response monitoring. The BESPOKE IO study is designed to investigate the clinical utility of serial ctDNA testing to assess treatment response using a tumour-informed, bespoke ctDNA assay (Signatera) and to determine its impact on clinical decision-making with respect to continuation/discontinuation, or escalation/de-escalation of immunotherapy in patients with advanced solid tumours.
Methods and analysis
The BESPOKE IO is a multicentre, prospective, observational study with a goal to enroll over 1500 patients with solid tumours receiving IO in up to 100 US sites. Patients will be followed for up to 2 years with serial ctDNA analysis, timed with every other treatment cycle. The primary endpoint is to determine the percentage of patients who will have their treatment regimen changed as guided by post-treatment bespoke ctDNA results along with standard response assessment tools. The major secondary endpoints include progression-free survival, overall survival and overall response rate based on the ctDNA dynamics.
Ethics and dissemination
The BESPOKE IO study was approved by the WCG Institutional Review Board (Natera-20–043-NCP BESPOKE Study of ctDNA Guided Immunotherapy (BESPOKE IO)) on 22 February 2021. Data protection and privacy regulations will be strictly observed in the capturing, forwarding, processing and storing patients’ data. Natera will approve the publication of any study results in accordance with the site-specific contract.
Trial registration number
Keywords: oncology, dermatological tumours, gastrointestinal tumours, respiratory tract tumours
Strengths and limitations of this study.
BESPOKE IO is a large, prospective, multicentre, observational study designed to investigate the clinical utility of the personalised, tumour-informed circulating tumour DNA (ctDNA) assay in assessing early treatment response in patients with advanced solid tumours receiving immunotherapy (IO).
This clinical study might potentially inform if the pretreatment ctDNA level can serve as a predictive biomarker for response to IO and prognosis early into treatment course.
This study might help with early identification of non-responders to IO based on the ctDNA dynamics that can inform the treating physicians to discontinue, intensify or switch treatment, thereby avoiding unnecessary treatment-related toxicities and costs.
This study might inform if ctDNA can distinguish between pseudoprogression and true tumour progression.
Given the non-interventional nature of the study, therapy is physician directed and not dictated by the trial.
Introduction
Immune-checkpoint inhibitors (ICIs) targeting programmed cell death-1 (PD-1)/its ligand-1 (PD-L1) and cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) have transformed treatment paradigms in patients with advanced cancer.1 A plethora of clinical trials have demonstrated significant antitumour activity with ICIs, often leading to durable and potentially curable responses in a wide variety of solid tumours. ICIs have shown superior survival outcomes compared with conventional chemotherapy in multiple advanced malignancies including melanoma, lung and subsets of colorectal with mismatch repair deficient tumours, breast and bladder cancers and have been integrated into the standard treatment algorithms for these tumour types.1 2 One of the anti-PD1 antibodies (pembrolizumab) hold two of the four currently approved tissue-agnostic Food and Drug Administration (FDA) approvals. In addition to the metastatic setting, these drugs are now making their way into the clinic for a number of adjuvant indications.
As ICIs have gained a prominent place in the routine clinical care, response assessment to ICIs has become of paramount importance. The tumour response patterns to ICIs vary widely and are often markedly different from the response pattern observed with cytotoxic chemotherapy, limiting the usefulness of the conventional radiological studies. Variations, for example, iRECIST and repeat follow-up scans, are often recommended.3 Around 10% of patients with solid tumours on ICI experience pseudoprogression, defined as an enlargement of existing tumours or the appearance of a new lesion followed by tumour regression that can be misinterpreted as true progression, leading to the premature discontinuation of a potentially effective treatment.4 5 Furthermore, the staggering cost of immunotherapy (~$10 000/dose) adds significant financial stress on patients and the health system,6 underscoring the importance of identifying non-responders early to avoid the cost and the toxicity burden. Although biomarkers including PD-L1 expression, microsatellite instability-high/deficient mismatch repair (MSI-H/dMMR) status and tumour mutational burden (TMB) have shown clinical utility for selecting patients suitable for immunotherapy, the predictive capability of these biomarkers is limited.7 8 Recently, the use of PD-1 blockade in combination with other therapies was approved, for example, combination immunotherapy with a CTLA-4 inhibitor, or combination immunotherapy in addition to chemotherapy. However, it is unclear who should get PD-1 blockade alone, and who would potentially benefit from the combination approach. Some investigators have described the potential benefit of using CTLA-4 rescue strategy.9 10 While the combination approaches bring the promise of better response rates and survival, they also incur added risk of severe adverse events (SAEs), as well as financial toxicity. Taken together, the variable treatment efficacy, toxicities, cost, the lack of predictive biomarkers and difficulty in interpreting radiologic response patterns underscore the urgent need for a tool that can identify treatment response and disease progression early.
