

Abstract
SARS-CoV-2 is the foremost culprit of the novel coronavirus disease 2019 (nCoV-19 and/or simply COVID-19) and poses a threat to the continued life of humans on the planet and create pandemic issue globally. The 3-chymotrypsin-like protease (MPRO or 3CLPRO) is the crucial protease enzyme of SARS-CoV-2, which directly involves the processing and release of translated non-structural proteins (nsps), and therefore involves the development of virus pathogenesis along with outbreak the forecasting of COVID-19 symptoms. Moreover, SARS-CoV-2 infections can be inhibited by plant-derived chemicals like amentoflavone derivatives, which could be used to develop an anti-COVID-19 drug. Our research study is designed to conduct an in silico analysis on derivatives of amentoflavone (isoginkgetin, putraflavone, 4′′′′′′-methylamentoflavone, bilobetin, ginkgetin, sotetsuflavone, sequoiaflavone, heveaflavone, kayaflavone, and sciadopitysin) for targeting the non-structural protein of SARS-CoV-2, and subsequently further validate to confirm their antiviral ability. To conduct all the in silico experiments with the derivatives of amentoflavone against the MPRO protein, both computerized tools and online servers were applied; notably the software used is UCSF Chimera (version 1.14), PyRx, PyMoL, BIOVIA Discovery Studio tool (version 4.5), YASARA (dynamics simulator), and Cytoscape. Besides, as part of the online tools, the SwissDME and pKCSM were employed. The research study was proposed to implement molecular docking investigations utilizing compounds that were found to be effective against the viral primary protease (MPRO). MPRO protein interacted strongly with 10 amentoflavone derivatives. Every time, amentoflavone compounds outperformed the FDA-approved antiviral medicine that is currently underused in COVID-19 in terms of binding affinity (− 8.9, − 9.4, − 9.7, − 9.1, − 9.3, − 9.0, − 9.7, − 9.3, − 8.8, and − 9.0 kcal/mol, respectively). The best-selected derivatives of amentoflavone also possessed potential results in 100 ns molecular dynamic simulation (MDS) validation. It is conceivable that based on our in silico research these selected amentoflavone derivatives more precisely 4′′′′′′-methylamentoflavone, ginkgetin, and sequoiaflavone have potential for serving as promising lead drugs against SARS-CoV-2 infection. In consequence, it is recommended that additional in vitro as well as in vivo research studies have to be conducted to support the conclusions of this current research study.
Graphical abstract

Keywords: nCoV-19, Amentoflavone derivatives, MPRO protein, Putraflavone, ADMET, YASARA, Molecular dynamics study
Background
COVID-19 or Coronavirus disease-19 has caused by severe acute respiratory virus-2 (SARS-CoV-2) in December 2019 and has been spreading rapidly from Wuhan, China, to globally. World Health Organization (WHO) observed all circumstances and declared the COVID-19 as a global pandemic [1–3]. At present (16 December 2021), according to the worldometer (https://www.worldometers.info/coronavirus/) estimated that 27,28,11,578 infected; 53,49,729 deaths; 24,51,13,998 recovered-covid-19 patients. However, SARS-CoV-2 also known as novel coronavirus 2019 or nCoV-19belongs to the Betacoronavirus genus similar to SARS-CoV and MERS-CoV [4, 5]. The SARS-CoV-2 holds the positive-sense single-stranded RNA viruses with approximately 30 kbp genomic length. Structural and non-structural proteins are characterized in SARS-CoV-2 where structural proteins (i.e., Spike (S), Envelope (E), Nucleocapsid (N), and Matrix (M)) and non-structural proteins, namely-Proteases 3-chymotrypsin-like protease (3CLPRO), RNA-dependent RNA polymerase (RdRp), and Papain-like protease (PLPRO) [6]. The SARS-CoV-2 Polyprotein encodes two main protease enzymes (i.e., 3CLPRO, PLPRO), which directly involve the processing and release of translated non-structural proteins (nsps), the main protease is known as 3-chymotrypsin-like protease (MPRO or 3CLPRO) [7]. The S protein, present on the outer surface of the virion, helps in viral attachment and entry to the host cells [8, 9] (Fig. 1).
Fig. 1.

Schematic illustration of the in silico study of the interaction of the amentoflavone results with the SARS-CoV-2 MPRO
For minimizing the pandemic conditions, clinicians and researchers have recommended the combinational therapy of hydroxychloroquine and azithromycin and also proposed various antiviral drugs, antibody therapy, and so on therapeutic agents [10, 11]. Importantly, Food and Drug Administration (FDA) approved camostat mesylate as a standard antiviral agent [12]. But researchers are trying to find out ideal plant-derived natural phytocompounds which address the diverse diseases conditions, and these bioactive compounds will hasten the drug discovery process [13–15]. Amentoflavone (i.e., biflavonoid in nature) is extremely found all over the world [2, 3, 16]. Clusiaceae, Caryophyllaceae, Euphorbiaceae, Cupressaceae, Calophyllum, and Selaginellaceae families are the major sources of amentoflavone [17]. It’s a complex compound where two apigenin structurally are bound with each other by C3′–C8′′ linkage [17]. It possesses numerous potential biological activities as an antivirus [18], anti-tumor [19], anti-senescence [20], antioxidant [21], anti-inflammatory [22], antibacterial [23], antifungal [24], neuroprotective [25], cardioprotective [26], antidiabetic [27], and so on. Diverse research findings suggest that it is effectively acting toward different types of viruses among them dengue [18], human immunodeficiency virus (HIV) [28], Coxsackievirus B-3 (CVB-3) [29], herpes simplex virus-1 (HSV-1), respiratory syncytial virus (RSV) [30], and acyclovir (ACV)-resistant strains (e.g., HSV-1/106, HSV-1/153, and HSV-1/Blue) [31]. Moreover, it has many derivatives (10) for example isoginkgetin (D1), putraflavone (D2), 4′′′′′′-methylamentoflavone (D3), bilobetin (D4), ginkgetin (D5), sotetsuflavone (D6), sequoiaflavone (D7), heveaflavone (D8), kayaflavone (D9), and sciadopitysin (D10) [32] (Fig. 2; Table 1). A study conducted by Ryu et al. 2010 [33] reported that amentoflavone inhibited the SARS-CoV-2 infection via mediating the inhibition of 3CLPRO with the significant IC50 value, i.e., 8.3 µM. Therefore, 3CLPRO has gained much more attention for the design, discovery, and development of drugs for the SARS-CoVs as a valuable target. It is also termed as ‘the Achilles’ or ‘heel of coronavirus’ [34].
Fig. 2.
