

Abstract
Background
Bone morphogenetic protein (BMP) is a phylogenetically conserved signaling pathway required for development that is aberrantly expressed in several age-related diseases including cancer, Alzheimer’s disease, obesity, and cardiovascular disease. Aberrant BMP signaling in mice leads to obesity, suggesting it may alter normal metabolism. The role of BMP signaling regulating cancer metabolism is not known.
Methods
To examine BMP regulation of metabolism, C. elegans harboring BMP gain-of-function (gof) and loss-of-function (lof) mutations were examined for changes in activity of catabolic and anabolic metabolism utilizing Western blot analysis and fluorescent reporters. AMP activated kinase (AMPK) gof and lof mutants were used to examine AMPK regulation of BMP signaling. H1299 (LKB1 wild-type), A549 (LKB1 lof), and A549-LKB1 (LKB1 restored) lung cancer cell lines were used to study BMP regulation of catabolic and anabolic metabolism. Studies were done using recombinant BMP ligands to activate BMP signaling, and BMP receptor specific inhibitors and siRNA to inhibit signaling.
Results
BMP signaling in both C. elegans and cancer cells is responsive to nutrient conditions. In both C. elegans and lung cancer cell lines BMP suppressed AMPK, the master regulator of catabolism, while activating PI3K, a regulator of anabolism. In lung cancer cells, inhibition of BMP signaling by siRNA or small molecules increased AMPK activity, and this increase was mediated by activation of LKB1. BMP2 ligand suppressed AMPK activation during starvation. BMP2 ligand decreased expression of TCA cycle intermediates and non-essential amino acids in H1299 cells. Furthermore, we show that BMP activation of PI3K is mediated through BMP type II receptor. We also observed feedback signaling, as AMPK suppressed BMP signaling, whereas PI3K increased BMP signaling.
Conclusion
These studies show that BMP signaling suppresses catabolic metabolism and stimulates anabolic metabolism. We identified feedback mechanisms where catabolic induced signaling mediated by AMPK negatively regulates BMP signaling, whereas anabolic signaling produces a positive feedback regulation of BMP signing through Akt. These mechanisms were conserved in both lung cancer cells and C. elegans. These studies suggest that aberrant BMP signaling causes dysregulation of metabolism that is a potential mechanism by which BMP promotes survival of cancer cells.
Keywords: BMP, AMPK, PIK3, Akt, mTOR, Lung cancer, Metabolism, LKB1, BMPR2
Background
Bone morphogenetic proteins (BMP) are phylogenetically conserved, orchestrating essential developmental processes from metazoans to mammals [1]. BMP signaling is increased in several age-related diseases including obesity [2], Alzheimer’s disease [3–6], cardiovascular disease [7] and cancer [8, 9]. Although a causative role for aberrant BMP activity in age-related diseases has been reported, a common mechanism has not been established.
There are over 20 BMP ligands that are divided into four distinct subtypes based on structure similarity and function [10]. BMP ligands signal through receptor serine/threonine kinases. BMP ligands bind to type 1 BMP receptors (BMPR1) (ALK2, ALK3, or ALK6) promoting phosphorylation by the constitutively active BMP type 2 receptors (BMPR2) (BMPR2, ActR-IIA, ActRIB) [11], which then phosphorylate Smad- 1/5, inducing transcriptional activation of downstream target genes. Transcriptional targets of BMPR1-Smad-1/5 include the inhibitor of differentiation proteins (ID1-ID4), which regulate survival, migration, proliferation, and self-renewal of stem cells and cancer cells [12, 13]. BMP2/4 and BMP7 also signal through BMPR2 without activating Smad-1/5 signaling. BMPR2 Smad-independent signaling includes an upregulation of anti-apoptotic proteins, X chromosome-linked inhibitor of apoptosis protein (XIAP) and transforming growth factor beta (TGFβ) activated kinase 1 (TAK1), which is also mediated in both embryonic development and survival of cancer cells [14–16].
Genetic and evolutionary analysis suggests that the increased expression of BMPR2 mRNA in adipose tissue of obese patients has been selected because it is a metabolic “thrifty” gene that is contributing to present day obesity [17]. Recent studies in mice showed that enhanced BMP activity causes obesity, hyperglycemia, and insulin resistance [18, 19]. The mechanism by which overactive BMP signaling contributes to obesity is not known. BMP ligands have been shown to activate PI3K during development as well as to induce cell migration [20–23]. PI3K promotes fat production by activating Akt, which promotes lipid and fatty acid synthesis [24]. Since BMP has had a significant developmental role for over 700 million years [25], and its overexpression promotes obesity in humans, we addressed whether BMP signaling has a broader role in cellular metabolism.
Utilizing lung cancer cells that have aberrant BMP signaling and C. elegans, we examined BMP signaling for a role in catabolic and anabolic metabolism. We find that in addition to promoting anabolic signaling through BMPR2 activation of PI3K, BMP signaling also suppresses AMP activated kinase (AMPK), the master regulator of catabolism. Furthermore, feed-back regulation by AMPK suppresses BMP signaling, whereas feed-forward regulation by Akt enhances it. BMP2 ligand suppressed expression of tricarboxylic acid cycle (TCA) intermediates and non-essential amino acids. These studies provide evidence that aberrant BMP signaling alters normal metabolic signaling events, which has implications as a potential contributing cause of the metabolic dysfunction in cancer and other age-related diseases.
Methods
General methods and strains
All C. elegans strains are derived from the Bristol strain N2 [26] and were grown at 20 °C on standard nematode growth media plates seeded with OP50 E. coli. Nematode cultures, genetic crosses, and other C. elegans husbandry were performed according to standard protocols [26].
Imaging
C. elegans at L4 stage were mounted on 2.5% agarose pads with 10 µM Levamisole. Fluorescence images were captured on a standard epifluorescent microscope. Quantification was carried out using the Fiji suite [27]. At least 30 animals per condition/genotype/treatment were used for quantification and statistics.
RNA isolation and qRT-PCR
Total RNA was extracted from C. elegans at 48 h after the L4 stage. C. elegans were synchronized by bleaching [28]. Total RNA was extracted by the freeze cracking method as previously described [33]. After RNA isolation, 2 µg of total RNA was primed with oligo(dT) and reverse transcribed to yield cDNA using the SuperScript III reverse transcriptase kit as per manufacturer’s protocol (Invitrogen). Real-time PCR was performed on QuantStudio3 (Applied Biosystems by Thermo Fisher Scientific) instrument using the PowerUP SYBR Green master mix (Applied Biosystems) according to manufacturer’s instructions. The experiments were performed in three technical replicates for each condition. Primer sequences for nhr-49 were forward primer TCCGAGTTCATTCTCGACG and reverse primer GGATGAATTGCCAATGGAGC [29]. Primer sequences for spp-9 were forward primer GTTCTCTTTCTGGTTGCGGT and reverse primer GCTCTACAAACATCTTCTGGTGCA. Primer sequences for act-1 were forward primer CCATCATGAAGTGCGACATTG and reverse primer CATGGTTGATGGGGCAAGAG [30]. QuantStudio Design and Analysis Software v1.5.1 was used to calculate raw Ct values and to normalize the values for dbl-1 and spp-9 to the housekeeping gene act-1 (Actin) (Applied Biosystems by Thermo Fisher Scientific). Fold-change in gene expression between experimental sample and the wild-type control was determined by this software using the formula: 2(−ΔΔCt). Experimental ΔCt values were compared with wild-type ΔCt values using the unpaired t test.