Accumulating data suggest that circulating tumour DNA (ctDNA), a non-invasive, quantitative and dynamic biomarker, can monitor treatment response in patients with advanced/metastatic cancer.11–16 The kinetics of ctDNA brings in several advantages for early response assessment.17 Previous studies in patients with advanced solid tumours have demonstrated that a decrease in the ctDNA level with treatment reflects a response to immunotherapy.12 16 18–20 Furthermore, undetectable or low ctDNA levels after treatment have been associated with better clinical outcomes with ICIs across multiple advanced stage cancers.15 19 21–24 Several recent studies in lung cancer have shown that ctDNA dynamics can predict disease progression and response to immunotherapy, weeks to months ahead of conventional radiological imaging.16 18–20 Existing evidence in literature supports that ctDNA can clearly differentiate pseudoprogression from true progression with high sensitivity and specificity, potentially assisting in interpreting ambiguous imaging findings.24 25 Despite this, ctDNA is currently not used in clinical practice. Some of the reasons for this include, data available from studies with small patient population12 14 22 26 27 and/or use of static panels focusing on a limited number of somatic variants,22 23 28 which restrict the applicability and generalisability of their findings. This need prompted the development of the BESPOKE IO observational study. Herein, we present a clinical study protocol of a prospective, longitudinal, multicentre observational study to investigate the clinical utility of a personalised, tumour-informed multiplex PCR (mPCR)-NGS ctDNA assay (Signatera) for treatment response monitoring in patients with advanced solid tumours receiving immunotherapy. The study will also examine the impact of ctDNA detection on clinical decision-making regarding continuation/discontinuation or escalation/de-escalation of immunotherapy.
Methods
Overall study design
The BESPOKE IO (clinicaltrials.gov NCT04761783) is a prospective, longitudinal, multicentre clinical study that uses a personalised mPCR-NGS assay (Signatera), designed to track somatic single nucleotide variants (SNVs) in patients with advanced cancer receiving ICIs. The study started in March 2021 and is actively recruiting. The study is composed of three cohorts representing three unique advanced cancer types: lung, melanoma and colorectal cancer (dMMR/MSI-H). Each cohort has two arms: a prospective arm in which serial ctDNA testing will be performed while patients receive immunotherapy (prospective Signatera arm) and a historical control arm. The data collected from the prospective arm will be compared with the outcomes in the historical control groups to evaluate the role of molecular response reflected by ctDNA levels in the management of patients with advanced cancers receiving IO.
Prospective Signatera arm
A total of 1539 patients with advanced solid tumours (lung, melanoma and dMMR/MSI-H colorectal cancer) undergoing treatment with IO will be enrolled in up to 100 study sites in the USA and patients will be followed up for up to 2 years with serial blood collection for ctDNA analysis. A whole blood (20 mL) sample will be collected for the Signatera assay at baseline and at subsequent time points and frequency determined by the healthcare provider (HCP). The sponsor recommends subsequent blood collection for the Signatera testing every 2 cycles, timed according to the immunotherapy treatment regimen (table 1). Optional blood sample collections for the ctDNA assay between week 2 and week 4 of therapy initiation and 4–6 weeks after the end of treatment/disease progression will be carried out (figure 1). All enrolled patients will be evaluated for immune-related adverse events (iRAEs). Written informed consent will be obtained from all patients. Study inclusion/exclusion criteria are detailed in table 2.
Table 1.
Signatera blood draw frequency based on immunotherapy treatment regimen (Prospective Signatera arm)
| Immunotherapy treatment regimen |
Immunotherapy treatment dose | Treatment frequency (every # weeks) |
Signatera blood draw Frequency*† (every # weeks) |
| Atezolizumab (Tecentriq) | 840 mg | 2 | 8 |
| Atezolizumab (Tecentriq) | 1200 mg | 3 | 6 |
| Atezolizumab (Tecentriq) | 1680 mg | 4 | 8 |
| Avelumab (Bavencio) | 800 mg | 2 | 8 |
| Cemiplimab (Libtayo) | 350 mg | 3 | 6 |
| Durvalumab (Imfinzi) | 10 mg/kg | 2 | 8 |
| Durvalumab (Imfinzi) | 1500 mg | 4 | 8 |
| Durvalumab (Imfinzi) | 1500 mg | 3 | 6 |
| Ipilimumab (Yervoy) | 3 mg/kg | 3 | 6 |
| Nivolumab (Opdivo) | 240 mg | 2 | 8 |
| Nivolumab (Opdivo) | 480 mg | 4 | 8 |
| Nivolumab (Opdivo) and ipilimumab (Yervoy) | 1 mg/kg 3 mg/kg |
3 3 |
6 |
| Nivolumab (Opdivo) and ipilimumab (Yervoy) | 360 mg 1 mg/kg |
3 6 |
6 |
| Nivolumab (Opdivo) and ipilimumab (Yervoy) | 3 mg/kg 1 mg/kg |
3 3 |
6 |
| Nivolumab (Opdivo) and ipilimumab (Yervoy) | 3 mg/kg 1 mg/kg |
2 6 |
8 |
| Pembrolizumab (Keytruda) | 200 mg | 3 | 6 |
| Pembrolizumab (Keytruda) | 400 mg | 6 | 6 |
*Signatera blood draw should coincide with every other treatment cycle.
†Additional optional Signatera blood draws are recommended on weeks 2–4 of immunotherapy, and 4–6 weeks after the end of treatment or disease progression.
Figure 1.
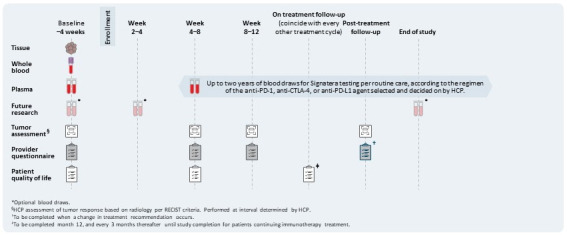
Overview of the BESPOKE IO study design: Samples (whole blood, FFPE tissue, plasma) will be collected, and questionnaires (physician assessment, quality of life (QoL) will be completed at the indicated times (weeks/months). FFPE, Formalin-Fixed Paraffin-Embedded; HCP, healthcare provider; PD-1, programmed cell death-1; PD-L1, programmed cell death ligand-1.