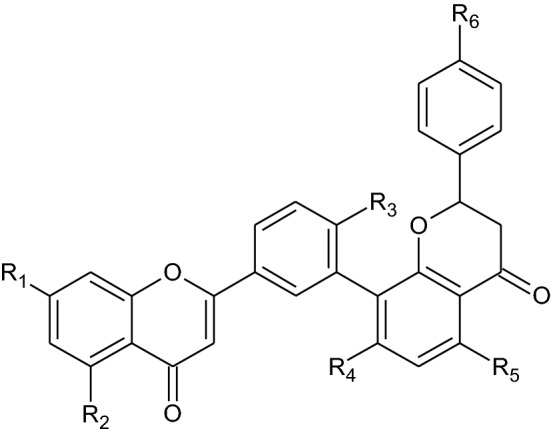
Diagrammatic representation of the chemical structure of amentoflavone and its derivatives
Table 1.
Tabular representation of the amentoflavone derivatives (D1–D10) chemical structure
| Sl/No. | Name | R1 | R2 | R3 | R4 | R5 | R6 |
|---|---|---|---|---|---|---|---|
| D1 | Isoginkgetin | OH | OH | OCH3 | OH | OH | OCH3 |
| D2 | Putraflavone | OCH3 | OH | OH | OH | OH | OCH3 |
| D3 | 4′′′′′′-Methylamentoflavone | OH | OH | OH | OH | OH | OCH3 |
| D4 | Bilobetin | OH | OH | OCH3 | OH | OH | OH |
| D5 | Ginkgetin | OCH3 | OH | OCH3 | OH | OH | OH |
| D6 | Sotetsuflavone | OH | OH | OH | OCH3 | OH | OH |
| D7 | Sequoiaflavone | OCH3 | OH | OH | OH | OH | OH |
| D8 | Heveaflavone | OCH3 | OH | OH | OCH3 | OH | OCH3 |
| D9 | kayaflavone | OH | OH | OCH3 | OCH3 | OH | OCH3 |
| D10 | Sciadopitysin | OCH3 | OH | OCH3 | OH | OH | OCH3 |
Generally, several strategies are applied for the development of new drugs against coronavirus. Here, focuses on two main strategies: initially, blocking the virus entry site into the host cells; and therefore, viral transcription and replication prevention. The main protease (MPRO) of SARS-CoV-2, which plays a significant role in mediating viral replication complex and transcription, is a particularly attractive target for anti-SARS drug design [35–38]. Therefore, MPRO is an important target for the design of potential anti-CoV-2 inhibitors.
In this current workflow, we selected 10 amentoflavone derivatives based on their in silico pharmacokinetics properties including ADMET (absorption, distribution, metabolism, elimination, and toxicity) via the Swiss ADME and pkCSM online server. Additionally, to find out the best leads (i.e., phytochemicals), we conducted the molecular docking study against the SARS-CoV-2 main protease (MPRO) which is provided with an inhibition site. To validate the results from the docking study, the compounds were subjected to the molecular dynamics simulations using the YASARA simulator software package; and lastly, build up the network between chosen drugs, and their targets, and also pathways were constructed based on the Cytoscape platform.
Computational methodology
Macromolecule (protein) preparation
The three-dimensional crystal structure of the SARS-CoV-2 MPRO (PDB ID: 6LU7) was retrieved from the Protein Data bank (PDB) in PDB format [39]. Importantly, PDB is a trusted source for the crystal structure of biological macromolecules, worldwide [40]. For the preparation of the protein removal, unwanted things namely all the heteroatoms, H2O molecules, ligands, and metal ions, extra chains from the protein by using UCSF Chimera (version 1.14) package were made. As part of the macromolecule preparation applied “Gasteiger’s methods”, the protonation states for “Histidine” and conserver as PDB format for further analysis [41, 42].
Lead molecules optimization
Selected amentoflavone’s derivatives (D1–D10) and control antiviral drugs (i.e., camostat mesylate) were retrieved from the most popular database PubChem in the “structure data files (SDF)” format, and their 2D structures are represented in Fig. 3. For the ligand (s) optimization “Gasteiger’s methods” were used, where net charge at “Zero” and the optimization process was conducted by the UCSF Chimera (version 1.14) package, and therefore the optimized ligands were saved as “mol2 format” for molecular docking analysis [2, 41].
Fig. 3.

Diagrammatic (2D) representation of chemical structures of amentoflavone derivatives and control antiviral drugs
Molecular docking and post-docking protocol
To find out the best drug candidates molecular docking is an excellent method [3]. This method is used for predicting the best drug candidates based on the scoring. For molecular docking operation, PyRx virtual screening software (version 0.8) was used which works based on the Auto Dock vina configuration [43]. Docking results determine the measure of ligand interaction to the active site of the targeted protein.
BIOVIA Discovery Studio tool (version 4.5) was applied to determine the interactions between “protein–ligand complex,” where mainly evaluate the diverse interactions (hydrogen-, hydrophobic-, pi-alkali-bond, and so on), and most importantly here used was the combined file which was collected from the PyMOL visualizer tool [44].
ADMET profiling
To predict the physiochemical with pharmacokinetics properties of the selected D1-D10 used the Swiss ADME and pKCSM online server. The Swiss ADME server was employed to accurately interpret the physiochemical properties, and the lead molecules must follow Lipinski’s 5 rules (L5 rule). Besides, the absorption, distribution, metabolism, excretion, and toxicity parameters of these ten derivatives were mainly analyzed via the pkCSM online portal [45, 46].
Molecular dynamics (MD) simulation and MM-PBSA analysis
The molecular dynamics simulation study was conducted into the YASARA dynamics software package [47] with the aid of the AMBER14 force field [48]. The complexes were initially cleaned, optimized and hydrogen bond networks were optimized. The TIP3P solvation model was used in a cubic simulation cell with a periodic boundary condition [49]. The physiological conditions of the simulation cells were set as 310 K, pH 7.4, and 0.9% NaCl [50]. The initial energy minimization process was conducted by the simulated annealing method by steepest gradient algorithms (5000 cycles). The long-range electrostatic interactions were calculated by Particle Mesh Ewalds algorithms by a cut-off radius of 8.0 Å [51]. The time step of the simulations systems was set as 2.0 fs [50]. The simulation trajectories were saved after every 100 ps. By following constant pressure and Berendsen thermostat, the simulations were extended for 100 ns where trajectories were utilized to analyze the root mean square deviations (RMSD), root means square fluctuations (RMSF), the radius of gyration (Rg), solvent-accessible surface area (SASA), and hydrogen bonds [52–56].
Therefore, the YASARA trajectories were utilized for the calculation of the binding free energy from MM-PBSA methods where the positive energy indicates better energy [53].
