

Abstract
Deep vein thrombosis results from the cooperative action of leukocytes, platelets, and endothelial cells. The proline-rich tyrosine kinase Pyk2 regulates platelet activation and supports arterial thrombosis. In this study, we combined pharmacological and genetic approaches to unravel the role of Pyk2 in venous thrombosis. We found that mice lacking Pyk2 almost completely failed to develop deep venous thrombi upon partial ligation of the inferior vena cava. Pyk2-deficient platelets displayed impaired exposure of phosphatidylserine and tissue factor expression by endothelial cells and monocytes was completely prevented by inhibition of Pyk2. In human umbilical vein endothelial cells (HUVEC), inhibition of Pyk2 hampered IL-1β-induced expression of VCAM and P-selectin, and von Willebrand factor release. Pyk2-deficient platelets showed defective adhesion on von Willebrand factor and reduced ability to bind activated HUVEC under flow. Moreover, inhibition of Pyk2 in HUVEC strongly reduced platelet adhesion. Similarly, Pyk2-deficient neutrophils were unable to efficiently roll and adhere to immobilized endothelial cells under venous flow conditions. Moreover, platelets and neutrophils from Pyk2-knockout mice showed defective ability to form heterogeneous aggregates upon stimulation, while platelet monocyte interaction occurred normally. Consequently, platelet neutrophil aggregates, abundant in blood of wild-type mice upon inferior vena cava ligation, were virtually undetectable in Pyk2-knockout mice. Finally, we found that expression of Pyk2 was required for NETosis induced by activated platelets. Altogether our results demonstrate a critical role of Pyk2 in the regulation of the coordinated thromboinflammatory responses of endothelial cells, leukocytes and platelets leading to venous thrombosis. Pyk2 may represent a novel promising target in the treatment of deep vein thrombosis.
Introduction
Deep vein thrombosis (DVT) is a consequence of impaired hemostasis and increased inflammation.1 Thrombi formed in the lower limbs frequently embolize and reach the lungs causing pulmonary thromboembolism.2 DVT results from the cooperative action of leukocytes and platelets that interact with activated endothelial cells (EC).3 The vascular endothelium can be activated by multiple insults, such as hypercholesterolemia, high blood pressure, and reactive oxygen species (ROS).4 Stasis of venous blood is a crucial determinant for DVT and facilitates the development of a pro-inflammatory phenotype in EC characterized by the expression of cell adhesion molecules (CAM) and P-selectin and associated to the release of von Willebrand factor (VWF).5,6 EC recruit blood borne leukocytes. Tissue factor (TF) expression by monocytes initiates the extrinsic pathway of the coagulation cascade,7 whereas thrombus-resident neutrophils propagate DVT through the activation of coagulation factor XII. Neutrophils also release neutrophil extracellular traps (NET) and engage circulating platelets8 which, in turn, recruit additional neutrophils forming platelet neutrophil aggregates in a vicious circle reinforcing thrombosis and inflammation.3
The activation of different vascular cells involved in the propagation of DVT is regulated by signaling molecules and key enzymes. Among them, protein tyrosine kinases play a pivotal role in cellular activation and are often directly linked to membrane receptors.9 In particular, Src family kinases mediate neutrophil adhesion to adherent platelets and modulate platelet neutrophil aggregate formation.10,11 Additional tyrosine kinases act as second line signal transducers and, among them, focal adhesion kinases (FAK) have attracted increasing attention over the past few years.
Proline-rich tyrosine kinase Pyk2 is a cytosolic non-receptor tyrosine kinase of 1,009 aminoacids that belongs to the FAK family.12 Pyk2 is highly expressed in hematopoietic cells.13 In macrophages, Pyk2 mediates adhesion, migration, and complement-mediated phagocytosis14,15 and in neutrophils it controls adhesion, migration, degranulation, ROS production and host responses to bacterial infection.16,17 These observations highlight a critical role of Pyk2 in innate immunity and inflammation.
We have previously shown that Pyk2 is also an important regulator of platelet activation.18 Pyk2 gets phosphorylated and thus activated in platelets stimulated by thrombin, collagen, ADP and VWF19-21 and it is also recruited in platelets adherent to collagen and fibrinogen.22,23 Pyk2 is required for TxA2 formation, integrin αIIbβ3 activation and platelet aggregation.18 Mice lacking Pyk2 show delayed formation of occlusive thrombi in a model of photochemical induced arterial thrombosis and increased bleeding time, showing that Pyk2 is a critical regulator of arterial thrombosis and primary hemostasis. Interestingly, Pyk2-knockout (Pyk2-KO) mice are also protected against platelet pulmonary embolism.18
The role of Pyk2 in the development of DVT however remains unexplored. In this study we show that Pyk2 is an important regulator of DVT and orchestrates the interplay between platelets, neutrophils, and EC. Our results disclose a novel potential therapeutic target for the pharmacological prevention of venous thrombosis by targeting Pyk2.
Methods
Materials
Pyk2-KO mice generation was described previously.14 All the procedures involving the use of mice were approved by the Ethics Committee (University of Pavia), by the Committee on Ethics of Animal Experiments (University of Perugia), and by the Italian Ministry of Public Health (authorization numbers 561/2015 PR; 8/2018 PR; 259/2018 PR). HUVEC were from Promocell, and THP-1 cells were a gift from Prof. Lanni, University of Pavia. PE-conjugated anti-VCAM-1 (CD106) (PE), FITC-conjugated anti-P-selectin and anti-mouse immunoglobulin G (IgG), as well as FITC-labeled Annexin V kit were from Beckam Coulter. Anti-tissue factor antibody (CD142), anti-Ly6G and anti-CD115 were from eBioscience. FITC-labeled anti-CD41/CD61 (Leo.D2) was from Emfret Analytics. Peridin chlorophyl protein (PerCP)-conjugated anti-mouse CD45 (clone 30-F11) was from Beckton Dickinson. Pyk2 inhibitor PF-4594755 was from Pfizer. Asserachrom VWF:Ag Kit was from Diagnostica Stag; IL-1β from PeproTech: CellTracker™ Red CMTPX Dye from Thermo Fisher Scientific. VWF was from Calbiochem.