Table 2.
Eligibility criteria
| Category | Inclusion criteria | Exclusion criteria |
| Demographics |
|
Female patients that are pregnant |
| Clinical presentation |
|
Patients who have initiated immunotherapy |
| Medical history |
|
Patients with a history of bone marrow or organ transplant, a medical condition that would place the patient at risk as a result of blood donation, such as bleeding disorder, or a serious medical condition that may adversely affect the ability to participate in the study |
| Provider-based criteria |
|
ctDNA, circulating tumour DNA; CTLA-4, cytotoxic T‐lymphocyte associated antigen-4; HCP, healthcare provider; PD-1, programmed cell death-1; PD-L1, programmed cell death ligand 1.
Historical control arm
Approximately 513 historical control cases will be enrolled retrospectively, at an approximate ratio of 1 patient to every 3 prospective patients who had previously received treatment with an ICI and had minimum 2 years of follow-up data after initiation of immunotherapy or death. Furthermore, the control patients will have to meet all study inclusion criteria as listed in table 2. Data on patients in the control arm will be abstracted retrospectively from the electronic medical records. No written informed consent will be required for the patients in the control arm since they will have completed treatment and/or deceased at the time of enrolment. No biological samples for the study will be collected from the patients in the control arm.
Immunotherapy treatment
Patients scheduled to receive an ICI in the prospective Signatera arm or those who previously received an ICI in the historical control arm will be eligible.
Study objectives/endpoints
Primary endpoint
The primary study objective is to examine the impact of the bespoke ctDNA assay on tumour assessment after initiation of immunotherapy, that is, the percentage of patients who have their immunotherapy treatment regimen changed due to post-treatment bespoke ctDNA assay result along with standard clinical assessments and care.
Secondary endpoints
The main secondary endpoints include progression-free survival (PFS) and overall survival (OS) according to change in ctDNA levels from baseline, wherein ctDNA change is defined as: (1) 50% increase or decrease from baseline, (2) an analytically significant increase or decrease from baseline, (3) ctDNA clearance or no clearance or (4) a cut-off as determined in exploratory analysis. Other secondary endpoints include determination of response rate (partial or complete response), response duration, percentage of patients with at least 6 months of durable clinical response and the impact of Signatera on informing immunotherapy treatment decisions and patient-reported outcomes (PROs).
Exploratory endpoints
Exploratory endpoints include evaluating the performance of ctDNA dynamics in detecting pseudoprogression, determining ctDNA cut-offs that predict durable clinical response for 6–12 months in patients who achieve stable disease or partial response, or determining PFS on the subsequent scan. Specifically, the sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV) and area under the curve will be analysed.
Data collection
Demographic, medical history, disease status, immunotherapy regimen and outcomes, pathological diagnosis including immunotherapy markers, biomarkers, and comorbidities, and imaging scans will be collected as part of the protocol and recorded (tables 3 and 4). At different time points, questionnaires pertaining to PROs and HCP will be completed by patients and HCPs, respectively.
Table 3.
Schedule of events prospective Signatera arm(s)
| Enrolment | Week following immunotherapy initiation | |||||||
| Baseline up to 4 weeks prior to immunotherapy Initiation* | Weeks 2–4 |
Weeks 4–8 |
Weeks 8–12 |
Weeks 12–16 |
On treatment follow-up |
Post-treatment follow-up† |
End of study or early termination |
|
| Informed consent | X | |||||||
| Confirmation of inclusion/exclusion criteria and enrolment |
X | |||||||
| Optional future research blood collection (Streck)‡ |
X | X | X | |||||
| Observational/data collection pieces | ||||||||
| Demographics and medical history height |
X | |||||||
| X | ||||||||
| Weight§ | X | X | X | X | X | X | X | |
| Prior and current concomitant medications |
X | |||||||
| Current cancer diagnosis details |
X | |||||||
| Prior and current comorbidities |
X | |||||||
| Laboratory results | X | X | X | X | X | X | X | X |
| Physician assessment of response (RECIST)¶ | X | X | X | X | X | X | ||
| Radiology§§ | X | X | X | X | X | X | X | |
| Pathology results | X | X | X | X | X | X | X | |
| Immunotherapy treatment regimen** | X | X | X | X | X | X | X | |
| Disease status and survival |
X | X | X | X | X | X | X | |
| Cancer treatment procedures | X | X | X | X | X | X | X | |
| Adverse event reporting | X | X | X | X | X | X | X | X |
| Patient disposition | X | |||||||
| Patient-reported outcomes | X | X | X†† | |||||
| HCP questionnaire | X | X | X | X‡‡ | ||||
*Baseline visit may occur the same day as immunotherapy initiation.
†Patients who experience disease progression and those who complete or discontinue immunotherapy treatment will be followed up to 2 years from the date of consent. Data will be collected when available in the medical record.
‡Optional blood collection kit.
§Collect at baseline. For subsequent treatment visits, the weight will be collected from the patient’s medical record, if available.
¶Healthcare provider (HCP) assessment of tumour response based on radiology per RECIST criteria. Performed at an interval determined by HCP.
**Collected at every visit and/or if there is a change in treatment or regimen.
††Patient-reported outcomes are completed at: (1) baseline; (2) after second SIGNATERA blood draws (expected week 4–8) and tumour assessment are complete; (3) month 12, and every 3 months thereafter until study completion for patients continuing immunotherapy treatment.
