

Abstract
Background:
Angiogenesis is a dynamic process that involves expansion of a pre-existing vascular network that can occur in a number of physiologic and pathologic settings. Despite its importance, the origin of the new angiogenic vasculature is poorly defined. In particular, the primary subtype of endothelial cells (capillary, venous, arterial) driving this process remains undefined.
Methods:
Endothelial cells were fate-mapped using genetic markers specific to arterial, capillary cells. In addition, we identified a novel venous endothelial marker gene (Gm5127) used it to generate inducible venous endothelial-specific Cre and Dre driver mouse lines. Contributions of these various types of endothelial cells to angiogenesis were examined during normal postnatal development and in disease-specific setting.
Results:
Using a comprehensive set of endothelial subtype-specific inducible reporter mice, including tip-, arterial- and venous- endothelial reporter lines, we showed that venous endothelial cells are the primary endothelial subtype responsible for the expansion of an angiogenic vascular network. During physiologic angiogenesis, venous endothelial cells proliferate, migrating against the blood flow, and differentiating into tip, capillary and arterial endothelial cells of the new vasculature. Using intravital 2-photon imaging, we observed venous endothelial cells migrating against the blood flow to form new blood vessels. Venous endothelial cell migration also plays a key role in pathologic angiogenesis. This was observed both in formation of arterio-venous malformations in mice with inducible endothelial-specific Smad4 deletion mice and in pathologic vessel growth seen in oxygen-induced retinopathy.
Conclusions:
Our studies establish venous endothelial cells are primary endothelial subtype responsible for the normal expansion of vascular networks, formation of arterio-venous malformations and pathologic angiogenesis. These observations highlight the central role of the venous endothelium in normal development and disease pathogenesis.
Keywords: venous endothelial cells, angiogenesis, arteriogenesis, vascular remodeling, lineage tracing, fate mapping, 2-photon intravital imaging, arterio-venous malformations, neovascular tuft formation
Summary:
Venous endothelial cells migrate against blood flow to form new capillaries and arteries
INTRODUCTION
Angiogenesis is a dynamic process responsible for the expansion and remodeling of vascular network that involves endothelial proliferation and migration. When tissues such as the retina or the brain are exposed to hypoxia, various cell types including macrophages, astrocytes and neurons, respond by upregulating a hypoxia-inducible factor (HIF1α) that, in turn, induces expression of a number of angiogenic genes. Among them is a family of vascular endothelial growth factors (VEGFs)1. VEGFs, and in particular VEGF-A, are thought to drive angiogenesis by stimulating endothelial proliferation and migration. Yet what type of endothelial cells respond to this VEGF stimulation has remained undefined. In the traditional model of angiogenesis, endothelial cells (ECs) in the angiogenic niche respond to angiogenic signals by migrating towards the signal with the proximal most ECs becoming tip cells (a transient EC fate state) and then proliferating, forming new vascular structures and then differentiating into capillary (CapiECs), arterial (AECs), and venous (VECs) endothelial subtypes2. But the identity of the cell population primarily driving this process has remained elusive. Prior studies, using a tip endothelial cell (TipEC)-specific reporter line3, suggested that this endothelial subtype contributes to formation of arteries, in process becoming AECs. However, since TipECs are a transient endothelial subtype, it remains to be established which of the “stable” EC subtypes (e.g. arterial, venous or capillary) contribute to neovascularization via the tip cell stage. Studies in zebrafish tailfin regeneration model suggested that VECs migrate against blood flow and differentiate into TipECs and AECs4. Whether this process occurs in a mammalian vasculature and plays a role in normal developmental angiogenesis and in disease pathogenesis has not been described to date.
Arteriovenous malformations (AVM) are a common vascular disorder characterized by abnormal connection between arteries and veins and known to be associated with misdirected endothelial alignment against blood flow. A number of genes have been linked to AVM development including Endoglin (Eng), Smad-4 (Smad4), Alk1 and Notch-1/4. Interestingly, these are largely arterial endothelial genes or genes showing enriched AEC expression5, 6 suggesting that that is the type of endothelial cells involved in its development. Misdirected ECs alignment has been thought to be the key process responsible for AVMs. For instance, in Endoglin (Eng) mutant zebrafish, altered EC shape associated with misdirected endothelial alignment leads to AVM formation between dorsal aorta and cardinal vein7. Smad4-deficient ECs which is known to be associated with AVMs also showed misdirected EC alignment and migration against fluid flow8, and arterioles have been proposed to be the initial site of AVM formation9. Yet only venous/capillary specific deletion of Eng using inducible Apj Cre, but not an AEC specific deletion, leads to AVM formation6.
A key difficulty in addressing this issue has been the lack of a VEC-specific reporter line. To address this problem, we have identified and characterized a VEC-specific gene and used it to fate map venous endothelium to study its role in normal developmental and pathologic angiogenesis. We find that only venous, but nor capillary or arterial endothelial cells, respond to VEGF signaling, undergo proliferation and reverse migration, form the new vasculature and differentiate into capillary and arterial endothelium. VEC is also the primary EC subtype responsible for formation of AVMs and pathologic angiogenesis in the setting of oxygen-induced retinopathy. Taken together, our data highlight on unexpected role of VECs in developmental and pathological angiogenesis and identifies venous endothelium as a potential therapeutic target.
METHODS
Mice
Cdh5(PAC)CreERT2 (pan-endothelial Cre line)10, Bmx(PAC)CreERT2 (arterial-endothelial Cre line)11, Esm1(PAC)CreERT2 (tip-endothelial Cre line)4 and Rosa26RC::RG (Dre reporter line)12 animals have been previously described. Rosa26mTmG and Rosa26nTnG animals were obtained from Jackson Laboratory13. All experiments were performed using littermates on a C57BL/6 background unless otherwise indicated. For endothelial subtype lineage tracing experiments utilizing the pan-endothelial (Cdh5(PAC)CreERT2), tip-endothelial (Esm1(PAC)CreERT2), arterial-endothelial (Bmx(PAC)CreERT2) and venous-endothelial (Gm5127(BAC)CreERT2) specific inducible Cre mice, the animals were intraperitoneally injected with 4-hydroxytamoxifen (4-OHT, Sigma-Aldrich) (15ug for pups and 150ug for adult mice) dissolved in 10% Ethanol (Sigma-Aldrich) with corn oil (Sigma-Aldrich) (v/v) by a 30-gauge needle (Sigma-Aldrich). Mice were then harvested, as previously described14, at specified time points. The P0 time point was defined by checking experimental breeding pairs every 12hr for new litters. The following day was counted as postnatal day 1(P1). All mouse protocols and experimental procedures were conducted with approval of the Yale University Institutional Animal Care and Use Committees.
BAC clone modification for transgenic construct.
The BAC clone (RP23–113I19) derived from RPCI-23 mouse genomic library was identified using the ENSEMBL database of the Wellcome Trust Sanger Institute (http://www.ensembl.org). The BAC clone was obtained from Source Bioscience (Berlin, Germany) and used for labda Red recombinase mediated recombination to introduce an inducible Cre recombinase cassette (CreERT2)15 or inducible Dre recombinase cassette (DreERT2). The CreERT2 cassette construct was obtained from Addgene (pCAG-CreERT2)7 and the DreERT2 cassette was subcloned from pCAG-NLS-HA-Dre construct (Addgene)16. Briefly, the replacement is achieved via homologous recombination between a CreERT2/DreERT2 cassette construct and a locus on the BAC clone containing the start codon of transgene. The CreERT2/DreERT2 cassette constructs for recombination were generated by PCR amplification incorporating the coding regions of CreERT2/DreERT2 and FRT site-flanked Neo/Kan cassette. Those PCR amplificons were designed to harbor 50bp overhangs at either ends that mediate integration into the BAC at the intended position. The primes used for PCR reaction were: CreForward : TTTACTCCAGGAATCCTGCCCAACTTCACATTTTAAAAAATCGAGACCATGCCACCATGGCCAATTTACTGA, CreReverse : TGACTGTTTGACAAAACAACGGGGCCATTATATCTCACAGGCCTGAGAAAATCCGAAGTTCCTATACTTTC, DreForward : TTTACTCCAGGAATCCTGCCCAACTTCACATTTTAAAAAATCGAGACCATCGCCACCATGTCTGAGCTGA and DreReverse : TTTACTCCAGGAATCCTGCCCAACTTCACATTTTAAAAAATCGAGACCATTGTAATACGACTCACTATAG, respectively. The homologous recombination was performed in E. coli transformed with the BAC clone using the pRed/ET system17 (Gene Bridges GmbH). Cells harboring pRed/ET were cultured aerobically in 1.4 mL LB medium at a temperature of 30°C. At an OD600 of approximately 0.3, expression of Red recombinase protein was induced by the addition of 50 μL 10% (w/v) L-arabinose (Sigma-Aldrich). At the same time, the temperature was increased to 37°C to ensure maximal expression and activity of recombination proteins. After 18hr plating on LB plate with kanamycin, colonies were picked up and genotyped with PCR reaction using an AccuPower PCR PreMix (Bioneer). After selection, the Kan cassette was removed by inducing the Flp recombinase (707-FLP, Gene Bridges GmbH). To remove an open reading frame for Cysltr1, which is located on the opposite strand of same clone, one more recombination step was performed. Then, modified DNA sequence were retrieved into low copy plasmid (pHW) using gap repair homologous recombination.
Transgenesis and genotyping.
BAC DNA was purified using NucleoBond BAC 100 (Takara). It was linearized by SgrDI digestion, then purified by phenol/chloroform extraction (Sigma-Aldrich) and the quality of the linearized DNA was checked by pulse-field gel electrophoresis (Bio-Rad). The linearized DNA was then injected at a concentration of 5 ng/uL into fertilized eggs of B6;SJLF2 hybrid mice (Jackson Laboratories) at the Yale Genome Editing Center. Transgenic animals were identified by performing PCR on genomic DNA purified from tail biopsies using the Phire Animal Tissue Direct PCR Kit (Thermo Scientific). The primers used for genotyping were: CreForward : GTTTCACTGGTTATGCGGCG and CreReverse : GGTGCTAACCAGCGTTTTCG. DreForward : TGGTGGATTCCTGCGAAACA and DreReverse : GCTACGAACAGGAAAGCCCT, respectively.
Edu staining.
