

Abstract
Today the importance of autophagy in physiological processes and pathological conditions is undeniable. Initially autophagy merely was described as an evolutionarily conserved mechanism to maintain metabolic homeostasis in times of starvation, however in recent years it is now apparent that autophagy is a powerful regulator of many facets of cellular metabolism, that its deregulation contributes to various human pathologies, including cancer and neurodegeneration, and its modulation has considerable potential as a therapeutic approach. Different lipid species, including sphingolipids, sterols, and phospholipids play important roles in the various steps of autophagy. In particular, there is accumulating evidence indicating the minor group of phospholipids called the phosphoinositides as key modulators of autophagy, including the signaling processes underlying autophagy initiation, autophagosome biogenesis and maturation. In this review we discuss the known functions to date of the phosphoinositides in autophagy and attempt to summarize the kinases and phosphatases that regulate them as well as the proteins that bind to them throughout the autophagy program. We will also provide examples of how the control of phosphoinositides and their metabolizing enzymes is relevant to understanding many human diseases.
Keywords: phosphoinositide, phosphoinositide kinase, phosphoinositide phosphatase, autophagy, lysosome, mTORC1
Introduction
Phosphoinositides are a small group of cellular phospholipids that are derived by the phosphorylation of the third, fourth and fifth positions of the inositol headgroup of phosphatidylinositol (PI), resulting in the generation of seven distinct phosphoinositide derivatives (Figure 1). In eukaryotic cells, these seven phosphoinositide species are interconverted into each other through the activity of phosphoinositide kinases and phosphatases (Figure 1). Although phosphoinositides only represent a minor percentage of phospholipids they are integral in the lion’s share of subcellular processes including membrane dynamics, trafficking and cellular signaling [1]. To this end, recently many reports have implicated the importance of phosphoinositides in the various steps of autophagy, including the signaling processes underlying autophagy initiation, autophagosome biogenesis and maturation [2].
Figure 1. Phosphoinositide cycle.
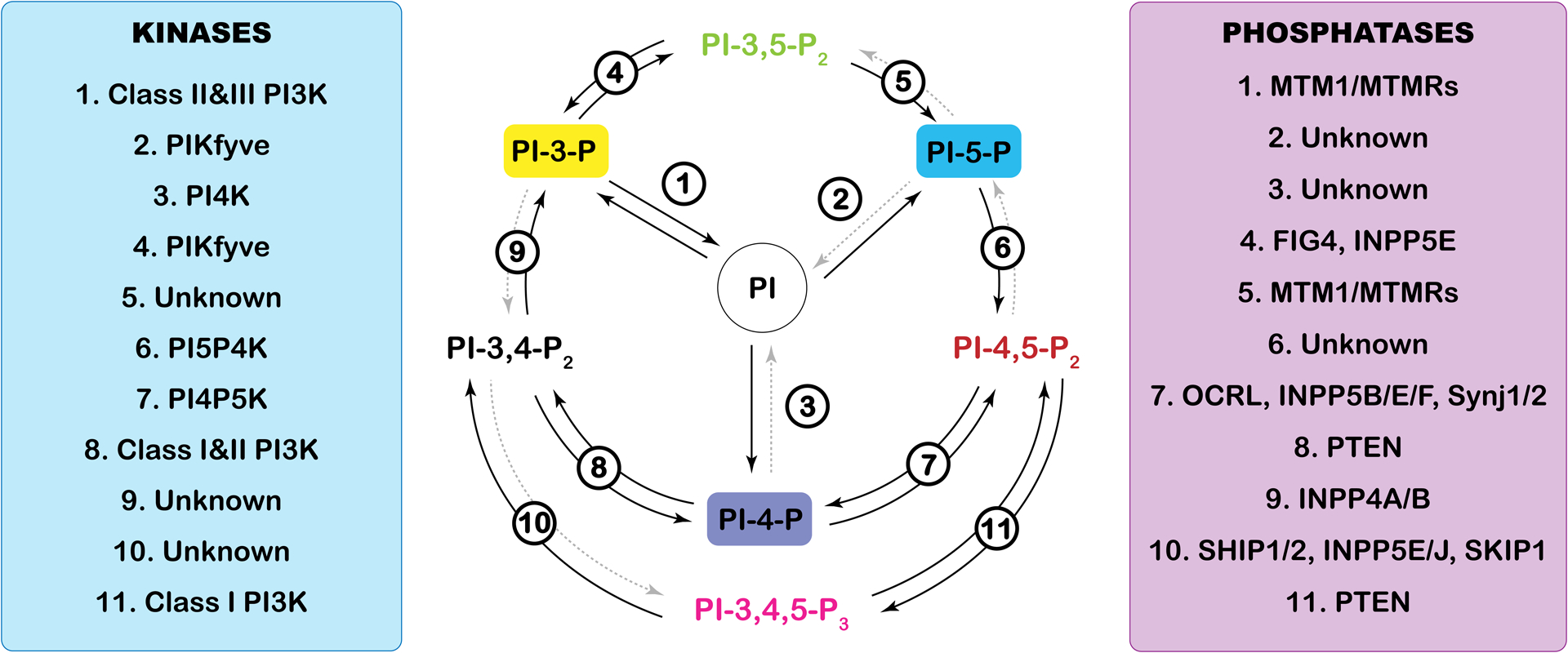
Phosphoinositides are a small group of phospholipids generated by the phosphorylation of the third, fourth and fifth positions of the inositol headgroup of phosphatidylinositol. The seven resulting phosphoinositides are cycled and regulated by a complex network of kinases and phosphatases that add or remove phosphate groups at specific positions. The precision of these enzymes is what keeps the phosphoinositide pools within a cell in perfect balance. The kinases (left) and phosphatases (right) are numbered, and the indicated number corresponds to the enzymes that allow the cycling between each of the phosphoinositide species starting with phosphatidylinositol (PI) at the center. Dashed grey arrows indicate enzymes that have not yet been characterized in vivo (Unknown).
Autophagy Overview
Proper balance between synthesis and degradation is paramount for cellular homeostasis. Autophagy (“self-eating”) is a fundamental degradative pathway for cytosolic components, such as proteins, RNA, DNA, lipids, and organelles. Although, autophagy is activated at the basal level, it may be induced by various cellular stresses, including nutrient deprivation, growth factor depletion, infection and hypoxia, thereby promoting cell survival. There are three main types of autophagy, macroautophagy [3, 4], microautophagy [5] and chaperone mediated autophagy [6] that have been identified in mammalian cells. To date, macroautophagy (hereafter autophagy) is the best characterized pathway and can further be divided into selective and non-selective (bulk) autophagy. Selective autophagy is the autophagy of aggregated proteins, damaged or over-abundant organelles. Such examples are, mitophagy [7], lipophagy [8], pexophagy [9], and ribophagy [10].
Autophagy is a multi-step process, including induction, isolation membrane formation, autophagosome formation/maturation, and cargo degradation in the autolysosome (Figure 2). Once autophagy is induced, the isolation membrane or phagophore emerges from different sources within the membrane, such as the endoplasmic reticulum (ER), mitochondria, the Golgi apparatus, or the plasma membrane (PM) in the cytosol. The phagophore elongates and expands to form the autophagosome, a double-membrane bound vesicle structure, which engulfs cytosolic components. After the closure of the autophagosome membrane, its outer membrane fuses with the lysosome to form the autolysosome for cargo degradation.
Figure 2. Phosphoinositides in autophagy.
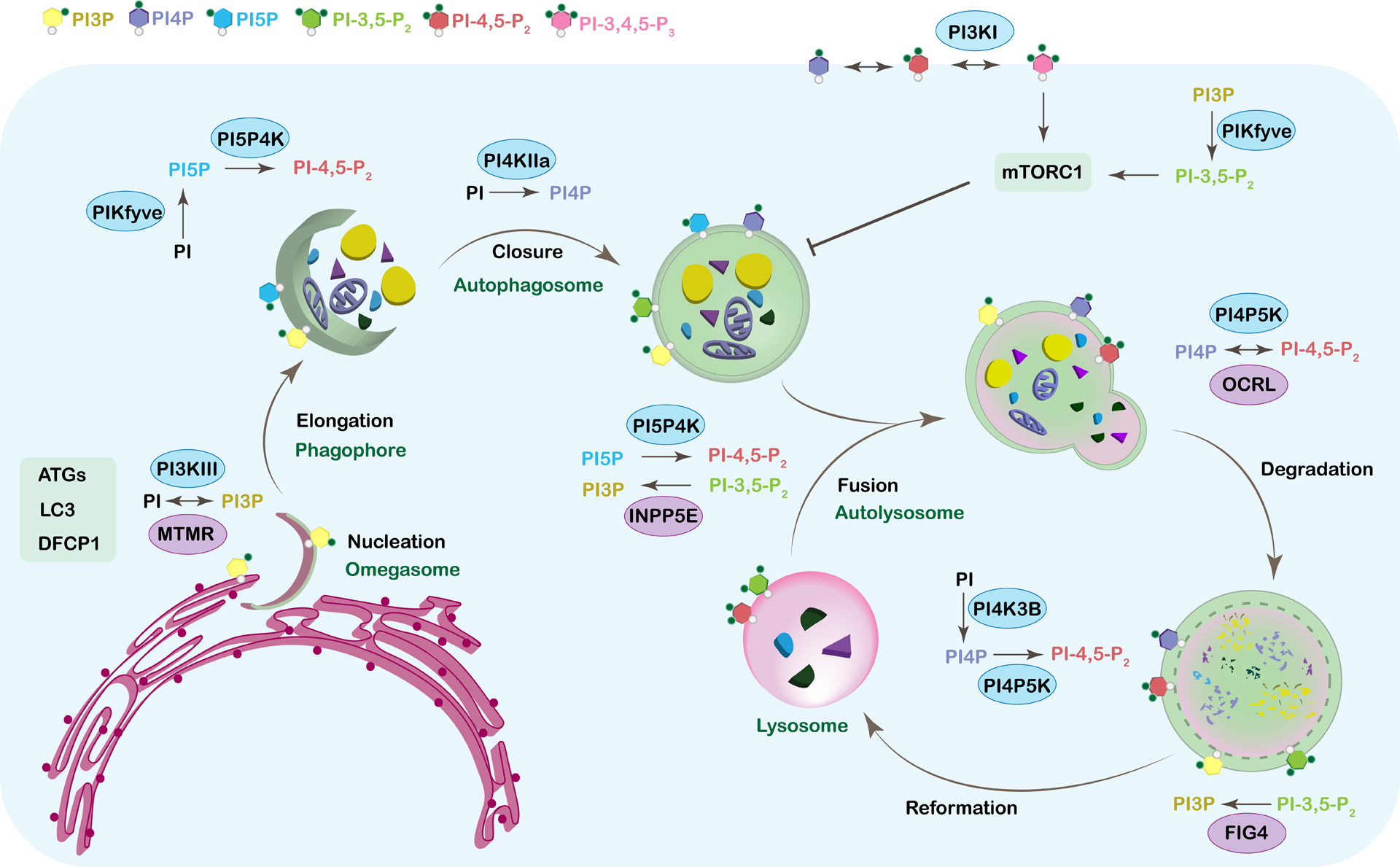
Autophagy is a highly regulated multistep process that is essential for recycling cytosolic components. Too much or too little autophagy leads to severe human diseases from neurodegeneration to cancer. Phosphoinositides play key roles in every step of this process from nucleation and the biogenesis of the phagophore to the reformation of the lysosome. Distinct pools of phospholipids characterize each membrane of the autophagic organelles. This phospholipid identity is ensured by specific kinases and phosphatases and their binding partners. The figure shows all the individual steps of autophagy starting with the nucleation and ending with the reformation of the lysosome. The corresponding phosphoinositide species that are known to play a role in regulating each step are shown on the membrane of the structure they are associated with and are color-coded. The kinases (blue oval) and phosphatases (purple oval) that are associated with regulating the phosphoinositide species at each step are also included. For simplicity, this model does not show all known binding partners, but these are discussed within the text.
