

Purpose
Children with histologically diagnosed high-risk medulloblastoma, supratentorial primitive neuroectodermal tumor of the CNS (CNS-PNET), and pineoblastoma (PBL) have had poor survival despite intensive treatment. We included these patients in this Children’s Oncology Group trial. Molecular profiling later revealed tumor heterogeneity that was not detectable at protocol inception. Enrollment of patients with CNS-PNET/PBL was subsequently discontinued, and outcomes for this part of the study are reported here.
Patients and Methods
In this phase III, four-arm prospective trial, consenting children age 3-22 years with newly diagnosed CNS-PNET were randomly assigned (1:1) to receive carboplatin during radiation and/or adjuvant isotretinoin after standard intensive therapy. Primary outcome measure was event-free survival (EFS) in the intent-to-treat population. Molecular tumor classification was retrospectively completed using DNA methylation profiling.
Results
Eighty-five participants with institutionally diagnosed CNS-PNETs/PBLs were enrolled. Of 60 patients with sufficient tissue, 31 were nonpineal in location, of which 22 (71%) represented tumors that were not intended for trial inclusion, including 18 high-grade gliomas (HGGs), two atypical teratoid rhabdoid tumors, and two ependymomas. Outcomes across tumor types were strikingly different. Patients with supratentorial embryonal tumors/PBLs exhibited 5-year EFS and overall survival of 62.8% (95% CI, 43.4% to 82.2%) and 78.5% (95% CI, 62.2% to 94.8%), respectively, whereas patients with molecularly classified HGG had EFS and overall survival of 5.6% (95% CI, 0% to 13.0%) and 12.0% (95% CI, 0% to 24.7%), respectively. Neither carboplatin, nor isotretinoin significantly altered outcomes for all patients. Survival for patients with HGG was similar to that of historic studies that avoid craniospinal irradiation and intensive chemotherapy.
Conclusion
For patients with CNS-PNET/PBL, prognosis is considerably better than previously assumed when molecularly confirmed HGGs are removed. Identification of molecular HGGs may spare affected children from unhelpful intensive treatment. This trial highlights the challenges of a histology-based diagnosis for pediatric brain tumors and indicates that molecular profiling should become a standard component of initial diagnosis.
INTRODUCTION
Supratentorial primitive neuroectodermal tumors of the CNS (CNS-PNETs) and pineoblastomas (PBLs) have historically been treated as a single group as a result of their rarity, characterization as poorly differentiated high-grade neuroepithelial/embryonal tumors, and similar responses to treatment1-3. These tumors have poor outcomes, with 5-year overall survival (OS) of 48.3% to 58% for patients with CNS-PNET/PBL2-5 and 50% to 75% for patients with high-risk medulloblastoma (HR-MB).6,7 Thus, the Children’s Oncology Group (COG) study ACNS0332 was designed to test two approaches for treatment intensification for these patients: addition of carboplatin during irradiation and/or addition of isotretinoin to the adjuvant regimen. Carboplatin has demonstrated preclinical and clinical efficacy in these tumors and is well tolerated in children,2,8-10 whereas isotretinoin crosses the blood-brain barrier efficiently and is effective against preclinical models of medulloblastoma (MB).11,12
Concurrent with the closure of the CNS-PNET/PBL portion of this trial, WHO classification removed CNS-PNET as a diagnostic entity1; however, this work will use CNS-PNETs and PBLs as terms to encompass tumors that were intended for treatment in this study on the basis of the 2007 WHO classification definitions that applied at the time.
In recent years, molecular profiling using genome-wide DNA methylation analysis has been demonstrated to more accurately predict treatment outcome than standard histopathologic diagnosis in pediatric CNS tumors, such as MB and ependymoma (EP).6,13 A previous analysis of institutionally diagnosed CNS-PNET revealed a wide spectrum of discrepant molecular entities, including high-grade glioma (HGG), embryonal tumor with multilayered rosettes (ETMR), atypical teratoid rhabdoid tumor (ATRT), and EP, among others.5 Although these were retrospective findings in heterogeneously treated cohorts, these results highlight the importance of molecular investigation. In this work, we report the outcomes of patients with CNS-PNET/PBL in the ACNS0332 trial and, more importantly, the critical impact of molecular profiling on interpreting efficacy in a prospective clinical trial.
PATIENTS AND METHODS
Patients and Eligibility
Children age 3 to 22 years were eligible with newly diagnosed primary CNS-PNET or PBL. Patients had minimum functional scores (Lansky/Karnofsky performance score) of 30; adequate renal, marrow, and hepatic function; and were staged with CSF cytology and brain/spine magnetic resonance imaging. Institutional review board approval and individual informed consent were obtained before enrollment.
ACNS0332 was constructed to ensure that patients with CNS-PNET/PBL and HR-MB were evenly distributed across the four arms of this 2 × 2 trial and was planned with 90% power to detect a 16% to 17% improvement in event-free survival (EFS) on the basis of 300 eligible patients. After 200 patients were enrolled, emerging genomic data indicated that the biologies of CNS-PNET/PBL and HR-MB were different; thus, in June 2013, accrual was discontinued for patients with CNS-PNET/PBL. Participants continue to enroll on the MB strata, and outcomes for those patients will be reported separately.
Institutional Tumor Classification
Patients in the CNS-PNET/PBL strata were deemed eligible by institutional pathologists according to the 2007 WHO classification with poorly differentiated tumors with the capacity for divergent differentiation, including embryonal tumor with abundant neuropil and true rosettes, ependymoblastoma, and medulloepithelioma, and pineoblastoma.
