

Summary:
Individual hosts within populations often show inter-individual variation in their susceptibility to bacterial pathogen-related disease. Utilizing Drosophila, we highlight that phenotypic variation in host-pathogen susceptibility within populations is driven by energetic trade-offs, facilitated by infection-mediated changes in glutamate metabolism. Furthermore, host-pathogen susceptibility is conditioned by life-history, which adjusts immune-metabolic sensing in muscle to direct vitamin-dependent re-allocation of host energy substrates from adipose (muscle-adipose axis). Life-history conditions inter-individual variation in the activation strength of intra-muscular NF-kB/innate immune signaling. Limited intra-muscular NF-kB/innate immune signaling activity allows for infection-mediated increases in mitochondrial biogenesis and function, which stimulates glutamate dehydrogenase (Gdh)-dependent synthesis of glutamate. Muscle-derived glutamate acts as systemic metabolite to promote lipid mobilization through modulating vitamin B enzymatic cofactor transport and function in adipose. This energy substrate re-allocation improves pathogen clearance and boosts host survival. Finally, life-history events that adjust energetic trade-offs can shape inter-individual variation in host-pathogen susceptibility after infection.
Keywords: Innate immunity, bacterial infection, lipid metabolism, muscle, immune-metabolic, mitochondria, glutamate, glutamate dehydrogenase, vitamin, Smvt, life-history
Introduction:
Across metazoan taxa, constant adaptation to nutrient availability and infectious agents has led to the co-evolution of metabolic and immune responses, such that pathogen sensing systems are often facilitated by metabolic responses in order to adjust organism-wide energy homeostasis and manage infections. (Buchon et al., 2014; Gaber et al., 2017; Lee and Lee, 2018). This fine-tuning of immune-metabolic interactions is likely critical to tightly control host defense responses and enhance survival outcomes during pathogenic infection (Lercher et al., 2020). However, individual hosts within the same population often display inter-individual variation in their susceptibility to identical infectious agents, highlighting heterogeneity in immune-metabolic responses (Nedelec et al., 2016; Satija and Shalek, 2014). This variation among hosts may represent an ancestral strategy to enhance population survival against a diversity of microorganisms that all impinge on similar defense mechanisms. However, the underlying immune-metabolic mechanisms that shape these variances remain largely unexplored.
Ancestral innate immune defense mechanisms throughout metazoans are energetically costly, and lipids (specifically neutral lipids like triglycerides [TAG]) represent the majority of stored energy used to support metabolic demand (Ganeshan et al., 2019; Wang et al., 2019). Through the dynamic coordination of anabolism and catabolism, adipose (and the liver) mobilize stored TAG and additional lipids from lipid droplets that is consumed by other tissues (Kuhnlein, 2012). Host innate immune responses to pathogenic bacterial infection can shift lipid/TAG metabolism across the organism in order to re-allocate energy resources and modulate the host’s ability to fight infection (Ayres and Schneider, 2009; Clark et al., 2013; Dionne et al., 2006; Hang et al., 2014; Lee et al., 2018). The evolution of complex organ systems in metazoans has dictated that the maintenance of metabolic homeostasis requires coordinating local and systemic energy demands between organs with specialized functions (Zhao and Karpac, 2020). This inter-tissue coordination is also required for systemic innate immune responses (Buchon et al., 2014; Lee and Lee, 2018). Thus, unique immune-metabolic sensing mechanisms in distinct tissue types, and the co-evolution of both pathogen and nutrient sensing responses in these tissues, are likely to coordinate host allocation of lipid-based energy stores and infection-mediated immune-metabolic responses.
Energy substrate (lipid) resource allocation during infection underlies competition, or energy trade-offs, with other critical functions, like growth or reproduction (Ganeshan et al., 2019; Wang et al., 2019). Furthermore, hosts employ multiple defense strategies to fight pathogenic infections. This includes resistance (which promotes pathogenic bacterial clearance) and tolerance (which limits both pathogen damage and self-damage caused by rigid innate immune responses, (Ayres and Schneider, 2012)). Metazoan hosts must thus balance pathogen clearance/damage, self-damage, and energy trade-offs to mitigate infections. This delicate balance also encompasses life history theory. For example, in order to maximize reproductive success, organisms must optimize the distribution of environment-specific energy resources (such as triglycerides) into key life history traits, including fecundity, growth, and maintenance programs (survival mechanisms such as innate immunity, ((Van Noordwijk and Dejong, 1986; Wang et al., 2019)). Thus, immune-metabolic sensing mechanisms likely evolved from a life history ‘symmetry’ between innate immunity and other key fitness traits, such as growth and reproduction (Wang et al., 2019). It is also plausible that life history evolution conditions phenotypic diversity, and individual host-pathogen susceptibility (Duneau et al., 2017; Nedelec et al., 2016), of bacterial infection outcomes within populations. Phenotypic diversity in host infection responses often encompasses inter-individual variation in activation strength, and/or spatio-temporal regulation, of innate immune signaling (Brodin and Davis, 2017). Exploring the integration of life history events, species-specific life history traits, and stochasticity in infection responses is likely key to understanding the functional integration of innate immune and metabolic responses.
Invertebrate models provide unique advantages to explore the ancient mechanistic integration of systemic innate immune-metabolic responses that are encumbered by life history traits and energy substrate resource allocation. Here, we exploited the fruit fly Drosophila melanogaster as a model and uncovered that systemic innate immune-metabolic sensing uniquely in muscle tissue shapes mitochondrial-derived glutamate levels, which in turn drives lipid mobilization through alterations in vitamin cofactor metabolism in lipid storage tissues (adipose) to impact host infection outcomes. Muscle is a mitochondria-dense tissue that can dictate systemic energy substrate allocation, and while mitochondria are essential for cellular respiration, lipid usage, and energy metabolism, these organelles are also central to life history evolution, innate immunity, and immune-metabolic integration across taxa (Cui et al., 2019; Mills et al., 2017; Tiku et al., 2020; West et al., 2011). Overall, our findings reveal a distinct mechanism by which muscle-derived glutamate, acting as a systemic metabolite to control inter-tissue communication, shapes phenotypic variation in host-pathogen susceptibility through energetic trade-offs and re-allocation of host energy substrates. This immune-metabolic integration is fine-tuned by life-history events among individuals, and subsequently enhances population survival after infection.
Results:
Phenotypic Variation in Host Infection Outcomes Correlates with Changes in Host Energy Substrate Resource Allocation
In order to rigorously explore inter-individual variation of host immune-metabolic responses to infectious agents, we analyzed phenotypic variation of host infection outcomes and host metabolism among individuals within adult Drosophila populations after Pseudomonas entomophila (P.e.) oral infection. P.e. is a natural entomopathogen (Liehl et al., 2006) that leads to robust systemic NF-kB (Drosophila Relish) innate immune activation in response to natural infection of the gastrointestinal tract. Relish is similar to mammalian p100/p105 NF-kB proteins, and stimulates transcription of anti-microbial peptides (AMPs), such as Drosocin (Buchon et al., 2014). Using a Drosocin-GFP transgenic reporter (Dro-GFP) to monitor systemic defense mechanisms, we found stochastic activation of systemic NF-kB/innate immune responses (characterized as Dro-GFP positive or negative among sibling female hosts) after P.e. oral infection (Figure 1A). The ratio of Dro-GFP positive-to-negative flies within populations is approximately 1:1 at 8 hours, and approximately 4:1 at 20 hours after P.e. infection (Figure 1B). Dro-GFP-negative flies elicit a mild or limited systemic NF-kB/innate immune response, compared to Dro-GFP positive flies, based on AMP transcription in the carcass and thoracic cavity (transcriptomics analysis (RNA-seq., Figure 1C–D) and S1A–B)). This differential Dro-GFP activation among individuals is not due to variability in oral ingestion of P.e., as pathogen abundance (CFU) in midguts is consistently high among all flies at 4 hours (Figure 1E), and there is no change in food intake/feeding (Figure S1C). However, Dro-GFP negative flies show significant decreases in CFUs at 20 hours, suggesting enhanced clearance (Figure 1E). This variation in P.e. clearance correlates with survival outcomes, as nearly all Dro-GFP positive flies succumb to oral infection while nearly all Dro-GFP negative flies survive (Figure 1F), highlighting enhanced infection resistance (impacting host-pathogen susceptibility) in Dro-GFP negative flies. Transcriptomics analysis (RNA-seq.) also revealed that while systemic NF-kB/Relish signaling is diminished in Dro-GFP negative flies, other stress-response genes still present a robust induction (Figure 1G–H and Table S2). This suggests that Dro-GFP negative flies are generally responsive to P.e. infection, and are protected by other host defense mechanisms.
Figure 1: Phenotypic Variation in Host Infection Outcomes Correlates with Changes in Host Energy Substrate Resource Allocation.
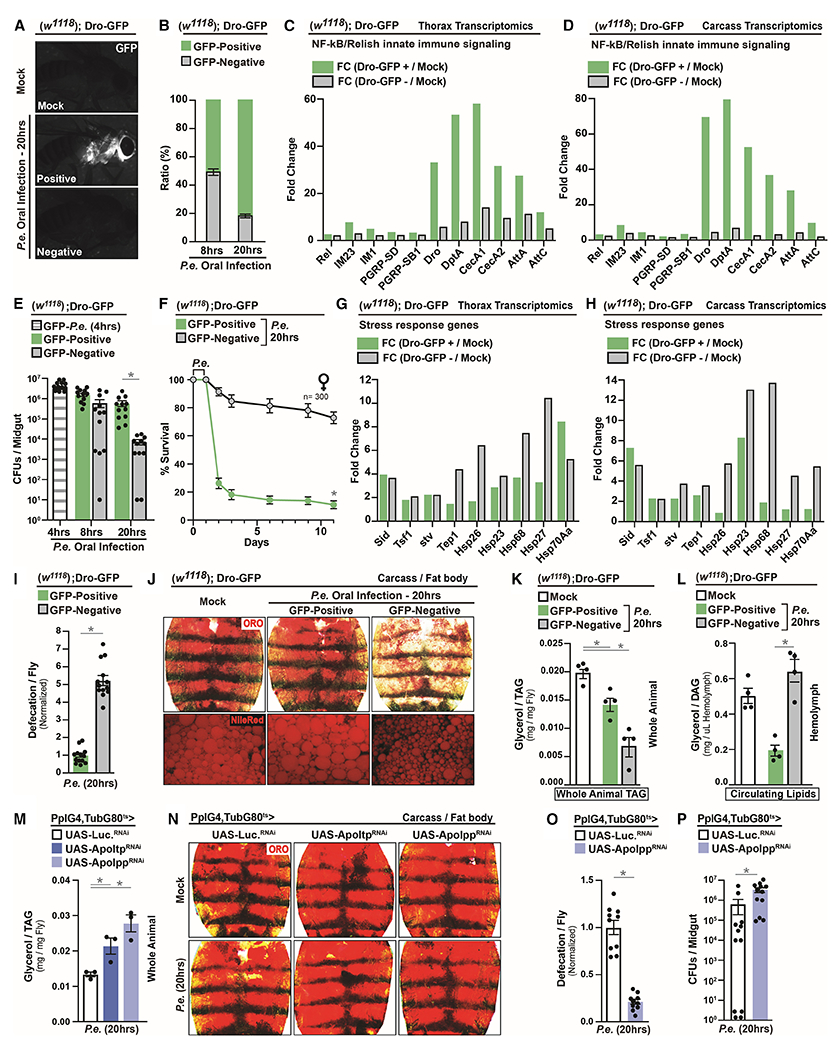
(A) Systemic induction of Dro-GFP after P.e. oral infection.
(B) Ratios of Dro-GFP flies after P.e. oral infection at 8hrs and 20hrs; n=400.
(C-D) RNA-seq. transcriptomics analysis of NF-kB/Relish target genes in dissected (C) thoraces or (D) carcass (adipose) of Dro-GFP flies.
(E-F) Infection resistance of Dro-GFP flies; (E) pathogen abundance (CFUs) at 4hrs, 8hrs and 20hrs; n=12, (F) survival rates; n=300.
(G-H) RNA-seq. transcriptomics analysis of stress responses genes in dissected (G) thoraces or (H) carcass of Dro-GFP flies.
(I) Measurement of defecation in Dro-GFP flies after P.e. oral infection; n=260.
(J-L) Changes in systemic lipid metabolism of Dro-GFP flies. (J) Neutral lipid storage; Oil Red O (ORO) stain and Nile-Red stain (lipid droplets) were assayed in dissected carcass/fat body; (K) Total TAG levels of whole flies and (L) Circulating lipids levels in isolated hemolymph; n=4.
(M-N) Changes in systemic lipid metabolism in PplGal4, tubGal80ts>UAS-Apo-ltpRNAi or UAS-Apo-lppRNAi flies. (M) Total TAG levels of whole flies; n=3, and (N) ORO stain of neutral lipid storage.
(O-P) Infection resistance. (O) Measurement of defecation; n=200. (P) Measurement of CFUs per midgut; n=12.
Error bars represent mean±SE, *P<0.01.
Defecation, regulated by host nutrient status (Cognigni et al., 2011), promotes pathogen expulsion and represents another important defense mechanism (Du et al., 2016). To this end, we discovered that gastrointestinal defecation responses are also robustly enhanced in Dro-GFP negative flies after P.e. infection (Figure 1I). Since defecation used to expel pathogens is likely energetically expensive, we investigated infection-mediated phenotypic variation of host metabolic processes. Within a population, only Dro-GFP negative flies uniquely display decreases in neutral lipid storage and lipid droplet size in adipose (carcass fat body) after P.e. infection (Figure 1J), which is not due to deficiencies in food intake (Figure S1C). This infection-mediated change in lipid storage in Dro-GFP negative flies correlates with decreases in grouped organismal triglyceride (TAG) levels and increases in circulating/transported lipids in hemolymph (diacylglycerol [DAG] (Palm et al., 2012), Figure 1K–L), highlighting lipid mobilization uniquely in these flies. Circulating lipids (DAG) that are mobilized from adipose are significant energy substrates for the gastrointestinal tract (Palm et al., 2012; Zhao and Karpac, 2017), thus we explored the consequences of blocking lipid mobilization in response to pathogenic bacteria. Inhibiting lipid mobilization/transport from adipose (attenuating Drosophila apoB lipoproteins) leads to enhanced TAG storage, but significant decreases in defecation responses and increases in bacterial abundance after P.e. infection (Figure 1M–P). These data suggest that infection-mediated lipid mobilization (and re-allocation of host energy resources) is likely required for host gastrointestinal defecation responses that adjust individual host-pathogen susceptibility.