Western blotting and quantification
C. elegans were synchronized by alkaline bleaching and arrested at L1 stage on unseeded NGM plates overnight. Arrested L1s were transferred to seeded plates and grown on standard NGM plates until L4 stage at 20 °C. Fifty synchronized L4 stage nematodes for each strain were placed in 15 μl M9 buffer and 15 μl NextGel protein sample loading buffer (4x) (VWR, catalog # M260-5.0 ML) was added, flash frozen in liquid nitrogen and stored at − 80 °C until used for western blotting. Protein samples were boiled for 5 min with sample buffer (Bio-Rad), then centrifuged at 13,000 rpm for 1 min. Western blot analysis were performed as previously reported [8]. Briefly, proteins from cells were extracted using RIPA buffer, protein determined using BCA method, separated on polyacrylamide gel by SDS-PAGE and transferred to nitrocellulose. Primary antibodies were rabbit monoclonal anti-ID1 (Calbioreagents, San Mateo, CA), rabbit anti-Actin, an affinity isolated antigen specific antibody (Sigma, Saint Louis, MO), rabbit monoclonal anti-pAMPKα (T173), rabbit monoclonal AMPKα, rabbit monoclonal pLKB1 (Ser428), rabbit monoclonal LKB1, rabbit BMPR2, rabbit monoclonal pACC (Ser79), rabbit monoclonal pAkt (S473), rabbit monoclonal Akt, rabbit monoclonal p-p70S6 kinase (Thr389), rabbit monoclonal p70S6 (Cell Signaling). Mouse anti-actin, Clone C4 was for Western blot analysis in C. elegans studies (Sigma-Aldrich, MAB1501). Quantification of band intensity was measured using ImageJ Software (NIH) and statistical comparisons were made using one-way ANOVA.
Cell culture and reagents
A549 and H1299 cell lines obtained from patients with non-small cell lung cancer (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Sigma Aldrich, Saint Louis, MO, USA) with 5% fetal bovine serum. Normal Human Primary Bronchial/Tracheal Epithelial Cells were from ATCC (PCS-300-010) and cultured in Airway Epithelial Cell Basal Medium (PCS-300-030) supplemented with Bronchial Epithelial Cell Growth Kit (PCS-300-040). JL5 was synthesized by John Gilleran and Jacques Roberge from Rutgers Molecular Design and Synthesis, and DMH1 (S7146) was purchased from Selleckchem. AICAR (A9978) was purchased from Sigma-Aldrich.
Immunofluorescence imaging of lung cancer cells
Immunofluorescence imaging was performed as previously described [31]. Briefly, cells were plated onto cover slips and the next day treated for the designated time and washed. For starvation studies, cells were incubated overnight with regular media without fetal calf serum (FCS, R&D Systems # S11550) then incubated in DPBS with glucose (Gibco # 14,287,080) for 120 min (Fig. 1). In studies using human insulin (100 ng/ml, Sigma-Aldrich # I9278) treatment began 20 min after adding Dulbecco’s phosphate-buffered saline (DPBS) and continued for 60 min. MK-2206 (1 μM, Selleckchem # S1078) was give 1 h. prior to adding insulin. Cells were then stained with polyclonal rabbit anti-BMPR2 antibody (Sigma-Aldrich # HPA049014), which recognizes an extracellular epitope (Sigma-Aldrich). Cells were washed then incubated Alexa Flour 488 conjugated secondary antibody (Invitrogen # A-11008) for 1 h at room temperature. Cells were then washed with PBS and counterstained with DAPI (Sigma-Aldrich) for 10 min. Cells were then examined using 60X oil lens using a fluorescence microscope (Nikon eclipse TE300). Studies were performed at least 4 times.
Fig. 1.

Nutrient stress suppresses BMP signaling. A Fluorescence quantification of C. elegans expression BMP transcriptional reporter spp-9::GFP after 8 h of starvation (n = 30 animals). B Fluorescence quantification of C. elegans BMP transcriptional reporter spp-9::GFP treated with 50 and 4.5 µM Metformin and Phenformin, respectively (n = 30 animals). Drugs were included in media for 3 days post-egg laying. C Western blot analysis of starved A549 cells with quantification. D Immunofluorescence image and quantification for BMPR2 of starved A549 cells. FCS indicates fetal calf serum. DPBS indicates Dulbecco’s phosphate buffered saline
Treating lung cancer cells the BMP2
For studies examining response to BMP2 ligand (R&D Systems # 355-BM), lung cancer cells were incubated overnight with media without FCS. In the morning media was replaced with fresh media without FCS then treated with recombinant BMP2 (20 ng/ml) for designated times. In starved experiments, DPBS was added in the morning for 20 min then treated with BMP2 (20 ng/ml) or media with FCS for 20 min (Fig. 6C). In starved studies (Fig. 3G), following overnight incubation without FCS, cells were placed in DPBS cells and immediately treated with BMP2.
Fig. 6.

BMP activation of PI3k requires BMPR2. A, B Immunoblot of (A) A549 and (B) H1299 cells treated with BMP2 ligand with pAkt expression normalized to Akt, n = 2. C Immunoblot of A549 cell treated with BMP2 following starvation with DPBS or in regular media without FCS. D Immunoblot for BMPR2 expression of normal human bronchial epithelial cells (BEC) and lung cancer cell lines. E Immunoblot of BEC treated with BMP2 for 20 min. F, G Immunoblot of H1299 cells treated with BMP inhibitors JL5 2.5 μM or DMH1 2.5 μM for 24 h. H Fluorescence quantification of DAF-16::GFP positive nuclei in the indicated in insulin/DAF-2 daf-2 (1370) and BMPR2/DAF-4 daf-4 (e1364) at L4 stage grown at 20 °C
Fig. 3.

BMP suppresses LKB1 in lung cancer cells. A, B Western blot analysis of H1299 cells treated with JL5 or DMH1 demonstrating an increase in pACC and LKB1 at 3 h. Graph represents the mean pLKB1 expression normalized to Actin, n = 3. B Representative immunoblot. Mean percent increase in pLKB1/Actin ratio compared to control at 3 h following treatment with DMH1, n = 3. C Immunoblot of H1299 cells treated with DMH1 for 24 h. Mean percent increase in normalized pAMPK expression compared to control at 24 h following treatment with DMH1, n = 3 D Immunoblot of A549 cells treated with DMH1 for 3 and 24 h, (n = 3) demonstrating no significant change in pACC or pAMPK. The pLKB1, pAMPK, and Actin bands scanned were the 2.5 µM concentration except for one 1.25 µM peak value band obtained from H1299 cells treated with DMH1 for 3 h. E Western blot analysis of A549 cells stably expressing LKB1 and A549 control transfected cells treated with JL5 for 24 h. Both Actin and spectrin were used as loading controls. Graph represents mean pACC/spectin ratio, n = 2. F Immunoblot of H1299 cells following siRNA knockdown of BMPR2. Graph represents the mean of 4 independent studies. G Western blot analysis of starved H1299 cells treated with and without BMP2 ligand. Graph represent the mean change in pLKB1/pLKB1 ratio compared to control over time, n = 3
Lung cancer cells treated with insulin were incubated with media overnight without FCS, starved with DPBS for 20 min then treated with insulin (100 ng/ml) for 60 min (Fig. 6A, B). Cell treated with the Akt inhibitor MK-2206. MK-2206 were pre-treated 1 h. prior to adding insulin. Starvation studies using ligands were performed 3 times.