Pathology and Neuroimaging Central Review
Formalin-fixed, paraffin-embedded tumor slides were retrospectively centrally reviewed, although this review did not affect eligibility or treatment. Histopathologic criteria required a densely cellular tumor with evidence of neuronal differentiation. Malignant gliomas were defined as highly cellular glial fibrillary acidic protein (GFAP)–positive tumors with absent or only focally present synaptophysin staining. H3K27 and H3G34 HGGs had not yet been described, although differentiation into ATRT, ependymoblastoma, and embryonal tumor with abundant neuropil and true rosettes was considered.
Imaging was retrospectively centrally reviewed by neuroradiologists who were blinded to outcome to confirm site(s) of disease, degree of resection, and time points for response and progression.
Study Design
Primary objectives were to determine whether carboplatin radiosensitization or adjuvant isotretinoin increased EFS. In brief, all patients underwent surgical resection, followed by irradiation (36 Gy craniospinal, tumor bed boost to 55.8 Gy) with vincristine (1.5 mg/m2 per week), and were randomly assigned to the addition of carboplatin during irradiation. Subsequently, patients received six cycles of maintenance chemotherapy—cisplatin, vincristine, cyclophosphamide—and were randomly assigned to receive isotretinoin (Data Supplement). Tissue for central pathologic review was mandatory. Patients were asked to allow optional tumor biology analysis.
Random Assignment and Masking
Random assignment (Data Supplement) was stratified between disease groups and based on dissemination (M0 v M+) and degree of postoperative residual disease (> 1.5 cm2 v ≤ 1.5 cm2). Patients were randomly allocated (1:1) to receive one of four regimens: regimen A, no carboplatin/no isotretinoin; regimen B, plus carboplatin/no isotretinoin; regimen C, no carboplatin/plus isotretinoin; and regimen D, plus carboplatin/plus isotretinoin.
Molecular Analysis
DNA methylation profiling was performed for cases with sufficient tumor DNA using the Infinium HumanMethylation450(450k) or the EPIC BeadChip arrays (Illumina, San Diego, CA) as described previously.5,14 Five to 10 formalin-fixed, paraffin-embedded sections per tumor were used for DNA isolation. Tumors were classified using the recently developed brain diagnostic classifier algorithm (www.molecularneuropathology.org)24 on the basis of unsupervised cluster analyses of the 60 methylation profiles, together with the methylation profiles of 216 reference cases that represented 27 distinct molecular brain tumor entities.14 All analyses were performed in R version 3.3.0 (https://www.R-project.org).
For unsupervised hierarchical clustering, we selected the 10,000 most variably methylated probes across the data set (standard deviation > 0.283). Distance between samples was calculated using the 1-Pearson correlation coefficient. Average linkage was used to generate dendrograms. The same distance matrix was used to perform the t-distributed stochastic neighbor embedding (Rtsne version 0.11) analysis. The following nondefault parameters were used: θ = 0 is distance = T, pca = F, max_iter = 10,000. Methylation probes in the heatmap representation were reordered by hierarchical clustering using Euclidean distance and average linkage.
Statistical Analysis
CNS-PNET/PBL enrollment was closed early as described, acknowledging that the statistical power to detect the benefit of carboplatin or isotretinoin for these patients would be limited, particularly if genomics revealed diagnostic heterogeneity. EFS was defined as the interval from the date of enrollment to the first event—disease progression/recurrence, second malignant neoplasm, or death from any cause—or to the date of the last follow-up for patients without events. OS was defined as the interval from enrollment to the date of death or last follow-up. Survival estimates were reported using the Kaplan-Meier method with 95% CIs. Standard errors were calculated using the Peto and Pike method.15 Log-rank test was used to compare outcomes among groups, and Cox proportional hazards regression analysis was used to examine associations between outcomes and age at enrollment. For the primary analyses that examined the effects of carboplatin and isotretinoin for all patients, one-sided stratified log-rank tests were used per the study design. Tests were stratified by study stratum (M+ v M0 with < 1.5 cm2 residual disease v M0 with > 1.5 cm2 residual disease) and other randomized treatment assignments. All other subgroup analyses were unplanned and reported P values were based on two-sided, unstratified exact tests. Data current as of December 31, 2016, were used.
RESULTS
Between March 2007 and July 2013, 87 patients with institutionally diagnosed CNS-PNET/PBLs were enrolled and randomly assigned between regimens A (n = 23), B (n = 19), C (n = 21), and D (n = 22). Two patients had tumors that were located in the posterior fossa on central radiographic review and were removed from the data analysis. No patients were ineligible on the basis of other inclusion/exclusion criteria. Of the analyzed 85 patients (median age, 9.7 years; range, 3.0 to 18.1 years; 47 females), 22 (26%) had metastatic disease and 26 (31%) had significant local residual disease. Other clinical details are included in the Data Supplement.
Outcomes for the Overall Patient Population
Forty-eight (56%) of 85 patients were alive at last follow-up, with median follow-up of 5.9 years (range, 2.9 to 9.1 years). First events included relapsed/progressive disease in 44 patients, second malignancy in one patient (nerve sheath tumor [regimen D]), and death in one patient (infection-related cardiac arrest [regimen C]). Median time to failure for patients who experienced relapse was 1.4 years (n = 44; range, 0.2 to 6.2 years), and median time to death for the 37 patients who died was 2.1 years (range, 0.3 to 7.7 years). OS and EFS for the entire group of patients were 58.6% (95% CI,46.4% to 70.9%) and 47.3% (95% CI, 34.8% to 59.8%) at 5 years (Data Supplement). Differences in EFS and OS among the four regimens were not statistically significant at the nominal .05 level (P = .087 and P = .065, respectively); however, carboplatin demonstrated a somewhat improved EFS and a trend toward significance for OS in the overall population (P = .045 and P = .056, respectively; Data Supplement), whereas no difference in outcome was observed on the basis of receipt of isotretinoin (Data Supplement). Grade ≥ 3 toxicity was most commonly myelosuppression in regimens that contained carboplatin or during the continuation phase with isotretinoin. Other notable toxicities included hearing impairment and weight loss, which were higher in carboplatin-containing regimens, and peripheral neuropathy, which was higher in regimen D (Data Supplement).