Network pharmacology of chosen drugs
The network pharmacology assessment includes three principal steps: (a) prediction targets of chosen drugs, (b) enrichment analysis for predicted targets, and (c) construction of network between chosen drugs, targets, and pathways and its analysis. Briefly, targets of chosen drugs were predicted using DIGEP-Pred [57] at the pharmacological activity (Pa) of 0.5. Next and the predicted proteins of chosen drugs were enriched using STRING [58] ver.11.0 to generate the protein–protein interaction. And all proteins which are obtained from the STRING database were submitted to the Enricher database (https://maayanlab.cloud/Enrichr/) to enrich their biological processes, molecular function cellular components, and KEGG pathways. Finally, the network between chosen drugs, their targets, and pathways was constructed using Cytoscape V. 3.8.2. [59].
Research findings
Molecular docking and interaction with the MPRO
Amentoflavone derivatives such as Isoginkgetin (D1), putraflavone (D2), 4′′′′′′-methylamentoflavone (D3), bilobetin (D4), ginkgetin (D5), sotetsuflavone (D6), sequoiaflavone (D7), heveaflavone (D8), kayaflavone (D9), and sciadopitysin (D10) exhibited promising binding affinity with the main protease of SARS-CoV-2 (MPRO). However, the binding energies are − 8.9, − 9.4, − 9.7, − 9.1, − 9.3, − 9.0, − 9.7, − 9.3, − 8.8, and − 9.0 kcal/mol, respectively, and tabulated in Table 2. The best binding affinity (−9.7 kcal/mol) was demonstrated by D3 relative to other compounds with the MPRO. BIOVIA Discovery Studios Software was employed to calculate the non-covalent interactions which established that both hydrogen and hydrophobic bonds had roles to play in promoting the binding affinity along with improving their binding specificity.
Table 2.
Interpretation of molecular docking of the amentoflavone’s derivatives with control drug (camostat mesylate) and their interactions against the MPRO protein
| Ligand–protein interaction | Binding energy (kcal/mol) | H-bond residues | H-bond length (Å) | No. of H-bonds | Other bond residues |
|---|---|---|---|---|---|
| D1-MPRO | − 8.9 | Gly143 | 2.31 | 3 | Pro168, Ser144, Glu166 |
| Phe140 | 3.06 | ||||
| Thr190 | 2.27 | ||||
| D2-MPRO | − 9.4 | His163 | 3.02 | 2 | Gly 143, Cys145, Glu166 |
| Thr26 | 2.10 | ||||
| D3-MPRO | − 9.7 | His163 | 2.40 | 3 | Asp187, Asn142, Cys145, Met165, Met 49, His41 |
| Glu166 | 2.64 | ||||
| Thr26 | 2.23 | ||||
| D4-MPRO | − 9.1 | Glu166 | 2.94 | 1 | Gly143, Cys145 |
| D5-MPRO | − 9.3 | Glu166 | 3.03 | 1 | Gly143, Cys145 |
| D6-MPRO | − 9.0 | – | – | 0 | Gly143, Cys145, Glu166 |
| D7-MPRO | − 9.7 | Asn142 | 2.62 | 2 | Thr45, Gln189, Cys145, Glu166, His 41 |
| His164 | 2.32 | ||||
| D8-MPRO | − 9.3 | Thr 26 | 2.21 | 1 | Gly143, Met 165, Glu166, Cys 145 |
| D9-MPRO | − 8.8 |
Gln189 Phe140 |
2.59 | 2 | Glu166, Met49, Cys145 |
| 2.08 | |||||
| D10-MPRO | − 9.0 | Asn142 | 2.97 | 2 | Glu166, Phe140, Cys145, Met 165 |
| Gly143 | 2.31 | ||||
| Camostat mesylate (references drug) | − 7.9 | Asp153 | 2.48 | 4 | Ile106, Phe294 |
| Asn151 | 2.84 | ||||
| Ile249 | 2.56 | ||||
| Ser158 | 2.72 |
D1 exhibited strong binding affinity with Gly143, Phe140, Thr190, Pro168, Ser144, Glu166 residues, while the D2 exhibited strong binding affinity with His163, Thr26, Gly 143, Cys145, Glu166 residues of MPRO active site. Besides this, D3 bonded with His163, Glu166, Thr26, Asp187, Asn142, Cys145, Met165, Met 49, His41 residues, and also Glu166, Gly143, and Cys145 have participated in the both D4-MPRO and D5-MPRO interaction. On the other hand, D6-MPRO forms a complex among Gly143, Cys145, Glu166 residues, where Asn142, His164, Thr45, Gln189, Cys145, Glu166, His 41 have interacted with D7. D8 directly involves the interactions among Thr 26, Gly143, Met 165, Glu166, Cys 145. Gln189, Phe140, Glu166, Met49, Cys145 bonded with D9, and therefore, Asn142, Gly143, Glu166, Phe140, Cys145, Met 165 also involve the complex with sciadopitysin (D10). The camostat mesylate (reference drug) fitted by the Asp153, Asn151, Ile249, Ser158, Ile106, and Phe294 residues. The 2D and 3D structures of non-bond interactions of amentoflavone’s derivatives with MPRO are shown in Fig. 4. The standard drugs such as camostat mesylate showed binding affinity toward MPRO by − 7.9 kcal/mol (Table 2). The 2D and 3D structures of non-bond interactions of camostat mesylate with MPRO are shown in Fig. 4.
Fig. 4.

Interactions among D1–D10, and control drug with targeted MPRO protein
ADMET profiling analysis
pkCSM and SwissADME were web-based tools employed for analysis together with optimization of pharmacokinetic properties of the selected therapeutic agents. The amentoflavone derivatives such as D1, D2, D3, D4, D5, D6, D7, D8, D9, and D10 have molecular weights of 566.518, 566.518, 552.491, 552.48, 566.51, 552.48, 552.491, 580.545, 580.545, and 580.54 gm/mol, respectively. All the compounds conferred the same no. of Lipinski rule violation (1) and have no AMES toxicity and same bioavailability score (0.55). The same topological polar surface area (159.80 Å2) was displayed by three amentoflavone derivatives (D1, D2, and D5). The other four derivatives of amentoflavone (D3, D4, D6, and D7) also possess the same topological polar surface area as 170.80 Å2. Conversely, D8, D9, and D10 have a topological polar surface area such as 148.80 Å2. After carrying out the pharmacokinetics analysis in pkCSM, the generated results indicated that some of the amentoflavone derivatives (D1, D2, D8, D9, and D10) have been shown positive results of hepatotoxicity, while others did not (D3, D4, D5, D6, and D7). Oral rat acute toxicity (LD50) for the amentoflavone derivatives is found in the range 2.535–3.06, while the maximum tolerated dose for humans (log mg/kg/day) is in the range 0.295–0.437. Also, the values range between 0.571 and 0.833 (log ml/min/kg) was observed for the total clearance (TC), and the predicted octanol/water partition coefficient (LogP) fell in the range of 5.3886–6.043 for all derivatives. Additionally, Table 3 represents the no. of hydrogen bond acceptors, no. of hydrogen bond donors, and no. of rotatable bonds.
