

Summary
Recent advances in single-cell sequencing technologies have enabled simultaneous measurement of multiple cellular modalities, but the combined detection of histone post-translational modifications and transcription at single-cell resolution has remained limited. Here, we introduce EpiDamID, an experimental approach to target a diverse set of chromatin types by leveraging the binding specificities of single-chain variable fragment antibodies, engineered chromatin reader domains, and endogenous chromatin-binding proteins. Using these, we render the DamID technology compatible with the genome-wide identification of histone post-translational modifications. Importantly, this includes the possibility to jointly measure chromatin marks and transcription at the single-cell level. We use EpiDamID to profile single-cell Polycomb occupancy in mouse embryoid bodies and provide evidence for hierarchical gene regulatory networks. In addition, we map H3K9me3 in early zebrafish embryogenesis, and detect striking heterochromatic regions specific to notochord. Overall, EpiDamID is a new addition to a vast toolbox to study chromatin states during dynamic cellular processes.
Keywords: single-cell genomics, multi-modal omics, histone post-translational modifications, chromatin, epigenetics, gene regulation, DamID, embryo development
Graphical abstract
Highlights
-
•
EpiDamID extends the use of DamID-based protocols to epigenetic chromatin marks
-
•
Histone PTM-specific binding domains target the Dam enzyme to the mark of interest
-
•
A single-cell implementation offers joint epigenetic and transcriptomic readouts
Rang and de Luca et al. develop EpiDamID, an addition to the DamID toolkit that enables the study of histone post-translational modifications. EpiDamID can be implemented in various biological systems and methodological approaches, including simultaneous single-cell measurement of epigenetic state and transcription during embryogenesis.
Introduction
Histone post-translational modifications (PTMs) contribute to chromatin structure and gene regulation. The addition of PTMs to histone tails can modulate the accessibility of the underlying DNA and form a binding platform for myriad downstream effector proteins. As such, histone PTMs play key roles in a multitude of biological processes, including lineage specification (e.g., Juan et al., 2016; Nicetto et al., 2019; Pengelly et al., 2013), cell cycle regulation (e.g., Hirota et al., 2005; W. Liu et al., 2010), and response to DNA damage (e.g., Rogakou et al., 1998; Sanders et al., 2004).
Over the past decade, antibody-based DNA-sequencing methods have provided valuable insights into the function of histone PTMs in a variety of biological contexts. Most studies employ ChIP-seq (chromatin immunoprecipitation after formaldehyde fixation [Solomon and Varshavsky, 1985]), or strategies based on in situ enzyme tethering such as chromatin immunocleavage (ChIC) (Schmid et al., 2004), and its derivative cleavage under targets and release using nuclease (CUT&RUN) (Skene and Henikoff, 2017). However, the requirement of high numbers of input cells consequently provides a population-average view, which disregards the complexity of most biological systems. As a result, several low-input methods have been developed that can assay histone PTMs in individual cells, including but not limited to Drop-ChIP (Rotem et al., 2015), ChIL-seq (Harada et al., 2019), ACT-seq (Carter et al., 2019), single-cell ChIP-seq (Grosselin et al., 2019), single-cell ChIC-seq (Ku et al., 2019), single-cell adaptation of CUT&RUN (Hainer et al., 2019), CUT&Tag (Kaya-Okur et al., 2019), CoBATCH (Wang et al., 2019), single-cell itChIP (Ai et al., 2019), and sortChIC (Zeller et al., 2021). While these techniques offer an understanding of the epigenetic heterogeneity between cells, they do not provide a direct link to other measurable outputs. Recently, however, three methods have been developed that jointly profile histone modifications and gene expression: Paired-Tag (parallel analysis of individual cells for RNA expression and DNA from targeted tagmentation by sequencing) (Zhu et al., 2021), CoTECH (combined assay of transcriptome and enriched chromatin binding) (Xiong et al., 2021), and SET-seq (same cell epigenome and transcriptome sequencing) (Sun et al., 2021). These techniques thus enable linking of gene regulatory mechanisms to transcriptional output and cellular state. Of note, all three methods rely on antibody binding for detection of histone modifications and Tn5-mediated tagmentation for sequencing library preparation. As can be expected from its implementation in ATAC-seq (assay for transposable-accessible chromatin using sequencing) (Buenrostro et al., 2013), the Tn5 transposase has a high affinity for exposed DNA in open chromatin. While approaches exist to mitigate this bias (Kaya-Okur et al., 2020), a recent systematic analysis of Tn5-based studies has provided preliminary indications that accessibility artifacts persist (Zhang et al., 2021).
We recently developed scDam&T-seq, a method that measures DNA-protein contacts and transcription in single cells by combining single-cell DamID and CEL-Seq2 (Rooijers et al., 2019). DamID-based techniques attain specificity by tagging a protein of interest (POI) with the E. coli Dam methyltransferase, which methylates adenines in a GATC motif in the proximity of the POI (Filion et al., 2010; van Steensel and Henikoff, 2000; Vogel et al., 2007). The approach is especially suited for single-cell studies, because DNA-protein contacts are recorded directly on the DNA in the living cell, and downstream sample handling is limited. However, Dam cannot be tethered directly to post-translationally modified proteins by genetic engineering, which has precluded the use of DamID for studying histone PTMs.
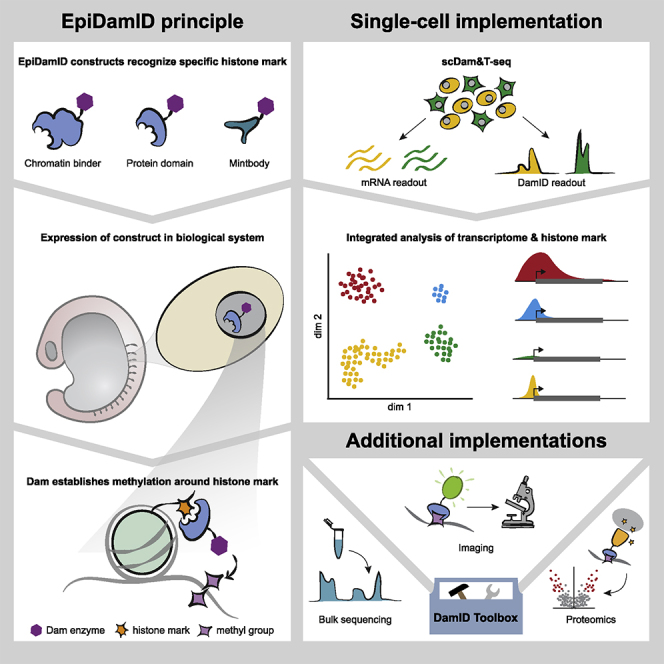
Here, we present EpiDamID, an extension of existing DamID protocols, based on the fusion of Dam to chromatin-binding modules for the detection of various types of histone PTMs. We validate the specificity of EpiDamID in population (Figure 1) and single-cell samples (Figure 2). Subsequently, we leverage its single-cell resolution to study the Polycomb mark H3K27me3 and its relationship to transcription in mouse embryoid bodies (EBs) (Figure 3) and identify distinct Polycomb-regulated and Polycomb-independent hierarchical TF networks (Figure 4). Finally, we implement a protocol to assay cell type-specific patterns of the heterochromatic mark H3K9me3 in the zebrafish embryo and discover broad domains of heterochromatin specific to the notochord (Figure 5). Together, these results show that EpiDamID provides a versatile tool that can be implemented in diverse biological settings to obtain single-cell histone PTM profiles.
Figure 1.

Targeting domains specific to histone modifications mark distinct chromatin types with EpiDamID
(A) Schematic overview of EpiDamID concept compared to conventional DamID.
(B) UMAP of DamID samples colored by targeting construct, and ChIP-seq samples of corresponding histone modifications. MB: mintbody; PD: protein domain; F: full protein.
(C) UMAPs as in (B), colored by correlation with selected ChIP-seq samples (H3K9ac, H3K9me3, and H3K27me3). Correlation values reflect the Pearson’s correlation coefficient of Dam-normalized samples with the indicated ChIP-seq sample. Control constructs (Dam, H3K27me3mut) are excluded from the UMAP. DamID samples are circles; ChIP-seq samples are squares.
(D) Left: three genome browser views of ChIP-seq (gray) and DamID (colored) enrichment. Data represent the combined signal of all samples of each targeting domain. Right: average DamID and ChIP-seq enrichment plots over genomic regions of interest. Signal is normalized for untethered Dam or input, respectively. Regions are the TSS (−10/+15 kb) of the top 25% H3K9ac-enriched genes for the active marks (top), and ChIP-seq domains (−/+ 10 kb) for H3K27me3 (middle), and H3K9me3 (bottom).
(E) Confocal images of nuclear chromatin showing DAPI (top), immunofluorescent staining against an endogenous histone modification (middle), and its corresponding EpiDamID construct visualized with m6A-Tracer (bottom). Left: H3K9ac, right: H3K9me3. Scale bar: 3 μm.
See also Figure S1.
Figure 2.

Detection of histone PTMs in single mouse embryonic stem cells with EpiDamID
(A) UMAP based on the single-cell DamID readout of all single-cell samples. MB: mintbody; PD: protein domain; F: full protein.
(B–D) DamID UMAP as in (A), colored by the enrichment of counts within H3K27me3 ChIP-seq domains (B), H3K9ac ChIP-seq peaks (C), and H4K20me1 ChIP-seq domains (D).
(E) Average signal over H3K27me3 ChIP-seq domains of CBX7 and H3K27me3 targeting domains and full-length RINGB1B protein.
(F) Average H4K20me1 signal over the TSS of the top 25% active genes (based on H3K9ac ChIP-seq signal).
(E and F) Top: in silico populations normalized for Dam; Bottom: five of the best single-cell samples (bottom) normalized only by read depth.
(G and H) Signal of various marks over the HoxD cluster and neighboring regions. ChIP-seq data is normalized for input control. DamID tracks show the Dam-normalized in silico populations of the various Dam-fusion proteins, DamID heatmaps show the depth-normalized single-cell data of the fifty richest cells. The HoxD cluster is indicated in red in (G) (bar) and (H) (RefSeq); additional RefSeq genes are shown (H).
See also Figure S2.
Figure 3.

Joint profiling of Polycomb chromatin and gene expression in mouse embryoid bodies
(A) Schematic showing the experimental design.
(B) UMAP of samples based on transcriptional readout, colored by cluster.
(C and D) UMAP of samples based on DamID readout, colored by construct (C) and cluster (D).
(E) Transcriptomic UMAP (left) and DamID UMAP (right), colored by expression of pluripotency marker Dppa5a.
(F) Transcriptomic UMAP (left) and DamID UMAP (right), colored by expression of hematopoietic regulator Tal1.
(G) Genomic tracks of H3K27me3 and RING1B DamID signal per cluster at the Tal1 locus.
(H) Heatmaps showing the H3K27me3 (left) and RING1B (right) DamID signal of all identified PRC targets for transcriptional clusters 3, 0, 1, 6, and 4. PRC targets are ordered based on hierarchical clustering.
(I) Fold-change in expression of Polycomb targets between clusters where the gene is PRC-associated and clusters where the gene is PRC-free. The significance was tested with a two-sided Wilcoxon’s signed rank test (p = 2.6 × 10−185).
See also Figure S3.
Figure 4.

Polycomb-regulated transcription factors form separate regulatory networks
(A) Heatmap showing SCENIC regulon activity per single cell. Cells (columns) are ordered by transcriptional cluster; regulon (rows) are ordered by hierarchical clustering. The black and white bar on the left indicates whether the regulon TF is a PRC target (black) or not (white).
(B) Example of the relationship between expression and Polycomb regulation for the MSX1 regulon. Pie chart indicates the percentages of Polycomb-controlled (blue) or Polycomb-independent (gray) target genes. Left: boxplots showing target gene expression per cluster for all target genes. Middle and right: boxplots showing the H3K27me3 and RING1B DamID signal at the TSS per cluster for the Polycomb-controlled target genes. The expression and DamID signal of Msx1 is indicated with a red circle.
(C) Genomic tracks of H3K27me3 and RING1B DamID signal per cluster at the Fgf10 locus, one of the target genes of MSX1. Arrow head indicates the location of the TSS; shaded area indicates −5kb/+3kb around the TSS.
(D) Boxplots showing the fraction of Polycomb-controlled target genes, split by whether the TF itself is Polycomb-controlled. The significance was tested with a two-sided Mann-Whitney U test (p = 2.8 × 10−20). Error bars indicate the data range within 1.5 times the inter-quartile range.
(E) Schematic of the regulatory network, indicating the relationship between a regulon TF (white hexagon), its upstream regulators (colored hexagons), and its downstream targets (colored hexagons/circles).
(F) Boxplots showing the fraction of Polycomb-controlled upstream regulators, split by whether the regulon TF is Polycomb-controlled. The significance was tested with a two-sided Mann-Whitney U test (p = 6.6 × 10−19). Error bars indicate the data range within 1.5 times the inter-quartile range.
(G) Scatterplot showing the relationship between the fraction of Polycomb-controlled targets and regulators of a regulon TF. Regulon TFs that are PRC controlled are indicated in blue; regulon TFs that are PRC independent are indicated in gray. Correlation was computed using Pearson’s correlation (p = 2.9 × 10−29).
See also Figure S4.
Figure 5.

Notochord-specific H3K9me3 enrichment in the zebrafish embryo
(A) Schematic representation of the experimental design and workflow.
(B) UMAP based on the transcriptional readout of all single-cell samples passing CEL-Seq2 thresholds (n = 3902).
(C) UMAP based on the genomic readout of all single-cell samples passing DamID thresholds (n = 2833). Samples are colored by transcriptional cluster (left) and Dam-targeting domain (right).
(D) Expression of the hatching gland marker he1.1 (left) and the notochord marker col9a2 (right) projected onto the DamID UMAP.
(E) Genomic H3K9me3 signal over chromosome 17. Top track: H3K9me3 ChIP-seq signal of 6-hpf embryo. Remaining tracks: combined single-cell Dam-MPHOSPH8 data for clusters 0–2. Heatmaps show the depth-normalized Dam-MPHOSPH8 data of the 50 richest cells.
(F) Heatmap showing the cluster-specific average H3K9me3 enrichment over all domains called per ChromHMM state. Per state, domains were clustered using hierarchical clustering.
(G) Genomic H3K9me3 signal over a part of chromosome 8 for clusters 0–2. The colored regions at the bottom of each track indicate the ChromHMM state.
(H) Gene density of all genes (top) and expressed genes (bottom) per state.
(I) Enrichment of repeats among the ChromHMM states. Example repeats are indicated.
(J) Representative images of DAPI staining in cryosections of zebrafish embryos at 15-somite stage. Scale bars represent 4 μm.
See also Figures S5 and S6.
Design
The conventional DamID approach involves genetically engineering a protein of interest (POI) to the bacterial methyltransferase Dam (Figure 1A). In this study, we adapted the DamID method to detect histone PTMs by fusing Dam to one of the following: (1) full-length chromatin proteins, (2) tuples of well-characterized reader domains (Kungulovski et al., 2016, 2014; Vermeulen et al., 2007), or (3) single-chain variable fragments (scFv) also known as mintbodies (Sato et al., 2016, 2013; Tjalsma et al., 2021) (Figure 1A, Methods). Similar strategies have been successfully applied in microscopy, proteomics and ChIP experiments (Sato et al., 2021, 2016, 2013; Tjalsma et al., 2021; Villaseñor et al., 2020). Our approach is henceforth referred to as EpiDamID, and the construct fused to Dam as the targeting domain. Since this approach can be applied to any existing DamID method, EpiDamID makes all these protocols available to the study of chromatin modifications. This includes the possibility to perform (live) imaging of Dam-methylated DNA (Altemose et al., 2020; Borsos et al., 2019; Kind et al., 2013), tissue-specific study of model organisms without cell isolation via Targeted DamID (TaDa) (Southall et al., 2013), DamID-directed proteomics (Wong et al., 2021), (multi-modal) single-cell (Altemose et al., 2020; Borsos et al., 2019; Kind et al., 2015; Rooijers et al., 2019; Pal et al., 2021) and single-molecule (Cheetham et al., 2021) sequencing studies, and the processing of samples with little material (Borsos et al., 2019; Pal et al., 2021).
Results
Targeting domains specific to histone modifications mark distinct chromatin types with EpiDamID
We categorized the various targeting domains into the following chromatin types: accessible, active, heterochromatin, and Polycomb. We generated various expression constructs for each of the different targeting domains, testing promoters (HSP, PGK), orientations (Dam-POI, POI-Dam), and two versions of the Dam protein (DamWT, Dam126) (Table S1). The choice of promoter influences the expression level of the Dam-POI, whereas the orientation may affect target binding. In the Dam126 mutant, the N126A substitution diminishes off-target methylation (Park et al., 2019; Szczesnik et al., 2019). We introduced the Dam constructs by viral transduction in hTERT-immortalized RPE-1 cells and performed DamID2 followed by high-throughput sequencing (Markodimitraki et al., 2020). To validate our data with an orthogonal method, we generated ChIP-seq samples for various histone modifications.