Transient knockdown
Validated siRNAs (Life Technologies, ID: s2044) were used for BMPR2 knockdown. Negative control was Silencer Select negative control siRNA (4,390,843). H1299 lung cancer cells were transfected with siRNA using Lipofectamine® RNAiMAX Reagent (Invitrogen, Carlsbad, CA, USA). 750,000 cells were plated in a 6 well plate and were grown overnight. Next morning the cells were transfected with 6 nM BMPR2 or 6 nM of siRNA control for 48 h. according to manufacturer’s protocol. Knockdown experiments were performed 4 times.
Metabolomic analysis by LC–MS
H1299 and A549 cells were incubated overnight in media without FCS. Cells were then treated with DMSO or BMP2 20 ng/ml for 40 min in triplicate. Cells were harvested with a 40:40:20 buffer of methano: acetonitrile: water and 0.5% formic acid. LC − MS analysis of the cellular metabolites was performed on the Q Exactive PLUS hybrid quadrupole-orbitrap mass spectrometer (Thermo Scientific) coupled to hydrophilic interaction chromatography (HILIC) as previously reported [32, 33]. The LC separation was performed on UltiMate 3000 UHPLC system with an XBridge BEH Amide column (150 mm × 2.1 mm, 2.5 μM particle size, Waters, Milford, MA) with the corresponding XP VanGuard Cartridge. The liquid chromatography used a gradient of solvent A (95%:5% H2O: acetonitrile with 20 mM ammonium acetate, 20 mM ammonium hydroxide, pH 9.4), and solvent B (20%:80% H2O: acetonitrile with 20 mM ammonium acetate, 20 mM ammonium hydroxide, pH 9.4). The gradient was 0 min, 100% B; 3 min, 100% B; 3.2 min, 90% B; 6.2 min, 90% B; 6.5 min, 80% B; 10.5 min, 80% B; 10.7 min, 70% B; 13.5 min, 70% B; 13.7 min, 45% B; 16 min, 45% B; 16.5 min, 100% B. The flow rate was 300 μl/min. Injection volume was 5 μL and column temperature 25 °C. The MS scans were in negative ion mode with a resolution of 70,000 at m/z 200. The automatic gain control (AGC) target was 3 × 106 and the scan range was 75 − 1000. Metabolite features were extracted in MAVEN [34].
Statistical analysis
In lung cancer studies, paired student t-test assuming unequal variances was used to compare means. The mean of control was compared with the mean of each treated group. Differences with p values < 0.05 were considered statistically significant. The following signified *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Results
Nutrient stress suppresses BMP signaling
To identify whether nutrient stress regulates BMP signaling, we subjected C. elegans to two different nutrient stress paradigms and monitored the expression of a BMP-regulated gene, spp-9 [35]. This gene is negatively regulated by BMP signaling, and thus an increase in the GFP signal of a spp-9p::GFP transcriptional reporter would indicate suppression of BMP signaling [35–37]. We find that starvation of C. elegans for 8 h leads to a decrease in BMP signaling (increase of SPP-9::GFP, Fig. 1A). Metformin and phenformin have been shown to induce a dietary restriction-like physiology in C. elegans [38], and we tested the effect of these treatments on BMP signaling. We find that both metformin- and phenformin-dependent nutrient stresses decrease BMP signaling (Fig. 1B). Importantly, this regulation of BMP signaling by nutrient deprivation is conserved in humans. A549 lung cancer cells exhibit a marked decrease in expression of ID1, a transcriptional target of BMP signaling, whereas ID1 levels are restored on addition of nutrients post-starvation (Fig. 1C).
Work in C. elegans has previously identified receptor trafficking as a mechanism by which BMP signaling might be regulated. Recycling of DAF-4, the nematode BMPR2, from the plasma membrane back to the surface after internalization is regulated via the recycling endosome, and BMPR2/DAF-4 levels are controlled by degradation within the lysosome [39]. We find that reduced nutrients by removal of fetal calf serum (FCS) decreased BMPR2 levels at the plasma membrane, and that this decrease was exacerbated by complete nutrient starvation (Fig. 1D).
Taken together, these studies demonstrate the conserved regulation of BMP signaling by nutrient stresses.
BMP signaling suppresses the energy sensor and regulator AMPK in C. elegans
AMP Associated Kinase (AMPK) is a conserved, central regulator of nutrient sensing and energy homeostasis in many organisms. In C. elegans, AMPK, encoded by the aak-2 gene, has a critical role in regulating the physiological response to metformin and phenformin, as well as to other dietary restriction paradigms [38, 40]. Upon nutrient stress, AMPK is activated by phosphorylation at Thr172 (humans) [41] and Thr243 (C. elegans) [42]. Phosphorylated AMPK then stimulates downstream signaling pathways that regulate organismal energy homeostasis.
Given that nutrient stress suppresses BMP signaling, we asked whether BMP signaling mutants themselves experienced nutrient stress and whether aak-2 was activated in these animals. Western blot analysis identified that AMPK phosphorylation is enhanced in various BMP loss-of-function (lof) mutants of BMPR2 daf4(e1364), BMPR1 (SMA-6), and Smad transcription factor (SMA-2) compared to wild-type animals, even in well-fed conditions (Fig. 2A). AMPK lof mutants aak-2 (ok524) were utilized as a control. If AMPK were activated, we would expect that downstream targets of AMPK would also be upregulated in BMP mutants. One such target is the nuclear hormone receptor-49 (nhr-49). We show that a lof BMP mutant SMA-6 exhibits increased expression of an nhr-49p::nhr-49::GFP transgene, whereas gain-of-function (gof) BMP mutants lon-1(e185) and lon2 (e678) exhibit decreased expression of this AMPK target (Fig. 2B). Taken together, these studies show that in C. elegans, BMP signaling inhibits AMPK activation during periods of ‘well-fed’ conditions, and that loss of signaling activates AMPK even with ample nutrition.
Fig. 2.

BMP signaling suppresses AMPK and AMPK negative feedback regulation of BMP in C. elegans. A Western blot analysis for pAMPK of C. elegans with BMP loss-of function (lof) mutants of BMPR2 daf4(e1364) and Smad transcription factors (sma-6 and sma-2) compared to wild-type worms. AMPK lof mutants aak-2 (ok524) were utilized as a control. Nematodes were grown at 20 °C until L4 stage. Each lane represents 25 animals. B Fluorescence images and quantification of C. elegans expressing the AMPK downstream reporter nhr-49p::nhr-49::GFP with genotypes harboring BMP lof (SMA-6) and gof lon-1( e185),lon-2 ( e678) (n = 30 animals). Images taken at L4 stage animals under well-fed conditions at 20 °C. Scale bar indicates 50 microns. C Fluorescence quantification of C. elegans expression of the BMP transcriptional reporter spp-9::GFP in AMPK wild-type and AMPK lof mutant aak-2 (ok524) in either well-fed animals or after 8 h of starvation (n = 30 animals). D Body-length measurements of indicated genotypes at L4 stage grown at 20 °C (n = 30 animals). E Relative quantification of mRNA levels of indicated genes in wild-type and AMPK gof mutant aak-2 animals at L4 stage grown at 20 °C. All alleles are loss of function except for aak-2(gof), which is a transgene that expresses a truncated, constitutively active kinase
AMPK negative feedback regulation of BMP signaling
We next examined whether feedback between AMPK and BMP signaling exists in C. elegans. The lof AMPK mutation aak-2(ok542) was crossed to BMP transcriptional reporter spp-9p::GFP to determine how BMP signaling is regulated when AMPK is absent. In fed conditions, we find that BMP signaling is suppressed in AMPK lof mutants, suggesting that AMPK promotes BMP signaling during nutrient-stable conditions (Fig. 2C). Importantly, we find that AMPK is required for the suppression of BMP signaling as aak-2(ok542) animals fail to increase spp-9::GFP expression under nutrient-deprived conditions when animals are starved for 8 h (Fig. 2C). Taken together, these results point a feedback regulation whereby BMP and AMPK signaling regulate each other to maintain energy homeostasis during periods of nutrient stress.