‡‡HCP questionnaires are completed at: (1) baseline; (2) after second SIGNATERA blood draws (expected week 4–8), imaging and tumour assessment are complete, and all results are discussed with the patient (tumour assessment 1); (3) after the third SIGNATERA blood draw (expected week 8–12), imaging and tumour assessment are complete, and all results are discussed with the patient (tumour assessment 2); (4) any time there is a change in the treatment regimen, indeterminate image finding, or treatment decision to hold or discontinue treatment due to a suspected side effect of immunotherapy.
§§Radiology scans are to be submitted and performed at intervals per standard of care determined by HCP. Reports are collected if available.
Table 4.
Schedule of events for control arm
| Within 2 months of cancer diagnosis | For each clinic visit 1–24 months from time of immunotherapy treatment | |
| Confirmation of inclusion/exclusion criteria and enrolment | X | |
| Demographics and medical history | X | |
| Height | X | |
| Weight | X | X* |
| Prior and current concomitant medications | X | |
| Current cancer diagnosis details | X | |
| Prior and current comorbidities | X | |
| Immunotherapy treatment regimen | X | X |
| ECOG performance status | X | |
| Cancer treatment procedures | X | |
| Laboratory results | X | X |
| Radiology† | X | X |
| Physician assessment of Response (RECIST) | X | |
| Pathology results | X | X |
| Patient disposition | X | |
| Disease status | X | X |
| Side effects‡ | X |
*If available in patient’s medical record.
†Radiology scans are to be submitted. Reports are collected if available.
‡Side effects related to immunotherapy treatment.
Follow-up data collection
Patients who experience disease progression, complete or discontinue their immunotherapy treatment will enter the follow-up period of up to 2 years from the date of patient’s consent. The following data will be collected:
Disease status and survival.
Results of any imaging studies performed since the prior visit (a deidentified copy of the report and images will be provided to the sponsor).
Immunotherapy treatment discontinuation, change in the treatment regimen or the initiation of steroids due to side effects.
Tumour markers (CEA, LDH, CA27-29, CA15-3) laboratory results, if available.
Description of any procedures performed to treat this cancer, including surgery, additional chemotherapy or immunotherapy, or radiation therapy.
If additional surgery is performed, results of any pathology testing (a deidentified copy of the report will be provided).
Signatera blood and tissue collection
First blood draw and tissue collection will be done at baseline during the study enrolment period in the prospective Signatera arm. For subsequent time points, up to 20 mL of whole blood will be collected at intervals as determined by the HCP (table 3, figure 1).
Future research blood collection
Up to three optional blood samples for future research may be collected for patients who agree and are enrolled in the prospective Signatera arm: at baseline, weeks 4–8 and at the end of the study (figure 1). Complete instructions for blood collection can be found in the laboratory manual using research collection kits provided by Natera. Venipuncture will be performed using the standard technique with a collection of up to 20 mL of whole blood. All blood must be deidentified and include a study ID number, and the Signatera case number for each time point must be recorded on the electronic Case Report Form.
Data management/organisation
All data will be collected and stored in a secure, Health Insurance Portability and Accountability Act (HIPAA)-compliant database and applicable regulatory requirements appropriate for each clinical site. Before enrolment, signed informed consent will be received from all patients except for the control arm, wherein a consent-waiver will be requested for data collection purposes. Data associated with the samples will be deidentified to maintain patient privacy. Access to the final trial dataset will be with Natera; each site will have access to their own site dataset.
Sample size and statistical considerations
The sample size for this study is based on a ±5% margin of error and 95% CI for the percentage of patients with a change in the treatment regimen. The expected percentage of treatment change is unknown and likely to vary by histological indication. Using a normal approximation to the binomial distribution, the worst-case scenario for reducing the width of the CI is when the probability is 0.5. Assuming this value is observed in the study, a minimum of 385 samples are needed to produce a 95% CI ±0.05. Similarly, the minimum number of patients per cohort in each arm will be calculated as shown in table 5.
Table 5.
Sample size calculations
| Assumption | Prospective Signatera arm | Historical control arm | ||
| Attrition rate | Total number of patients | Minimum number of patients per cohort | Total number of patients | Minimum number of patients per cohort |
| *25% | 1539 | 513 | 513 | 171 |
*Assumption based on patients lost to follow-up, non-compliance, non-evaluable circulating tumour DNA results, etc.
Primary analysis
For analysis of the primary endpoint, the point estimate and a 95% Agresti-Coull CI for the proportion of patients who underwent a change in immunotherapy treatment regimen will be calculated separately for the Lung, Melanoma and Colorectal cohorts.
General statistical methods
Dichotomous (eg, change in postsurgical treatment regimen) and ordinal (eg, adverse event severity) data will be tabulated by category, expressed as proportions and percentages. The mean, SD, median, maximum and minimum will be tabulated for continuous data (eg, age), which may be presented graphically (eg, box plots). Pairwise comparisons of continuous data will be performed using a t-test if the data distribution appears normal; otherwise, a non-parametric rank test will be used. Comparisons of independent binomial data will be performed using Fisher’s exact test, and comparisons of dependent binomial data will be performed using McNemar’s test. Survival endpoints will be assessed using Kaplan-Meier analysis or Cox proportional hazards model; binary endpoints will generally be assessed using logistic regression.