The DamID samples were filtered on sequencing depth and information content (IC), a metric for determining signal-to-noise levels (Figures S1A and S1B) (STAR Methods). IC additionally showed that tuples of reader domains fused to Dam typically perform better than single domains (p < 0.05 for three out of four domains, Figure S1B), in agreement with a recent study employing similar domains for proteomics purposes (Villaseñor et al., 2020). Therefore, only data from the triple reader domains were included in further analyses.
Visualization of all filtered samples by uniform manifold approximation and projection (UMAP) shows that EpiDamID mapping identifies distinct chromatin types and that samples consistently group with their corresponding ChIP-seq datasets (Figure 1B). Genome-wide DamID signal also correlates well with ChIP-seq signal (mean Pearson’s correlation coefficients from 0.40–0.64 for active marks, 0.58–0.61 for heterochromatin marks, and 0.56-0.60 for Polycomb marks) (Figures 1C and S1C). Importantly, DamID samples do not group based on construct type, promoter, Dam type, sequencing depth, or IC (Figure S1D and S1E), indicating that those properties do not influence target specificity. All targets display the expected patterns of enrichment along the linear genome (Figure 1D, left), as well as genome-wide on-target signal (Figure 1D, right). To further explore the specificity of constructs that target active chromatin, we compared signal of Dam-H3K9ac and Dam-TAF3 at H3K9ac ChIP-seq peaks with high and low H3K4me3 ChIP-seq levels. Dam-H3K9ac shows enrichment in both categories, while Dam-TAF3 is enriched specifically in the high-H3K4me3 category (Figure S1F). This confirms that, while the untethered Dam protein preferentially marks accessible chromatin, targeting it to active regions of the genome yields specific methylation patterns.
Next, we quantified the spreading of Dam signal from its binding location to determine the resolution for all chromatin types. We found that DamID signal decays to 50% (from 100% at peak center or domain border) across a distance that extends ∼1 kb past the ChIP-seq 50% decay point (Figure S1G), implying a resolution of ∼1–2 kb, similar to earlier studies with transcription factors (Cheetham et al., 2018; Tosti et al., 2018). It was previously reported that the Dam126 mutant improves signal quality compared to DamWT (Szczesnik et al., 2019). Indeed, this mutant markedly improved sensitivity and reduced background methylation (mean IC increase of 0.07–0.21 per construct) (Figures S1H and S1I).
We further validated the correct nuclear localization of Dam-marked chromatin with microscopy, by immunofluorescent staining of endogenous histone PTMs and DamID visualization using m6A-Tracer protein (Kind et al., 2013; van Schaik et al., 2020) (Figure 1E).
Together, these results show that EpiDamID specifically targets histone PTMs and enables identification of their genomic distributions by next-generation sequencing.
Detection of histone PTMs in single mouse embryonic stem cells with EpiDamID
We next established EpiDamID for single-cell sequencing. To this end, we generated clonal, inducible mESC lines for the following targeting domains fused to Dam: H4K20me1 mintbody, H3K27me3 mintbody, and the H3K27me3-specific CBX7 protein domain (3x tuple). While H4K20me1 is enriched over the gene body of active genes (Shoaib et al., 2021), the heterochromatic mark H3K27me3 is enriched over the promoter of developmentally regulated genes (Boyer et al., 2006; Riising et al., 2014). As controls, we included an H3K27me3mut mintbody construct whose antigen-binding ability is abrogated by a point mutation in the third complementarity determining region of the heavy chain (Y105F), and a published mESC line expressing untethered Dam (Rooijers et al., 2019). We performed scDam&T-seq to generate 442–1,402 single-cell samples per construct, retaining 283–855 samples after filtering on the number of unique GATCs and IC (10,417–45,067 median unique counts per construct and median IC of 2.0–2.9) (Figure S2A–S2C and Table S2). For subsequent analyses, we also included a published dataset of Dam fused to RING1B (Rooijers et al., 2019) as an example of a full-length chromatin reader targeting Polycomb chromatin. All constructs contained DamWT, as the Dam126 methylation levels were found insufficient to produce high-quality single-cell signal (data not shown).
Dimensionality reduction of the single-cell datasets revealed that the samples primarily separated on chromatin type (Figure 2A). To further confirm the specificity of the constructs, we used mESC H3K27me3 (ENCSR059MBO) and H3K9ac (ENCSR000CGP) ChIP-seq datasets from the ENCODE portal (Davis et al., 2018) and generated our own for H4K20me1. For all single cells, we computed the enrichment of counts within H3K27me3, H3K9ac, and H4K20me1 ChIP-seq domains. These results show a strong enrichment of EpiDamID counts within domains for the corresponding histone PTMs (Figures 2B–2D and S2D), indicating that the methylation patterns are specific for their respective chromatin targets, even at the single-cell level. The combined single-cell data also showed the expected enrichment over H3K27me3 ChIP-seq domains (Figure 2E) and active gene bodies (Figure 2F) for the Polycomb-targeting constructs and H4K20me1, respectively. Contrary to the H3K27me3 construct, H3K27me3mut showed little enrichment over H3K27me3 ChIP-seq domains (Figure S2E). The specificity of the signal is also evident at individual loci in both the in silico populations and single cells (Figures 2G, 2H, and S2F).
These results demonstrate that mintbodies and protein domains can be used to map histone PTMs in single cells with EpiDamID.
Joint profiling of Polycomb chromatin and gene expression in mouse embryoid bodies
To exploit the benefits of simultaneously measuring histone PTMs and transcriptome, we profiled Polycomb chromatin in mouse EBs. We targeted the two main Polycomb repressive complexes (PRC) with EpiDamID using the full-length protein RING1B and H3K27me3-mintbody fused to Dam. RING1B is a core PRC1 protein that mediates H2AK119 ubiquitylation (de Napoles et al., 2004; Wang et al., 2004), and H3K27me3 is the histone PTM deposited by PRC2 (Cao et al., 2002; Czermin et al., 2002; Kuzmichev et al., 2002; Müller et al., 2002). Both PRC1 and PRC2 have key roles in gene regulation during stem cell differentiation and early embryonic development (see [Piunti and Shilatifard, 2021] and [Blackledge and Klose, 2021] for recent reviews on this topic).
To assay a diversity of cell types at various stages of differentiation, we harvested EBs for scDam&T-seq at day 7, 10, and 14 post aggregation, next to ESCs grown in 2i/LIF (Figure 3A). We used Hoechst incorporation in combination with fluorescence-activated cell sorting (FACS) to deposit live, single cells into 384-well plates and record their corresponding cell cycle phase (STAR Methods). In addition to RING1B and H3K27me3-mintbody, we included the untethered Dam protein for all time points as a control for chromatin accessibility. Collectively, we obtained 2,943 cells after filtering (Figures S3A and S3B), in a similar range as CoTECH (∼7,000 cells), higher than SET-seq (∼500 cells) and lower than Paired-Tag (∼65,000 nuclei). The number of unique genomic and transcriptomic counts per cell was similar or higher compared to the other methods (Figures S3A and S3B). Based on the transcriptional readout, we identified eight distinct clusters across time points (Figure 3B). We integrated the EB transcriptome data with the publicly available mouse embryo atlas (Pijuan-Sala et al., 2019) to confirm the correspondence of cell types with early mouse development and guide cluster annotations (Figures S3C and S3D). This indicated the presence of pluripotent and more differentiated cellular states, including epiblast, endoderm, and mesoderm lineages. Notably, the DamID readout alone was sufficient to consistently separate cells on chromatin type (Figure 3C) and to distinguish between the pluripotent and more lineage-committed cells (Figures 3D and 3E). Thus, the EpiDamID profiles display cell type-specific patterns of chromatin accessibility and Polycomb association. Prompted by this observation, we trained a linear discriminant analysis (LDA) classifier to assign an additional 1,543 cells with poor transcriptional data to cell type clusters, based on their DamID signal (Figure S3E and Table S2).
Next, we defined the set of genes that is Polycomb-regulated in the EB system. First, we determined the H3K27me3 and RING1B signal at the promoter region of all genes and compared these two readouts across the clusters. This confirmed good correspondence between H3K27me3 and RING1B profiles (Pearson’s r = 0.60-0.82, p = 0 between profiles of the same cluster) (Figures S3F and S3G), albeit with a slightly higher signal amplitude for RING1B (Figure S3G). This difference between RING1B and H3K27me3 may be biological (e.g., differential binding sites or kinetics) and/or technical (e.g., the use of a full-length protein versus a mintbody to target Dam). Nonetheless, because of the overall similarity, we decided to classify high-confidence Polycomb targets as having both H3K27me3 and RING1B enrichment in at least one of the EB clusters (excluding cluster 7 due to the relatively low number of cells) or in the previous ESC dataset. We identified 9,159 Polycomb-regulated targets across the dataset, in good concordance with previous work in mouse development (4,059 overlapping genes out of a total of 5,986; p = 9.5 × 10−135, Chi-square test) (Gorkin et al., 2020) (Figure S3H).
Next, we intersected the cluster-specific transcriptome and DamID data to relate gene expression patterns to Polycomb associations. Based on the role of Polycomb in gene silencing, differential binding of PRC1/2 to genes is expected to be associated with changes in expression levels. As exemplified in Figures 3F and 3G, the cell type-specific expression of Tal1, a master regulator in hematopoiesis, is indeed inversely related to Polycomb enrichment. This negative association is apparent for all PRC targets that are upregulated in the hematopoietic cluster (Figures S3I and S3J). In addition, unsupervised clustering of H3K27me3 and RING1B promoter occupancy shows variation in signal between target genes as well as between cell types, indicating dynamic regulation of these targets in EBs (Figure 3H). In line with this, Polycomb targets with variable PRC occupancy are typically more highly expressed in those clusters where Polycomb is absent (Wilcoxon’s signed-rank test, p = 2.6 × 10−185, Figure 3I). Since the negative relationship between Polycomb occupancy and transcription is not perfect, we were interested to see whether an additional layer of epigenetic regulation could further explain the observed transcriptional changes. To this end, we integrated our data with a publicly available scNMT-seq dataset (Argelaguet et al., 2019), also generated in EBs (Figure S3K). This resulted in sufficient scNMT-seq samples in four clusters to compare CpG methylation profiles with Polycomb occupancy. The integrated profiles indeed revealed a complementary relationship between the two marks, where genes with either CpG methylation or Polycomb at their promoter tend to be expressed at lower levels (Figure S3L). This was also apparent for CpG methylation and expression of genes with variable Polycomb enrichment between the clusters (Figure S3M). The observed trends are in line with the known repressive effects of both marks and their largely mutually exclusive localizations (Brinkman et al., 2012; Hagarman et al., 2013; Li et al., 2018).
Collectively, these data illustrate the strength of EpiDamID to jointly capture transcription and chromatin dynamics during differentiation, as well as the potential to integrate the results with datasets derived from different techniques.
Polycomb-regulated transcription factors form separate regulatory networks
We next focused on the Polycomb targets based on their function and found that TF genes are over-represented within the Polycomb target genes (Figure S4A), in line with previous observations (Boyer et al., 2006). Nearly half of all TF genes in the genome (761/1,689) is bound by Polycomb in at least one cluster. In addition, genes encoding TFs generally accumulate higher levels of H3K27me3 and RING1B compared to other protein-coding genes (Figure S4B). Consistent with an important role in lineage specification, Polycomb-controlled TFs are expressed in a cell type-specific pattern, as opposed to the more constitutive expression across cell types for Polycomb-independent TFs (Figures S4C and S4D). Accordingly, the Polycomb-controlled TFs are enriched for Gene Ontology (GO) terms associated with animal development (Figure S4E).
The high Polycomb occupancy at developmentally regulated TF genes prompted further investigation into the role of Polycomb in TF network hierarchies. We used SCENIC to systematically identify target genes that are associated with the expression of TFs (Aibar et al., 2017; Van de Sande et al., 2020). SCENIC employs co-expression patterns and binding motifs to link TFs to their targets, together henceforth termed “regulons” (per SCENIC nomenclature). We identified 285 “activating” regulons after filtering (Figure 4A and STAR Methods). While regulons and their activity were found independently of RNA-based cluster annotations, regulon activity trends clearly matched the annotated clusters (Figure 4A).
We first determined how overall regulon activity identified by SCENIC correlates to Polycomb binding. As illustrated for the homeobox TF gene Msx1, we found that regulon activity is generally inversely related to Polycomb association of both the TF gene (red dot) and its Polycomb-controlled targets (boxplots, 65% of all MSX1 targets) (Figures 4B and 4C). We wondered whether there is a general preference for Polycomb-controlled TFs to target genes that themselves are regulated by Polycomb. Indeed, while Polycomb-controlled TFs have a similar number of target genes compared to other TFs (Figure S4F), the expression of the targets is much more frequently controlled by Polycomb than expected by chance (Mann-Whitney-U test p = 2.8 × 10−20, Figure 4D). This effect is even stronger when considering the subset of targets that is exclusively regulated by Polycomb TFs (Chi-square test p = 0, Figure S4G). Similarly, upstream TFs controlling the regulon TFs (Figure 4E) also tend to be Polycomb-controlled (Mann-Whitney-U test, p = 6.6 × 10−19, Figure 4F). Moreover, the fractions of Polycomb-controlled upstream regulators and downstream targets are correlated (Pearson’s r = 0.61, p = 2.9 × 10−29, Figure 4G), indicating consistency in the level of Polycomb regulation across at least three layers of the TF network. This trend is especially strong for the lineage-specific genes (Pearson’s r = 0.48, p = 9.2 × 10−8), but also holds for other, unspecific genes (Pearson’s r = 0.41, p = 4.0 × 10−4) (Figures S4H and S4I). These results suggest that Polycomb-associated hierarchies exist, forming relatively separate networks isolated from other gene regulatory mechanisms, and that this phenomenon extends beyond lineage-specific genes alone.
Together, the above findings demonstrate that single-cell EpiDamID can be successfully applied in complex developmental systems to gather detailed information on cell type-specific Polycomb regulation and its interaction with transcriptional networks.
Implementation of EpiDamID during zebrafish embryogenesis
Next, we applied EpiDamID in an in vivo system to study the heterochromatic mark H3K9me3 during zebrafish development. To bypass the need for genetic engineering, we employed microinjection of mRNA into the zygote (Figure 5A), a strategy successfully applied in the mouse embryo (Borsos et al., 2019). H3K9me3 is reprogrammed during the early stages of development in several species (Laue et al., 2019; Mutlu et al., 2018; Rudolph et al., 2007; Santos et al., 2005; Wang et al., 2018) and the deposition of this mark coincides with decreased developmental potential (Ahmed et al., 2010). It was previously shown that H3K9me3 is largely absent before the maternal-to-zygotic transition (MZT) (Laue et al., 2019), but it remains unclear whether the H3K9me3 distribution undergoes further remodeling after this stage, and whether its establishment differs across cell types during development.
We injected mRNA encoding the H3K9me3-specific construct Dam-Mphosph8 and untethered Dam into the yolk at the one-cell stage and collected embryos at the 15-somite stage (Figure 5A), which comprises a wide diversity of cell types corresponding to all germ layers. We generated 2,127 single-cell samples passing both DamID and CEL-Seq2 thresholds (Figure S5A and Table S2). Comparing the DamID data of an in silico whole-embryo sample to published H3K9me3 ChIP-seq data of 6-hpf embryos (Laue et al., 2019) showed good concordance (Pearson’s r = 0.72, p = 0; Figure S5B).
Broad domains of notochord-specific H3K9me3 enrichment revealed by scDam&T-seq
Analysis of the single-cell transcriptome data resulted in 22 clusters of diverse cell types (Figure 5B), which we annotated according to expression of known marker genes (Figure S5C). After dimensionality reduction based on the DamID signal, we observed a clear visual separation of cells in accordance with their Dam construct, and to a lesser extent with their cell type (Figures 5C and 5D). Cluster-specific DamID profiles allowed us to employ the LDA classifier to assign a further 705 cells with poor transcriptional readout to a cluster (Figure S5D and Table S2). Notably, the MPHOSPH8 samples of hatching gland (cluster 1, he1.1 expression) and notochord (cluster 2, col9a2 expression) segregated strongly from the other cell types (Figure 5D), implying differences in their single-cell H3K9me3 profiles. In particular, we observed the appearance of large domains of H3K9me3 enrichment in the notochord, and seemingly lower levels of H3K9me3 in the hatching gland (Figures 5E and S5E).