C. elegans BMP signaling governs growth and body-size [43]; BMP lof mutants have smaller body lengths compared with wild-type, whereas gof mutants exhibit longer body lengths. Our above results suggests that activation of AMPK inhibits BMP signaling; we wondered whether the ‘small’ body might be a result of activated AMPK in lof BMP mutants and vice versa in gof BMP mutants. We utilized an aak-2(gof) (AMPK) transgenic strain in which a truncated version of the AAK-2 protein has a gain-of-function phenotype [44]. We find that these aak-2(gof) mutants are smaller in body-size compared to wild-type animals (Fig. 2D). However, they fail to suppress the ‘long’ body phenotype of lon-2(e678) gof mutants. These results suggest that the regulation of body-size and energy homeostasis by BMP-AMPK cross-talk is uncoupled; other BMP-regulated genes are likely responsible for the body size regulation.
Finally, using a qRT-PCR approach, we show that AMPK inhibition of BMP signaling is through post-transcriptional mechanisms (Fig. 2E). In aak-2(gof) animals, nhr-49 mRNA levels are upregulated as would be expected for a downstream target of AMPK activation. However, Type 1 BMP receptor (sma-6) and Type 2 BMP receptor (daf-4) mRNA levels are unchanged compared to wild-type, but spp-9 mRNA levels are increased, showing that AMPK regulation of BMP signaling is post-transcriptional.
Taken together, these data show that nutrient-dependent energy homeostasis is regulated by BMP-AMPK cross-talk.
BMP suppresses LKB1 in lung cancer cells
We examined whether BMP regulates AMPK/LKB1 signaling in lung cancer cells, which have aberrant expression of BMP signaling [45, 46]. Full activation of AMPK requires phosphorylation at Thr173, which in most cells is mediated by LKB1 [47]. H1299 cells treated the BMP receptor inhibitor JL5 [8], demonstrated an increased phosphorylation of the AMPK downstream target acetyl-CoA carboxylase (ACC) Ser79 within 3 h (Fig. 3A). LKB1 demonstrated an increase in phosphorylation at its Ser428 activation site at 3 h following treatment with JL5 without a change in phosphorylation of AMPK (Fig. 3A). This suggests that LKB1 activation occurs prior to that of AMPK following inhibition of BMP signaling. H1299 cells treated with the BMP receptor inhibitor DMH1 [48] also showed an increase in pACC Ser79 and pLKB1 Ser428 levels after 3 h but not in pAMPK levels (Fig. 3B). An increase in pAMPK at Thr173 was seen 24 h after treatment with DMH1 (Fig. 3C). A549 cells, which do not express LKB1, did not demonstrate an increase in expression in either pACC (Ser79) or pAMPK (Thr172) after 3 and 24 h of treatment with DMH1 (Fig. 3D). In A549-LKB1 cells, which are A549 cells with restored LKB1 from a stably expressed transgene, JL5 did not show an increase phosphorylation of AMPK at this time point but did enhance phosphorylation of ACC (Ser79) (Fig. 3E). These pharmacological studies suggest that BMP signaling suppresses AMPK signaling through the regulation of LKB1 in lung cancer cells.
BMPR2 siRNA was used to further evaluate the role of BMP signaling suppressing LKB1. BMPR2 siRNA knockdown in H1299 cells did not change the levels of pAMPK (Thr172) but did significantly increase pLKB1(Ser428) levels (Fig. 3F). BMPR2 knockdown also increased the phosphorylation of ACC (Ser79) (Fig. 3F). Moreover, in starved H1299 cells, BMP2 ligand significantly decreased pLKB1 (Ser428) and pACC (Ser79) levels over time while it increased in controls (Fig. 3G). These molecular studies complement our studies using BMPR2 inhibitors, further supporting the hypothesis that BMP signaling suppresses AMPK signaling.
BMP signaling decreases TCA intermediates and non-essential amino acids in H1299 cells
Next, we examined whether BMP signaling regulated metabolism in lung cancer cells by examining changes in tricarboxylic acid cycle (TCA) intermediates and amino acids following treatment with BMP2 ligand. We examined TCA cycle intermediates and amino acids because suppression of AMPK would decrease TCA cycle activity [49], which would negatively effect production of essential amino acids [50, 51]. BMP2 treated H1299 but not A549 cells had significantly lower TCA intermediates compared to controls (Fig. 4A). BMP2 treated H1299 cells but not A549 cells also had significantly lower AMP levels (Fig. 4B). Although ADP and ATP levels trended lower in H1299 cells treated with BMP2, these differences did not reach statistical significance (Fig. 4B). Several non-essential amino acids derived from TCA cycle intermediates were also significantly decreased in BMP2 treated H1299 cells but not in A549 cells (Fig. C, D). These studies suggest that BMP2 has a suppressive effect on metabolic intermediates of the TCA cycle.
Fig. 4.

BMP2 ligand decreases expression of TCA cycle intermediates and non-essential amino acids. A549 and H1299 cells were treated with BMP2 ligand for 40 min and metabolomic analysis performed by LC-MC. Data represents mean of 3 experiments for A TCA cycle intermediates, B ATP intermediates, and C, D amino acids
AMPK suppresses BMP signaling in lung cancer cells
Our prior studies showed that JL5 induces the mislocalization of BMPR2 from the plasma membrane to the cytosol [31, 52]. Since JL5 activates AMPK, we examined if AMPK affected the localization and expression of BMPR2. To activate AMPK, we used the allosteric activator AICAR. In both H1299 and A549 cells, AICAR caused a decrease protein expression of the long and short form of BMPR2 (Fig. 5A, B). Immunofluorescence imaging demonstrated in both H1299 and A549 that AICAR decreases the expression of BMPR2 at the plasma membrane (Fig. 5C). A549 cells express more ID1 then H1299 (Fig. 5D). A549 cells with LKB1 restored (A549-LKB1) have lower expression of ID1 (Fig. 5D) and BMPR2 (Fig. 5E). These studies suggest that AMPK decreases BMP signaling in lung cancer cells by downregulating BMPR2.
Fig. 5.

AMPK suppress BMP signaling in lung cancer cells. A, B Immunoblot showing long form (LF) and short form (SF) of lung cancer cells in regular media treated with increasing doses of the AMPK inhibitor AICAR for 24 h. AMPK expression was normalized to Actin. C Immunofluorescence imaging for BMPR2 at the cell surface of lung cancer cells treated with AICAR (500 μM) for 24 h. Graph of mean fluorescence intensity, n = 4. D, E Representative immunoblot of ID1 and BMPR2 in lung cancer cells with and without LKB1 expression. D, E quantification of normalized ID1 and BMPR2 expression, n = 3
Taken together, these results show the conserved crosstalk between BMP and AMPK across phyla.