Phosphoinositides are involved in virtually every step in the autophagy process, whether it be the lipid itself, the phosphoinositide-binding proteins or the phosphoinositide enzymes that generate them, thereby underscoring the importance of their balance for functional autophagy (Figure 2 and Table 1). Overall, this review will focus on how phosphoinositides impact autophagy, from the induction to cargo degradation via the autolysosome. Further, how these tiny yet significant lipids’ equilibrium is essential for the autophagic process and when altered or imbalanced contribute to pathogenesis of human diseases, such as cancer and neurodegeneration, will be discussed.
Table 1. Phosphoinositide roles and localization during autophagy.
An organized list of each phosphoinositide known to be involved in autophagy as illustrated in Figure 2.
| Phosphoinositide | Autophagy step | Localization |
|---|---|---|
| PI-3-P |
|
|
| PI-4-P |
|
|
| PI-5-P |
|
|
| PI-3,5-P2 |
|
|
| PI-4,5-P2 |
|
|
| PI-3,4,5-P3 |
|
|
Roles of Phosphoinositides in Autophagy:
Autophagy induction/initiation
In mammalian cells, signal to suppress or initiate autophagy relies on the activation or inactivation of the mTORC1 (mammalian target of rapamycin complex 1) pathway, which occurs based on the availability of nutrients [11]. For the purpose of this review, we will focus only on the phosphoinositide players that are known to directly regulate the autophagy pathway. Presence of abundant nutrients or growth factor mediated signaling, class I phosphatidylinositol 3-kinase (PI3KI) is recruited to the PM where it binds to and phosphorylates phosphatidylinositol 4,5-bisphosphate (PI-4,5-P2) at the 3’ position leading to the formation of phosphatidylinositol 3,4,5-triphosphate (PI-3,4,5-P3 or PIP3) (Figure 2). PIP3 recruits PH-domain containing proteins such as phosphoinositide-dependent kinase-1 (PDK1) and protein kinase B (PKB, also known as Akt), where phosphorylation of Akt by PDK1 leads to its activation [12]. This in turn activates a signaling cascade where mTORC1 gets activated, leading to phosphorylation of ULK1 and destabilization of the ULK-mAtg13-FIP200-Atg101 complex (induction complex) and inhibition of autophagy [13]. PTEN, a 3-phosphatase, dephosphorylates PIP3, leading to inactivation of the PI3K-Akt-mTOR pathway, thereby initiating the autophagy process [14].
Bridges et al., have shown that PIKfyve and PI3K-CIIα (Class II PI3K) is essential for mTORC1 activation and localization to the PM in an insulin-dependent manner and phosphatidylinositol 3,5-bisphosphate (PI-3,5-P2) directly interacts with Raptor which is a component of the mTORC1 complex [15]. A similar study in yeast showed that PI-3,5-P2 localization to the vacuole (mammalian lysosome) was essential for activation of TORC1 signaling and inhibition of autophagy downstream in a nutrient-regulated manner. PI-3,5-P2 is also suggested to have an mTORC1-independent role in regulating the transcription factor EB (TFEB) under nutrient starvation [16]. Phosphatidylinositol 3-monophosphate (PI-3-P) has been shown to be necessary for lysosome translocation to the cell periphery, leading to activation of mTORC1. Under nutrient replete conditions, presence of amino acids stimulates the recruitment of FYCO1 (FYVE and coiled-coiled domain containing 1), a PI-3-P binding protein, to the lysosomes. The translocation of lysosomes and mTORC1 activation occurs due to interaction of FYCO1 containing lysosomes with the ER, which have a PI-3-P effector protein, Protrudin. This indicates that PI-3-P can also negatively regulate autophagy in addition to its major role in autophagosome biogenesis, as discussed in the next section [17].
Isolation membrane formation (nucleation) and elongation
Once the autophagic process is induced, autophagosome membrane formation begins. The organelle formed from this is called the phagophore. In mammals, the phagophore membrane formation primarily is initiated at the ER (Figure 2). These can also be derived from the trans-Golgi, the late endosomes and sometimes even the PM [18, 19]. Beclin1 is a key protein that kicks off this process. Upon activation, either by phosphorylation or ubiquitination, Beclin1 dissociates from an inhibitory complex and serves as platform on which other proteins assemble [20–22]. Along with its co-factors, it promotes the formation of the core complex containing Beclin1-Vps34-Vps15. Vps34, a lipid kinase found in yeast and its mammalian ortholog class III PI3K (PI3KC3), phosphorylates phosphatidylinositol PI at the 3’ position to form PI-3-P. ATG14L has binding domains for ER as well as Beclin1 and hence recruits the core complex to ER to initiate autophagosome nucleation [23, 24]. While PI3KC3 has a major role to play in the generation of PI-3-P during autophagy and accounts for approximately 50% of the protein degradation via autophagy, studies have shown that class II PI3K also contributes to a pool of PI-3-P that is involved in autophagosome biogenesis [25]. PI-3-P pools at the ER lead to recruitment of various PI-3-P-binding proteins such as the FYVE-domain containing protein DFCP1 [26, 27]. These PI-3-P-DFCP1 sites on the ER are called the omegasomes (Figure 2) and are the membranous regions that elongate to give rise to phagophores [27]. The transition from omegasomes to phagophores occurs due to sequential reactions involving ATG (autophagy-related genes) proteins. The WIPI family of proteins contain PROPPIN, another PI-3-P-binding domain [28]. WIPI2 bind to PI-3-P at the omegasomes and recruit the ATG16L1 complex, that consists of ATG16L-ATG5-ATG12, which acts as an E3-like ligase in the LC3 lipidation process [29–31]. ATG3 acts as an E2-enzyme and recruits LC3 to the ATG16L1 complex and leads to conjugation of phosphatidylethanolamine to LC3 (LC3-PE or LC3 II) [32]. This lipidation process is essential for expansion and closure of the phagophore [33, 34]. Downstream of LC3, association of WIPI4 with ATG2 and their recruitment of ATG9 is also essential for this process. A recent study showed that at PI-3-P abundant sites, WIPI4-ATG2 tethers ER to the expanding phagophore as well as other membranes, allowing for lipid transfers required for the expansion [35, 36]. ATG9 shuttling between membranes can also provide lipids and membranes for the expansion of the phagophore [37].
While generation of PI-3-P is necessary for the initiation of the autophagic process, its clearance is equally important in keeping the machinery running smoothly. The myotubularin family of phosphatases (MTMRs) are known to dephosphorylate PI-3-P and PI-3,5-P2 (Figure 2). There are 15 members in this family out of which only 8 of them have an active phosphatase domain [38]. Further, only four of them that act on PI-3-P have been identified to play a role in autophagy, namely Jumpy, MTMR6, MTMR7 [39] and MTMR3 [40]. Depletion of these phosphatases have been shown to have effects on the regulation of autophagy and lead to accumulation of LC3-positive autophagosomes in the cell as well as are associated with various diseases which will be discussed later in the review. Jumpy is mainly required for regulating the WIPI2-mediated ATG9 retrieval process [39]. In yeast, Rab5-related Vps21 has been shown to be important for the closure of the autophagosome prior to its fusion with the vacuole and deletion of Vps21 leads to accumulation of PI-3-P containing autophagosome clusters. This indicates that Vps21 also plays a role in PI-3-P clearance from the autophagosome membrane [41].
Further, a recent study elucidates the role of ATG9A containing vesicles and the lipids delivered by these during the autophagosome initiation process [42]. During starvation, ATG9A predominantly resides in the vesicular-tubular ATG9 compartment, which transiently interacts with the ER to bind with ATG13 to take part in the autophagosome nucleation [43]. The study shows that ATG9A-vesicles contain Arfaptins such as ARFIP1 and ARFIP2. ARFIP2 is possibly required for exit of ATG9A from the Golgi and leads to the formation of the vesicles themselves. These vesicles also have PI4KIIIβ, which is a binding partner of ATG9A. This recruitment possibly leads to activation of PI4KIIIβ by protein kinase D (PKD) and localized generation of PI-4-P on this compartment or at the site of autophagosome biogenesis through the recruitment of ATG13 and ULK1 complex [42].