For detection of proliferating cells in vivo, a stock of 50mg 5-ethynyl-2-deoxyuridine (EdU) (Sigma-Aldrich) dissolved in 1mL of DMSO. This stock solution was diluted with PBS to make a working solution (10mg/mL ). Edu solution were injected intraperitoneally (200mg/kg) 4hr before the animals were sacrificed. Retinas were isolated and fixed as previously described14, and the EdU-positive cells were detected according to the user manual of the Click-iT EdU Alexa Fluor- 647 or 488 Imaging Kit (Invitrogen). The EdU-stained tissues were visualized by Leica confocal laser scanning microscope SP8 (Leica, Wetzlar, Germany).
Retina whole mount preparation.
After 4-OHT administration, mice were euthanized at specific timepoint for analysis of the retinal vasculature as previously described6. Briefly, whole mount retinas were fixed by immersion in freshly made 4% paraformaldehyde (Sigma-Aldrich) in PBS buffer for overnight at 4°C, permeabilized with 1% Triton X-100 (Sigma-Aldrich) in TBS (TBS-T), and then exposed to 1% bovine serum albumin in PBS-T for antigen blocking before immunostaining.
Vibratome section.
Brain tissues were fixed in 4% paraformaldehyde (Sigma-Aldrich) in phosphate buffer for overnight at 4°C. Brains were cut using Leica VT1000 S vibratome (150μm thickness) and washed with 1% Triton X-100 (Sigma-Aldrich) in PBS 3 times for further immunostaining.
Frozen Tissue Section.
Embryonic tissues were fixed in freshly made 4% paraformaldehyde overnight at 4°C, rinsed with PBS at room temperature, incubated in 15% sucrose overnight at 4°C, and transferred to 30% sucrose at 4°C until the tissue sank. Fixed tissues were infiltrated with Tissue-Tek O.C.T. embedding medium for 30 minutes at room temperature, transferred to an embedding mold filled with OCT, frozen on dry ice, and stored at −70°C. Frozen sections (10μm-thick) were cut at −20°C, and slides were kept at −70°C until use for Immunohistochemistry.
Immunohistochemistry.
In brief, the tissues/sections were permeabilized in ice-cold 70% ethanol (Sigma-Aldrich) for 10min at −20°C and washed with 1% Triton X-100 (Sigma-Aldrich) in TBS (TBS-T) 3 times for further immunostaining. The specimens were first incubated in blocking buffer (1% FBS, 3% BSA, 0.5% Triton-X100, 0.01% sodium deoxycholate in TBS) for 30min at room temperature and subsequently incubated in blocking buffer containing antibodies for overnight at 4°C. Next day, specimens were washed 3 times with TBS-T (10 min/wash), incubated in secondary antibody at room temperature for 1hr. After they were washed three times with TBS-T in the dark (10 min/wash), the specimens were mounted on glass slides with anti-fading mounting medium (10% 0.1M Tris buffer (pH 9.0), 0.2mg/mL p-phenylenediamine hydrochloride (Sigma-Aldrich) and 4mM sodium azide (Sigma-Aldrich) in glycerol). Confocal microscopy was performed with Nikon ECLIPSE 80i fluorescence microscope, Leica spinning disk confocal microscope or a Leica SP8 confocal microscope. ImageJ (NIH) was used for the data analysis
CLARITY tissue clearing.
Dissected brain tissues were treated using CLARITY tissue clearing protocol as previously described.18 Brains were dissected in PBS buffer and fixed in 4% PFA consequently. After 24hr fixation, the brains were sliced into 500μm section by Leica VT1000 S Vibratome, followed by 3 times washing step with PBS buffer for 10mins at room temperature. Next, brain sections were embedded with hydrogel monomers (acrylamide/bisacrylamide) (Sigma-Aldrich) 24hr at 4°C, followed by oil laid over the top of hydrogel solution and shaken at 37 °C for 4hr. The brain sections then washed with 8% sodium dodecyl sulfate (SDS) for 4 consecutive days at 37°C. After SDS washing step the brain sections became transparent, the brain sections then were washed for 2hr with a changing 1% PBS-T every 15minutes at room temperate. The brain sections were subsequently incubated with the primary antibody(1:200) in 1% PBS-T and 5% donkey serum overnight at 4°C. The sections were then washed with 1% PBS-T for one day 6 times at room temperature followed by a 4°C washing step with the same buffer. Next, brain sections incubated with a second antibody in 1% PBS-T and 5% donkey serum) for 24hr at 4°C and washed with PBS buffer for 2 consequence days at room temperate, with the washing buffer changed 6 times per day. The brain sections were finally re-cleaned with RIMS (88% Histodenz wt/vol in 0.02M phosphate buffer). The whole mount images were obtained by Leica confocal laser scanning microscope SP8 using navigator mode (Leica, Wetzlar, Germany).
Cranial window installation and 2-photon imaging.
Postnatal mice (Cdh5(PAC)CreERT2Rosa26+/nTnG) were used for cranial window installation. At P1, pups were intraperitoneally injected with 15 μg of 4-OHT. On the following day (P2), a cranial window was installed in anesthetized mice for in vivo multiphoton imaging. Briefly, P2 mice were intraperitoneally injected with 150uL of glucose solution (0.2M glucose in saline) and anesthetized using 2% isoflurane anesthesia. A tail-pinch test was used to check the level of anesthesia before surgery. Then, the skin covering the skull was sterilized by wiping it with 70% ethanol. Under a surgical microscope (Stereo Microscope Fluorescent Leica M165-FC), approximately 25mm2 of the skin covering the skull was removed using sterilized surgical scissors and the fascia of the skull was carefully removed using sterilized forceps. A sterile custom-made titanium bar was attached to the head using dental cement and saline solution was applied via pipette to the exposed tissue to prevent dehydration during the experimental process. Then, the handle of titanium bar was attached to a goniometer (GN05/M Goniometer, Thorlab) and mice were placed under a water immersion, long-working distance Olympus 20X objective lens (NA 0.95) of the 2-photon microscope. During imaging, mice were anesthetized with 0.9% isoflurane and kept on a heating pad (SmartStage, BioTherm) to maintain normal body temperature. Images were acquired using an Olympus BX61WI fluorescence microscope with a ×20, 0.95NA water immersion Olympus objective and dedicated single beam LaVision TriM scanning laser (LaVision Biotec) that controlled by Imspector software. The microscope was outfitted with a Chameleon Vision II Ti:Sapphire Laser (Coherent) with pulse precompensation. Emission wavelengths of 390–480 nm (blue), 500–550 nm (green, GFP), and 565–665 nm (orange-red) were collected with an array of 3 photomultiplier tubes (Hamamatsu). Volocity software (Improvision) was used for image acquisition and Adobe Premiere Pro software was used for the video edit and annotation.
Analysis of Publicly Available scRNAseq databases.
To check the expression of Gm5127 in various tissues and different developmental stages, we used publicly available processed data and corresponding metadata of BrainEC, Tabula Muris, MOCA and EC atlas to avoid any discrepancies between processing and annotation of the data. For BrainEC atlas, pre-processed and normalized data as well as the accompanying metadata were used from the Vanlandewijck et al. For Tabula Muris atlas, tSNE plot and processed Robjects were acquired from their website (https://tabula-muris.ds.czbiohub.org/ and https://figshare.com/projects/Tabula_Muris_Transcriptomic_characterization_of_20_organs_and_tissues_from_Mus_musculus_at_single_cell_resolution/27733) For MOCA (Mouse Organogenesis Cell Atlas), processed data for 100,000 cell from filtered data set was extracted from their website (https://oncoscape.v3.sttrcancer.org/atlas.gs.washington.edu.mouse.rna/downloads). For EC atlas, normalized dataset was downloaded from EndoDB (https://endotheliomics.shinyapps.io/ec_atlas/). The R (R-3.6.3) implementation of the ggplot2 and Seurat 3.0 software was used for t-SNE, UMAP and bar graph visualization.
Antibodies.
The following primary antibodies were used for immunostaining: NR2F2 (Invitrogen, Cat: PA5-35141); Cre (Cell Signaling, Cat: 15036); CD31 (RND, Cat: AF3628); GM130 (BDbiosciences, Cat: 610822); SOX17 (RND, Cat: AF1924) and Isolectin-B4 (Invitrogen); The secondary antibodies used were anti-mouse, rabbit or goat Alexa antibodies from the Invitrogen.
Statistics.
Data are presented as the mean ± SEM. A Student unpaired, two-tailed t test and one-way ANOVA followed by a multiple comparison procedure for pairwise comparisons were used for statistical analysis using GraphPad Prism 8.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Custom-written scripts used in this study are available from the corresponding authors upon reasonable request.
RESULT
Endothelial subtype-specific behavior during angiogenesis
To study endothelial cell migration in the developing vasculature, we first set to determine their polarization. Tio this end, we immunostained P7 retina with an antibody against an endothelial nucleus marker (ERG1/2/3) and a Golgi marker (GM130). As expected, this demonstrated EC polarization in the direction of blood flow (Figure 1A). Given that EC polarization is an important factor determining the direction of EC migration (ref), we sought to investigate the direction of EC migration in different vascular beds.
Figure 1. VECs differentiate into tip, capillary and arterial ECs in the retinal vasculature.