BMP activation of PI3K requires BMPR2
Our prior studies showed that BMP2 ligand activated mTORC1 (p-p70S6) signaling in lung cancer cells through phosphatidylinositol-3-kinase (PI3K) [22]. Whether BMP2 ligand signals through BMP type 1 or type 2 receptors in lung cancer cells has not been elucidated. In A549 cells, BMP2 activates both p-p70S6 and pAkt S473 (Fig. 6A). In the H1299 cells, BMP2 activated p70S6 but not Akt (Fig. 6B). During starvation there is a significant decrease in the expression of BMPR2 at cell surface (Fig. 1D). When A549 cells were starved for 20 min they were no longer responsive to BMP2 activation of Akt (Fig. 6C). By Western blot analysis, lung cancer cell lines express BMPR2 (Fig. 6D). BMPR2 expression was not detected in normal bronchial epithelial cells (BEC) (Fig. 6D). BMP2 did not active either p70S6 or Akt in BEC cells (Fig. 5E). BMP inhibitors of the type 1 receptors, JL5 and DMH1, did not inhibit either Akt or p70S6 in H1299 cells (Fig. 6F, G), suggesting the regulation is specific to type 2 receptors. These data suggest that BMPR2 is required for PI3K activation in lung cancer cells.
To assess if BMPR2 signaling through PI3K is conserved, we tested FoxO/daf-16 nuclear localization in C. elegans in BMPR2/daf-4 lof mutants. Insulin-like proteins signal through DAF-2 receptor activating PI3K/Akt signaling. The DAF-2/PI3K/Akt signaling pathway phosphorylates FoxO/DAF-16, inhibiting its entry into the nucleus thereby decreasing its activity [53]. As expected, wild-type animals show little-to-no DAF-16::GFP nuclear localization, whereas daf-2(e1370) lof mutants exhibit strong DAF-16::GFP nuclear localization (Fig. 6H). Interestingly, loss of BMPR2/daf-4 significantly increases DAF-16::GFP nuclear localization compared to wild type, although this activation is not as strong as loss of insulin signaling (Fig. 6H). These studies suggest that BMPR2 regulation of PI3K is conserved in C. elegans.
Insulin increase BMPR2 expression in lung cancer cells during starvation
Prior studies have shown that BMP7 enhances insulin signaling through PI3K, suggesting an interaction between BMPR2 and insulin signaling. We examined if insulin regulated BMPR2 in lung cancer cells. Unlike BMP2, insulin activated Akt in lung cancer cells during starvation (Fig. 7A). Insulin prevented the decrease in BMPR2 protein expression during starvation (Fig. 7A). To assess if there is feedback regulation from Akt, we used the specific Akt inhibitor MK-2206. In nutrient conditions, MK-2206 caused a decrease in protein expression of BMPR2 in H1299 cells but not A549 cells (Fig. 7B). In starved A549 cells, MK-2206 attenuated insulin’s increase in protein expression of BMPR2 and its activation of pAkt (Fig. 7C). Immunofluorescence imaging showed that insulin prevented the decreased expression of BMPR2 at plasma membrane during starvation in both H1299 and A549 cells (Fig. 7D, E). The inhibition of Akt with MK-2206 attenuated insulin’s increase in BMPR2 expression at the cell surface during starvation (Fig. 7D, E). These studies suggest that activated Akt causes a positive feedback regulation of BMPR2.
Fig. 7.

Akt increases BMPR2 expression during starvation. A Immunoblot of starved A549 cells or starved then treated with insulin for 60 min. BMPR2 expression was normalized to Actin, n = 2. B Immunoblot of lung cancer cells in regular media with FCS treated with Akt inhibitor MK-2206 for 24 h. C Immunoblot of starved A549 cells or starved then treated with insulin for 60 min with and without the Akt inhibitor MK-2206. D Immunofluorescence imaging for BMPR2 at the cell surface of starved cancer cells treated with insulin for 60 min with and without MK-2206. E Mean fluorescence of 4 experiments of starved cells treated with insulin for 60 min with and without MK-2206
Fig. 8 demonstrates the integration of BMP with anabolic and catabolic signaling.
Fig. 8.
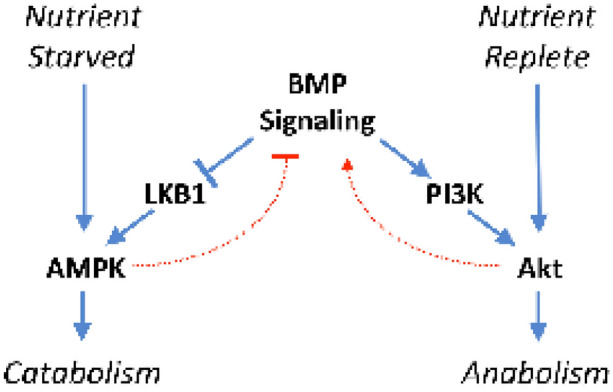
Schematic demonstrating integration of BMP signaling with anabolic and catabolic metabolism. Arrows indicate positive signaling interactions, whereas T-bars indicate negative signaling interactions. Solid blue arrows and T-bars indicate the primary effects of nutrients, nutrient starvation, and BMP signaling on metabolic signaling. BMP signaling promotes PI3K/Akt signaling and anabolism in response to abundant nutrients. By contrast, BMP signaling reduces AMPK signaling and catabolism in response to nutrient starvation. Dotted red arrows and T-bars indicate regulatory feedback by AMPK signaling and feedforward by PI3K/Akt signaling on BMP signaling
Discussion
BMP ligand and receptor expression is increased in cancer and several other age-related diseases. Normally there is little signaling BMP in the mature lung [54]; however, BMP signaling is reactivated with inflammation and cancer [45, 54]. Although there is little to no BMP2 expression in normal lung tissue or benign tumors, BMP2 is highly expressed in 98% of non-small lung carcinoma (NSLC) [46]. During normal aging, BMP ligand expression increases tenfold in the dentate gyrus [3, 55]. BMP ligand expression is increased further in patients with Alzheimer’s disease [4]. The expression of BMP ligands and receptors are also increased in adipocytes in obese humans and mice [2, 17].
Aberrant BMP signaling is pathological. Studies in cancer show that BMP signaling enhances cell survival, migration, cancer stem cell self-renewal, and metastasis, and increased BMP expression is associated with a worse survival [8, 45, 56–60]. BMP induces the differentiation of neural stem cells into astrocytes and inhibits neuronal differentiation. Decreasing BMP signaling with the BMP inhibitor noggin or with exercise in mice promotes neurogenesis and improves cognition in mice [6, 61]. Adipose specific knockout of noggin enhances BMP activity, causing significant increase in white adipose tissue, fatty liver, and glucose intolerance in mice fed a high-fat diet [18]. BMP over-activity has also been shown to cause anemia of chronic disease [62] and heterotopic bone formation from a gain-of-function mutation in alk2 [63].
AMPK is a master regulator of catabolism, and like BMP, is phylogenetically conserved in metazoans [64]. AMPK catabolic responses are essential to maintain nutrient and energy availability. AMPK decreases anabolic signaling to conserve energy utilization, and activated AMPK also increases ATP production by stimulating mitochondrial biogenesis. The ability to activate AMPK has been shown to improved longevity across multiple species [65], while its suppression has been implicated in disease including diabetes, obesity, increased tumorigenicity, and a decrease in neurogenesis and synaptic activation [47, 66]. The mechanisms by which AMPK signaling is suppressed in disease are poorly understood.