Next, to more systematically identify and characterize regions of differential H3K9me3 enrichment between cell clusters, we performed ChromHMM (Ernst and Kellis, 2017, 2012). The approach uses the H3K9me3 signal per cluster to annotate genomic segments as belonging to different H3K9me3 states. We included the 12 cell clusters containing > 30 cells per construct and identified five H3K9me3 states across the genome. These represented: A) three states of constitutive H3K9me3 with different enrichment levels [A1-A3], B) notochord-specific H3K9me3 enrichment, and C) constitutive depletion of H3K9me3 (Figures 5F and 5G). While all 12 clusters had the highest H3K9me3 enrichment in state A1, cells belonging to the hatching gland (cluster 1) tended to have lower signal in these regions compared to other cell types (Figure S5F). Notochord cells (cluster 2), conversely, displayed somewhat higher enrichment in state A1 and dramatically higher enrichment in state B compared to the others. State A (A1-3) chromatin forms broad domains (Figure S5G) that together comprise 27% of the genome (Figure S5H) and, as expected for H3K9me3-associated chromatin regions, are characterized by sparser gene density and lower gene activity compared to the H3K9me3-depleted state C (Figure 5H). Moreover, state A1 is strongly enriched for zinc-finger transcription factors (Figure S5I), which are known to be demarcated by H3K9me3 in other species (Hahn et al., 2011; Vogel et al., 2006). The notochord-specific state B has similar characteristics to states A1-A3 (Figures 5H, S5G, and S5H) yet exhibits broader consecutive regions of H3K9me3 enrichment (Figures 5G and S5G) and an even lower active gene density (Figure 5H). However, we did not find a notable increase in H3K9me3 at genes downregulated in notochord (Figure S5J), implying that these domains do not play a role in gene expression regulation.
One of the known functions of H3K9me3 chromatin is the repression of transposable elements (Bulut-Karslioglu et al., 2014; Liu et al., 2014; Mosch et al., 2011). Indeed, it was previously observed in zebrafish that nearly all H3K9me3 domains in early embryos are associated with repeats (Laue et al., 2019). We determined whether distinct repeat classes were over-represented in each H3K9me3 ChromHMM state (Figure S6A) and found a strong enrichment of several repeat classes in state A1, including LTR and tRNA. Further discrimination within the classes showed a high frequency of pericentromeric satellite repeats SAT-1 and BRSATI in state A1 (Figure 5I), in line with the known occupancy of H3K9me3 at pericentromeric regions. Inspection of the DamID patterns showed a clear increase of signal centered on specific repeat regions in state A1, and to lesser extents in other states (Figure S6B). In addition, we found that state B harbors specific enrichment of certain repeats (Figures 5I and S6C), although further study is required to determine whether H3K9me3 is involved in cell type-specific repression of repetitive genomic regions in the notochord.
Altered expression of chromatin proteins and pronounced nuclear compartmentalization in notochord
Finally, we evaluated cluster-specific expression of known chromatin proteins in relation to the differential H3K9me3 patterns. Expression levels of histone methyltransferases, demethylases and other chromatin factors did not show an upregulation of known H3K9 methyltransferases (setdb2, setdb1a/b, suv39 h11a/b, ehmt2) nor demethylases (kdm4aa/ab/b/c, phf8) in notochord (Figure S6D). However, the H3K9- and H3K36-specific demethylase kdm4c was exclusively upregulated in hatching gland, which could explain the low H3K9me3 levels in this cluster. Notably, the notochord cluster showed significant upregulation of lmna, the gene encoding nuclear lamina protein Lamin A/C that associates with heterochromatin (Gruenbaum and Foisner, 2015) and plays an important structural role in the nucleus (Donnaloja et al., 2020; Gruenbaum and Foisner, 2015). This could be relevant in relation to the structural role of the notochord and the resulting mechanical forces the cells are subjected to (Corallo et al., 2015). To more directly investigate chromatin state and nuclear organization in these embryos, we performed confocal imaging of H3K9me3 and DAPI stainings in notochord, brain, and skeletal muscle. H3K9me3-marked chromatin displayed a typical nuclear distribution in all tissues, including heterochromatin foci as previously reported (Laue et al., 2019) (Figure S6E). DAPI staining showed more structure in the notochord compared to the other tissues (Figure 5J), visible as a clear rim along the nuclear periphery and denser foci within the nuclear interior. This indicates a stronger separation between euchromatin and heterochromatin, although it remains to be elucidated whether these features are related to the notochord-specific H3K9me3 domains in the genome.
The implementation of EpiDamID in zebrafish embryos shows that this strategy provides a flexible and accessible approach to generate high-resolution single-cell information on the epigenetic states that underlie biological processes during organismal development.
Discussion
Advantages of DamID for single-cell multi-modal omics during embryo development
The DamID workflow involves few enzymatic steps and is thus especially suitable for integration with other single-cell protocols to achieve multi-modal measurements (Markodimitraki et al., 2020). Minimal sample handling prior to molecular processing results in a high recovery rate of collected cells (Borsos et al., 2019); for example, scDam&T-seq with EpiDamID constructs could be used to individually assay all cells of a single preimplantation mouse embryo and examine epigenetic and transcriptomic differences that may point toward cell fate commitment, while tracking intra-embryonic variability. Further, DamID genomic marks are stable upon deposition, offering the possibility to track ancestral EpiDamID signatures through mitosis to study inheritance and spatial distribution of epigenetic states in daughter cells (Kind et al., 2013; Park et al., 2019).
Comparison to other single-cell transcriptome and chromatin profiling techniques
In the past year, three other techniques have been published that are capable of simultaneously measuring chromatin modifications and transcription: Paired-Tag (Zhu et al., 2021), CoTECH (Xiong et al., 2021), and SET-seq (Sun et al., 2021). One major conceptual difference between above methods and DamID-based techniques is the manner of capturing DNA in proximity of the chromatin mark of interest. Strategies leveraging CUT&Tag obtain a readout of chromatin by targeting protein A fused to transposase Tn5 (pA-Tn5) to antibody-bound regions and integrating barcoded adapters into the surrounding DNA. DamID deposits signal in living cells over time; consequently, it represents a historic record of chromatin state over a period of multiple hours up to a full cell cycle, while antibody-based techniques provide a snapshot view. In DamID, regions that are only transiently bound by the mark of interest will thus be represented more strongly in the signal relative to CUT&Tag-based methods. Another key difference is the extent to which chromatin accessibility affects the data. DamID techniques are known to have an accessibility signature due to extended exposure to free-floating Dam protein (discussed in more detail under Limitations), which is controlled for by performing experiments with untethered Dam. While CUT&Tag- and CUT&RUN-based methods have reported less of such an accessibility bias and do not customarily include explicit control experiments, early results (Zhang et al., 2021) suggest that such a bias may indeed be present. The question of data interpretation and normalization in light of this bias should be carefully considered among all existing single-cell genomics techniques. With regard to the transcriptional readout, the four techniques also employ different approaches: Paired-Tag exclusively amplifies the nuclear fraction of mRNA, SET-seq separates and measures total RNA in the cytoplasm, while CoTECH and scDam&T-seq both amplify the total mRNA. Finally, the Paired-Tag and CoTECH protocols have been adapted for combinatorial indexing and consequently have a higher throughput compared to scDam&T-seq and SET-seq.
Limitations
EpiDamID requires the expression of a construct encoding for the Dam-fusion protein in the system of interest. This may involve a substantial time investment depending on the system of choice and conditions generally need to be optimized for each Dam-fusion protein to reach high signal quality. DamID techniques are also limited in their resolution by the distribution of GATC motifs in the genome (median inter-GATC distance: 263 bp in mouse, 265 bp in human). In addition, we and others (Cheetham et al., 2018, Tosti et al., 2018, Szczesnik et al., 2019) have found that the methylation spreads ∼1 kb from the site of binding (Figure S1G), thus yielding an empirical resolution of 1–2 kb. This is sufficient to study the localization of many chromatin factors but may be restrictive when exact binding sites are required. Finally, due to the in vivo expression and consequent roaming of the Dam-POI in the nucleus, spurious methylation gradually accumulates in unspecific, mostly accessible, chromatin regions. The degree of accumulated background signal differs substantially between different Dam-POIs yet interferes most with proteins that reside within active chromatin. This can be overcome through computational normalization to the untethered Dam protein. In the case of single-cell experiments, this requires the grouping of similar cells into in silico populations. While this strategy yields good results, it does not provide a way to eliminate the accessibility component in individual cells, and the signal in single cells should therefore be interpreted as convolution of on-target and accessibility signal. Computational imputation of accessibility signal based on transcriptional similarity between targeted samples and Dam control samples could provide a solution to this problem, similar to current single-cell transcriptional imputation methods (see Hou et al., [2020] for an overview). We explored one experimental strategy to reduce off-target effects by implementing Dam mutants with decreased affinity for DNA, which yielded promising results in population data but insufficient m6A-events for single-cell profiling. Further adaptation of the Dam protein to engineer an enzyme with high enzymatic activity and reduced DNA-binding affinity may further improve the quality of EpiDamID profiles in single cells. Alternatively, molecular processing could be extended to facilitate an orthogonal accessibility readout from the same sample.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-H3K4me3 | Abcam | Cat#ab8580; RRID:AB_306649 |
| Rabbit polyclonal anti-H3K9ac | Abcam | Cat#ab4441; RRID:AB_2118292 |
| Rabbit polyclonal anti-H3K9me3 | Abcam | Cat#ab8898; RRID:AB_306848 |
| Rabbit polyclonal anti-H3K27me3 | Merck Millipore | Cat#07-449; RRID:AB_310624 |
| Rabbit polyclonal anti-H3K36me3 | Active Motif | Cat#61902; RRID:AB_2615073 |
| Rabbit polyclonal anti-H4K20me1 | Abcam | Cat#ab9051; RRID:AB_306967 |
| Mouse monoclonal anti-V5 | Invitrogen | Cat#R960-25; RRID:AB_2556564 |
| Chicken anti-GFP | Aves Labs | Cat#GFP-1020; RRID:AB_10000240 |
| Alexa Fluor 488 goat anti-chicken | Invitrogen | Cat#A-11039; RRID:AB_142924 |
| Alexa Fluor 647 goat anti-Rabbit | Invitrogen | Cat#A-21245; RRID:AB_2535813 |
| Bacterial and Virus Strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Thermo Fisher Scientific | Cat#C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Formaldehyde 37% | Sigma | Cat#F8775-500ml; CAS: 50-00-0 |
| Glycine | Sigma | Cat#50046-250 g; CAS: 56-40-6 |
| RNase A | Promega | Cat#A7973 |
| Proteinase K | Roche | Cat#3115879001; CAS: 39450-01-6 |
| Protein G beads | Thermo Fisher Scientific | Cat#88847 |
| Bovine Serum Albumin | Sigma | Cat#A2153-50G; CAS: 9048-46-8 |
| DMEM/F12, GlutaMAX™ supplement | GIBCO | Cat#31331028 |
| Fetal Bovine Serum | Sigma | Cat#F7524 lot BCBW6329 |
| Penicillin/Streptomycin (10,000 U/mL) | GIBCO | Cat#5140122 |
| Glasgow’s MEM | GIBCO | Cat#21710025 |
| MEM non-essential amino acids solution (100x) | GIBCO | Cat#1140035 |
| 100 mM Sodium Pyruvate | GIBCO | Cat#11360039 |
| GlutaMAX supplement (100 × ) | GIBCO | Cat#5050038 |
| TrypleE Express Enzyme | GIBCO | Cat#12605010 |
| ESGRO mLIF Medium Supplement | EMD Millipore | Cat#ESG1107; 10,000,000 U/mL |
| 1M Β-mercaptoethanol | Sigma | Cat#M3148; CAS: 60-24-2 |
| Indole-3-acetic acid sodium salt | Sigma | Cat#I5148; CAS: 6505-45-9 |
| Polybrene | Sigma | Cat#TR-1003-G; CAS: 28728-55-4 |
| Wizard® Genomic DNA Purification Kit | Promega | Cat#A1620 |
| MyTaq Red DNA Polymerase, 5000 units | Bioline | Cat#BIO-21110 |
| Lipofectamine3000 | Thermo Fisher Scientific | Cat#L3000008 |
| Puromycin dihydrochloride | Sigma | Cat#P9620; CAS:58-58-2 |
| 50mg/mL Hygromycin B | Thermo Fisher Scientific | Cat#10687010; CAS: 31282-04-9 |
| 10mg/mL Blasticidin S HCl | Thermo Fisher Scientific | Cat#A1113903; CAS: 2079-00-7 |
| Geneticin (G418 sulfate) | Thermo Fisher Scientific | Cat#11811031; CAS: 108321-42 |
| Neurobasal medium | GIBCO | Cat#21103049 |
| N2 supplement (100x) | GIBCO | Cat#17502048 |
| B27 supplement (50x) | GIBCO | Cat#A3582801 |
| CHIR99021 | Tocris | Cat#SML1046-5MG; CAS: 252917-06-9 |
| PD0325901 | Axon Medchem | Cat#PZ0162-5MG; CAS: 391210-10-9 |
| 5mg/mL 4-Hydroxytamoxifen | Sigma | Cat#SML1666; CAS: 68392-35-8 |
| 0.4%Trypan Blue solution | Sigma | Cat#T8154; CAS: 72-57-1 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | Cat#D1306; CAS: 28718-91-4 |
| Hoechst 34580 | Sigma | Cat#63493; CAS: 911004-45-0 |
| Propidium iodide | Sigma | Cat#P4864; CAS: 25535-16-4 |
| Nuclease-free water | Invitrogen | Cat#1097035 |
| Filtered Mineral Oil | Sigma | Cat#69794 |
| 1M Magnesium Acetate solution | Sigma | Cat#63052 |
| 5M Potassium Acetate solution | Sigma | Cat#95843 |
| Tween 20 | Sigma | Cat#P1379; CAS:9005-64-5 |
| ERCC RNA Spike-In mix 1 | Ambion | Cat#4456740 |
| Igepal | Sigma | Cat#I8896: CAS:9036-19-5 |
| dNTPs set (100 mM each) | Invitrogen | Cat#10297018 |
| SuperScript II | Thermo Fisher Scientific | Cat#18064014 |
| RNaseOUT Recombinant Ribonuclease Inhibitor | Invitrogen | Cat#10777019 |
| 5 × second-strand buffer | Thermo Fisher Scientific | Cat#10812014 |
| E. coli DNA ligase | Invitrogen | Cat#18052019 |
| DNA polymerase I | Thermo Fisher Scientific | Cat#18010025 |
| Ribonuclease H | Thermo Fisher Scientific | Cat#18021071 |
| 10 × CutSmart buffer | New England Biolabs | Cat#B7204S |