DNA Methylation Profiling Identified Varying Molecular Tumor Types in the Trial Cohort
DNA methylation profiling demonstrated that CNS-PNET consists of a range of molecularly distinct tumor entities5; therefore, we analyzed all tumors in the trial cohort by DNA methylation profiling to differentiate molecular entities and compare outcomes between these groups. Of the 85 patients, 77 consented to molecular phenotyping, of whom 73 also had at least five slides available for DNA extraction. For 60 cases, this yielded sufficient DNA; molecularly defined outcomes are based on these cases.
Profiling revealed a spectrum of molecular diagnoses that was broader than that found on institutional and central pathology review (Table 1). Most pineal region tumors were molecularly classified as PBL (26 [90%] of 29). The three other molecular diagnoses in this region were two ATRTs and one MB. However, CNS-PNETs comprised a heterogeneous mixture of molecular entities, including HGG (n = 18), ATRT (n = 2), CNS neuroblastoma with FOXR2 activation (n = 3), ependymoma, RELA positive (n = 2), MB (n = 1), ETMR (n = 1), PBL (n = 1), and high-grade neuroepithelial tumor with MN1 alteration (n = 1). Two remaining tumors could not be classified and were designated as embryonal tumor, not otherwise specified (Fig 1). Overall, 22 of the CNS-PNETs (71%) represented molecularly defined tumor entities that were not intended for trial inclusion. Initial central neuropathologic review demonstrated concordance with molecular diagnosis more reliably than institutional analysis, identifying three of four molecularly defined ATRTs, one of two EPs, one of one ETMR, and five of 18 HGGs (Table 1).
Table 1.
Methylation Profiling Results With Subsequent Grouping by Molecular Identification (n = 60) and Central Pathology Review Concordance
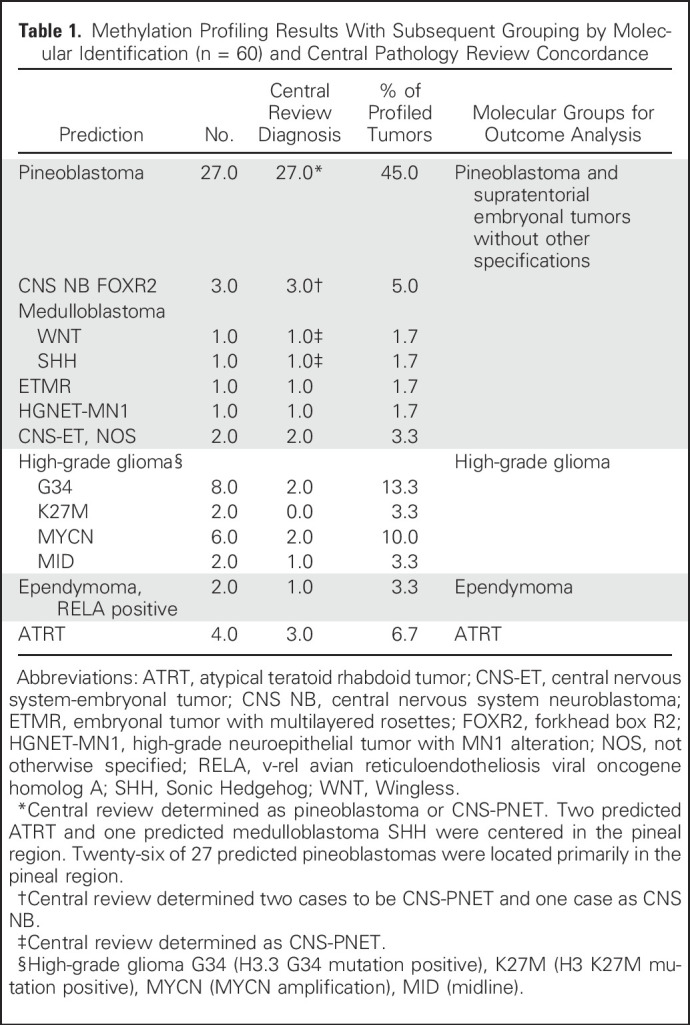
Fig 1.

Methylation profiling identifies disparate molecular diagnoses in tumors pathologically classified institutionally as supratentorial primitive neuroectodermal tumor of the CNS (CNS-PNET) and pineoblastoma (PBL). Tumors were molecularly classified on the basis of unsupervised cluster analyses of the 60 CNS-PNET/PBL methylation profiles, together with the methylation profiles of 216 reference cases that represented 27 distinct molecular brain tumor entities. AD, adult; ATRT, atypical teratoid rhabdoid tumor; BCOR, bcl6 corepressor; CNS EFT-CIC, Ewing sarcoma family tumor with CIC alteration; CIC, capicua transcriptional repressor; CNS NB-FOXR2, forkhead box R2; CPC, choroid plexus carcinoma; CPP, choroid plexus papilloma; EPN, ependymoma: ET, embryonal tumor; GBM, glioblastoma multiforme; HGNET-MN1, high-grade neuroepithelial tumor with MN1 alteration; INF, infant; MB, medulloblastoma; MELCYT, melanocytoma; MNG, meningioma; NOS, not otherwise specified; PB, pineoblastoma; PXA, pleomorphic xanthoastrocytoma; RELA, v-rel avian reticuloendotheliosis viral oncogene homolog A; SHH, Sonic Hedgehog; TSNE, stochastic neighbor embedding (tSNE) method; WNT, Wingless; YAP, yes-associated protein 1.