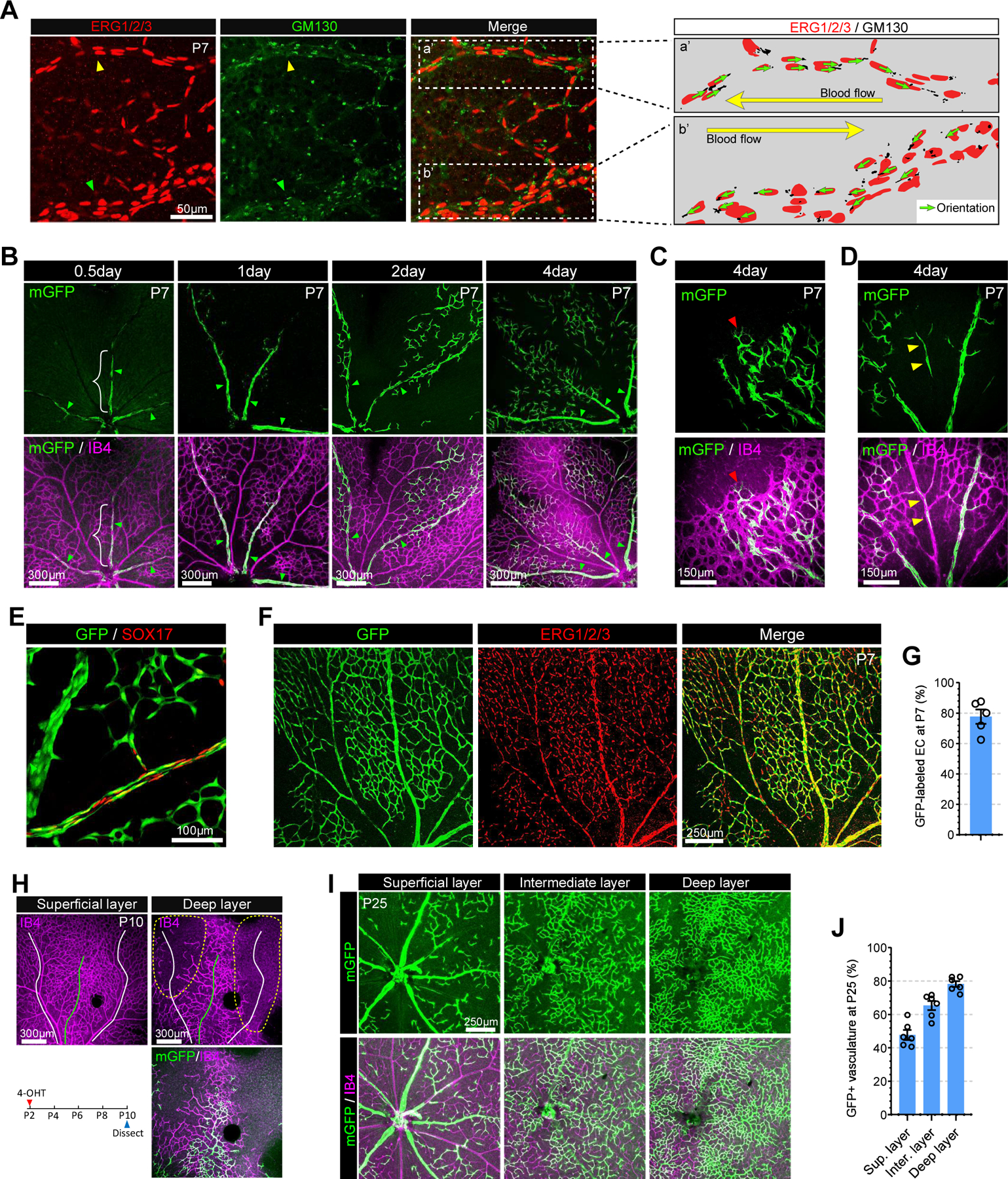
A. Confocal images showing ERG1/2/3 (red, EC nucleus marker) and GM130 (green, golgi marker) immunostaining in P7 retinal vasculature. Right panel represents endothelial nucleus and golgi in the area with white dotted rectangle (a’ : artery and b’ : vein). Note that the orientation (green arrows) of EC polarization is against blood flow (yellow arrow). B. Tracing of mGFP-expressing VECs (green) in the retinal vasculature (Isolectin B4, purple) of VECmReporter mice. Note that mGFP expressing VECs are restricted to proximal veins (white brace) at 12hr after induction. These cells are incorporated into capillaries (2days after induction) and arteries (4 days after induction). C. Immunostaining for Isolectin B4 (purple) at 4 days after induction showing mGFP-positive ECs (green) in the vascular front of P7 retinal vasculature. Red arrowhead indicates mGFP-positive TipEC D. Immunostaining for Isolectin B4 (purple) at 4 days after induction showing mGFP-positive ECs (green) in a retinal artery (yellow arrowheads). E. Confocal images showing GFP (green) and SOX17 (red) immunostaining in P7 retinal vasculature of VECiDreReporter mice when 4-OHT was administrated at P0, P1 and P2. F. Confocal images showing GFP (green) and ERG1/2/3 (red) immunostaining in P7 retinal vasculature of VECiDreReporter mice when 4-OHT was administrated at P0, P1 and P2. G. Quantification of GFP+ ECs in P7 retinal vasculature of VECiDreReporter mice. (n=5) H. P10 retinal whole mount Immunostaining of VECmReporter mice for Isolectin B4 (purple) and mGFP (green) at 8 days after induction. Upper panel shows vasculature in superficial (left) and deep layer (right). Lower panel shows 4-OHT administration strategy (left) and mGFP expression (green) in the vasculature (Isolectin B4, purple) of deep layer (right). Note that angiogenic sprouts are mainly located under a vein (green line) while the yellow dotted area under arteries and capillaries (white lines) is avascular. I. P25 retinal whole mount Immunostaining of VECmReporter mice showing vasculature (purple) and mGFP+ (green) ECs in superficial, intermediate and deep retinal layers after 4-OHT administration at P2.5. J. Quantification of GFP+ ECs in each retinal layer of VECmReporter mice. (n=6)
To trace migration of different EC subtypes during angiogenesis, we crossed TipEC specific (Esm1(PAC)CreERT2) 4 and AEC specific (Bmx(PAC)CreERT2) 11 inducible Cre lines with Rosa26mTmG Cre reporter line, and generated inducible TipEC (Esm1(PAC)CreERT2Rosa26+/mTmG (hereafter denoted as TipECmReporter)) and AEC (Bmx(PAC)CreERT2 (hereafter denoted as AECmReporter)) reporter lines. Irreversible mGFP labelling of various endothelial cell subsets was induced by treating reporter mice lines with 4-hydroxytamoxyfen (4-OHT) at various time points prior to sacrifice (Supplemental Figure I A).
In agreement with previous publications3, 4, mGFP+ TipECs in TipECmReporter mice were observed giving rise to arterial ECs 96hr after Cre activation (Supplemental Figure I B–C). At the same time, analysis of AECmReporter mice showed that mGFP+ aECs were detected only in arteries and did not give appear in other vascular beds even 3 weeks after labeling (Supplemental Figure I D–E). Since there is no available VEC reporter line, the behavior of VECs during developmental or pathological angiogenesis remains undefined. Interestingly, VECs are highly proliferative during postnatal retinal angiogenesis (P4~P11) while AECs show minimal proliferative activity (Supplemental Figure II A–B). Yet, despite this intense proliferative activity, the diameter of venous vessels was not altered during development (Supplemental Figure II C), suggesting that some of these VECs do not remain in their parent vessel but migrate to other vascular beds.
A novel gene (Gm5127) as VEC specific marker
To test this hypothesis, we set out to generate a VEC-specific reporter mouse strain. While a number of genes have been suggested to be vein-specific, such as EphB410, Vegfr319, Nr2f220 and Nrp221, recent single cell RNA-seq (scRNAseq) studies showed that these genes are also expressed in other subtype of endothelial cells and even in non-endothelial cells 22, 23. Therefore, we carried out in silico screening of candidates for VEC-specific gene expression using a publicly available scRNAseq database (BrainEC atlas)22. One gene, Gm5127, stood out for its VEC specific expression based on vascular zonation analysis (Supplemental Figure III). However, because Gm5127 has not been studied due to the lack of a corresponding gene in other species, its expression pattern in mice has not been reported. To identify the endogenous Gm5127expression pattern in various organ and different developmental stages in mice, we first took advantage of other publicly available scRNAseq datasets. Tabula Muris24, a single cell atlas of 20 mouse organs, shows Gm5127 expression in endothelial cells of the brain, spleen and skin (Supplemental Figure IV A–B) as well as in a small subset of myeloid cells in the brain (Supplemental Figure IV C–D). We could not find any endothelial expression of Gm5127 in other organs (Supplemental Figure IV A,B,E and F).
To investigate the expression of Gm5127 during embryonic development, we checked MOCA scRNAseq atlas of mouse embryonic tissues gene expression between E9.5 and E13.5 25 (Supplemental Figure V A–F). During this time interval, Gm5127 is mainly expressed in non-endothelial cells including chondrocyte progenitors, jaw and tooth progenitors and early mesenchyme cells (Supplemental Figure V A–H). Indeed, endothelial cells ranked as the 18th cell type among the 38 major cell types for Gm5127 expression between E9.5 and E13.5 (Supplemental Figure V F). Finally, since endothelial gene expression is very tissue-dependent, we examined expression of Gm5127 in the endothelium of various tissues using a recently published endothelial scRNAseq atlas of 11 mouse organs23. The atlas data indicated that Gm5127 mRNA is present only in ECs from the brain and colon (Supplemental Figure VI A–B). Furthermore, in agreement with the Brain EC atlas, Gm5127 expression in the brain is tightly restricted to VECs (Supplemental Figure VI C–D), while it is mainly expressed in capillary endothelium in the colon (Supplemental Figure VI E–F). Taken together, this distinct Gm5127 expression pattern suggests that it can be used as VEC-specific marker in the CNS vasculature. To generate a VEC-specific reporter line, we engineered a BAC clone (RP23-113I19) carrying 27kb of 5’-upstream and 70kb of 3’-downstream sequence of Gm5127 genomic locus (Supplemental Figure VII A) by replacing its open reading frame with a CreERT2-containing cassette (Supplemental Figure VII B–C). This BAC clone was then used to establish a transgenic mouse line Gm5127(BAC)CreERT2. To explore the expression of the Gm5127-driven Cre recombinase in this line, the mice were crossed with the Rosa26mTmG Cre reporter line13 to generate Gm5127(BAC)CreERT2;Rosa26+/mTmG strain (hereafter denoted as VECmReporter). Induction of Cre activity in VECmReporter mice was examined in various tissues at 12hr after 4-OHT administration. Analysis of both postnatal (P7) and adult (P60) brain vasculature showed that mGFP expression was restricted to the venous endothelium (Supplemental Figure VIII A–B). While Tabula muris data suggested the existence of a small population of GM5127-expressing myeloid cells in the brain, we were unable to find any such cells. Examination of the postnatal (P7) retinal vasculature showed that only endothelial cells in proximal, but not distal, veins were labelled (Figure 1B and Supplemental Figure VIII C). This was in agreement with antibody staining for the Cre recombinase that also demonstrated expression restricted to the proximal venous endothelium (Supplemental Figure VIII D). In agreement with scRNAseq data, we could not find any αSMA and GFP double positive ECs in P7 retinal vasculature (Supplemental Figure VIII E). In the adult (P60) retinal vasculature mGFP expression was still restricted to VEC but was evident both in first and second order veins (Supplemental Figure VIII F). In the head and tail skin vasculatures mGFP expression was largely limited to VECs except for a small subset of basal cells of the epidermis (Supplemental Figure VIII G) and a subset of smooth muscle cells (Supplemental Figure VIII H–I). While the majority of VECs in these tissues expressed mGFP, some VECs showed no mGFP expression, indicating incomplete labeling (Supplemental Figure VIII H–I). Consistent with the MOCA atlas, we could not find VEC-specific expression of mGFP in embryos (E11.5) of the VECmReporter mice (Supplemental Figure VIII J–K). We also screened mGFP expression in various organs including small intestine, diaphragm, bone, kidney, heart, lung and liver, but failed to find VEC-specific expression in those organs (data not shown). Taken together, the distinct Cre recombinase activity in VECmReporter mice suggests that this transgenic line can be used for lineage tracing of VECs in the brain, retinal and dermal vasculature in postnatal and adult mice stages.