Experimental procedures
Experimental procedures were performed as previously described.24-31 Additional details are included in the Online Supplementary Appendix.
Results
Pyk2 promotes venous thrombosis
We have previously demonstrated that Pyk2 deficiency protects mice from platelet pulmonary embolism triggered by the injection of collagen plus epinephrine.18 In order to investigate the role of Pyk2 in the development of DVT in mice, we applied a model of partial inferior vena cava (IVC) ligation, in which thrombosis is driven by a flow disturbance without physical disruption of the endothelial lining.5,32 Mice were sacrificed 24- and 48-hours post IVC ligation and formed thrombi were extracted and analyzed. All the WT mice developed an evident vena cava thrombus both at 24 and 48 hours upon IVC ligation (Figure 1A). By contrast, thrombus formation was observed only in four of 15 Pyk2-KO mice at 24 hours, and in six of 16 Pyk2-KO mice 48 hours after IVC ligation (Figure 1A). When present, thrombi formed in Pyk2-KO mice were very small compared to those formed in WT mice or were, in some animals, nearly undetectable (Figure 1B). Thrombus weight and length were strikingly and significantly reduced in Pyk2-KO compared to WT mice (Figure 1C and D). At 24 hours, the mean weight of thrombi in WT mice was 6.92±4.89 mg, while that of thrombi observed in Pyk2-KO was 1.09±2.4 mg. The thrombi length was about 3.06±1.97 mm and 0.63±1.14 mm in WT and Pyk2-KO mice, respectively. At 48 hours, the average weight of the thrombi further increased in WT mice to 10.45±5 mg and the size to 5.69±2.2 mm but remained unchanged in Pyk2-deficient mice (0.63± 1.1 mg and 0.67±1.0 mm). These data clearly show that the absence of Pyk2 protects mice from venous thrombosis upon IVC ligation. In order to discriminate the contribution of Pyk2 expressed in platelets versus endothelial cells or leukocytes we performed cross-transfusion experiments in which platelets from Pyk2-KO mice were reinfused into recipient thrombocytopenic WT mice and vice versa. Figure 1E shows that WT mice receiving Pyk2-deficient platelets were still able to form thrombi upon partial IVC ligation. However, both the length and the weight of these thrombi were slightly reduced when compared to those formed in WT mice (P<0.05). By contrast, transfusion of WT platelets into Pyk2-KO mice did not restore at all the ability to form venous thrombi, as thrombus formation was never observed in all the analysed animals. These results indicate that Pyk2 in endothelial cells and/or leukocytes is essential for initiation of DVT and that Pyk2 in platelets has a secondary but evident role in thrombus growth.
Figure 1.
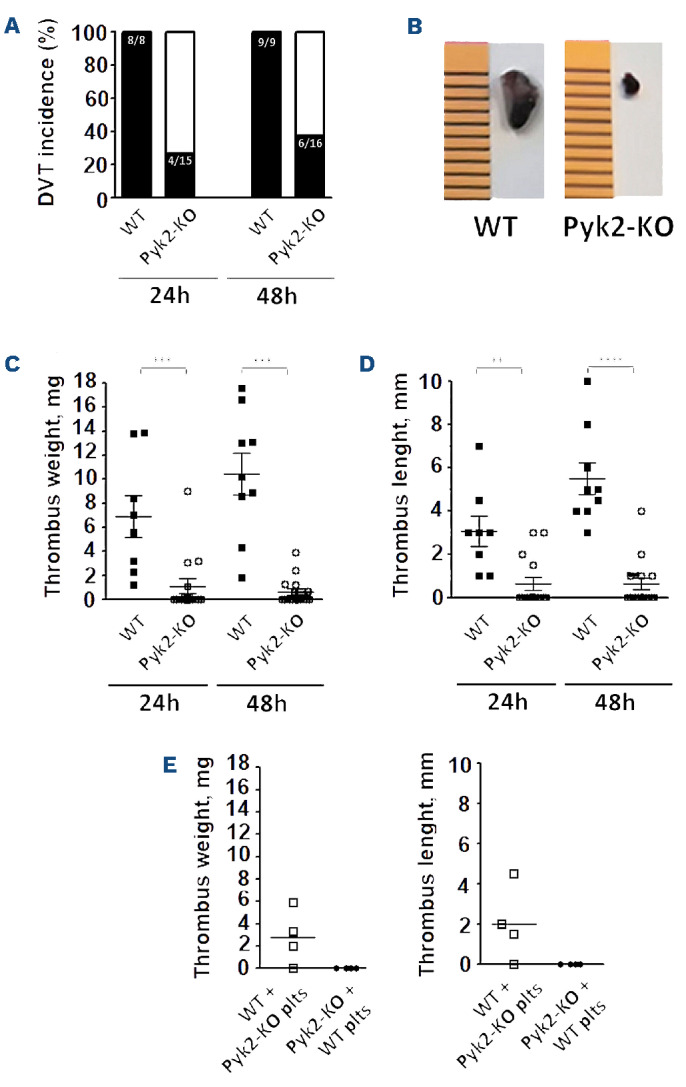
IVC-ligation-induced deep vein thrombosis in wild-type and Pyk2-knockout mice. Thrombus formation in the inferior vena cava (IVC) was induced by partial vein ligation. Thrombi were extracted 24 or 48 hours (h) after surgery. (A) Incidence of thrombus formation in wild-type (WT) vs. Pyk2-knockout (Pyk2-KO) mice is reported as percentage of the analyzed animals (black bars: mice developing thrombus; white bars: mice with no detectable thrombus). (B) Comparative representative images showing the different size of thrombi from WT and Pyk2-KO mice after 24 hours of IVC ligation. (C and D) Thrombus weight and length at 24 and 48 hours, respectively. **P≤0.01; ***P≤0.005; ****P≤0.001 (17 WT and 31 Pyk2-KO mice were analyzed). (E) Thrombus formation in cross-transfused mice. Pyk2-deficient platelets were re-infused in recipient thrombocytopenic WT mice and WT platelets were re-infused in recipient Pyk2-KO mice (4 animals/group), as indicated. Thrombi were isolated and measured 48 hours upon IVC ligation.