Activation Strength of Intra-muscular NF-kB/Innate Immune Signaling Directs Re-allocation of Host Energy Substrates to Alter Host-Pathogen Susceptibility
We next assessed whether host NF-kB/Relish-mediated systemic innate immune signaling activity was directly shaping this phenotypic variation in host metabolic responses. First, it was essential to determine which unique peripheral tissue(s) is driving these infection-mediated changes in lipid mobilization. We uncovered that systemic defense responses in thoracic muscle, and not adipose (Figure 2 and S1D–H), direct energy substrate re-allocation to adjust individual host-pathogen susceptibility (infection resistance). Similar to numerous vertebrate somatic muscle types, Drosophila indirect flight muscle (thoracic IFM) is a cross-striated fibrillar muscle, containing longitudinal and ventral segments. Muscle is central to the control of lipid-dependent energy homeostasis across taxa, and is an emerging pathogen-responsive tissue (Chatterjee et al., 2016; Yang and Hultmark, 2017; Zhao and Karpac, 2017). First, we confirmed inter-individual variation of NF-kB/Relish innate immune signaling activation in muscle after P.e. infection (Dro-GFP reporter induction in dorsal longitudinal muscle [DLM] is shown, Figure 2A). The NF-kB/Relish pathway is triggered by activation of peptidoglycan recognition proteins (such as receptors/sensors PGRP-LC and PGRP-LE) that recognize bacterial derived peptidoglycans and host-generated metabolites (Buchon et al., 2014). To this end, Dro-GFP reporter induction in muscle can be eliminated by attenuating NF-kB/innate immune signaling pathway activity autonomously (using Act88FGal4, (Zhao and Karpac, 2017)) at the level of the transcription factor (Act88FGal4>UAS-RelishRNAi) or receptor (Act88FGal4>UAS-PGRP-LERNAi) (Figure 2B). P.e. induction of NF-kB/Relish activity in thoracic muscle also appears dependent on adult blood cells (hemocytes, Figure S1I) as previously described for other peripheral tissues (Bosch et al., 2019; Yang et al., 2019). Furthermore, differential systemic activation of the Dro-GFP reporter is not caused by midgut barrier dysfunction or the presence of bacteria in the hemolymph (Figure S1J–K).
Figure 2: Activation Strength of Intra-muscular NF-kB/Innate Immune Signaling Directs Re-allocation of Host Energy Substrates to Alter Host-Pathogen Susceptibility.
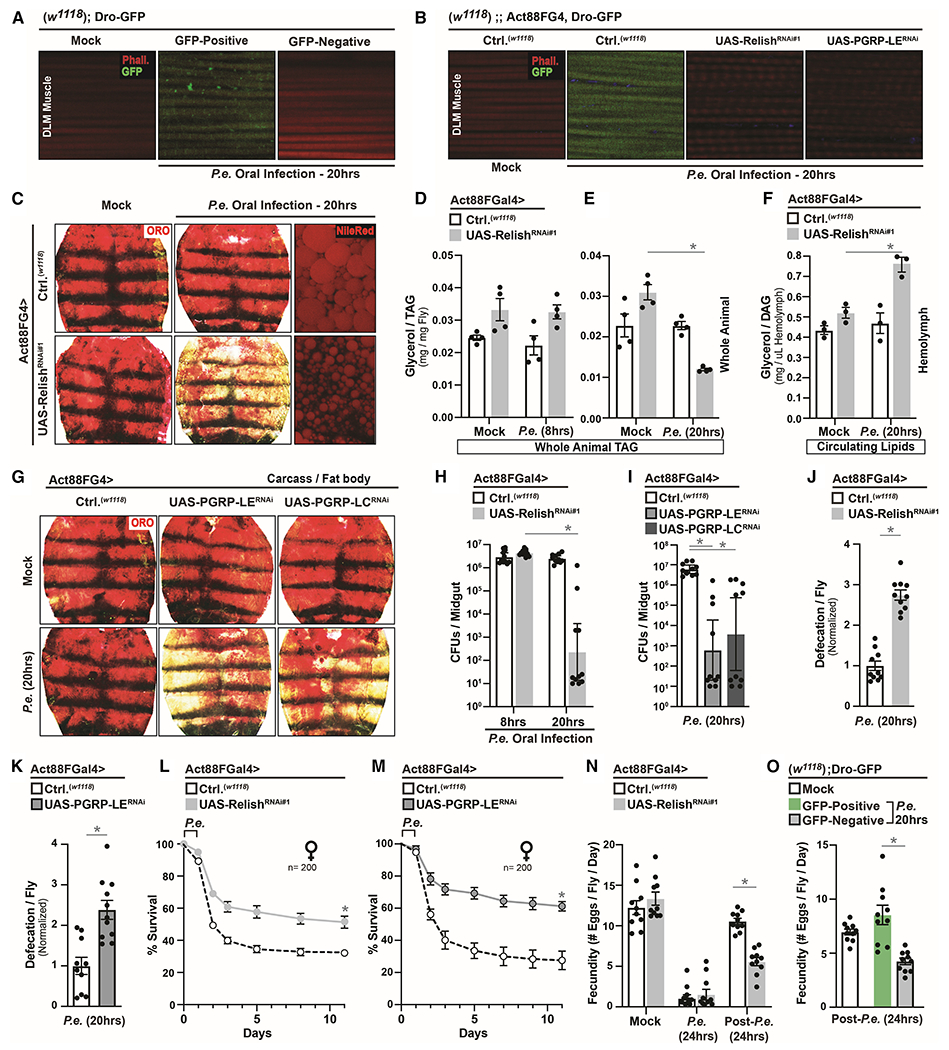
(A-B) Induction of Dro-GFP after P.e. oral infection in (A) Dro-GFP flies, (B) control flies (w1118; Dro-GFP; Act88FGal4) or flies with muscle-specific attenuation of the NF-kB innate immune pathway (w1118; Dro-GFP; Act88FGal4>UAS-RelishRNAi#1 or UAS-PGRP-LERNAi).
(C-G) Changes in systemic lipid metabolism in Act88FGal4>UAS-RelishRNAi#1 or UAS-PGRP-LERNAi or UAS-PGRP-LCRNAi flies after P.e. oral infection. (C and G) Neutral lipid ORO stain and (C) Nile-red stain of dissected carcass/fat body. (D-E) Total TAG levels of whole flies after infection at 8hrs (D) and 20hrs (E); n=4. (F) Circulating lipids levels from isolated hemolymph; n=3.
(H-M) Infection outcomes in Act88FGal4>UAS-RelishRNAi#1 or UAS-PGRP-LERNAi flies after P.e. oral infection. (H-I) Measurement of CFUs (8hrs or 20hrs); n=10. (J-K) Measurement of defecation; n=200. (L-M) Survival rates; n=200.
(N-O) Measurement of fecundity (egg laying) of (N) Act88FGal4>UAS-RelishRNAi#1 flies and (O) Dro-GFP flies after (during) or post P.e. oral infection.
Error bars represent mean±SE, *P<0.01.
We subsequently found that limiting systemic NF-kB/Relish-dependent innate immune signaling in muscle allows for organism-wide re-allocation of host energy substrates after pathogenic bacterial infection. Genetically attenuating NF-kB/Relish specifically in thoracic muscle (Act88FGal4>UAS-RelishRNAi) leads to marked decreases in adipose neutral lipid storage and lipid droplet size after P.e. infection (Figure 2C and S1D–H), which correlates with temporally distinct decreases in organismal TAG levels and increases in circulating lipids (Figure 2D–F, and confirmed with an independent transgenic RNAi line [Figure S1L–N]). This infection-mediated lipid mobilization can be phenocopied by attenuating PGRP receptors/sensors (Act88FGal4>PGRP-LERNAi/PGRP-LCRNAi) in host muscle (Figure 2G and S2A). Additionally, attenuating systemic NF-kB/Relish activity in thoracic muscle after P.e. oral infection also enables enhanced bacterial clearance and survival outcomes (temporally correlated with lipid mobilization at 20 hrs after infection), which corresponds to significant increases in gastrointestinal defecation responses (Figure 2H–M). These data were confirmed (highlighting reproducibility and specificity) using an independent muscle driver (MHCGal4, Figure S2B–D), numerous additional RNAi control experiments (Figure S2E–H), and additional transgenic RNAi lines targeting other genes essential for PGRP/IMD-dependent activation of NF-kB/Relish (Kenny [key, the homolog of mammalian IKKϒ/NEMO] and the apical caspase DREDD, Figure S2I–N). There is also no change in developmental morphology and no change in food intake/feeding in Act88FGal4>UAS-RelishRNAi flies without infection (mock), during infection, or post-infection (Figure S3A–E).
Thus, genetically limiting infection-mediated and NF-kB-dependent innate immune responses in muscle can direct adipose lipid mobilization (muscle-adipose axis) and adjust individual host-pathogen susceptibility, essentially ‘shifting’ Drosophila populations towards a majority Dro-GFP negative phenotype (Figure 1). To further highlight the biological relevance of this functional integration of immune and metabolic responses, we explored energy substrate re-allocation after oral infection with Ecc15 (Erwina carotovora carotovora strain 15), a non-pathogenic bacterium (Figure S3F) that elicits mild, but consistent, systemic defense mechanisms in Drosophila hosts. Ecc15 is an isolate of the Erwina carotovora phytopathogen, which uses insect hosts as vectors (Basset et al., 2000). As expected, Ecc15 oral infection induced differential activation of systemic innate immune responses among individuals opposite to that induced by P.e., with the vast majority of individual flies classified as Dro-GFP negative and displaying reduced organismal TAG levels (Figure S3G–H). Corroborating these observations, Act88FGal4>w1118 control flies present with decreased organismal TAG levels and adipose neutral lipid storage after infection (different from P.e.), and attenuating NF-kB/Relish in muscle has no additive effect over controls (Figure S3I–K). Additionally, Ecc15 promotes defecation responses (independent of genotype, Figure S3L). This suggests that other microorganisms like phytopathogens may co-opt these immune-metabolic responses to induce energy substrate re-allocation and defecation (ensuring host/vector survival) to either employ the insect as alternative host or improve plant host infection, both through enhanced bacterial dissemination.
Taken together, these data show that systemic NF-kB/innate immune signaling activation strength in muscle can directly shape host metabolic responses in Drosophila. Mild or limited systemic innate immune responses in muscle allow for re-allocation of energy substrates from adipose to enhance the percentage of host survival after infection by shifting inter-individual variation in host-pathogen susceptibility (Figure S3M). Since strong intra-muscular innate immune responses seemingly block energetic trade-offs after bacterial infection, host self-damage from elevated innate immune signaling is also likely to encompass metabolic dysfunction as well. Phenotypic variation in individual host-pathogen responses within a population therefore underlies distinct re-allocation of host energy substrate and likely energetic trade-offs. To this end, female flies that mobilize lipids to enhance infection resistance (Dro-GPF negative or Act88FGal4>UAS-RelishRNAi flies) also suffer from reduced fecundity post-infection, while flies that maintain lipid stores (and suffer the consequences of infection) maintain fecundity. Thus, re-allocation of energy substrate and improved infection resistance comes at the cost of normal reproduction, a key life history trait driving natural selection (Figure 2N–O).
Intra-muscular NF-kB/Innate Immune Signaling Adjusts Mitochondrial Dynamics After Infection
We next wanted to characterize both immune-metabolic sensing in muscle, as well as the mechanisms that drive organism-wide energetic trade-offs, in response to infection. To this end, we used thoraces/muscle transcriptomics to explore unique infection-mediated changes in gene expression networks when NF-kB/innate immune signaling activity is genetically attenuated in muscle (Act88FGal4>UAS-RelishRNAi). Transcriptomic analysis showed expected decreases of defense responses genes (Figure S4A and Table S3), but conversely, genes associated with general mitochondrial function are up-regulated uniquely in Act88FGal4>UAS-RelishRNAi thoraces/muscle after P.e. infection (Figure 3A–C and Table S4). This includes proteins required for mitochondrial electron transport chain (ETC) function and oxidative phosphorylation (OXPHOS) of energy substrates, as well as crucial TCA cycle enzymes (Figure 3D–E and Table S4). Muscle is a mitochondria-dense tissue which utilizes these organelles for rapid breakdown/usage of various energy substrates to generate ATP (Figure 3F).
Figure 3: Intra-muscular NF-kB/Innate Immune Signaling Adjusts Mitochondrial Dynamics After Infection.
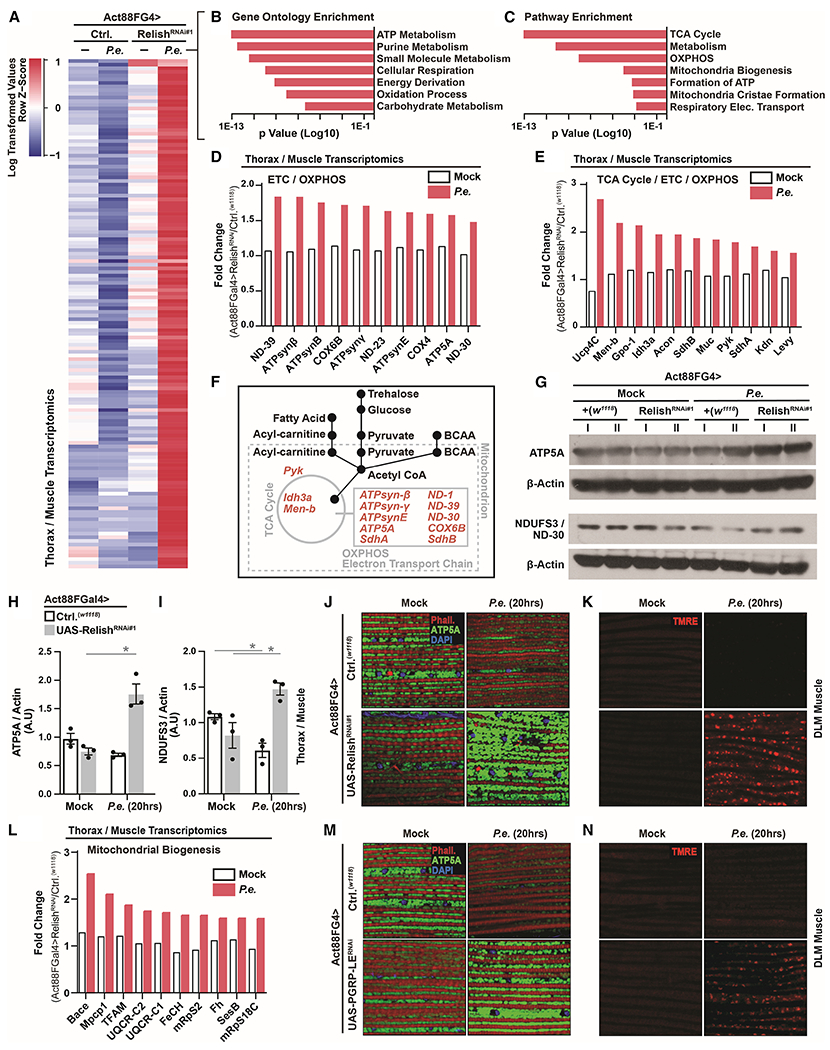
(A) RNA-seq. transcriptomics analysis (dissected thoraces/muscle) in Act88FGal4>UAS-RelishRNAi#1 flies after P.e. oral infection (±P.e.).