| DpnI | New England Biolabs | Cat#R0176L |
| Tris pH 7.5 | Roche | Cat#10708976001 |
| 5M NaCl | Sigma | Cat#S5150 |
| 0.5M EDTA pH 8 | Invitrogen | Cat#15575020 |
| T4 ligase 5 U/μl | Roche | Cat#10799009001 |
| PEG8000 | Merck | Cat#1546605 |
| SPRI beads | CleanNA | Cat#CPCR-0050 |
| Phusion High-Fidelity PCR Master Mix HF Buffer | New England Biolabs | Cat#M0531S |
| MyTaq Red DNA Polymerase, 5000 units | Bioline | Cat#BIO-21110 |
| VECTASHIELD Antifade mounting medium | Vector Laboratories | Cat#H-1000-10 |
| ProLong Gold Antifade Mountant | Thermo Fisher Scientific | Cat#P36930 |
| Collagenase type II from Cl. Histolyticum | GIBCO | Cat#17101015 |
| Hanks’ Balanced Salt Solution without Mg2+/Ca2+ | Thermo Fisher Scientific | Cat#88284 |
| Purified m6A-Tracer protein | Bas van Steensel lab | Van Schaik et al., 2020 |
| Critical Commercial Assays | ||
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat#Q33230 |
| Wizard® Genomic DNA Purification Kit | Promega | Cat#A1620 |
| Agilent RNA 6000 Pico Kit + chips | Agilent | Cat#50671513 |
| Agilent High Sensitivity DNA Kit + chips | Agilent | Cat#50674627 |
| mMESSAGE mMACHINE™ SP6 Transcription Kit | Invitrogen | Cat#AM1340 |
| Deposited Data | ||
| hTERT-RPE1 - DamID H3K9ac (mintbody) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID H4K20me1 (mintbody) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID POLR2F (full protein) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID TAD3 (protein domain tuple) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID CBX7 (protein domain tuple) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID H3K27me3 (mintbody) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID RING1B (full protein) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID CBX1 (protein domain tuple) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID CBX1 (full protein) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID MPHOSPH8 (protein domain tuple) | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID untethered Dam | This manuscript | GSE184036 |
| hTERT-RPE1 - DamID H3K27me3MUT (Y105F) | This manuscript | GSE184036 |
| hTERT-RPE1 - ChIP-seq H3K4me3 | This manuscript | GSE184036 |
| hTERT-RPE1 - ChIP-seq H3K9ac | This manuscript | GSE184036 |
| hTERT-RPE1 - ChIP-seq H3K36me3 | This manuscript | GSE184036 |
| hTERT-RPE1 - ChIP-seq H4K20me1 | This manuscript | GSE184036 |
| hTERT-RPE1 - ChIP-seq H3K27me3 | This manuscript | GSE184036 |
| hTERT-RPE1 - ChIP-seq H3K9me3 | This manuscript | GSE184036 |
| F1 hybrid mESC - scDam&T-seq H3K27me3 (mintbody) | This manuscript | GSE184036 |
| F1 hybrid mESC - scDam&T-seq CBX7 (protein domain tuple) | This manuscript | GSE184036 |
| F1 hybrid mESC - scDam&T-seq untethered Dam | This manuscript | GSE184036 |
| F1 hybrid mESC - scDam&T-seq H3K27me3MUT (Y105F) | This manuscript | GSE184036 |
| F1 hybrid mESC - scDam&T-seq RING1B (full protein) | Rooijers et al., 2019 | GSE108639 |
| F1 hybrid mESC - ChIP-seq H4K20me1 | This manuscript | GSE184036 |
| ES-E14TG2a.4 - ChIP-seq H3K27me3 | ENCODE | ENCSR059MBO |
| ES-E14 - ChIP-seq H3K9ac | ENCODE | ENCSR000CGP |
| F1 hybrid EB - scDam&T-seq H3K27me3 (mintbody) | This manuscript | GSE184036 |
| F1 hybrid EB - scDam&T-seq RING1B (full protein) | This manuscript | GSE184036 |
| F1 hybrid EB - scDam&T-seq untethered Dam | This manuscript | GSE184036 |
| EB - scNMT-seq | Argelaguet et al., 2019 | ftp://ftp.ebi.ac.uk/pub/databases/scnmt_gastrulation |
| Mouse Gastrulation Atlas - scRNA-seq | Pijuan-Sala et al., 2019 | https://github.com/MarioniLab/EmbryoTime course2018 |
| Zebrafish 15-somite embryo - scDam&T-seq MPHOSPH8 (protein domain tuple) | This manuscript | GSE184036 |
| Zebrafish 15-somite embryo - scDam&T-seq untethered Dam | This manuscript | GSE184036 |
| hTERT-RPE1 – unprocessed microscopy data | This manuscript | https://doi.org/10.17632/sp7hsw68c4.1 |
| Experimental Models: Cell Lines | ||
| human TERT-immortalized RPE-1 | ATCC | Cat#CRL-4000 |
| HEK293T | ATCC | Cat#CRL-3216 |
| BRL 3A | ATCC | Cat#CRL-1442 |
| F1 hybrid mESC | Joost Gribnau lab | Cast/EiJ x 129SvJae; RRID: CVCL_XY63 |
| F1 hybrid ESC EF1a-Tir1-IRES-neo | Rooijers et al., 2019 | N/A |
| F1 hybrid mESC EF1a-Tir1/AID-Dam-scFv-H4K20me1 | This manuscript | N/A |
| F1 hybrid mESC EF1a-Tir1/AID-Dam-scFv-H3K27me3 | This manuscript | N/A |
| F1 hybrid mESC EF1a-Tir1/AID-Dam-scFv-H3K27me3MUT(Y105F) | This manuscript | N/A |
| F1 hybrid mESC EF1a-Tir1/AID-Dam | This manuscript | N/A |
| F1 hybrid mESC EF1a-Tir1/AID-Dam-(PD-CBX7)3 | This manuscript | N/A |
| F1 hybrid mESC EF1a-Tir1/AID-Dam-RING1B | This manuscript | N/A |
| F1 hybrid mESC Tir1-TIGRE/Rosa26 knock-in AID-Dam-scFv-H3K27me3 | This manuscript | N/A |
| F1 hybrid mESC Tir1-TIGRE/Rosa26 knock-in AID-Dam | This manuscript | N/A |
| F1 hybrid mESC Tir1-TIGRE/knock-in AID-Dam-RING1B | This manuscript | N/A |
| Experimental Models: Organisms/Strains | ||
| Danio rerio Tüpfel long fin | EZRC or ZIRC | ZDB-GENO-990623-2 |
| Oligonucleotides | ||
| “AdRt” for adaptor ligation, top: CTAATACGACTCACTATA GGGCAGCGTGGTCGCGG CCGAGGA |
Vogel et al., 2007 | N/A |
| “AdRb” for adaptor ligation, bottom: TCCTCGGCCGCG |
Vogel et al., 2007 | N/A |
| “AdR_PCR” for m6A-PCR: GGTCGCGGCCGAGGATC |
Vogel et al., 2007 | N/A |
| RandomhexRT primer: GCCTTGGCACCCGAG AATTCCANNNNNN |
Follow Illumina design | N/A |
| RNA PCR primer 1: AATGATACGGCGACCACCGAGAT CTACACGTTCAGAGTTCTACAGTCCG∗A |
Follow Illumina design | N/A |
| RNA PCR index primer (example): CAAGCAGAAGACGGCATACGAGATCGTGATGT GACTGGAGTTCCTTGGCACCCGAGAATTCC∗A |
Follow Illumina design | N/A |
| Tir1-5′ Fw: cctctgctaaccatgttcatg | This manuscript | N/A |
| Tir1-5 Rev:tccttcacagctgatcagcacc | This manuscript | N/A |
| Tir1-3′ Fw:gggaagagaatagcaggcatgct | This manuscript | N/A |
| Tir1-3′ Rev:accagccacttcaaagtggtacc | This manuscript | N/A |
| Dam Fw:ttcaacaaaagccaggatcc | This manuscript | N/A |
| Dam Rev:gacagcggtgcataaggcgg | This manuscript | N/A |
| sgRNA RING1B: gctttttattcctagaaatgtctc |
This manuscript | N/A |
| sgRNA scFv-H3K27me3: gtccagtctttctagaagatgggc |
This manuscript | N/A |
| sgRNA ROSA26: gtccagtctttctagaagatgggc | This manuscript | N/A |
| Ring1Bki fw-gaacaacaagcgcatctggc | This manuscript | N/A |
| Ring1Bki rev:tcctcccctaacctgcttttgg | This manuscript | N/A |
| Ring1Bwt fw:tcctcccctaacctgcttttgg | This manuscript | N/A |
| Ring1Bwt+ rev:gccttgcctgcttggtttg | This manuscript | N/A |
| scFv-H3K27me3ki fw:gaactccatatatgggctatg | This manuscript | N/A |
| scFv-H3K27me3ki rev:cttggtgcgtttgcgggga | This manuscript | N/A |
| Primers for SORT-seq / CEL-Seq2 | Markodimitraki et al., 2020 | N/A |
| Adapters for DamID2, top and bottom oligonucleotides | Markodimitraki et al., 2020 | N/A |
| Recombinant DNA | ||
| pCCL.sin.cPPT.ΔLNGFR.Wpre | Bas van Steensel lab | (Amendola et al., 2005) |
| pCCL.PGK-Dam-(PD-CBX1)3x | This manuscript | N/A |
| pCCL.HSP-Dam-(PD-CBX1)2x | This manuscript | N/A |
| pCCL.HSP-CBX1-Dam | This manuscript | N/A |
| pCCL.PGK-(PD-CBX7)3x | This manuscript | N/A |
| pCCL.HSP-(PD-CBX7)3x | This manuscript | N/A |
| pCCL.PGK-Dam | This manuscript | N/A |
| pCCL.HSP-Dam | This manuscript | N/A |
| pCCL.PGK-Dam126 | This manuscript | N/A |
| pCCL.PGK-Dam-scFv-H3K27me3 | This manuscript | N/A |
| pCCL.PGK-Dam126-scFv-H3K27me3 | This manuscript | N/A |
| pCCL.HSP-Dam-scFv-H3K27me3 | This manuscript | N/A |
| pCCL.PGK-scFv-H3K27me3-Dam | This manuscript | N/A |
| pCCL.HSP-scFvH-3K27me3-Dam | This manuscript | N/A |
| pCCL.PGK-Dam-scFv-H3K27me3MUT(Y105F) | This manuscript | N/A |
| pCCL.PGK-Dam126-scFv-H3K27me3MUT(Y105F) | This manuscript | N/A |
| pCCL.PGK-scFv-H3K27me3MUT-Dam | This manuscript | N/A |
| pCCL.PGK-Dam-scFv-H3K9ac | This manuscript | N/A |
| pCCL.PGK-Dam-scFv-H4K20me1 | This manuscript | N/A |
| pCCL.PGK-Dam126-scFv-H4K20me1 | This manuscript | N/A |
| pCCL.HSP-Dam-scFv-H4K20me1 | This manuscript | N/A |
| pCCL.HSP-scFv-H4K20me1-Dam | This manuscript | N/A |
| pCCL.HSP-Dam-(PD-MPHOSPH8)3x | This manuscript | N/A |
| pCCL.PGK-Dam-POLR2F | This manuscript | N/A |
| pCCL.HSP-Dam-RING1B | This manuscript | N/A |
| pCCL.PGK-Dam-(PD-TAF3)3x | This manuscript | N/A |
| pCCL.HSP-Dam-(PD-TAF3)3x | This manuscript | N/A |
| pCCL-EF1a-Tir1-IRES-puro | This manuscript | N/A |
| pCCL-EF1a-Tir1-IRES-neo | Rooijers et al., 2019 | N/A |
| pCCL-hPGK-AID-Dam-scFv-H4K20me1 | This manuscript | N/A |
| pCCL-hPGK-AID-Dam-scFv-H3K9ac | This manuscript | N/A |
| pCCL-hPGK-AID-Dam-scFv-H3K27me3 | This manuscript | N/A |
| pCCL-hPGK-AID-Dam-scFv-H3K27me3MUT | This manuscript | N/A |
| pCCL-hPGK-AID-Dam-(PD-CBX7)3x | This manuscript | N/A |
| pCCL-hPGK-AID-Dam-RING1B | This manuscript | N/A |
| pHomRING1B-BSD-p2A-HA-mAID-Dam | This manuscript | N/A |
| pHomROSA26-ER-mAID-V5-Dam-scFv_H3K27me3-P2A-BSD-Hom | This manuscript | N/A |
| pHomROSA26-ER-mAID-V5-Dam-P2A-BSD-Hom | This manuscript | N/A |
| p225a-ROSA26spCas9-RNA | This manuscript | N/A |
| p225a-RING1BspCas9-gRNA | This manuscript | N/A |
| pX330-EN1201 | Zeng et al., 2008 | Addgene plasmid #92144 |
| pEN396-pCAGGS-Tir1-V5-2A-PuroR-TIGRE | Nora et al., 2017 | Addgene plasmid #92142 |
| SP6-GFP-T2A-HA-AID-Dam-V5-pA | This manuscript | N/A |
| SP6-HA-AID-Dam-V5-(MPHOSPH8-PD)3x-pA | This manuscript | N/A |
| Software and Algorithms | ||
| Tophat2 (v. 2.1.1) | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| DeepTools (v. 3.3.2) | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| MACS2 (v. 2.1.1.20160309) | Zhang et al., 2008 | N/A |
| Information Content | This manuscript | https://github.com/KindLab/EpiDamID2022 |
| MUSIC | Harmanci et al., 2014 | N/A |
| Seurat (v. 3.2.2) | Stuart et al., 2019 | https://satijalab.org/seurat/ |
| Harmony (v. 1.0) | Korsunsky et al., 2019 | https://portals.broadinstitute.org/harmony/articles/quickstart.html |
| SCENIC (v. 0.11.2) | van de Sande et al., 2020 | https://github.com/aertslab/pySCENIC |
| ChromHMM (v. 1.22) | Ernst and Kellis., 2012, 2017 | https://ernstlab.biolchem.ucla.edu/ChromHMM/ |
| LDA classifier | This manuscript | https://github.com/KindLab/EpiDamID2022 |
| Pipeline for DamID and scDam&T-seq data | Rooijers et al., 2019 | https://github.com/KindLab/scDamAndTools |
| Bowtie2 (v. 2.3.3.1) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Imaris 9.3 | Bitplane | https://imaris.oxinst.com/packages |
| Other | ||
| Bioruptor sonicator | Diagenode | N/A |
| 2100 Bioanalyzer platform | Agilent | N/A |
| BD FACSJazz Cell Sorter system | BD Biosciences | N/A |
| BD FACSInflux Cell Sorter system | BD Biosciences | N/A |
| Nanodrop II liquid handling platform | Innovadyne Technologies | N/A |
| mosquito LV liquid handling platform | SPT Labtech | N/A |
| Freedom EVO liquid handling platform | Tecan Life Sciences | N/A |
| Illumina NextSeq500 and/or Illumina NextSeq2000 hardware and sequencing reagents | Illumina | N/A |
| TCS SP8 laser scanning confocal microscope | Leica Microsystems | N/A |
| LSM900 confocal with AiryScan2 | Zeiss | N/A |
| Type F oil immersion liquid | Leica Microsystems | Cat#11513859; CAS: 195371-10-9 |
| Falcon™ Round-Bottom Polystyrene Test Tubes with Cell Strainer Snap Cap, 5mL | Thermo Fisher Scientific | Cat#08-771-23 |
| Falcon™ Round-Bottom Polypropylene Test Tubes with Cap, 5 mL | Thermo Fisher Scientific | Cat#14-959-11A |
| 384-well hard-shell plates | BioRad | HSP3801 |
| Amicon Ultra-15 centrifugal filter units | Merck | Cat#UFC910024 |
| 70-μm cell strainer | Greiner Bio-One | Cat#542070 |
| 40-μm cell strainer | Greiner Bio-One | Cat#542070 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jop Kind (j.kind@hubrecht.eu).
Materials availability
Unique/stable materials generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell lines
All cell lines were grown in a humidified chamber at 37 °C in 5% CO2, and were routinely tested for mycoplasma. Human TERT-immortalized RPE-1 cells were cultured in DMEM/F12 (GIBCO) containing 10% FBS (Sigma F7524 lot BCBW6329) and 1% Pen/Strep (GIBCO). This cell line does not contain a Y chromosome. Human HEK293T cells were cultured in DMEM (GIBCO) containing 10% FBS and 1% Pen/Strep (GIBCO). This cell line does not contain a Y chromosome. Mouse F1 hybrid Cast/EiJ x 129SvJae embryonic stem cells (mESCs; a kind gift from the Joost Gribnau laboratory) were cultured on irradiated primary mouse embryonic fibroblasts (MEFs), in mESC culture media CM+/+ defined as follows: G-MEM (GIBCO) supplemented with 10% FBS (Sigma F7524 lot BCBW6329), 1% Pen/Strep (GIBCO), 1x GlutaMAX (GIBCO), 1x non-essential amino acids (GIBCO), 1x sodium pyruvate (GIBCO), 0.1 mM β-mercaptoethanol (Sigma) and 1000 U/mL ESGROmLIF (EMD Millipore ESG1107). Cells were split every 3 days and medium was changed every other day. Expression of the Dam-POI constructs was suppressed by addition of 0.5 mM indole-3-acetic acid (IAA; Sigma, I5148). This cell line does not contain a Y chromosome.
Zebrafish
All experiments were conducted under the guidelines of the animal welfare committee of the Royal Netherlands Academy of Arts and Sciences (KNAW). Adult Tüpfel long fin (wild type) zebrafish (Danio rerio) were maintained and embryos raised and staged as previously described (Aleström et al., 2019; Westerfield, 2000).
Method details
ChIP-seq
ChIP-seq was performed as described previously (Collas, 2011), with the following adaptations. Cells were harvested by trypsinization, and chemically crosslinked with fresh formaldehyde solution (1% in PBS) for 8 min while rotating at room temperature. Crosslinking was quenched with glycine on ice and sample was centrifuged at 500 g for 10 min at 4°C. Pellet was then resuspended in lysis buffer for 5 min on ice and sonicated as follows: 16 cycles of 30 s on / 30 s off at max power (Bioruptor Diagenode), and centrifuged at 14,000 rpm at 4°C for 10 min. The chromatin in supernatant was treated with RNase A for 30 min at 37°C, and Proteinase K for 4 h at 65°C to reverse crosslinks, then cleared using DNA purification columns and eluted in nuclease-free water. Chromatin was incubated with antibodies (see below), after which Protein G beads (ThermoFisher #88847) were added for antibody binding. After successive washing, samples were cleared using DNA purification columns, eluted in nuclease-free water, and measured using a Qubit fluorometer. Libraries were prepared according to the Illumina TruSeq DNA LT kit and sequenced on the Illumina HiSeq 2500 following manufacturer’s protocols. Up to 50 ng of immunoprecipitated chromatin was used as input for library preparation. Antibodies used were: anti-H3K4me3 Abcam ab8580, anti-H3K9ac Abcam ab4441, anti-H3K9me3 Abcam ab8898, anti-H3K27me3 Merck Millipore 07-449, anti-H3K36me3 Active Motif 61902, anti-H4K20me1 Abcam ab9051.