Pyk2 is required for endothelial cell activation
DVT results from a complex interplay between enzymatic and cellular processes in which EC, platelets and leukocytes orchestrate a proinflammatory condition that culminates in clot formation. FAK have been implicated in vascular inflammation induced by TNFα and IL-1β in human aortic EC.33 We investigated the involvement of Pyk2 in EC activation using HUVEC and a pharmacological Pyk2 inhibitor, PF-4594755, whose selectivity and potency have been previously characterized.34 HUVEC were pre-incubated with PF-4594755 and then stimulated with IL-1β for 4 hours. Surface expression of the adhesive molecules P-selectin and VCAM was significantly increased by IL-1β in stimulated HUVEC, and this response was completely suppressed by the Pyk2 inhibitor PF-4594755 (Figure 2A). We also analyzed the contribution of Pyk2 to the secretion of VWF, which plays a critical role in hemostasis and inflammation, as well as in DVT.35 We found that the constitutive release of VWF was unchanged upon pre-incubation with PF-4594755. However, VWF secretion stimulated by IL-1β was significantly affected by inhibition of Pyk2 activity (Figure 2B).
Clot formation is initiated by TF that can be expressed by stimulated EC as well as by monocytes. Figure 2C(i) shows that stimulation of HUVEC with IL-1β induced a marked expression of TF, which was completely prevented upon Pyk2 inhibition by PF-4594755. By measuring the increase of TF mRNA in IL-1β-stimulated HUVEC treated with PF-4594755, we obtained clear evidence that Pyk2 regulates TF expression mainly at the transcriptional level (Figure 2Cii). Similarly, we found that also in monocytes TF exposure stimulated by treatment with LPS was completely prevented upon Pyk2 inhibition (Figure 2D). Altogether, these results indicate that TF exposure by pro-inflammatory stimulation of EC and monocytes is strictly regulated by Pyk2.
Pyk2 regulates neutrophil rolling over endothelial cells
In order to unravel the contribution of Pyk2 expressed by neutrophils in the early steps of DVT, we compared the ability of neutrophils isolated from the bone marrow of WT and Pyk2-KO mice to adhere over immobilized EC under flow. Isolated WT and Pyk2-deficient neutrophils were perfused over a layer of HUVEC stimulated with IL-
1β at a shear stress of 250 sec-1, which mimics venous blood flow. Neutrophils from WT mice adhered to stimulated HUVEC in a time-dependent manner and maximal, firm adhesion was observed after 300 seconds. Conversely, Pyk2-deficient neutrophils almost completely failed to stop on endothelial cells and rolled away in few seconds (Figure 3A and B). Consequently, the number of firmly adherent neutrophils remained very low over time. In a separated set of experiments, we analyzed whether inhibition of Pyk2 in HUVEC could affect neutrophil rolling. Figure 3C shows that WT neutrophils almost completely failed to roll and adhere over IL-1β-stimulated HUVEC in which Pyk2 is inhibited by PF-4594755. Thus, Pyk2 activity in neutrophil as well as in EC is necessary for a correct recruitment of neutrophils on the activated endothelium.
Figure 2.

Pyk2 supports a pro-inflammatory and procoagulant phenotype in endothelial cells. Human umbilical vein endothelial cells (HUVEC) were pre-incubated with dimethyl sulfoxide (DMSO) as vehicle (black bars) or with 10 µM Pyk2 inhibitor PF-4594755 (white bars) for 30 minutes and then stimulated with IL-1β for 4 hours (IL-1β) or left untreated (none). (A) Surface expression of P-selectin and VCAM-1, as assessed by flow cytometry, expressed as % of positive cells. (B) Measurement of von Willebrand factor (VWF) release in the supernatant of HUVEC. (C) Surface expression of tissue factor (TF), analyzed by flow cytometry, is reported in panel (i), while reverse transcription polymerase chain reaction (RT-PCR) analysis of TF mRNA expression in vehicle (none, black bars) or PF-4594755 (white bars)-treated HUVEC stimulated with IL-1β is reported in panel (ii), as fold increase over unstimulated cells. (D) THP-1 cells were pre-incubated with DMSO as vehicle (black bars) or with 10 µM Pyk2 inhibitor PF-4594755 (white bars) for 30 minutes and stimulated with 10 µg/ml lipopolysaccharide (LPS) for 3 hours (LPS) or left untreated (none) and surface expression of TF was analyzed by flow cytometry. Results are the mean ± standard error of the mean of 3 different experiments. *P≤0.05; **P≤0.01; ***P≤0.005; ****P≤0.001.