(B-C) Unique up-regulated genes analyzed for Gene Ontology (GO) Terms and Pathway Enrichment (enrichment p value<0.001).
(D-E) Fold change (RelishRNAi#1/Ctrl., ±P.e.) of select genes involved in mitochondria function (OXPHOS/TCA Cycle/Electron transport chain [ETC]).
(F) Model depicting the function of select genes involved in mitochondria function/OXPHOS/TCA Cycle/Electron transport chain (ETC) related to energy substrate usage in muscle.
(G-L) Changes in mitochondrial dynamics in Act88FGal4>UAS-RelishRNAi#1 flies after P.e. oral infection. (G) ATP5A and NDUF3/ND-30 protein levels (assayed by Western blot) in dissected thoraces/muscle; independent samples l and II are shown. (H-I) Quantification of three independent samples of (H) ATP5A and (I) NDUF3/ND-30, A.U.–arbitrary units. (J) Immunostaining to detect mitochondrial morphology (anti-ATP5A, green) in dissected DLM muscle. F-actin filaments (Phalloidin, Red), and nuclei (DAPI, blue). (K) TMRE fluorescent histochemistry (red) to detect mitochondrial membrane potential in dissected DLM muscle. (L) Fold change (RelishRNAi#1/Ctrl.) of select genes involved in mitochondria biogenesis.
(M-N) Immunostaining to detect (M) mitochondrial morphology (anti-ATP5A [green], F-actin filaments [Phalloidin, Red], and nuclei [DAPI, blue]) and (N) TMRE fluorescent histochemistry (red) in Act88FGal4>UAS-PGRP-LERNAi flies.
Error bars represent mean±SE, *P<0.01.
To confirm these putative changes in mitochondrial dynamics, we monitored protein levels of mitochondrial ATP5A (ATP synthase F1 alpha) and ND-30 (NDUFS3, and NADH dehydrogenase) in thoraces/muscle before and after P.e. infection. We found that attenuating NF-kB/Relish in muscle autonomously enhances the levels of these mitochondrial proteins only after infection (Figure 3G–I and S4B). Furthermore, attenuating NF-kB/Relish in muscle leads to infection-mediated increases in both intra-muscular mitochondrial size/number (Figure 3J) and mitochondrial membrane potential/function (TMRE, Figure 3K), as well as overall enhanced muscle mitochondrial biogenesis/function after P.e. infection (Figure 3J–L). These infection-mediated changes in intra-muscular mitochondrial dynamics can be phenocopied by attenuating PGRP receptors/sensors in host muscle (Figure 3M–N). Thus, systemic NF-kB/innate immune signaling activity in Drosophila muscle is capable of adjusting mitochondrial function/biogenesis in response to bacterial pathogens. Accordingly, changes in intra-muscular mitochondrial dynamics also correlate with phenotypic variation in host infection outcomes within populations. Only Dro-GFP negative flies are capable of maintaining or enhancing both thoraces/muscle ATP5A and NDUFS3 protein levels (Figure 4A–C and S4C), as well as intra-muscular mitochondrial size/number and membrane potential after P.e. infection (Figure 4D). Furthermore, attenuating mitochondrial function in thoracic muscle (utilizing RNAi against ATP synthase ß) decreases the number of infection-induced Dro-GFP negative individuals within a population (Figure 4E and S4D), revealing an antagonism between mitochondrial function and systemic NF-kB-mediated defense responses in muscle. Collectively, these data show that mitochondria are central to systemic immune-metabolic sensing, and that intra-muscular NF-kB/innate immune signaling activation strength can adjust mitochondrial dynamics and host metabolic responses (Figure 4F). Strong intra-muscular innate immune signaling activity limits or even represses mitochondrial function/biogenesis, while mild activation enhances mitochondrial dynamics. To this end, non-pathogenic Ecc15 infection can also generally enhance intra-muscular mitochondrial size/number (Figure S4E).
Figure 4: Life-history Events Influence Phenotypic Diversity of Host Immune-Metabolic Responses by Modulating Mitochondrial Dynamics and Host Energetic Trade-offs.
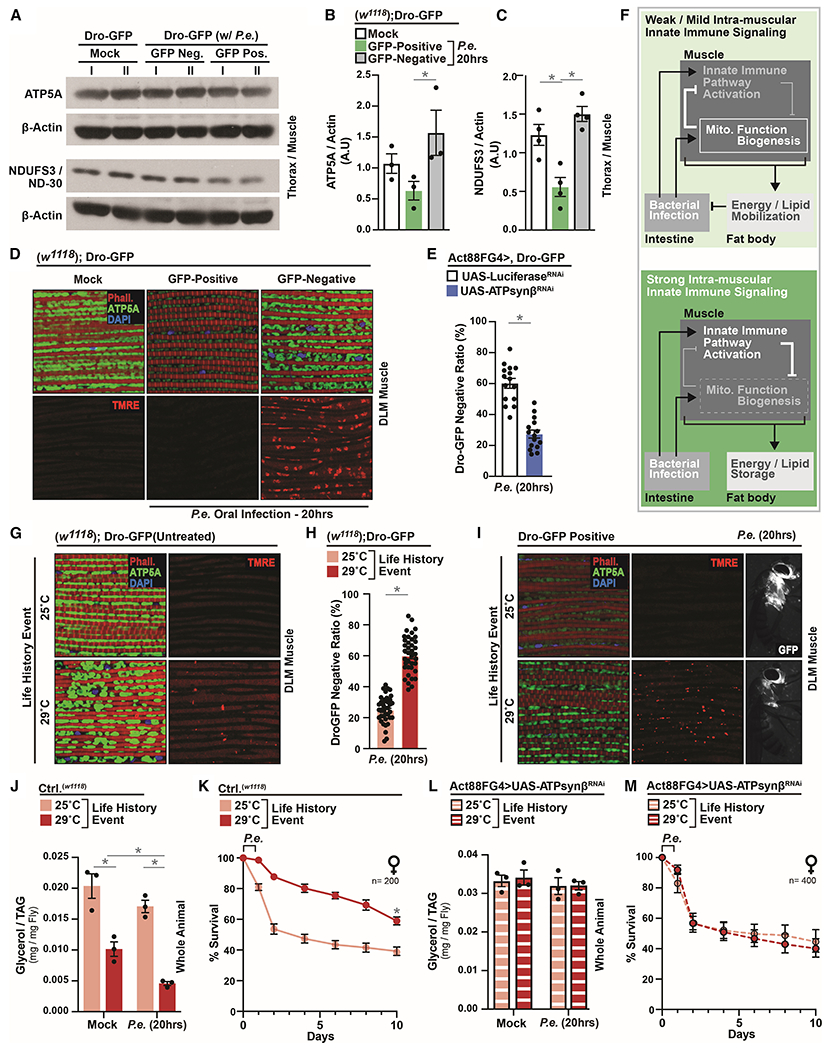
(A-D) Changes in mitochondrial dynamics in Dro-GFP flies after P.e. oral infection. (A) ATP5A and NDUF3/ND-30 protein levels (assayed by Western blot) in dissected thoraces/muscle; independent samples l and II are shown. (B-C) Quantification of three independent samples of (B) ATP5A and (C) NDUF3/ND-30, A.U.–arbitrary units. (D) Immunostaining to detect mitochondrial morphology (anti-ATP5A [green], F-actin filaments [Phalloidin, Red], and nuclei [DAPI, blue]) and TMRE fluorescent histochemistry (red).
(E) Ratios of Dro-GFP negative flies (percentage) within populations; n=300.
(F) Model summarizing conclusions.
(G-I) ATP5A immunostaining and TMRE fluorescent histochemistry (red) in dissected DLM muscle (G) before and (I) after infection; anti-ATP5A (green), F-actin filaments (Phalloidin, Red), and nuclei (DAPI, blue). (I) Right panel; corresponding induction of systemic drosocin (Dro-GFP). (H) Ratio (percentage) of Dro-GFP negative flies after P.e. oral infection; n=760.
(J) Total TAG levels of w1118 flies after P.e. oral infection; n=3.
(K) Survival of w1118 control flies after P.e. oral infection; n=200.
(L) Total TAG levels of Act88FGal4>UAS-ATPsynβRNAi flies after P.e. oral infection; n=3.
(M) Survival of Act88FGal4>UAS-ATPsynβRNAi flies after P.e. oral infection; n=400.
Error bars represent mean±SE, *P<0.01.
Life-history Events Influence Host-Pathogen Susceptibility by Modulating Mitochondrial Dynamics and Host Energetic Trade-offs
Since mitochondria appear central to life history evolution, we subsequently explored whether life history events that shift muscle mitochondrial dynamics also influence phenotypic diversity of host-pathogen responses within populations. We used developmental-specific temperature shifts (25°C–29°C, Figure S4F), as rearing insects at higher temperatures can modulate mitochondrial function while negatively impacting other key life history traits, such as growth rates and fecundity (Klepsatel et al., 2019; Pichaud et al., 2010). Developmental-rearing flies at 29°C enhances intra-muscular mitochondria size/number and membrane potential before infection (Figure 4G). These temperature-dependent adjustments in mitochondrial dynamics appear to drive significant increases in the number of Dro-GFP negative individuals within a population after P.e. infection (Figure 4H). Additionally, even Dro-GFP positive flies within populations reared at 29°C display enhanced mitochondrial dynamics in muscle, and tempered Dro-GFP reporter induction, in response to bacterial pathogens (Figure 4I). Corresponding to these shifts in differential activation of systemic innate immune responses among individuals, control flies developmentally reared at 29°C display elevated re-allocation of energy substrates (Figure 4J and S4G), increases in defecation responses (Figure S4H), and, consequently, improved individual infection resistance (Figure S4I) and enhanced survival (Figure 4K). This shift in phenotypic variation of host infection outcomes phenocopies the attenuation of intra-muscular NF-kB/Relish, which again suggest a ‘shaping’ of Drosophila populations towards the Dro-GFP negative phenotype. Conversely, genetically attenuating intra-muscular mitochondrial function eliminates this ‘shaping’ of populations towards Dro-GFP negative individuals (Figure S4J), and thus blocks developmental temperature-mediated changes in host immune-metabolic responses, individual host-pathogen susceptibility (infection resistance), and survival (Figure 4L–M and S4K–M). These developmental temperature-dependent effects on metabolism, infection resistance, and survival are not due to changes in developmental morphology or food intake/feeding without infection (mock), during infection, or post-infection (Figure S5A–F). Therefore, enhanced mitochondrial dynamics are required for temperature-dependent changes in phenotypic variation that condition populations towards infection resistance.
Furthermore, other life history events that enhance muscle mitochondrial function/biogenesis (such as pre-infection with Ecc15, Figure S4E) also lead to elevated mitochondrial-dependent lipid mobilization after pathogenic P.e. infection, as well as differential activation of systemic innate immune responses and improved infection resistance/survival (Figure S5G–L).
Taken together, these results highlight that life-history events which adjust mitochondrial dynamics can govern phenotypic variation in host-pathogen responses within populations, likely through inter-tissue metabolic responses that alter host energy substrate re-allocation and energetic trade-offs.
Muscle- and Mitochondrial-derived Glutamate is Integral for Host Immune-Metabolic Responses that Shape Host-Pathogen Susceptibility
In order to uncover NF-kB/Relish- and mitochondrial-centric mechanisms that may direct host energy substrate re-allocation after infection, we again turned to our thoraces/muscle transcriptomic libraries. Exploring these libraries, we identified genes that are essential for mitochondrial-dependent amino acid/nucleotide metabolism uniquely up-regulated in muscle of Act88FGal4>UAS-RelishRNAi after P.e. infection (Figure 5A and Table S4). This included glutamate dehydrogenase (Gdh), an ancient enzyme that catalyzes the reversible conversion of alpha-ketoglutarate and ammonia to the amino acid glutamate in mitochondria (Figure 5A and S5M, (Plaitakis et al., 2017)). Gdh-dependent glutamate metabolism is generally utilized in eukaryotic amino acid/nitrogen disposal, so we explored the function of Gdh in host immune-metabolic responses to pathogenic infection. We found that NF-kB/Relish can directly bind to the promoter of Gdh and limit the inducibility of Gdh transcription after P.e. infection in muscle (Figure 5B and S5N). Thus, attenuating intra-muscular NF-kB/innate immune signaling activity enables infection-mediated enrichment of the Gdh enzyme in muscle mitochondria (Figure 5C and S5O–P). Muscle Gdh is subsequently required for infection-mediated host metabolic responses, as attenuating Gdh function in muscle completely blocks intra-muscular NF-kB/Relish-mediated re-allocation of host energy stores after P.e. infection (assaying adipose neutral lipid storage, organismal TAG levels, and circulating lipids, Figure 5D–F). This tissue-specific Gdh function is also indispensable for changes in survival and individual host-pathogen susceptibility promoted by intra-muscular NF-kB/Relish activity (Figure 5G–I). These data were confirmed using an independent Gdh transgenic RNAi line (Figure S6A–D).
Figure 5: Muscle- and Mitochondrial-derived Glutamate is Integral for Host Immune-Metabolic Responses that Shape Host-Pathogen Susceptibility.