For outcome analysis, we grouped patients with molecularly diagnosed supratentorial embryonal tumors (ET) without other specification and PBLs together (n = 36; including PBL, CNS neuroblastoma, MB, ETMR, high-grade neuroepithelial tumor, and embryonal tumor, not otherwise specified). ATRT was excluded from this group as it is been considered a unique tumor with ATRT-specific treatment algorithms and was not intended for trial inclusion. Patients with a molecular diagnosis of HGG were analyzed separately and were disproportionately represented in regimen C (eight of 18; Data Supplement).
Secondary Central Pathology Review
Ten of the 60 methylation-profiled cases were selected for a second central pathology review, given the discrepancies between profiling and centrally reviewed histopathologic diagnoses. Of these 10 tumors, six had a molecular diagnosis of glioblastoma multiforme (GBM) H3F3A/G34. Central review diagnosis was PNET (n = 4), neuroblastoma with large-cell/anaplastic features (n = 1), and malignant neoplasm (n = 1). These cases were stained immunohistochemically for Olig2, ATRX, p53, GFAP, and synaptophysin within the limits of available tissue. An additional tumor—originally considered PNET—was profiled as a GBM H3F3A/K27 category and was stained for H3F3A/K27M. One case, called PNET, profiled as RELA-positive ependymoma and was stained with epithelial membrane antigen (EMA) on rereview. One case, called PBL by central review, was ATRT by profiling. This undifferentiated tumor did not have available tissue for INI1 staining. Finally, one tumor, originally called PNET, was embryonal, not otherwise specified on profiling.
Three of the G34-profiled tumors had focal/predominant PNET/neuroblastic architecture in hematoxylin and eosin sections, with one having Homer Wright rosettes. Of these three, two were at focally positive for synaptophysin. One was negative, but the stain was not considered interpretable. GFAP staining was questionable in one and focally positive in two. One of the two GFAP-positive cases had separate areas of synaptophysin-positive small-cell PNET-like tissue and GFAP-positive pleomorphic, phenotypically glial tissue. Olig2 staining was negative, as expected, in four of five, but was unexpectedly diffusely positive in one. ATRX staining was lost in four and retained in the Olig2-positive tumor. One of the three cases stained for p53 was strongly positive. The remaining two were negative, but a false negative could not be excluded. The RELA-positive tumor was positive on EMA staining and considered an anaplastic ependymoma with focal clear cell features on reanalysis. The last tumor, although called embryonal tumor and not otherwise specified on profiling, pathologically was a clear-cell ependymoma with many EMA-positive dots.
The single H3F3A/K27 profiled tumor was consistent histologically with glioblastoma and immunopositive for H3F3A/K27M. Thus, in the 10 cases with discrepancy between original central pathologic review and methylation-driven diagnoses, subsequent directed reanalysis was generally concordant. Although all of the G34-mutated GBMs were retrospectively considered to be consistent with that entity with variable proportions of malignant glioma and neuroblastic/PNET areas, molecular confirmation would be appropriate. The single ATRT would likely have been confirmed if tissue had existed for appropriate immunostaining.
Outcomes Were Strikingly Different Between Patients With Supratentorial ET/PBLs Versus HGG
Patients with supratentorial ET/PBL tumors (n = 36) had 5-year EFS and OS of 62.8% (95% CI, 43.4% to 82.2%) and 78.5% (95% CI, 62.2% to 94.8%), respectively (Fig 2). Within this group, there was no significant outcome difference between patients with PBL versus those with supratentorial ET (Data Supplement). This finding contrasts with the perception that patients with pineal CNS-ETs fare better than those with nonpineal CNS-ETs, which was based on three prior studies that did not have the molecular tools to exclude glioblastomas and other diagnoses from the nonpineal ET cohort.2,16,17 Patients with molecularly defined HGG (n = 18) had dramatically worse outcomes with 5-year EFS and OS of 5.6% (95% CI, 0% to 13.0%) and 12.0% (95% CI ,0% to 24.7%), respectively (Fig 2). There were no significant differences in patient characteristics between those in whom tumors were analyzed by methylation profiling and those that were not (Data Supplement).
Fig 2.