Recently, phosphatidylinositol 5-monophosphate (PI-5-P) was identified as the key lipid in Vps34-independent autophagosome biogenesis [44]. Vicinanza et al., showed that PIKfyve, a 5-kinase, was present on the phagophore membranes and was required for generation of PI-5-P from PI (Figure 2). The PI-5-P present in these regions is able to bind both DFCP1 and WIPI2 for autophagosome biogenesis. Further, it has been demonstrated that overexpression of the 4-kinase, phosphatidylinositol-5-phosphate 4-kinase (PI5P4K) leads to rapid conversion of PI-5-P to PI-4,5-P2 which impaired autophagosome biogenesis [44]. While this study suggests a negative correlation between PI-4,5-P2 and autophagosome biogenesis, other studies have shown that PI-4,5-P2 can be responsible for PM contributing to precursor autophagosome structures. This process is dependent on the binding of ATG16L1-clathrin with the PM [45]. SNX18, a PX-domain containing protein was shown to have the ability to interact with PI-4,5-P2 and was responsible for the ATG16L1 recruitment to early endosomes. SNX18 can also bind to LC3-I. The authors hypothesized that SNX18 binding of ATG16L1 and LC3-I is important for lipidation of the latter and allowing fusion with the growing phagophore membrane [46]. Arf6, a small GTPase, is known to activate phosphatidylinositol-4-phosphate 5-kinase (PI4P5K), a 5-kinase necessary for the formation of PI-4,5-P2 at the PM and the PI-4,5-P2 generated by this method has been implicated in stimulating the PM uptake into the growing phagophores [47]. Another study has also shown that PI-4,5-P2 generation by PIPKIγi5 is necessary for autophagy initiation upstream of PI-3-P. PI-4,5-P2 generated by this enzyme interacts with the BATS (Barkor/ATG14L autophagosome targeting sequence) domain on ATG14L and leads to stabilization of ATG14L and Beclin1, allowing for the assembly of Beclin1-ATG14L-Vps34 complex, which then generates PI-3-P [48]. This indicates a role for PI-4,5-P2 in autophagy that is not limited to its role at the PM and is discussed in detail in the review by Tan et. al. (2016) [49]
Fusion (autolysosome formation) and degradation
Upon autophagosome closure, these structures are now ready for degradation. For this, the autophagosome fuses with endosomes leading to formation of amphisomes [50], which then fuse with the lysosomes or the autophagosomes directly fuse with the lysosomes leading to maturation of the autophagosome and to the formation of structures known as the autolysosomes [51] (Figure 2). While autophagosomes are formed randomly throughout the cytoplasm, endosomes and lysosomes are seen predominantly in the perinuclear region of the cell. For this fusion event to occur, the autophagosomes have to be transported towards the endosomes/lysosomes. This process is carried out through the association of autophagosomes with motor proteins, such as kinesin which transports the autophagosome towards the periphery of the cells and dynein, which transports the autophagosome towards the perinuclear region. Dynein has been shown to be essential for efficient delivery of the autophagosomes to the lysosomes [52]. PI-3-P has been implicated as an important player in this process. The FYCO1 protein has binding domains for LC3, PI-3-P and Rab7 as well as one to bind microtubules. This allows the protein to tether autophagosomes with PI-3-P and LC3 on the surface to microtubules for plus-end dependent transport of autophagosomes towards the cell periphery. FYCO1-mediated autophagosome transport is necessary mainly for autophagosome maturation under basal conditions and not upon nutrient starvation [53, 54].
Another PI-3-P binding protein TECPR1 localizes to the lysosomal membrane, where it recruits the ATG5-ATG12 complex and this allows TECPR1 binding to PI-3-P, facilitating the autophagosome-lysosome fusion. It has been shown that depletion of TECPR1 leads to accumulation of LC3-II, autophagic vacuoles and substrates and thus a defect in the autophagic flux [55]. In yeast, PI-3-P recruits a Rab7-like GTPase, Ypt7 to the autophagosome, which in turn is necessary for recruitment of the HOPS (Homotypic fusion and protein sorting) complex. The HOPS complex allows for the assembly of SNARE proteins which are important for the autophagosome-vacuole fusion process [56]. This study also shows that presence of R-SNARE, Ykt6, on the autophagosome and Q-SNAREs Vam3, Vam7 and Vti1, on the vacuole are integral to this process [56]. While no 3-phosphatases have been shown to regulate this process in the mammalian system, in yeast, the clearance of PI-3-P on the autophagosomes by Ymr1 is necessary for them to fuse with the vacuoles [57].
PI-4-P generation on the autophagosome is also important for the autophagosome-lysosome fusion process. This requires the recruitment of palmitoylated PI4KIIα to the autophagosome by Gamma-aminobutyric acid receptor-associated protein (GABARAP). Wang et al., reported that depletion of either PI4KIIα or GABARAP resulted in enlarged autophagosomes, and accumulation of LC3 and p62 [58]. The data also shows that GABARAP is upstream of PI4KIIα, as PI4KIIα depletion did not result in a change in the distribution of GABARAP on the autophagosome membrane. The defective phenotype could only be rescued by PI-4-P and not PI-4,5-P2, indicating that it is the PI-4-P generation and not its downstream products that are essential for the fusion event [58]. The PI-4-P present on the lysosomes are also sites for Rab7 accumulation which has been shown to be important for the autophagosome-lysosome fusion [59] along with the HOPS complex. A Rab7 effector protein, PLEKHM1 (Pleckstrin homology domain containing protein family member 1), has now been shown to interact with HOPS as well as LC3, via an LC3-interacting region (LIR) and hence mediating the tethering of lysosomes with LC3-positive autophagosomes [60]. A recent study by Baba et al., showed that PI-4,5-P2 generation from PI-4-P by PI4P5Kγ is essential for Rab7 inactivation and release of PLEKHM1, a regulator of autophagosome-lysosome fusion, from the membrane[61]. The authors hypothesize that since PLEKHM1 has been shown to be necessary for tethering autophagosomes with the lysosomes, it is the cycling of this protein that is essential for the fusion process. They also suggest the possibility that there are other effector proteins of PI-4,5-P2 and Rab7 that might be involved in this process [61]. OCRL, a 5-phosphatase, that converts PI-4,5-P2 to PI-4-P, is activated at the lysosome. Depletion of this 5-phosphatase is associated with a defective autophagosome-lysosome fusion [62]. This indicates that, it is not just the cycling of Rab7 but PI-4,5-P2 itself that is necessary to control this process. This is consistent with the study that shows that loss of Synaptojanin 1 (SynJ1) in zebrafish during the photoreceptor development process leads to the accumulation of autophagosomes [63].
PI-4,5-P2 can also be generated by the phosphorylation of PI-5-P by the PI5P4Ks (Figure 2). These kinases have three isoforms (α,β,γ) out of which PI5P4Kα and PI5P4Kβ have been studied as being necessary for the autophagosome-lysosome fusion event, especially under stress conditions such as loss of p53 and under starvation conditions in mouse liver [64].
Similar to PI-4,5-P2, various studies involving PI-3,5-P2 indicate that it is the cycling of this lipid that is essential for the fusion. Depletion of PIKfyve or its yeast counterpart Fab1 decreases the levels of PI-3,5-P2 at the lysosomes, preventing their fusion with the autophagosomes [65]. Proving that it is indeed the cycling of PI-3,5-P2 that is important. Further, the loss of INPP5E, a 5-phosphatase, resulted in a similar phenotype. The dephosphorylation of PI-3,5-P2 to PI-3-P is required for phosphorylation of cortactin. This leads to polymerization of actin filaments and autophagosome-lysosome fusion [66]. Fig4, another 5-phosphatase important for cycling of PI-3,5-P2, has been shown to be necessary for the autophagosome degradation. Fig4 mutant tissues do not show accumulation of autophagosomes and p62 co-localizes with LAMP2 indicating that autophagosomes are able to fuse with the lysosomes but are unable to undergo further degradation. It might also be responsible for regeneration of lysosomes post-degradation [67].
Reformation of lysosome
Following the degradation process, the structural components of the autolysosome are recycled back into new lysosomes. Under physiological conditions, it is thought that small vesicle carriers bud off to form new lysosomes. Whereas under starvation, a process called ALR or autophagic lysosome reformation (Figure 2) occurs due to the tubulation of autolysosomes, resulting in the formation of new lysosomes [68]. ALR is said to be modulated by clathrin and PI-4,5-P2. Rong et al. showed that PI4P5K1B is essential for phosphorylation of PI-4-P to PI-4,5-P2 and this event is important in extrusion of the tubules from the autolysosomes [69]. While PI4P5K1A was shown to be important in the later stages of ALR leading to the pinching off of the proto-lysosomes from the tubules. Depletion of PI4P5K1B results in enlarged autolysosomes and depletion of PI4P5K1A results in elongated tubules on the autolysosomes. The PI-4,5-P2 present on the tubules are also essential for the recruitment of clathrin to these structures, leading to the budding of the proto-lysosomes [69]. PI4KIIIβ is essential for lysosome regeneration under starvation. It phosphorylates PI to generate PI-4-P and hence provides the substrate required by PI4P5K1B and PI4P5K1A to mediate their functions in lysosome reformation. This study also showed that PI4KIIIβ plays a role independent of the PI4P5Ks, to allow for the sorting of the materials into the tubular structures during ALR [70]. The motor protein KIF5B also associates with PI-4,5-P2 on the autolysosomes and drives the tubulation event by generating a pulling force on these structures [71].
Technical Challenges: Studying Phosphoinositides and Autophagy in the Laboratory
To understand the specific roles of phosphoinositides involved in regulating the autophagic process, identifying their subcellular localization during the different stages and their kinetics becomes paramount to furthering our knowledge. With technology becoming more advanced, we have better tools available to aid us in this process. To be able to accurately identify the phosphoinositides associated with this process, rapid isolation of the various compartments involved is the first challenge. Lysosome immunoprecipitation or LysoIP described by the Sabatini group, utilizes a tagged, lysosome-specific protein, which allows rapid isolation of lysosomes [72], and these can then be subjected to further processing as such lipidomics, metabolomics etc. Various studies have identified a subset of proteins that preferentially localize to autophagosomes at specific stages of maturation. By tagging these proteins, we can isolate the various autophagosomal compartments with ease, thereby allowing us to identify phosphoinositides that localize to them. Immunoprecipitation techniques like this will also be essential in identifying proteins that bind specific phosphoinositides and regulate autophagy. Not only are these important to understand the induction and progression of autophagic process under physiological conditions, but the lipid-binding domains of these proteins can also be used for the purpose of imaging phosphoinositides with accuracy in cells as discussed later in this section.