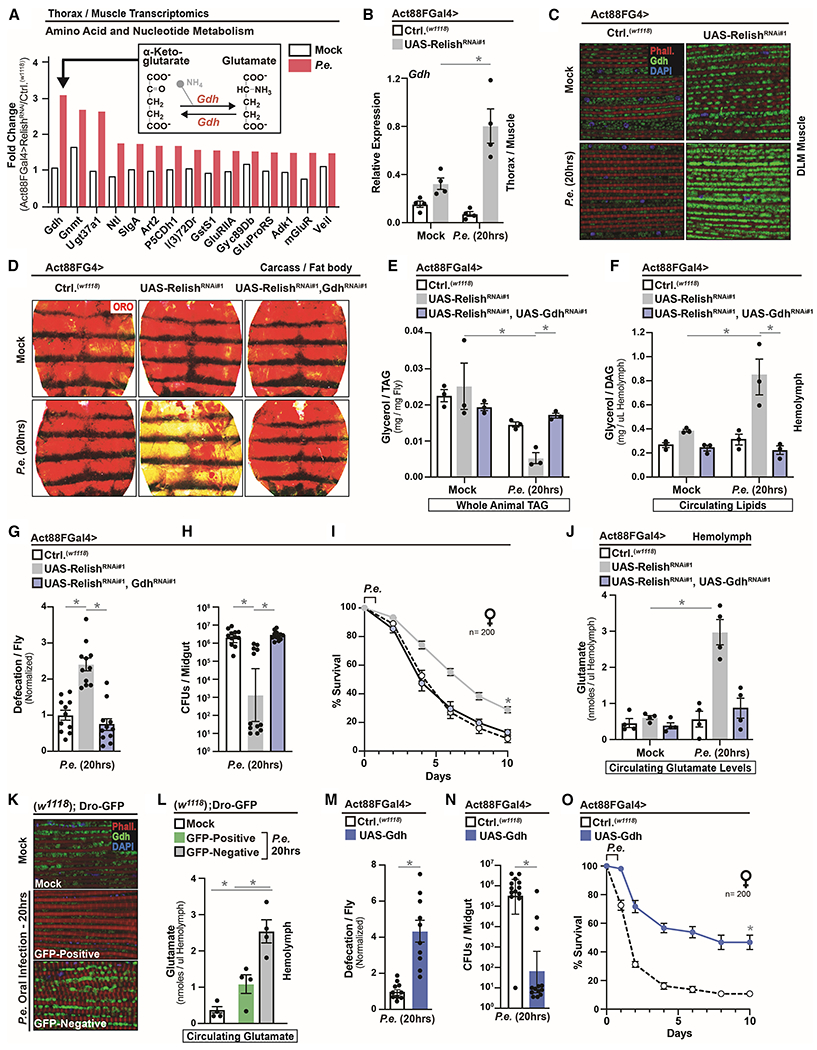
(A) Fold change (RelishRNAi#1/Ctrl.) of select genes involved in amino acid and nucleotide metabolism.
(B-C) Drosophila Gdh regulation in Act88FGal4>UAS-RelishRNAi#1 flies after P.e. oral infection. (B) Gdh transcription from dissected thoraces (measured by qRT-PCR, plotted as relative expression); n=4. (C) Gdh immunostaining in dissected DLM muscle; anti-Gdh (green), F-actin filaments (Phalloidin, red), and nuclei (DAPI, blue).
(D-F) Changes in systemic lipid metabolism of Act88FGal4>UAS-RelishRNAi#1, UAS-GdhRNAi#1 flies after P.e. oral infection. (D) ORO stain of dissected carcass/fat body. (E) Total TAG levels of whole flies and (F) Circulating lipids levels from isolated hemolymph; n=3.
(G-I) Infection outcomes of Act88FGal4>UAS-RelishRNAi#1, UAS-GdhRNAi#1 flies after P.e. oral infection. (G) Measurement of defecation; n=220, (H) measurement of CFUs; n=12, and (I) survival rates; n=200.
(J) Circulating glutamate levels from isolated hemolymph of Act88FGal4>UAS-RelishRNAi#1, UAS-GdhRNAi#1 flies after P.e. oral infection; n=4.
(K-L) Drosophila Gdh regulation in Dro-GFP flies after P.e. oral infection. (K) Gdh immunostaining in dissected DLM muscle; anti-Gdh (green), F-actin filaments (Phalloidin, red), and nuclei (DAPI, blue), and (L) circulating glutamate levels from isolated hemolymph in Dro-GFP transgenic flies after P.e. oral infection; n=4.
(M-O) Infection outcomes of Act88FGal4>UAS-Gdh flies after P.e. oral infection. (M) Measurement of defecation; n=200, (N) CFUs; n=13, and (O) survival rates; n=200.
Error bars represent mean±SE, *P<0.01.
The ability of Gdh function in muscle to control organism-wide metabolic responses suggests that mitochondrial- and Gdh-derived systemic metabolites (glutamate, alpha-ketoglutarate, or amino acid metabolism in general) may be acutely required for infection-mediated changes in host energy resource allocation. NF-kB/Relish attenuation in thoracic muscle enables tissue-autonomous shifts in glutamate/alpha-ketoglutarate levels after P.e. infection, with higher levels of glutamate coupled with lower levels of alpha-ketoglutarate (Figure S6E–G). This suggests that Gdh function favors glutamate production under these conditions, potentially due to elevated intra-muscular alpha-ketoglutarate production after infection (Figure S6F–G). To this end, these infection-mediated metabolite shifts in muscle underlie significant increases in circulating (hemolymph) levels of glutamate when attenuating intra-muscular NF-kB/Relish, and elevated levels of systemic glutamate require muscle-specific Gdh function (Figure 5J).
Additionally, changes in intra-muscular Gdh function and systemic glutamate also correlate with phenotypic variation in host infection outcomes within populations (Figure 5K–L). Only Dro-GFP negative flies are capable of maintaining or enriching the Gdh enzyme in muscle mitochondria after P.e. infection (Figure 5K), as well as elevating circulating glutamate levels (Figure 5L).
Finally, directly over-expressing Gdh in thoracic muscle (Act88FGal4>UAS-Gdh), similar to Dro-GFP negative flies or attenuating systemic NF-kB/Relish function, drives organism-wide energy substrate re-allocation after pathogenic bacterial infection (Figure S6H–M). These adjustments in host metabolism promote defecation responses, and enhance population survival by shifting inter-individual variation in host-pathogen susceptibility (infection resistance, Figure 5M–O). Thus, muscle and mitochondrial-derived glutamate, regulated by NF-kB/Relish-dependent control of mitochondrial dynamics and Gdh transcription, can likely act as a systemic metabolite to communicate infection-mediated energy substrate demands (muscle-adipose axis) within the host. Glutamate metabolism is therefore integral to host immune-metabolic responses that determine infection outcomes (Figure S6N).
Glutamate Adjusts Smvt-mediated Vitamin Metabolism in Adipose to Direct Re-allocation of Host Energy Substrates and Alter Host-Pathogen Susceptibility
As a secretory molecule, glutamate is primarily described as a neurotransmitter with expanded function in non-excitable cells (Hansen and Caspi, 2010; Nedergaard et al., 2002), suggesting that circulating glutamate acts an unexpected metabolite that can shape host immune-metabolic responses. In order to further characterize the role of muscle-derived systemic glutamate in regulating organism-wide energy substrate re-allocation, we performed adipose (carcass fat body) RNA sequencing, after infection, in Act88FGal4>UAS-RelishRNAi flies. Transcriptomic analysis revealed that genes enriched for fatty acid metabolism and small molecule (including amino acid) transport are significantly increased after infection (Figure 6A–C and Table S5), while most down-regulated genes are related to defense responses as expected (Figure S6O and Table S6). Included in these up-regulated genes was Smvt (Sodium-dependent multivitamin transporter, Figure 6D), a transmembrane protein previously shown to transport cellular biotin (vitamin B7) and pantothenic acid (PA, vitamin B5) (Vadlapudi et al., 2012). Biotin and PA are essential to balance cellular lipid synthesis, mobilization and usage through adjusting non-esterified fatty metabolism as enzymatic cofactors (Figure 6E). Thus, we explored the role of Smvt in shaping host immune-metabolic responses and energy substrate resource allocation after infection. Smvt transcription is significantly induced in carcass/fat body when NF-kB/Relish is attenuated in muscle after P.e. infection, and this induction requires intra-muscular Gdh function (Figure 6F and S7A). Smvt transcription also underlies infection-mediated phenotypic variation within populations, as only Dro-GFP negative flies up-regulate gene expression in carcass/fat body (Figure 6G). Utilizing ex vivo carcass/fat body cultures, we also found that Smvt transcription increases dose-dependently in response to glutamate (Figure 6H). dmGlut (dietary and metabolic glutamate transporter) in adipose is required for this glutamate-dependent regulation of Smvt (Figure 6I and S7B). We also identified a multitude of glutamate metabolism genes that are up-regulated in adipose after infection when attenuating intra-muscular NF-kB/Relish activity (Figure S7C). Genetically changing expression of dmGlut in adipose does not affect developmental morphology of flies (Figure S7D–E).
Figure 6: Muscle-derived Glutamate Adjusts Smvt-mediated Vitamin Metabolism in Adipose to Shape Phenotypic Diversity of Host Infection Outcomes.
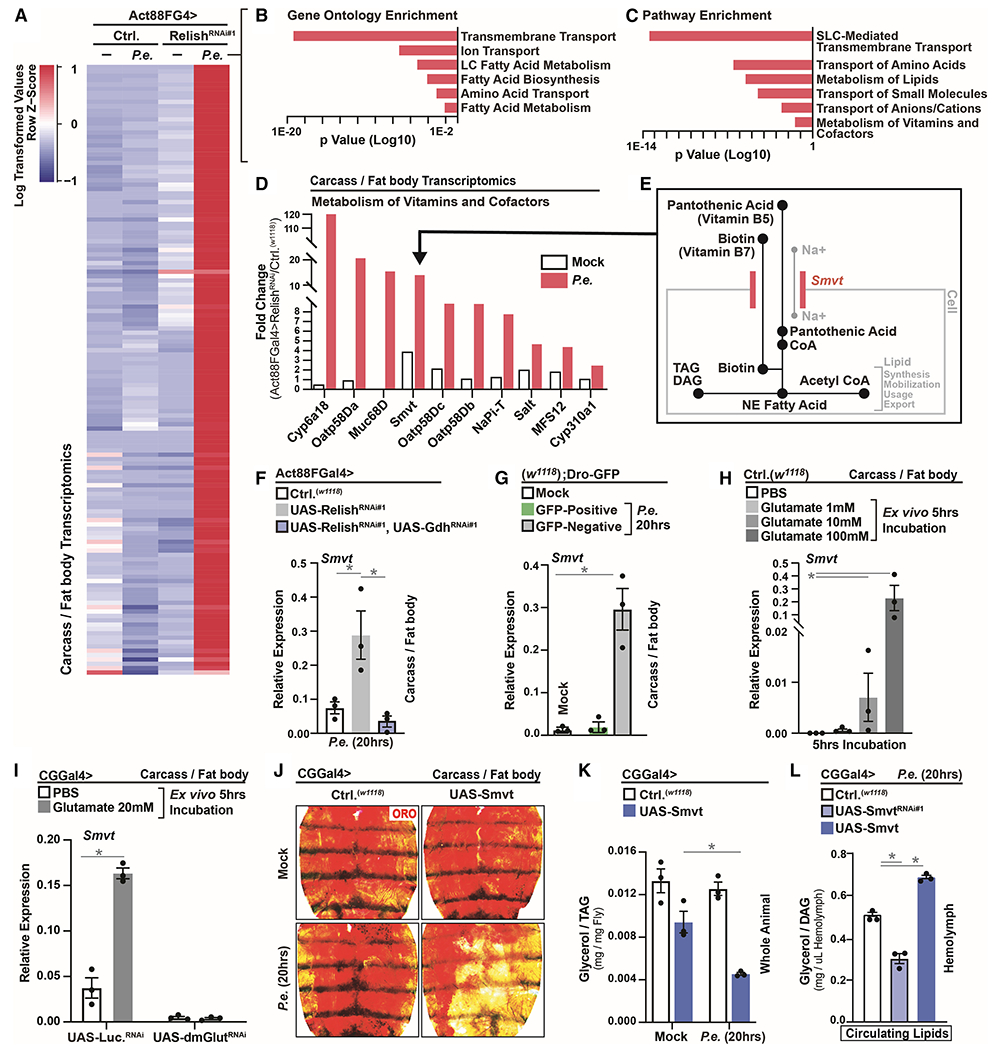
(A) RNA-seq. transcriptomics analysis (dissected carcass/fat body) of Act88FGal4>UAS-RelishRNAi#1 flies after P.e. oral infection (±P.e.).
(B-C) Unique up-regulated genes analyzed for Gene Ontology (GO) Terms and Pathway Enrichment (enrichment P<0.001).
(D) Fold change (RelishRNAi#1/Ctrl.) of select genes involved in cellular transport and co-factor biology.
(E) Model depicting the function of Smvt in transport and uptake of pantothenic acid (vitamin B5) and biotin (vitamin B7).
(F-G) Smvt transcription (qRT-PCR) in dissected carcass/fat body of (F) Act88FGal4>UAS-RelishRNAi#1, UAS-GdhRNAi#1 flies or (G) in Dro-GFP flies after P.e. oral infection; n=3.
(H) Smvt transcription (measured by qRT-PCR) in dissected w1118 carcass/fat body after ex vivo incubation with the indicated concentration of glutamate for 5 hrs; n=3.
(I) Smvt transcription (measured by qRT-PCR) in dissected carcass/fat body of CGGal4>UAS-dmGlutRNAi flies after ex vivo incubation with 20mM glutamate for 5 hrs; n=3.
(J-K) Changes in systemic lipid metabolism of CGGal4>UAS-Smvt flies after P.e. oral infection. (J) ORO stain of dissected carcass/fat body. (K) Total TAG levels of whole flies; n=3.
(L) Circulating lipids levels from isolated hemolymph of CGGal4>UAS-Smvt or CGGal4>UAS-SmvtRNAi#1 flies after P.e. oral infection; n=3.
Error bars represent mean±SE, *P<0.01.