Table 3.
ADMET properties of amentoflavone derivatives
| Compounds | MW | H-Ac | H-Do | N.rot | TPSA (Å2) | LogP | B.S | LD50 | HT | AT | MTD | NLV | TC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D1 | 566.51 | 10 | 4 | 5 | 159.80 | 5.74 | 0.55 | 2.723 | Yes | No | 0.43 | 1 | 0.710 |
| D2 | 566.51 | 10 | 4 | 5 | 159.80 | 5.74 | 0.55 | 2.854 | Yes | No | 0.423 | 1 | 0.723 |
| D3 | 552.49 | 10 | 5 | 4 | 170.80 | 5.3886 | 0.55 | 2.96 | No | No | 0.295 | 1 | 0.619 |
| D4 | 552.48 | 10 | 5 | 4 | 170.80 | 5.437 | 0.55 | 2.56 | No | No | 0.437 | 1 | 0.571 |
| D5 | 566.51 | 10 | 4 | 5 | 159.80 | 5.74 | 0.55 | 2.733 | No | No | 0.427 | 1 | 0.646 |
| D6 | 552.48 | 10 | 5 | 4 | 170.80 | 5.437 | 0.55 | 2.548 | No | No | 0.436 | 1 | 0.617 |
| D7 | 552.49 | 10 | 5 | 4 | 170.80 | 5.437 | 0.55 | 2.535 | No | No | 0.437 | 1 | 0.488 |
| D8 | 580.54 | 10 | 3 | 6 | 148.80 | 6.043 | 0.55 | 2.997 | Yes | No | 0.412 | 1 | 0.791 |
| D9 | 580.54 | 10 | 3 | 6 | 148.80 | 6.043 | 0.55 | 2.84 | Yes | No | 0.419 | 1 | 0.794 |
| D10 | 580.54 | 10 | 3 | 6 | 148.80 | 6.043 | 0.55 | 3.06 | Yes | No | 0.419 | 1 | 0.833 |
MW molecular weight (g/mol), H-Ac no. of hydrogen bond acceptors, H-Do no. of hydrogen bond donors, N.rot no. of rotatable bonds, TPSA Topological polar surface area (Å2), LogP predicted octanol/water partition coefficient, BS bioavailability score, TC total clearance (log ml/min/kg), LD50 oral rat acute toxicity, HT hepatotoxicity, AT ames toxicity, MTD The maximum tolerated dose for human (log mg/kg/day), NLV no. of lipinski rule violation, TC total clearance
Molecular dynamics (MD) simulation
To establish the structural deviation of the docked macromolecule and phytochemical complex, the molecular dynamics trajectories were analyzed where the root means square deviation of the docked complex molecules was assessed to find out the deviations. Figure 5A illustrates that the complex initially raised its RMSD profile which might be due to the presence of a high level of flexibility among the other complexes. After the course of the 30 ns simulation, all the docked complexes displayed straight lines and they possessed lower levels of deviations. Although some fluctuations were observed for some docked complexes, they did not exceed 3.0 Å in RMSD analysis.
Fig. 5.

Molecular dynamics (MD) simulation of the amentoflavone derivatives with MPRO which have shown RMSD (A), SASA (B), Rg (C), hydrogen bond (D), RMSF (E), and MM-PBSA (F)
The solvent-accessible surface area of the complexes was analyzed to check the change in the surface area. The higher SASA value defines the expansion of the surface area, whereas the lower SASA value relates to the truncation of the surface area. D4 had a high level of fluctuation which might occur due to instability. The other docked complexes had a better profile as they did not change too much Fig. 5B. The radius of gyration of the simulation trajectories defines the labile nature of the protein. The camostat mesylate complex molecules had a higher Rg profile which denotes the loose packaging systems of the molecules. The other docked complexes had lower deviations in the Rg profile in Fig. 5C. The hydrogen bond systems define the structural stability of the complexes where any change may lead to the hydrogen-bonding systems defining the instability. Figure 5D states that all docked complexes had stable hydrogen-bonding patterns. The root means square fluctuations of the docked complexes were analyzed to find out flexibility across the amino acid residues. Figure 5E states that maximum amino acid residues of the docked complexes had lower RMSF than 2.5 Å which indicates a lower level of flexibility of the complexes. The binding free energy of the docked complexes was calculated through the MM-PBSA model where positive energy indicates more favorable binding. The docked complexes had a comparative stable binding score in Fig. 5F which indicates more favorable binding.
It is well known that the higher solvent-accessible surface area (SASA) value defines the expansion of the surface area, whereas the lower SASA value relates to the truncation of the surface area. Due to its instability, D4 had a high level of fluctuation. The other docked complexes had a more stable profile since they encountered fewer changes (Fig. 5B). The radius of gyration (RG) values of the simulation trajectories determines the labile nature of the protein. The control drug namely, camostat mesylate complex molecules, had a higher RG profile, whereas other docked complexes had lower deviations in the RG profile in Fig. 5C. The hydrogen bond systems define the structural stability of the complexes where any change may lead to disorder and disruption in the hydrogen bond system. Figure 5D illustrates that all docked complexes had stable hydrogen-bonding patterns. The root means square fluctuations of the docked complexes were analyzed to find out flexibility across the amino acid residues. On the contrary, Fig. 5E states that maximum amino acid residues of the docked complexes had lower RMSF than 2.5 Å demonstrating the complexes had a lower level of flexibility. The binding free energy of the docked complexes was calculated through the MM-PBSA, and model favorable binding was dictated by positive energy. The docked complexes had a comparative stable binding score in Fig. 5F which indicates a more favorable and efficient binding between the macromolecule and phytochemical.
Construction of “drug-target-pathway” network
Based on the network pharmacology, D1–D10 collectively downregulates nine genes as follows: TNNB1, KLK3, CASP8, FLT1, NOS2, MMP2, MMP7, FE2L2, and MDM2. The selected compounds, predicted gene targets, and the top 10 pathways were imported into Cytoscape 3.8.2 software, and the “Drug-Target-Pathway” network was obtained as shown in Fig. 6. Light green hexagonal symbols represent the chosen drugs, the blue circle represents potential targets, and the red triangle represents pathways. Through the software, the node is visualized by degree value, and the node size is proportional to degree value. According to the requirements of topological parameters, the key nodes were determined by degree values greater than twice the median to obtain potential active components for later molecular docking tests. As recorded in KEGG, the MDM2 pathway is networked with 8 different pathways such as cancer, prostate cancer, proteoglycans in cancer, human cytomegalovirus infection, bladder cancer, human papillomavirus infection, p53 signaling pathway, and C-type lectin receptor signaling pathway.