VECs differentiate into tip, capillary and arterial endothelial cells in the developing vasculature
To trace the fate of VEC during angiogenesis in retinal vasculature, 4-OHT was administered to VECmReporter mice at various timepoints and the presence and location of mGFP+ ECs were analyzed at P7. Twelve hours (0.5 day) after labeling, mGFP+ ECs were observed only in veins while two days later they were also observed in capillaries (Figure 1B) and four days later in TipECs (Figure 1C) and AECs (Figure 1D), indicating these ECs were derived from VECs. GFP+ ECs in arteries also expressed arterial marker gene (Sox17) indicating that VEC migrated, differentiated and acquired AEC identity (Figure 1E). When 4-OHT was administered for three consecutive days at P0, P1 and P2, mGFP+ ECs accounted for ~80% of the total ECs in the P7 vasculature, indicating VECs are the major source of endothelial expansion during postnatal angiogenesis (Figure 1F–G).
After P8, the expanding vascular network on the surface of the retina reaches its periphery and begins to dive to form deep retinal blood vessel layers. Examination of the newly forming downward-directed angiogenic sprouts in the superficial layer, a step necessary for the deep layers vasculature to form, showed that these were only observed beneath veins, but not arteries or capillaries (Figure 1H, upper panel). This indicates that venous endothelium is the primary EC subtype initiating angiogenic sprouting that will form the deep layer vasculature. Indeed, ECs in these angiogenic sprouts were mainly mGFP+ in VECmReporter mice, thus demonstrating their venous origin. (Figure 1H, low panel). However, we also observed mGFP-negative sprouts (Figure 1H, low panel) beneath veins indicating that not only Gm5127-expressing VECs in proximal vein, but also VECs in distal vein (or venules) could initiate angiogenic sprouting.
To explore the origin of endothelial cells in the fully developed retinal vascular network, VECmReporter and AECmReporter mice were treated with 4-OHT at P2.5 to label, respectively, venous and arterial endothelial cells and the presence of mGFP+ ECs in various parts of the retinal vasculature was studied at P25 (Figure 1I–J and Supplemental Figure I E). As shown in Figure 1I–J, the majority (~80%) of ECs in the intermediate and deep layers were mGFP+ in VECmReporter, but not in AECmReporter mice (Supplemental Figure I E), confirming that VEC is the primary EC subtype supporting angiogenic expansion.
We also traced fate-mapped VECs in the developing postnatal brain vasculature. 12hr after labeling, only ECs in large veins and some venules in the cerebral cortex were mGFP labeled (Supplemental Figure IX A). Similarly to retinal VECs, these mGFP+ VECs then started spreading throughout the brain vasculature over time (Supplemental Figure IX A). As the brain vasculature develops, branches from arteries and veins in the cerebral cortex dive into deeper cortical layers to form a deep vascular network26. To investigate whether VECs contribute to this inner network, cleared brain tissues (Supplemental Figure IX B) were subjected to confocal microscopy followed by a 3-D reconstruction (Supplemental Figure IX C). While there were no mGFP+ ECs in deeper layers of the brain cortex 0.5 day after labelling, there were numerous mGFP-postive ECs at 4 days later, indicating that these were VECs that have migrated from superficial veins (Supplemental Figure IX C). As was the case with VECs in the retinal vasculature, VECs in the brain vasculature also differentiated into capillary and arterial endothelial cells (Supplemental Figure IX D). Finally, tracing of VECs in the skin vasculature also showed their differentiation into CapiECs (Supplemental Figure X).
VECs migrate against blood flow in the developing but not quiescent vasculature
The appearance of mGFP+ ECs in capillary and arterial positions suggests that VECs migrate against the blood flow. To observe if this reverse migration indeed occurs, we utilized a time-lapse 2-photon microscopy. Since intravital 2-photon imaging of the brain vasculature is challenging due to strong scattering caused by the skull and skin, we installed a cranial window into neonatal (P2) mice (Figure 2A) carrying the endothelial nuclear-localizing GFP (nGFP) expressing transgene, Cdh5(PAC)CreERT2Rosa26+/nTnG; hereafter denoted as panECnReporter, thereby enabling visualization of individual endothelial cells migration. The mice were subjected to 2-photon imaging 24hr after treatment with 4-OHT at P1 (Figure 2B). Time-lapse imaging demonstrated the reverse migration of VECs while ECs in other vascular beds and nRFP-labelled non-endothelial cells remained immobile during the 5hr observation period (Figure 2C, Supplemental Movie. I). All VECs showed a uniform direction of migration against blood flow (Figure 2D) moving at 8.71± 0.78 μm/hr in the P2 vasculature of the brain cortex (Figure 2E). While VECs in the postnatal stage actively proliferate, proliferation ceases once the vascular development is complete by about P30 (Supplemental Figure II A). To see if VECs migration also ceases in the quiescent vasculature, VECmReporter mice were treated with 4-OHT at P60 and P61, and the distribution of mGFP+ endothelial cells in the retinal vasculature was studied at various time points thereafter (Figure 3A). 24hr after labeling, mGFP+ VECs were only observed in main veins and venules (Figure 3B). Prolonged observation, up to 16 days after Cre activation, failed to demonstrate any migration of these mGFP+ VECs to any part of the vascular tree (Figure 3B). Similarly, tracing VECs in the brain vasculature of adult (P60) mice failed to show any VEC migration (Figure 3C).
Figure 2. VECs migrate against blood flow.
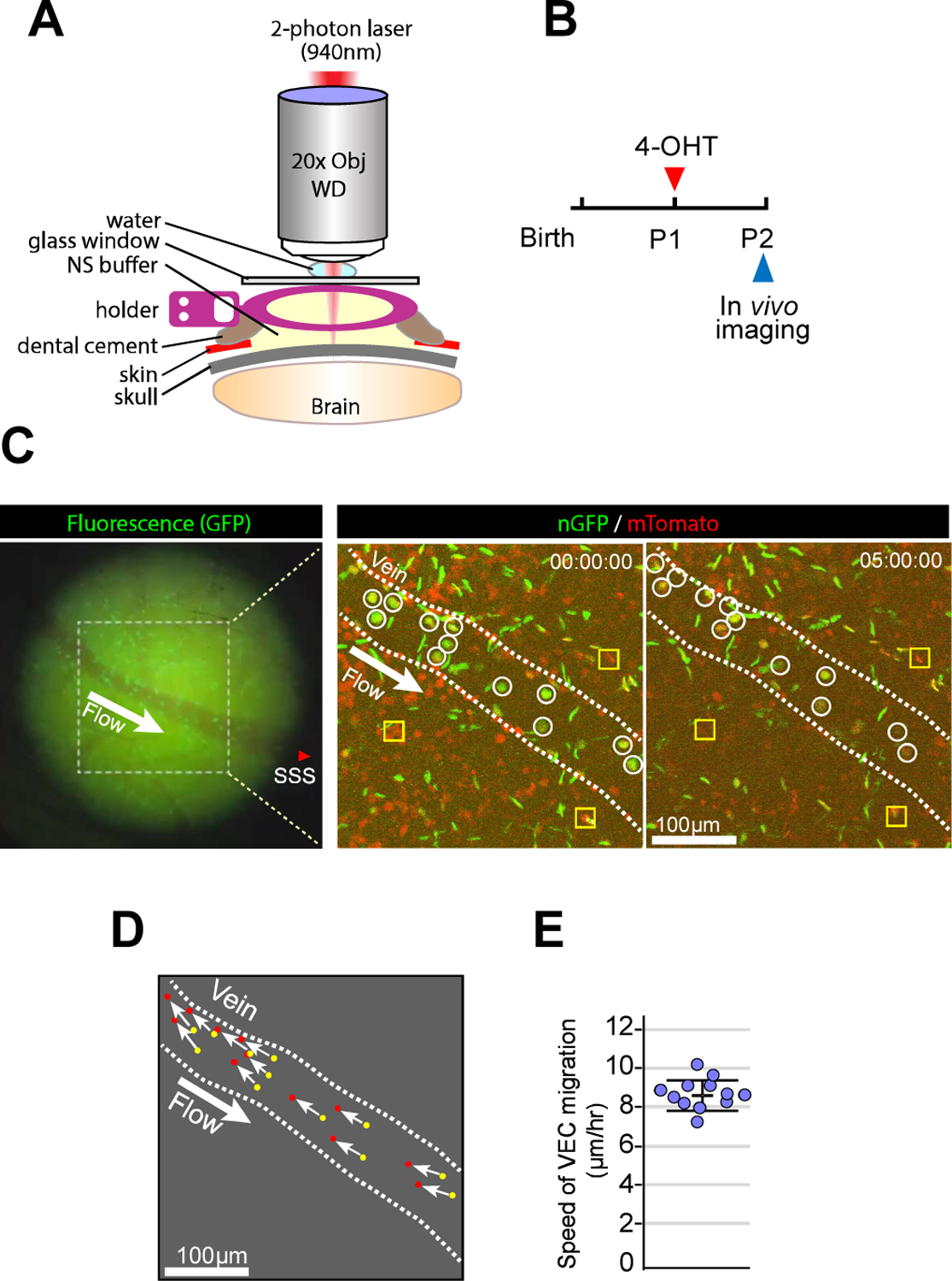
A. Schematic illustration of in vivo 2-photon imaging procedure. B. Timeline of the experimental procedure for 4-OHT administration and imaging using pan-ECnReporter mice. C. Fluorescence image from GFP channel before 2-photon microscopy (left panel) showing a first branch of superior sagittal sinus at cortex surface. The fluorescence image also provides the structural information of vessels and indicates the location of superior sagittal sinus(SSS) and the blood flow (from distal (small diameter) to proximal (larger diameter) vein). Area with white dotted rectangle in left panel was observed with 2-photon microscopy (right panel). 2-photon images from two different time points (0hr and 5hr) showing the position of individual ECs (mGFP, green) and non-ECs (mRFP, red). Note that ECs (white circles) in the vein (inner space of white dotted line) migrate against the blood flow. Yellow rectangles represent immobile cells throughout the observation. D. Trajectories (white arrow) of individual ECs in the vein from 0hr (yellow dots) to 5hr(red dots). (see Supplemental Movie. I). E. Quantification of speed of VEC migration. (n=12)
Figure 3. VECs on quiescent stage shows lack of migration.