Directly over-expressing Smvt in adipose (CGGal4>UAS-Smvt) promotes autonomous lipid mobilization after P.e. infection (Figure 6J–L and S7F–I), similar to attenuating systemic innate immune signaling activity or activating Gdh in muscle. These data were confirmed using independent adipose/fat body drivers (PplGal4 and LppGal4, Figure S7J–K). The ability of Smvt to mobilize lipids is maximized during infection (Figure 6J–L and S7F, J–K), suggesting that other infection-mediated changes in host physiology (potentially adjustments in vitamin homeostasis) are required for its capacity to initiate re-allocation of energy substrates. We subsequently found that treating ex vivo carcass/fat body cultures with biotin or PA can dose-dependently decrease TAG levels, neutral lipid storage, and reduce lipid droplet size (Figure 7A–B and S7L), phenocopying in vivo results targeting the Smvt in adipose (Figure 6J–L and S7J–K). Furthermore, glutamate can synergistically enhance biotin and PA vitamin-dependent changes in TAG levels and neutral lipid storage (ex vivo) when vitamin availability is more limited (Figure 7C–E). Smvt is required, specifically, for biotin and PA-mediated changes in lipid storage (Figure 7F and S7M–N).
Figure 7: Smvt-mediated Vitamin Metabolism Directs Re-allocation of Host Energy Substrates to Alter Host-Pathogen Susceptibility.
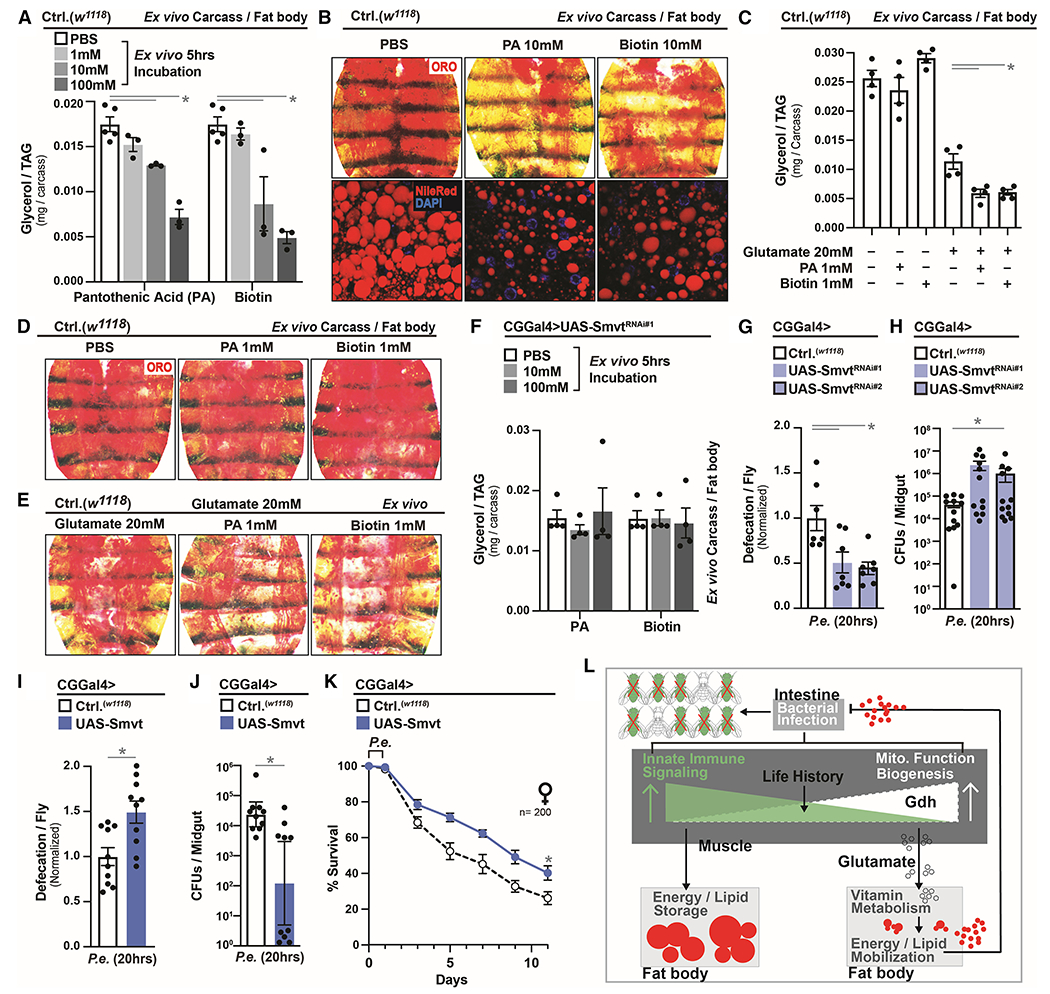
(A-B) Changes in lipid metabolism in dissected w1118 carcass/fat body after ex vivo incubation with the indicated concentration of pantothenic acid (PA) or biotin for 5 hrs. (A) Total TAG levels of carcass/fat body; n=3. (B) Neutral lipid (ORO) stain.
(C-E) Changes in lipid metabolism in dissected w1118 carcass/fat body after ex vivo incubation with the indicated concentration of glutamate, PA or biotin for 5 hrs. (C) Total TAG levels of carcass/fat body; n=3. (D-E) Neutral lipid (ORO) stain.
(F) Total TAG levels in dissected carcass/fat body of CGGal4>UAS-SmvtRNAi#1 flies after ex vivo incubation with the indicated concentration of PA or biotin for 5 hrs; n=4.
(G-H) Infection resistance of CGGal4>UAS-SmvtRNAi#1/UAS-SmvtRNAi#2 flies after P.e. oral infection. Measurement of (G) defecation; n=140, and (H) CFUs; n=12.
(I-K) Infection outcomes of CGGal4>UAS-Smvt flies after P.e. infection. Measurement of (I) defecation; n=200, (J) CFUs; n=10, and (K) survival rates; n=200.
(L) Model summarizing conclusions.
Error bars represent mean±SE, *P<0.01.
Smvt-regulated vitamin metabolism therefore plays a critical role in regulating infection-mediated energy substrate re-allocation from adipose in response to changes in systemic innate immune responses and muscle-derived glutamate. Thus, we next wanted to assess the role of adipose Smvt function in shaping host-pathogen susceptibility, as well as survival. Genetically attenuating Smvt in adipose (using multiple transgenic RNAi lines) leads to inhibition of gastrointestinal defecation responses and increases in bacterial abundance after P.e. infection (Figure 7G–H). Conversely, directly over-expressing Smvt in adipose, similar to attenuating NF-kB/Relish or activating Gdh in muscle, promotes infection resistance, and consequently, enhanced survival (Figure 7I–K and S7O–R). In totality, Smvt-regulated vitamin B cofactor function therefore plays a critical role in regulating infection-mediated energetic trade-offs in response to mitochondrial-derived glutamate, which shapes phenotypic variation in host infection outcomes within populations.
Discussion:
In totality, our findings reveal a distinct mechanism by which muscle-derived glutamate metabolism directs host energetic trade-offs and energy substrate re-allocation after infection to condition individual host-pathogen susceptibility (Figure 7L). This functional integration of immune-metabolic responses is fine-tuned by gene-environment interactions (life history events) among individuals, and subsequently shapes phenotypic diversity of host immune-metabolic responses within Drosophila populations. Thus, our study provides mechanistic insight into how metazoan hosts balance bacterial clearance, self-damage, and energy trade-offs to alleviate infections.
Muscle tissue is a central cog in the control of energy substrate balance and allocation (Baskin et al., 2015; Zhao and Karpac, 2017). To this end, muscle is highly enriched with mitochondria, which support efficient energy substrate usage. We uncovered that muscle mitochondria also play a central role in systemic immune-metabolic sensing, which is limited by intra-muscular NF-kB innate immune signaling. Previous studies have also revealed a putative role for NF-kB in the regulation of mitochondrial dynamics in various contexts (Laforge et al., 2016; Mauro et al., 2011; Zhong et al., 2016). Innate immune and/or infection-mediated changes in mitochondrial dynamics within immune cells can act cell-autonomously to control immune responses through release of mitochondrial metabolites, such as succinate and 2-hydroxyglutarate (Mills et al., 2016; Tyrakis et al., 2016). However, we found that intra-muscular mitochondrial-derived glutamate, mediated by glutamate dehydrogenase (Gdh) function, acts an unexpected systemic metabolite to direct re-allocation of host energy substrates after infection. Gdh is an ancestral enzyme that is conserved across biological taxonomic domains (Plaitakis et al., 2017). However, little is known about this mitochondrial enzyme’s regulation aside from eukaryotic nitrogen disposal in amino acid metabolism. Our data suggest that Gdh may additionally function as a key innate immune target gene, in mitochondrial-dense cell types, that is required to manage infection.
Intra-cellular mitochondrial metabolites can also function as cell-autonomous signaling molecules to regulate gene expression through various modifications of histones (chromatin) or DNA within various cell types (Carey et al., 2015; Chakraborty et al., 2019; Sciacovelli et al., 2016; Wellen et al., 2009). Distinctly, we uncovered that muscle and mitochondrial-derived glutamate can act as systemic metabolite to control Smvt transcription in adipose through a muscle-adipose communication axis. Previous findings indicate that Smvt directs the cellular transport of vitamin B5 and vitamin B7, and these B-family vitamins are co-factors/co-enzymes in several metabolic pathways involved in fatty acid metabolism (Yoshii et al., 2019). Although both Smvt and vitamins B5/B7 have not previously been reported to directly promote lipid mobilization, Gdh-dependent glutamate oxidation has been shown to reshape peripheral energy stores by dictating mobilization of energy substrates (Karaca et al., 2015). Our data indicate that glutamate and vitamin B cofactor transport/function drive organism-wide energy substrate re-allocation, which likely represents a unique strategy to remodel host energetic trade-offs after infection. We also found that infection-mediated lipid mobilization may be crucial to drive gastrointestinal defecation responses and bacterial expulsion, a plausible version of ‘immune cell activation’ in insects of which the intestine acts as a primary barrier to oral infection. Indeed, recent studies have highlighted that excretion/defecation mechanisms are critical for host-pathogen responses in Drosophila, including the removal toxic oxidized molecules that amass in the host (Li et al., 2020).
Limitation of Study:
Glutamate is primarily described as a major excitatory neurotransmitter within the nervous system (including control of locomotion). We identified an unexpected systemic function of glutamate to direct re-allocation of host energy substrates after infection. The unique functional and regulatory differences between infection-driven circulating glutamate and intra-neuronal or glial glutamate still need to be explored. To this end, we found that a dietary and metabolic glutamate transporter (dmGlut) is required for circulating glutamate function in adipose, which may be unique to peripheral metabolic tissues (different from glutamate regulation in neuronal/glial cell types).
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jason Karpac (karpac@tamu.edu).
Materials Availability
UAS-Smvt transgenic flies generated in this study are available from the lead contact upon request.
Data and Code Availability
Raw data files for the RNA sequencing analysis have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE160652.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila husbandry and strains
The following strains were obtained from Bloomington Drosophila Stock Center: w1118 (Hazelrigg et al., 1984); Act88FGal4 (38461) (Gajewski and Schulz, 2010); MhcGal4 (55133) (Klein et al., 2014); PplGal4 (58768) (Zinke et al., 1999); LppGal4 (84317) (Brankatschk and Eaton, 2010); tubP-GAL80ts (65406) (Ferris et al., 2006); UAS-ApoltpRNAi (51937) (Perkins et al., 2015); UAS-ApolppRNAi (33388, 28946) (Perkins et al., 2015); UAS-LuciferaseRNAi (31603) (Perkins et al., 2015); UAS-GdhRNAi#2 (53255) (Perkins et al., 2015); UAS-Gdh (20165) (Bellen et al., 2004); UAS-ATPsynbetaRNAi (28056) (Perkins et al., 2015); UAS-dmGlutRNAi (36724) (Perkins et al., 2015). The following strains were obtained from Vienna Drosophila RNAi Center (Dietzl et al., 2007): UAS-RelRNAi#1 (49413), UAS-RelRNAi#2 (108469); UAS-KeyRNAi (7723); UAS-DreddRNAi (104726); UAS-PGRP-LCRNAi (101636); UAS-PGRP-LERNAi (23664); UAS-GdhRNAi#1 (22059); UAS-SmvtRNAi#1 (40650), UAS-SmvtRNAi#2 (102662). UAS-LD-GFP was kindly provided by M. Welte (Yu et al., 2011). CGGal4 was kindly provided by C. Thummel (Hennig et al., 2006). TubGeneSwitch was kindly provided by H. Jasper (Sykiotis and Bohmann, 2008). Drosocin-GFP was kindly provided by John Gerard Tower (Chatterjee et al., 2016). UAS-Rpr was kindly provided by JC Billeter (Aplin and Kaufman, 1997). Hml∆G4, UAS-GFP was kindly provided by K. Bruckner (Sinenko and Mathey-Prevot, 2004). UAS-Smvt transgenic flies were generated for this study.
All flies were reared on standard yeast and cornmeal-based diet at 25°C and 65% humidity on a 12 hr light/dark cycle, unless otherwise indicated. The standard lab diet (cornmeal-based) was made with the following protocol: 14g Agar/165.4g Malt Extract/ 41.4g Dry yeast/ 78.2g Cornmeal/ 4.7ml propionic acid/ 3g Methyl 4-Hydroxybenzoate/ 1.5L water. In order to standardize metabolic results, fifty virgins were crossed to 10 males and kept in bottles for 2–3 days to lay enough eggs. Wet folded filters (GE healthcare, CAT No.10311843) were inserted in bottles after parental flies removed. Progeny of crosses was collected for 3–4 days after initial eclosion. Collected progeny were then transferred to new bottles to allow them mate for 2 days (representing unique populations). All these flies were reared on a standard lab diet at 25°C and 65% humidity on a 12 hr light/dark cycle, unless otherwise indicated. Around 20 female flies were then separated into each vial (before mock or oral infection treatment) for 10 days at 25 °C and 65% humidity on a 12 hr light/dark cycle. Post-mated female flies were used for all experiments due to elevated lipid content, sensitivity to Pseudomonas infections, and ability to rapidly assay fecundity.
The UAS-RelRNAi (VDRC:49413, 108469); UAS-KeyRNAi (VDRC: 7723); UAS-DreddRNAi (VDRC: 104726); UAS-PGRP-LCRNAi (VDRC: 101636); UAS-PGRP-LERNAi (VDRC: 23664); UAS-GdhRNAi (VDRC: 22059); UAS-SmvtRNAi (VDRC: 102662, 40650), UAS-Smvt, UAS-Gdh, CGGal4, PplGal4 and Act88FGal4 transgenic lines were backcrossed 10x into the w1118 background that was used as a control strain, with continued backcrossing every 6–8 months to maintain isogeneity. All experimental genotypes were assayed for developmental defects (developmental timing, growth, fly size, and organ size), and no gross anatomical deficiencies were noted in any genotype represented in the results.