Fig. 6.

Drug-targets-pathways network
Discussion
Nowadays, the world is going through a pandemic situation due to COVID-19 [60]. Researchers all over the world are trying to find out the inhibitory agents, but they have hardly been discovered. There are some antiviral drugs such as ritonavir, lopinavir, indinavir, remdesivir, and camostat mesylate are that have been applied for animal trials to confirm the potentiality of these drugs as anti-coronavirus agents [61]. Although the pH of the protease is tried to elevate and reported as a potent inhibitor of SARS-CoV-2 in the event of an emergency, hydroxychloroquine and azithromycin are being provided to the infected persons [62–64]. Despite having potentiality, these drugs have not been allowed as candidate drugs due to an enormous number of hazardous and drastic side effects to the human body. Therefore, researchers are searching for some tremendous natural compounds that would be the more efficient candidates as therapeutics against the COVID-19 as well as they will show fewer side effects. Also, this research was conducted to discover medicinal plant-derived compounds and their derivatives that have the ability for becoming the lead compound to combat the nCoV-19 infection [65].
Amentoflavone (biflavonoid) is one of the plant derivatives compounds that were isolated by Okigawa et al. [16], and due to a wider range of bioactivities, it has drawn much attention among the scientific community [3, 66, 67]. A compound should have some fundamental properties for becoming an ideal anti-COVID-19 drug candidate [44] such as (i) restriction ability of viral entrance, thereby inhibiting cellular attachment; (ii) inhibition of viral replication inside the host cells; (iii) cytotoxic effects on the existing viruses; and (iv) protection of the host normal cells from the viral origin oxidative stress and inflammatory responses. And the compound amentoflavone is the evidence that followed all of these pathways and performed as potential anti-COVID-19 therapeutics; even not only this compound alone but also its derivatives showed the antioxidant [68, 69] and anti-inflammatory [3, 70–72] activities. Moreover, the MPRO should have the ability to break host polyproteins as well as induce the formation of protein for viral replication [73], and amentoflavone has the interaction capability with MPRO [28]. So, it can be possible that derivatives of amentoflavone will bind to the MPRO and inhibit the viral infection.
Derivatives of amentoflavone such as D1–D10 displayed better binding affinities with COVID-19 main protease (MPRO) − 8.6, − 9.1, − 9.9, − 9.1, − 9.0, − 9.1, − 8.7, − 9.3, − 8.2, and − 8.9 kcal/mol, respectively, whereas camostat mesylate (control drug) has been shown − 7.9 kcal/mol. Furthermore, some flavonoids and polyphenolic plant-derived compounds such as kaempferol, quercetin, demethoxycurcumin, curcumin, catechin, epicatechin gallate, gingerol, and gingerol are studied to inhibit the main protease of SARS-CoV-2. These compounds have significant docking scores as − 9.41, − 8.58, − 8.17, − 7.31, − 7.05, − 7.24, − 6.67, and − 5.40 kcal/mol, respectively, from the Khaerunnisa et al. [13], which claimed that the derivative of amentoflavone has better properties than other proposed inhibitors. Here, the control drug (camostat mesylate) displayed good binding interactions with MPRO protein, and also the derivatives of amentoflavone showed better binding interactions with MPRO protein, and the significant data are tabulated in Table 2 with illustration in Fig. 4.
Swiss ADME and pkCSM calculated the pharmacokinetic parameters of the selected drugs based on their expected values (Table 3), we have observed that amentoflavone derivatives are non-toxic and can be administered as a medication with the level of tolerance specified for human consumption, as anticipated by the website. Furthermore, additional research should be conducted to determine the toxicogenicity (including toxicity, cytotoxicity, genotoxicity, and mutagenicity) of the substances using plant-derived eukaryotic and/or animal models.
Molecular dynamic simulation (MDS) is a versatile technique that analyzes biomolecular interactions and the contact between the arrangement and activity of proteins to aid in modern drug discovery, as well as performance data from dynamic trajectory analysis [2]. In this study, the YASARA dynamics simulator package software has been conducted to run molecular dynamic simulation (MDS) with the selected physiological and physicochemical parameters. This simulation trajectory of the simulation tool has also been used to perfectly analyze the root mean square deviation (RMSD), solvent-accessible surface area (SASA), the radius of gyration (Rg), hydrogen bond number, root mean square fluctuation (RMSF), and molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) [74]. All the parameters possess more significant results in the 100 ns dynamics simulation process with the drug-protein complex. Additionally, the selected compounds, predicted gene targets under the study of network pharmacology, and the top 10 pathways were imported into Cytoscape 3.8.2 software, and the “Drug-Target-Pathway” network was obtained as shown in Fig. 6. Even though additional in vivo studies are required to confirm the findings of this study, our findings will be useful for future non-clinical, pre-clinical, and clinical investigations including these compounds in the future. Finally, this study will be able to motivate medicinal experts to do enough research on this potentially beneficial natural lead molecule and its laboratory derivatives, as demonstrated in the previous section.
Conclusion and future directions
Amentoflavone (biflavonoid) compounds as D1–D10 have antiviral activity against a variety of viruses. According to this molecular docking and dynamic simulation assessment, derivatives of amentoflavone exhibit a favorable interaction with the MPRO protein. Amentoflavone compounds had higher binding energies than clinical trial antiviral medicines (camostat mesylate), which are commonly utilized in numerous countries to treat COVID-19. Aside from that, pharmacokinetic studies show that these amentoflavone derivatives are superior to camostat mesylate in terms of effectiveness. In consideration of the findings of this study, it is possible to observe amentoflavone derivatives more specifically D3, D5, and D7 (4′′′′′′-methylamentoflavone, ginkgetin, and sequoiaflavone) for serving as potential lead drugs against SARS-CoV-2 infection. Although additional in vivo testing is required to confirm our findings which will help future non-clinical, pre-clinical, and clinical studies with these compounds, while also inspiring medicinal chemistry researchers to conduct appropriate research on this promising natural lead compound and its derivatives.
The treatment of SARS-CoV-2 infection has rapidly advanced from poorly tolerated oral and injectable medication combinations to all oral, well-tolerated drug combinations with virologic cures for over 90% of patients. During the next generation of treatments, COVID-19 treatment will continue to advance. Several novel therapeutic techniques for the treatment of COVID-19 have to be examined in the following section; however, they are only a few examples of the numerous current development programs and novel tactics for the treatment of COVID-19. For future generation antiviral drugs, several practical concerns need to be addressed, including drug-drug interactions, side effects, and overlapping safety issues, dose, as well as futility restrictions, which now prevent them from being used in certain situations. Therefore, our selected amentoflavone derivatives would be addressed all major challenges and act as a major medicative option.