DamID construct design
The constructs for mintbodies, chromatin binding domains, and full-length protein constructs were fused to Dam in both possible orientations under the control of the auxin-inducible degron (AID) system (Kubota et al., 2013; Nishimura et al., 2009) with either the hPGK or HSP promoter, and cloned into the pCCL.sin.cPPT.ΔLNGFR.Wpre lentiviral construct (Amendola et al., 2005) by standard cloning procedures.
The linkers used for the triple fusion domains are, in order of appearance:
Dam; V5 linker [GKPIPNPLLGLDST]; 1st domain (e.g., chromo); GSAGSAAGSGEF; 2nd domain; linker [KESGSVSSEQLAQFRSLD]; 3rd domain. All other POIs are linked to Dam via a V5 linker, which has been commonly used in DamID constructs (Meuleman et al., 2013; Peric-Hupkes et al., 2010; Vogel et al., 2006). The Gly- and Ser-rich flexible linker, GSAGSAAGSGEF, was designed to express GFP-fusion proteins for rapid protein-folding assay (Waldo et al., 1999). The KESGSVSSEQLAQFRSLD flexible linker was previously used for the construction of a bioactive scFv (Bird et al., 1988). For context: the Gly and Ser residues in the linker were designed to provide flexibility, whereas Glu and Lys were added to improve the solubility (Chen et al., 2013).
Bulk DamID2
hTERT-RPE1 cells were grown as described above. At 30% confluence in 6-well plates, cells were transduced with 1500 μL total volume unconcentrated lentivirus, amounts ranging between 20-1500 μL unconcentrated lentivirus (or 0.1-40 μL concentrated) in the presence of 10 μg/mL polybrene. Cells were collected for genomic DNA isolation (Wizard, Promega) 48 h after transduction. Dam methylation levels were checked by m6A-PCR as previously described (de Luca & Kind, 2021; Vogel et al., 2007) and sequenced following the DamID2 protocol (Markodimitraki et al., 2020).
Immunofluorescent staining and confocal imaging of RPE-1 cells
Viral transduction was performed as described above for bulk DamID2, with the exception that RPE-1 cells were grown on glass coverslips. Two days after transduction, cells were washed with PBS and chemically crosslinked with fresh formaldehyde solution (2% in PBS) for 10 min at RT, then permeabilized (with 0.5% IGEPAL® CA-630 in PBS) for 20 min and blocked (with 1% bovine serum albumin (BSA) in PBS) for 30 min. All antibody incubations were performed in final 1% BSA in PBS followed by three PBS washes at RT. Incubation with primary antibody against the endogenous histone modification as well as purified m6A-Tracer protein (Van Schaik et al., 2020) (recognizing methylated DNA) was performed at 4°C for 16 h (overnight), followed by anti-GFP (against m6A-Tracer protein) incubation at RT for 1 h, and secondary antibody incubations at RT for 1 h. The final PBS wash was simultaneously an incubation with DAPI at 0.5 μg/mL for 2 min, followed by a wash in MilliQ and sample mounting on glass slides using VECTASHIELD Antifade mounting medium (Vector Laboratories). Primary antibodies: anti-H3K9ac abcam ab4441 (rabbit) at 1:1000, anti-H3K9me3 abcam ab8898 (rabbit) at 1:300, anti-GFP Aves GFP-1020 (chicken) at 1:1000. Secondary antibodies: AlexaFluor anti-chicken 488 at 1:500 and anti-rabbit 647 at 1:500. Purified m6A-Tracer protein (used at 1:1000) was a kind gift from the Bas van Steensel laboratory. Imaging was performed on a Leica TCS SP8 laser scanning confocal microscope with a 63X (NA 1.40) oil-immersion objective. Images were processed in Imaris 9.3 (Bitplane) by baseline subtraction. Additional background correction was done with a 1-μM Gaussian filter for the images of Dam-CBX1 m6A-Tracer and H3K9me3 stainings.
Generation of mouse embryonic stem cell lines
The various stable clonal F1 hybrid mESC lines for the initial single cell experiments were created by lentiviral co-transduction of pCCL-EF1α-Tir1-IRES-puro and pCCL-hPGK-AID-Dam-POI constructs with a 4:1 ratio in a EF1α-Tir1-IRES-neo mother line (Rooijers et al., 2019), after which the cells were selected for 10 days on 0.1% gelatine coated 10-cm dishes in 60% Buffalo Rat Liver (BRL)-conditioned medium containing 0.8 μg/mL puromycin (Sigma P9620), 250 μg/mL G418 (ThermoFisher 11811031) and 0.5 mM IAA. Individual puromycin resistant colonies were handpicked and tested for the presence of the constructs by PCR using Dam-specific primers fw-ttcaacaaaagccaggatcc and rev-gacagcggtgcataaggcgg.
The clonal F1 hybrid knock-in cell lines were CRISPR targeted in a mother line carrying Tir1-Puro in the TIGRE locus (Zeng et al., 2008). For all CRISPR targeting, cells were cultured on gelatin-coated 6-wells in 60% BRL conditioned medium to 70%–90% confluency and transfected with Lipofectamin3000 (Invitrogen L3000008) according to the supplier protocol with 2 μg donor vector and 1 μg Cas9/guide vector. At 24 h after transfection the cells were split to a gelatin-coated 10-cm dish and antibiotic selection of transfected cells is started 48 h after transfection. Cells were selected with 60% BRL conditioned medium containing 0.8 μg/mL puromycin for the Tir1 knock-in and 2.5 μg/mL blasticidin (Invivogen) for the AID-Dam knock-in lines. After 5-10 days of selection, individual colonies were manually picked and screened by PCR for the correct genotype.
All CRISPR knock-in lines were made in a Tir1-TIGRE mother line that was generated by co-transfection of Cas9-gRNA plasmid pX330-EN1201(Addgene plasmid #92144) and donor plasmid pEN396-pCAGGS-Tir1-V5-2A-PuroR TIGRE (Addgene plasmid #92142) (Nora et al., 2017). The Tir1-puro clones were screened for the presence of Tir1 by PCR from the CAGG promoter to Tir1 with the primers fw-cctctgctaaccatgttcatg and rev-tccttcacagctgatcagcacc, followed by screening for correct integration in the TIGRE locus by PCR from the polyA to the TIGRE locus with primers fw-gggaagagaatagcaggcatgct and rev-accagccacttcaaagtggtacc. The Tir1 expression was further confirmed by Western blot using a V5 antibody (Invitrogen R960-25).
A knock-in of AID-Dam in the N terminus of the RING1B locus was made by co-transfection of a donor vector carrying the blasticidin-p2A-HA-mAID-Dam cassette flanked by two 500-bp homology arms of the endogenous RING1B locus (pHom-BSD-p2A-HA-mAID-Dam) and p225a-RING1B spCas9-gRNA vector (sgRNA: 5′gctttttattcctagaaatgtctc3′) as described above. Picked clones were screened for correct integration by PCR with primers from Dam to the RING1B locus outside the targeting construct; fw-gaacaacaagcgcatctggc and rev-tcctcccctaacctgcttttgg. Presence of the RING1B wildtype allele was checked by PCR with primers fw-tcctcccctaacctgcttttgg and rev-gccttgcctgcttggtttg. The H3K27me3 mintbody coupled to ER-mAID-Dam was knocked into the Rosa26 locus by co-transfection of pHom-ER-mAID-V5-Dam-scFv_H3K27me3-P2A-BSD-Hom donor vector and p225a-Rosa26 spCas9-RNA vector (sgRNA: gtccagtctttctagaagatgggc) as described above. Picked clones were screened for correct integration by PCR from a sequence adjacent to the Rosa homology arm to the Rosa26 locus with primers fw-gaactccatatatgggctatg and rev-cttggtgcgtttgcgggga. The untethered mAID-Dam was knocked into the Rosa26 locus by co-transfection with the pHom-ER-mAID-V5-Dam-P2A-BSD-Hom donor vector and p225a-Rosa26 spCas9-RNA vector (sgRNA: gtccagtctttctagaagatgggc) as described above. Picked clones were screened for correct integration by PCR with the same primers as for the Dam-H3K27me3 mintbody knock-in line.
All clones with correct integrations were furthermore screened for their level of induction upon IAA removal by m6A-PCR evaluated by gel electrophoresis (de Luca and Kind, 2021; Vogel et al., 2007), followed by DamID2 sequencing in bulk (Markodimitraki et al., 2020), to select the clone with a correct karyotype and the best signal-to-noise ratio of enrichment over expected regions or chromatin domains. Finally, the best 3-4 clones were selected for testing of IAA removal timing in single cells by DamID2.
Mouse embryonic stem cell culture and induction of Dam-fusion proteins
When plated for targeting or genomics experiments, cells were passaged at least 2 times in feeder-free conditions, on plates coated with 0.1% gelatin, grown in 60% BRL-conditioned medium, defined as follows and containing 1 mM IAA: 40% CM+/+ medium and 60% of CM+/+ medium conditioned on BRL cells. For timed induction of the constructs the IAA was washed out at different clone-specific times before single-cell sorting.
Embryoid body differentiation and induction of Dam-fusion proteins
For EB differentiation, the stable knock-in F1ES lines were cultured for 2 weeks on plates coated with 0.1% gelatin, grown in 2i+LIF ES cell culture medium defined as follows: 48% DMEM/F12 (GIBCO) and 48% Neurobasal medium (GIBCO), supplemented with 1x N2 (GIBCO), 1x B27 supplement + vitamin A (GIBCO), 1x non-essential amino acids, 1% FBS, 1% Pen/Strep, 0.1mM β-mercaptoethanol, 1 μM PD0325901 (Axon Medchem, PZ0162-5MG), 3 μM CHIR99021 (Tocris, SML1046-5MG), 1000 U/mL ESGRO mLIF. EB differentiation was performed according to ATCC protocol. On day 1 of differentiation, 2x10∧6 cells were grown in suspension on a non-coated bacterial 10-cm dish with 15 mL CM+/− (with β-mercaptoethanol, without LIF) and 0.5 mM IAA. On day 2, half the cell suspension was divided over five non-coated bacterial 10-cm dishes each containing 15 mL CM+/− medium and 0.5 mM IAA. Plates were refreshed every other day. EBs were harvested at day 7, 10, and 14. Two days before single-cell sorting, the EBs were grown in CM+/− medium containing 1 mM IAA, and induced as follows: 6 h without IAA (RING1B); 20 h without IAA and 7 h with 1 μM 4OHT (Sigma SML1666) (Dam-H3K27me3-mintbody); 7 h without IAA and 4 h with 1 μM 4OHT (untethered Dam). The EBs were evaluated by brightfield microscopy and hand-picked for further handling (see below).
FACS for single-cell experiments
FACS was performed on BD FACSJazz or BD FACSInflux Cell Sorter systems with BD Sortware. mESCs and EBs were harvested by trypsinization, centrifuged at 300 g, resuspended in medium containing 20 μg/mL Hoechst 34580 (Sigma 63493) per 1x106 cells and incubated for 45 min at 37°C. Prior to sorting, cells were passed through a 40-μm cell strainer. Propidium iodide (1 μg/mL) was used as a live/dead discriminant. Single cells were gated on forward and side scatters and Hoechst cell cycle profiles. Index information was recorded for all sorts. One cell per well was sorted into 384-well hard-shell plates (Biorad, HSP3801) containing 5 μL of filtered mineral oil (Sigma #69794) and 50 nL of 0.5 μM barcoded CEL-Seq2 primer (Markodimitraki et al., 2020; Rooijers et al., 2019). In the EB experiment, the knock-in mESC lines were cultured alongside on 2i+LIF medium and included as a reference at each time point.
Single-cell Dam&T-seq
The scDam&T-seq protocol was performed as previously described in detail (Markodimitraki et al., 2020), with the adaptation that all volumes were halved to reduce costs. Liquid reagent dispension steps were performed on a Nanodrop II robot (Innovadyne Technologies / BioNex). Addition of barcoded adapters was done with a mosquito LV (SPT Labtech). In short, after FACS, 50 nL per well of lysis mix (0.07% IGEPAL, 1 mM dNTPs, 1:50,000 ERCC RNA spike-in mix (Ambion, 4456740)) was added, followed by incubation at 65 °C for 5 min. 100 nL of reverse transcription mix (1 × First Strand Buffer and 10 mM DTT (Invitrogen, 18064-014), 2 U RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen, 10777019), 10 U SuperscriptII (Invitrogen, 18064-014)) was added, followed by incubation at 42 °C for 2 h, 4 °C for 5 min and 70 °C for 10 min. Next, 885 nL of second strand synthesis mix (1 × second strand buffer (Invitrogen, 10812014), 192 μM dNTPs, 0.006 U E. coli DNA ligase (Invitrogen, 18052019), 0.013 U RNase H (Invitrogen, 18021071), 0.26 U E. coli DNA polymerase (Invitrogen)) was added, followed by incubation at 16 °C for 2 h. 250 nL of protease mix was added (1 × NEB CutSmart buffer, 1.0cmg/mL Proteinase K (Roche, 000000003115836001)), followed by incubation at 50 °C for 10 h and 80 °C for 20 min. Next, 115 nL of DpnI mix (1 × NEB CutSmart buffer, 0.1 U NEB DpnI) was added, followed by incubation at 37 °C for 6 h and 80 °C for 20 min. Finally, 50 nL of 0.5 uM DamID2 adapters were dispensed (final concentrations 25 nM), followed by 400 nL of ligation mix (1 × T4 Ligase buffer (Roche, 10799009001), 0.13 U T4 Ligase (Roche, 10799009001)) and incubation at 16 °C for 16 hr and 65 °C for 10 min. Contents of all wells were pooled and the aqueous phase was recovered by centrifugation and transferred to clean tubes. Samples were purified by incubation for 10 min with 0.8 volumes magnetic beads (CleanNA, CPCR-0050) diluted 1:7 with bead binding buffer (20% PEG8000, 2.5 M NaCl), washed twice with 80% ethanol and resuspended in 8 μL of nuclease-free water before in vitro transcription at 37 °C for 14 h using the MEGAScript T7 kit (Invitrogen, AM1334). Library preparation was done as described in the CEL-Seq2 protocol with minor adjustments (Hashimshony et al., 2016). Amplified RNA (aRNA) was purified with 0.8 volumes beads as described above, and resuspended in 20 μL of nuclease-free water, and fragmented at 94 °C for 90 s with the addition of 0.25 volumes fragmentation buffer. Fragmentation was stopped by addition of 0.1 volumes of 0.5 M EDTA pH 8 and quenched on ice. Fragmented aRNA was purified with beads as described above, and resuspended in 12 μL of nuclease-free water. Thereafter, library preparation was done as previously described (Hashimshony et al., 2016) using up to 7 μL or approximately 150 ng of aRNA, and 8-10 PCR cycles depending on input material. Libraries were sequenced on the Illumina NextSeq500 (75-bp reads) or NextSeq2000 (100-bp reads) platform.
Collection of zebrafish samples and FACS
Tüpfel long fin (wild type) pairs were set up and the following morning, approximately 1 nL of 1 ng/μL Dam-Mphosph8 mRNA or 0.5 ng/μL Dam-Gfp mRNA was injected into the yolk at the 1-cell stage. Embryos were slowed down overnight at 23 °C and the following morning all embryos were manually dechorionated. At 15-somite stage, embryos were transferred to 2-mL Eppendorf tubes and digested with 0.1% Collagenase type II from Cl. Histolyticum (GIBCO) in Hanks Balanced Salt Solution without Mg2+/Ca2+ (Thermofisher) for 20-30 min at 32 °C with constant shaking. Once embryos were noticeably digested, cell solution was spun at 2000 g for 5 min at room temperature and the supernatant was removed. Cell pellet was resuspended with TrypLE Express (Thermofisher) and digested for 10 min at 32 °C with constant shaking. Cell solution was inactivated with 10% Fetal Bovine Serum (Thermofisher) in Hanks Balanced Salt Solution without Mg2+/Ca2+ and filtered through a 70-μm cell strainer (Greiner Bio-One). Cells were pelleted at 2000 g for 5 min at room temperature and washed twice with 10% Fetal Bovine Serum (Thermofisher) in Hanks Balanced Salt Solution without Mg2+/Ca2+. Hoechst 34580 at a final concentration of 16.8 μg/mL was added to the cell solution and incubated for 30 min at 28 °C in the dark. Solution was then filtered through a 40-μm cell strainer (Greiner Bio-One), and propidium iodide was added at a final concentration of 5 μL/mL. FACS was performed on BD FACSInflux as described above, retaining only cells in G2/M phase based on Hoechst DNA content. Plates were processed for scDam&T-seq as described above.