Figure 3.
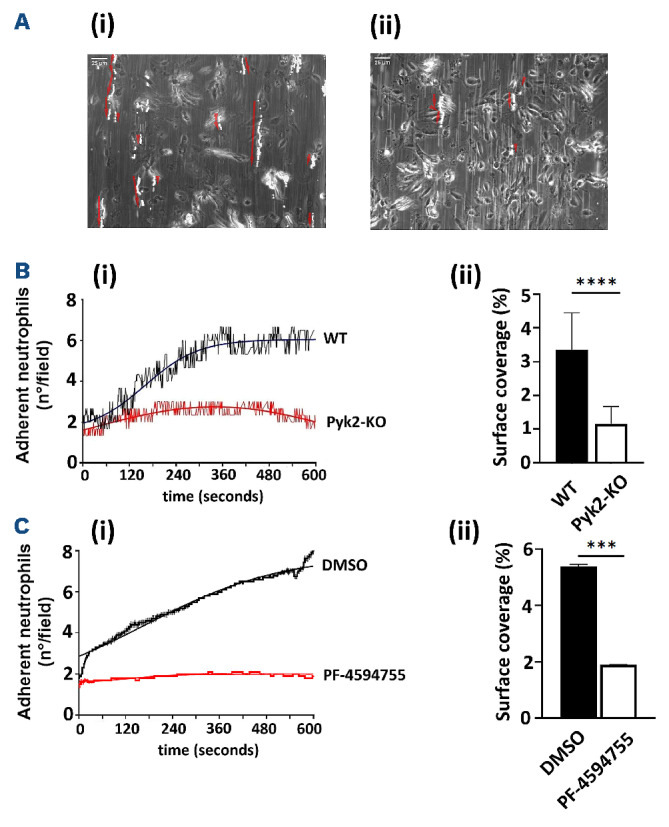
Pyk2 mediates neutrophil rolling over human umbilical vein endothelial cells. Neutrophils were isolated from the bone marrow of wild-type (WT) and Pyk2-knockout (Pyk2-KO) mice and perfused over IL-1β-activated human umbilical vein endothelial cells (HUVEC) at 250 sec-1, a shear stress that mimics venous blood flow. Number of adherent neutrophils was acquired at a rate of 1 frame/second. (A) Representative phase contrast microscopy images of rolling neutrophils (bright spots), obtained by overlapping of all the acquired frames are shown in panels (i) for WT neutrophils, and (ii) for Pyk2-deficient neutrophils, respectively. Arrows indicate the trajectory of rolling neutrophils over time. (B) The number of adherent neutrophils/field acquired at the different time points over a period of 10 minutes is reported in panel (i), while the percentage of surface coverage is shown in panel (ii). ****P≤0.001. (C) Rolling and adhesion of WT neutrophils perfused at 250 sec-1 over IL-1β-activated HUVEC pretreated with dimethyl sulfoxide (DMSO) or with this is 10 µM Pyk2 inhibitor PF-4594755 for 30 minutes (panel i) and the percentage of surface coverage (panel ii). The reported data are representative of 3 different experiments. ***P≤0.005.
Pyk2 modulates the interactions between platelets and neutrophils and neutrophil extracellular trap formation
Circulating platelet leukocyte aggregates (PLA) in WT and Pyk2-deficient mice were similar in basal conditions, but they were significantly reduced in Pyk2-KO mice upon stimulation with thrombin (Figure 4A). Similar results were also obtained upon stimulation with the GPVI agonist convulxin (data not shown). Using antibodies against Ly6G or CD115, we found that Pyk2 deficiency selectively affected interaction of platelets with neutrophils, but not with monocytes (Figures 4B and C). The number of circulating PLA in whole blood collected 48 hours after DVT induced by IVC partial ligation was dramatically reduced in Pyk2KO mice compare to WT littermates (Figure 4D).
We next evaluated in vitro platelet-stimulated NET extrusion. Neutrophils were left untreated or mixed with thrombin-activated platelets for 30 minutes, and NET formation was evaluated by DNA staining with Hoechst. Intact neutrophils showed the typical nucleus with multilobulated shape, that lost its compactness and shape and extruded DNA and histones upon NET formation (Figure 4E). In the absence of added platelets, no differences in NET formation were observed between WT and Pyk2-deficient neutrophils. However, upon addition of thrombinstimulated WT platelets a significant increase of NET formation was observed with WT but not with Pyk2-deficient neutrophils, indicating that, in neutrophils Pyk2 is required to elaborate a functional response to activated platelets. Interestingly, addition of thrombin-stimulated platelets from Pyk2-KO mice failed to induce NET extrusion by both WT and Pyk2-deficient neutrophils (Figures 4E and F), demonstrating a role also for platelet Pyk2 in the development of a neutrophil NET phenotype. Importantly, upon neutrophil stimulation with PMA, that bypasses early signal transduction events, NET extrusion was comparable in WT and Pyk2-deficient cells (Figure 4G).
Pyk2 modulates platelet adhesion to von Willebrand factor
During DVT, platelets also bind to the activated endothelium and to the released VWF.36 Figure 5A shows that, in the presence of botrocetin, platelet adhesion to VWF under static conditions, was significantly impaired in the absence of Pyk2 (Figure 5A).