We show in both C. elegans and lung cancer cells that BMP suppresses AMPK. Suppressing BMP signaling in lung cancer cells and C. elegans led to the activation of AMPK. These data demonstrate that pharmaceutical targeting of BMP receptors can induce AMPK activation. Inhibition of either BMP type 1 or BMP type 2 receptors led to the activation of AMPK. This suggests that inhibition of BMP type 1 receptors is required, but AMPK activation can be achieved by inhibiting BMPR2. Further studies are needed to determine whether it is best to target BMP type 1 or type 2 to promote activation of AMPK. Our studies suggest that BMP signaling inhibits AMPK by inhibiting LKB1. The regulation might be reciprocal, as prior studies have shown that LKB1 binds to BMP type 1 receptors, inhibiting their signaling by inducing degradation [67]. LKB1 has also been shown to phosphorylate Smad-4 on Thr77 inhibiting its binding to transcriptional targets of BMP and TGFβ [68]. Further studies are needed to determine the mechanism by which the interactions of BMP receptors and/or Smad-1/5 inhibit LKB1.
AMPK induced negative feedback regulation of BMP signaling in both lung cancer cells and C. elegans. Activated AMPK suppressed BMPR2 in both H1299 and A549 cells. Since A549 cells that do not express LKB1, it suggests that AMPK and not LKB1 mediates the downregulation of BMPR2. Although LKB1 expression may not be required to suppress BMPR2, it is required for full activation of AMPK. Therefore lung tumors with LKB1 lof would have less AMPK activity and less suppression of BMP signaling. LKB1 is mutated in 20–30% of NSCLC and is associated with a more aggressive phenotype. LKB1 mutated tumors having a less suppressive effect on BMP signaling in cancer is suggested by a prior report demonstrating that NSCLC with LKB1 lof had higher expression of BMP2 [67].
Prior studies have suggested that BMP signaling activates PI3K through BMPR2 independent of Smad activation. In mesenchymal progenitors, BMP2 mediates chemotaxis by inducing binding of BMPR2 to PI3K regulatory p55γ subunit [21]. Our studies in lung cancer cells and C. elegans are consistent with BMP signaling activating PI3K and downstream anabolic signaling pathways Akt and mTOR by BMPR2 Smad-independent mechanisms. BMP induction of anabolic signaling pathways was responsive to nutrient conditions. During starvation BMPR2 expression is decreased and lung cancer cells were no longer responsive to BMP2 ligand. Furthermore, Akt-mediated insulin signaling prevented the decrease in BMPR2 expression during starvation. Since a number of tyrosine kinases and oncogenic mutations, including K-ras, mediate cell growth through PI3K/Akt signaling, multiple oncogenes may utilize Akt to enhance BMPR2 signaling.
Conclusion
We demonstrate that BMP signaling is integrated with the master regulators of anabolic and catabolic metabolism in cancer, which is conserved in C. elegans. Aberrant BMP signaling in lung cancer is growth promoting and has been shown to be pathological in other age-related diseases. These studies suggest the aberrant BMP signaling causes metabolic dysfunction, which has implications in causing pathological responses in cancer and potentially other age-related diseases.
Acknowledgements
We would like to thank Vrushank Bhatt and Jessie Yanxiang Guo from the Rutgers Cancer Institute of New Jersey for their assistance with the metabolomics studies
Abbreviations
- BMP
Bone morphogenetic protein
- BMPR2
Bone morphogenetic protein receptor 2
- AIF
Apoptosis inducing factor
- XIAP
X-linked inhibitor of apoptosis protein
- TGFβ
Transforming growth factor beta
- TAK1
Activated kinase 1
- NSCLC
Non-small cell lung carcinomas
- AMPK
AMP activated kinase
- LKB1
Liver kinase B1
- lof
Loss-of-function
- gof
Gain-of-function
- DPBS
Dulbecco’s phosphate-buffered saline
- nhr-49
Nuclear hormone receptor-49
- ACC
Acetyl-CoA carboxylase
- kD
Kilodalton
- PI3K
Phosphatidylinositol-3-kinase
Author contributions
MV contributed to writing of the manuscript, created strains of C elegans, designed and performed C. elegans experiments; PG created FoxO/daf-4 strain of C elegans; AM did immunofluoresent imaging studies, Western blot analysis, and edited the manuscript, tabulating data and preparing figures; DJ and ME treated cells and performed Western blots; CR assisted with the design and interpretation of C elegans studies; DG and JR synthesized JL5 used for these studies; JL designed experiments, interpreted data, and wrote the body of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from National Institute of Health (NIH) R01 CA225830 and 3R01CA225830-01S1.
Availability of data and materials
The datasets obtained and analyzed for this study will be made available from the corresponding author in a reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Feiner N, Motone F, Meyer A, Kuraku S. Asymmetric paralog evolution between the "cryptic" gene Bmp16 and its well-studied sister genes Bmp2 and Bmp4. Sci Rep. 2019;9:3136. doi: 10.1038/s41598-019-40055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blázquez-Medela AM, Jumabay M, Boström KI. Beyond the bone: bone morphogenetic protein signaling in adipose tissue. Obes Rev. 2019;20:648–658. doi: 10.1111/obr.12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yousef H, Morgenthaler A, Schlesinger C, Bugaj L, Conboy IM, Schaffer DV. Age-associated increase in BMP signaling inhibits hippocampal neurogenesis. Stem Cells. 2015;33:1577–1588. doi: 10.1002/stem.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crews L, Adame A, Patrick C, Delaney A, Pham E, Rockenstein E, Hansen L, Masliah E. Increased BMP6 levels in the brains of Alzheimer’s disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J Neurosci. 2010;30:12252–12262. doi: 10.1523/JNEUROSCI.1305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen HL, Lein PJ, Wang JY, Gash D, Hoffer BJ, Chiang YH. Expression of bone morphogenetic proteins in the brain during normal aging and in 6-hydroxydopamine-lesioned animals. Brain Res. 2003;994:81–90. doi: 10.1016/j.brainres.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 6.Diaz-Moreno M, Armenteros T, Gradari S, Hortiguela R, Garcia-Corzo L, Fontan-Lozano A, Trejo JL, Mira H. Noggin rescues age-related stem cell loss in the brain of senescent mice with neurodegenerative pathology. Proc Natl Acad Sci USA. 2018;115:11625–11630. doi: 10.1073/pnas.1813205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derwall M, Malhotra R, Lai CS, Beppu Y, Aikawa E, Seehra JS, Zapol WM, Bloch KD, Yu PB. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:613–622. doi: 10.1161/ATVBAHA.111.242594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newman JH, Augeri DJ, NeMoyer R, Malhotra J, Langenfeld E, Chesson CB, Dobias NS, Lee MJ, Tarabichi S, Jhawar SR, et al. Novel bone morphogenetic protein receptor inhibitor JL5 suppresses tumor cell survival signaling and induces regression of human lung cancer. Oncogene. 2018;37:3672–3685. doi: 10.1038/s41388-018-0156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiramongkolchai P, Owens P, Hong CC. Emerging roles of the bone morphogenetic protein pathway in cancer: potential therapeutic target for kinase inhibition. Biochem Soc Trans. 2016;44:1117–1134. doi: 10.1042/BST20160069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rahman MS, Akhtar N, Jamil HM, Banik RS, Asaduzzaman SM. TGF-β/BMP signaling and other molecular events: regulation of osteoblastogenesis and bone formation. Bone Res. 2015;3:15005. doi: 10.1038/boneres.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nickel J, Sebald W, Groppe JC, Mueller TD. Intricacies of BMP receptor assembly. Cytokine Growth Factor Rev. 2009;20:367–377. doi: 10.1016/j.cytogfr.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 12.Katagiri T, Imada M, Yanai T, Suda T, Takahashi N, Kamijo R. Identification of a BMP-responsive element in Id1, the gene for inhibition of myogenesis. Genes Cells. 2002;7:949–960. doi: 10.1046/j.1365-2443.2002.00573.x. [DOI] [PubMed] [Google Scholar]