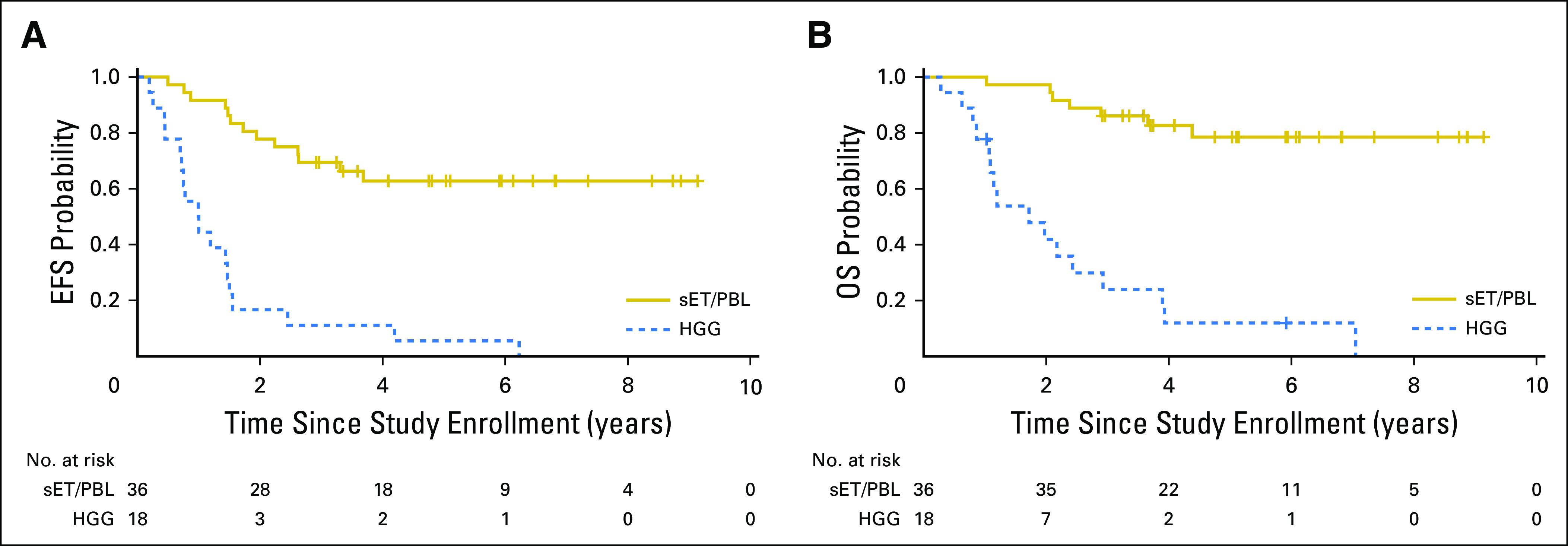
Outcome of molecularly classified high-grade glioma versus supratentorial embryonal tumor (sET)/pineoblastoma (PBL) tumors. (A) Event-free survival (EFS). Estimated EFS at 5 years for molecularly defined high-grade glioma (HGG) was 5.6% (95% CI, 0% to 13.0%)%, whereas 5-year EFS for supratentorial ET/PBL tumors was 62.8% (95% CI, 43.4% to 82.2%; P < .001). (B) Overall survival (OS). Estimated OS at 5 years for molecularly defined high-grade glioma was 12.0% (95% CI, 0% to 24.7%), whereas 5-year OS for supratentorial ET/PBL tumors was 78.5% (95% CI, 62.2% to 94.8%; P < .001).
Patterns of Failure for Defined Groups
All 18 patients with HGG experienced recurrence, most only locally (n = 10), although some exhibited distant failure alone (n = 2) or combined local and distant recurrence (n = 6). For patients with supratentorial ET/PBLs, recurrences were more evenly distributed among local relapse alone (n = 5), local and distant (n = 3), and distant recurrence only (n = 5).
Neither Carboplatin, Nor Isotretinoin Altered Outcomes for Molecularly Defined Classes
Analyses of OS and EFS for patients with supratentorial ET/PBLs as a function of the receipt of carboplatin (Figs 3A and 3B) or isotretinoin (Figs 3C and 3D) demonstrated no significant difference. Patients with HGG were not intended to be treated in this study and the number of patients with molecularly defined HGG enrolled was insufficient for conclusions; however, the survival curves (Data Supplement) may provide guidance for the design of larger trials with patients with molecularly confirmed HGG.
Fig 3.
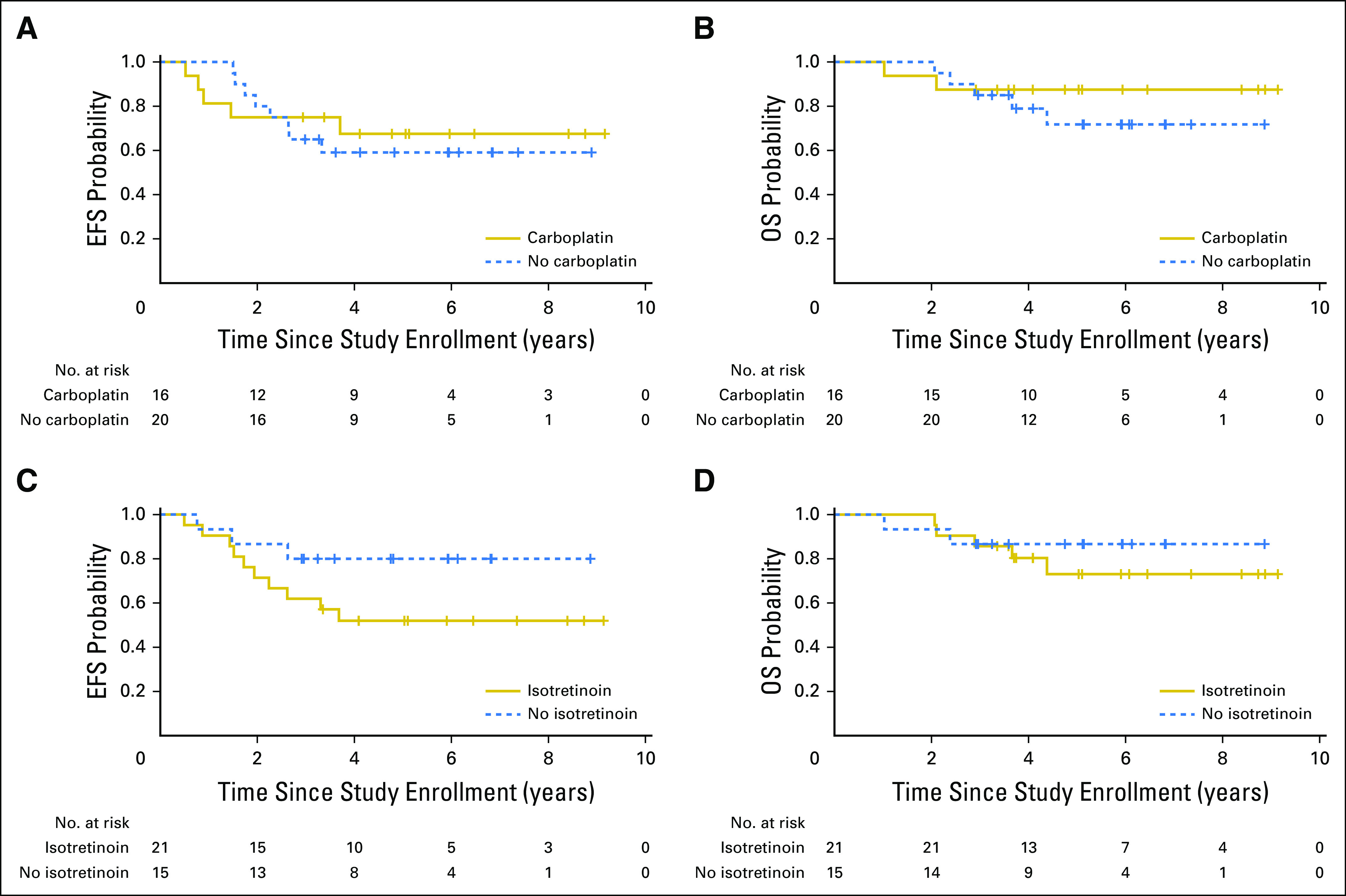
Outcome of patients with molecularly classified supratentorial embryonal tumor (ET)/pineoblastoma (PBL) tumors randomly assigned to receipt of (A and B) carboplatin (n = 36) and (C and D) isotretinoin. (A) Event-free survival (EFS) at 5 years for patients with molecularly defined supratentorial ET/PBL who received carboplatin was 67.5% (95% CI, 40.8% to 94.2%), whereas patients who did not receive carboplatin had 5-year EFS of 59.1% (95% CI, 32.8% to 85.4%) at 5 years, which was not significantly different from those who received carboplatin (P = .73). (B) Overall survival (OS) at 5 years for patients with molecularly defined supratentorial ET/PBL tumors who received carboplatin was 87.5% (95% CI, 67.3% to 100%), whereas patients who did not receive carboplatin had 5-year OS of 71.8% (95% CI, 49.3% to 94.3%; P = .46). (C) EFS at 5 years for patients with molecularly defined supratentorial ET/PBL tumors who received isotretinoin was 51.9% (95% CI, 28.4% to 75.4%), whereas patients who did not receive isotretinoin had 5-year EFS of 80.0% (95% CI, 53.5% to 100%; P = .15). (D) OS at 5 years for patients with molecularly defined supratentorial ET/PBL tumors who received isotretinoin was 73.1% (95% CI, 50.8% to 95.4%), whereas lack of isotretinoin conferred 5-year OS of 86.7% (95% CI, 66.1% to 100%; P = .63).