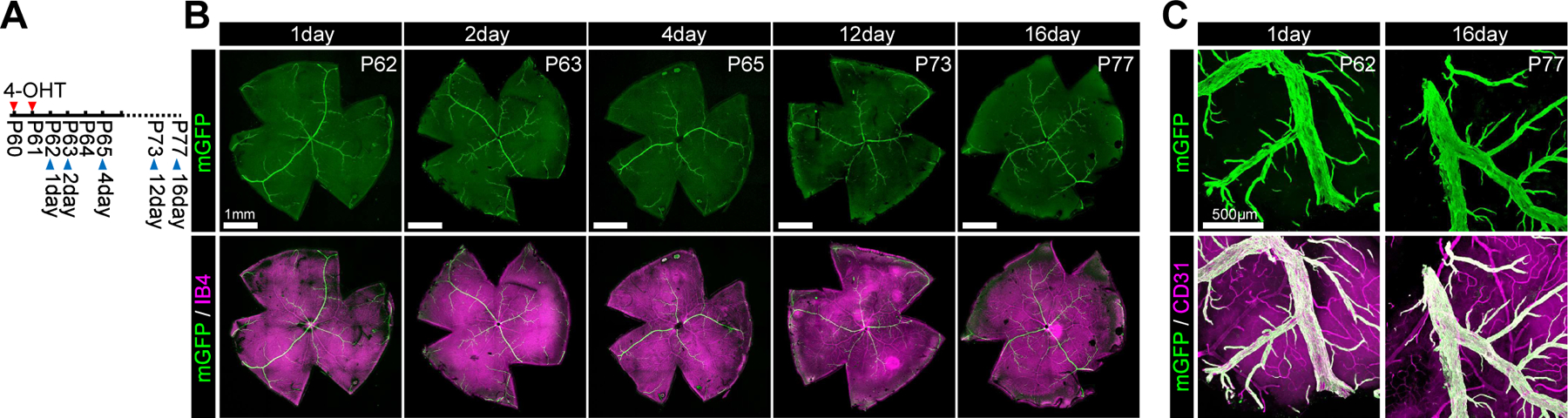
A. Timeline of the experimental procedure for 4-OHT administration and imaging. B. Confocal images of the whole mount retina immunohistochemistry showing mGFP-positive VECs (green) and vasculature (Isolectin B4, purple) on adulthood Gm5127imGFP mice as indicated timepoints after 4-OHT administration. C. Confocal images of the brain (cerebral cortex) immunohistochemistry showing mGFP-positive VECs (green) and vasculature (CD31, purple) on adulthood Gm5127imGFP mice as indicated timepoints after 4-OHT administration.
VECs are major source of endothelium in arterio-venous malformations
We next set out to explore if VECs contribute to neovascularization occurring in pathological settings such as arteriovenous malformations (AVMs). AVMs are characterized by abnormal direct connections between arteries and veins (AV shunt) and the source of ECs responsible for this abnormality has never been reported. Mice with a pan-endothelial-specific Smad4 deletion develop AVMs in various organs, including the retina27. To study the contribution of VECs to AVM formation, we set out to make a mouse line with fate-mapped VECs and an EC-specific deletion of Smad4. While the Gm5127-Cre was sufficiently strong to activate mGFP expression in the Rosa26mTmG Cre reporter line as shown above, it was not strong enough to induce the AVM phenotype in Smad4fx/fx mice. Therefore, we generated Gm5127 promoter-driven inducible Dre (iDre) recombinase expressing mice (Gm5127(BAC)DreERT2) and crossed it with a GFP-expressing Dre reporter line12 (Gm5127(BAC)DreERT2;Rosa26+/RC::RG, hereafter denoted as VECiDreReporter). By crossing panEC-specific Smad4 deletion mice (Smad4fx/fx;Cdh5(BAC)CreERT2, hereby denoted as Smad4iECKO) with the VECiDreReporter line, we could label VECs with inducible Dre recombinase and delete Smad4 in panECs with inducible Cre recombinase (Figure 4A).
Figure 4. Misdirected migration of VECs results in AV shunt formation.
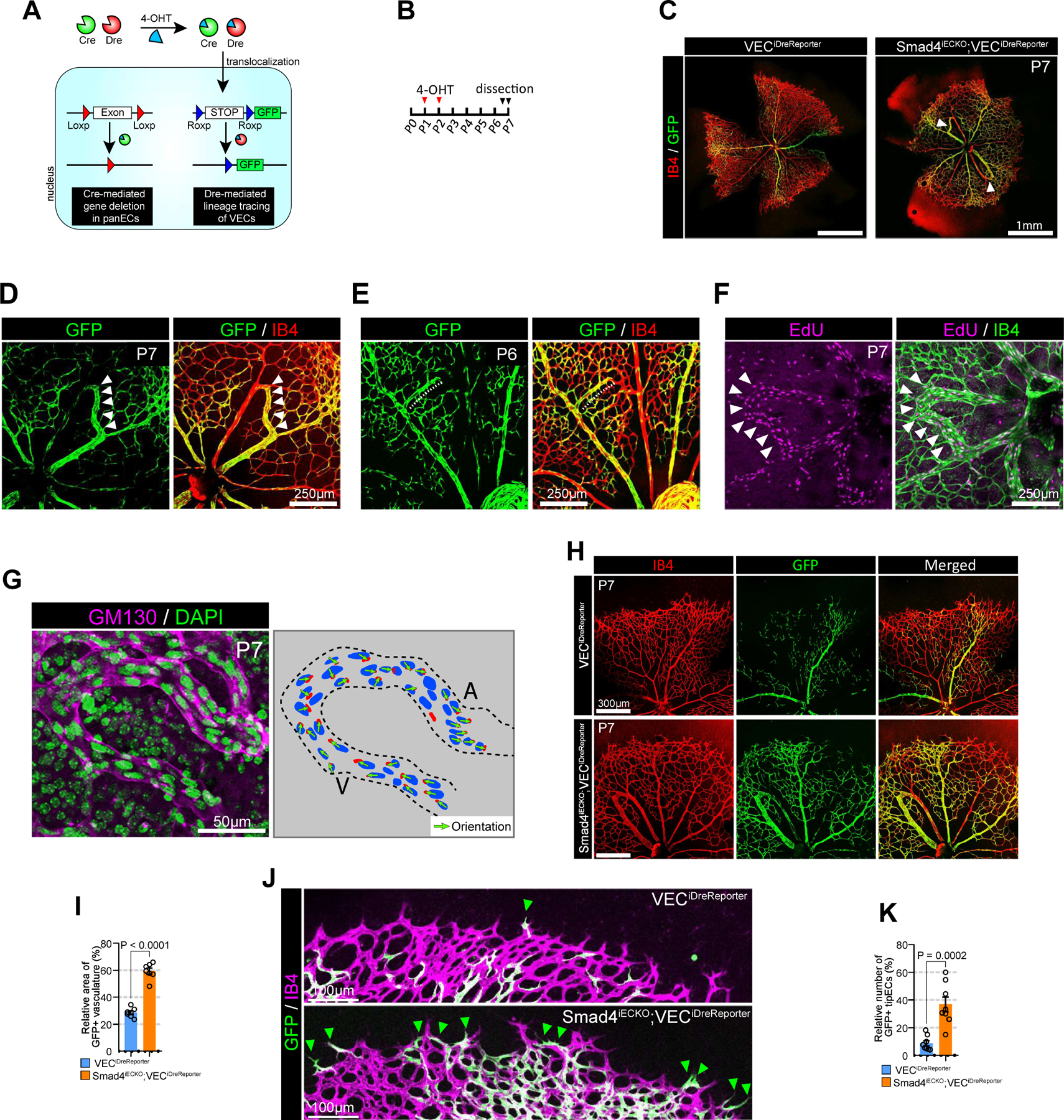
A. Schematic diagram showing a strategy for generation of Smad4iECKO;VECiDrereporter mice. Smad4 is deleted by inducible panEC specific Cre (Cdh5(PAC)CreERT2) while VECs is labeled with GFP using VEC-specific inducible Dre (Gm5127(BAC)DreERT2). B. Strategy for 4-OHT administration and analysis. C. Representative images showing the vascular phenotype (Isolectin B4 : red and GFP: green, respectively) of Smad4iECKO;VECiDrereporter (right) mice and WT littertmate(left, VECiDrereporter). White arrowheads indicate AVMs. D. Representative images showing the P7 retinal vasculature (Isolectin B4 : red, GFP: green) of Smad4iECKO;VECiDrereporter mice. White arrowheads indicate AV shunt. E. Representative images showing the P6 retinal vasculature (Isolectin B4 : red, GFP: green) of Smad4iECKO;VECiDrereporter mice. Note that vein starts to form abnormal branch (white dotted line) toward the nearby artery and ECs in the sprout are labeled with GFP. F. Confocal images showing EdU and IB4 staining in P7 retinal vasculature of Smad4iECKO mice. Note that ECs in AV shunt are highly proliferative. G. Representative images showing GM130 (cyan) and DAPI (green) immunostaining in P7 retinal vasculature (Isolectin B4 : red, GFP: green) of Smad4iECKO mice. Right panel represents endothelial nucleus and golgi in AV shunt. Note that endothelial golgi orientation in AV shunt is toward artery. H. Representative images showing the P7 retinal vasculature (Isolectin B4 : red, GFP: green) of Smad4iECKO;VECiDrereporter mice and WT littermate at low magnification. I. Quantification of GFP+ area in the P7 retinal vasculature of VECiDrereporter (blue) and Smad4iECKO;VECiDrereporter (orange) mice. (n=7, unpaired two-samples t-test) J. Representative images showing the vascular front area of P7 Smad4iECKO;VECiDrereporter mice (bottom) and WT littermate (top). (Isolectin B4 : purple, GFP: green). Green arrowheads indicate GFP+ TipECs. K. Quantification of GFP+ TipECs in the P7 retinal vasculature of VECiDrereporter (blue) and Smad4iECKO;VECiDrereporter (orange) mice. (n=8, unpaired two-samples t-test)
After Smad4 deletion and VEC fate mapping using 4-OHT administration at P1 and P2, we studied the retinal vasculature of Smad4iECKO;VECiDreReporter and VECiDreReporter (littermate control) mice (Figure 4B). In agreement with a previous report28, by P7 Smad4iECKO;VECiDreReporter mice showed the presence of AV shunts in the retinal vasculature (Figure 4C). Strikingly, most endothelial cells in these AV shunts were GFP-positive (Figure 4D), suggesting that VEC is the main endothelial subtype contributing to AV shunt formation. To better visualize this process, we examined the P6 retinal vasculature of Smad4iECKO;VECiDreReporter mice. During normal retinal vessel development, veins do not form branches toward the nearby artery. But, in P6 Smad4iECKO;VECiDreReporter mice, we could observe an abnormal GFP+ vein branch going towards the artery (Figure 4E). At P7, this GFP+ branch was directly connected to an artery thereby forming an AV shunt (Figure 4D). ECs in the AV shunt showed strong proliferation activity (Figure 4F) and were polarized towards the artery (Figure 4G) indicating that highly proliferative VECs migrate towards the arterial side of the shunt. In addition to AV shunt formation, the area of GFP+ vasculature (Figure 4H–I) and the number of GFP+ TipECs (Figure 4J–K) were increased in Smad4iECKO;VECiDreReporter mice compared to control littermates indicating altered VEC migration in the vasculature of Smad4iECKO;VECiDreReporter mice.