- 13.Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem. 2002;277:4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- 14.Augeri DJ, Langenfeld E, Castle M, Gilleran JA, Langenfeld J. Inhibition of BMP and of TGFbeta receptors downregulates expression of XIAP and TAK1 leading to lung cancer cell death. Mol Cancer. 2016;15:27. doi: 10.1186/s12943-016-0511-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z, Shen J, Pu K, Katus HA, Ploger F, Tiefenbacher CP, Chen X, Braun T. GDF5 and BMP2 inhibit apoptosis via activation of BMPR2 and subsequent stabilization of XIAP. Biochim Biophys Acta. 2009;1793:1819–1827. doi: 10.1016/j.bbamcr.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi K, Nagai S, Ninomiya-Tsuji J, Nishita M, Tamai K, Irie K, Ueno N, Nishida E, Shibuya H, Matsumoto K. XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1-TAK1 in the BMP signaling pathway. Embo J. 1999;18:179–187. doi: 10.1093/emboj/18.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schleinitz D, Klöting N, Böttcher Y, Wolf S, Dietrich K, Tönjes A, Breitfeld J, Enigk B, Halbritter J, Körner A, et al. Genetic and evolutionary analyses of the human bone morphogenetic protein receptor 2 (BMPR2) in the pathophysiology of obesity. PLoS ONE. 2011;6:e16155. doi: 10.1371/journal.pone.0016155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blázquez-Medela AM, Jumabay M, Rajbhandari P, Sallam T, Guo Y, Yao J, Vergnes L, Reue K, Zhang L, Yao Y, et al. Noggin depletion in adipocytes promotes obesity in mice. Mol Metab. 2019;25:50–63. doi: 10.1016/j.molmet.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Zhao C, Xu Y, Huang K, Wang Y, Wang X, Zhou X, Pang W, Yang G, Yu T. Adipose-specific BMP and activin membrane-bound inhibitor (BAMBI) deletion promotes adipogenesis by accelerating ROS production. J Biol Chem. 2021;296:100037. doi: 10.1074/jbc.RA120.014793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perron JC, Dodd J. Structural distinctions in BMPs underlie divergent signaling in spinal neurons. Neural Dev. 2012;7:16. doi: 10.1186/1749-8104-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiepen C, Benn A, Denkis A, Lukonin I, Weise C, Boergermann JH, Knaus P. BMP2-induced chemotaxis requires PI3K p55γ/p110α-dependent phosphatidylinositol (3,4,5)-triphosphate production and LL5β recruitment at the cytocortex. BMC Biol. 2014;12:43. doi: 10.1186/1741-7007-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langenfeld EM, Kong Y, Langenfeld J. Bone morphogenetic protein-2-induced transformation involves the activation of mammalian target of rapamycin. Mol Cancer Res. 2005;3:679–684. doi: 10.1158/1541-7786.MCR-05-0124. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Liao J, Lu Y, Duan X, Sun W. Activation of the PI3K/Akt pathway mediates bone morphogenetic protein 2-induced invasion of pancreatic cancer cells Panc-1. Pathol Oncol Res. 2011;17:257–261. doi: 10.1007/s12253-010-9307-1. [DOI] [PubMed] [Google Scholar]
- 24.Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14:1483–1496. doi: 10.7150/ijbs.27173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pang K, Ryan JF, Baxevanis AD, Martindale MQ. Evolution of the TGF-β signaling pathway and its potential role in the ctenophore Mnemiopsis leidyi. PLoS ONE. 2011;6:e24152. doi: 10.1371/journal.pone.0024152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stiernagle T. Maintenance of C. elegans. WormBook. 2006 doi: 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goh GYS, Winter JJ, Bhanshali F, Doering KRS, Lai R, Lee K, Veal EA, Taubert S. NHR-49/HNF4 integrates regulation of fatty acid metabolism with a protective transcriptional response to oxidative stress and fasting. Aging Cell. 2018;17:e12743. doi: 10.1111/acel.12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zugasti O, Ewbank JJ. Neuroimmune regulation of antimicrobial peptide expression by a noncanonical TGF-beta signaling pathway in Caenorhabditis elegans epidermis. Nat Immunol. 2009;10:249–256. doi: 10.1038/ni.1700. [DOI] [PubMed] [Google Scholar]
- 31.Mondal A, NeMoyer R, Vora V, Napoli L, Syed Z, Langenfeld E, Jia D, Peng Y, Gilleran J, Roberge J, Rongo C, Jabbour S, Langenfeld., J, Bone morphogenetic protein receptor 2 inhibition destabilizes microtubules promoting the activation of lysosome and cell death of lung cancer cells. Cell Commun Signal. 2021 doi: 10.1186/s12964-021-00743-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhatt V, Khayati K, Hu ZS, Lee A, Kamran W, Su X, Guo JY. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev. 2019;33:150–165. doi: 10.1101/gad.320481.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melamud E, Vastag L, Rabinowitz JD. Metabolomic analysis and visualization engine for LC-MS data. Anal Chem. 2010;82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts AF, Gumienny TL, Gleason RJ, Wang H, Padgett RW. Regulation of genes affecting body size and innate immunity by the DBL-1/BMP-like pathway in Caenorhabditis elegans. BMC Dev Biol. 2010;10:61. doi: 10.1186/1471-213X-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lakdawala MF, Madhu B, Faure L, Vora M, Padgett RW, Gumienny TL. Genetic interactions between the DBL-1/BMP-like pathway and dpy body size-associated genes in Caenorhabditis elegans. Mol Biol Cell. 2019;30:3151–3160. doi: 10.1091/mbc.E19-09-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madhu B, Lakdawala MF, Issac NG, Gumienny TL. Caenorhabditis elegans saposin-like spp-9 is involved in specific innate immune responses. Genes Immun. 2020;21:301–310. doi: 10.1038/s41435-020-0108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS ONE. 2010;5:e8758. doi: 10.1371/journal.pone.0008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gleason RJ, Akintobi AM, Grant BD, Padgett RW. BMP signaling requires retromer-dependent recycling of the type I receptor. Proc Natl Acad Sci USA. 2014;111:2578–2583. doi: 10.1073/pnas.1319947111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vora M, Shah M, Ostafi S, Onken B, Xue J, Ni JZ, Gu S, Driscoll M. Deletion of microRNA-80 activates dietary restriction to extend C. elegans healthspan and lifespan. PLoS Genet. 