The next challenge in furthering this understanding lies in being able to identify the lipids present in various compartments involved in autophagy with accuracy. Phosphoinositides mainly differ in the number and position of phosphates on their inositol moiety. Phosphoinositides also vary in the abundance with which each of the 7 species are found in the compartments and overall in the cell (Figure 1). We also have to keep in mind that while we are focusing on the phosphoinositides, these compartments also have other lipids, and the added variations such as length of the fatty acyl chains and double bonds, complicate this process further. Not only that, isobaric species within the phosphoinositides add another layer of complexity. For example, PI-5-P is one of the least abundant phosphoinositides in the cell and is structurally similar to the more abundant PI-4-P, making it hard to determine its presence and pinpoint its location in the cell. Taking these factors into consideration, identifying the phosphoinositides with accuracy is not a small feat. Modifications to the extraction method, such as using perchloric acid instead of the commonly used chloroform : methanol to precipitate lipids, has led to a decrease in fluctuations in the levels of the lipids being extracted. The precipitates can be treated with methylamine to deacylate the lipids and subsequently the water soluble glycero-phospho-inositols can be extracted [73]. Using this technique, followed by chromatography using an ion exchange column has allowed for separation of all 7 phosphoinositide species in neuronal cultures [74]. Recent and past studies have built upon mass spectrometry (MS) or tandem MS (MS/MS) and coupled it with other techniques to make differentiating the phosphoinositides from one another as well as enriching the lower abundance ones more plausible. The ‘shotgun’ method which utilizes electron spray ionization (ESI) coupled with MS/MS is used widely for lipidomics [75] but falls short in accurately differentiating isobaric species. Coupling this (ESI-MS/MS) with high performance liquid chromatography (HPLC) makes it feasible to identify lipids with similar masses [76]. Various studies have used HPLC-ESI-MS in combination with chemical modifications of the lipid groups to differentiate the phosphoinositides from one another [77, 78]. Malek et al. have shown that methylating the acidic phosphate groups of phosphoinositides with trimethylsilyl (TMS)-diazomethane followed by ozonolysis aids in separating the isobaric species PI-3,4-P2 from PI-4,5-P2 [79]. This allowed for identification of a new role for PTEN as a 3-phosphatase for PI-3,4-P2. While being highly sensitive, these techniques come at the cost of losing the high throughput nature of the experiment [79]. To overcome this, various modifications have been suggested. One such method is the Concerted Tandem and traveling wave ion mobility mass spectrometry or CTS analysis. This is a non-targeted approach that generates a four-dimensional data set which includes: nominal precursor ion mass, product ion mobility, accurate mass of product ion, and ion abundance. Further, it has been suggested that this 4D-data set acquisition can be a powerful tool in the identifying lipids based on their structure in a complex biological sample. [80].
While the previous strategy involved identifying lipids in processed samples, imaging techniques allow us to visualize lipid localization ‘in situ’. These techniques primarily involve lipid-binding domains of proteins tagged with fluorescent labels. Pleckstrin homology (PH) domains found in many membrane-targeted proteins have been thought to do so through their ability to bind phosphoinositides. It has been shown that though not all PH domain-containing proteins have high affinity for phosphoinositides, some proteins do have PH-domains with high specificity for a particular species of phosphoinositides [81]. The PH domain of phospholipase C δ1 (PLCδ1-PH), for example, binds PI-4,5-P2 specifically and does not interact with any other phosphoinositide [82]. With PI-4,5-P2 being predominantly present at the PM and to a much lesser extent on the internal membranes, fluorescent tagged PLCδ1-PH domain has been mainly shown to localize to the PM, making it difficult to identify PI-4,5-P2 on internal membranes due to the lack of sufficient amount of probe in the cytosol. A recent study has shown that release of the probe from the PM using angiotensin II stimulation, which causes the hydrolysis of PI-4,5-P2 at the PM, leads to marking of the internal membranes [61]. Detailed description on how to accurately quantify the lipids in the cells using this method is discussed in the review by Varnai et. al., 2017 [83]. Phosphoinositides can also be labelled using commercially available antibodies. Hammond et. al., have described the immunofluorescence technique by which the cellular membranes can be preserved while being able to detect different pools of PI-4-P and PI-4,5-P2 in the cell [84]. There are also a variety of labelled phosphoinositides available that can be extrinsically added, and the localization of the lipids tracked in a live cell. One also has to keep in mind that using lipid-binding domains, fixing the cells and extrinsically adding the lipids come with the possibility that the localization, dynamics and functions might be altered. Nonetheless, visualizing phosphoinositide dynamics in cells using fluorescent labels has become a very powerful tool in the understanding of the autophagic process. Another technique that can be used to visualize lipids is the freeze-fracture replica method [85]. This involves rapid freezing of the cells to stop the movement of the lipids within the cells, followed by deposition of evaporated platinum and carbon layers to stabilize the membrane in a freeze-fracture replica. These are then labelled with probes which will allow for imaging via an electron microscope at a nanometer scale [85]. This technique has been successfully used to observe the presence of PI-3-P in the luminal leaflet of the yeast, the cytoplasmic leaflet of the mammalian autophagosomes [86] and PI-4-P on the cytoplasmic leaflet of both the inner and outer membrane of the autophagosome [87]. To overcome the disadvantages of fluorescence imaging we can also use another imaging technique that is ‘non-invasive’. Coherent Anti-Stokes Raman Scattering (CARS) microscopy and Stimulated Raman Scattering (SRS) microscopy makes use of the intrinsic vibrational properties of the different chemical groups on the lipids for high-resolution imaging. These techniques are discussed in detail in these two reviews [88, 89].
In the end, every technique discussed comes with its own caveats while being able to answer important questions about lipid signaling and dynamics in relation to autophagy. Thus, it is important to use combinatorial approaches to get a correct and complete picture.
Understanding the Phosphoinositides Function in the Pathophysiology of Human Diseases: Development of Animal Models and Therapeutic Approaches
The dynamics and subcellular localization of phosphoinositides play a pivotal role in cell metabolism and homeostasis, membrane function and dynamics. Alongside lipids themselves, the intricate network of kinases and phosphatases regulate their clockwork turnover (Figure 1). It is therefore not surprising that any deregulation has dramatic effects and, as such, phosphoinositides have been linked to a many human diseases from cancer to neurodegeneration. The pathophysiological mechanisms of these small lipids are complex and not fully understood. Mutations and dysfunctions of the kinases and phosphatases regulating phosphoinositides leads to impaired cycling of these lipids and are often accompanied by the disruption of autophagy. However, the actual link between phosphoinositides, autophagy and disease development remains elusive.
Mytotubularins (MTMs) are a family of 3-phosphatases that preferentially dephosphorylate PI-3-P and PI-3,5-P2. Myotubular myopathy 1 (MTM1) was the first protein of this class to be identified [90] followed by 13 MTM-related proteins (MTMRs 1–13). Of note, not all MTMRs are catalytically active, as some play a scaffolding or activity-enhancing role [91]. Members of the MTM family are essential for muscle maintenance [92] and their mutations are associated with neuromuscular diseases. The best characterized are MTM1, often mutated in X-linked myotubular myopathy (XLMTM) [90], and MTMR2, mutated in Charcot-Marie-Tooth disease [93]. MTM1 and MTMR2 are essential for membrane remodeling at various sites, including the PM and the ER [94], maturation of early endosomes and autophagosomes. MTM1 mutant mice are a model for muscular dystrophy disorder and are characterized by PI-3-P accumulation in their muscle tissue. Their muscle tissue shows an increased number of autophagosomes, increased expression of p62 and LC3 early in disease development, suggesting that autophagy dysfunction takes part in disease progression [95]. Knocking out kinases responsible for generating PI-3-P also leads to muscle impairment. For instance, muscle specific deletion of class III PIK3C, leads to muscular dystrophy associated with significant alteration of the autophagolysosomal pathway [96]. In the PIK3C mutant animals Reifler et al. show ultrastructural aberrations consistent with defective autophagy as well as increased expression of autophagy marker-genes and accumulation of p62, LC3 and LAMP1 positive structures [96]. Interestingly, the same group also showed that inhibiting class II PIK3C2B (but not PIK3C) is sufficient to rescue the phenotype in the MTM1 knockout (KO) mouse and a zebra fish model of MTM, even after symptom onset [97]. The authors speculate that the MTM1-PIK3C2B interplay regulates a specific pool of PI-3-P. PI3KC modulates a different pool of PI-3-P that when disrupted in the MTM1 KO mouse leads to an increase in the severity of the disease and decreased lifespan. These observations support the hypothesis that the balance between kinases and phosphatases maintain adequate levels but also distinct, membrane-specific pools of PI-3-P in the cell to support homeostasis and promote health. Other studies show an important role for MTMRs in autophagy-dependent clearance of dysfunctional proteins, making them attractive targets for neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s disease. For instance, inhibiting MTMR14/Jumpy using the small molecules such as AUTEN67 (autophagy enhancer 67) and AUTEN99 [98, 99] restore autophagic flux and reduces symptoms in Drosophila models for Parkinson’s and Huntington’s diseases (Table 2).
Table 2. Targeting autophagy through the phosphoinositide network.
The role of phosphoinositides in human diseases is intimately linked to the key roles they hold in autophagy. Efforts to identify specific molecules have demonstrated that targeting autophagy through the kinases and phosphatases that regulate phosphoinositides is a valid therapeutic strategy of relevance for many human diseases. This table includes some of the most promising molecules that modulate autophagy parallel to a reduction in disease-related phenotypes.