Author contributions
Conceptualization: D.D., R.H., P.B., and M.T.I.; validation, investigation, data curation, and manuscript writing: all authors; review and editing: D.D., R.H., P.B., M.M.H., M.T.I., and S.M.; Reviewing-Writing-editing: S.M., D.D., S.B.; Supervision: M.A.R. and B.K.; Funding: B.K. All authors read and approved the final version of the manuscript.
Funding
This research was supported by Korea Institute of Oriental Medicine (Grant No. KSN2021240), Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2020R1I1A2066868), the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1A5A2019413), a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant No.: HF20C0116), and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant No.: HF20C0038).
Data availability
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Declarations
Conflict of interest
The authors declared no conflict of interest.
Consent for publication
Not applicable.
Ethical approval
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Partha Biswas, Email: partha_160626@just.edu.bd.
Bonglee Kim, Email: bongleekim@khu.ac.kr.
References
- 1.Biswas P, Hasan MM, Dey D, Dos Santos Costa AC, Polash SA, Bibi S, Ferdous N, Kaium MA, Rahman MDH, Jeet FK, et al. Candidate antiviral drugs for COVID-19 and their environmental implications: a comprehensive analysis. Environ Sci Pollut Res Int. 2021 doi: 10.1007/s11356-021-16096-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharif MA, Hossen MS, Shaikat MM, Mashuk F, Haidary TIE, Dey D, Paul PK, Al Azad S, Al Mazid MF, Badal MNU. Molecular optimization, docking and dynamic simulation study of selective natural aromatic components to block E2-CD81 complex formation in predating protease inhibitor resistant HCV influx. Int J Pharm Res. 2021;13:3511–3525. [Google Scholar]
- 3.Rahman M, Zilani M, Hasan N, Islam M, Hasan M, Yasmin F, Biswas P, Hirashima A, Kim B. In vivo neuropharmacological potential of Gomphandra Tetrandra (wall) Sleumer and in-silico study against β-amyloid precursor protein. Processes. 2021 doi: 10.3390/pr9081449. [DOI] [Google Scholar]
- 4.Chan JF, Lau SK, To KK, Cheng VC, Woo PC, Yuen KY. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev. 2015;28:465–522. doi: 10.1128/cmr.00102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dey D, Paul PK, Al Azad S, Al Mazid MF, Khan AM, Sharif MA, Rahman MH. Molecular optimization, docking, and dynamic simulation profiling of selective aromatic phytochemical ligands in blocking the SARS-CoV-2 S protein attachment to ACE2 receptor: an in silico approach of targeted drug designing. J Adv Vet Anim Res. 2021;8:24–35. doi: 10.5455/javar.2021.h481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dipta D, Tanzila Ismail E, Partha Biswas SA, Shoeba Islam URR, Firoz M, Ahmed SZ, Salauddin A, Rahman A, Afrin S, Mahedi RA. Antiviral effects of bacteriocin against animal-to-human transmittable mutated SARS-COV-2: a systematic review. Front Agric Sci Eng. 2021;8:603–622. doi: 10.15302/J-FASE-2021397. [DOI] [Google Scholar]
- 7.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ibrahim IM, Abdelmalek DH, Elfiky AA. GRP78: a cell’s response to stress. Life Sci. 2019;226:156–163. doi: 10.1016/j.lfs.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samad A, Ahammad F, Nain Z, Alam R, Imon RR, Hasan M, Rahman MS. Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J Biomol Struct Dyn. 2022;40:14–30. doi: 10.1080/07391102.2020.1792347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/s0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perimal-Lewis L, Williams PA, Mudd G, Gunasekara G. Telehealth innovations in remote healthcare services delivery. Netherlands: IOS Press; 2021. Virtual care: the future for telehealth; pp. 106–113. [Google Scholar]
- 12.Lam S, Lombardi A, Ouanounou A. COVID-19: a review of the proposed pharmacological treatments. Eur J Pharmacol. 2020 doi: 10.1016/j.ejphar.2020.173451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khaerunnisa S, Kurniawan H, Awaluddin R, Suhartati S, Soetjipto S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints. 2020;2020:2020030226. [Google Scholar]
- 14.Dey D, Quispe C, Hossain R, Jain D, Ahmed Khan R, Janmeda P, Islam MT, Ansar Rasul Suleria H, Martorell M, Daştan SD, et al. Ethnomedicinal use, phytochemistry, and pharmacology of Xylocarpus granatum J. Koenig. Evid Based Complement Altern Med. 2021 doi: 10.1155/2021/8922196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul P, Biswas P, Dey D, Saikat ASM, Islam M, Sohel M, Hossain R, Mamun AA, Rahman M, Hasan M. Exhaustive plant profile of “dimocarpus longan lour” with significant phytomedicinal properties: a literature based-review. Processes. 2021 doi: 10.3390/pr9101803. [DOI] [Google Scholar]
- 16.Okigawa M, Hwa CW, Kawano N, Rahman W. Biflavones in selaginella species. Phytochemistry. 1971;10:3286–3287. doi: 10.1016/S0031-9422(00)97392-8. [DOI] [Google Scholar]
- 17.Yu S, Yan H, Zhang L, Shan M, Chen P, Ding A, Li SF. A review on the Phytochemistry, pharmacology, and pharmacokinetics of amentoflavone, a naturally-occurring biflavonoid. Molecules. 2017 doi: 10.3390/molecules22020299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coulerie P, Nour M, Maciuk A, Eydoux C, Guillemot JC, Lebouvier N, Hnawia E, Leblanc K, Lewin G, Canard B, et al. Structure-activity relationship study of biflavonoids on the dengue virus polymerase DENV-NS5 RdRp. Planta Med. 2013;79:1313–1318. doi: 10.1055/s-0033-1350672. [DOI] [PubMed] [Google Scholar]
- 19.Park NH, Lee CW, Bae JH, Na YJ. Protective effects of amentoflavone on Lamin a-dependent UVB-induced nuclear aberration in normal human fibroblasts. Bioorg Med Chem Lett. 2011;21:6482–6484. doi: 10.1016/j.bmcl.2011.08.067. [DOI] [PubMed] [Google Scholar]
- 20.Shen X, Niu X, Li G, Deng X, Wang J. Amentoflavone ameliorates streptococcus suis-induced infection in vitro and in vivo. Appl Environ Microbiol. 2018 doi: 10.1128/aem.01804-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abdallah HM, Almowallad FM, Esmat A, Shehata IA, Abdel-Sattar EA. Anti-inflammatory activity of flavonoids from Chrozophora tinctoria. Phytochem Lett. 2015;13:74–80. doi: 10.1016/j.phytol.2015.05.008. [DOI] [Google Scholar]