Generation of transgenic flies
UAS-Smvt flies were generated by PCR amplification of Smvt coding sequence from adult Drosophila (w1118) cDNA, with specific primers, and then cloned into the pUASt plasmid. This plasmid was injected into w1118 embryos (Rainbow Transgenic Flies, Inc).
METHOD DETAILS
Oral infection of adult flies
Bacterial strains Pseudomonas entomophila (P.e) and Erwinia carotovora carotovora 15 (Ecc15) were grown in LB medium at 29°C, shaking at 200 rpm overnight. Fresh bacterial cultures were generated daily. The liquid cultures were poured into a sterile centrifuge flask and centrifuged at 4000 g at 4°C for 15 minutes. The liquid LB medium was removed from the centrifuge flask, and the bacterial pellet was resuspended in a small amount of LB medium. The final bacteria concentration (of the resuspended pellet, OD600=50-60 for P.e and OD600=300 for Ecc15) was adjusted by diluting with additional LB medium. Bacterial cultures were routinely genotyped for accuracy and reproducibility of experiments (using PCR and primers specific for P.e. or Ecc15, Table S1).
Next, 2.5% and 5% sucrose (in sterile water) were prepared fresh. The 5% sucrose solution was mixed with an equal volume of bacteria solution (at OD600=50-60 for P.e and OD600=300 for Ecc15) to create the solution used for oral infection (feeding). NOTE: These OD were used for all experiments except for survival analysis. 10 day-old mated female flies (20 per vial) were transferred into a fly food vial containing a filter paper that totally covers the food and was soaked with a solution consisting of 185 μL either bacterial oral infection mix (for infections) or 2.5% sucrose, for unchallenged (mock) controls. Drosophila were always infected at 3:00-4:00pm to ensure diurnal reproducibility, and subsequently incubated at 25°C and 65% humidity on a 12 hours light/dark cycle for required infection times (16-20 hours for P.e and 48 hours for Ecc15) while feeding.
Measuring bacterial load (CFUs) in dissected adult midguts
Mock-treated or infected flies, dissecting forceps and dissecting dish were surface sterilized with 70% ethanol and washed with sterile 1× PBS. Midguts from flies were then dissected individually in 1xPBS and each single gut was homogenized using a sterile pestle in 1.5 ml Eppendorf tube containing 200 μL 1xPBS. The homogenate was diluted to 1:1000 with 1xPBS. 50 μl of 1000-fold dilutions of the homogenate was plated onto LB plates and incubated at 29°C overnight. The number of colonies in each plate were counted (only counting separated, well defined single colonies). At least 10 individual guts were measured in each treatment. Similar to bacterial cultures, these colonies derived from midgut dissections were routinely genotyped for accuracy and reproducibility of experiments (using PCR and primers specific for P.e. or Ecc15, Supplementary Information). All plates from mock-treated plate controls were negative for bacterial colonies.
CFU/fly for every plate was calculated using the following formulas:
CFU/ml = ((total number of colonies on a plate) * (dilution factor (1000)) / (plated volume: 0.05ml)
CFU/fly = ((CFU/ml) * total volume Eppendorf tube: 0.2ml)) / (number of flies per condition: 1 gut)
Measuring bacterial load (CFUs) in hemolymph
Mock-treated or infected flies, dissecting forceps and dissecting dish were surface sterilized with 70% ethanol and washed with sterile 1× PBS. Hemolymph from flies was collected individually using a sterile capillary (VWR, #53432–706), transferred to 1.5 ml Eppendorf tube containing 200 μL 1xPBS, and then plated 1:1000 dilution with 1xPBS onto LB plates and incubated at 29°C overnight.
Measuring defecation in adult flies
Defecation was measured by counting defecation ‘spots’ left dried on the inner wall of standard lab rearing vials. For the convenience of measuring defecation frequencies, 20 infected flies were transferred from vials containing bacterial solution (infection) to vials containing standard lab food with 1% Brilliant Blue FCF (no bacteria). The dried blue spots left on the inner wall of vials were counted after 8 hours. Experiments were repeated with at least 10 independent biological replicates/samples with 20 flies per replicate.
Survival analysis (population)
Flies were infected with P.e at OD600=30-40 as described above and kept at 25°C for 16 hours. After overnight feeding (flies were always orally infected at 3:00-4:00pm), bacterial infected flies were transferred from bacterial infection vials to vials containing standard lab food. Flies were transferred every day to a fresh vial for the first two days, and every two days after, and dead flies were counted (and removed) when changing vials.
Hemolymph metabolite measurements in adult flies
To collect fly hemolymph, the thoraces of 40 flies (per sample) were carefully pierced with a sterile tungsten needle (under the wing connection by the ubithorax) and then placed in a perforated 0.5 ml Eppendorf tube (using 0.0005 mm syringe) within a 1.5 ml Eppendorf tube. Then, the pierced flies were centrifuged at 4000 rpm for 3 min at 4 °C. The supernatant was carefully collected to avoid debris. Collected hemolymph was centrifuged again at 4000 rpm for 3 min to precipitate debris. The collected hemolymph was used measure metabolites immediately. For TAG/DAG assays, 1 μl of collected hemolymph was diluted with 10 μl of PBST (PBS, 0.1% Tween 20) and used to measure triglycerides/diacylglycerides (StanBio Liquicolor Triglycerides) according to the manufacturer instructions. Note: The kit measures glycerol cleaved from TAG and DAG, as well as minimal amounts of free glycerol; neutral lipids extracted from hemolymph are comprised of mainly DAG, as well as some TAG. For hemolymph glutamate assays, 1 μl of collected hemolymph was used to measure glutamate using Glutamate Assay Kit (Sigma-Aldrich MAK004) according to the manufacturer instructions.
Food intake/feeding measurements
The CAFE assay was done as described previously (Deshpande et al., 2014). Briefly, a single fly was transferred from SY standard food to vials filled with 5 ml of 1.5% agar that maintains internal humidity and serves as a water source. Flies were fed with P.e. or sucrose solution maintained in 5 ml capillaries (VWR, #53432-706). After two hours habituation, the old capillaries were replaced with a new one at the start of the assay. The amount of liquid food consumed was recorded after 24 hr and corrected on the basis of the evaporation (typically < 10% of ingested volumes) observed the identical vials without flies. 5 flies were weighed in order to normalize samples.
Feeding assays on blue dye-labeled food were done as described previously (Deshpande et al., 2014). 20 flies were transferred to vials filled with identical medium containing 0.5% brilliant blue (with or without bacterial infection mix [P.e. or Ecc15]). Feeding was interrupted and 5 flies each were transferred to 200 μl 1 x PBS containing 0.1% Triton X-100 (PBST) and homogenized immediately. Blue dye consumption was quantified by measuring absorbance of the supernatant at 630 nm (A630).
Conditional expression of UAS-linked transgenes
The TARGET system was used in combination with PplGal4 to conditionally express UAS-linked transgenes in fat body (PPLGal4, tub-Gal80ts). Flies were developed at 19°C, then shifted to 29°C to induce transgene expression post-eclosion (Day 5).
Analysis of gene expression
Total RNA from intact fly thorax (containing mostly muscle), carcass (containing mostly fat body/adipose) and intestines were extracted using Trizol and complementary DNA synthesized using Superscript III (Invitrogen). Real-time PCR was performed using SYBR Green, the Applied Biosystems StepOnePlus Real-Time PCR systems, and the primers pairs described in the extended experimental procedures (Table S1). Results are average ± standard error of at least three independent samples, and quantification of gene expression levels calculated using the ΔCt method and normalized to actin5C expression levels.
Western blot analysis
Thoraces from 10 flies (per sample) were dissected in PBS and then homogenized in a protein sample buffer; proteins were separated by SDS-PAGE and transferred to nitrocellulose membrane using standard procedures. The following antibodies were used: anti-ATP5A (ab14748, 1:10,000) and anti-NDUFS3 (ab14711, 1:10,000) were incubated with the membrane for 1 hour at 4 °C or 30 min at room temperature. Secondary anti-mouse (BIO-RAD, 1:10,000) was incubated for 30 min at room temperature. For beta-actin loading controls, anti-beta-actin (Cell Signaling; rabbit, 1:1000) was incubated at 4 °C overnight and then incubated with Secondary anti-rabbit (BIO-RAD, 1:5,000) for 1 hour at room temperature. Signal was detected using HRP-conjugated anti-rabbit and ECL Western Blotting Substrate (Pierce), according to manufacturer instructions. Quantification of signal was measured with Image J; densitometry (normalized to beta-actin).
Triglycerides (TAG) measurements in adult flies
For TAGs assays, five flies (without head, per sample) were homogenized in 200 μl of PBST (PBS, 0.1% Tween 20) and heated at 70°C for 5 min to inactivate endogenous enzymes. Samples were centrifuged at 4000 rpm for 3 min at 4 °C and ten microliters of cleared extract was used to measure triglycerides (StanBio Liquicolor Triglycerides Kit) according to the manufacturer instructions. TAG levels were normalized to weight. Note: The kit measures glycerol cleaved from TAG and diacylglycerol (DAG), as well as minimal amounts of free glycerol; the majority of neutral lipids extracted from whole flies are TAG.
Glutamate and α-ketoglutarate measurements in adult flies
Glutamate and α-ketoglutarate concentrations were measured using the Glutamate Assay Kit (Sigma-Aldrich MAK004) and α-Ketoglutarate Assay Kit (Sigma-Aldrich MAK054) according to the manufacturer’s instructions. Briefly, dissected thoraces from 15 flies (per sample) were homogenized in 100 μL of ice-cold Glutamate Assay Buffer or α-KG Assay Buffer. The samples were centrifuged at 13,000g for 10 min to remove insoluble material. The supernatant was deproteinized with 10 kDa MWCO spin filter (Life Technologies) to remove enzymes. Samples were brought to 50 μL per well with Glutamate Assay Buffer or α-KG Assay Buffer. Reaction mix was added to each well and incubated for 30 min at 37°C. Absorbance (450 nm [A450] for Glutamate Assay and 570 nm [A570] for α-Ketoglutarate Assay) was obtained using an Epoch Microplate Spectrophotometer (BioTek Instruments). Glutamate and α-Ketoglutarate levels were normalized to total protein levels using the BCA Protein Assay Kit (Pierce).
Oil Red O staining
Intact carcasses (with all ovaries and intact intestines removed) were dissected in PBS and fixed in 4% paraformaldehyde for 20 min, then washed twice with PBS, incubated for 30 min in fresh Oil Red O solution (6 ml of 0.1% Oil Red O in isopropanol and 4 ml distilled water, and passed through a 0.45 μm syringe), followed by rinsing with distilled water. Bright light images were collected using a Leica M125 stereoscope and processed using Adobe Photoshop.
Nile Red staining
Fat body/carcasses were dissected in PBS (with all of the eggs and intact intestines removed) and then incubated for 2 hours in fresh Nile Red solution with DAPI (1μl of 0.004% Nile Red Solution in 500 μl PBS), followed by rinsing with PBS. Confocal images were immediately collected using a Nikon Eclipse Ti confocal system (utilizing a single focal plane) and processed using the Nikon software and Adobe Photoshop.
Immunostaining and microscopy
For muscle immunostaining, dorsal longitudinal thorax muscle segments were dissected in PBS and fixed with 4% paraformaldehyde for 20 min at room temperature, washed 3 times with PBS containing 0.2% Triton X-100 (PBST) and then block in blocking buffer (5% BSA in PBST) for 1 h. Primary antibodies; anti-ATP5A from Abcam (ab14748, 1:500) and anti-GDH from Sigma (Anti-GLUD1, HPA061369, 1:500) were applied overnight at 4°C. Alexa Flour-conjugated secondary (Jackson Immunoresearch, 1:500) antibodies, Alexa Fluor 555 Phalloidin (Thermo Fisher Scientific, 1:500) and Hoechst (DAPI; 1:500) were incubated overnight at 4°C.
Confocal images were collected using a Nikon Eclipse Ti confocal system (utilizing a single focal plane) and processed using the Nikon software and Adobe Photoshop.
TMRE (mitochondrial membrane potential) staining
For muscle TMRE (tetramethylrhodamine, ethyl ester) staining, dorsal longitudinal thorax muscle segments were dissected in 1XPBS and incubated in 200nm TMRE staining solution (ab113852, Abcam) for 20 min at room temperature. After staining, samples were then rinsed once in wash solution (25nm TMRE) for 30s. Samples were quickly mounted in the wash solution onto a slide, kept overnight at 4°C and imaged using identical setting on the confocal microscope. Confocal images were collected using a Nikon Eclipse Ti confocal system (utilizing a single focal plane) and processed using the Nikon software and Adobe Photoshop.
RNA-seq analysis
Intact fly thoraces (8) or carcass/fat body (8) were dissected in PBS. Total RNA was extracted using Trizol reagent and used as template to generate sample libraries for RNA sequencing (using the TruSeq Stranded Total RNA Library Prep Kit). Sample libraries were sequenced using the Illumina NextSeq 500. Sequence cluster identification, quality pre-filtering, base calling and uncertainty assessment were done in real time using Illumina’s HCS and RTA software with default parameter settings. Between 8 and 10 million (2X150) base pair reads were generated per library and mapped to the Drosophila genome (Release 6). Expression was recorded as TPKM (transcripts per kbp per million reads) followed by Log2 transformation. Gene Ontology clustering analysis was performed using FlyMine (http://www.flymine.org). Expression-based heat maps were performed using Heatmapper (http://www.heatmapper.ca/expression/). FASTQ data files representing unique libraries were deposited in the NCBI Gene Expression Omnibus database (GSE160652).
Chromatin immunoprecipitation (ChIP) analysis
Approximately 200 adult flies (per sample) were ground in liquid nitrogen then homogenized and cross-linked for 10 minutes at room temperature in 600 mL of 1x PBS containing 1% formaldehyde, 1 mM PMSF and 1x Protease Inhibitor cocktail (Thermo Scientific). The homogenate was then centrifuged for 20 min at 12000x rpm at 4°C. The pellet was washed twice by resuspending in 600 mL of 1xPBS containing 1 mM PMSF and 1x Protease Inhibitor cocktail and centrifuged at 12000x rpm for 20 min at 4°C. To lyse tissue and cells, the pellet was resuspended in 600 mL of RIPA buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.1% SDS, 0.1% Na-Deoxycholate, 1% Triton X-100, containing 1 mM PMSF and 1x Protease Inhibitor Cocktail) and incubated at room temperature for 30 min.