VEC is the primary endothelial subtype initiating neovascularization in OIR
We also evaluated the role of VECs in pathologic neovascularization occurring in the oxygen-induced retinopathy (OIR) model. In this model, high oxygen environment (75% oxygen from P7 to P12) 29 (Figure 5A–B) induces formation of an avascular vaso-obliteration (VO) zone. A return to normoxia at P12 initiates angiogenic sprouting to revascularize the VO zone, a process that involves formation of neovascular tufts at P17 with the VO zone is fully recovering its vasculature by P25 (Figure 5B). Since the VO zone at P12 is surrounded by three different endothelial subtypes (arterial, venous, and capillary) (Figure 5C) and the neovascularization process to recover the VO zone is known to be mediated by hypoxia-induced VEGF signaling30, observation of the VO zone would indicate which EC subtype is the most responsive. Examination of the forming VO vasculature showed that angiogenic sprouts at P14 were observed only arising from veins (Figure 5D–E) where most of proliferating ECs were located. At the same time, ew proliferating ECs were found in arteries (Figure 5D and F), suggesting VEC is the primary EC subtype initiating neovascularization in OIR.
Figure 5. Vein is the primary site for neoangiogenic spout initiation in OIR-induced neovascularization.
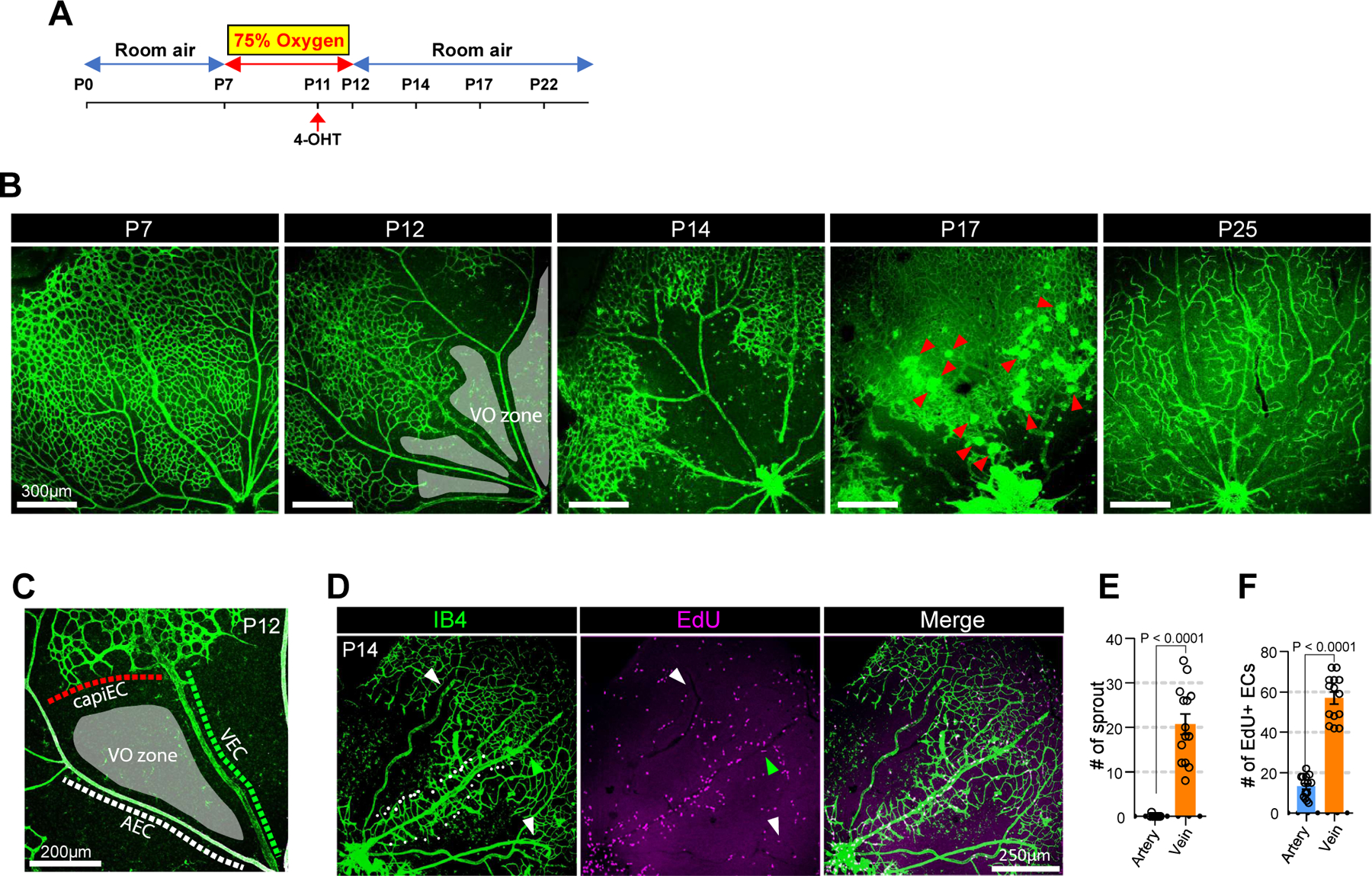
A. schematic diagram showing the experimental design of the OIR model. Postnatal pups along with their nursing female were exposed to 75% oxygen from P7 to P12 to induce vaso-obliteration and returned to room air from P12 until further analysis. 4-OHT was injected at P11 to label VECs in VECiDrereporter mice before dissection at indicated time points. B. Time course observation of vasculature (Isolectin B4, green) during OIR at indicated time points (P12, P14, P17 and P22, respectively). At P12, the vasculature forms vaso-obliteration zone (VO zone, gray-colored area). At P14, the vasculature initiates neovascularization. At P17, the vasculature shows strong angiogenic activity and forms neovascular tuft (red arrowheads). At P25, the VO zone is fully recovered. C. Representative image showing the endothelial environment of vaso-obliteration zone. (VO zone) Note that VO zone is surrounded by three different endothelial subtypes including AEC, VEC and CapiEC. D. Representative images showing EdU staining (purple) in the P14 vasculature (Isolectin B4, green) of OIR model. Note that only vein (green arrowhead) initiates angiogenic sprouting (white dots), but not artery (white arrowheads). E. Quantification of angiogenic sprouts from artery (blue) and vein (orange) at P14 OIR retina. (n=14, unpaired two-samples t-test) F. Quantification of Edu-positive ECs in artery (blue) and vein (orange) at P14 OIR retina. (n=14, unpaired two-samples t-test)
To further test the origin of ECs during neovascularization in VO zones, we used VECiDreReporter mice. 4-OHT was administered to VECiDreReporter mice at P11 (a day before normoxia was restored) and the retinal vasculature was analyzed at indicated timepoints (Fig 6). As expected, only VECs were labeled in the P12 OIR retinal vessels (Figure 6A). In agreement with observations noted above, the neovascularization process in this model was exclusively initiated by VECs (Figure 6A). About 80% of TipECs in the P17 VO zone were GFP+ (Figure 6B–C) and all neovascular tufts were GFP-positive (Figure 6D) indicating their origin from the venous endothelium. By P21, whilst the avascular area around veins largely recovered due to ingrowth of GFP+ sprouts initiating from veins, areas around AECs and CapiECs remained avascular (Figure 6E) showing that AECs and CapiECs do not respond to hypoxia-induced VEGF signaling. At P25 OIR retinal vasculature, the VO zone is completely recovered and the majority (>80%) of ECs in those area was GFP positive (Figure 6F–H) showing VEC is the primary EC subtype involved in the de novo angiogenesis.
Figure 6. VEC is the primary source of ECs in OIR-induced neovascularization.