| Target | Molecule | Disease relevance | Autophagy phenotype | In vivo validation | Reference |
|---|---|---|---|---|---|
| Phosphatase inhibitors | |||||
| MTMR14 | Auten 67 | Aging, neuroprotection | Increased autophagy flux | Mouse model for Alzheimer’s | Papp et al. 2016 |
| MTMR14 | Auten 99 | Aging, neuroprotection | Increased autophagy flux | Drosophila models for Parkinson’s and Huntington’s | Kovács et al. 2017 |
| OCRL INPP5B | YU142670 | Lowe syndrome | Accumulation of autophagosomes, decreased autophagy flux | PTCs derived from patients |
Pirruccello et al. 2014
De Leo et al. 2016 |
| Kinase inhibitors | |||||
| PIK3C3 | SAR405 | Cancer | Late endosome defects, blocks autophagy | Renal tumor cells | Ronan et al. 2014 |
| PIK3C3 | PIK-III | N/A | Inhibits autophagy; stabilization of autophagy substrates; | N/A | Dowdle et al. 2014 |
| PIKfyve | Apilimod | Cancer/Non-Hodgkin lymphoma | Inhibits TFEB, lysosomal genes | Mouse xenografts | Gayle et al. 2017 |
| PIKfyve | WX8-family | Cancer/melanoma | Inhibit lysosome fission, traffic into the lysosome, autophagosome formation | Tumor derived cell lines | Sharma et al. 2019 |
| PI5P4Kγ | NCT-504 | Huntington’s disease | Increased autophagic flux | Rat primary cortical neurons, Drosophila HD model | Al-Ramahi et al. 2017 |
| PI5P4K | I-OME Tryphostin AG-538 | Cancer | Predicted | N/A | Davis et al. 2013 |
| PI5P4K | SAR088 | Diabetes | Predicted | Obese SDF mice | Voss et al. 2014 |
The balance between PI-3-P/PI-3,5-P2/PI-5-P plays an important role in various steps in autophagy especially at the late endosome and lysosome biology (Figure 2 and Table 1). Furthermore, PI-3,5-P2 has been shown to alter lysosomal ion channels, leading to arrested recycling of lysosomes and accumulation of inclusion bodies. Although MTMRs can dephosphorylate PI-3,5-P2 in vitro, in vivo FIG4-PIKfyve interplay and their scaffold VAC31, is the key to supporting this balance. FIG4, the PI-3,5-P2 5-phosphatase, is mutated in several neuropathies such as the severely demyelinating Charcot-Marie-Tooth type 4J (CMT4J) neuropathy, Yunnis-Varon syndrome [100], familial epilepsy with polymicrogyria, and neurodegenerative disorders such as Amyotrophic Lateral Sclerosis. The documented mutations lead to the destabilization of the FIG4-PIKfyve-VAC31 complex contributing to disease development [101] in a mouse model for CMT4J. The spontaneous Fig4-null mouse model (pale tremor mouse) presents with depigmentation, a dramatic loss of sensory neurons as well as reduction of myelin in the brain and the spinal cord [102]. These symptoms have been related to the function of the lysosome. The MEFs isolated from this Fig4paletremor mouse present with enlarged LAMP2 positive vacuoles. The brains of these animals show enlarged endosomes and lysosomes as well as the accumulation of LC3 and p62. Interestingly, FIG4 has recently been proposed as a neuroprotector. Deletion of Fig4 in adult mice leads to severe neurological impairment and reduced life span, in part due to defective myelin repair [103]. These mice recapitulate most of the Fig4paletremor phenotype, including the accumulation of LAMP1 positive vacuoles. The PIKfyve-mutant mice are not viable, however fibroblasts isolated from their embryos, present similar accumulation of LAMP2 positive vacuoles. Interestingly, inhibitors developed against PIKfyve such as Apilimod [104] or the WX8-family [105] dramatically inhibit autophagy by disrupting lysosomal function (Table 2). Further, emerging evidence also supports a role for PI-3,5-P2 and the PIKfyve-FIG4-VAC14 complex beyond the nervous system. PIKfyve depletion in platelets correlates with defective maturation and excessive storage of lysosomal enzymes, leading to inflammation, thrombosis and multi-organ defects in mice [106]. PIKfyve depletion specifically in mouse intestine leads to intestinal inflammation and fibrosis, accompanied by shorter life span. Accumulation of vacuoles was also observed in the enterocytes of these conditional KO animals [107].
PI-4,5-P2 has been shown to accumulate in kidney tissues and cells collected from Lowe syndrome patients. This disease presents with renal proximal tubule degeneration, cataracts and neurological symptoms and is caused by mutations in various regions of the 5-phosphatase OCRL (OculoCerebroRenal of Lowe). PI-4,5-P2 regulation is essential for various steps in autophagy from early endosome to autophagosome-lysosome fusion and autophagy flux (Figure 2 and Table 1). OCRL acts preferentially on PI-4,5-P2 regulating its accumulation at the lysosome. The authors of this study [62] show that in the absence of OCRL, PI-4,5-P2 has an inhibitory effect on autophagic flux. De Leo et al. have shown that OCRL inhibition leads to PI-4,5-P2 accumulation around the lysosome and subsequent inhibition of MCOLN1 and autophagy flux [62]. As a result, autophagosomes accumulate in OCRL-depleted cells or in kidney tissue of patients with Lowe syndrome. Animal models for Lowe syndrome are not perfect, as mere deletion of OCRL does not fully recapitulate the disease. The INPP5B phosphatase (Figure 1) can in fact compensate for OCRL leading to a mild symptomatology in mice. A kidney tubule-specific KO of INPP5B in OCRL deficient mice recapitulates the human disease to a better extent. While these mice show defective endocytosis, the autophagy status has not been yet described [108]. Similarly, in a zebrafish model for Lowe disease, endocytosis and early endosome formation was shown to be deficient in the nervous tissue [109] and the pronephric tube [110]. A new family of 5-phosphatase inhibitors has been recently characterized (Table 2) using a novel high throughput assay [111]. The OCRL/INPP5B inhibitor (YU142670) identified through this screen also led to the accumulation of autophagosomes and a reduction in autophagy flux in proximal tubule cells derived from patients with Lowe syndrome [62].
SynJ1 is another phosphatase with a key role at the synapse where it regulates synaptic vesicle recycling and trafficking. This phosphatase is mutated in patients with early onset Parkinson’s disease and is unique as it has two different domains. The 5-phosphatase domain targets specifically PI-4,5-P2, while the SAC domain dephosphorylates PI-3-P, PI-4-P and PI-3,5-P2 indiscriminately. The 5-phosphatase regulates endocytic vesicle uncoating. The SAC domain plays an essential role in autophagosome formation at synaptic vesicles. Mutations in the SAC domain, leads to increased PI-3-P or PI-3,5-P2 accumulation at the developing autophagosome, leading to increased ATG18 binding, blocking further development. These observations in the D. melanogaster nervous system, were confirmed in human iPSCs derived from Parkinson’s disease patient brains [112].
Phosphoinositide regulating enzymes are also frequently mutated in various cancers, with phosphoinositides playing an essential part in cancer biology. PI3K, and its counterpart PTEN, are often mutated in cancers, and are essential for the dynamics between PI-4,5-P2 to PI-3,4,5-P3, at the PM. This event triggers signaling cascades that have been central to understanding tumor metabolism and developing new therapies. Small molecules targeting PI3K have been shown to inhibit autophagy [113,114] (Table 2) and contribute to cancer cell death [113] (Table 2). This central signaling axis and its therapeutic potential and hurdles have been extensively covered in comprehensive reviews [115–118]. It is worth reiterating that these mutations are essential for cancer cell survival by providing alternative routes to nutrient availability and ensuring exquisite metabolic plasticity and stress resistance. While PI3K/PTEN have been intensively studied in cancer biology, other phosphoinositide kinases are currently emerging as key players tumor development. For example, inhibition of the PI5P4Ks have been shown to promote cancer cell “starvation” by inhibiting autophagy in p53 mutant cancer cells [64, 119]. However, inhibition of PI5P4Kβ has also been shown to enhance EMT in MCF10A cells, and low expression of PI5P4Kβ correlates with poor prognosis in breast cancer patients [120]. PI5P4Ks seem to play an important role in keeping metabolic homeostasis but the full extent of the mechanisms they employ to fulfill this function is still largely uncharacterized. Small molecules that target PI5P4Ks are being developed and tested (Table 2) [121–123] and some showed positive results in a mouse model for diabetes [122]. Interestingly, a specific PI5P4Kg inhibitor (NCT-504) has been shown to increase autophagic flux and delay symptoms in a Drosophila model for Huntington’s disease [123].
From the studies discussed here, cellular pathophysiological processes associated with imbalances in phosphoinositide cycling stem from membrane remodeling defects, and regulation of ion trafficking at various steps of autophagy. These defects lead to a blocked autophagic flux and accumulation of inclusion bodies and large vacuoles. Autophagy is tightly regulated by what has been defined as “membrane identity” [124], that is the specific pool of phosphoinositides and the kinase/phosphatase signature that characterizes specific membranes within the cell. However, we have to specify that while the focus of this review is autophagy, phosphoinositides and their regulating enzymes and binding partners contribute to other signaling events within the cell as well. Indeed, several studies demonstrate that even when lacking the catalytically active site, the kinases and phosphatases still play an important role in cellular metabolism. For instance, kinase-dead MTM1 has been shown to significantly improve muscle function in MTM1-null mice [125]. This added layer of complexity makes it difficult to date to assess the extent to which phosphoinositide-related autophagy contributes to disease development. Despite these difficulties, efforts to develop high throughput screens to identify inhibitors for kinases and phosphatases are bringing the field closer to clinical relevance (Table 2) [121]. The manipulation of these enzymes for therapeutical purposes has to proceed with caution as depending on the genetic and metabolic status of the cell, the effects can be both beneficial and deleterious. Understanding the full complexity of phosphoinositide-related pathophysiology is paramount for the development of future therapeutic approaches.
Concluding Remarks
Understanding the role of phosphoinositides and how they regulate the autophagy process is undoubtedly an arduous undertaking. In this review we aimed to highlight the most recent findings with regard to the phosphoinositides, their metabolizing enzymes, as well as their known effectors in the tightly regulated multistep process of autophagy (Figure 2 and Table 1), thus providing a working framework that can be updated as the field and techniques evolve.
Autophagy is a complex membrane shaping process including induction, nucleation, maturation, and fusion, therefore it is not surprising that there is an extensive crosstalk between phosphoinositides and the autophagic protein machinery. Importantly, phosphoinositide species and the various kinases and phosphatases that regulate their turnover play essential roles in modulating metabolic pathways, including autophagy. As mentioned here in this review, in order to fully comprehend and appreciate the role of phosphoinositides in autophagy, the lipid composition of autophagosome precursors, completed autophagosomes, and lysosomes will need to be elucidated. To this end, methods to purify such organelles to a high degree and subsequent lipidomics are essential. Further, improvement of imaging techniques allowing the visualization of lipids using super-resolution microscopy will also aid to study the distribution and dynamics of lipids throughout the autophagy process.
Finally, there is intense debate about the potential of autophagy modulation as a therapeutic tool. Thus, understanding the role of phosphoinositides as well as the phosphoinositide kinases and phosphatases may be meaningful as many of the phosphoinositide enzymes are druggable and the phosphoinositides themselves may serve as potential biomarkers. In the end, exploiting or targeting the phosphoinositide signaling pathways that regulate autophagy may prove to be a beneficial therapeutic strategy in future years.
Acknowledgements
We thank Ryan Loughran and Joanna Triscott for their critical reading and insightful comments on the manuscript. This work is supported by a grant from the Department of Defense (W81XWH-19-1-0614) to BME.