The chromatin was sheared to 250-500 bp DNA fragments using a Diagenode sonicator (20 min sonication, highest power, 30 s sonication, 30 s rest). After sonication, the sheared chromatin was centrifuged for 20 min at 12000x rpm, 4°C. The supernatant was collected, aliquoted, snap-frozen, and stored at 80°C.
For immunoprecipitation, 10 uL of Rabbit anti-Relish (RayBiotech, RB-14-0004), or 2 uL of anti-Histone H3 (acetyl 9) antibody (abcam, ab4441) was incubated with 100 mL of chromatin diluted 1:10 with dilution buffer (20 mM Tris-HCl pH 8, 2 mM EDTA, pH 8, 150 mM NaCl, 1% Triton X-100) overnight at 4°C with rotation. 40 mL protein A magnetic beads were added the following morning and incubated at 4°C for 4 hr with rotation. Beads were then washed with the following buffers at 4°C, for 10 min each: 2x with 1 mL of RIPA Buffer + 1mM PMSF + 1x Protease Inhibitor, 2x with 1 mL RIPA buffer + 0.3 M NaCl, 2x with 1 mL of LiCl buffer (0.25 M LiCl, 0.5% Triton X-100, 0.5% NADOC), 1x with 1 mL of 1x TE + 0.2% Triton X-100, 1x with 1 mL of 1x TE.
To reverse crosslinking, beads were re-suspended in 100 mL of 1x TE + 3 mL 10% SDS + 5 mL of 20 mg/mL Proteinase K (VWR) and incubated at 65°C overnight. Beads were applied to the magnet and DNA was purified from the supernatant using a QIAGEN PCR Purification kit. To prepare input, 100 mL of chromatin extract was incubated overnight at 65°C with 3 mL 10% SDS + 5 mL of 20 mg/mL Proteinase K, DNA was then precipitated from 10 mL of the initial sample using a QIAGEN PCR Purification kit. For all Immunoprecipitated (IP) and Input samples, DNA was eluted in 20 mL of water, and 2 mL was used as a template for qRT-PCR (Table S1).
To assess enrichment, %Input was calculated between ChIP DNA and input DNA for each primer set, and then fold change in Input was calculated by dividing the %Input of each primer set to the %Input of a negative control primer set designed for Drosophila (Drosophila Negative Control primer set 1, Active Motif).
Epithelial barrier assay (‘smurf’ assay)
The barrier defect assay (‘smurf’ assay) was conducted as described previously (Rera et al., 2012). Dyed medium was prepared using P.e infection medium with dyes added at a concentration of Blue dye no. 1 at 2.5% (wt/vol). Dro-GFP transgenic flies were maintained on dyed medium for 24 h. A fly was counted as a Smurf when dye coloration could be observed outside of the digestive tract. Dro-GFP positive and negative flies were then monitored for the ‘Smurf’ phenotype as an indicator for epithelial barrier dysfunction.
Fecundity assay
10 day-old mated female flies (20 per vial) were transferred into a fly food vial containing a filter paper that totally covers the food and was soaked with a solution consisting of 185 μL of either P.e. bacterial oral infection mix (for infections) or 2.5% sucrose (for mock controls) for 24 hrs. All flies were then flipped onto fresh (standard) food vials every 24 hrs for the indicated number of days. The number of eggs after (during) or post P.e. bacterial oral infection were counted daily, and fecundity was calculated by dividing the total number of eggs by the number of flies in each cage.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all quantifications, n represents the number of biological replicates, and error bar represents SEM. Statistical significance was determined using either the unpaired t test or one-way ANOVA with Tukey post hoc test where multiple comparisons were necessary, in GraphPad Prism Software, and expressed as P values. (*) denotes values whose difference was significant, and (n) denotes values whose difference was not significant. Exact values of all n’s can be found in Figure legends and individual data points are represented in all histograms.
Supplementary Material
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse polyclonal anti-ATP5A | abcam | ab14748 |
| Mouse polyclonal anti-NDUFS3 | abcam | ab14711 |
| Rabbit polyclonal anti-GLUD1 | Sigma-Aldrich | HPA061369 |
| Rabbit polyclonal Anti-Histone H3 (acetyl K9) | abcam | Ab4441 |
| Rabbit polyclonal Anti-Relish | RayBiotech | RB-14-0004 |
| Rabbit polyclonal anti-beta-actin | Cell Signaling | 4967 |
| Goat Anti-Rabbit IgG HRP-conjugate | BioRad | 1706515 |
| Goat Anti-Mouse IgG HRP-conjugate | BioRad | 1706516 |
| Alexa Flour 488-conjugated Anti-Rabbit IgG | Jackson Immunoresearch | 119191 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alexa Fluor 555 Phalloidin | Thermo Fisher Scientific | A34055 |
| Alexa Fluor 488 Phalloidin | Thermo Fisher Scientific | A12379 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Thermo Fisher Scientific | D1306 |
| Drosophila Agar, Type II | Genesee | 66-103 |
| Malt Extract | Genesee | 62-110 |
| Inactive Dry yeast | Genesee | 62-106 |
| Cornmeal | Genesee | 62-101 |
| Propionic acid | VWR | TCP0500-500mL |
| Methyl 4-Hydroxybenzoate | VWR | 97061-946 |
| Sucrose | VWR | 97063-788 |
| Trizol | Life Technologies | 15596018 |
| Superscript III Reverse Transcriptase | Life Technologies | 2129278 |
| iTaq Universal SYBR Green Supermix | BioRad | 1725121 |
| Nitrocellulose membrane | VWR | 10600006 |
| Oil Red O | abcam | ab150678 |
| Nile Red | Life Technologies | N1142 |
| Phosphoric acid | VWR | 97064-780 |
| Brilliant blue | Sigma-Aldrich | B0149 |
| ECL Western Blotting Substrate | Pierce | 32106 |
| L-Glutamate | abcam | Ab120049 |
| Biotin | Sigma-Aldrich | B4639,14400 |
| D-Pantothenic acid | Sigma-Aldrich | P5155 |
| L-Ascorbic acid | Sigma-Aldrich | A7506 |
| DreamTaq PCR Master Mix | Thermo Fisher Scientific | K1081 |
| Xho I | New England BioLab | R0146S |
| NotI-HF | New England BioLab | R3189S |
| T4 DNA Ligase | New England BioLab | M0202T |
| RNase A | QIAGEN | 19101 |
| Protease Inhibitor Cocktail | Thermo Fisher Scientfic | 78440 |
| Proteinase K | VWR | 0706 |
| LiCl | Amresco | 0416-100G |
| KCl | J.T.Baker | 3052-01 |
| CaCl2 | Macron | 4160-12 |
| PMSF | Thermo Fisher Scientific | 36978 |
| Bovine Serum Albumin | VWR | 97061 |
| Critical Commercial Assays | ||
| StanBio Liquicolor Triglycerides Kit | Fisher | SB-2100-430 |
| Bio-Rad Protein Assay | Bio-Rad | 5000006 |
| TMRE-Mitochondrial Membrane Potential Assay Kit | abcam | Ab113852 |
| QIAquick PCR purification Kit | QIAGEN | 28104 |
| QIAquick Gel extraction Kit | QIAGEN | 28704 |
| QIAprep spin miniprep Kit | QIAGEN | 27104 |
| Plasmid midi Kit | QIAGEN | 12143 |
| Glutamate Assay Kit | Sigma-Aldrich | MAK004 |
| α-Ketoglutarate Assay Kit | Sigma-Aldrich | MAK054 |
| Deposited Data | ||
| Raw and analyzed RNA sequencing data | This paper | GEO: GSE160652 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: w1118 | Bloomington Drosophila Stock Center and (Hazelrigg et al., 1984) | BDSC: 3605; FlyBase: FBst0003605 |
| D. melanogaster: Act88FGal4 (w*; P{Act88F-GAL4.1.3}3) | Bloomington Drosophila Stock Center and (Gajewski and Schulz, 2010) | BDSC: 38461; FlyBase: FBst0038461 |
| D. melanogaster: MhcGal4 (w*; P{Mhc-GAL4.K}2/TM3, Sb1) | Bloomington Drosophila Stock Center and (Klein et al., 2014) | BDSC: 55133; FlyBase: FBst0055133 |
| D. melanogaster: PplGal4 (w*; P{ppl-GAL4.P}2) | Bloomington Drosophila Stock Center and (Zinke et al., 1999) | BDSC: 58768; FlyBase: FBst0058768 |
| D. melanogaster: LppGal4 (w[*]; P{w[+mC]=Lpp-GAL4.B}3) | Bloomington Drosophila Stock Center and (Brankatschk and Eaton, 2010) | BDSC: 84317; FlyBase: FBst0084317 |
| D. melanogaster: tubP-Gal80ts (P{tubP-GAL80ts}20) | Bloomington Drosophila Stock Center and (Ferris et al., 2006) | BDSC: 65406; FlyBase: FBst0065406 |
| D. melanogaster: UAS-Apoltp RNAi (y1sc*v1; P{TRiP.HMC03294}attP2) | Bloomington Drosophila Stock Center and (Perkins et al., 2015) | BDSC:51937; FlyBase: FBst0051937 |
| D. melanogaster: UAS-Rfabg/Apolpp RNAi (y1v1; P{TRiP.HM05157}attP2) | Bloomington Drosophila Stock Center and (Perkins et al., 2015) | BDSC:28946; FlyBase: FBst0028946 |
| D. melanogaster: UAS-Rfabg/Apolpp RNAi (y1sc* v1; P{TRiP.HMS00265}attP2/TM3) | Bloomington Drosophila Stock Center and (Perkins et al., 2015) | BDSC:33388; FlyBase: FBst0033388 |
| D. melanogaster: UAS-ATPsynbeta RNAi (y [1] v [1]; P{y[+t7.7] v[+t1.8] =TRiP.JF02892} attP2) | Bloomington Drosophila Stock Center and (Perkins et al., 2015) | BDSC: 28056; FlyBase:FBgn0010217 |
| D. melanogaster: UAS-luciferase RNAi (y1v1; P{TRiP.JF01355}attP2) | Bloomington Drosophila Stock Center and (Perkins et al., 2015) | BDSC: 31603; FlyBase: FBst0031603 |
| D. melanogaster: UAS-Gdh RNAi (y [1] sc[*] v[1] sev[21]; P{y[+t7.7] v[+t1.8]=TRiP.GLC01815}attP2) | Bloomington Drosophila Stock Center and (Perkins et al., 2015) | BDSC: 53255; FlyBase:FBgn0001098 |
| D. melanogaster: UAS-Gdh (y [1] w[67c23]; P{y[+mDint2] w[+mC] =EPgy2} Gdh[EY07150]) | Bloomington Drosophila Stock Center and (Bellen et al., 2004) | BDSC: 20165; FlyBase:FBgn0001098 |
| D. melanogaster: UAS-dmGlut RNAi (y1 sc* v1 sev21; P{TRiP.HMS01615}attP2/TM3, Sb) | Bloomington Drosophila Stock Center and (Perkins et al., 2015) | BDSC: 36724; FlyBase:FBgn0010497 |
| D. melanogaster: UAS-Rel RNAi (w1118; P{GD1199} v49413) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 49413; FlyBase: FBst0468440 |
| D. melanogaster: UAS-Rel RNAi (w1118; P{KK109851} VIE-260B) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 108469; FlyBase: FBst0480279 |
| D. melanogaster: UAS-Key RNAi (w1118; P{GD1249} v7723) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 7723; FlyBase: FBst0470808 |
| D. melanogaster: UAS-Dredd RNAi (w1118; P{KK110428} VIE-260B) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 104726; FlyBase: FBst0476565 |
| D. melanogaster: UAS-PGRP-LC RNAi (w1118; P{KK105287} VIE-260B) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 101636; FlyBase: FBst0473509 |
| D. melanogaster: UAS-PGRP-LE RNAi (w1118; P{GD14089} v23664) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 23664; FlyBase: FBst0455134 |
| D. melanogaster: UAS-White RNAi (GD) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 30033; FlyBase: FBgn0026792 |
| D. melanogaster: UAS-Gdh RNAi (w1118; P{GD11605} v22059) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 22059; FlyBase:FBgn0001098 |
| D. melanogaster: UAS-Smvt RNAi (w1118; P{KK105033} VIE-260B) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 102662; FlyBase:FBgn0039873 |
| D. melanogaster: UAS-Smvt RNAi (w1118; P{GD12439} v40650/TM3) | Vienna Drosophila RNAi Center and (Dietzl et al., 2007) | VDRC: 40650; FlyBase:FBgn0039873 |
| D. melanogaster: UAS-Smvt | This paper | N/A |
| D. melanogaster: UAS-LD-GFP | (Yu et al., 2011) | N/A |
| D. melanogaster: CGGal4 (w*; P{CG-GAL4.A}) | (Hennig et al., 2006) | N/A |
| D. melanogaster: TubGSGal4 (y1w*; TubGeneSwitch/CyO) | (Sykiotis and Bohmann, 2008) | N/A |
| D. melanogaster: Drosocin-GFP | (Chatterjee et al., 2016b) | N/A |
| D. melanogaster: UAS-Rpr | (Aplin and Kaufman, 1997) | N/A |
| D. melanogaster: Hml∆G4, UAS-GFP | (Sinenko and Mathey-Prevot, 2004) | N/A |
| Oligonucleotides | ||
| Primers for Dipt, see Table S1 | This paper | N/A |
| Primers for Dro, see Table S1 | This paper | N/A |
| Primers for AttA, see Table S1 | This paper | N/A |
| Primers for CecA, see Table S1 | This paper | N/A |
| Primers for Gdh, see Table S1 | This paper | N/A |
| Primers for Smvt, see Table S1 | This paper | N/A |
| Primers for Actin 5c, see Table S1 | This paper | N/A |
| Primers for P. entomophila, see Table S1 | This paper | N/A |
| Primers for Erwinia carotovora carotovora15, see Table S1 | This paper | N/A |
| Primers for Smvt-pUAST, see Table S1 | This paper | N/A |
| Primers for R1 (Gdh locus), see Table S1 | This paper | N/A |
| Primers for R2 (Gdh locus), see Table S1 | This paper | N/A |
| Primers for R3 (Gdh locus), see Table S1 | This paper | N/A |
| Primers for R4 (Gdh locus), see Table S1 | This paper | N/A |
| Primers for R5 (Gdh locus), see Table S1 | This paper | N/A |
| Primers for Act5cP (promoter region), see Table S1 | This paper | N/A |
| Primers for DroP (promoter region), see Table S1 | This paper | N/A |
| Primers for DiptP (promoter region), see Table S1 | This paper | N/A |
| Drosophila Negative Control (NC) Primer Set 1 | Active Motif | 71028 |
| Software and Algorithms | ||
| ImageJ | NIH Image | https://imagej.net/ImageJ |
| Heatmapper | University of Alberta | http://www.heatmapper.ca/expression/ |
| FlyMine | Cambridge University | https://www.flymine.org//flymine/begin.do |
| FlyBase | N/A | https://flybase.org/ |
| DNA Sequencing | Eton Bioscience | https://www.etonbio.com/ |
| Generation of Transgenic Flies | Rainbow Transgenic Flies | https://www.rainbowgene.com/ |
| JASPAR Database | N/A | http://jaspar.genereg.net/ |
| Other | ||
| Illumina HiSeq 2500 | Illumina | N/A |
| Leica M165FC system | Leica | N/A |
| StepOnePlus Real-Time PCR systems | Applied Biosystems | N/A |
| Nikon Eclipse Ti confocal system | Nikon | N/A |
| Capillaries | VWR | 53432-706 |
| Bioruptor/sonicator | Diagenode | UCD-200 |
| Pierce Protein A magnetic beads | Thermo Fisher Scientific | 88845 |
| QUBIT 4 Fluorometer | Thermo-Fisher Scientific | N/A |
| Epoch Microplate | BioTek | N/A |
| Ecc15 Bacteria | Basset et al., 2000 | N/A |
| Pseudomonas entomophila (P.e.) | Liehl et al., 2006 | N/A |
Acknowledgments:
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (grant R01 DK108930 to J.K.)