Bulky Residual Disease Correlated With Outcome for Molecularly Defined Subgroups
In supratentorial ET/PBL, an increased degree of residual disease in patients with M0 disease (< 1.5 cm2 v > 1.5 cm2) was associated with significantly worse OS (Fig 4). Differences in EFS for both patients with supratentorial ET/PBL and those with HGG did not reach significance (Data Supplement). In the molecularly defined HGG subset, there was some evidence that age was a significant predictor of OS (P = .037), with younger age conferring worse outcome.
Fig 4.

Impact of residual disease on overall survival (OS) for patients with molecularly classified supratentorial embryonal tumor (ET)/pineoblastoma (PBL) tumors. Estimated OS at 3 years for patients with molecularly defined supratentorial ET/PBL tumors who had minimal residual disease was 93.8% (95% CI, 81.5% to 100%) compared with and OS of 75.0% (95% CI, 47.2% to 100%) for patients with > 1.5 cm2 of residual disease (P = .012).
Metastatic status (M0 v M+) did not predict EFS (P = .62) or OS (P = .57) for either all enrolled patients or patients with supratentorial ET/PBL tumors (Data Supplement). This analysis was not performed for HGG as only two patients had M+ disease. No significant differences in outcome were observed on the basis of gender.
DISCUSSION
The current trial reveals the challenges of histology-based classification for supratentorial CNS-PNETs and indicates that upfront molecular profiling should become a standard component of initial diagnosis, complementing traditional diagnostic analyses. Rates of discordance between central and site pathologic reviews have been reported to range between 28% and 38%.2,18 Even more meaningfully, recent publications have confirmed that histopathologic diagnosis in general in CNS-PNET may be discrepant from molecular diagnosis.5,19 Despite this information, the extent of inconsistency between histopathology-based and molecularly based diagnoses in this trial was striking. Approximately three fourths of histologically diagnosed CNS-PNETs, but also some PBLs, were reclassified into different molecular entities after methylation analysis. As different tumor diagnoses trigger markedly dissimilar treatment strategies and result in widely varying outcomes, an objective diagnostic test is essential for both the interpretation of clinical trial results as well as best treatment planning for patients.