Adhesion of platelets to activated EC in function of time was investigated under flow conditions over IL-1β activated HUVEC. Figure 5B shows that, when compared to WT platelets, Pyk2-KO platelets displayed a modestly reduced ability to adhere to a monolayer of activated HUVEC. However, quantitative analysis of collected images showed that the percentage of total area covered by adherent platelets was slightly, but significantly higher for WT than for Pyk2-KO platelets (0.17±0.01% vs. 0.15±0.02%, respectively, P<0.05) (Figure 5Bii). Importantly, however, the ability of platelets to adhere over IL-1β-activated HUVEC was almost completely prevented when endothelial Pyk2 was pharmacological inhibited by PF-4594755 (Figure 5C, panels i and ii). These results indicate that Pyk2 activity in the endothelium, rather than in platelets, plays a predominant role in the regulation of platelet adhesion.
Figure 4.

Pyk2 regulates platelets-neutrophils interaction and is required for neutrophil extracellular traps formation. (A) Platelet-leukocyte, (B) platelet-monocyte and (C) platelet-neutrophil aggregates in whole blood from wild-type (WT) (black bars) and Pyk2-knockout (Pyk2-KO) mice (white bars) before (basal) of upon stimulation with 0.1 U/mL thrombin were measured by flow cytometry using specific antibodies: CD45 for leukocytes, CD115 for monocytes, Ly6G for neutrophils and CD42 for platelets. Results are the mean ± standard error of the mean (SEM) of 3 different experiments. *P≤0.05; ***P≤0.005. (D) Platelet-neutrophil aggregates in whole blood of WT (black bar) or Pyk2-KO mice (white bar) measured 48 hours after partial inferior vena cava (IVC). ***P≤0.005. (E to G) Neutrophils were isolated from bone marrow and platelets from whole blood of WT and Pyk2-KO mice. Neutrophils (from either genotypes) were left untreated (none) or incubated with thrombin-activated platelets from WT mice or from Pyk2-KO mice, as indicated in (E and F), or with 100 nM phorbol 12-myristate 13-acetate (PMA) (G). Neutrophil extracellular trap (NET) formation was analyzed by fluorescent microscopy upon staining of nuclei with Hoechst. (E) Representative images at 400X magnification, while quantification of NET formation, expressed as % of NET-extruding neutrophils, is reported in panels (F and G). Results are the mean ± SEM of 3 different experiments. **P≤0.01
Finally, we investigated the role of Pyk2 in the expression of platelet procoagulant activity, by measuring the surface exposure of PS, through annexin V binding. Figure 5D shows that platelet PS exposure increased in both genotypes when platelets were stimulated with low or high doses of thrombin plus convulxin, but the percentage of annexin V-positive cells remained significantly lower in Pyk2-deficient platelets.
Discussion
In this study we have shown that the proline-rich tyrosine kinase Pyk2 is a critical regulator of venous thrombosis in mice. This novel function of Pyk2 results from its multifaceted role in the pro-inflammatory responses of several blood cells involved in DVT development, including platelets, neutrophils and EC. Pyk2-KO mice failed to develop thrombi in the lower limbs upon IVC ligation or formed, occasionally, thrombi very small in size and weight. Specifically, we found that Pyk2: i) regulates the expression of adhesive molecules by activated EC; ii) modulates the expression of TF by activated monocytes and EC and the expression of pro-coagulant activity by platelets; iii) mediates neutrophil and platelet adhesion to EC under flow; iv) supports the formation of platelet neutrophil aggregates and stimulates NETosis.
Figure 5.

Pyk2 regulates platelet adhesion to von Willebrand factor, rolling over human umbilical vein endothelial cells and phosphatidylserine expression. (A) Washed platelets from wild-type (WT) or Pyk2-knockout (Pyk2-KO) mice were let to adhere over immobilized von Willebrand afcator (VWF) in the presence of 0.5 mg/mL botrocetin for 60 minutes. Adherent platelets were stained with TRITC-conjugated phalloidin and counted with the software Image J. A representative image is reported in (i), while quantification of the results is reported in the histograms (ii) (black bar: WT platelets; white bar: Pyk2-deficient platelets). Results are the mean ± standard error of the mean (SEM) of 4 different experiments. ***P≤0.005. (B) Fluorescent-labeled platelets were perfused at a shear rate of 200 sec-1 over a monolayer of IL-1β (1 ng/mL)-activated human umbilical vein endothelial cells (HUVEC) and rolling platelets were evaluated by measuring fluorescent spots moving on the HUVEC surface. The relative area covered by platelets was calculated with ImageJ at 1 FPS steps. The plots in (i) show the covered area and quantitative analysis of are presented in (ii) as mean ± SEM of 3 different experiments. *P≤0.05; ***P≤0.005. (C) Fluorescent-labeled platelets were perfused over PF-4594755-treated HUVEC and the number of adherent cells was acquired at a rate of 1 frame/second (panel i). Quantification of the percentage of surface coverage is reported in panel (ii). ****P≤0.001. (D) Phosphatidylserine (PS) exposure on activated WT and Pyk2-KO mouse platelets. Murine platelets were left untreated (basal) or were treated with 0.5 U/mL thrombin plus 250 ng/mL convulxin (low dose) or with 1 U/mL thrombin plus 500 ng/mL convulxin (high dose) for 10 minutes. Cells were then stained with annexin V-FITC and analyzed by flow cytometry. Data are expressed as the percentage of positive cells and are the mean ± SEM of 3 different experiments. *P≤0.05; ****P≤0.001.