Immunofluorescent staining and confocal imaging of zebrafish embryos
Embryos at 15-somite stage were fixed in 4% PFA (Sigma) for 2 h at RT, followed by washes in PBS. Embryos were then washed three times in 4% sucrose/PBS and allowed to equilibrate in 30% sucrose/PBS at 4°C for 3-5 h. Embryos were suspended in Tissue Freezing Medium (Leica) orientated in the sagittal plane and frozen with dry ice. Blocks were sectioned at 8 μm and slides were rehydrated in PBS, treated with −20°C pre-cooled acetone for 7 min at −20°C, washed three times with PBS and digested with Proteinase K (Promega) at a final concentration of 10 μg/mL for 3 min, washed 1x PBS and incubated in blocking buffer (10% Fetal Bovine Serum, 1% DMSO, 0.1% Tween20 in PBS) for 30 min. Primary antibody was diluted in blocking buffer and slides incubated overnight at 4°C. Slides were washed the following day and incubated with the appropriate AlexaFluor secondary antibodies (1:500), DAPI (0.5 μg/mL) and Phalloidin-TRITC (1:200) diluted in blocking buffer for 1 h at RT. Slides were washed, covered with glass coverslips with ProLong Gold Antifade Mountant (Thermofisher) and imaged at 63X with a LSM900 confocal with AiryScan2 (Zeiss). Images were viewed and processed in Imaris 9.3 (Bitplane) and Adobe Creative Cloud (Adobe). Primary antibody: anti-H3K9me3 abcam ab8898 at 1:500 (Chandra et al., 2012).
Processing DamID and scDam&T-seq data
Data generated by the DamID and scDam&T-seq protocols was largely processed with the workflow and scripts described in (Markodimitraki et al., 2020) (see also https://www.github.com/KindLab/scDamAndTools). The procedure is described in short below.
Demultiplexing
All reads are demultiplexed based on the barcode present at the start of R1 using a reference list of barcodes. In the case of scDam&T-seq data, the reference barcodes contain both DamID-specific and CEL-Seq2-specific barcodes and zero mismatches between the observed barcode and reference are allowed. In the case of the population DamID data, the reference barcodes only contain DamID-specific barcodes and one mismatch is allowed. The UMI information, also present at the start of R1, is appended to the read name.
DamID data processing
DamID reads are aligned using bowtie2 (v. 2.3.3.1) (Langmead and Salzberg, 2012) with the following parameters: “--seed 42 --very-sensitive -N 1.” For human samples, the hg19 reference genome is used; for mouse samples, the mm10 reference genome; and for zebrafish samples the GRCz11 reference genome. The resulting alignments are then converted to UMI-unique GATC counts by matching each alignment to known strand-specific GATC positions in the reference genome. Any reads that do not align to a known GATC position or have a mapping quality smaller than 10 are removed. In the case of bulk DamID samples, up to 64 unique UMIs are allowed per GATC position, while up to 4 unique UMIs are allowed for single-cell samples to account for the maximum number of alleles in G2. Finally, counts are binned at the desired resolution.
CEL-Seq2 data processing
CEL-Seq2 reads are aligned using tophat2 (v. 2.1.1) (Kim et al., 2013) with the following parameters: “--segment-length 22 --read-mismatches 4 --read-edit-dist 4 --min-anchor 6 --min-intron-length 25 --max-intron-length 25000 --no-novel-juncs --no-novel-indels --no-coverage-search --b2-very-sensitive --b2-N 1 --b2-gbar 200.” For mouse samples, the mm10 reference genome and the GRCm38 (v. 89) transcript models are used. For zebrafish samples, the GRCz11 reference genome and the adjusted transcript models published by the Lawson lab (Lawson et al., 2020) are used. Alignments are subsequently converted to transcript counts per gene with custom scripts that assign reads to genes similar to HTSeq’s (Anders et al., 2015) htseq-count with mode “intersection_strict.”
Processing of ChIP-seq data
External ChIP-seq datasets were downloaded from the NCBI GEO repository and the ENCODE database (Davis et al., 2018). The external ChIP-seq data used in this manuscript consists of: H3K9ac ChIP-seq in mESC (ENCSR000CGP), H3K27me3 ChIP-seq in mESC (ENCSR059MBO), and H3K9me3 ChIP-seq in 6-hpf zebrafish embryos (Laue et al., 2019) (GSE113086). Internal and external ChIP-seq data were processed in an identical manner. First reads were aligned using bowtie2 (v. 2.3.3.1) with the following parameters: “--seed 42 --very-sensitive -N 1.” Indexes for the alignments were then generated using “samtools index” and genome coverage tracks were computed using the “bamCoverage” utility from DeepTools (v. 3.3.2) (Ramírez et al., 2016) with the following parameters: “--ignoreDuplicates --minMappingQuality 10.” For marks that exist in broad domains in the genome, domains were called using MUSIC (Harmanci et al., 2014) according to the suggested workflow (https://github.com/gersteinlab/MUSIC). For marks that form narrow peaks in the genome, peaks were called using MACS2 (v. 2.1.1.20160309) (Zhang et al., 2008) using the “macs2 callpeak” utility with the following parameters: “-q 0.05.”
Computing the Information Content (IC) of DamID samples
The Information Content (IC) of a DamID sample is a measure of how much structure is in the detected methylation signal. It is essentially an adaptation of the RNA-seq normalization strategy called PoissonSeq (Li et al., 2012). Its goal is to compare the obtained signal to a background signal (the density of mappable GATCs), identify regions where the signal is similar to background, and finally compare the amount of total signal (i.e., total GATC counts) to the total signal in background regions. The IC is the ratio of total signal over background signal and can be used to filter out samples that contain little structure in their data. The code used to compute the IC is available online (https://github.com/KindLab/EpiDamID2022) and the procedure is explained below.
As an input, we use the sample counts binned at 100-kb intervals, smoothened with a 250-kb Gaussian kernel. The large bin size and smoothing are necessary when working with single-cell samples that have very sparse and peaky data and would otherwise be difficult to match to the background signal. As a control, we use the number of mappable GATCs in the same 100-kb bins, similarly smoothened. We subsequently remove all genomic bins that do not have any observed counts in the sample. Our starting data is then , a matrix with size , where is the number of genomic bins and is the number of samples. Since we are comparing one experimental sample with the control, is always 2. denotes the number of counts observed in the th bin of the th sample. We first compute the expected number of counts for each based on the marginal probabilities of observing counts in each bin and in each sample:
Where is the total sum of ; is the marginal probability of observing counts in bin ; is the marginal probability of observing counts in sample ; and is the matrix of size where entry is the expected number of counts in bin for sample , computed as .
We subsequently compute the goodness of fit of our predictions compared to the actual counts per bin:
Where is the measure of how well the predictions of match the observed counts in in bin . The better the prediction, the closer is to zero, indicating that the signal of the experimental sample closely resembles the background in bin . Next, an iterative process is performed where in each step a subset of the original bins is chosen that exclude bins with extreme values of . Specifically, all bins with a goodness of fit in the top and bottom 5th percentiles are excluded to progressively move toward a stable set of bins where the sample resembles the background. After each iteration, the chosen bins are compared to the previous set of bins and when this has stabilized, or when the maximum number of iterations is reached, the procedure stops. In practice, convergence is usually reached after only a couple of iterations. The IC is then computed for the experimental sample as the ratio of its summed total counts to the sum of counts observed in the final subset of bins.
Population DamID data filtering and analyses
The population DamID samples were filtered based on a depth threshold of 300,000 UMI-unique GATC counts and an IC of at least 1.1. Per Dam-construct, the best samples based on the IC were maintained. Samples were normalized for the total number of counts using reads per kilobase per million (RPKM). Normalization for Dam controls was performed by adding a pseudo count of 1, taking the per bin fold-change with Dam, and performing a log2-transformation, resulting in log2 observed-over-expected (log2OE) values. The UMAP presented in Figure 1B was computed by performing principal component analysis (PCA) on the RPKM-normalized samples (20-kb bins) and using the top components for UMAP computation in python with custom scripts. For the correlations presented in Figures 1C and S1C, the RPKM-normalized DamID values were normalized for the density of mappable GATCs and log-transformed. The Spearman’s rank correlation was then computed with the input-normalized ChIP-seq values of the various marks.
Resolution analysis on RPE-1 samples
To evaluate the resolution of EpiDamID signal compared to ChIP-seq, we wanted to determine the spread of the signal around regions of known enrichment. To this end, we used ChIP-seq peaks for H3K9ac and H3K4me3, and domains for H3K27me3 and H3K9me3. We computed the average ChIP-seq signal and DamID signal around these regions, using a resolution (i.e., bin size) of 200 bp. The resulting signal was mildly smoothed to get a better representation of the trends. For each sample, we then determined the distance over which the signal measured at the reference point decayed to 50% relative to the background. As a reference point, we chose the center of H3K9ac and H3K4me3 peaks, or the boundary of H3K27me3 and H3K9me3 domains. The spread of the DamID signal can then be determined as the increase in this distance relative to the corresponding ChIP-seq sample.
Single-cell DamID data filtering and analyses
Filtering and normalizing scDamID data
Single-cell DamID samples were filtered based on a depth and an IC threshold. For the mouse samples, these thresholds were 3,000 unique GATCs and an IC within the range of 1.5 to 7 (the upper threshold removes samples with very sparse profiles); for zebrafish, these thresholds were 1,000 unique GATCs and an IC within the range of 1.2 to 7. For the zebrafish samples, chromosome 4 was excluded when determining depth and IC (and in all downstream analyses) since the reference assembly of this chromosome is poor and alignments unreliable. The quality of scDam&T-seq samples is determined separately for the DamID readout and the CEL-Seq2 readout. To preserve as much of the data as possible, we used all samples passing DamID thresholds for analyses that relied exclusively on the DamID readout. Wherever single-cell data was used, samples were normalized for their total number of GATCs, scaled by a factor 10,000, and log-transformed with a pseudo-count of 1, equivalent to the normalizations customarily performed for single-cell RNA-seq samples. To generate in silico populations based on single-cell samples, the binned UMI-unique counts of all single-cells were combined and normalization was performed equivalent to population DamID samples.
scDamID UMAPs
The UMAPs presented in Figures 2A, 3C, and 5C were computed by performing PCA on the depth-normalized single-cell samples and using the top components for UMAP computation. Since in EBs inactivation of chromosome X can coincides with a strong enrichment of H3K27me3/RING1B on that chromosome, we depth-normalized these samples using the total number of GATCs on somatic chromosomes. For the zebrafish samples, chromosome 4 was completely excluded from the analysis. For the mouse UMAPs, the single-cell data were binned at a resolution of 10-kb intervals, while for the zebrafish UMAPs, the resolution was 100 kb. Notably, when the first principal components showed a strong correlation to sample depth, it was excluded.
Single-cell count enrichment
Figures 2B–2D show the enrichment of counts in ChIP-seq domains for all single-cell mESC samples; Figure S5F shows the enrichment of counts for all MPHOSPH8 zebrafish samples. The count enrichment is equivalent to the more well-known Fraction Reads in Peaks (FRiP) metric, but has been normalized for the expected fraction of counts within the domains based on the total number of mappable GATCs covered by these domains. In other words, if the domains cover 50% of the mappable GATCs in the genome and we observe that 70% of a sample’s counts fall within these domains, the count enrichment is 0.7 / 0.5 = 1.4.
Single-cell CEL-Seq2 data filtering and analyses
Filtering CEL-Seq2 data
Single-cell datasets were evaluated with respect to the number of unique transcripts, percentage mitochondrial reads, percentage ERCC-derived transcripts and the percentage of reads coming from unannoted gene models (starting with “AC” or “Gm”) and appropriate thresholds were chosen. For the EB data, the used thresholds were 1,000 UMI-unique transcripts, < 7.5% mitochondrial transcripts, < 1% ERCC-derived transcripts, and < 5% transcripts derived from unannotated gene models. In addition, a small group of cells (29/6,554 ≈ 0.4%) from different time points, which formed a cluster that could not be annotated and was characterized by high expression of ribosomal genes, was removed from further analyses. For the zebrafish data, the used thresholds were 1,000 UMI-unique transcripts and < 5% ERCC-derived transcripts. Only genes observed in at least 5 samples across the entire dataset were maintained in further analyses. The quality of scDam&T-seq samples is determined separately for the DamID readout and the CEL-Seq2 readout. To preserve as much of the data as possible, we used all samples passing CEL-Seq2 thresholds (independent of DamID quality) for transcriptome-based analyses.
Analysis of CEL-Seq2 data with Seurat and Harmony
Single-cell transcription data was processed using Seurat (v3) (Stuart et al., 2019). First, samples were processed using the “NormalizeData,” “FindVariableFeatures,” “ScaleData,” and “RunPCA” commands with default parameters. Subsequently, batch effects relating to processing batch and plate were removed using Harmony (Korsunsky et al., 2019) using the “RunHarmony” command, using a theta = 2 for the batch variable and theta = 1 for the plate variable. Clustering and dimensionality reduction were subsequently performed with the “FindNeighbors,” “FindClusters” and “RunUMAP” commands. Differentially expressed genes per cluster were found using the “FindAllMarkers” command.
Integration with external single-cell datasets
The EB data was integrated with part of the single-cell mouse embryo atlas published by (Pijuan-Sala et al., 2019) and with the transcription data from the scNMT-seq EB dataset published by (Argelaguet et al., 2019). In the case of the mouse embryo atlas, the data was loaded directly into R via the provided R package “MouseGastrulationData.” One dataset per time point was included (datasets 18, 14, 19, 16, 17, corresponding to embryonic stages E6.5, E7.0, E7.5, E8.0, E8.5, respectively). In the case of the scNMT-seq dataset, the transcript count tables were downloaded from the repository provided in the publication. Only cells derived fom wild type embryos were included. The external data and our own data was integrated using the SCTransform (Hafemeister and Satija, 2019) and the anchor-based intregration (Stuart et al., 2019) functionalities from Seurat. First, all data was normalized per batch using the “SCTransform” command. Datasets were then integrated using the “SelectIntegrationFeatures,” “PrepSCTIntegration,” “FindIntegrationAnchors,” and “IntegrateData,” as per Seurat documentation. To assign scNMT-seq samples to the previously determined EB clusters, we used Seurat’s “TransferData” command.
SCENIC
We used SCENIC (Aibar et al., 2017) on the command line according to the documentation provided for the python-based scalable version of the tool (pySCENIC) (Van de Sande et al., 2020). Specifically, we ran “pyscenic grn” with the parameters “--method grnboost2”; “pyscenic ctx” with the parameters “--all_modules”; and “pyscenic aucell” with the default parameters. We used the transcription factor annotation and the transcription factor motifs (10 kb ± of the TSS) provided with SCENIC. This yielded 414 activating regulons. We subsequently filtered regulons based on the expression of the regulon as a whole (at least 50% of cells having an AUCell score > 0 within at least one Seurat cluster) and based on the expression of the regulon transcription factor (detected in at least 5% of cells in at least one cluster) to retain only high confidence regulons. This resulted in 285 remaining activating regulons. However, repeating all analyses with the unfiltered set of regulons yielded the same trends and relationships.
Linear Discriminant Analysis (LDA) classifier to assign samples to transcriptional clusters based on DamID signal
In both the EB results and the zebrafish results, we noticed that there was a substantial number of cells that passed DamID thresholds, but that had a poor CEL-Seq2 readout. Since most of our analyses rely on the separation of cells in transcriptional clusters (i.e., cell types) and cells with a poor CEL-Seq2 readout cannot be included in the clustering, these cells cannot be used in downstream DamID-based analyses. However, we noticed that the separation of different cell types was recapitulated to a considerable extent in low-dimensionality representations of the DamID readout (see the DamID-based UMAPs in Figures 3 and 5D). Since cell-type information is captured in the DamID readout, we reasoned that a classifier could be trained based on cells with both good DamID and CEL-Seq2 readouts to assign cells with a poor CEL-Seq2 readout to transcriptional clusters based on their DamID readout.
To this end, we implemented a Linear Discriminant Analysis (LDA) classifier as described below. In addition, the code is available online (https://github.com/KindLab/EpiDamID2022).
Data input and preprocessing
As in input for the classifier, we used the binned DamID data of all samples passing DamID thresholds and the transcriptional cluster labels of these samples (samples with a poor CEL-Seq2 readout had the label “unknown”). The DamID data was depth-normalized (as described above) and genomic bins that contained fewer than 1 mappable GATC motif per kb were excluded, resulting in a matrix of size N x M, where N is the number of samples and M is the number of remaining genomic bins. For the EB data, a bin size of 10 kb was used, while a bin size of 100 kb was used for the zebrafish data. Subsequently, the pairwise correlation was computed between all samples, resulting in a correlation matrix of size N x N. This transformation had two reasons: First, it served as a dimensionality reduction, since N < < M. Second, it resulted in a data type that effectively describes the similarity of a sample with all other samples, including samples without a cluster label. Consequently, during the training phase, the classifier can indirectly use the information of these unlabeled samples to learn about the overall data structure. We found that using the correlation matrix (N x N) as an input for the classifier yielded much better results than using the original matrix (N x M).