Adjudication of the discrepancies between central review and methylation profiling was in favor of the latter when reviewed and, in some cases, after additional immunostaining. It should be recognized that the histone-mutated tumors, K27 and G34, were not identified during most of this trial’s conduct. The utility of profiling was particularly apparent in G34 lesions as they can easily be classified as PNETs, although additional stains—Olig2, ATRX, and p53—are being used to identify this malignant CNS tumor.20,21 A high degree of suspicion—and additional studies—would be necessary for diagnosis in many of these cases, which may also be aided by the use of additional expert neuropathologic reviews.
Overall outcomes reported for all enrolled patients closely approximated historical trials,2,3 which suggests that the discrepant biologic diagnoses found here were likely also present in previous trials. Metastatic disease did not confer worse outcomes for either all enrolled patients or those with supratentorial ET/PBL tumors, as has been observed in some2 but not all previous reports16,22; however, the degree of residual disease was found to be prognostic in predicting OS in patients with molecularly confirmed supratentorial ET/PBL, which confirms similar conclusions in previous studies. These results emphasize the importance of extensive resection in supratentorial ET/PBLs with either primary or second-look surgery.2,16
Despite the general perception that children with CNS-PNETs/PBLs tend to have extremely poor prognoses, this study demonstrates that outcomes for these patients are considerably better when molecular HGGs are appropriately removed from outcome analysis. When patients with PBL are considered separately, outcomes seem to closely approximate historical PBL-specific outcomes, although direct comparisons are difficult given age and treatment heterogeneity.2,22 Ultimately, prospective trials may need to focus on PBL as a separate entity.
Conversely, children with molecularly diagnosed HGG treated with intensive craniospinal radiotherapy and chemotherapy fared poorly, and, with a 3-year EFS of < 12%, did no better than historical reports that used temozolomide and focal irradiation alone.23 Thus, children with molecularly defined HGG may have received potentially debilitating excess therapy and experienced unnecessarily diminished quality of life.
In conclusion, molecular analysis provides an element of objectivity in differentiating entities within a group of tumors that are sometimes difficult to distinguish histologically. Of importance, patients here were often reclassified into different diagnoses and exhibited strikingly disparate outcomes on the basis of these distinctions, which highlights the importance of accurate, upfront molecular diagnosis. Multiple sites in the United States are beginning to offer College of American Pathologists (CAP)/Clinical Laboratory Improvement Amendments (CLIA)-accredited diagnostic methylation studies. To our knowledge, our study is the first prospective trial in pediatric CNS malignancies to detail this discrepancy and underscores the critical importance of genomic-based diagnoses in the planning of appropriate clinical treatment of patients as well as in the design of future clinical trials.
ACKNOWLEDGMENT
We thank the patients and their families, as well as the institutional study teams involved in the trial for making this study possible. We also thank Charles Eberhart, MD, for assistance in the pathology reviews; Susan Conway for coordination of the trial; Richard Sposto, MD, and Shaoyu Li, MD, for statistical assistance; Natalie Beeler, Brittney Schlagenhaft, and Thomas Silver for coordination of specimen collection and analysis; Sandy Kessel for coordination of the imaging review; and Nancy Heideman for initial conceptual input. We thank the Microarray unit of the Genomics and Proteomics Core Facility, German Cancer Research Center, especially Nadja Wermke and Anja Schramm-Glück, for providing excellent methylation services.
Supported in part by National Institutes of Health Grants No. U10-CA180886 (National Clinical Trials Network [NCTN] Operations Center Grant), U10-CA180899 (NCTN Statistics and Data Center), U10-CA098543 (Chair’s Grant), U10-CA098413 (Statistics and Data Center Grant), and R01- CA114567 (to J.M.O.), as well as grants from the St. Baldrick’s Foundation, the German Childhood Cancer Foundation (“Neuropath 2.0 – Increasing diagnostic accuracy in pediatric neurooncology” [DKS 2015.01]), and the German Cancer Consortium joint funding project, “Next Generation Molecular Diagnostics of Malignant Gliomas.”
Presented at the Society of Neuro-Oncology Annual Meeting, San Francisco, CA, November 16-19, 2017.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the US National Institutes of Health.
Clinical trial information: NCT00392327.