Previous studies have recognized the functional relevance of Pyk2 in vascular cells, but none have assessed its role in the mechanisms triggering venous thrombosis. We have previously shown that Pyk2 is essential for platelet activation and adhesion,22,23 granule secretion, and arterial thrombosis.18 In neutrophils, Pyk2 regulates adhesion, motility and inflammatory responses,15,17,37,38 while in EC, Pyk2 was shown to regulate adhesion dynamics and to promote vascular inflammation.33 Platelets, neutrophils, and EC cooperate in the development of venous thrombosis.39 The present study documents for the first time that Pyk2 coordinates the activation of these vascular cells and regulates multiple events which converge toward the development of a pro-inflammatory, hypercoagulable, and prothrombotic phenotype. This articulated contribution of Pyk2 to EC, platelet, and leukocyte functions provides a molecular explanation for its essential role in DVT. Importantly, cross-transfusion experiments indicate that the role of Pyk2 in the different vascular cells implicated in DVT is not redundant but may regulate specific steps of thrombus formation. In particular, the inability of transfused WT platelets to restore DVT in Pyk2-KO mice indicates that Pyk2 in EC or leukocytes is essential for the initiation of thrombus formation. By contrast, transfusion of Pyk2-deficient platelets into WT mice only caused a reduction of thrombus length and weight, suggesting that Pyk2 in platelets is not required for the initial formation of venous thrombi, but contributes to its development and growth. This is in line with the notion that EC and leukocytes act in an early phase during DVT while platelets may then contribute to recruit leukocytes and support fibrin formation by enhancing neutrophil-dependent coagulation.3
The contribution of Pyk2 on DVT is demonstrated using both genetic and pharmacologic approaches. Pyk2-KO mice have been used for in vivo studies and for the investigation of the molecular mechanisms involved at the level of isolated cells. Some experiments have been performed with human cells and the role of Pyk2 has been determined using the pharmacological inhibitor PF-4594755. The use of this inhibitor was necessary under all those circumstances in which it was impossible to obtain a significant number of primary cells from Pyk2-KO mice, mainly in the case of the studies performed on EC and monocytes. The combined use of murine and human cells does not represent a limitation of the study, but rather provides reliability to the translation of the in vivo observations made in mice to humans. In this regard, it is noteworthy that the inhibitor used in our study, PF-4594755, is selective and specific for Pyk2, as previously demonstrated by the analysis of its effects on Pyk2-deficient cells.34 We also found that PF-4594755 was able to prevent Pyk2 phosphorylation in activated platelets34 and well as in LPS-stimulated monocytes (data not shown). In the present study, the use of PF-4594755 was limited to in vitro experiments, because previous analysis of the pharmacokinetic properties of this compound highlighted its low bioavailability and unsuitability to be used in vivo probably because of its high hydrophobicity.40,41
A pro-inflammatory phenotype deriving in part from the prolonged disturbance of blood flow represents a common first trigger of DVT. Our results highlight a precise role for Pyk2 in the development of such a pro-inflammatory phenotype and are in line with previous works, based on the use of less selective inhibitors of FAK. For instance, Murphy and collaborators showed that dual FAK and Pyk2 inhibition by PF-271 reduced cytokine-induced expression of inflammatory adhesion molecules by HUVEC.33 Here, we found that the selective inhibition of Pyk2 by PF-4594755 is sufficient to completely prevent IL-1β-induced expression of P-selectin, VCAM and TF, as well as VWF release by HUVEC, suggesting that Pyk2 is an important regulator of the pro-inflammatory phenotype of human EC. Accordingly, we found that, inhibition of Pyk2 in EC strongly prevented neutrophil rolling and adhesion under venous flow conditions. Moreover, also Pyk2-deficient neutrophils rolled on and adhered to HUVEC less efficiently than Pyk2-expressing cells. These data show that Pyk2 is an important regulator of the cell activation pathways supporting the leukocyte endothelium interactions. We also collected evidence that Pyk2 is required for the initiation of blood clotting. In fact, we have documented that expression of TF in stimulated monocytes and EC was completely prevented by the inhibition of Pyk2 by PF-4594755. We also suggest that, at least in ECs, Pyk2 regulate TF expression mainly at a transcriptional level. Moreover, our results indicate that also Pyk2 expressed in platelets may contribute to clot formation, as the expression of procoagulant PS was significantly, albeit not completely, reduced in Pyk2-deficient platelets. These results point to a role for Pyk2 in the regulation of the extrinsic blood coagulation pathway. Actually, we did measure a prolonged aPTT in Pyk2-KO mice compared to WT littermates (data not shown). However, the specific mechanism underlying this observation remains to be elucidated, as the current aPTT assay implies the addition of a certain amount of exogenous TF, thus making difficult to appreciate the precise contribution of endogenous TF. Neutrophil and platelet activation are closely intertwined in the development of thromboinflammation. Stimulated neutrophils release NET which activate platelets promoting the release of pro-inflammatory cytokines and triggering clot formation.8 Platelet neutrophil interactions thus support the development of DVT. We found that platelet neutrophil aggregates were abundant in blood of WT mice upon induction of DVT by IVC ligation but were almost undetectable in Pyk2-KO mice which failed to develop DVT. Although it cannot be excluded that the reduced number of platelet neutrophil aggregates in Pyk2KO mice may be consequence of the failure to develop DVT, it is reasonable that this represents one of the causes of the lack of thrombus formation. Indeed, we found that the expression of Pyk2 both in platelets and neutrophils is required for the formation in vitro of heterologous aggregates upon stimulation with thrombin. These observations integrate and extend previous findings by Evangelista et al. showing that Src family kinases mediate neutrophil adhesion to platelets through the recruitment of Pyk2.10 P-selectin mediates platelet interaction with neutrophils, and it is noteworthy that the lack of Pyk2 impairs P-selectin exposure on activated platelets.18 The reduced ability of platelets and neutrophils to form heterologous aggregates may provide an additional mechanistic explanation for the markedly reduced DVT in Pyk2KO mice, since circulating platelet neutrophil aggregates have been recognized as a risk factor for DVT.42 NET formation by neutrophils is stimulated by the interaction with activated platelets and represents a triggering event for DVT. Src and MAP kinases which are commonly indicated as upstream and downstream effectors of Pyk2, respectively, are regulators of NETosis.43 In our study we show that Pyk2-deficient neutrophils form less NET in vitro when stimulated with either Pyk2-expressing or Pyk2-deficient platelets. We also verified that the lack of Pyk2 in platelets hampers NET formation by WT neutrophils in manner comparable to that produced by the lack of Pyk2 in neutrophils. Thus, Pyk2 elicits a biological effect by controlling the contribution of both cell types involved in this response.