Abbreviations
- PI
phosphatidylinositol
- ER
endoplasmic reticulum
- PM
plasma membrane
- mTORC1
mammalian target of rapamycin complex 1
- PI3KI
class I phosphatidylinositol 3-kinase
- PI-4,5-P2
phosphatidylinositol 4,5-bisphosphate
- PI-3,4,5-P3 or PIP3
phosphatidylinositol 3,4,5-triphosphate
- PDK1
phosphoinositide-dependent kinase-1
- PKB
protein kinase B
- PI-3,5-P2
phosphatidylinositol 3,5-bisphosphate
- TFEB
transcription factor EB
- PI-3-P
phosphatidylinositol 3-monophosphate
- FYCO1
FYVE and coiled-coiled domain containing 1
- LC3-PE or LC3 II
phosphatidylethanolamine to LC3
- MTMRs
myotubularin family of phosphatases
- PKD
protein kinase D
- PI-5-P
phosphatidylinositol 5-monophosphate
- PI5P4K
phosphatidylinositol-5-phosphate 4-kinase
- PI4P5K
phosphatidylinositol-4-phosphate 5-kinase
- BATS
Barkor/ATG14L autophagosome targeting sequence
- HOPS
homotypic fusion and protein sorting
- GABARAP
gamma-aminobutyric acid receptor-associated protein
- PLEKHM1
pleckstrin homology domain containing protein family member 1
- LIR
LC3-interacting region
- SynJ1
synaptojanin 1
- ALR
autophagic lysosome reformation
- LysoIP
lysosome immunoprecipitation
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- ESI
electron spray ionization
- HPLC
high performance liquid chromatography
- CTS analysis
Concerted Tandem and traveling wave ion mobility mass spectrometry
- PH
pleckstrin homology
- PLCδ1-PH
PH domain of phospholipase C δ1
- CARS
Coherent Anti-Stokes Raman Scattering microscopy
- SRS
Stimulated Raman Scattering microscopy
- MTMs
Mytotubularins
- MTM1
Myotubular myopathy 1
- XLMTM
X-linked myotubular myopathy
- KO
knockout
- AUTEN67
autophagy enhancer 67
- AUTEN99
autophagy enhancer 99
- CMT4J
Charcot-Marie-Tooth type 4J
- OCRL
OculoCerebroRenal of Lowe
Footnotes
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Balla T (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation, Physiol Rev. 93, 1019–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dall’Armi C, Devereaux KA & Di Paolo G (2013) The role of lipids in the control of autophagy, Curr Biol. 23, R33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klionsky DJ (2005) The molecular machinery of autophagy: unanswered questions, J Cell Sci. 118, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yorimitsu T & Klionsky DJ (2005) Autophagy: molecular machinery for self-eating, Cell Death Differ. 12 Suppl 2, 1542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kunz JB, Schwarz H & Mayer A (2004) Determination of four sequential stages during microautophagy in vitro, J Biol Chem. 279, 9987–96. [DOI] [PubMed] [Google Scholar]
- 6.Majeski AE & Dice JF (2004) Mechanisms of chaperone-mediated autophagy, Int J Biochem Cell Biol. 36, 2435–44. [DOI] [PubMed] [Google Scholar]
- 7.Ding WX & Yin XM (2012) Mitophagy: mechanisms, pathophysiological roles, and analysis, Biol Chem. 393, 547–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu K & Czaja MJ (2013) Regulation of lipid stores and metabolism by lipophagy, Cell Death Differ. 20, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Till A, Lakhani R, Burnett SF & Subramani S (2012) Pexophagy: the selective degradation of peroxisomes, Int J Cell Biol. 2012, 512721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An H & Harper JW (2018) Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy, Nat Cell Biol. 20, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rabanal-Ruiz Y, Otten EG & Korolchuk VI (2017) mTORC1 as the main gateway to autophagy, Essays Biochem. 61, 565–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shanware NP, Bray K & Abraham RT (2013) The PI3K, metabolic, and autophagy networks: interactive partners in cellular health and disease, Annu Rev Pharmacol Toxicol. 53, 89–106. [DOI] [PubMed] [Google Scholar]
- 13.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M & Kim DH (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery, Mol Biol Cell. 20, 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P & Ogier-Denis E (2001) The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway, J Biol Chem. 276, 35243–6. [DOI] [PubMed] [Google Scholar]
- 15.Bridges D, Ma JT, Park S, Inoki K, Weisman LS & Saltiel AR (2012) Phosphatidylinositol 3,5-bisphosphate plays a role in the activation and subcellular localization of mechanistic target of rapamycin 1, Mol Biol Cell. 23, 2955–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin N, Mao K, Jin Y, Tevzadze G, Kauffman EJ, Park S, Bridges D, Loewith R, Saltiel AR, Klionsky DJ & Weisman LS (2014) Roles for PI(3,5)P2 in nutrient sensing through TORC1, Mol Biol Cell. 25, 1171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong Z, Pedersen NM, Wang L, Torgersen ML, Stenmark H & Raiborg C (2017) PtdIns3P controls mTORC1 signaling through lysosomal positioning, J Cell Biol. 216, 4217–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlsson SR & Simonsen A (2015) Membrane dynamics in autophagosome biogenesis, J Cell Sci. 128, 193–205. [DOI] [PubMed] [Google Scholar]
- 19.Tooze SA & Yoshimori T (2010) The origin of the autophagosomal membrane, Nat Cell Biol. 12, 831–5. [DOI] [PubMed] [Google Scholar]
- 20.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R & Kimchi A (2009) DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy, EMBO Rep. 10, 285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei Y, Pattingre S, Sinha S, Bassik M & Levine B (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy, Mol Cell. 30, 678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi CS & Kehrl JH (2010) TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy, Sci Signal. 3, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itakura E, Kishi C, Inoue K & Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG, Mol Biol Cell. 19, 5360–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baskaran S, Carlson LA, Stjepanovic G, Young LN, Kim DJ, Grob P, Stanley RE, Nogales E & Hurley JH (2014) Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex, Elife. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Devereaux K, Dall’Armi C, Alcazar-Roman A, Ogasawara Y, Zhou X, Wang F, Yamamoto A, De Camilli P & Di Paolo G (2013) Regulation of mammalian autophagy by class II and III PI 3-kinases through PI3P synthesis, PLoS One. 8, e76405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheung PC, Trinkle-Mulcahy L, Cohen P & Lucocq JM (2001) Characterization of a novel phosphatidylinositol 3-phosphate-binding protein containing two FYVE fingers in tandem that is targeted to the Golgi, Biochem J. 355, 113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G & Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum, J Cell Biol. 182, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baskaran S, Ragusa MJ, Boura E & Hurley JH (2012) Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy, Mol Cell. 47, 339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI & Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1, Mol Cell. 55, 238–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F & Ohsumi Y (2007) The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy, J Biol Chem. 282, 37298–302. [DOI] [PubMed] [Google Scholar]
- 31.Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ & Tooze SA (2010) Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation, Autophagy. 6, 506–22. [DOI] [PubMed] [Google Scholar]
- 32.Fujita N, Itoh T, Omori H, Fukuda M, Noda T & Yoshimori T (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy, Mol Biol Cell. 19, 2092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakatogawa H, Ichimura Y & Ohsumi Y (2007) Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion, Cell. 130, 165–78. [DOI] [PubMed] [Google Scholar]
- 34.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V & Elazar Z (2010) LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis, EMBO J. 29, 1792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chowdhury S, Otomo C, Leitner A, Ohashi K, Aebersold R, Lander GC & Otomo T (2018) Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex, Proc Natl Acad Sci U S A. 115, E9792–E9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kotani T, Kirisako H, Koizumi M, Ohsumi Y & Nakatogawa H (2018) The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation, Proc Natl Acad Sci U S A. 115, 10363–10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM & Tooze SA (2012) Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy, Mol Biol Cell. 23, 1860–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vergne I & Deretic V (2010) The role of PI3P phosphatases in the regulation of autophagy, FEBS Lett. 584, 1313–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vergne I, Roberts E, Elmaoued RA, Tosch V, Delgado MA, Proikas-Cezanne T, Laporte J & Deretic V (2009) Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy, EMBO J. 28, 2244–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taguchi-Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T & Noda T (2010) Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy, Traffic. 11, 468–78. [DOI] [PubMed] [Google Scholar]
- 41.Zhou F, Zou S, Chen Y, Lipatova Z, Sun D, Zhu X, Li R, Wu Z, You W, Cong X, Zhou Y, Xie Z, Gyurkovska V, Liu Y, Li Q, Li W, Cheng J, Liang Y & Segev N (2017) A Rab5 GTPase module is important for autophagosome closure, PLoS Genet. 13, e1007020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Judith D, Jefferies HBJ, Boeing S, Frith D, Snijders AP & Tooze SA (2019) ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIbeta, J Cell Biol. 218, 1634–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karanasios E, Walker SA, Okkenhaug H, Manifava M, Hummel E, Zimmermann H, Ahmed Q, Domart MC, Collinson L & Ktistakis NT (2016) Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles, Nat Commun. 7, 12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vicinanza M, Korolchuk VI, Ashkenazi A, Puri C, Menzies FM, Clarke JH & Rubinsztein DC (2015) PI(5)P regulates autophagosome biogenesis, Mol Cell. 57, 219–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ravikumar B, Moreau K, Jahreiss L, Puri C & Rubinsztein DC (2010) Plasma membrane contributes to the formation of pre-autophagosomal structures, Nat Cell Biol. 12, 747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knaevelsrud H, Soreng K, Raiborg C, Haberg K, Rasmuson F, Brech A, Liestol K, Rusten TE, Stenmark H, Neufeld TP, Carlsson SR & Simonsen A (2013) Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation, J Cell Biol. 202, 331–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moreau K, Ravikumar B, Puri C & Rubinsztein DC (2012) Arf6 promotes autophagosome formation via effects on phosphatidylinositol 4,5-bisphosphate and phospholipase D, J Cell Biol. 196, 483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan X, Thapa N, Liao Y, Choi S & Anderson RA (2016) PtdIns(4,5)P2 signaling regulates ATG14 and autophagy, Proc Natl Acad Sci U S A. 113, 10896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan X, Thapa N, Choi S & Anderson RA (2015) Emerging roles of PtdIns(4,5)P2--beyond the plasma membrane, J Cell Sci. 128, 4047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berg TO, Fengsrud M, Stromhaug PE, Berg T & Seglen PO (1998) Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes, J Biol Chem. 273, 21883–92. [DOI] [PubMed] [Google Scholar]