Patient and public involvement
The protocol was designed and discussed with the patient advocacy group and academic community (GI oncology). Patients and general public were not involved in the design, conduct, reporting or dissemination plans of this protocol. Patients will receive ctDNA test results from their provider, according to the current evidence-informed schedule, as part of routine practice.
Ethics and dissemination
This study will be conducted in accordance with Good Clinical Practice (GCP), International Conference on Harmonisation (ICH), the Declaration of Helsinki, and US FDA guidelines. Prior to enrolment, written informed consent will be obtained from all patients and compliance with all inclusion and exclusion criteria will be verified and documented. The protocol (Natera-20-043-NCP BESPOKE Study of ctDNA Guided Immunotherapy (BESPOKE IO)) was approved by the WCG Institutional Review Board on 22 February 2021. Publication of any study results in papers, abstracts, posters or other material presented at scientific meetings or published in professional journals will be approved by Natera in accordance with the site-specific study contract.
Discussion
The BESPOKE IO study is one of the first and large prospective, observational study designed to investigate the utility of ctDNA in guiding treatment response assessment along with standard clinical tools in patients with advanced solid tumours receiving immunotherapy. ctDNA is a highly specific and dynamic blood-based cancer biomarker that provides a real-time snapshot of the tumour burden. Its short half-life of approximately 2 hours puts it in a unique position for assessing early treatment response.29 Previous studies have demonstrated the ability of ctDNA to detect molecular residual disease, identify cancer recurrence early and monitor treatment response across multiple cancers and treatment modalities, including immunotherapy.11 13 15 21 24 28 30–35
The use of ctDNA kinetics to predict response to immunotherapy has been described across tumour types, using various assays.17 Timely identification of non-responders from responders based on the ctDNA status can guide further treatment decisions, wherein non-responders can be switched to alternative treatment and spared of the toxicities associated with IO treatment. Alternatively, it can help inform decisions of escalation to combination immunotherapy, for example, addition of a CTLA-4 inhibitor, or addition of chemotherapy in addition to immunotherapy in malignancies that have these agents approved.10 Currently, there are no dynamic real-time biomarkers to help aid in this decision-making or early response assessment. The commonly used biomarkers used in the IO setting include, PD-L1,25 36–38 TMB39 40 and MSI.41 Although these biomarkers may help select patients who are most likely to respond ICI, most of these patients may still never respond to treatment. Thus, these biomarkers have limited predictive accuracy and specificity and are unsuitable for early response assessment (table 6).
Table 6.
Limitations with existing predictive biomarkers
| Predictive biomarkers | Limitations |
| PD-L1 expression—IHC assay25 36–38 |
|
| Tissue-based TMB39 40 |
|
| MSI41 |
|
ICI, immune-checkpoint inhibitor; IHC, immunohistochemistry; MSI, microsatellite instability; PD-L1, programmed cell death ligand 1; TMB, tumour mutational burden.
The bespoke tumour-informed (Signatera) ctDNA assay used in this study tracks tumour-specific somatic, SNVs in patients’ plasma based on the upfront whole-exome sequencing of the patient’s tumour tissue and matched normal blood. As described previously,42 the bespoke ctDNA assay can detect clonal variants with high sensitivity (down to 0.01% tumour fraction) and high specificity (>99.8%), which has been validated across numerous studies.11 13 15 33 43 44 More importantly, the assay filters out clonal hematopoiesis of indeterminate potential and germline-derived variants from analysis, thereby reducing false positives.42
In this study, ctDNA levels will be evaluated at baseline (immediately before starting treatment) and during treatment with IO, with serial ctDNA analysis planned every two cycles during the 2-year long follow-up in all cohorts. Several studies demonstrated that patients with declining ctDNA levels on-treatment had better survival outcomes, suggesting that the decline in the ctDNA level with treatment reflected a favourable response to IO.11 12 15 21–23 25 28 In a recent study by Bratman et al, the bespoke ctDNA assay was used in a cohort of 94 patients with 25 different types of solid tumours.11 In the study, the bespoke assay identified immunotherapy non-responders (eg, disease progression) with a 98% PPV. Among patients whose ctDNA levels increased after 6 weeks of treatment, PFS at 6 months was only 7.5%, compared with 54.5% in patients whose ctDNA levels decreased at the same time point. In conjunction with increasing tumour volume on a CT scan, bespoke ctDNA assay demonstrated 100% PPV for detecting non-responders. The study also found that complete clearance of ctDNA was associated with exceptionally durable response (100% OS with a median follow-up period of 25.4 months (range 10.8–29.5)).11
By contrast, the OS among patients who did not clear their ctDNA was 42.5% and 17.5% at 12 and 24 months, respectively. These data suggest that ctDNA clearance at any time point during treatment is highly predictive of long-term durable response. This finding is consistent with the results of an independent study in patients with hepatocellular carcinoma (n=48) undergoing treatment with atezolizumab and bevacizumab that used bespoke ctDNA assay and showed longer PFS in patients whose ctDNA level was undetectable with treatment.45 Not only did ctDNA changes predict the responses, all patients who had their ctDNA cleared were alive till the last date of follow-up. The study by Bratman et al also demonstrated that 55% of patients experienced molecular progression (ctDNA increase) at 6 weeks, and those patients received on average 2 cycles (6 weeks) of additional immunotherapy guided by radiologic study, which could have been avoided.11 Thus, bespoke ctDNA assay can enable an earlier switch to an alternative treatment that may have a higher chance of success and lower financial and toxicity burden.