In conclusion, our study highlights a novel multifaceted role of Pyk2 in the cross-talk between platelets, neutrophils and EC and identifies this kinase as a novel player in the different steps involved in the development of DVT. Pyk2, therefore, could represent a novel potential therapeutic target for the prevention of DVT associated with inflammatory conditions.
Supplementary Material
Acknowledgments
The authors thank Prof. Cristina Lanni for providing THP-1.
Funding Statement
Funding: This research was supported by the Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022), Department of Biology and Biotechnology “L. Spallanzani,” University of Pavia, and by Fondazione CARIPLO (2018-0483 to MT and PG).
References
- 1.Schulz C, Engelmann B, Massberg S. Crossroads of coagulation and innate immunity: the case of deep vein thrombosis. J Thromb Haemost. 2013;11(1):233-241. [DOI] [PubMed] [Google Scholar]
- 2.Di Nisio M, van Es N, Büller HR. Deep vein thrombosis and pulmonary embolism. Lancet. 2016;388(10063):3060-3073. [DOI] [PubMed] [Google Scholar]
- 3.von Brühl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scioli MG, Storti G, D'Amico F, et al. Oxidative stress and new pathogenetic mechanisms in endothelial dysfunction: potential diagnostic biomarkers and therapeutic targets. J Clin Med. 2020;9(6):1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brill A, Fuchs TA, Chauhan AK, et al. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117(4):1400-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Budnik I, Brill A. Immune factors in deep vein thrombosis initiation. Trends Immunol. 2018;39(8):610-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cimmino G, Cirillo P. Tissue factor: newer concepts in thrombosis and its role beyond thrombosis and hemostasis. Cardiovasc Diagn Ther. 2018;8(5):581-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012;32(8):1777-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124(13):2013-2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evangelista V, Pamuklar Z, Piccoli A, et al. Src family kinases mediate neutrophil adhesion to adherent platelets. Blood. 2007;109(6):2461-2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Totani L, Amore C, Di Santo A, et al. Roflumilast inhibits leukocyte-platelet interactions and prevents the prothrombotic functions of polymorphonuclear leukocytes and monocytes. J Thromb Haemost. 2016;14(1):191-204. [DOI] [PubMed] [Google Scholar]
- 12.Guidetti GF, Torti M, Canobbio I. Focal adhesion kinases in platelet function and thrombosis. Arterioscler Thromb Vasc Biol. 2019;39(5):857-868. [DOI] [PubMed] [Google Scholar]
- 13.Avraham S, Avraham H. Characterization of the novel focal adhesion kinase RAFTK in hematopoietic cells. Leuk Lymphoma. 1997;27(3-4):247-256. [DOI] [PubMed] [Google Scholar]
- 14.Okigaki M, Davis C, Falasca M, et al. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc Natl Acad Sci U S A. 2003;100(19):10740-10745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paone C, Rodrigues N, Ittner E, et al. The Tyrosine kinase Pyk2 contributes to complement-mediated phagocytosis in murine macrophages. J Innate Immun. 2016;8(5):437-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamen LA, Schlessinger J, Lowell CA. Pyk2 is required for neutrophil degranulation and host defense responses to bacterial infection. J Immunol. 2011;186(3):1656-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canino J, Guidetti GF, Galgano L, et al. The proline-rich tyrosine kinase Pyk2 modulates integrin-mediated neutrophil adhesion and reactive oxygen species generation. Biochim Biophys Acta Mol Cell Res. 2020;1867(10):118799. [DOI] [PubMed] [Google Scholar]
- 18.Canobbio I, Cipolla L, Consonni A, et al. Impaired thrombin-induced platelet activation and thrombus formation in mice lacking the Ca(2+)-dependent tyrosine kinase Pyk2. Blood. 2013;121(4):648-657. [DOI] [PubMed] [Google Scholar]
- 19.Canobbio I, Lova P, Sinigaglia F, et al. Proline-rich tyrosine kinase 2 and focal adhesion kinase are involved in different phases of platelet activation by vWF. Thromb Haemost. 2002;87(3):509-517. [PubMed] [Google Scholar]
- 20.Manganaro D, Consonni A, Guidetti GF, et al. Activation of phosphatidylinositol 3-kinase β by the platelet collagen receptors integrin α2β1 and GPVI: The role of Pyk2 and c-Cbl. Biochim Biophys Acta. 2015;1853(8):1879-1888. [DOI] [PubMed] [Google Scholar]
- 21.Canobbio I, Cipolla L, Guidetti GF, et al. The focal adhesion kinase Pyk2 links Ca2+ signalling to Src family kinase activation and protein tyrosine phosphorylation in thrombin-stimulated platelets. Biochem J. 2015;469(2):199-210. [DOI] [PubMed] [Google Scholar]