- 51.Fader CM, Sanchez D, Furlan M & Colombo MI (2008) Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells, Traffic. 9, 230–50. [DOI] [PubMed] [Google Scholar]
- 52.Kimura S, Noda T & Yoshimori T (2008) Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes, Cell Struct Funct. 33, 109–22. [DOI] [PubMed] [Google Scholar]
- 53.Olsvik HL, Lamark T, Takagi K, Larsen KB, Evjen G, Overvatn A, Mizushima T & Johansen T (2015) FYCO1 Contains a C-terminally Extended, LC3A/B-preferring LC3-interacting Region (LIR) Motif Required for Efficient Maturation of Autophagosomes during Basal Autophagy, J Biol Chem. 290, 29361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjorkoy G & Johansen T (2010) FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport, J Cell Biol. 188, 253–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen D, Fan W, Lu Y, Ding X, Chen S & Zhong Q (2012) A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate, Mol Cell. 45, 629–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bas L, Papinski D, Licheva M, Torggler R, Rohringer S, Schuschnig M & Kraft C (2018) Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome-vacuole fusion, J Cell Biol. 217, 3656–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cebollero E, van der Vaart A, Zhao M, Rieter E, Klionsky DJ, Helms JB & Reggiori F (2012) Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion, Curr Biol. 22, 1545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang H, Sun HQ, Zhu X, Zhang L, Albanesi J, Levine B & Yin H (2015) GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion, Proc Natl Acad Sci U S A. 112, 7015–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gutierrez MG, Munafo DB, Beron W & Colombo MI (2004) Rab7 is required for the normal progression of the autophagic pathway in mammalian cells, J Cell Sci. 117, 2687–97. [DOI] [PubMed] [Google Scholar]
- 60.McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P & Dikic I (2015) PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins, Mol Cell. 57, 39–54. [DOI] [PubMed] [Google Scholar]
- 61.Baba T, Toth DJ, Sengupta N, Kim YJ & Balla T (2019) Phosphatidylinositol 4,5-bisphosphate controls Rab7 and PLEKMH1 membrane cycling during autophagosome-lysosome fusion, EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Leo MG, Staiano L, Vicinanza M, Luciani A, Carissimo A, Mutarelli M, Di Campli A, Polishchuk E, Di Tullio G, Morra V, Levtchenko E, Oltrabella F, Starborg T, Santoro M, Di Bernardo D, Devuyst O, Lowe M, Medina DL, Ballabio A & De Matteis MA (2016) Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL, Nat Cell Biol. 18, 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.George AA, Hayden S, Stanton GR & Brockerhoff SE (2016) Arf6 and the 5’phosphatase of synaptojanin 1 regulate autophagy in cone photoreceptors, Bioessays. 38 Suppl 1, S119–35. [DOI] [PubMed] [Google Scholar]
- 64.Lundquist MR, Goncalves MD, Loughran RM, Possik E, Vijayaraghavan T, Yang A, Pauli C, Ravi A, Verma A, Yang Z, Johnson JL, Wong JCY, Ma Y, Hwang KS, Weinkove D, Divecha N, Asara JM, Elemento O, Rubin MA, Kimmelman AC, Pause A, Cantley LC & Emerling BM (2018) Phosphatidylinositol-5-Phosphate 4-Kinases Regulate Cellular Lipid Metabolism By Facilitating Autophagy, Mol Cell. 70, 531–544 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rusten TE, Vaccari T, Lindmo K, Rodahl LM, Nezis IP, Sem-Jacobsen C, Wendler F, Vincent JP, Brech A, Bilder D & Stenmark H (2007) ESCRTs and Fab1 regulate distinct steps of autophagy, Curr Biol. 17, 1817–25. [DOI] [PubMed] [Google Scholar]
- 66.Hasegawa J, Iwamoto R, Otomo T, Nezu A, Hamasaki M & Yoshimori T (2016) Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome, EMBO J. 35, 1853–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferguson CJ, Lenk GM & Meisler MH (2010) PtdIns(3,5)P2 and autophagy in mouse models of neurodegeneration, Autophagy. 6, 170–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH & Lenardo MJ (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR, Nature. 465, 942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rong Y, Liu M, Ma L, Du W, Zhang H, Tian Y, Cao Z, Li Y, Ren H, Zhang C, Li L, Chen S, Xi J & Yu L (2012) Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation, Nat Cell Biol. 14, 924–34. [DOI] [PubMed] [Google Scholar]
- 70.Sridhar S, Patel B, Aphkhazava D, Macian F, Santambrogio L, Shields D & Cuervo AM (2013) The lipid kinase PI4KIIIbeta preserves lysosomal identity, EMBO J. 32, 324–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Du W, Su QP, Chen Y, Zhu Y, Jiang D, Rong Y, Zhang S, Zhang Y, Ren H, Zhang C, Wang X, Gao N, Wang Y, Sun L, Sun Y & Yu L (2016) Kinesin 1 Drives Autolysosome Tubulation, Dev Cell. 37, 326–336. [DOI] [PubMed] [Google Scholar]
- 72.Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG & Sabatini DM (2017) mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient, Cell. 171, 642–654 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonangelino CJ, Nau JJ, Duex JE, Brinkman M, Wurmser AE, Gary JD, Emr SD & Weisman LS (2002) Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p, J Cell Biol. 156, 1015–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCartney AJ, Zolov SN, Kauffman EJ, Zhang Y, Strunk BS, Weisman LS & Sutton MA (2014) Activity-dependent PI(3,5)P2 synthesis controls AMPA receptor trafficking during synaptic depression, Proc Natl Acad Sci U S A. 111, E4896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koivusalo M, Haimi P, Heikinheimo L, Kostiainen R & Somerharju P (2001) Quantitative determination of phospholipid compositions by ESI-MS: effects of acyl chain length, unsaturation, and lipid concentration on instrument response, J Lipid Res. 42, 663–72. [PubMed] [Google Scholar]
- 76.Godzien J, Ciborowski M, Martinez-Alcazar MP, Samczuk P, Kretowski A & Barbas C (2015) Rapid and Reliable Identification of Phospholipids for Untargeted Metabolomics with LC-ESI-QTOF-MS/MS, J Proteome Res. 14, 3204–16. [DOI] [PubMed] [Google Scholar]
- 77.Clark J, Anderson KE, Juvin V, Smith TS, Karpe F, Wakelam MJ, Stephens LR & Hawkins PT (2011) Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry, Nat Methods. 8, 267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kielkowska A, Niewczas I, Anderson KE, Durrant TN, Clark J, Stephens LR & Hawkins PT (2014) A new approach to measuring phosphoinositides in cells by mass spectrometry, Adv Biol Regul. 54, 131–41. [DOI] [PubMed] [Google Scholar]
- 79.Malek M, Kielkowska A, Chessa T, Anderson KE, Barneda D, Pir P, Nakanishi H, Eguchi S, Koizumi A, Sasaki J, Juvin V, Kiselev VY, Niewczas I, Gray A, Valayer A, Spensberger D, Imbert M, Felisbino S, Habuchi T, Beinke S, Cosulich S, Le Novere N, Sasaki T, Clark J, Hawkins PT & Stephens LR (2017) PTEN Regulates PI(3,4)P2 Signaling Downstream of Class I PI3K, Mol Cell. 68, 566–580 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berry KA, Barkley RM, Berry JJ, Hankin JA, Hoyes E, Brown JM & Murphy RC (2017) Tandem Mass Spectrometry in Combination with Product Ion Mobility for the Identification of Phospholipids, Anal Chem. 89, 916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lemmon MA (2004) Pleckstrin homology domains: not just for phosphoinositides, Biochem Soc Trans. 32, 707–11. [DOI] [PubMed] [Google Scholar]
- 82.Garcia P, Gupta R, Shah S, Morris AJ, Rudge SA, Scarlata S, Petrova V, McLaughlin S & Rebecchi MJ (1995) The pleckstrin homology domain of phospholipase C-delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes, Biochemistry. 34, 16228–34. [DOI] [PubMed] [Google Scholar]
- 83.Varnai P, Gulyas G, Toth DJ, Sohn M, Sengupta N & Balla T (2017) Quantifying lipid changes in various membrane compartments using lipid binding protein domains, Cell Calcium. 64, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hammond GR, Schiavo G & Irvine RF (2009) Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2), Biochem J. 422, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fujita A, Cheng J & Fujimoto T (2010) Quantitative electron microscopy for the nanoscale analysis of membrane lipid distribution, Nat Protoc. 5, 661–9. [DOI] [PubMed] [Google Scholar]
- 86.Cheng J, Fujita A, Yamamoto H, Tatematsu T, Kakuta S, Obara K, Ohsumi Y & Fujimoto T (2014) Yeast and mammalian autophagosomes exhibit distinct phosphatidylinositol 3-phosphate asymmetries, Nat Commun. 5, 3207. [DOI] [PubMed] [Google Scholar]
- 87.Kurokawa Y, Yoshida A, Fujii E, Tomioku K, Hayashi H, Tanabe K & Fujita A (2019) Phosphatidylinositol 4-phosphate on Rab7-positive autophagosomes revealed by the freeze-fracture replica labeling, Traffic. 20, 82–95. [DOI] [PubMed] [Google Scholar]
- 88.Yu Y, Ramachandran PV & Wang MC (2014) Shedding new light on lipid functions with CARS and SRS microscopy, Biochim Biophys Acta. 1841, 1120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zumbusch A, Langbein W & Borri P (2013) Nonlinear vibrational microscopy applied to lipid biology, Prog Lipid Res. 52, 615–32. [DOI] [PubMed] [Google Scholar]
- 90.Laporte J, Jia Hu L, Kretz C, Mandel Group J-L, Kioschis P, Coy JF, Klauck SM & Poustka A (1996). A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. [DOI] [PubMed]