2013;9:e1003737. doi: 10.1371/journal.pgen.1003737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017;66:789–800. doi: 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee H, Cho JS, Lambacher N, Lee J, Lee SJ, Lee TH, Gartner A, Koo HS. The Caenorhabditis elegans AMP-activated protein kinase AAK-2 is phosphorylated by LKB1 and is required for resistance to oxidative stress and for normal motility and foraging behavior. J Biol Chem. 2008;283:14988–14993. doi: 10.1074/jbc.M709115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savage-Dunn C, Padgett RW. The TGF-beta Family in Caenorhabditis elegans. Cold Spring Harb Perspect Biol. 2017;9:a022178. doi: 10.1101/cshperspect.a022178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burkewitz K, Morantte I, Weir HJM, Yeo R, Zhang Y, Huynh FK, Ilkayeva OR, Hirschey MD, Grant AR, Mair WB. Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell. 2015;160:842–855. doi: 10.1016/j.cell.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langenfeld EM, Calvano SE, Abou-Nukta F, Lowry SF, Amenta P, Langenfeld J. The mature bone morphogenetic protein-2 is aberrantly expressed in non-small cell lung carcinomas and stimulates tumor growth of A549 cells. Carcinogenesis. 2003;24:1445–1454. doi: 10.1093/carcin/bgg100. [DOI] [PubMed] [Google Scholar]
- 46.Langenfeld EM, Bojnowski J, Perone J, Langenfeld J. Expression of bone morphogenetic proteins in human lung carcinomas. Ann Thorac Surg. 2005;80:1028–1032. doi: 10.1016/j.athoracsur.2005.03.094. [DOI] [PubMed] [Google Scholar]
- 47.Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48:e245. doi: 10.1038/emm.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hao J, Ho JN, Lewis JA, Karim KA, Daniels RN, Gentry PR, Hopkins CR, Lindsley CW, Hong CC. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem Biol. 2010;5:245–253. doi: 10.1021/cb9002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai Z, Li CF, Han F, Liu C, Zhang A, Hsu CC, Peng D, Zhang X, Jin G, Rezaeian AH, et al. Phosphorylation of PDHA by AMPK drives TCA cycle to promote cancer metastasis. Mol Cell. 2020;80:263–278.e267. doi: 10.1016/j.molcel.2020.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lieu EL, Nguyen T, Rhyne S, Kim J. Amino acids in cancer. Exp Mol Med. 2020;52:15–30. doi: 10.1038/s12276-020-0375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Victor WR, Kathleen MB, Peter JK, Anthony Weil P. Biosynthesis of the nutritionally nonessential amino acids. In: Victor WR, editor. Chapter 27: Harpers illustrated biochemistry. 31. New York: Mcgraw Hill Medical; 2018. [Google Scholar]
- 52.NeMoyer R, Mondal A, Vora M, Langenfeld E, Glover D, Scott M, Lairson L, Rongo C, Augeri DJ, Peng Y, et al. Targeting bone morphogenetic protein receptor 2 sensitizes lung cancer cells to TRAIL by increasing cytosolic Smac/DIABLO and the downregulation of X-linked inhibitor of apoptosis protein. Cell Commun Signal. 2019;17:150. doi: 10.1186/s12964-019-0469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dorman JB, Albinder B, Shroyer T, Kenyon C. The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics. 1995;141:1399–1406. doi: 10.1093/genetics/141.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sountoulidis A, Stavropoulos A, Giaglis S, Apostolou E, Monteiro R, de Sousa C, Lopes SM, Chen H, Stripp BR, Mummery C, Andreakos E, Sideras P. Activation of the canonical bone morphogenetic protein (BMP) pathway during lung morphogenesis and adult lung tissue repair. PLoS ONE. 2012;7:e41460. doi: 10.1371/journal.pone.0041460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyers EA, Gobeske KT, Bond AM, Jarrett JC, Peng CY, Kessler JA. Increased bone morphogenetic protein signaling contributes to age-related declines in neurogenesis and cognition. Neurobiol Aging. 2016;38:164–175. doi: 10.1016/j.neurobiolaging.2015.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langenfeld E, Deen M, Zachariah E, Langenfeld J. Small molecule antagonist of the bone morphogenetic protein type I receptors suppresses growth and expression of Id1 and Id3 in lung cancer cells expressing Oct4 or nestin. Mol Cancer. 2013;12:129. doi: 10.1186/1476-4598-12-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langenfeld EM, Langenfeld J. Bone morphogenetic protein-2 stimulates angiogenesis in developing tumors. Mol Cancer Res. 2004;2:141–149. [PubMed] [Google Scholar]
- 58.Le Page C, Puiffe ML, Meunier L, Zietarska M, de Ladurantaye M, Tonin PN, Provencher D, Mes-Masson AM. BMP-2 signaling in ovarian cancer and its association with poor prognosis. J Ovarian Res. 2009;2:4. doi: 10.1186/1757-2215-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ye L, Mason MD, Jiang WG. Bone morphogenetic protein and bone metastasis, implication and therapeutic potential. Front Biosci. 2011;16:865–897. doi: 10.2741/3725. [DOI] [PubMed] [Google Scholar]
- 60.Langenfeld EM, Kong Y, Langenfeld J. Bone morphogenetic protein 2 stimulation of tumor growth involves the activation of Smad-1/5. Oncogene. 2006;25:685–692. doi: 10.1038/sj.onc.1209110. [DOI] [PubMed] [Google Scholar]
- 61.Gobeske KT, Das S, Bonaguidi MA, Weiss C, Radulovic J, Disterhoft JF, Kessler JA. BMP signaling mediates effects of exercise on hippocampal neurogenesis and cognition in mice. PLoS ONE. 2009;4:e7506. doi: 10.1371/journal.pone.0007506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gangat N, Wolanskyj AP. Anemia of chronic disease. Semin Hematol. 2013;50:232–238. doi: 10.1053/j.seminhematol.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 63.Bagarova J, Vonner AJ, Armstrong KA, Borgermann J, Lai CS, Deng DY, Beppu H, Alfano I, Filippakopoulos P, Morrell NW, et al. Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol Cell Biol. 2013;33:2413–2424. doi: 10.1128/MCB.01595-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roustan V, Jain A, Teige M, Ebersberger I, Weckwerth W. An evolutionary perspective of AMPK-TOR signaling in the three domains of life. J Exp Bot. 2016;67:3897–3907. doi: 10.1093/jxb/erw211. [DOI] [PubMed] [Google Scholar]
- 65.Burkewitz K, Zhang Y, Mair WB. AMPK at the nexus of energetics and aging. Cell Metab. 2014;20:10–25. doi: 10.1016/j.cmet.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marinangeli C, Didier S, Ahmed T, Caillerez R, Domise M, Laloux C, Bégard S, Carrier S, Colin M, Marchetti P, et al. AMP-activated protein kinase is essential for the maintenance of energy levels during synaptic activation. iScience. 2018;9:1–13. doi: 10.1016/j.isci.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raja E, Tzavlaki K, Vuilleumier R, Edlund K, Kahata K, Zieba A, Morén A, Watanabe Y, Voytyuk I, Botling J, et al. The protein kinase LKB1 negatively regulates bone morphogenetic protein receptor signaling. Oncotarget. 2016;7:1120–1143. doi: 10.18632/oncotarget.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morén A, Raja E, Heldin CH, Moustakas A. Negative regulation of TGFβ signaling by the kinase LKB1 and the scaffolding protein LIP1. J Biol Chem. 2011;286:341–353. doi: 10.1074/jbc.M110.190660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets obtained and analyzed for this study will be made available from the corresponding author in a reasonable request.