- 22.Camero M, Marinaro M, Lovero A, Elia G, Losurdo M, Buonavoglia C, Tempesta M. In vitro antiviral activity of Ficus carica latex against caprine herpesvirus-1. Nat Prod Res. 2014;28:2031–2035. doi: 10.1080/14786419.2014.918120. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Yang X, Zhang H, Zhang J, Zhou Y, Wang T, Hu N, Deng X, Bai X, Wang J. Amentoflavone attenuates Clostridium perfringens gas gangrene by targeting alpha-toxin and perfringolysin O. Front Pharmacol. 2020 doi: 10.3389/fphar.2020.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang IS, Lee J, Jin HG, Woo ER, Lee DG. Amentoflavone stimulates mitochondrial dysfunction and induces apoptotic cell death in Candida albicans. Mycopathologia. 2012;173:207–218. doi: 10.1007/s11046-011-9503-x. [DOI] [PubMed] [Google Scholar]
- 25.Rong S, Wan D, Fan Y, Liu S, Sun K, Huo J, Zhang P, Li X, Xie X, Wang F, et al. Amentoflavone affects epileptogenesis and exerts neuroprotective effects by inhibiting NLRP3 inflammasome. Front Pharmacol. 2019 doi: 10.3389/fphar.2019.00856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng XK, Liu CX, Zhai YY, Li LL, Wang XL, Feng WS. Protection effect of amentoflavone in selaginella tamariscina against TNF-alpha-induced vascular injury of endothelial cells. Yao Xue Xue Bao Acta Pharm Sin. 2013;48:1503–1509. [PubMed] [Google Scholar]
- 27.Su C, Yang C, Gong M, Ke Y, Yuan P, Wang X, Li M, Zheng X, Feng W. Antidiabetic activity and potential mechanism of amentoflavone in diabetic mice. Molecules. 2019 doi: 10.3390/molecules24112184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin YM, Anderson H, Flavin MT, Pai YH, Mata-Greenwood E, Pengsuparp T, Pezzuto JM, Schinazi RF, Hughes SH, Chen FC. In vitro anti-HIV activity of biflavonoids isolated from Rhus succedanea and Garcinia multiflora. J Nat Prod. 1997;60:884–888. doi: 10.1021/np9700275. [DOI] [PubMed] [Google Scholar]
- 29.Wilsky S, Sobotta K, Wiesener N, Pilas J, Althof N, Munder T, Wutzler P, Henke A. Inhibition of fatty acid synthase by amentoflavone reduces coxsackievirus B3 replication. Arch Virol. 2012;157:259–269. doi: 10.1007/s00705-011-1164-z. [DOI] [PubMed] [Google Scholar]
- 30.Ma SC, But PP, Ooi VE, He YH, Lee SH, Lee SF, Lin RC. Antiviral amentoflavone from Selaginella sinensis. Biol Pharm Bull. 2001;24:311–312. doi: 10.1248/bpb.24.311. [DOI] [PubMed] [Google Scholar]
- 31.Li F, Song X, Su G, Wang Y, Wang Z, Jia J, Qing S, Huang L, Wang Y, Zheng K, et al. Amentoflavone inhibits HSV-1 and ACV-resistant strain infection by suppressing viral early infection. Viruses. 2019 doi: 10.3390/v11050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogunwa TH, Ayenitaju FC. An insight into the precise molecular interaction and inhibitory potential of amentoflavone and its substituted derivatives on human α-amylase. Arch Curr Res Int. 2017;10:1–14. doi: 10.9734/ACRI/2017/35759. [DOI] [Google Scholar]
- 33.Ryu YB, Jeong HJ, Kim JH, Kim YM, Park JY, Kim D, Nguyen TT, Park SJ, Chang JS, Park KH, et al. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CL(pro) inhibition. Bioorg Med Chem. 2010;18:7940–7947. doi: 10.1016/j.bmc.2010.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu K, Chen L, Peng G, Zhou W, Pennell CA, Mansky LM, Geraghty RJ, Li F. A virus-binding hot spot on human angiotensin-converting enzyme 2 is critical for binding of two different coronaviruses. J Virol. 2011;85:5331–5337. doi: 10.1128/jvi.02274-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gagneur A, Sizun J, Vallet S, Legr MC, Picard B, Talbot PJ. Coronavirus-related nosocomial viral respiratory infections in a neonatal and paediatric intensive care unit: a prospective study. J Hosp Infect. 2002;51:59–64. doi: 10.1053/jhin.2002.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, Wertheim-van Dillen PM, Kaandorp J, Spaargaren J, Berkhout B. Identification of a new human coronavirus. Nat Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siddell SG, Ziebuhr J, Snijder EJ. Topley and Wilson’s microbiology and microbial infections. London: Edward Arnold; 2005. Coranaviruses, toroviruses and arteriviruses; pp. 823–856. [Google Scholar]
- 38.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005;79:884–895. doi: 10.1128/jvi.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 40.Burley SK, Berman HM, Kleywegt GJ, Markley JL, Nakamura H, Velankar S. Protein data bank (PDB): the single global macromolecular structure archive. Methods Mol Biol. 2017;1607:627–641. doi: 10.1007/978-1-4939-7000-1_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biswas P, Dey D, Rahman A, Islam MA, Susmi TF, Kaium MA, Hasan MN, Rahman MDH, Mahmud S, Saleh MA, et al. Analysis of SYK gene as a prognostic biomarker and suggested potential bioactive phytochemicals as an alternative therapeutic option for colorectal cancer: an in-silico pharmaco-informatics investigation. J Pers Med. 2021 doi: 10.3390/jpm11090888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jayaraman S, Veeraraghavan V, Sreekandan RN, Mohan SK, Suga SSD, Kamaraj D, Mohandoss S, Koora S. Molecular docking analysis of the BRCA1 protein with compounds from justica adhatoda. Bioinformation. 2020;16:888–892. doi: 10.6026/97320630016888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain D, Hossain R, Khan RA, Dey D, Toma TR, Islam MT, Janmeda P, Hakeem KR. Computer-aided evaluation of anti-SARS-CoV-2 (3-chymotrypsin-like protease and transmembrane protease serine 2 Inhibitors) activity of cepharanthine: an in silico approach. Biointerface Res Appl Chem. 2021;12:768–780. doi: 10.33263/BRIAC121.768780. [DOI] [Google Scholar]