Footnotes
Declaration of Interests: The authors declare no competing interests.
References:
- Aplin AC, and Kaufman TC (1997). Homeotic transformation of legs to mouthparts by proboscipedia expression in Drosophila imaginal discs. Mech Dev 62, 51–60. [DOI] [PubMed] [Google Scholar]
- Ayres JS, and Schneider DS (2009). The Role of Anorexia in Resistance and Tolerance to Infections in Drosophila. Plos Biology 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS, and Schneider DS (2012). Tolerance of Infections. Annual Review of Immunology, Vol 30 30, 271–294. [DOI] [PubMed] [Google Scholar]
- Baskin KK, Winders BR, and Olson EN (2015). Muscle as a “Mediator” of Systemic Metabolism. Cell Metabolism 21, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basset A, Khush RS, Braun A, Gardan L, Boccard F, Hoffmann JA, and Lemaitre B (2000). The phytopathogenic bacteria Erwinia carotovora infects Drosophila and activates an immune response. P Natl Acad Sci USA 97, 3376–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, et al. (2004). The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 167, 761–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch PS, Makhijani K, Herboso L, Gold KS, Baginsky R, Woodcock KJ, Alexander B, Kukar K, Corcoran S, Jacobs T, et al. (2019). Adult Drosophila Lack Hematopoiesis but Rely on a Blood Cell Reservoir at the Respiratory Epithelia to Relay Infection Signals to Surrounding Tissues. Developmental Cell 51, 787-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankatschk M, and Eaton S (2010). Lipoprotein particles cross the blood-brain barrier in Drosophila. J Neurosci 30, 10441–10447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P, and Davis MM (2017). Human immune system variation. Nature Reviews Immunology 17, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchon N, Silverman N, and Cherry S (2014). Immunity in Drosophila melanogaster--from microbial recognition to whole-organism physiology. Nat Rev Immunol 14, 796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty AA, Laukka T, Myllykoski M, Ringel AE, Booker MA, Tolstorukov MY, Meng YJ, Meier SR, Jennings RB, Creech AL, et al. (2019). Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363, 1217-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Roy D, Patnaik E, and Nongthomba U (2016). Muscles provide protection during microbial infection by activating innate immune response pathways in Drosophila and zebrafish. Disease Models & Mechanisms 9, 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RI, Tan SWS, Pean CB, Roostalu U, Vivancos V, Bronda K, Pilatova M, Fu JQ, Walker DW, Berdeaux R, et al. (2013). MEF2 Is an In Vivo Immune-Metabolic Switch. Cell 155, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cognigni P, Bailey AP, and Miguel-Aliaga I (2011). Enteric Neurons and Systemic Signals Couple Nutritional and Reproductive Status with Intestinal Homeostasis. Cell Metabolism 13, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui R, Medeiros T, Willemsen D, Iasi LNM, Collier GE, Graef M, Reichard M, and Valenzano DR (2019). Relaxed Selection Limits Lifespan by Increasing Mutation Load. Cell 178, 385–399 e320. [DOI] [PubMed] [Google Scholar]
- Deshpande SA, Carvalho GB, Amador A, Phillips AM, Hoxha S, Lizotte KJ, and Ja WW (2014). Quantifying Drosophila food intake: comparative analysis of current methodology. Nat Methods 11, 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156. [DOI] [PubMed] [Google Scholar]
- Dionne MS, Pham LN, Shirasu-Hiza M, and Schneider DS (2006). Akt and foxo dysregulation contribute to infection-induced wasting in Drosophila. Current Biology 16, 1977–1985. [DOI] [PubMed] [Google Scholar]
- Du EJ, Ahn TJ, Kwon I, Lee JH, Park JH, Park SH, Kang TM, Cho H, Kim TJ, Kim HW, et al. (2016). TrpA1 Regulates Defecation of Food-Borne Pathogens under the Control of the Duox Pathway. PLoS Genet 12, e1005773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duneau D, Ferdy JB, Revah J, Kondolf H, Ortiz GA, Lazzaro BP, and Buchon N (2017). Stochastic variation in the initial phase of bacterial infection predicts the probability of survival in D. melanogaster. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris J, Ge H, Liu L, and Roman G (2006). G(o) signaling is required for Drosophila associative learning. Nat Neurosci 9, 1036–1040. [DOI] [PubMed] [Google Scholar]
- Gaber T, Strehl C, and Buttgereit F (2017). Metabolic regulation of inflammation. Nat Rev Rheumatol 13, 267–279. [DOI] [PubMed] [Google Scholar]
- Gajewski KM, and Schulz RA (2010). CF2 represses Actin 88F gene expression and maintains filament balance during indirect flight muscle development in Drosophila. PLoS One 5, e10713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganeshan K, Nikkanen J, Man K, Leong YA, Sogawa Y, Maschek JA, Van Ry T, Chagwedera DN, Cox JE, and Chawla A (2019). Energetic Trade-Offs and Hypometabolic States Promote Disease Tolerance. Cell 177, 399-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang SY, Purdy AE, Robins WP, Wang ZP, Mandal M, Chang S, Mekalanos JJ, and Watnick PI (2014). The Acetate Switch of an Intestinal Pathogen Disrupts Host Insulin Signaling and Lipid Metabolism. Cell Host & Microbe 16, 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AM, and Caspi RR (2010). Glutamate joins the ranks of immunomodulators. Nat Med 16, 856–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelrigg T, Levis R, and Rubin GM (1984). Transformation of white locus DNA in drosophila: dosage compensation, zeste interaction, and position effects. Cell 36, 469–481. [DOI] [PubMed] [Google Scholar]
- Hennig KM, Colombani J, and Neufeld TP (2006). TOR coordinates bulk and targeted endocytosis in the Drosophila melanogaster fat body to regulate cell growth. J Cell Biol 173, 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca M, Frigerio F, Migrenne S, Martin-Levilain J, Skytt DM, Pajecka K, Martin-Del-Rio R, Gruetter R, Tamarit-Rodriguez J, Waagepetersen HS, et al. (2015). GDH-Dependent Glutamate Oxidation in the Brain Dictates Peripheral Energy Substrate Distribution. Cell Reports 13, 365–375. [DOI] [PubMed] [Google Scholar]
- Klein P, Muller-Rischart AK, Motori E, Schonbauer C, Schnorrer F, Winklhofer KF, and Klein R (2014). Ret rescues mitochondrial morphology and muscle degeneration of Drosophila Pink1 mutants. EMBO J 33, 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepsatel P, Wildridge D, and Galikova M (2019). Temperature induces changes in Drosophila energy stores. Sci Rep-Uk 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnlein RP (2012). Thematic Review Series: Lipid Droplet Synthesis and Metabolism: from Yeast to Man Lipid droplet-based storage fat metabolism in Drosophila. J Lipid Res 53, 1430–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforge M, Rodrigues V, Silvestre R, Gautier C, Weil R, Corti O, and Estaquier J (2016). NF-kappaB pathway controls mitochondrial dynamics. Cell Death Differ 23, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KA, Cho KC, Kim B, Jang IH, Nam K, Kwon YE, Kim M, Hyeon DY, Hwang D, Seol JH, et al. (2018). Inflammation-Modulated Metabolic Reprogramming Is Required for DUOX-Dependent Gut Immunity in Drosophila. Cell Host & Microbe 23, 338-+. [DOI] [PubMed] [Google Scholar]
- Lee KA, and Lee WJ (2018). Immune-metabolic interactions during systemic and enteric infection in Drosophila. Curr Opin Insect Sci 29, 21–26. [DOI] [PubMed] [Google Scholar]
- Lercher A, Baazim H, and Bergthaler A (2020). Systemic Immunometabolism: Challenges and Opportunities. Immunity 53, 496–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Rommelaere S, Kondo S, and Lemaitre B (2020). Renal Purge of Hemolymphatic Lipids Prevents the Accumulation of ROS-Induced Inflammatory Oxidized Lipids and Protects Drosophila from Tissue Damage. Immunity 52, 374–387 e376. [DOI] [PubMed] [Google Scholar]
- Liehl P, Blight M, Vodovar N, Boccard F, and Lemaitre B (2006). Prevalence of local immune response against oral infection in a Drosophila/Pseudomonas infection model. Plos Pathogens 2, 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, Moretti M, De Smaele E, Beg AA, Tergaonkar V, et al. (2011). NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol 13, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I, et al. (2016). Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, and O’Neill LAJ (2017). Mitochondria are the powerhouses of immunity. Nat Immunol 18, 488–498. [DOI] [PubMed] [Google Scholar]
- Nedelec Y, Sanz J, Baharian G, Szpiech ZA, Pacis A, Dumaine A, Grenier JC, Freiman A, Sams AJ, Hebert S, et al. (2016). Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell 167, 657-+. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Takano T, and Hansen AJ (2002). Beyond the role of glutamate as a neurotransmitter. Nat Rev Neurosci 3, 748–755. [DOI] [PubMed] [Google Scholar]
- Palm W, Sampaio JL, Brankatschk M, Carvalho M, Mahmoud A, Shevchenko A, and Eaton S (2012). Lipoproteins in Drosophila melanogaster-assembly, function, and influence on tissue lipid composition. PLoS Genet 8(7), e1002828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins LA, Holderbaum L, Tao R, Hu Y, Sopko R, McCall K, Yang-Zhou D, Flockhart I, Binari R, Shim HS, et al. (2015). The Transgenic RNAi Project at Harvard Medical School: Resources and Validation. Genetics 201, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichaud N, Chatelain EH, Ballard JWO, Tanguay R, Morrow G, and Blier PU (2010). Thermal sensitivity of mitochondrial metabolism in two distinct mitotypes of Drosophila simulans: evaluation of mitochondrial plasticity. Journal of Experimental Biology 213, 1665–1675. [DOI] [PubMed] [Google Scholar]
- Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, and Spanaki C (2017). The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology (Basel) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rera M, Clark RI, and Walker DW (2012). Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila. Proc Natl Acad Sci U S A 109, 21528–21533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, and Shalek AK (2014). Heterogeneity in immune responses: from populations to single cells. Trends in Immunology 35, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa ASH, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MGB, et al. (2016). Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinenko SA, and Mathey-Prevot B (2004). Increased expression of Drosophila tetraspanin, Tsp68C, suppresses the abnormal proliferation of ytr-deficient and Ras/Raf-activated hemocytes. Oncogene 23, 9120–9128. [DOI] [PubMed] [Google Scholar]
- Sykiotis GP, and Bohmann D (2008). Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell 14, 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiku V, Tan MW, and Dikic I (2020). Mitochondrial Functions in Infection and Immunity. Trends in Cell Biology 30, 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, You J, Chia GS, Sim J, Doedens A, et al. (2016). S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature 540, 236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlapudi AD, Vadlapatla RK, and Mitra AK (2012). Sodium Dependent Multivitamin Transporter (SMVT): A Potential Target for Drug Delivery. Curr Drug Targets 13, 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Noordwijk AJ, and Dejong G (1986). Acquisition and Allocation of Resources: Their Influence on Variation in Life History Tactics. Am Nat 128, 137–142. [Google Scholar]
- Wang A, Luan HH, and Medzhitov R (2019). An evolutionary perspective on immunometabolism. Science 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, and Ghosh S (2011). Mitochondria in innate immune responses. Nature Reviews Immunology 11, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HR, and Hultmark D (2017). Drosophila muscles regulate the immune response against wasp infection via carbohydrate metabolism. Sci Rep-Uk 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Zhao Y, Yu J, Fan Z, Gong ST, Tang H, and Pan L (2019). Sugar Alcohols of Polyol Pathway Serve as Alarmins to Mediate Local-Systemic Innate Immune Communication in Drosophila. Cell Host Microbe 26, 240–251 e248. [DOI] [PubMed] [Google Scholar]
- Yoshii K, Hosomi K, Sawane K, and Kunisawa J (2019). Metabolism of Dietary and Microbial Vitamin B Family in the Regulation of Host Immunity. Front Nutr 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YV, Li Z, Rizzo NP, Einstein J, and Welte MA (2011). Targeting the motor regulator Klar to lipid droplets. BMC Cell Biol 12, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, and Karpac J (2017). Muscle Directs Diurnal Energy Homeostasis through a Myokine-Dependent Hormone Module in Drosophila. Current Biology 27, 1941-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, and Karpac J (2020). The Drosophila midgut and the systemic coordination of lipid-dependent energy homeostasis. Current Opinion in Insect Science 41, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, et al. (2016). NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 164, 896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinke I, Kirchner C, Chao LC, Tetzlaff MT, and Pankratz MJ (1999). Suppression of food intake and growth by amino acids in Drosophila: the role of pumpless, a fat body expressed gene with homology to vertebrate glycine cleavage system. Development 126, 5275–5284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data files for the RNA sequencing analysis have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE160652.