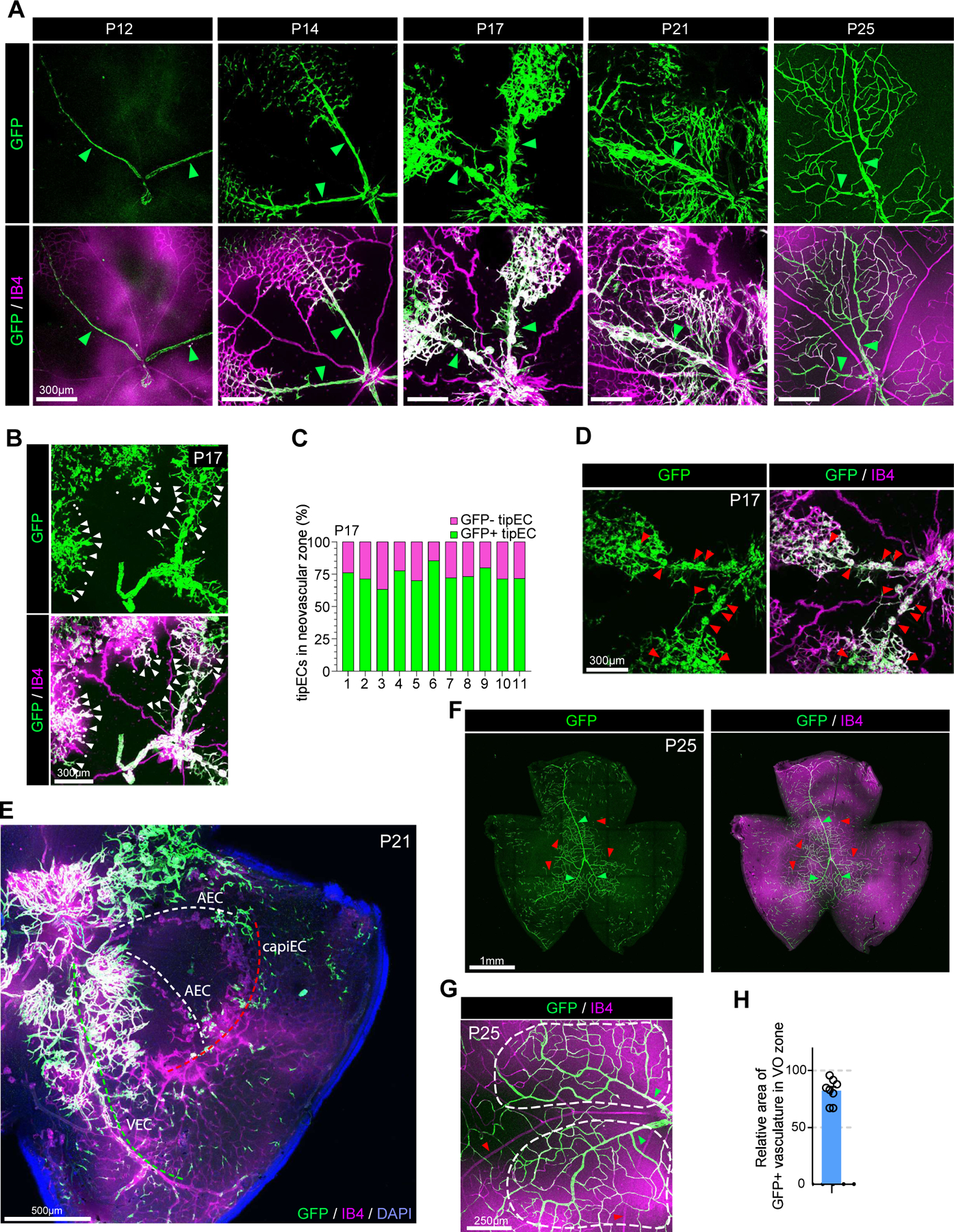
A. Time course observation of the vasculature (Isolectin B4, purple) and GFP+ VECs (green) in VECiDrereporter mice during OIR at indicated time points (P12, P14, P17, P21 and P25, respectively). Green arrowheads indicate veins. At P12, only veins express GFP showing VEC specific labeling in VECiDrereporter mice. At P14, GFP+ VECs start migration from veins. At P17, veins, but not arteries, initiate angiogenic sprouting. At P21, the majority of ECs in VO zone of P21 retinal vasculature are GFP+. At p25, the VO zone is completely recovered by GFP+ ECs showing that they are originated from VECs. B. Representative images showing GFP+ TipECs in P17 retinal vasculature of VECiDrereporter mice. (white arrowheads : GFP-positive TipEC, white dots: GFP-negative TipECs) C. Quantification of relative number of GFP+ TipECs versus GFP- TipECs in the neovascular zone of VECiDrereporter mice (P17). X-axis shows individual sample (n=11). D. Representative images showing neovascular tufts (red arrowheads) and GFP expression in P17 retinal vasculature of VECiDrereporter mice. Note that all neovascular tufts are GFP+ showing ECs in the tuft are originated from VECs. E. Representative images showing vasculature in P21 OIR of VECiDrereporter mice (DAPI: blue, GFP: green, Isolectin B4 : purple) Note that the VO zone around vein (green dotted line) is recovered by neovascularization, but not artery (white dotted line) and capillary (red dotted line). F. Representative images showing the vasculature (Isolectin B4 : purple) and GFP-expressing ECs (green) in P25 OIR of VECiDrereporter mice. Note that the majority of VO zone is recovered by GFP+ ECs. Red and green arrowheads indicate arteries and veins, respectively. G. Representative images showing the VO zone (the area in white dotted lines) in P25 OIR of VECiDrereporter mice (Isolectin B4 : purple, GFP: green). Red and green arrowheads indicate arteries and veins, respectively. H. Quantification of GFP+ area in the vasculature of VO zone in P25 OIR. (n=8)
Discussion
The long-unanswered question in the field of angiogenesis has been the source of endothelial cells responsible for the new vessel growth postnatally and in adult tissues. Here, using new venous-specific mapping with Cre- and Dre-reporter mice, we provide evidence that venous endothelial cells are the primary EC subtype responsible for angiogenic expansion of the vasculature through active proliferation, reverse migration and differentiation into other EC subtypes. Fate-mapping studies demonstrated that as the new vasculature is forming, venous endothelial cells move against the blood flow to the capillary bed, then the vascular front, transiently acquitting tip cells characteristics (TipECs) and, finally, to the arterial vasculature (Supplemental Figure XI). During normal postnatal vascular development, ~80% of ECs are of the venous endothelium origin in both of P7 and P25 retinal vasculatures. Similarly to the developmental angiogenesis, ~80% of ECs are of venous origin in pathological neovascularization settings. The presence of EdU+ CapiECs (supplemental Figure II B) indicates that some ECs in capillary plexus are immediate descendants of CapiECs. However, these EdU+ CapiECs might also be of venous origin since fate mapping demonstrates that VEC is the predominant source of CapiECs. The source of unlabeled ECs around arteries in VEC reporter mice (Fig. 1F) has not been determined, but the lack of migration AECs seen in AEC reporter mice (supplemental Figure I D) rules out that cell type for their origin leaving reverse-migrating capillary or tip ECS as the only possibility.
A 2-photon intravital imaging of the brain vasculature showed that VEC migrate against the blood flow at a speed of 8.71± 0.78 μm/hr. Compared to previously reported migratory cell in vivo including B cells (~5 μm/min)31, leukemia (~2 μm/min)32 and T cells (~13 μm/min)33, VECs migrate approximately 50 times slower. Importantly, VEC migration occurs against the blood flow. As VECs are the main type of proliferative cells in the angiogenic vasculature, this process of reverse migration provides the expanding capillary vasculature with the source of endothelial cell needed to form new vessels. The reverse migration of endothelial cells has also been reported during fin regeneration in zebrafish 4 and in the extraembryonic yolk sac model34 suggesting endothelial reverse migration is a conserved feature in different species and developmental stages. Importantly, VECs migrate and proliferate only in the postnatal stages but not in the quiescent adult vasculature.
VECs may be particularly sensitive to VEGF stimulation: vascular expansion in the retina into deep layers is driven by VEGF secreted by Müller cells35. Given that initial angiogenic sprouts from the pre-existing superficial vasculature into deep layer were almost exclusively observed arising from VEC but not capillary or AEC populations, this suggests heightened sensitivity of VECs to this signal. Similarly, in the OIR model, neoangiogenic sprouts, which are driven by tissue hypoxia-induced VEGF signaling36, arise exclusively from veins. We thus propose a model of angiogenesis that different from a traditional model where hypoxia-induced VEGF gradient is thought to stimulate pre-existing capillary endothelial cells to induce tip cell differentiation and migration2. Given that various key molecules in the VEGF signaling cascade, including VEGFR237, VE-CAD38 and ERK1/239, are rapidly phosphorylated under shear stress, the predominantly venous angiogenic initiation might be the result of a distinct microfluidic environment in veins or different responses to shear stress in venous vs. arterial endothelial cells. For instance, it has been known arterial high shear stress inhibits endothelial proliferation and migration40 and induces proinflammatory genes expression while venous low shear stress reduces proinflammatory genes expression41.
The origin of arterio-venous malformations (AVM),a relatively common vascular pathology, has been traced to mutations in TGFβ signaling cascade components, including Alk142, Eng9 and Smad427, 28. Its hallmark is an abnormal direct connection between an artery and a vein (AV shunt) that bypasses the capillary bed. Tracing of lesions development in fate-mapped mice showed essentially all endothelial cells forming AVMs are of the venous origin. Time course analysis of AVM showed that AVM formation is initiated by abnormal sprouting from veins. This agrees with a prior observation of an exuberant proliferation of VECs, but not AECs in Smad4iECKO mice 28 and abnormal endothelial cell migration in the absence of SMAD4 8. While under laminar flow endothelial cells migrate against the flow, SMAD4 knockdown inhibits EC alignment along the flow axis and results in undirected random migration8 (Supplemental Figure XII). Taken together with our observations, these data demonstrate that AVMs result from undirected migration of excessively proliferating VECs.
Given that developmental and pathological angiogenesis share many molecular signaling pathways, targeting VEC migration and proliferation could be a more selective therapeutic strategy to control angiogenesis. A recent study of the lung tumor vasculature shows increased proportion of VECs in the tumor compared to normal endothelium43. This highlights VECs importance in pathological angiogenesis and supports the concept of targeting this endothelial population for therapeutic purposes.
Supplementary Material
Supplemental Figures I - XII
Supplemental Movie I
Clinical Perspective.
1. What’s new:
The source of endothelial cells, the key cell type necessary for growth of new blood vessels, has long remained undetermined.
This study identifies venous endothelial cells as key contributes to the growth and expansion of the capillary bed (angiogenesis) and arteries (arteriogenesis).
Venous endothelial cells migrate against the blood flow, form new vascular structures and acquire identity of the formed vessels (e.g. capillaries, arteries).
Abnormal migration of venous endothelial cells leads to the development of arterio-venous malformations.
2. What are the clinical implications:
Venous endothelial cells are genetically and molecularly distinct from arterial and capillary endothelium
Specific targeting of venous endothelial cells may be more effective in inducing new blood vessel growth than indiscriminate stimulation of the entire endothelium.