- 44.Hossain R, Al-Khafaji K, Khan RA, Sarkar C, Islam M, Dey D, Jain D, Faria F, Akbor R, Atolani O. Quercetin and/or ascorbic acid modulatory effect on phenobarbital-induced sleeping mice possibly through gabaa and gabab receptor interaction pathway. Pharmaceuticals. 2021 doi: 10.3390/ph14080721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017 doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pires DE, Blundell TL, Ascher DB. pkCSM: predicting small-Molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Land H, Humble MS. Yasara: a tool to obtain structural guidance in biocatalytic investigations. Methods Mol Biol. 2018;1685:43–67. doi: 10.1007/978-1-4939-7366-8_4. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 49.Harrach MF, Drossel B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J Chem Phys. 2014 doi: 10.1063/1.4872239. [DOI] [PubMed] [Google Scholar]
- 50.Krieger E, Vriend G. New ways to boost molecular dynamics simulations. J Comput Chem. 2015;36:996–1007. doi: 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krieger E, Nielsen JE, Spronk CA, Vriend G. Fast empirical pKa prediction by Ewald summation. J Mol Graph Model. 2006;25:481–486. doi: 10.1016/j.jmgm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Adji A, Niode NJ, Memah VV, Posangi J, Wahongan GJP, Ophinni Y, Idroes R, Mahmud S, Emran TB, Nainu F, et al. Designing an epitope vaccine against dermatophagoides pteronyssinus: an in silico study. Acta Trop. 2021 doi: 10.1016/j.actatropica.2021.106028. [DOI] [PubMed] [Google Scholar]
- 53.Dash R, Ali MC, Dash N, Azad MAK, Hosen SMZ, Hannan MA, Moon IS. Structural and dynamic characterizations highlight the deleterious role of SULT1A1 R213H polymorphism in substrate binding. Int J Mol Sci. 2019 doi: 10.3390/ijms20246256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dutta M, Tareq AM, Rakib A, Mahmud S, Sami SA, Mallick J, Islam MN, Majumder M, Uddin MZ, Alsubaie A, et al. Phytochemicals from leucas zeylanica targeting main protease of SARS-CoV-2: chemical profiles, molecular docking, and molecular dynamics simulations. Biology (Basel) 2021 doi: 10.3390/biology10080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahmud S, Biswas S, Paul GK, Mita MA, Afrose S, Hasan MR, Shimu MSS, Uddin MAR, Uddin MS, Zaman S. Antiviral peptides against the main protease of SARS-CoV-2: a molecular docking and dynamics study. Arab J Chem. 2021 doi: 10.1016/j.arabjc.2021.103315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mahmud S, Biswas S, Paul GK, Mita MA, Promi MM, Afrose S, Hasan MR, Zaman S, Uddin MS, Dhama K, et al. Plant-based phytochemical screening by targeting main protease of SARS-CoV-2 to design effective potent inhibitors. Biology (Basel) 2021 doi: 10.3390/biology10070589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lagunin A, Ivanov S, Rudik A, Filimonov D, Poroikov V. DIGEP Pred: web service for in silico prediction of drug-induced gene expression profiles based on structural formula. Bioinformatics. 2013;29:2062–2063. doi: 10.1093/bioinformatics/btt322. [DOI] [PubMed] [Google Scholar]
- 58.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. String v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou S, Wang Y, Zhu T, Xia L. CT features of coronavirus disease 2019 (COVID-19) pneumonia in 62 patients in Wuhan, China. AJR Am J Roentgenol. 2020;214:1287–1294. doi: 10.2214/ajr.20.22975. [DOI] [PubMed] [Google Scholar]
- 61.Babadaei MMN, Hasan A, Vahdani Y, Bloukh SH, Sharifi M, Kachooei E, Haghighat S, Falahati M. Development of remdesivir repositioning as a nucleotide analog against COVID-19 RNA dependent RNA polymerase. J Biomol Struct Dyn. 2021;39:3771–3779. doi: 10.1080/07391102.2020.1767210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, Seidah NG, Nichol ST. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005 doi: 10.1186/1743-422x-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heald-Sargent T, Gallagher T. Ready, set, fuse! the coronavirus spike protein and acquisition of fusion competence. Viruses. 2012;4:557–580. doi: 10.3390/v4040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jassim SA, Naji MA. Novel antiviral agents: a medicinal plant perspective. J Appl Microbiol. 2003;95:412–427. doi: 10.1046/j.1365-2672.2003.02026.x. [DOI] [PubMed] [Google Scholar]
- 66.Islam MA, Zilani MNH, Biswas P, Khan DA, Rahman MH, Nahid R, Nahar N, Samad A, Ahammad F, Hasan MN. Evaluation of in vitro and in silico anti-inflammatory potential of some selected medicinal plants of Bangladesh against cyclooxygenase-II enzyme. J Ethnopharmacol. 2022 doi: 10.1016/j.jep.2021.114900. [DOI] [PubMed] [Google Scholar]
- 67.Sohel M, Biswas P, Al Amin M, Hossain M, Sultana H, Dey D, Aktar S, Setu A, Khan M, Paul P. Genistein, a potential phytochemical against breast cancer treatment-insight into the molecular mechanisms. Processes. 2022 doi: 10.3390/pr10020415. [DOI] [Google Scholar]
- 68.Saroni Arwa P, Zeraik ML, Ximenes VF, da Fonseca LM, Bolzani Vda S, Siqueira Silva DH. Redox-active biflavonoids from Garcinia brasiliensis as inhibitors of neutrophil oxidative burst and human erythrocyte membrane damage. J Ethnopharmacol. 2015;174:410–418. doi: 10.1016/j.jep.2015.08.041. [DOI] [PubMed] [Google Scholar]
- 69.Li X, Wang L, Han W, Mai W, Han L. Amentoflavone protects against hydroxyl radical-induced DNA damage via antioxidant mechanism. Turk J Biochem/Turk Biyokim Derg. 2014 doi: 10.1186/1999-3110-55-16. [DOI] [Google Scholar]
- 70.Ishola IO, Chaturvedi JP, Rai S, Rajasekar N, Adeyemi OO, Shukla R, Narender T. Evaluation of amentoflavone isolated from Cnestis ferruginea vahl ex DC (connaraceae) on production of inflammatory mediators in LPS stimulated rat astrocytoma cell line (C6) and THP-1 cells. J Ethnopharmacol. 2013;146:440–448. doi: 10.1016/j.jep.2012.12.015. [DOI] [PubMed] [Google Scholar]
- 71.Oh J, Rho HS, Yang Y, Yoon JY, Lee J, Hong YD, Kim HC, Choi SS, Kim TW, Shin SS, et al. Extracellular signal-regulated kinase is a direct target of the anti-inflammatory compound amentoflavone derived from Torreya nucifera. Mediat Inflamm. 2013 doi: 10.1155/2013/761506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Padilla F, Rincón A, Bou Rached L, Suárez A. Protium neglectum, Podocalyx loranthoides, and Brosimum utile source of flavonoids and other phenolic compounds with antioxidant activity. Rev Fac Farm. 2008;71:8–14. [Google Scholar]
- 73.Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA–a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.