The predictive value of ctDNA was illustrated in a post hoc analysis of IMvigor010 trial, a randomised, phase III study comparing adjuvant atezolizumab to observation after radical cystectomy for urothelial cancer.15 The study showed that ctDNA detection after radical cystectomy in both arms was associated with reduced disease-free survival (DFS) (atezolizumab arm, HR=3.36, 95% CI 2.44 to 4.62; observation arm, HR=6.3, 95% CI 4.45 to 8.92; p<0.0001) as well as reduced OS (atezolizumab arm, HR=3.63, 95% CI 2.34 to 5.64; observation arm, HR=8.0, 95% CI 4.92 to 12.99), compared with patients with undetectable postoperative ctDNA. In addition, ctDNA-positive patients in the adjuvant atezolizumab arm had an improved OS (HR=0.59, 95% CI 0.41 to 0.86; median DFS 25.8 vs 15.8 months in the observation arm), while ctDNA-negative patients showed no difference in survival if they received adjuvant atezolizumab. Furthermore, patients who cleared ctDNA with adjuvant atezolizumab had dramatically better survival outcomes compared with those who did not clear ctDNA (DFS, HR=0.26, 95% CI 0.12 to 0.56; p=0.0014; median DFS: 5.7 months vs not reached; and OS, HR=0.41, 95% CI 0.1 to 1.70).15 Overall, this study demonstrated that postoperative ctDNA could predict benefit from adjuvant immunotherapy in resected patients with urothelial cancer. Furthermore, patients can be stratified based on the presence/absence of ctDNA after resection, and the ctDNA-negative patients may be spared of adjuvant immunotherapy.15
Pseudoprogression poses a unique challenge in patients with solid tumour receiving immunotherapy as validated methods that differentiate between true progression and pseudoprogression are lacking. Limited studies have shown the potential of ctDNA in distinguishing pseudoprogression from true progression.11 24 26 In the study reported by Bratman et al, seven patients showed pseudoprogression (tumour progression on scans but decreasing ctDNA level at 6 weeks). Of these, four patients exhibited a better OS >18 months (range 19–27) when compared with patients who showed true progression (n=30, increasing ctDNA and progressive disease on scan).11 Further, the bespoke ctDNA assay was able to detect pseudoprogression 5 months earlier than the imaging studies.11 In the present study, as one of the exploratory endpoints, we plan to evaluate the association of ctDNA dynamics with pseudoprogression. ctDNA clearance or decline in such patients could help differentiate and direct patients with true progression to alternative treatment.
Taken together, the studies described above provide preliminary evidence that ctDNA can help in immunotherapy response monitoring. However, most of these studies included a small patient population.14 18 20 22 23 26 Additionally, several of these studies have used targeted panels to select the variants and tracked the variants with droplet digital PCR (ddPCR). However, the use of a targeted gene panel can result in suboptimal variant selection and decreased ctDNA sensitivity (43%–73%).12 23 25 46 By contrast, the bespoke ctDNA assay selects clonal variants from a whole-exome analysis of the tumour (approximately 20 000 genes), minimising suboptimal variant selection potential.
The predictive role of ctDNA is currently being studied in several ongoing clinical trials investigating the role of immunotherapy across multiple cancer types (NCT03512847, NCT04636047, NCT04053725, NCT03712566, NCT04589845, NCT04853017, NCT03409848, NCT03178552). Although most of these trials are designed to include small to moderate sample sizes and employ variable assay designs, these trials would be instrumental in establishing ctDNA’s role as a surrogate endpoint for immunotherapy treatment efficacy.
The limitation of our study is that it is purely observational. Therapy is physician directed and not dictated by the trial given the non-interventional nature of the study. However, the prospective design of the study, a large sample size, and the 2-year long follow-up period will allow us to compare the sensitivity, specificity, PPV, NPV and clinical utility within as well as among different study cohorts. Of note, our study design includes a retrospectively enrolled control group for adequate comparisons, which will further help in determining the clinical utility of the personalised, tumour-informed ctDNA assay in guiding treatment monitoring in patients receiving immunotherapy. Another limitation is the fewer tumour types being considered in this clinical study, which may limit the generalisability of ctDNA-based treatment response monitoring in patients with other tumour types getting IO therapy.
We believe this study will also help generate the relevant data required to allow for future prospective interventional studies. We expect that our study will help establish the real-world evidence of ctDNA’s utility in monitoring treatment response to immunotherapy in patients with solid tumours and support its integration into clinical practice and guidelines, leading to meaningful improvements in patient outcomes and quality of life.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the clinical project management and data management support provided by Worldwide Clinical Trials.
Footnotes
Twitter: @doctorC369
Contributors: Study coordinator: SS. Site identification: MK, SS. Design and writing of the protocol: AA, SS, MK, PMK, JE, AR, SE. Data collection: SS, SE, MK. Data analysis: JE. Data interpretation: JE, MK, AA, AR, SS. Writing of the manuscript: MiM, MeM, PMK, SC. Statistical setting of the study design and data analysis: JE. All authors reviewed and approved the final manuscript: PMK, SC, SS, MK, AP, GA, MiM, MeM, JE, LG, ZE, SE, PB, AR, AA.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: PMK acknowledges role as a consultant/advisor for Taiho Oncology, Ipsen, Natera, Foundation Medicine, Research/Trial Support (to institution): BMS, Celgene, AstraZeneca, BTG, Advanced Accelerator Applications, Array Biopharma. SC acknowledges membership of the Natera’s speakers’ bureau. AP acknowledges membership of the Bristol Myers Squibb speakers’ bureau, role as a consultant for Novartis, participatory role in a Natera Ad Board, and role as a speaker for Natera and Grail. GA has nothing to disclose. LG is a federal employee and reports no conflict of interest. ZE: Research Support: Pfizer, Novartis; Consultancy: Pfizer, Eisai, Natera Inc., OncoSec, Genentech. All other authors are employees of Natera, Inc. with stock/options to own stock on the company. This study is being sponsored by Natera, Inc.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting or dissemination plans of this research.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Not applicable.