Reverse migration of the venous endothelium presents an important therapeutic target for both stimulation of therapeutic blood vessel growth (angiogenesis, arteriogenesis) and for prevention of A-V malformations development.
Acknowledgements
We thank R. Adams for Cdh5(PAC)CreERT2, Bmx(PAC)CreERT2 and Esm1(PAC)CreERT2 mice and P. Jensen for Rosa26RC::RG mice, respectively. We also thank K. Boyé for useful advice on OIR experiment.
Funding sources:
Supported, in part, by NIH grants HL135582, HL062289 and HL107205 all to MS
Abbreviation:
- VECs
Venous endothelial cells
- AECs
Arterial endothelial cells
- TipECs
Tip endothelial cells
- CapiECs
Capillary endothelial cells
- VECmReporter
Gm5127(BAC)CreERT2Rosa26+/mTmG
- TipECmReporter
Esm1(PAC)CreERT2Rosa26+/mTmG
- AECmReporter
Bmx(PAC)CreERT2Rosa26+/mTmG
- panECnReporter
Cdh5(PAC)CreERT2Rosa26+/nTnG
- Smad4iECKO
Smad4fx/fx;Cdh5(BAC)CreERT2
- VECiDreReporter
Gm5127(BAC)DreERT2;Rosa26+/RC::RG
- iCre
Inducible Cre recombinase
- mGFP
Membrane localizing GFP
- nGFP
Nuclear localizing GFP
- 4-OHT
4-hydroxy-tamoxifen
- AVM
Arteriovenous malformation
- OIR
Oxygen-induced retinopathy
- VO
vaso-obliteration
Footnotes
Disclosures: None.
REFERENCES
- 1.Simons M, Gordon E and Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 2016;17:611–25. [DOI] [PubMed] [Google Scholar]
- 2.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D and Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 2003;161:1163–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D, Rocha SF et al. Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nature Cell Biology 2017;19:915–927. [DOI] [PubMed] [Google Scholar]
- 4.Xu C, Hasan SS, Schmidt I, Rocha SF, Pitulescu ME, Bussmann J, Meyen D, Raz E, Adams RH and Siekmann AF. Arteries are formed by vein-derived endothelial tip cells. Nature Communications 2014. Dec 15;5:5758.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atri D, Larrivee B, Eichmann A and Simons M. Endothelial signaling and the molecular basis of arteriovenous malformation. Cell Mol Life Sci 2013. Sep 28;10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh E, Redgrave RE, Phillips H and Arthur HM. Arterial endoglin does not protect against arteriovenous malformations. Angiogenesis 2020;23:559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugden WW, Meissner R, Aegerter-Wilmsen T, Tsaryk R, Leonard EV, Bussmann J, Hamm MJ, Herzog W, Jin Y, Jakobsson L et al. Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat Cell Biol 2017;19:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poduri A, Chang AH, Raftrey B, Rhee S, Van M and Red-Horse K. Endothelial cells respond to the direction of mechanical stimuli through SMAD signaling to regulate coronary artery size. Development 2017;144:3241–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin Y, Muhl L, Burmakin M, Wang Y, Duchez AC, Betsholtz C, Arthur HM and Jakobsson L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat Cell Biol 2017;19:639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang YD, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD and Adams RH. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010;465:483–U108. [DOI] [PubMed] [Google Scholar]
- 11.Ehling M, Adams S, Benedito R and Adams RH. Notch controls retinal blood vessel maturation and quiescence. Development 2013;140:3051–3061. [DOI] [PubMed] [Google Scholar]
- 12.Plummer NW, Evsyukova IY, Robertson SD, de Marchena J, Tucker CJ and Jensen P. Expanding the power of recombinase-based labeling to uncover cellular diversity. Development 2015;142:4385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muzumdar MD, Tasic B, Miyamichi K, Li L and Luo L. A global double-fluorescent Cre reporter mouse. Genesis 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- 14.Yu PC, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS, Xie Y, Zhu J, Chen ZH, De Smet F et al. FGF-dependent metabolic control of vascular development. Nature 2017;545:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuda T and Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A 2007;104:1027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hermann M, Stillhard P, Wildner H, Seruggia D, Kapp V, Sanchez-Iranzo H, Mercader N, Montoliu L, Zeilhofer HU and Pelczar P. Binary recombinase systems for high-resolution conditional mutagenesis. Nucleic Acids Res 2014;42:3894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angrand PO, Daigle N, van der Hoeven F, Scholer HR and Stewart AF. Simplified generation of targeting constructs using ET recombination. Nucleic Acids Res 1999;27:e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee H, Park JH, Seo I, Park SH and Kim S. Improved application of the electrophoretic tissue clearing technology, CLARITY, to intact solid organs including brain, pancreas, liver, kidney, lung, and intestine. BMC Dev Biol 2014;14:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008;454:656–60. [DOI] [PubMed] [Google Scholar]
- 20.You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ and Tsai SY. Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature 2005;435:98–104. [DOI] [PubMed] [Google Scholar]
- 21.Fantin A, Schwarz Q, Davidson K, Normando EM, Denti L and Ruhrberg C. The cytoplasmic domain of neuropilin 1 is dispensable for angiogenesis, but promotes the spatial separation of retinal arteries and veins. Development 2011;138:4185–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanlandewijck M, He LQ, Mae MAA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018;554:475–480. [DOI] [PubMed] [Google Scholar]
- 23.Kalucka J, de Rooij L, Goveia J, Rohlenova K, Dumas SJ, Meta E, Conchinha NV, Taverna F, Teuwen LA, Veys K et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020;180:764–779. [DOI] [PubMed] [Google Scholar]
- 24.Schaum N, Karkanias J, Neff NF, May AP, Quake SR, Wyss-Coray T, Darmanis S, Batson J, Botvinnik O, Chen MB et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, Trapnell C and Shendure J. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019;566:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams DL, Piserchia V, Economides JR and Horton JC. Vascular Supply of the Cerebral Cortex is Specialized for Cell Layers but Not Columns. Cereb Cortex 2015;25:3673–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ola R, Kunzel SH, Zhang F, Genet G, Chakraborty R, Pibouin-Fragner L, Martin K, Sessa W, Dubrac A and Eichmann A. SMAD4 Prevents Flow Induced Arteriovenous Malformations by Inhibiting Casein Kinase 2. Circulation 2018;138:2379–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crist AM, Lee AR, Patel NR, Westhoff DE and Meadows SM. Vascular deficiency of Smad4 causes arteriovenous malformations: a mouse model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis 2018;21:363–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stahl A, Connor KM, Sapieha P, Chen J, Dennison RJ, Krah NM, Seaward MR, Willett KL, Aderman CM, Guerin KI, Hua J, Lofqvist C, Hellstrom A and Smith LE. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci 2010;51:2813–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J, Kim KE, Choi DK, Jang JY, Jung JJ, Kiyonari H, Shioi G, Chang W, Suda T, Mochizuki N, Nakaoka Y, Komuro I, Yoo OJ and Koh GY. Angiopoietin-1 guides directional angiogenesis through integrin alphavbeta5 signaling for recovery of ischemic retinopathy. Sci Transl Med 2013;5:203ra127. [DOI] [PubMed] [Google Scholar]
- 31.Reimer D, Meyer-Hermann M, Rakhymzhan A, Steinmetz T, Tripal P, Thomas J, Boettcher M, Mougiakakos D, Schulz SR, Urbanczyk S et al. B Cell Speed and B-FDC Contacts in Germinal Centers Determine Plasma Cell Output via Swiprosin-1/EFhd2. Cell Rep 2020;32:108030. [DOI] [PubMed] [Google Scholar]
- 32.Duarte D, Amarteifio S, Ang H, Kong IY, Ruivo N, Pruessner G, Hawkins ED and Lo Celso C. Defining the in vivo characteristics of acute myeloid leukemia cells behavior by intravital imaging. Immunol Cell Biol 2019;97:229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Megrelis L, El Ghoul E, Moalli F, Versapuech M, Cassim S, Ruef N, Stein JV, Mangeney M and Delon J. Fam65b Phosphorylation Relieves Tonic RhoA Inhibition During T Cell Migration. Front Immunol 2018;9:2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Udan RS, Vadakkan TJ and Dickinson ME. Dynamic responses of endothelial cells to changes in blood flow during vascular remodeling of the mouse yolk sac. Development 2013;140:4041–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stone J, Itin A, Alon T, Pe’er J, Gnessin H, Chan-Ling T and Keshet E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci 1995;15:4738–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mowat FM, Luhmann UF, Smith AJ, Lange C, Duran Y, Harten S, Shukla D, Maxwell PH, Ali RR and Bainbridge JW. HIF-1alpha and HIF-2alpha are differentially activated in distinct cell populations in retinal ischaemia. PLoS One 2010;5:e11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD and Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res 2003;93:354–63. [DOI] [PubMed] [Google Scholar]
- 38.Caolo V, Peacock HM, Kasaai B, Swennen G, Gordon E, Claesson-Welsh L, Post MJ, Verhamme P and Jones EAV. Shear Stress and VE-Cadherin. Arterioscler Thromb Vasc Biol 2018;38:2174–2183. [DOI] [PubMed] [Google Scholar]
- 39.Sumpio BE, Yun S, Cordova AC, Haga M, Zhang J, Koh Y and Madri JA. MAPKs (ERK1/2, p38) and AKT can be phosphorylated by shear stress independently of platelet endothelial cell adhesion molecule-1 (CD31) in vascular endothelial cells. J Biol Chem 2005;280:11185–91. [DOI] [PubMed] [Google Scholar]
- 40.Ji Q, Wang YL, Xia LM, Yang Y, Wang CS and Mei YQ. High shear stress suppresses proliferation and migration but promotes apoptosis of endothelial cells co-cultured with vascular smooth muscle cells via down-regulating MAPK pathway. J Cardiothorac Surg 2019;14:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molins B, Mora A, Romero-Vazquez S, Pascual-Mendez A, Rovira S, Figueras-Roca M, Balcells M, Adan A and Martorell J. Shear stress modulates inner blood retinal barrier phenotype. Exp Eye Res 2019;187:107751. [DOI] [PubMed] [Google Scholar]
- 42.Tual-Chalot S, Mahmoud M, Allinson KR, Redgrave RE, Zhai Z, Oh SP, Fruttiger M and Arthur HM. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS One 2014;9:e98646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, Geldhof V, de Rooij L, Kalucka J, Sokol L et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 2020;37:21–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figures I - XII
Supplemental Movie I
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.