- 22.Consonni A, Cipolla L, Guidetti G, et al. Role and regulation of phosphatidylinositol 3-kinase β in platelet integrin α2β1 signaling. Blood. 2012;119(3):847-856. [DOI] [PubMed] [Google Scholar]
- 23.Cipolla L, Consonni A, Guidetti G, et al. The proline-rich tyrosine kinase Pyk2 regulates platelet integrin αIIbβ3 outside-in signaling. J Thromb Haemost. 2013;11(2):345-356. [DOI] [PubMed] [Google Scholar]
- 24.Canobbio I, Visconte C, Momi S, et al. Platelet amyloid precursor protein is a modulator of venous thromboembolism in mice. Blood. 2017;130(4):527-536. [DOI] [PubMed] [Google Scholar]
- 25.Pitchford SC, Momi S, Giannini S, et al. Platelet P-selectin is required for pulmonary eosinophil and lymphocyte recruitment in a murine model of allergic inflammation. Blood. 2005;105(5):2074-2081. [DOI] [PubMed] [Google Scholar]
- 26.Giannini S, Falcinelli E, Bury L, et al. Interaction with damaged vessel wall in vivo in humans induces platelets to express CD40L resulting in endothelial activation with no effect of aspirin intake. Am J Physiol Heart Circ Physiol. 2011;300(6):H2072-2079. [DOI] [PubMed] [Google Scholar]
- 27.Falcinelli E, Petito E, Becattini C, et al. Role of endothelial dysfunction in the thrombotic complications of COVID-19 patients. J Infect. 2020;82(5):186-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bosshart H, Heinzelmann M. THP-1 cells as a model for human monocytes. Ann Transl Med. 2016;4(21):438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petito E, Falcinelli E, Paliani U, et al. COVIR study investigators. Association of neutrophil activation, more than platelet activation, with thrombotic complications in Coronavirus disease 2019. J Infect Dis. 2021;223(6):933-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mócsai A, Zhang H, Jakus Z, et al. G-protein-coupled receptor signaling in Syk-deficient neutrophils and mast cells. Blood. 2003;101(10):4155-4163. [DOI] [PubMed] [Google Scholar]
- 31.Canobbio I, Visconte C, Oliviero B, et al. Increased platelet adhesion and thrombus formation in a mouse model of Alzheimer's disease. Cell Signal. 2016;28(12):1863-1871. [DOI] [PubMed] [Google Scholar]
- 32.Momi S, Caracchini R, Falcinelli E, et al. Stimulation of platelet nitric oxide production by nebivolol prevents thrombosis. Arterioscler Thromb Vasc Biol. 2014;34(4):820-829. [DOI] [PubMed] [Google Scholar]
- 33.Murphy JM, Jeong K, Rodriguez YAR, et al. FAK and Pyk2 activity promote TNF-α and IL-1β-mediated pro-inflammatory gene expression and vascular inflammation. Sci Rep. 2019;9(1):7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guidetti GF, Zarà M, Canobbio I, et al. Novel pharmacological inhibitors demonstrate the role of the tyrosine kinase Pyk2 in adhesion and aggregation of human platelets. Thromb Haemost. 2016;116(5):904-917. [DOI] [PubMed] [Google Scholar]
- 35.Kawecki C, Lenting PJ, Denis CV. von Willebrand factor and inflammation. J Thromb Haemost. 2017;15(7):1285-1294. [DOI] [PubMed] [Google Scholar]
- 36.Coenen DM, Mastenbroek TG, Cosemans JMEM. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood. 2017;130(26):2819-2828. [DOI] [PubMed] [Google Scholar]
- 37.Tse KW, Lin KB, Dang-Lawson M, et al. Small molecule inhibitors of the Pyk2 and FAK kinases modulate chemoattractant-induced migration, adhesion and Akt activation in follicular and marginal zone B cells. Cell Immunol. 2012;275(1-2):47-54. [DOI] [PubMed] [Google Scholar]
- 38.Cheung SM, Ostergaard HL. Pyk2 Controls integrin-dependent CTL migration through regulation of de-adhesion. J Immunol. 2016;197(5):1945-1956. [DOI] [PubMed] [Google Scholar]
- 39.Schrottmaier WC, Mussbacher M, Salzmann M, et al. Platelet-leukocyte interplay during vascular disease. Atherosclerosis. 2020;307:109-120. [DOI] [PubMed] [Google Scholar]
- 40.Bhattacharya SK, Aspnes GE, Bagley SW, et al. Identification of novel series of pyrazole and indole-urea based DFG-out PYK2 inhibitors. Bioorg Med Chem Lett. 2012;22(24):7523-7529. [DOI] [PubMed] [Google Scholar]
- 41.Grossi M, Bhattachariya A, Nordström I, et al. Pyk2 inhibition promotes contractile differentiation in arterial smooth muscle. J Cell Physiol. 2017;232(11):3088-3102 [DOI] [PubMed] [Google Scholar]
- 42.Zhou J, Xu E, Shao K, et al. Circulating platelet-neutrophil aggregates as risk factor for deep venous thrombosis. Clin Chem Lab Med. 2019;57(5):707-715. [DOI] [PubMed] [Google Scholar]
- 43.Nie M, Yang L, Bi X, et al. Neutrophil extracellular traps induced by IL8 promote diffuse large B-cell lymphoma progression via the TLR9 Signaling. Clin Cancer Res. 2019;25(6):1867-1879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.