References
- 1.Borcoman E, Kanjanapan Y, Champiat S, et al. Novel patterns of response under immunotherapy. Ann Oncol 2019;30:385–96. 10.1093/annonc/mdz003 [DOI] [PubMed] [Google Scholar]
- 2.Akinleye A, Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol 2019;12:92. 10.1186/s13045-019-0779-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seymour L, Bogaerts J, Perrone A, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 2017;18:e143–52. 10.1016/S1470-2045(17)30074-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borcoman E, Nandikolla A, Long G, et al. Patterns of response and progression to immunotherapy. Am Soc Clin Oncol Educ Book 2018;38:169–78. 10.1200/EDBK_200643 [DOI] [PubMed] [Google Scholar]
- 5.Thomas R, Somarouthu B, Alessandrino F, et al. Atypical response patterns in patients treated with nivolumab. AJR Am J Roentgenol 2019;212:1177–81. 10.2214/AJR.18.20938 [DOI] [PubMed] [Google Scholar]
- 6.Monthly and median costs of cancer drugs at the time of FDA approval 1965-2016. J Natl Cancer Inst 2017;109:djx173. 10.1093/jnci/djx173 [DOI] [PubMed] [Google Scholar]
- 7.Gjoerup O, Brown CA, Ross JS, et al. Identification and utilization of biomarkers to predict response to immune checkpoint inhibitors. Aaps J 2020;22:132. 10.1208/s12248-020-00514-4 [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Tong Z, Zhang W, et al. Fda-Approved and emerging next generation predictive biomarkers for immune checkpoint inhibitors in cancer patients. Front Oncol 2021;11:683419. 10.3389/fonc.2021.683419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kasi P, Chan C. 23 circulating tumor DNA (ctDNA) serial analysis during progression on PD-1 blockade and later CTLA4 rescue in patients with mismatch repair deficient metastatic colorectal cancer. J Immunother Cancer 2020;8:A12–13. 10.1136/jitc-2020-SITC2020.0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kooshkaki O, Derakhshani A, Hosseinkhani N, et al. Combination of ipilimumab and nivolumab in cancers: from clinical practice to ongoing clinical trials. Int J Mol Sci 2020;21:4427. 10.3390/ijms21124427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bratman SV, Yang SYC, Iafolla MAJ, et al. Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nat Cancer 2020;1:873–81. 10.1038/s43018-020-0096-5 [DOI] [PubMed] [Google Scholar]
- 12.Cabel L, Riva F, Servois V, et al. Circulating tumor DNA changes for early monitoring of anti-PD1 immunotherapy: a proof-of-concept study. Ann Oncol 2017;28:1996–2001. 10.1093/annonc/mdx212 [DOI] [PubMed] [Google Scholar]
- 13.Christensen E, Birkenkamp-Demtröder K, Sethi H, et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by Ultra-Deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J Clin Oncol 2019;37:1547–57. 10.1200/JCO.18.02052 [DOI] [PubMed] [Google Scholar]
- 14.Giroux Leprieur E, Herbretau G, Dumenil C, et al. Circulating tumor DNA evaluated by next-generation sequencing is predictive of tumor response and prolonged clinical benefit with nivolumab in advanced non-small cell lung cancer. Oncoimmunology 2018;7:e1424675. 10.1080/2162402X.2018.1424675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powles T, Assaf ZJ, Davarpanah N, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 2021;595:432–7. 10.1038/s41586-021-03642-9 [DOI] [PubMed] [Google Scholar]
- 16.Ricciuti B, Jones G, Severgnini M, et al. Early plasma circulating tumor DNA (ctDNA) changes predict response to first-line pembrolizumab-based therapy in non-small cell lung cancer (NSCLC). J Immunother Cancer 2021;9:e001504. 10.1136/jitc-2020-001504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.PM K. Kinetics of liquid biopsies in predicting response to immunotherapy: ASCO daily news. Available: https://dailynews.ascopubs.org/do/10.1200/ADN.20.200338/full/ [Accessed 14 Dec 2021].