- 91.Raess MA, Friant S, Cowling BS & Laporte J (2017) WANTED – Dead or alive: Myotubularins, a large disease-associated protein family, Advances in Biological Regulation. 63, 49–58. [DOI] [PubMed] [Google Scholar]
- 92.Buj-Bello A, Furling D, Tronchere H, Laporte J, Lerouge T, Butler-Browne GS & Mandel JL (2002) Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells, Hum Mol Genet. 11, 2297–307. [DOI] [PubMed] [Google Scholar]
- 93.Bolino A, Muglia M, Conforti FL, LeGuern E, Salih MAM, Georgiou D-M, Christodoulou K, Hausmanowa-Petrusewicz I, Mandich P, Schenone A, Gambardella A, Bono F, Quattrone A, Devoto M & Monaco AP (2000) Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2, Nature Genetics. 25, 17–19. [DOI] [PubMed] [Google Scholar]
- 94.Amoasii L, Hnia K, Chicanne G, Brech A, Cowling BS, Müller MM, Schwab Y, Koebel P, Ferry A, Payrastre B & Laporte J (2013) Myotubularin and PtdIns3P remodel the sarcoplasmic reticulum in muscle in vivo., Journal of cell science. 126, 1806–19. [DOI] [PubMed] [Google Scholar]
- 95.Al-Qusairi L, Prokic I, Amoasii L, Kretz C, Messaddeq N, Mandel J-L & Laporte J (2013) Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin-proteasome pathways, The FASEB Journal. 27, 3384–3394. [DOI] [PubMed] [Google Scholar]
- 96.Reifler A, Li X, Archambeau AJ, McDade JR, Sabha N, Michele DE & Dowling JJ (2014) Conditional knockout of pik3c3 causes a murine muscular dystrophy, Am J Pathol. 184, 1819–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sabha N, Volpatti JR, Gonorazky H, Reifler A, Davidson AE, Li X, Eltayeb NM, Dall’Armi C, Paolo GD, Brooks SV, Buj-Bello A, Feldman EL & Dowling JJ (2016) PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models, The Journal of Clinical Investigation. 126, 3613–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kovács T, Billes V, Komlós M, Hotzi B, Manzéger A, Tarnóci A, Papp D, Szikszai F, Szinyákovics J, Rácz Á, Noszál B, Veszelka S, Walter FR, Deli MA, Hackler L, Alfoldi R, Huzian O, Puskas LG, Liliom H, Tárnok K, Schlett K, Borsy A, Welker E, Kovács AL, Pádár Z, Erdős A, Legradi A, Bjelik A, Gulya K, Gulyás B & Vellai T (2017) The small molecule AUTEN-99 (autophagy enhancer-99) prevents the progression of neurodegenerative symptoms, Scientific Reports. 7, 42014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Papp D, Kovacs T, Billes V, Varga M, Tarnoci A, Hackler L Jr., Puskas LG, Liliom H, Tarnok K, Schlett K, Borsy A, Padar Z, Kovacs AL, Hegedus K, Juhasz G, Komlos M, Erdos A, Gulyas B & Vellai T (2016) AUTEN-67, an autophagy-enhancing drug candidate with potent antiaging and neuroprotective effects, Autophagy. 12, 273–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Campeau PM, Lenk GM, Lu JT, Bae Y, Burrage L, Turnpenny P, Roman Corona-Rivera J, Morandi L, Mora M, Reutter H, Vulto-van Silfhout AT, Faivre L, Haan E, Gibbs RA, Meisler MH & Lee BH (2013) Yunis-Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase, Am J Hum Genet. 92, 781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lenk GM, Ferguson CJ, Chow CY, Jin N, Jones JM, Grant AE, Zolov SN, Winters JJ, Giger RJ, Dowling JJ, Weisman LS & Meisler MH (2011) Pathogenic Mechanism of the FIG4 Mutation Responsible for Charcot-Marie-Tooth Disease CMT4J, PLoS Genetics. 7, e1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, Lupski JR, Weisman LS & Meisler MH (2007) Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J, Nature. 448, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mironova YA, Lin JP, Kalinski AL, Huffman LD, Lenk GM, Havton LA, Meisler MH & Giger RJ (2018) Protective role of the lipid phosphatase Fig4 in the adult nervous system, Hum Mol Genet. 27, 2443–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gayle S, Landrette S, Beeharry N, Conrad C, Hernandez M, Beckett P, Ferguson SM, Mandelkern T, Zheng M, Xu T, Rothberg J & Lichenstein H (2017) Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma., Blood. 129, 1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sharma G, Guardia CM, Roy A, Vassilev A, Saric A, Griner LN, Marugan J, Ferrer M, Bonifacino JS & DePamphilis ML (2019) A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis, Autophagy, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Min SH, Suzuki A, Stalker TJ, Zhao L, Wang Y, McKennan C, Riese MJ, Guzman JF, Zhang S, Lian L, Joshi R, Meng R, Seeholzer SH, Choi JK, Koretzky G, Marks MS & Abrams CS (2014) Loss of PIKfyve in platelets causes a lysosomal disease leading to inflammation and thrombosis in mice, Nat Commun. 5, 4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Takasuga S, Horie Y, Sasaki J, Sun-Wada G-H, Kawamura N, Iizuka R, Mizuno K, Eguchi S, Kofuji S, Kimura H, Yamazaki M, Horie C, Odanaga E, Sato Y, Chida S, Kontani K, Harada A, Katada T, Suzuki A, Wada Y, Ohnishi H & Sasaki T (2013) Critical roles of type III phosphatidylinositol phosphate kinase in murine embryonic visceral endoderm and adult intestine., Proceedings of the National Academy of Sciences of the United States of America. 110, 1726–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Inoue K, Balkin DM, Liu L, Nandez R, Wu Y, Tian X, Wang T, Nussbaum R, De Camilli P & Ishibe S (2017) Kidney Tubular Ablation of Ocrl/Inpp5b Phenocopies Lowe Syndrome Tubulopathy., Journal of the American Society of Nephrology : JASN. 28, 1399–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ramirez IB-R, Pietka G, Jones DR, Divecha N, Alia A, Baraban SC, Hurlstone AFL & Lowe M (2012) Impaired neural development in a zebrafish model for Lowe syndrome, Human Molecular Genetics. 21, 1744–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oltrabella F, Pietka G, Ramirez IB-R, Mironov A, Starborg T, Drummond IA, Hinchliffe KA & Lowe M (2015) The Lowe syndrome protein OCRL1 is required for endocytosis in the zebrafish pronephric tubule., PLoS genetics. 11, e1005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pirruccello M, Nandez R, Idevall-Hagren O, Alcazar-Roman A, Abriola L, Berwick SA, Lucast L, Morel D & De Camilli P (2014) Identification of inhibitors of inositol 5-phosphatases through multiple screening strategies., ACS chemical biology. 9, 1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vanhauwaert R, Kuenen S, Masius R, Bademosi A, Manetsberger J, Schoovaerts N, Bounti L, Gontcharenko S, Swerts J, Vilain S, Picillo M, Barone P, Munshi ST, de Vrij FM, Kushner SA, Gounko NV, Mandemakers W, Bonifati V, Meunier FA, Soukup SF & Verstreken P (2017) The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals, The EMBO Journal. 36, 1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, Bachelot M-F, Lamberton A, Mathieu M, Bertrand T, Marquette J-P, El-Ahmad Y, Filoche-Romme B, Schio L, Garcia-Echeverria C, Goulaouic H & Pasquier B (2014) A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy, Nature Chemical Biology. 10, 1013–1019. [DOI] [PubMed] [Google Scholar]
- 114.Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, Cantwell J, Luu C, Cornella-Taracido I, Harrington E, Fekkes P, Lei H, Fang Q, Digan ME, Burdick D, Powers AF, Helliwell SB, D’Aquin S, Bastien J, Wang H, Wiederschain D, Kuerth J, Bergman P, Schwalb D, Thomas J, Ugwonali S, Harbinski F, Tallarico J, Wilson CJ, Myer VE, Porter JA, Bussiere DE, Finan PM, Labow MA, Mao X, Hamann LG, Manning BD, Valdez RA, Nicholson T, Schirle M, Knapp MS, Keaney EP & Murphy LO (2014) Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo, Nat Cell Biol. 16, 1069–79. [DOI] [PubMed] [Google Scholar]
- 115.Wong K-K, Engelman JA & Cantley LC (2010) Targeting the PI3K signaling pathway in cancer, Current Opinion in Genetics & Development. 20, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dibble CC & Cantley LC (2015) Regulation of mTORC1 by PI3K signaling, Trends in Cell Biology. 25, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fruman DA & Rommel C (2014) PI3K and cancer: lessons, challenges and opportunities., Nature reviews Drug discovery. 13, 140–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Janku F, Yap TA & Meric-Bernstam F (2018) Targeting the PI3K pathway in cancer: are we making headway?, Nature Reviews Clinical Oncology. 15, 273–291. [DOI] [PubMed] [Google Scholar]
- 119.Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf GM, Bell EL, Shim HS, Lamia KA, Rameh LE, Bellinger G, Sasaki AT, Asara JM, Yuan X, Bullock A, Denicola GM, Song J, Brown V, Signoretti S & Cantley LC (2013) Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors, Cell. 155, 844–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Keune WJ, Sims AH, Jones DR, Bultsma Y, Lynch JT, Jirstrom K, Landberg G & Divecha N (2013) Low PIP4K2B expression in human breast tumors correlates with reduced patient survival: A role for PIP4K2B in the regulation of E-cadherin expression, Cancer Res. 73, 6913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Davis MI, Sasaki AT, Shen M, Emerling BM & Thorne N (2013) A Homogeneous, High-Throughput Assay for Phosphatidylinositol 5-Phosphate 4-Kinase with a Novel, Rapid Substrate Preparation, PLoS ONE. 8, 54127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Voss MD, Czechtizky W, Li Z, Rudolph C, Petry S, Brummerhop H, Langer T, Schiffer A & Schaefer H-L (2014) Discovery and pharmacological characterization of a novel small molecule inhibitor of phosphatidylinositol-5-phosphate 4-kinase, type II, beta, Biochemical and Biophysical Research Communications. 449, 327–331. [DOI] [PubMed] [Google Scholar]
- 123.Al-Ramahi I, Giridharan SSP, Chen Y-C, Patnaik S, Safren N, Hasegawa J, de Haro M, Wagner Gee AK, Titus SA, Jeong H, Clarke J, Krainc D, Zheng W, Irvine RF, Barmada S, Ferrer M, Southall N, Weisman LS, Botas J & Marugan JJ (2017) Inhibition of PIP4Kγ ameliorates the pathological effects of mutant huntingtin protein, eLife. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Di Paolo G & De Camilli P (2006) Phosphoinositides in cell regulation and membrane dynamics. [DOI] [PubMed]
- 125.Amoasii L, Bertazzi DL, Lè Ne Tronchè Re H, Hnia K, Tan Chicanne G, Rinaldi B, Cowling BS, Ferry A, Klaholz B, Payrastre B, Laporte J & Friant S (2012) Phosphatase-Dead Myotubularin Ameliorates X-Linked Centronuclear Myopathy Phenotypes in Mice. [DOI] [PMC free article] [PubMed]