To train the LDA classifier, we used two thirds (∼66%) of all samples with cluster labels (i.e., with a good CEL-Seq2 readout). Since the number of cells per cluster varied extensively, we randomly selected two thirds of the samples per cluster and thereby ensured that all clusters were represented in both training and testing. The training data thus consisted of the correlation matrix of size Ntrain x N and a list of sample labels of size Ntrain, where Ntrain is the number of samples used for training. Consequently, we retained one third (∼33%) of labeled samples to test the performance of the LDA classifier, consisting of the correlation matrix of size Ntest x N and a list of sample labels of size Ntest, where Ntest is the number of samples used for testing. In summary, this split the samples into three groups: one group for training, one group for testing, and the group of unlabeled samples.
Training the classifier
For the implementation of the LDA classifier, we used the “LinearDiscriminantAnalysis” function provided in the Python (v. 3.8.10) scikit-learn toolkit (v. 0.24.2). The number of components was set to the number of transcriptional clusters minus one and the LDA classifier was trained using the training samples.
Testing the performance
To test the performance, the trained LDA classifier was used to predict the labels of the training set of samples. Predictions with a probability larger than 0.5 were maintained, while predictions with a lower probability were discarded (and the corresponding cells were thus not labeled). The predicted labels were subsequently compared to the known labels (Figures S3E and S5D). In general, we found a very good performance for clusters with many cells, while the performance tended to be lower for clusters with few cells. This is as expected, since the number of samples for these clusters was also very low during training.
Predicting cluster labels for unlabeled samples
After establishing that the performance was satisfactory, the LDA was retrained, this time using all labeled samples. The actual performance on the unlabeled data is likely higher than the performance on the test data, since the number of samples used for the final training is notably higher. Finally, the cluster labels were predicted for the unlabeled samples. Once again, only predictions with a probability higher than 0.5 were maintained. Table S2 contains all annotations of predicted cluster labels.
Defining PRC targets
First, we identified for each gene the region of 5 kb upstream and 3 kb downstream of the TSS. Only protein-coding genes and genes for non-coding RNA were considered. When the TSS domains of two genes overlapped, they were merged if the overlap was > 4 kb, otherwise the two domains were split in the middle of the overlap. This resulted in 30,356 domains covering a total of 35,814 genes. Subsequently, for all single-cells, the number of observed GATC counts within each domain was determined. In silico populations per transcriptional cluster were generated by combining the counts of all cells belonging to each cluster per DamID construct. The in silico population counts were subsequently RPKM-normalized, using the total number of GATC counts on the somatic chromosomes of the combined single-cell samples as the depth (i.e., also counts outside the domains). Normalization for Dam controls was performed for the H3K27me3 and RING1B data per transcriptional cluster by adding a pseudo count of 1, taking the fold-change with Dam, and performing a log2-transformation, resulting in log2 observed-over-expected (log2OE) values. The correlation of the resulting H3K27me3 and RING1B values per cluster is shown in Figure S3F. We subsequently determined PRC targets as those genes that showed H3K27me3 and RING1B log2OE values > 0.35 in at least one cluster. PRC targets were defined based on the in silico population of the H3K27me3 and RING1B data of the mESCs (Figure 2) and the EB clusters, excluding cluster 7. Cluster 7 was excluded, because it consisted of relatively few cells and the combined data was consequently sparse.
Comparing EpiDamID and scNMT-seq data at transcription start sites
We downloaded the tables of single-cell CpG methylation values at regions ± 2 kb of gene TSS from the repository provided in the scNMT-seq publication (Argelaguet et al., 2019). We subsequently averaged the CpG methylation scores across cells per cluster to gain an average CpG methylation for all genes per cluster. This could be done for four out of eight transcriptional clusters to which sufficient scNMT-seq samples were attributed (cluster 3: 31 cells; cluster 5: 21 cells; cluster 1: 37 cells; cluster 4: 43 cells). We subsequently could integrate the CpG methylation scores with our own H3K27me3 and RING1B DamID data for all genes, for which the enrichment scores were computed as described in the previous section. The subsequent analyses were performed on genes that were represented in both datasets.
ChromHMM of zebrafish in silico populations
In order to determine regions that were characterized by H3K9me3-enrichment in specific (sets of) cell types in the zebrafish embryo, we made use of ChromHMM (v. 1.22) (Ernst and Kellis, 2017, 2012). As input, we used the in silico H3K9me3 signal (log2OE) of all clusters that had at least 30 cells passing DamID thresholds for both Dam and MPHOSPH8 (clusters 0-11). The genome-wide signal at a resolution of 50 kb was used and the values were binarized based on a threshold of log2OE > 0.35. Bins that had fewer than 1 mappable GATC per kb were given a value of 2, indicating that the data was missing. As in all other analysis, chromosome 4 was excluded. The binarized values of clusters 0-11 were provided as input for the ChromHMM and the results were computed using the “LearnModel” function using the following parameters: -b 50000 -s 1 -pseudo. The number of ChromHMM states was varied from 2 to 10 and for each result the differences between the states (based on the emission probabilities) were inspected. We found that a ChromHMM model with 5 states was optimal, since this yielded the most diverse states and increasing the number of states just added redundant states with similar emission probabilities.
Repeat enrichment in ChromHMM states
The RepeatMasker repeat annotations for GRCz11 were downloaded from the UCSC Genome Browser website (https://genome.ucsc.edu/). The enrichment of repeats within each ChromHMM state was computed either for repeat classes as a whole (Figure S6A) or for individual types of repeats (Figures 5I and S6C). To compute the enrichment of a repeat class/type in a ChromHMM state, the fraction of repeats belonging to that class/type that fell within the state was computed and normalized for the fraction of the genome covered by that state. In other words, if we observe that 70% of a certain repeat falls within state B and state B covers 7% of the genome, then the repeat enrichment is 0.7 / 0.07 = 10.
GO term and PANTHER protein class enrichment analysis
GO term and PANTHER (Mi et al., 2013) protein class enrichment analyses were performed via de Gene Ontology Consortium website (http://geneontology.org/). For Figure S4E, the list of PRC-regulated TFs was used as a query and the list of all TFs as a reference to determine enriched Biological Process GO terms. Only the top 10 most significant terms are shown. For Figure S5I, the list of genes in ChromHMM state A1 or B was used as a query and the list of genes in all ChromHMM states as a reference to determine enriched PANTHER protein classes. All hits are shown.
Quantification and statistical analysis
The number of n samples included in analyses is provided within each figure and/or accompanying figure legend. Statistical p values are associated with the significance test as described in the figure legends. The boxes of boxplots indicate the quartiles of the dataset, the middle shows the median, and the error bars indicate the data range falling within 1.5 times the inter-quartile range.
Acknowledgments
We would like to thank the members of the Kind laboratory for their helpful comments and suggestions. In particular, we thank Koos Rooijers for providing input and support on the computational work. This work was funded by an ERC Starting grant (ERC-StG 678423-EpiID) to J.K. The Oncode Institute is partially funded by the KWF Dutch Cancer Society. I.G. is supported by an EMBO Long-Term Fellowship ALTF1214-2016, Swiss National Science Fund grant P400PB_186758 and NWO-ENW Veni grant VI.Veni.202.073. P.D.N. is supported by an EMBO Long-Term Fellowship ALTF1129-2015, HFSPO Fellowship (LT001404/2017-L) and an NWO-ZonMW Veni grant (016.186.017-3). The laboratory of J.B. is supported by the Netherlands Cardiovascular Research Initiative, an initiative with support of the Dutch Heart Foundation and Hartekind, CVON2019-002 OUTREACH. The laboratory of H.K. is supported by MEXT/JSPS KAKENHI (JP18H05527, JP20K06484, and JP21H04764) and Japan Science and Technology Agency (JPMJCR16G1 and JPMJCR20S6). We additionally thank the Hubrecht Sorting Facility, the Hubrecht Imaging Center, and the Utrecht Sequencing Facility (USEQ), subsidized by the University Medical Center Utrecht.
Author contributions
Conceptualization: F.J.R., K.L.d.L., S.S.d.V., and J.K. Data curation & Validation: F.J.R., K.L.d.L., and S.S.d.V. Formal analysis & Software: F.J.R. Funding acquisition & Project administration: J.K. Investigation & Methodology: K.L.d.L. and S.S.d.V. designed and performed all experiments unless noted otherwise. C.V.Q. and E.B. designed and generated knock-in mouse ESC lines. P.D.N. performed all zebrafish experiments, with assistance from I.G. and S.S.d.V. Resources: Y.S. and H.K. Supervision: J.B. and J.K. Visualization: F.J.R. and K.L.d.L. Writing – original draft: F.J.R., K.L.d.L., and J.K. Writing – review & editing: all authors.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Published: April 1, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2022.03.009.
Supplemental information
Data and code availability
-
•
All sequencing data generated in this manuscript are deposited on the NCBI Gene Expression Omnibus (GEO) portal and are publicly available as of the data of publication under accession number GEO: GSE184036 (see Key resource table for further details). Imaging data are publicly available on Mendeley Data: https://doi.org/10.17632/sp7hsw68c4.1.
-
•
Key scripts are available at Zenodo: https://doi.org/10.5281/zenodo.6308373.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
References
- Ahmed K., Dehghani H., Rugg-Gunn P., Fussner E., Rossant J., Bazett-Jones D.P. Global chromatin architecture reflects pluripotency and lineage commitment in the early mouse embryo. PLoS ONE. 2010;5:e10531. doi: 10.1371/journal.pone.0010531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai S., Xiong H., Li C.C., Luo Y., Shi Q., Liu Y., Yu X., Li C., He A. Profiling chromatin states using single-cell itChIP-seq. Nat. Cell Biol. 2019;21:1164–1172. doi: 10.1038/s41556-019-0383-5. [DOI] [PubMed] [Google Scholar]
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleström P., D’Angelo L., Midtlyng P.J., Schorderet D.F., Schulte-Merker S., Sohm F., Warner S. Vol. 54. 2019. Zebrafish: Housing and husbandry recommendations; pp. 213–224.https://doi-org.proxy.library.uu.nl/10.1177/0023677219869037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altemose N., Maslan A., Rios-Martinez C., Lai A., White J.A., Streets A. μDamID: A Microfluidic Approach for Joint Imaging and Sequencing of Protein-DNA Interactions in Single Cells. Cell Syst. 2020;11:354–366.e9. doi: 10.1016/j.cels.2020.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola M., Venneri M.A., Biffi A., Vigna E., Naldini L. Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat Biotechnol. 2005;23:108–116. doi: 10.1038/nbt1049. [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argelaguet R., Clark S.J., Mohammed H., Stapel L.C., Krueger C., Kapourani C.A., Imaz-Rosshandler I., Lohoff T., Xiang Y., Hanna C.W., et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature. 2019;576:487–491. doi: 10.1038/s41586-019-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird R.E., Hardman K.D., Jacobson J.W., Johnson S., Kaufman B.M., Lee S.M., Lee T., Pope S.H., Riordan G.S., Whitlow M. Single-chain antigen-binding proteins. Science. 1988;242:423–426. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- Blackledge N.P., Klose R.J. The molecular principles of gene regulation by Polycomb repressive complexes. Nat. Rev. Mol. Cell Biol. 2021;22:815–833. doi: 10.1038/s41580-021-00398-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsos M., Perricone S.M., Schauer T., Pontabry J., de Luca K.L., de Vries S.S., Ruiz-Morales E.R., Torres-Padilla M.E., Kind J. Genome-lamina interactions are established de novo in the early mouse embryo. Nature. 2019;569:729–733. doi: 10.1038/s41586-019-1233-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer L.A., Plath K., Zeitlinger J., Brambrink T., Medeiros L.A., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., Bell G.W., Otte A.P., Vidal M., Gifford D.K., Young R.A., Jaenisch R. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Brinkman A.B., Gu H., Bartels S.J.J., Zhang Y., Matarese F., Simmer F., Marks H., Bock C., Gnirke A., Meissner A., Stunnenberg H.G. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012;22:1128–1138. doi: 10.1101/gr.133728.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulut-Karslioglu A., De La Rosa-Velázquez I.A., Ramirez F., Barenboim M., Onishi-Seebacher M., Arand J., Galán C., Winter G.E., Engist B., Gerle B., et al. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell. 2014;55:277–290. doi: 10.1016/j.molcel.2014.05.029. [DOI] [PubMed] [Google Scholar]
- Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Carter B., Ku W.L., Kang J.Y., Hu G., Perrie J., Tang Q., Zhao K. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq) Nat. Commun. 2019;10:3747. doi: 10.1038/s41467-019-11559-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T., Kirschner K., Thuret J.-Y., Pope B.D., Ryba T., Newman S., Ahmed K., Samarajiwa S.A., Salama R., Carroll T., et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell. 2012;47:203–214. doi: 10.1016/j.molcel.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham S.W., Gruhn W.H., van den Ameele J., Krautz R., Southall T.D., Kobayashi T., Surani M.A., Brand A.H. Targeted DamID reveals differential binding of mammalian pluripotency factors. Development. 2018;145 doi: 10.1242/dev.170209. dev170209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham S.W., Jafrani Y.M.A., Andersen S.B., Jansz N., Ewing A.D., Faulkner G.J. Single-molecule simultaneous profiling of DNA methylation and DNA-protein interactions with Nanopore-DamID. Preprint at bioRxiv. 2021 doi: 10.1101/2021.08.09.455753. [DOI] [Google Scholar]
- Chen X., Zaro J.L., Shen W.C. Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 2013;65:1357–1369. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collas P. A chromatin immunoprecipitation protocol for small cell numbers. Methods Mol. Biol. 2011;791:179–193. doi: 10.1007/978-1-61779-316-5_14. [DOI] [PubMed] [Google Scholar]
- Corallo D., Trapani V., Bonaldo P. The notochord: structure and functions. Cellular and Molecular Life Sciences. 2015;72:2989–3008. doi: 10.1007/s00018-015-1897-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermin B., Melfi R., McCabe D., Seitz V., Imhof A., Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K., et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46(D1):D794–D801. doi: 10.1093/NAR/GKX1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Luca K.L., Kind J. In: Methods in Molecular Biology. Bodega Beatrice, Lanzuolo Chiara., editors. Humana; New York, NY: 2021. Single-cell damid to capture contacts between dna and the nuclear lamina in individual mammalian cells; pp. 159–172. [DOI] [PubMed] [Google Scholar]
- de Napoles M., Mermoud J.E., Wakao R., Tang Y.A., Endoh M., Appanah R., Nesterova T.B., Silva J., Otte A.P., Vidal M., et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell. 2004;7:663–676. doi: 10.1016/j.devcel.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Donnaloja F., Carnevali F., Jacchetti E., Raimondi M.T. Lamin A/C Mechanotransduction in Laminopathies. Cells. 2020;9:1306. doi: 10.3390/cells9051306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017;12:2478–2492. doi: 10.1038/nprot.2017.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion G.J., van Bemmel J.G., Braunschweig U., Talhout W., Kind J., Ward L.D., Brugman W., de Castro I.J., Kerkhoven R.M., Bussemaker H.J., van Steensel B. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143:212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorkin D.U., Barozzi I., Zhao Y., Zhang Y., Huang H., Lee A.Y., Li B., Chiou J., Wildberg A., Ding B., et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature. 2020;583:744–751. doi: 10.1038/s41586-020-2093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosselin K., Durand A., Marsolier J., Poitou A., Marangoni E., Nemati F., Dahmani A., Lameiras S., Reyal F., Frenoy O., et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019;51:1060–1066. doi: 10.1038/s41588-019-0424-9. [DOI] [PubMed] [Google Scholar]
- Gruenbaum Y., Foisner R. Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Annu. Rev. Biochem. 2015;84:131–164. doi: 10.1146/annurev-biochem-060614-0341. [DOI] [PubMed] [Google Scholar]
- Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagarman J.A., Motley M.P., Kristjansdottir K., Soloway P.D. Coordinate regulation of DNA methylation and H3K27me3 in mouse embryonic stem cells. PLoS ONE. 2013;8:e53880. doi: 10.1371/JOURNAL.PONE.0053880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M.A., Wu X., Li A.X., Hahn T., Pfeifer G.P. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS ONE. 2011;6:e18844. doi: 10.1371/JOURNAL.PONE.0018844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainer S.J., Bošković A., McCannell K.N., Rando O.J., Fazzio T.G. Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell. 2019;177:1319–1329.e11. doi: 10.1016/j.cell.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A., Maehara K., Handa T., Arimura Y., Nogami J., Hayashi-Takanaka Y., Shirahige K., Kurumizaka H., Kimura H., Ohkawa Y. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat. Cell Biol. 2019;21:287–296. doi: 10.1038/s41556-018-0248-3. [DOI] [PubMed] [Google Scholar]
- Harmanci A., Rozowsky J., Gerstein M. MUSIC: identification of enriched regions in ChIP-Seq experiments using a mappability-corrected multiscale signal processing framework. Genome Biol. 2014;15:474. doi: 10.1186/s13059-014-0474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T., Senderovich N., Avital G., Klochendler A., de Leeuw Y., Anavy L., Gennert D., Li S., Livak K.J., Rozenblatt-Rosen O., et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016;17:77. doi: 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T., Lipp J.J., Toh B.-H., Peters J.-M. Histone H3 serinec10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- Hou W., Ji Z., Ji H., Hicks S.C. A systematic evaluation of single-cell RNA-sequencing imputation methods. Genome Biol. 2020;21:218. doi: 10.1186/S13059-020-02132-X/FIGURES/6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan A.H., Wang S., Ko K.D., Zare H., Tsai P.F., Feng X., Vivanco K.O., Ascoli A.M., Gutierrez-Cruz G., Krebs J., et al. Roles of H3K27me2 and H3K27me3 Examined during Fate Specification of Embryonic Stem Cells. Cell Rep. 2016;17:1369–1382. doi: 10.1016/J.CELREP.2016.09.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur H.S., Wu S.J., Codomo C.A., Pledger E.S., Bryson T.D., Henikoff J.G., Ahmad K., Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019;10:1930. doi: 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur H.S., Janssens D.H., Henikoff J.G., Ahmad K., Henikoff S. Efficient low-cost chromatin profiling with CUT&Tag. Nature Protocols. 2020;15:3264–3283. doi: 10.1038/s41596-020-0373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology. 2013;14:1–13. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J., Pagie L., Ortabozkoyun H., Boyle S., de Vries S.S., Janssen H., Amendola M., Nolen L.D., Bickmore W.A., van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013;153:178–192. doi: 10.1016/J.CELL.2013.02.028. [DOI] [PubMed] [Google Scholar]
- Kind J., Pagie L., de Vries S.S., Nahidiazar L., Dey S.S., Bienko M., Zhan Y., Lajoie B., de Graaf C.A., Amendola M., et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell. 2015;163:134–147. doi: 10.1016/j.cell.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku W.L., Nakamura K., Gao W., Cui K., Hu G., Tang Q., Ni B., Zhao K. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat. Methods. 2019;16:323–325. doi: 10.1038/s41592-019-0361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T., Nishimura K., Kanemaki M.T., Donaldson A.D. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell. 2013;50:273–280. doi: 10.1016/J.MOLCEL.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Kungulovski G., Kycia I., Tamas R., Jurkowska R.Z., Kudithipudi S., Henry C., Reinhardt R., Labhart P., Jeltsch A. Application of histone modification-specific interaction domains as an alternative to antibodies. Genome Res. 2014;24:1842–1853. doi: 10.1101/gr.170985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kungulovski G., Mauser R., Reinhardt R., Jeltsch A. Application of recombinant TAF3 PHD domain instead of anti-H3K4me3 antibody. Epigenetics Chromatin. 2016;9:11. doi: 10.1186/s13072-016-0061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/GAD.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue K., Rajshekar S., Courtney A.J., Lewis Z.A., Goll M.G. The maternal to zygotic transition regulates genome-wide heterochromatin establishment in the zebrafish embryo. Nat. Commun. 2019;10:1551. doi: 10.1038/s41467-019-09582-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson N.D., Li R., Shin M., Grosse A., Yukselen O., Stone O.A., Kucukural A., Zhu L. An improved zebrafish transcriptome annotation for sensitive and comprehensive detection of cell type-specific genes. eLife. 2020;9:1–76. doi: 10.7554/ELIFE.55792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Witten D.M., Johnstone I.M., Tibshirani R. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostatistics. 2012;13:523–538. doi: 10.1093/biostatistics/kxr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zheng H., Wang Q., Zhou C., Wei L., Liu X., Zhang W., Zhang Y., Du Z., Wang X., Xie W. Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol. 2018;19:18. doi: 10.1186/S13059-018-1390-8/TABLES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Tanasa B., Tyurina O.V., Zhou T.Y., Gassmann R., Liu W.T., Ohgi K.A., Benner C., Garcia-Bassets I., Aggarwal A.K., et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Brind’Amour J., Karimi M.M., Shirane K., Bogutz A., Lefebvre L., Sasaki H., Shinkai Y., Lorincz M.C. Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev. 2014;28:2041–2055. doi: 10.1101/gad.244848.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markodimitraki C.M., Rang F.J., Rooijers K., de Vries S.S., Chialastri A., de Luca K.L., Lochs S.J.A., Mooijman D., Dey S.S., Kind J. Simultaneous quantification of protein-DNA interactions and transcriptomes in single cells with scDam&T-seq. Nat. Protoc. 2020;15:1922–1953. doi: 10.1038/s41596-020-0314-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuleman W., Peric-Hupkes D., Kind J., Beaudry J.B., Pagie L., Kellis M., Reinders M., Wessels L., van Steensel B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013;23:270–280. doi: 10.1101/GR.141028.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Casagrande J.T., Thomas P.D. Large-scale gene function analysis with the PANTHER classification system. Nature Protocols. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosch K., Franz H., Soeroes S., Singh P.B., Fischle W. HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats. PLoS ONE. 2011;6:e15894. doi: 10.1371/JOURNAL.PONE.0015894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J., Hart C.M., Francis N.J., Vargas M.L., Sengupta A., Wild B., Miller E.L., O’Connor M.B., Kingston R.E., Simon J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/S0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- Mutlu B., Chen H.M., Moresco J.J., Orelo B.D., Yang B., Gaspar J.M., Keppler-Ross S., Yates J.R., 3rd, Hall D.H., Maine E.M., Mango S.E. Regulated nuclear accumulation of a histone methyltransferase times the onset of heterochromatin formation in C. elegans embryos. Sci. Adv. 2018;4:6224–6246. doi: 10.1126/SCIADV.AAT6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicetto D., Donahue G., Jain T., Peng T., Sidoli S., Sheng L., Montavon T., Becker J.S., Grindheim J.M., Blahnik K., et al. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science. 2019;363:294–297. doi: 10.1126/SCIENCE.AAU0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K., Fukagawa T., Takisawa H., Kakimoto T., Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nature Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Nora E.P., Goloborodko A., Valton A.L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell. 2017;169:930–944.e22. doi: 10.1016/J.CELL.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal M., Kind J., Torres-Padilla M.-E. DamID to Map Genome-Protein Interactions in Preimplantation Mouse Embryos. Methods Mol. Biol. 2021;2214:265–282. doi: 10.1007/978-1-0716-0958-3_18. [DOI] [PubMed] [Google Scholar]
- Park M., Patel N., Keung A.J., Khalil A.S. Engineering Epigenetic Regulation Using Synthetic Read-Write Modules. Cell. 2019;176:227–238.e20. doi: 10.1016/j.cell.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly A.R., Copur Ö., Jäckle H., Herzig A., Müller J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science. 2013;339:698–699. doi: 10.1126/SCIENCE.1231382. [DOI] [PubMed] [Google Scholar]
- Peric-Hupkes D., Meuleman W., Pagie L., Bruggeman S.W.M., Solovei I., Brugman W., Gräf S., Flicek P., Kerkhoven R.M., van Lohuizen M., et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijuan-Sala B., Griffiths J.A., Guibentif C., Hiscock T.W., Jawaid W., Calero-Nieto F.J., Mulas C., Ibarra-Soria X., Tyser R.C.V., Ho D.L.L., et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature. 2019;566:490–495. doi: 10.1038/s41586-019-0933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A., Shilatifard A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nature Reviews Molecular Cell Biology. 2021;22:326–345. doi: 10.1038/s41580-021-00341-1. [DOI] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44(W1):W160–W165. doi: 10.1093/NAR/GKW257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riising E.M., Comet I., Leblanc B., Wu X., Johansen J.V., Helin K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell. 2014;55:347–360. doi: 10.1016/J.MOLCEL.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/JBC.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rooijers K., Markodimitraki C.M., Rang F.J., de Vries S.S., Chialastri A., de Luca K.L., Mooijman D., Dey S.S., Kind J. Simultaneous quantification of protein-DNA contacts and transcriptomes in single cells. Nat. Biotechnol. 2019;37:766–772. doi: 10.1038/s41587-019-0150-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem A., Ram O., Shoresh N., Sperling R.A., Goren A., Weitz D.A., Bernstein B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature Biotechnology. 2015;33:1165–1172. doi: 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph T., Yonezawa M., Lein S., Heidrich K., Kubicek S., Schäfer C., Phalke S., Walther M., Schmidt A., Jenuwein T., Reuter G. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3-3. Mol. Cell. 2007;26:103–115. doi: 10.1016/J.MOLCEL.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Sanders S.L., Portoso M., Mata J., Bähler J., Allshire R.C., Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/J.CELL.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Santos F., Peters A.H., Otte A.P., Reik W., Dean W. Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev. Biol. 2005;280:225–236. doi: 10.1016/J.YDBIO.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Sato Y., Mukai M., Ueda J., Muraki M., Stasevich T.J., Horikoshi N., Kujirai T., Kita H., Kimura T., Hira S., et al. Genetically encoded system to track histone modification in vivo. Sci. Rep. 2013;3:2436. doi: 10.1038/srep02436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y., Kujirai T., Arai R., Asakawa H., Ohtsuki C., Horikoshi N., Yamagata K., Ueda J., Nagase T., Haraguchi T., et al. A Genetically Encoded Probe for Live-Cell Imaging of H4K20 Monomethylation. J. Mol. Biol. 2016;428:3885–3902. doi: 10.1016/j.jmb.2016.08.010. [DOI] [PubMed] [Google Scholar]
- Sato Y., Nakao M., Kimura H. Live-cell imaging probes to track chromatin modification dynamics. Microscopy (Oxf.) 2021;70:415–422. doi: 10.1093/JMICRO/DFAB030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M., Durussel T., Laemmli U.K. ChIC and ChEC: genomic mapping of chromatin proteins. Mol. Cell. 2004;16:147–157. doi: 10.1016/j.molcel.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Shoaib M., Chen Q., Shi X., Nair N., Prasanna C., Yang R., Walter D., Frederiksen K.S., Einarsson H., Svensson J.P., et al. Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes. Nat. Commun. 2021;12:4800. doi: 10.1038/s41467-021-25051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene P.J., Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. elife. 2017;6 doi: 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon M.J., Varshavsky A. Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc. Natl. Acad. Sci. USA. 1985;82:6470–6474. doi: 10.1073/pnas.82.19.6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southall T.D., Gold K.S., Egger B., Davidson C.M., Caygill E.E., Marshall O.J., Brand A.H. Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Dev. Cell. 2013;26:101–112. doi: 10.1016/j.devcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., Tang Y., Zhang Y., Fang Y., Jia J., Zeng W., Fang D. Joint single-cell multiomic analysis in Wnt3a induced asymmetric stem cell division. Nat. Commun. 2021;12:5941. doi: 10.1038/s41467-021-26203-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesnik T., Ho J.W.K., Sherwood R. Dam mutants provide improved sensitivity and spatial resolution for profiling transcription factor binding. Epigenetics Chromatin. 2019;12:36. doi: 10.1186/s13072-019-0273-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalsma S.J.D., Hori M., Sato Y., Bousard A., Ohi A., Raposo A.C., Roensch J., Le Saux A., Nogami J., Maehara K., et al. H4K20me1 and H3K27me3 are concurrently loaded onto the inactive X chromosome but dispensable for inducing gene silencing. EMBO Rep. 2021;22:e51989. doi: 10.15252/embr.202051989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosti L., Ashmore J., Tan B.S.N., Carbone B., Mistri T.K., Wilson V., Tomlinson S.R., Kaji K. Mapping transcription factor occupancy using minimal numbers of cells in vitro and in vivo. Genome Res. 2018;28:592–605. doi: 10.1101/gr.227124.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Sande B., Flerin C., Davie K., De Waegeneer M., Hulselmans G., Aibar S., Seurinck R., Saelens W., Cannoodt R., Rouchon Q., et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020;15:2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- van Schaik T., Vos M., Peric-Hupkes D., Hn Celie P., van Steensel B. Cell cycle dynamics of lamina-associated DNA. EMBO Rep. 2020;21:e50636. doi: 10.15252/EMBR.202050636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B., Henikoff S. Identification of in vivo DNA targets of chromatin proteins using tethered Dam methyltransferase. Nature Biotechnology. 2000;18:424–428. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- Vermeulen M., Mulder K.W., Denissov S., Pijnappel W.W.M.P., van Schaik F.M.A., Varier R.A., Baltissen M.P.A., Stunnenberg H.G., Mann M., Timmers H.T.M. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Villaseñor R., Pfaendler R., Ambrosi C., Butz S., Giuliani S., Bryan E., Sheahan T.W., Gable A.L., Schmolka N., Manzo M., et al. ChromID identifies the protein interactome at chromatin marks. Nat. Biotechnol. 2020;38:728–736. doi: 10.1038/s41587-020-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel M.J., Guelen L., de Wit E., Peric-Hupkes D., Lodén M., Talhout W., Feenstra M., Abbas B., Classen A.K., van Steensel B. Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res. 2006;16:1493–1504. doi: 10.1101/GR.5391806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel M.J., Peric-Hupkes D., van Steensel B. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat. Protoc. 2007;2:1467–1478. doi: 10.1038/nprot.2007.148. [DOI] [PubMed] [Google Scholar]
- Waldo G.S., Standish B.M., Berendzen J., Terwilliger T.C. Rapid protein-folding assay using green fluorescent protein. Nature Biotechnology. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- Wang H., Wang L., Erdjument-Bromage H., Vidal M., Tempst P., Jones R.S., Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- Wang C., Liu X., Gao Y., Yang L., Li C., Liu W., Chen C., Kou X., Zhao Y., Chen J., et al. Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat. Cell Biol. 2018;20:620–631. doi: 10.1038/s41556-018-0093-4. [DOI] [PubMed] [Google Scholar]
- Wang Q., Xiong H., Ai S., Yu X., Liu Y., Zhang J., He A. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol. Cell. 2019;76:206–216.e7. doi: 10.1016/j.molcel.2019.07.015. [DOI] [PubMed] [Google Scholar]
- Westerfield M. Fourth Edition. University of Oregon Press; Eugene: 2000. The Zebrafish Book : A Guide for the Laboratory Use of Zebrafish. [Google Scholar]
- Wong X., Cutler J.A., Hoskins V.E., Gordon M., Madugundu A.K., Pandey A., Reddy K.L. Mapping the micro-proteome of the nuclear lamina and lamina-associated domains. Life Sci Alliance. 2021;4 doi: 10.26508/lsa.202000774. e202000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H., Luo Y., Wang Q., Yu X., He A. Single-cell joint detection of chromatin occupancy and transcriptome enables higher-dimensional epigenomic reconstructions. Nat. Methods. 2021;18:652–660. doi: 10.1038/s41592-021-01129-z. [DOI] [PubMed] [Google Scholar]
- Zeller P., Yeung J., de Barbanson B.A., Gaza H.V., Florescu M., van Oudenaarden A. Hierarchical chromatin regulation during blood formation uncovered by single-cell sortChIC. Preprint at bioRxiv. 2021 doi: 10.1101/2021.04.26.440606. [DOI] [Google Scholar]
- Zeng H., Horie K., Madisen L., Pavlova M.N., Gragerova G., Rohde A.D., Schimpf B.A., Liang Y., Ojala E., Kramer F., et al. An inducible and reversible mouse genetic rescue system. PLoS Genet. 2008;4:e1000069. doi: 10.1371/JOURNAL.PGEN.1000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/GB-2008-9-9-R137/FIGURES/3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Wang M., Zhang Y. Tn5 transposase-based epigenomic profiling methods are prone to open chromatin bias. Preprint at bioRxiv. 2021 doi: 10.1101/2021.07.09.451758. [DOI] [Google Scholar]
- Zhu C., Zhang Y., Li Y.E., Lucero J., Behrens M.M., Ren B. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods. 2021;18:283–292. doi: 10.1038/s41592-021-01060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All sequencing data generated in this manuscript are deposited on the NCBI Gene Expression Omnibus (GEO) portal and are publicly available as of the data of publication under accession number GEO: GSE184036 (see Key resource table for further details). Imaging data are publicly available on Mendeley Data: https://doi.org/10.17632/sp7hsw68c4.1.
-
•
Key scripts are available at Zenodo: https://doi.org/10.5281/zenodo.6308373.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.