- 18.Anagnostou V, Forde PM, White JR, et al. Dynamics of tumor and immune responses during immune checkpoint blockade in non-small cell lung cancer. Cancer Res 2019;79:1214–25. 10.1158/0008-5472.CAN-18-1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017;7:1394–403. 10.1158/2159-8290.CD-17-0716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldberg SB, Narayan A, Kole AJ, et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin Cancer Res 2018;24:1872–80. 10.1158/1078-0432.CCR-17-1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cabel L, Proudhon C, Romano E, et al. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat Rev Clin Oncol 2018;15:639–50. 10.1038/s41571-018-0074-3 [DOI] [PubMed] [Google Scholar]
- 22.Gray ES, Rizos H, Reid AL, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015;6:42008–18. 10.18632/oncotarget.5788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herbreteau G, Vallée A, Knol A-C, et al. Quantitative monitoring of circulating tumor DNA predicts response of cutaneous metastatic melanoma to anti-PD1 immunotherapy. Oncotarget 2018;9:25265–76. 10.18632/oncotarget.25404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH, Long GV, Menzies AM, et al. Association between circulating tumor DNA and pseudoprogression in patients with metastatic melanoma treated with Anti-Programmed cell death 1 antibodies. JAMA Oncol 2018;4:717–21. 10.1001/jamaoncol.2017.5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JH, Long GV, Boyd S, et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann Oncol 2017;28:1130–6. 10.1093/annonc/mdx026 [DOI] [PubMed] [Google Scholar]
- 26.Guibert N, Mazieres J, Delaunay M, et al. Monitoring of KRAS-mutated ctDNA to discriminate pseudo-progression from true progression during anti-PD-1 treatment of lung adenocarcinoma. Oncotarget 2017;8:38056–60. 10.18632/oncotarget.16935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raja R, Kuziora M, Brohawn PZ, et al. Early reduction in ctDNA predicts survival in patients with lung and bladder cancer treated with Durvalumab. Clin Cancer Res 2018;24:6212–22. 10.1158/1078-0432.CCR-18-0386 [DOI] [PubMed] [Google Scholar]
- 28.Zhang Q, Luo J, Wu S, et al. Prognostic and predictive impact of circulating tumor DNA in patients with advanced cancers treated with immune checkpoint blockade. Cancer Discov 2020;10:1842–53. 10.1158/2159-8290.CD-20-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985–90. 10.1038/nm.1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia N, Sun Z, Gao X, et al. Serial monitoring of circulating tumor DNA in patients with metastatic colorectal cancer to predict the therapeutic response. Front Genet 2019;10:470. 10.3389/fgene.2019.00470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parikh AR, Mojtahed A, Schneider JL, et al. Serial ctDNA monitoring to predict response to systemic therapy in metastatic gastrointestinal cancers. Clin Cancer Res 2020;26:1877–85. 10.1158/1078-0432.CCR-19-3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pereira E, Camacho-Vanegas O, Anand S, et al. Personalized circulating tumor DNA biomarkers dynamically predict treatment response and survival in gynecologic cancers. PLoS One 2015;10:e0145754. 10.1371/journal.pone.0145754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coombes RC, Page K, Salari R, et al. Personalized detection of circulating tumor DNA Antedates breast cancer metastatic recurrence. Clin Cancer Res 2019;25:4255–63. 10.1158/1078-0432.CCR-18-3663 [DOI] [PubMed] [Google Scholar]
- 34.Goodall J, Mateo J, Yuan W, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov 2017;7:1006–17. 10.1158/2159-8290.CD-17-0261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moding EJ, Liu Y, Nabet BY, et al. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small cell lung cancer. Nat Cancer 2020;1:176–83. 10.1038/s43018-019-0011-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Büttner R, Gosney JR, Skov BG, et al. Programmed Death-Ligand 1 immunohistochemistry testing: a review of analytical assays and clinical implementation in non-small-cell lung cancer. J Clin Oncol 2017;35:3867–76. 10.1200/JCO.2017.74.7642 [DOI] [PubMed] [Google Scholar]
- 37.Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US food and drug administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer 2019;7:278. 10.1186/s40425-019-0768-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaderbhaï C, Tharin Z, Ghiringhelli F. The role of molecular profiling to predict the response to immune checkpoint inhibitors in lung cancer. Cancers 2019;11:201. 10.3390/cancers11020201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meléndez B, Van Campenhout C, Rorive S, et al. Methods of measurement for tumor mutational burden in tumor tissue. Transl Lung Cancer Res 2018;7:661–7. 10.21037/tlcr.2018.08.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rousseau B, Foote MB, Maron SB, et al. The spectrum of benefit from checkpoint blockade in hypermutated tumors. N Engl J Med 2021;384:1168–70. 10.1056/NEJMc2031965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marcus L, Lemery SJ, Keegan P, et al. Fda approval summary: pembrolizumab for the treatment of microsatellite Instability-High solid tumors. Clin Cancer Res 2019;25:3753–8. 10.1158/1078-0432.CCR-18-4070 [DOI] [PubMed] [Google Scholar]
- 42.Kasi PM, Sawyer S, Guilford J, et al. BESPOKE study protocol: a multicentre, prospective observational study to evaluate the impact of circulating tumour DNA guided therapy on patients with colorectal cancer. BMJ Open 2021;11:e047831. 10.1136/bmjopen-2020-047831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magbanua MJM, Swigart LB, Wu H-T, et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann Oncol 2021;32:229–39. 10.1016/j.annonc.2020.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reinert T, Henriksen TV, Christensen E, et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol 2019;5:1124–31. 10.1001/jamaoncol.2019.0528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu C-H, Lu S, Abbas A, et al. Longitudinal and personalized detection of circulating tumor DNA (ctDNA) for monitoring efficacy of atezolizumab plus bevacizumab in patients with unresectable hepatocellular carcinoma (HCC). Journal of Clinical Oncology 2020;38:3531–31. 10.1200/JCO.2020.38.15_suppl.3531 [DOI] [Google Scholar]
- 46.Keller L, Guibert N, Casanova A, et al. Early circulating tumour DNA variations predict tumour response in melanoma patients treated with immunotherapy. Acta Derm Venereol 2019;99:206–10. 10.2340/00015555-3080 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.