E.I.H. and M.K. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Chris Williams-Hughes, Linda Heier, Alok Jaju, Jeff Michalski, Yimei Li, Tianni Zhou, Ian F. Pollack, Amar Gajjar, Roger J. Packer, James M. Olson
Collection and assembly of data: Eugene I. Hwang, Marcel Kool, Peter C. Burger, Lukas Chavez, Chris Williams-Hughes, Catherine Billups, Tianni Zhou, Andreas von Deimling, David T.W. Jones, Stefan M. Pfister, James M. Olson
Data analysis and interpretation: Eugene I. Hwang, Marcel Kool, Peter C. Burger, David Capper, Lukas Chavez, Sebastian Brabetz, Catherine Billups, Linda Heier, Alok Jaju, Jeff Michalski, Yimei Li, Sarah Leary, Tianni Zhou, Andreas von Deimling, David T.W. Jones, Maryam Fouladi, Amar Gajjar, Roger J. Packer, Stefan M. Pfister, James M. Olson
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Extensive Molecular and Clinical Heterogeneity in Patients With Histologically Diagnosed CNS-PNET Treated as a Single Entity: A Report From the Children’s Oncology Group Randomized ACNS0332 Trial
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Eugene I. Hwang
Research Funding: Merck Sharp & Dohme
Marcel Kool
No relationship to disclose
Peter C. Burger
No relationship to disclose
David Capper
Patents, Royalties, Other Intellectual Property: Patent application for a DNA-methylation based method for classifying tumor species of the brain by the Deutsches Krebsforschungszentrum Stiftung des Öffentlichen Rechts und Ruprecht-Karls-Universität Heidelberg (EP 3067432 A1), IDH1 R132H and BRAF V600E mutation specific antibody
Lukas Chavez
No relationship to disclose
Sebastian Brabetz
No relationship to disclose
Chris Williams-Hughes
No relationship to disclose
Catherine Billups
No relationship to disclose
Linda Heier
No relationship to disclose
Alok Jaju
No relationship to disclose
Jeff Michalski
Honoraria: Augmenix
Consulting or Advisory Role: Augmenix
Travel, Accommodations, Expenses: Augmenix
Yimei Li
No relationship to disclose
Sarah Leary
No relationship to disclose
Tianni Zhou
No relationship to disclose
Andreas von Deimling
Patents, Royalties, Other Intellectual Property: Royalties for antibodies H09 (IDHqR132H), VE1 (BRAFV600E)
David T.W. Jones
Patents, Royalties, Other Intellectual Property: Patent WO 2013075237 A1
Maryam Fouladi
Research Funding: Merck (Inst)
Ian F. Pollack
No relationship to disclose
Amar Gajjar
Consulting or Advisory Role: Celgene
Research Funding: Genentech (Inst)
Roger J. Packer
No relationship to disclose
Stefan M. Pfister
Patents, Royalties, Other Intellectual Property: DNA methylation-based diagnosis of pediatric brain tumors
James M. Olson
Leadership: Blaze Bioscience
Stock or Other Ownership: Blaze Bioscience, Presage Biosciences
Patents, Royalties, Other Intellectual Property: Patents related to peptide drugs and intratumoral microdosing
REFERENCES
- 1.Louis DN, Perry A, Reifenberger G, et al. : The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol 131:803-820, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Jakacki RI, Burger PC, Kocak M, et al. : Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: A report from the Children’s Oncology Group. Pediatr Blood Cancer 62:776-783, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pizer BL, Weston CL, Robinson KJ, et al. : Analysis of patients with supratentorial primitive neuro-ectodermal tumours entered into the SIOP/UKCCSG PNET 3 study. Eur J Cancer 42:1120-1128, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Schwalbe EC, Hayden JT, Rogers HA, et al. : Histologically defined central nervous system primitive neuro-ectodermal tumours (CNS-PNETs) display heterogeneous DNA methylation profiles and show relationships to other paediatric brain tumour types. Acta Neuropathol 126:943-946, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sturm D, Orr BA, Toprak UH, et al. : New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164:1060-1072, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramaswamy V, Remke M, Bouffet E, et al. : Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol 131:821-831, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson EM, Hielscher T, Bouffet E, et al. : Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: A retrospective integrated clinical and molecular analysis. Lancet Oncol 17:484-495, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen JC, Walker R, Luks E, et al. : Carboplatin and recurrent childhood brain tumors. J Clin Oncol 5:459-463, 1987 [DOI] [PubMed] [Google Scholar]
- 9.Douple EB, Richmond RC, O'Hara JA, et al. : Carboplatin as a potentiator of radiation therapy. Cancer Treat Rev 12:111-124, 1985. (suppl A) [DOI] [PubMed] [Google Scholar]
- 10.Gaynon PS, Ettinger LJ, Baum ES, et al. : Carboplatin in childhood brain tumors. A Children’s Cancer Study Group phase II trial. Cancer 66:2465-2469, 1990 [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Guo L, Jun-Wei L, et al. : All-trans retinoic acid modulates fas expression and enhances chemosensitivity of human medulloblastoma cells. Int J Mol Med 5:145-149, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Hallahan AR, Pritchard JI, Chandraratna RAS, et al. : BMP-2 mediates retinoid-induced apoptosis in medulloblastoma cells through a paracrine effect. Nat Med 9:1033-1038, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Pajtler KW, Mack SC, Ramaswamy V, et al. : The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol 133:5-12, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. : Minfi: A flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363-1369, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peto R, Pike MC, Armitage P, et al. : Design and analysis of randomized clinical trials requiring prolonged observation of each patient. II. Analysis and examples. Br J Cancer 35:1-39, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi SH, Kim SH, Shim K-W, et al. : Treatment outcome and prognostic molecular markers of supratentorial primitive neuroectodermal tumors. PLoS One 11:e0153443, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerber NU, von Hoff K, Resch A, et al. : Treatment of children with central nervous system primitive neuroectodermal tumors/pinealoblastomas in the prospective multicentric trial HIT 2000 using hyperfractionated radiation therapy followed by maintenance chemotherapy. Int J Radiat Oncol Biol Phys 89:863-871, 2014 [DOI] [PubMed] [Google Scholar]
- 18.Fouladi M, Hunt DL, Pollack IF, et al. : Outcome of children with centrally reviewed low-grade gliomas treated with chemotherapy with or without radiotherapy on Children’s Cancer Group high-grade glioma study CCG-945. Cancer 98:1243-1252, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Picard D, Miller S, Hawkins CE, et al. : Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: An integrative genomic analysis. Lancet Oncol 13:838-848, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sturm D, Witt H, Hovestadt V, et al. : Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425-437, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Korshunov A, Capper D, Reuss D, et al. : Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol 131:137-146, 2016 [DOI] [PubMed] [Google Scholar]
- 22.Reddy AT, Janss AJ, Phillips PC, et al. : Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer 88:2189-2193, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Cohen KJ, Pollack IF, Zhou T, et al. : Temozolomide in the treatment of high-grade gliomas in children: A report from the Children’s Oncology Group. Neuro-oncol 13:317-323, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capper D, Jones DTW, Sill M, et al. : DNA methylation-based classification of central nervous system tumours. Nat Commun 555:469-474, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]